 Emma K. Wear1*
Emma K. Wear1* Matthew J. Church1,2
Matthew J. Church1,2 Beth N. Orcutt3
Beth N. Orcutt3 Christine N. Shulse4
Christine N. Shulse4 Markus V. Lindh5
Markus V. Lindh5 Craig R. Smith6
Craig R. Smith6- 1Flathead Lake Biological Station, University of Montana, Polson, MT, United States
- 2Department of Biological Sciences, University of Montana, Missoula, MT, United States
- 3Bigelow Laboratory for Ocean Sciences, East Boothbay, ME, United States
- 4Department of Biology, Los Medanos College, Pittsburg, CA, United States
- 5Swedish Meteorological and Hydrological Institute, Gothenburg, Sweden
- 6Department of Oceanography, University of Hawai’i at Mānoa, Honolulu, HI, United States
Bacteria and archaea are key contributors to deep-sea biogeochemical cycles and food webs. The disruptions these microbial communities may experience during and following polymetallic nodule mining in the Clarion-Clipperton Zone (CCZ) of the North Pacific Ocean could therefore have broad ecological effects. Our goals in this synthesis are to characterize the current understanding of biodiversity and biogeography of bacteria and archaea in the CCZ and to identify gaps in the baseline data and sampling approaches, prior to the onset of mining in the region. This is part of a large effort to compile biogeographic patterns in the CCZ, and to assess the representivity of no-mining Areas of Particular Environmental Interest, across a range of taxa. Here, we review published studies and an additional new dataset focused on 16S ribosomal RNA (rRNA) gene amplicon characterization of abyssal bacterial and archaeal communities, particularly focused on spatial patterns. Deep-sea habitats (nodules, sediments, and bottom seawater) each hosted significantly different microbial communities. An east-vs.-west CCZ regional distinction was present in nodule communities, although the magnitude was small and likely not detectable without a high-resolution analysis. Within habitats, spatial variability was driven by differences in relative abundances of taxa, rather than by abundant taxon turnover. Our results further support observations that nodules in the CCZ have distinct archaeal communities from those in more productive surrounding regions, with higher relative abundances of presumed chemolithoautotrophic Nitrosopumilaceae suggesting possible trophic effects of nodule removal. Collectively, these results indicate that bacteria and archaea in the CCZ display previously undetected, subtle, regional-scale biogeography. However, the currently available microbial community surveys are spatially limited and suffer from sampling and analytical differences that frequently confound inter-comparison; making definitive management decisions from such a limited dataset could be problematic. We suggest a number of future research priorities and sampling recommendations that may help to alleviate dataset incompatibilities and to address challenges posed by rapidly advancing DNA sequencing technology for monitoring bacterial and archaeal biodiversity in the CCZ. Most critically, we advocate for selection of a standardized 16S rRNA gene amplification approach for use in the anticipated large-scale, contractor-driven biodiversity monitoring in the region.
Introduction
Deep-sea bacteria and archaea (hereafter referred to as microbes) are some of the most abundant lifeforms below the sunlit ocean (Whitman et al., 1998; Kallmeyer et al., 2012) and constitute the greatest standing stocks of biomass inhabiting the seafloor at depths of abyssal plains and below (Wei et al., 2010; Danovaro et al., 2015), in part because of the variety of habitats in which they can survive. In the abyssal plains of the Clarion-Clipperton Zone (CCZ), microbial habitats include polymetallic nodules as well as the more cosmopolitan habitats of sediments and the overlying water column. The CCZ, an approximately 6,000,000 km2 region in international jurisdiction in the equatorial North Pacific (Wedding et al., 2013), contains the greatest observed concentrations of seabed nodule deposits (International Seabed Authority, 2010). Proposed polymetallic nodule mining operations, currently in the exploratory phase but expected to reach commercial feasibility within the coming decade (Hein et al., 2020) will therefore impact CCZ microbial habitats in a variety of ways, the most obvious being the physical removal of nodules from large swathes of the seafloor. Current mining technology will also necessitate removal of surface sediments and benthic boundary layer water along with nodules. This will result in a pronounced increase in sediment plumes from both direct seafloor disturbance and waste sediment disposal (Christiansen et al., 2020; Drazen et al., 2020), to which local biota – including microbes – are unlikely to be accustomed (Smith et al., 2020).
Microbes play critical and diverse roles in energy acquisition pathways and nutrient cycles in the abyss, including both chemoheterotrophic and chemoautotrophic metabolisms and substantial elemental transformations associated with nitrogen, phosphorus, sulfur, iron, and manganese cycling (Orcutt et al., 2011). There are therefore motivations beyond conservation of biodiversity to improve our understanding of the extant microbial communities in the abyssal CCZ prior to human-induced disturbance: in addition to critical ecological functions, mining activity may also detrimentally impact societally relevant ecosystem services provided by microbes in these regions (Thurber et al., 2014; Orcutt et al., 2020). Further, similar to the situation with other deep-sea organisms, abyssal microbes are poorly known to science – the putative metabolic functions and phylogeny of major groups of deep-sea bacteria and archaea are still actively being described in the literature (Baker et al., 2021): e.g., the little-known candidate order Woeseiales (Hoffmann et al., 2020) and putative heterotrophic deep-sea Thaumarchaeota, in contrast with the canonical autotrophic ammonia oxidizers (Aylward and Santoro, 2020).
This combination of factors suggests that, despite the likely importance of microbial communities to the overall biogeochemical and ecological function of the abyssal plains of the CCZ, we are unable to fully describe their roles in this ecosystem. And while bacteria and archaea in general have much shorter generation times than macro-organisms, current evidence indicates that deep-sea microbes impacted by mining will not recover quickly. A recent return visit to the site of the DISCOL simulated small-scale mining experiment in the Peru Basin found that portions of the disturbed areas retained persistently lower CO2 fixation rates, reduced hydrolytic extracellular enzyme activity, and reduced microbial cell abundances relative to nearby reference sites, 26 years after the sediments were initially disturbed (Vonnahme et al., 2020). Deep-sea nodules themselves have been estimated to accrete at rates on the order of millimeters per 106 years (Ku and Broecker, 1969; Hein et al., 2020); thus, their removal will functionally lead to the extirpation of any microbes that rely on them for habitat. It is therefore particularly important that the reserve areas set aside from mining impacts, such as the nine current Areas of Particular Environmental Interest (APEIs), which cover 24% of the CCZ planning region (Wedding et al., 2013; see Figure 1), are representative of the taxonomic structure and metabolic function of microbial communities of the surrounding habitats.
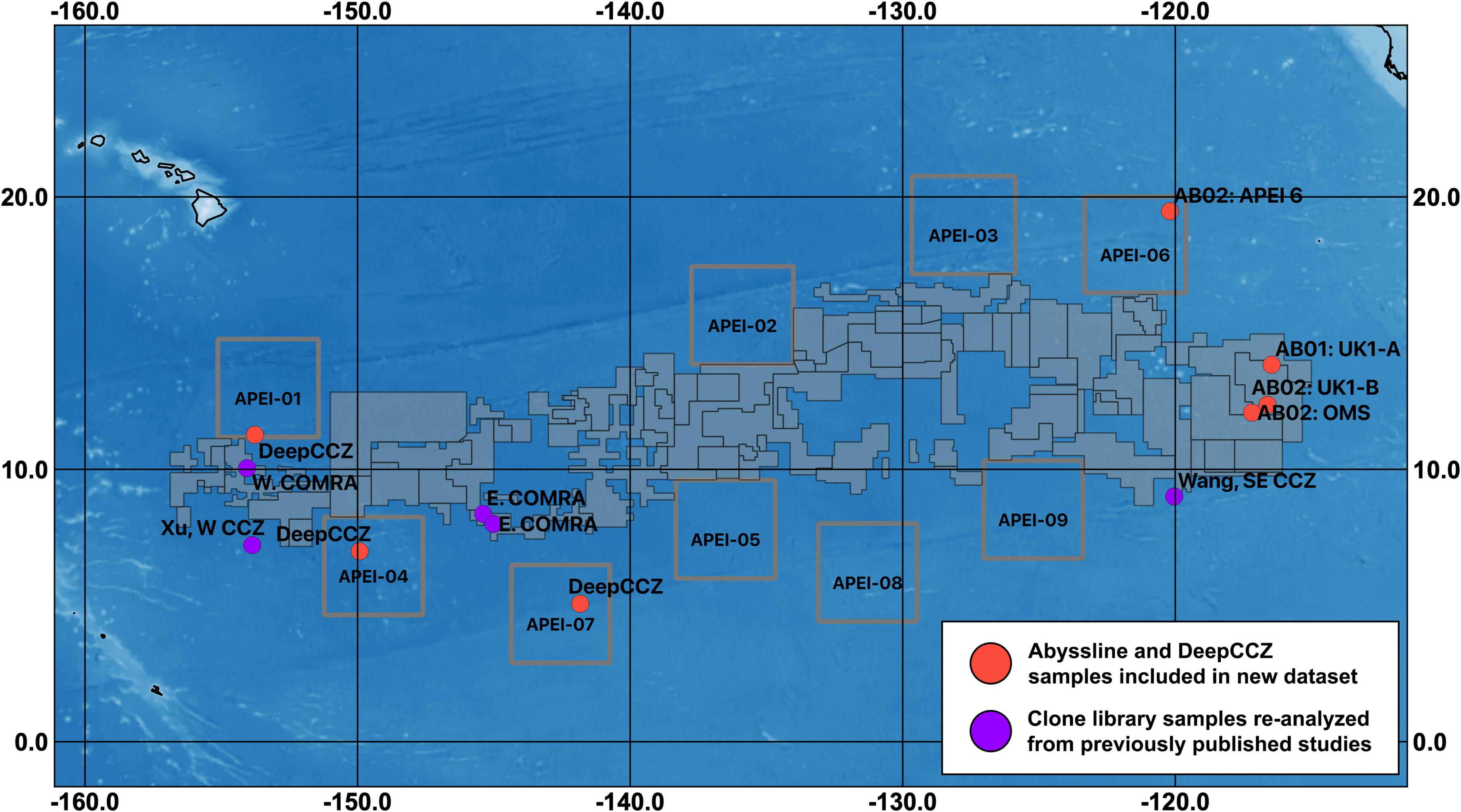
Figure 1. Map of sampling sites for datasets included in this synthesis and designated exploration license and APEI areas within the CCZ. Sampling regions are labeled with unique cruise and/or location identifiers (see Table 1). Solid gray polygons represent both reserved and exploration areas, as designated by the International Seabed Authority; squares outlined in gray are APEIs. COMRA, China Ocean Mineral Resources R&D Association contract area (which is divided into east and west regions); UK1, UK Seabed Resources Ltd contract area, with two sampling sites (A and B); OMS, Ocean Mineral Singapore contract area. The three west COMRA sampling sites in the western CCZ share one location dot. For fine-scale sample locations for Abyssline (sampled from 30 × 30 km areas in UK1 and OMS; Lindh et al., 2017) and DeepCCZ (samples collected within 15 km of one another at each site) cruises, see Supplementary Table 1.
Because the energy used by abyssal microbes at sites such as the CCZ that are distant from hydrothermal features ultimately derives from inputs from the surface ocean, the biogeochemical gradients in the upper water column of the south-eastern Pacific raise the possibility of similar, related habitat gradients at the seafloor. These biogeochemical gradients include particulate organic carbon flux to the sediments, which decreases northward from the equatorial upwelling region into the stratified gyre proper, as well as from east to west (Christian et al., 2002; McQuaid et al., 2020; Washburn et al., 2021). East-west variability in particulate organic carbon flux has been posited to explain spatial patterns in species richness of nodule-associated xenophyophores and metazoans (Veillette et al., 2007). The southeastern corner of the CCZ encompasses the pronounced oxygen minimum zone of the eastern tropical North Pacific (Talley, 2007), which may further impact the quantity and composition of organic matter reaching the benthos. More directly, nodule abundance and sediment composition also vary within the CCZ, inherently affecting habitat quality and availability for benthic microbes (International Seabed Authority, 2010; McQuaid et al., 2020).
To better understand the current state of knowledge of bacterial and archaeal microbes in the CCZ, here we synthesize a new dataset from polymetallic nodules, sediment, and bottom waters of the CCZ with published datasets from the CCZ and other regions. Our goals in this synthesis are to characterize the current understanding of biodiversity and biogeography of bacteria and archaea in the CCZ and to identify gaps in the baseline data and sampling approaches, prior to the onset of mining in the region. We describe observed biogeographic patterns as they relate both to habitat type (that is, nodules vs. sediments vs. bottom waters) and to spatial distribution. In addition, we highlight how gaps in available data hinder prediction in how these habitats might respond to mining disturbances. We focus here (1) on bacteria and archaea and not on eukaryotic microbes such as the protists or the fungi, as the former two groups have historically been sampled independently from the latter, and (2) on the abyssal water column and benthic habitats, as this is where the majority of published microbial datasets from the CCZ have concentrated their efforts. This is not to suggest that the shallower pelagic environments are expected to be spared the impact of nodule mining; on the contrary, the release of waste sediments into the water column will likely have substantial and still poorly understood impacts on the structure and activity of midwater pelagic communities of all taxa, including the microbes (Drazen et al., 2020). However, there are not at present sufficient, validly inter-comparable samples from the overlying water column of the CCZ to make a meaningful assessment of spatial heterogeneity.
Caveats in Assessing Microbial Biodiversity From DNA Sequencing Data
A substantial caveat limiting our ability to directly compare results across existing studies results from the rapid shifts in techniques used to characterize bacterial and archaeal communities over the past approximately 15 years. Marine bacteria and archaea are primarily identified through molecular techniques, in particular taxonomic profiling targeting various hypervariable regions of the 16S small subunit ribosomal RNA (rRNA) marker gene. The move from gene cloning and Sanger sequencing to next-generation sequencing (NGS) techniques such as Illumina sequencing (see review in Klindworth et al., 2013) has increased the number of sequences attainable per sample by several orders of magnitude, in turn increasing the detected richness and diversity of the microbial communities (e.g., Sogin et al., 2006), and this discrepancy only grows as technological advances continue to increase the sequencing depth per run and decrease the cost per read. Much of the early work on CCZ microbes was conducted with Sanger sequencing of cloned 16S rRNA gene amplicons, while many recent samples have been sequenced with NGS, complicating inter-cruise comparisons and detection of any potential temporal patterns in biodiversity.
Further, polymerase chain reaction (PCR) primers used to amplify 16S rRNA genes have changed substantially in recent years. Improvements in primer design have increased detection of abundant and cosmopolitan marine microbes such as the SAR11 clade in the water column (Apprill et al., 2015; Parada et al., 2016) and the Thaumarchaeota (Parada et al., 2016). Studies within the CCZ have also used PCR primers that target different combinations of 16S rRNA gene hypervariable regions, which can affect measures of richness and diversity in addition to the taxonomic composition of the resulting libraries (Klindworth et al., 2013; Wear et al., 2018; see also Supplementary Figures 1–3 for a direct example). These effects limit our ability to validly, directly compare results between studies conducted with different 16S rRNA gene primer sets at the level of taxonomic composition. Thus, where necessary, we qualify results with relevant caveats and in some cases omit comparisons between datasets that would otherwise be of interest. We conclude with recommendations that could help to minimize these issues in the future.
Materials and Methods
Generation of New Dataset
To address the inter-comparability issues introduced by variability in primer choice, we re-sequenced selected benthic boundary layer water, sediment, and nodule genomic DNA samples from the Abyssline cruise program (see Table 1; Lindh et al., 2017; Shulse et al., 2017). We compared these samples with a new dataset collected as part of the DeepCCZ cruise to the western CCZ in spring 2018. DeepCCZ aimed to characterize biodiversity within the three westernmost APEIs. We report here a subset of samples focused on bottom water [nominally 5 m above bottom (MAB)], upper sediments (surface to 5 cm depth), and nodules; additional samples will be described elsewhere.
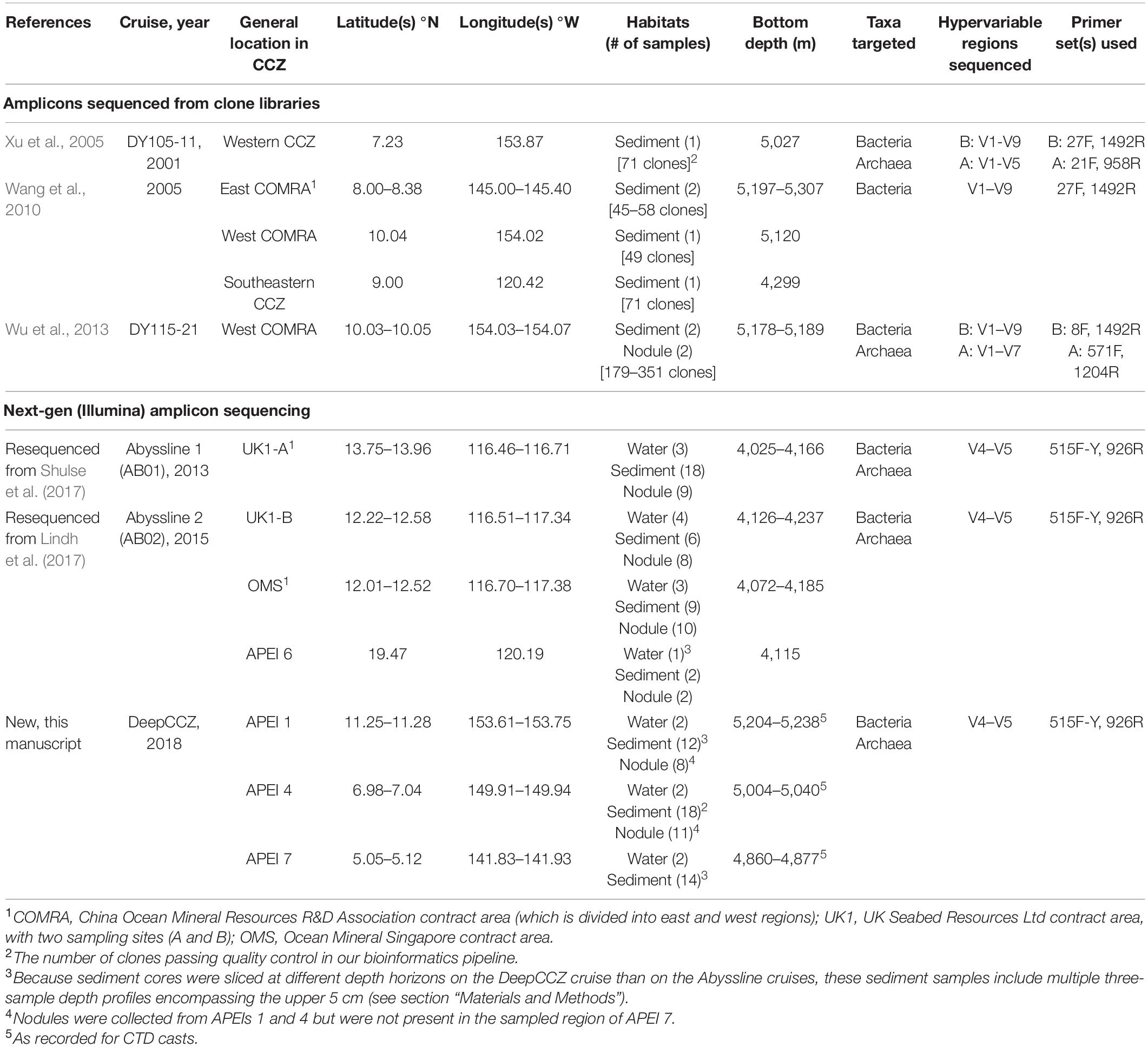
Table 1. 16S rRNA gene amplicon datasets included in synthesis.
On the DeepCCZ cruise, water column samples were collected via Niskin bottles affixed to a sampling rosette, with an altimeter allowing sampling of water ∼5 MAB. Seawater (ca. 7.5–8 L) was prefiltered via gentle peristaltic pressure through 3 μm pore size, 25 mm diameter polycarbonate prefilters (Millipore Isopore) to remove macro-organisms and large particles followed by 0.2 μm pore size, 25 mm diameter polyethersulfone filters (Supor) in series to concentrate microbes. Only results from 0.2 μm pore size filters are reported here. Filters were stored dry at −80°C. Sampling bottles, tubing, and filter holders were rinsed in Milli-Q water between casts.
Sediments were collected via the ROV Lu’ukai using a push corer. Sediment was extruded and sliced into horizons; we report here the upper horizons, from surface−0.5 cm depth; 0.5–1 cm; and 1–5 cm. The upper two sections were transferred to sterile Whirl-Pak bags and frozen at −80°C. For the larger 1–5 cm section, sterile plastic syringes with the tips cut off were used to subsample sediment from the center of the slices (∼20–30 mLs) for freezing in Whirl-Pak bags. Core tubes were washed with dish soap and soaked in 10% bleach, followed by a Milli-Q water rinse, between ROV deployments. Sampling gear such as slicing plates was rinsed with Milli-Q water between cores and soaked in 10% bleach between dives.
Nodules were collected either from push cores or by targeted sampling with the manipulator arm of the ROV. Nodules were gently rinsed with 0.2-μm filtered seawater to remove attached sediments, measured and photographed, and frozen whole at −80°C.
DNA from water column filters was extracted using the DNeasy Plant Mini Kit as described in Shulse et al. (2017). Nodule and sediment DNA was extracted using the FastDNA Spin Kit for Soil (MP Biomedical) with modifications as described in Shulse et al. (2017). Sediment samples were homogenized in the Whirl-Pak bag before DNA was extracted from ∼0.5 g subsamples. Whole nodules were broken apart within a Whirl-Pak bag using a ceramic pestle and resulting particles were mixed; when nodules were large enough, DNA was extracted from replicate ∼0.5 g subsamples and then pooled (For detailed methods for DNA extraction through bioinformatics, see Supplementary Information). All extracted samples were concentrated with the Zymo Clean & Concentrator-5 kit.
On the Abyssline cruises, benthic sediment and nodule samples were retrieved using box corers and Megacorers. Sediments were collected from the surface to 5 cm depth horizon. Deep water samples (8 L) were collected whole on a 0.2 μm pore size filter without prefiltering. DNA was extracted using similar methods.
16S rRNA gene amplicons were generated using primers targeting the V4-V5 hypervariable regions: 515F-Y (5′-GTGYC AGCMGCCGCGGTAA-3′) and 926R (5′-CCGYCAATTYM TTTRAGTTT-3′) as recommended by Parada et al. (2016) with a multiplexing index on the forward primer following the design of the Earth Microbiome Project (Caporaso et al., 2012). Triplicate amplifications were combined and cleaned using an ENZA Cycle Pure Kit (Omega Bio-tek) then pooled into two libraries. Libraries were sequenced on an Illumina MiSeq using paired-end 250 v2 chemistry. Amplicon sequence variants (ASVs) were generated using DADA2 v1.14.1 (Callahan et al., 2016). ASVs are error-corrected, discrete DNA sequences which can differ from one another by as little as a single base pair, in contrast with operational taxonomic units (OTUs), which cluster closely related sequences based on a pre-defined percent nucleotide dissimilarity (Callahan et al., 2016). ASVs were classified to the Silva v138 database (Quast et al., 2013). All code is available at https://github.com/ekwear/DeepCCZreview.
Individual samples were randomly subsampled to 6,900 sequences using the “rrarefy” function in the R package vegan (Oksanen et al., 2019). Singlet ASVs were removed (for final totals of 6,805–6,876 sequences per sample) and relative abundances were calculated. Weighted UniFrac distance was generated using the R package phyloseq (McMurdie and Holmes, 2013).
Non-metric multidimensional scaling ordinations and multivariate statistics were conducted, and diversity metrics were calculated, in Primer-E v6 (Clarke and Gorley, 2006). Analysis of Similarity (ANOSIM) tests were conducted using weighted UniFrac distances between communities that had been calculated based on ASV-level composition. Two outlier samples from AB02 were discarded based on ordination patterns. Differentially abundant ASVs were identified using the R package DESeq2 (Love et al., 2014). We considered as significantly differentially abundant those ASVs with a greater than three-fold difference between categories (that is, a log2-fold change between categories > 1.585) and a false discovery rate-adjusted p < 0.05. Throughout the text, the phrase “differential abundance” specifically refers to DESeq2 results that have met this significance threshold.
Literature Survey and Dataset Reanalysis
We aggregated datasets where community level, amplicon-based 16S rRNA gene sequences from the CCZ were publicly available, through a combination of literature surveys, public database reviews, and expert knowledge of cruises within the CCZ. Figure 1 and Table 1 highlight the locations of the datasets included in the analysis. We opted not to include microbial sequences from isolates or metagenomes in our spatial comparison of microbial community composition, as these approaches provide different types of information than 16S rRNA gene amplicon surveys; such studies from the CCZ are also numerically sparse and geographically clustered, limiting their utility for spatial analysis. A small number of published studies where raw sequence results were not available in a public database, or where the public database was missing metadata necessary for sample identification, were included in the discussion where relevant but not in the reanalysis. We also omitted those studies that sampled exclusively the upper water column.
To standardize taxonomic classifications to the greatest extent possible, we reanalyzed previously published sequences meeting the above qualifications (Table 1). Clone library sequences from Xu et al. (2005), Wang et al. (2010), and Wu et al. (2013) were aligned and classified to the Silva v138 database using mothur v1.42.3 (Schloss et al., 2009). Published datasets sequenced with the Illumina platform included Lindh et al. (2017) and Shulse et al. (2017). We have focused on resequenced samples rather than using original sequences from these two publications to avoid potential confounding effects of different primer sets (e.g., see Supplementary Figures 1–3).
Taxonomic Nomenclature
The phylogenetic relationships between, and taxonomic classifications of, marine microbes remain in flux. We have chosen herein to follow the nomenclature used in Silva v138, with the sole exception of the Thaumarchaeota, which are referred to as the Crenarchaeota in Silva v138.
Data Availability
Sequence files from DeepCCZ samples, and re-sequenced samples from the Abyssline cruises, are available from the Sequence Read Archive1 under project code PRJNA660809. Sample metadata are available in Supplementary Tables 1, 2, as well as archived on Zenodo2; analyzed sequence results are archived on Zenodo.
Results and Discussion
Our survey of the community-level microbial literature focused in the CCZ yielded three clone library studies that met our criteria for inclusion, encompassing two nodule samples and seven sediment samples, and two published NGS studies from the Abyssline cruise program, from which we resequenced 75 samples (Table 1) – 29 from nodules, 35 from sediment, and 11 from bottom waters. The DeepCCZ dataset added an additional 69 new samples – 19 from nodules, 44 from sediment, and 6 from bottom waters. Altogether, these studies include samples from three exploration license areas and four APEIs, as well as some samples from outside currently allocated areas (Table 1 and Figure 1).
Habitat Specificity Is the Primary Driver of Microbial Community Composition
Previous studies from the CCZ (Wu et al., 2013; Shulse et al., 2017; Lindh et al., 2017) have noted the selective effect of habitat – that is, water column vs. sediment vs. nodule – in determining overall microbial community composition as well as measures of diversity and richness, regardless of methodologies used (PCR primers, sequencing technique, etc.). Our literature survey and new dataset add further support to the assessment that habitat is the primary control of microbial community composition in the CCZ (Figures 2A, 3 and Supplementary Figure 4). Overall community composition across studies was similar (Figure 3): where archaeal 16S rRNA genes were measured, ASVs in the family Nitrosopumilaceae were dominant members of all habitat types. An assortment of members of the classes Alphaproteobacteria and Gammaproteobacteria were consistently abundant, with the composition varying by habitat: the SAR11 order was very abundant in the near-seabed water samples, while the sediment and nodule samples had high relative abundances of the alphaproteobacterial order Kiloniellales and the gammaproteobacterial genus Woeseia. Most samples also contained common deep-sea phyla including the Acidobacteria, members of the Bacteroidota, and the Planctomycetota. Where there were clear differences between studies at a broad taxonomic level (Figure 3), they were between habitat types rather than between geographic locations.
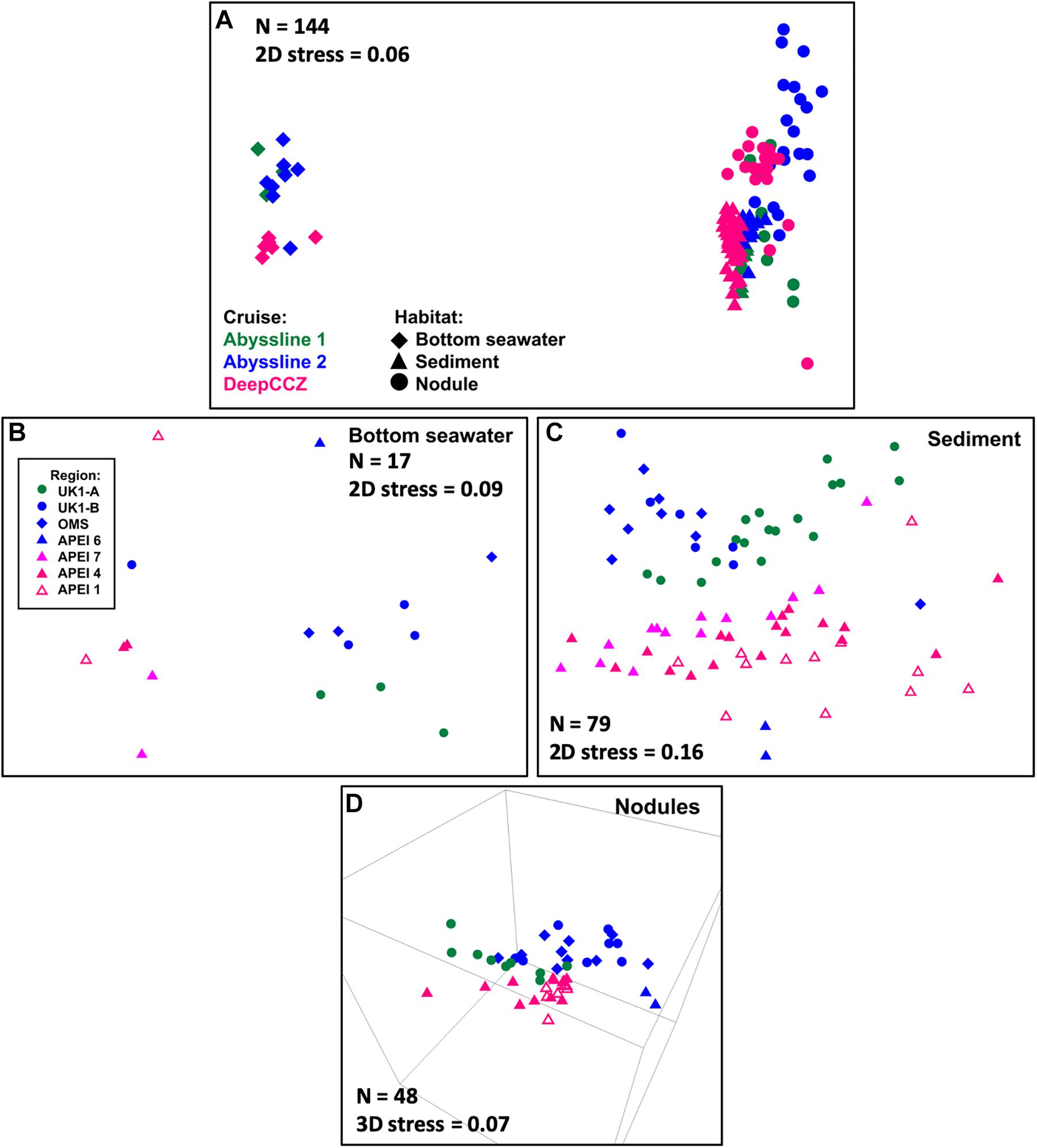
Figure 2. NMS ordinations of samples from the Abyssline and DeepCCZ cruises. (A) Has a different legend than (B–D). (A) All samples in the new dataset, with shape indicating benthic habitat and color indicating source cruise. For a further breakdown of sample numbers, see Table 1. (B) Bottom seawater samples. In this and subsequent panels, point color and shape indicate sample source region. (C) Sediment samples. This ordination is more correctly viewed as a 3D plot; see Supplementary Video 1 for a 3D animation. The most notable difference in the 3D plot is that the two extremes of the Y-axis in the 2D ordination wrap around to approach each other, such that the samples from APEI 6 are not in reality as distinct from the other AB02 samples as they appear to be in this plot. (D) Nodules.

Figure 3. Overview of prevalence of select clades across habitats within the CCZ. (A) Community composition from studies that sequenced amplicons from clone libraries, shown as presence/absence due to variable sequencing depths. “Not sampled” indicates archaeal primers were not used in the relevant study. Wang = sequences reanalyzed from Wang et al. (2010); Xu = Xu et al. (2005); and Wu = Wu et al. (2013). No clone library studies included in this synthesis reported results from water samples. (B) Community composition from the new dataset, shown as mean relative abundances within habitats and sites. Sites are arranged from roughly northeast to southwest, reading downward within a habitat; water samples are grouped within cruises due to limited sample numbers.
Within our new dataset of DeepCCZ and re-sequenced Abyssline samples, ANOSIM tests indicated that all habitat types were significantly different from one another, even the sediment and nodule communities that were in some cases collected in physical contact with one another (Supplementary Table 3). While sediment and nodule communities shared a high proportion (>85%) of abundant ASVs (with abundant ASVs defined here as those with a cumulative relative abundance within two orders of magnitude of the maximum cumulative relative abundance within a habitat; see Supplementary Figure 5), only about 16% of abundant ASVs were present in both water samples and at least one benthic habitat (Supplementary Figure 5D). In addition to the differences in ASV presence, there were strong patterns in differential abundance as identified by DESeq2 analysis between habitats, with 2,490 ASVs differentially abundant between nodules and water samples; 2,947 between sediment and water samples; and 1,607 between nodules and sediment (Supplementary Figure 6 and Supplementary Table 4). Some of these differences at the ASV level reflect the clear ecological trends seen at broader phylogenetic levels (Figure 3 and Supplementary Figure 4), such as significantly higher abundances of SAR11 and phylum Thermoplasmatota ASVs in water samples than in sediments and nodules.
Notably, many clades, even at taxonomic levels as low as genera, contained distinct ASVs that were enriched in each habitat (Supplementary Figure 6). This pattern included very common groups across CCZ habitats, such as the family Nitrosopumilaceae, the phylum Planctomycetota, and the gammaproteobacterial genus Woeseia. This ASV-level habitat selection adds an important nuance to the bulk views of microbial community composition, such as in Figure 3 and Supplementary Figure 4: while a clade may be present and even dominant within multiple habitats in the CCZ, that does not necessarily mean the same members of that clade are present across all habitats. Tully and Heidelberg (2013) observed similar fine-scale habitat preferences within Thaumarchaea MG-1 OTUs collected from nodules and sediment from the South Pacific Gyre, with some OTUs shared across habitats and others limited to one habitat. Ultimately, this suggests that, to most correctly assess microbial diversity in the CCZ, we need to work at an ASV-level resolution – that is, at the highest possible resolution for amplicon data, able to identify taxa with marker genes differing by as little as a single base pair after application of an error-correction model (Callahan et al., 2017). While broad summaries such as Figure 3 may be the best possible approach for combining currently available datasets that were sampled with different techniques, the fine-scale differential abundances within clades seen in this ASV-level analysis indicates that such an approach may overlook ecologically important levels of biodiversity.
Case Study: Eastern vs. Western CCZ Sites
The DeepCCZ and re-sequenced Abyssline samples allow for a direct comparison of microbial communities from the three westernmost APEIs with those from mining license exploration areas and an APEI in the northeastern CCZ. Based on non-metric multidimensional scaling (NMS) ordinations, spatial differences were present, though less pronounced than differences between spatially co-occurring habitats (e.g., sediments vs. seawater). The magnitude of spatial effects depended on habitat type. Spatial differences were most clear in the sediments (Figure 2C and Supplementary Video 1), where the samples from the western APEIs fell into a south-to-north gradient. The sediments from APEI 6 in the eastern CCZ most closely resembled those of the northernmost western APEI 1, while the sediments from the Abyssline license sites formed a distinct cluster. The bottom waters showed less spatial separation, with weak partitioning along the east-west divide (Figure 2B), while nodules appeared to vary more by cruise than by geographic location as such (Figure 2D). These biogeographic patterns are consistent with the broad patterns seen in physicochemical variables across the CCZ: bottom waters are similar across the region, while multiple parameters expected to affect benthic organisms (e.g., modeled particulate organic carbon flux to the seafloor, water depth, and sediment composition) vary both from the eastern to western CCZ and latitudinally within the western CCZ (Washburn et al., 2021).
ANOSIM comparisons indicated small but significant differences at the community level between nodules collected from the eastern CCZ versus the western CCZ (if APEI 6 is included: R = 0.123, p = 0.008; if APEI 6 is excluded: R = 0.158, p = 0.003). The majority of pairwise comparisons between nodule communities from specific eastern and western sampling sites were likewise significantly different, while nodules from sites sampled on the same cruises did not differ at the community level (that is, APEI 4 vs. APEI 1 in the west and the UK1-B vs. OMS sites in the east; Supplementary Table 5). ANOSIM comparisons also suggested significant east-west differences in both bottom water and sediment samples (Supplementary Table 5). However, geographic distance in these comparisons is confounded with sampling differences between the DeepCCZ and Abyssline cruises, including in pre-filtering of water samples and sampling of different sediment depth horizons, and thus these observed differences may not be solely ecological in origin.
Consistent with these relatively subtle spatial patterns, the majority of abundant ASVs (defined based on cumulative relative abundances, as described above) within each habitat type (84–94%) were shared between all three cruises (Supplementary Figure 5), and spatial differences originated in large part from shifts in relative abundances rather than from presence/absence dynamics. DESeq2 analysis of differential abundances of ASVs within nodules (the habitat type sampled comparably across all cruises in this dataset) from the eastern and western CCZ indicated there were 375 ASVs enriched in the nodules from the eastern sites and 204 in the nodules from the western sites, out of 17,510 total ASVs present across the nodule samples (Supplementary Figure 7 and Supplementary Table 4). However, these patterns were not driven by ASVs with high relative abundances, as only six differentially abundant ASVs were present at mean relative abundances of 0.5% or greater within a region (Supplementary Table 6). As was the case in the differential abundance analysis between habitats, several common taxonomic groups had ASVs that were enriched in each region, including the order Nitrosopumilales, the phylum Gemmatimonadota, the genus Woeseia in the Gammaproteobacteria, and the genus Nitrospina, suggesting some members of these groups may have preferred habitat niches across the CCZ when examined at a fine taxonomic resolution.
Diversity metrics varied between habitat types but not between geographic locations (Figure 4), with the exception of the benthic samples from APEI 6 (discussed below). Across cruises and stations, sediment samples had the highest observed ASV richness (mean of all samples: 1,774 ASVs), Pielou’s evenness (0.91), and Shannon diversity (6.77) values; bottom water samples had the lowest values of all metrics (634; 0.82; and 5.30, respectively); and nodules were in between (1,166; 0.87; and 6.15, respectively). Although sampled volumes differed between habitats, these diversity metrics were calculated on a standardized number of sequences per sample (see section “Materials and Methods”). The bottom water samples from DeepCCZ had lower mean ASV richness and Shannon diversity than the bottom water samples from the Abyssline cruises, but as the DeepCCZ samples were subjected to a 3 μm pore-size pre-filtration step and the Abyssline samples were not, we do not have confidence that this difference is ecological rather than methodological in origin.
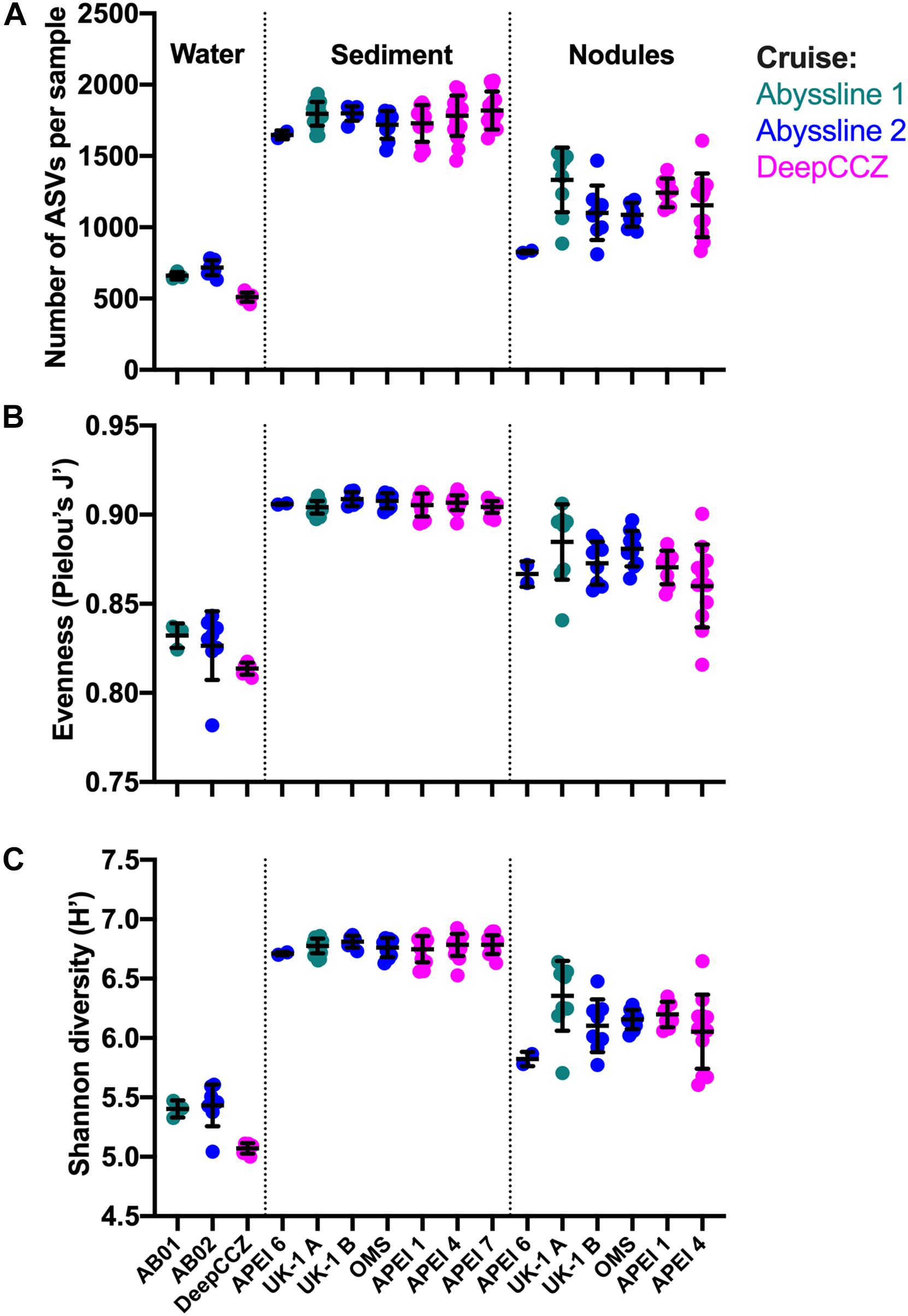
Figure 4. Richness, evenness, and diversity across habitat types and sampling locations from the new dataset. (A) Observed richness: number of observed ASVs (amplicon sequence variants) per sample, after each sample was randomly subsampled to 6,900 reads and singlet ASVs were removed. Within each habitat type, sampling locations are arranged from roughly northeast to southwest. Because of the much smaller number of water column samples, these were grouped by cruise; no nodules were collected in APEI 7. Lines indicate mean ± one standard deviation. (B) Evenness calculated as Pielou’s J′. (C) Shannon diversity, log base e.
An unexpected result of this analysis was the relatively pronounced intra-habitat difference in sediments and nodules between spatially adjacent but temporally separated (∼17 months) Abyssline license area sites. The differences between samples collected on AB01 and AB02 were, by several measures, roughly as large as the differences between samples collected across the approximately 1,500 km latitudinal gradient of the DeepCCZ cruise or the differences between the eastern and western CCZ sites (Figures 2, 5 and Supplementary Figure 8). Nodule communities from the UK1-A site sampled on AB01 were significantly different from those from both AB02 license area sites according to an ANOSIM analysis, and sediment communities differed between the UK1-A site and the AB02 OMS license site (Supplementary Table 5). The Abyssline cruises had overlapping personnel and protocols, making it unlikely that this difference is solely a sampling artifact. This raises the possibility that there could be temporal as well as spatial variability in benthic microbial communities in the CCZ, which would need to be considered in assessing the extent of mining-induced community shifts, particularly in light of the relatively subtle spatial differences we find in microbial communities across the CCZ. Despite the stereotypical view of the abyssal sea as a largely stable habitat, there is evidence this is not the case for some trophic groups – for example, abyssal sediment macrofaunal community composition and function were observed to correlate with seasonal to interannual variability in particulate organic carbon flux in a decade-long study (Ruhl et al., 2008). Alternatively, our findings may indicate that spatial variability within the CCZ can be non-linear over fairly short geographic distances. Distinguishing between these effects will require more extensive datasets than are currently available, but such a distinction will be critical to designing sampling strategies that fully characterize the extant microbial biodiversity in the CCZ.
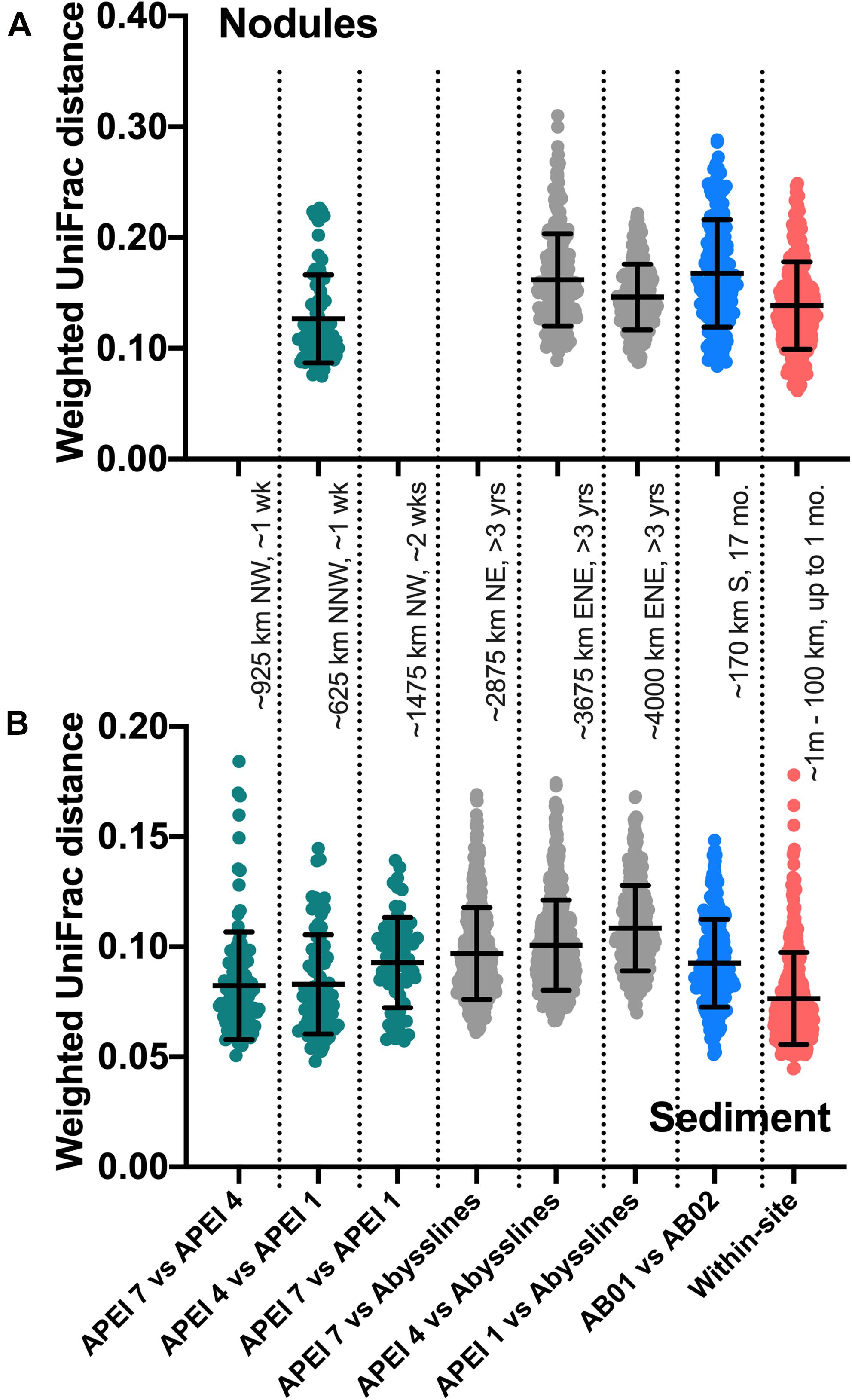
Figure 5. Weighted UniFrac distances between comparable samples across spatial and temporal distances. AB01 and AB02 are Abyssline cruises 1 and 2, respectively; APEI 6 was omitted from AB02 for this analysis as it is spatially distant from the other sampled regions. Approximate distances and times between sample collections are listed between panels; cardinal directions are reported as traveling from the first to the second location. Colors are visual guides to distinguish categories of comparisons only. (A) Nodule samples; category labels as in (B). (B) Sediment samples. For sediments, DeepCCZ samples were only compared within depth horizons between APEIs and within sites, as depth profiles are a distinct aspect of spatial ecology, but all depths were compared with Abyssline samples due to different depths sampled.
Case Study: APEI and Mining License Areas in the Eastern CCZ
Most studies of CCZ microbes have focused on either mining exploration license areas or on APEIs. An exception to this is Lindh et al. (2017), in which the authors sampled one box core and one rosette cast in an APEI (APEI 6) relatively close to the license areas that were the main focus of their work. However, given the large spatial extent of the CCZ, this still constituted an almost 900 km distance between the APEI sampling site and the closest license-area samples. Their sequencing of bacterial 16S rRNA gene amplicons indicated that community composition across all habitats – water column, sediment, and nodules – was significantly different between the APEI and corresponding samples from two nearby license areas.
Based on our spatially expanded analysis, it appears that the region of APEI 6 that Lindh et al. (2017) sampled may be an outlier relative to the CCZ in general, at least in sediments and nodules where we have enough samples for a confident assessment (see re-sequenced data in Figures 2B–D and Supplementary Video 1). Consistent with the observations of Lindh et al. (2017), the two nodules resequenced from APEI 6 had higher relative abundances of Actinobacteria (7.3 and 8.1%, vs. 0.4–6.1% in all other nodules) and Nitrospira (3.7 and 7.8%, vs. 0.1–2.8%) and lower relative abundances of Planctomycetota (3.1 and 3.2%, vs. 4.2–11.4%) than any other nodules included in the new dataset (Supplementary Figure 4C). The APEI 6 nodule samples also had low richness, evenness, and diversity metrics compared with those of the nearby license areas (Figure 4). These distinct benthic communities are perhaps not surprising given the location of APEI 6 on the far northeastern edge of the CCZ, and correspondingly we interpret this specific example as an illustration of the need for further comparisons between APEI and license area microbial communities. While our synthesis and new dataset both indicate that geographic variability within specific habitat types in the sampled portions of the CCZ is relatively low (that is, the absolute weighted UniFrac distances between samples are low (<0.3 between nodule samples and <0.2 between sediment samples; Figure 5) and the majority of ASVs are present within habitat types across the CCZ (Supplementary Figure 5), the current APEIs are generally located on the periphery of the CCZ, in some cases spanning the fracture zones (Figure 1), and thus could be prone to conditions promoting outlier microbial communities.
Nodule Communities Within the CCZ vs. Bordering Regions
While the CCZ is a particular target for potential mining activity due to the abundance and elemental composition of polymetallic nodules found there, microbial communities have also been studied from nodules collected from: the central South Pacific Gyre (Tully and Heidelberg, 2013); the Peru Basin, which is approximately 4,000 km to the southeast of the southeastern corner of the CCZ (Molari et al., 2020); the South China Sea (Zhang et al., 2015); and the western North Pacific Gyre (Jiang et al., 2020). The same issues with different sampling techniques that hinder direct comparisons between studies within the CCZ also arise in comparisons with bordering regions, yet several trends are clear despite these complications. Where both sediments and nodules have been examined, there are always pronounced overlaps in OTU or ASV presence between the two habitats, with habitat differences largely driven by differential relative abundances rather than unique organisms, consistent with the patterns we observed in the CCZ. Bacterial communities in nodules across sites have broadly similar composition when examined at the class to phylum levels, dominated by diverse assemblages of both Alphaproteobacteria and Gammaproteobacteria, in particular members of the Alteromonadales. A further suite of phyla was frequently present in nodules across sites in the general range of circa 1 to 10% relative abundances, including the Bacteroidota, Actinobacteria, Planctomycetota, and Verrucomicrobiota.
However, differences in archaeal abundance are substantial enough to suggest that nodules in the CCZ, or perhaps more broadly within the meso- to oligotrophic central gyres of the North and South Pacific (Smith and Demopoulos, 2003), may constitute a distinct microbial habitat than nodules from more productive oceanic regions. In our CCZ dataset, we observed that members of the family Nitrosopumilaceae made up between 12 and 35% of nodule communities (mean 22%). Molari et al. (2020) reported lower relative abundances of Nitrosopumilaceae in nodules sampled from the Peru Basin compared with the CCZ results reported by Shulse et al. (2017); they observed total archaeal sequences constituting at most 7% of nodule communities, a difference the authors attribute to the higher particulate matter fluxes in the Peru Basin and subsequent differential selective pressure on the Nitrosopumilaceae from increased ammonium availability [although bacteria and archaea were amplified with distinct primers in Molari et al. (2020), which may additionally confound this comparison]. Zhang et al. (2015) also observed very low (0.01%) relative abundances of an OTU related to Nitrosopumilus in nodules collected from a dormant hydrothermal vent on a seamount in the South China Sea. While Zhang et al. (2015) used a primer set (515F and 806R) that is known to underrepresent the relative abundance of the Thaumarchaeota, the reported primer effects in direct comparisons have not been severe enough to solely explain that discrepancy from the much higher relative abundances of Nitrosopumilaceae seen in nodules across the CCZ (Parada et al., 2016; see also Supplementary Figures 2, 3). In contrast, Tully and Heidelberg (2013) found a high relative abundance (in some cases, >50% of the community) and OTU-level diversity of Thaumarchaeota in nodules from the South Pacific Gyre, and Jiang et al. (2020) reported a high proportion of cloned archaeal 16S rRNA genes from the Thaumarchaeota, and particularly those closely related to Nitrosopumilus, in the western North Pacific Gyre. This variable relative abundance of Thaumarchaeota raises the possibility that polymetallic nodules underlying meso- to oligotrophic regions such as the CCZ may play more of a role in supplying their local community with chemoautotrophic production of organic matter, and thus nodule removal could have different trophic implications between regions.
Conclusion
Our results strongly support a number of previously posited and novel conclusions about microbial communities across the CCZ. First, habitat type is the strongest determinant of microbial community composition; bottom waters share relatively few high resolution, ASV-level taxa with the benthic habitats, while sediments and nodules share many ASV-level taxa that differ in relative abundances. Second, we observe an east-vs.-west distinction in microbial community composition in the CCZ, although the magnitude of this effect, at least with current sample sizes, is relatively subtle and driven by changes in relative abundances rather than ASV-level taxon turnover. However, we also observed community-level differences between sites within the same claim area but sampled ∼17 months apart that were comparable in magnitude to differences between the eastern and western CCZ, suggesting either temporal variability or non-linear spatial shifts in community structure may be important. Either of these patterns would necessitate a specialized monitoring plan, accounting for seasonality or empirically determined spatial factors, to better characterize microbial biodiversity in the region. Finally, the observation that nodules in the CCZ harbor different microbial communities than nodules from more productive regions to the east and west of the CCZ, specifically higher relative abundances of putative chemoautotrophic organisms in CCZ nodules, suggests a possible effect of mining on trophic ecology in this meso- to oligotrophic region, although functional studies are required to confirm this. We can also unambiguously conclude that the currently available datasets do not represent a thorough spatial sampling of the microbial communities of the CCZ. In particular, the central and southeastern regions of the CCZ are poorly represented, and comparisons between proximate license areas and APEIs are extremely limited. We are aware of a small number of research cruises that have occurred but have not yet published microbial biodiversity data, which will improve but not fully alleviate these deficits.
Knowledge Gaps Concerning Microbial Biodiversity and Ecology in Nodule Regions
There remain several major gaps in our knowledge of CCZ microbes, in particular of those inhabiting polymetallic nodules, that we cannot address with current datasets. This includes the basic issue of whether reserve areas provide similar habitats to nearby exploration license areas. As noted above, available studies have largely sampled either APEIs or exploration areas, with scant paired comparisons between neighboring sites. Given the anticipated lengthy recovery time of sediment microbial communities after mining exposure (Vonnahme et al., 2020), and the presumably much longer time before nodules could re-form to be colonized (Ku and Broecker, 1969; Hein et al., 2020), it is particularly important that these reserves of intact habitat be representative of the broader CCZ.
This synthesis has focused on habitat selection and basin-scale spatial distribution patterns, but we lack knowledge of the basic ecology of deep-sea nodule-associated bacteria and archaea at much finer scales as well. This lack of basic ecological understanding has monitoring implications, in that it may inhibit our ability to design sampling programs that are robust to local variability when the goal is detecting spatial effects (see for example, some samples presenting high within-site variability in weighted UniFrac distances in Figure 5). At this point, we cannot confidently answer basic questions, including: how do properties such as nodule size and metal composition affect microbial community composition? Is nodule density a factor in structuring small-scale microbial communities, as has been observed for megabenthic faunal composition (Simon-Lledó et al., 2019)? Does the frequent colonization of nodules by larger organisms including xenophyophores and sponges (e.g., Gooday et al., 2020) select for particular microbial communities, as has been observed with co-occurring epizoans in other marine habitats (e.g., on kelp blades: James et al., 2020)? Sediment-dwelling xenophyophores have been shown to contain enhanced and compositionally distinct bacterial fatty acids relative to surrounding sediments, suggesting some combination of bacterivory and/or bacterial growth on xenophyophore fecal pellets (Laureillard et al., 2004); both processes potentially could affect microbial community composition in a nodule to which a xenophyophore was attached. And like their shallow-water counterparts, deep-sea sponges host communities of bacteria and archaea that are distinct from those of the surrounding seawater, with deep-sea sponges particularly favorable habitats for members of the Nitrosopumilaceae (e.g., Steinert et al., 2020). Over the very long temporal duration of nodules (Ku and Broecker, 1969; Hein et al., 2020), attached macro-organisms would presumably episodically die; live vs. detrital epizoans would likely have different effects on microbes, and we speculate that changes in epizoan presence and health could potentially explain some nodule samples that have been observed to have outlier microbial communities, such as the three nodules from studies in the central Pacific that were each highly enriched in the chemoheterotrophic genus Colwellia (Tully and Heidelberg, 2013; Blöthe et al., 2015), which includes piezophilic members with chitin-degradation pathways (Peoples et al., 2020).
We also have an incomplete picture of how microbes use nodules as habitat. What are the proportions of microbes in surficial biofilms vs. shallow indentations vs. interior pore waters, and how do they differ? One study that compared the interior vs. exterior of a nodule found that microbial cell abundance was elevated at the nodule surface and decreased inward (Shiraishi et al., 2016). Studies that have compared interior and exterior bacterial and archaeal communities have seen differences in relative abundance and to a lesser extent in presence/absence of OTUs or higher clades between layers (Tully and Heidelberg, 2013; Shiraishi et al., 2016), while another observed higher OTU richness in layers closer to the exterior than in those closer to the nodule nucleus (Jiang et al., 2020). Other studies have found evidence for intra-nodule spatial heterogeneity, with differences in the microbial communities within the hydrogenetic (facing and formed by precipitation from the seawater) vs. diagenetic (facing the sediment and formed by precipitation from the pore waters) portions of individual nodules (Blöthe et al., 2015; Cho et al., 2018). The available nodule-associated microbial samples are insufficient to tease apart these potential factors, with the additional complication that different datasets have variable metadata that further reduce our ability to draw ecological conclusions.
Finally, what are the ecological and biogeochemical roles of bacteria and archaea inhabiting the benthos and near-seabed waters of the CCZ? While this information may be less important for designing sampling schemes, it could help us to better anticipate possible effects of mining on benthic community function. Rate measurements of key metabolic processes and molecular analyses of functional genes via metagenomics and metatranscriptomics would enable an accurate assessment of whether removing nodules and disturbing surface sediments will alter a key part of the abyssal elemental cycles in the CCZ. While rate measurements on deep-sea microbes are complicated by slow speeds and the need to maintain relevant hydrostatic pressures, with careful methodology such rates in the abyssal CCZ do appear to be quantifiable. For example, in situ tracer experiments in the eastern CCZ demonstrated that bacteria in nodule-containing sediments were active both in remineralization of added phytodetritus and in chemoautotrophic fixation of inorganic carbon, although the metabolic pathways underlying the latter process remain unknown (Sweetman et al., 2019).
Recommendations for Future Sampling
Beyond the limited number of extant microbial datasets from the CCZ, our ability to assess biodiversity and microbial ecosystem services is further hampered by the difficulties in directly comparing the samples that do exist. We make several recommendations for future microbial biodiversity work in the CCZ, contributing to the assessment guidelines recommended by the International Seabed Authority (2019), to facilitate future spatial and temporal inter-comparison efforts:
(1) Standardized sampling to the greatest extent practical, from field collection through DNA sequencing, will be key to enabling long-term monitoring of microbial diversity in response to mining activity – particularly in light of the very subtle spatial patterns within habitat types that we report in this analysis. International Seabed Authority guidelines already recommend next-generation (i.e., Illumina) amplicon-based sequencing. As the most pressing aspect of this standardization, we suggest adopting a single set of primers for consistent use, ideally one endorsed by a consortium, such as those validated by the Earth Microbiome Project3 (Walters et al., 2015). The re-analysis included in this manuscript demonstrates how extant sequence information can be reprocessed as necessary to be compatible with future changes in bioinformatics processing routines (e.g., the shift in identifying working amplicon sequences from OTUs to ASVs) and updated reference databases. However, if the physical regions of the 16S rRNA gene, and original generation biases from factors such as PCR primers, constituting those sequences are not inter-comparable, that cannot be corrected for in silico. Yet we recognize that a static option may not be possible if improved primers are introduced over time; thus recommendation 2:
(2) As full DNA sequencing standardization over timescales of decades will be impossible in the face of expected future technological improvements, we strongly encourage archiving of biological samples or extracted environmental DNA. This would enable the design of valid Before-After-Control-Impact studies (e.g., as are used to assess responses to the establishment of Marine Protected Areas or the effects of human-caused or natural perturbations; Osenberg and Schmitt, 1996) of the effects of mining processes on microbial communities, using the anticipated future sequencing approaches and instrumentation. Ideally there would be a central repository for such samples, but failing that, we encourage a strong commitment to long-term maintenance from individual research laboratories and contractors, as well as a willingness to share samples within the community when possible. Planned and coordinated archiving will likely be particularly important in the CCZ in light of the anticipated slow pace of post-disturbance community recovery and corresponding need for long-term monitoring (discussed in Jones et al., 2020).
(3) Collection of a standardized set of metadata for polymetallic nodules sampled for DNA will enable ecologically relevant comparisons and potentially help to identify those factors influencing microbial community composition. Beyond the basics of location and depth (both of the overlying water column and within the sediment), we encourage measures of nodule size and notes or photographs documenting attached organisms and unusual colors or features (e.g., see Supplementary Table 2). Additional information realistically may not always be feasible for all samples; for example, metal composition and nodule density could influence microbial communities associated with nodules and in nearby sediments. This list would likely expand or change as we better understand the ecology of nodule-inhabiting microbes.
(4) While a shift to non-targeted sequencing (i.e., shotgun metagenomics) could help to alleviate some of the primer-related inter-comparability issues and would have the additional benefit of providing functional information to address ecological questions, it is unlikely to be a panacea for biodiversity monitoring. This approach carries both technical and practical limitations. It too is affected by improvements in sequence yields and therefore changes in sequencing depth over timescales of decades. Functional gene analyses can be less precise for taxonomic identification than 16S rRNA genes, particularly in sparsely studied environments such as the deep sea. Finally, metagenomic analysis remains relatively expensive compared with 16S rRNA gene profiling and requires an arguably greater degree of specialist knowledge to interpret – it is highly unlikely to be accepted by commercial contractors as a component of routine monitoring activity in the near future. Metagenomics certainly has a place in furthering our understanding of the ecology of the CCZ in a research context, but for routine impact monitoring, we believe that the community should focus on optimizing amplicon-based approaches at this time.
We strongly encourage future sampling to better characterize the spatial and temporal biodiversity and activity of the microbial communities of the CCZ, to enable both more accurate management decisions and future detection of potential mining effects – particularly as our analysis here suggests that there are spatial differences in microbial communities within the CCZ. Careful planning of sampling techniques and target locations, to ensure inter-comparable datasets and alleviation of large areal gaps, will be key to obtaining an accurate understanding of microbial biodiversity in the CCZ prior to the onset of mining-related disturbances.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA660809; https://zenodo.org/record/4214934, 4214934; https://github.com/ekwear/DeepCCZreview.
Author Contributions
EW drafted the manuscript, based in part on a conference report authored by MC, EW, and BO with input from CRS. EW, MC, CNS, ML, and CRS collected and/or conducted the initial analysis of samples included in the analysis. CRS led the DeepCCZ and Abyssline cruises that collected the samples. EW conducted the bioinformatics analyses. All the authors revised and approved the manuscript.
Funding
The review portion of this work originated in, and expands upon, a section of the workshop report from the DeepCCZ Biodiversity Synthesis Workshop in October 2019 (Church et al., 2019), funded by the Gordon and Betty Moore Foundation, the Pew Charitable Trusts, and the International Seabed Authority. New data included here were funded by the Gordon and Betty Moore Foundation (Grant 5596 to MC and CRS). Funding for collection of Abyssline samples derived from contracts from the UK Seabed Resources, LTD. (UKSRL) to CRS and MC. UKSRL had no influence in the study design, data collection, data analyses, and data interpretation. BO acknowledges funding support from the Center for Dark Energy Biosphere Investigations (C-DEBI) funded by the US National Science Foundation (award OIA-0939564). This is C-DEBI contribution number 566. This is contribution 11328 from SOEST, University of Hawai’i at Mānoa.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jason Smith, Travis Washburn, C. Rob Young, other members of the DeepCCZ working groups, two reviewers, and members of the Church lab for conversations and input that contributed to and improved this manuscript. We also thank the officers and crew of the R/V Kilo Moana, the members of the Hawaii Undersea Research Lab, and the DeepCCZ science party, in particular Jen Durden, Erica Goetze, Oliver Kersten, Kirsty McQuaid, Regan Drennan, Astrid Leitner, and Jeff Drazen.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.634803/full#supplementary-material
Supplementary Figure 1 | Diversity parameters of microbial communities from Abyssline 1 samples sequenced by both this study and Shulse et al. (2017), using two different 16S rRNA gene primer sets.
Supplementary Figure 2 | Relative abundance of microbial communities from Abyssline 1 samples sequenced by both this study and Shulse et al. (2017), using two different 16S rRNA gene primer sets.
Supplementary Figure 3 | Log2-fold difference in relative abundances of selected taxa between Abyssline 1 samples sequenced in this study and in Shulse et al. (2017).
Supplementary Figure 4 | Microbial community composition, highlighting taxa of interest from genera to phylum level, in the new dataset.
Supplementary Figure 5 | Venn diagrams illustrating shared abundant ASVs between cruises and habitats.
Supplementary Figure 6 | Taxonomic distribution of differentially abundant ASVs between habitats, based on all samples from the new dataset.
Supplementary Figure 7 | Taxonomic distribution of differentially abundant ASVs in nodules between the eastern and western regions within the new dataset.
Supplementary Figure 8 | Cluster diagram of sediment samples from the new dataset, based on weighted UniFrac distance between samples.
Supplementary Table 1 | Metadata on samples included in new and re-sequenced dataset.
Supplementary Table 2 | Metadata on DeepCCZ nodule size and appearance.
Supplementary Table 3 | ANOSIM comparisons between habitats, based on weighted UniFrac distances.
Supplementary Table 4 | DESeq2 differential abundance analysis results of ASVs in nodules from the eastern vs. western CCZ sites and between different benthic habitat types.
Supplementary Table 5 | ANOSIM comparisons between eastern and western CCZ sites, based on weighted UniFrac distances.
Supplementary Table 6 | Highest relative abundance ASVs that are significantly different between eastern and western CCZ nodules.
Supplementary Video 1 | Animation showing the three-dimensional version of the sediment ordination in Figure 2C. 3D stress: 0.06.
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/sra
- ^ http://doi.org/10.5281/zenodo.4214934
- ^ https://earthmicrobiome.org
References
Apprill, A., McNally, S., Parsons, R., and Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol. 75, 129–137. doi: 10.3354/ame01753
Aylward, F. O., and Santoro, A. E. (2020). Heterotrophic Thaumarchaea with small genomes are widespread in the dark ocean. mSystems 5:e0415-20. doi: 10.1128/mSystems.00415-20
Baker, B. J., Appler, K. E., and Gong, X. (2021). New microbial biodiversity in marine sediments. Annu. Rev. Mar. Sci. 13, 161–175. doi: 10.1146/annrev-marine-032020-014552
Blöthe, M., Wegorzewski, A., Müller, C., Simon, F., Kuhn, T., and Schippers, A. (2015). Manganese-cycling microbial communities inside deep-sea manganese nodules. Environ. Sci. Technol. 49, 7692–7700. doi: 10.1021/es504930v
Callahan, B. J., McMurdie, P. J., and Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 11, 2639–2643. doi: 10.1038/ismej.2017.119
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Cho, H., Kim, K. H., Son, S. K., and Hyun, J. H. (2018). Fine-scale microbial communities associated with manganese nodules in deep-sea sediment of the Korea Deep Ocean Study Area in the northeast equatorial Pacific. Ocean Sci. J. 53, 337–353. doi: 10.1007/s12601-018-0032-0
Christian, J. R., Verschell, M. A., Murtugudde, R., Busalacchi, A. J., and McClain, C. R. (2002). Biogeochemical modelling of the tropical Pacific Ocean. I: seasonal and interannual variability. Deep Sea Res. II 49, 509–543. doi: 10.1016/S0967-0645(01)00110-2
Christiansen, B., Denda, A., and Christiansen, S. (2020). Potential effects of deep seabed mining on pelagic and benthopelagic biota. Mar. Policy 114:103442. doi: 10.1016/j.marpol.2019.02.014
Church, M. J., Wear, E. K., Orcutt, B. N., Young, C. R., and Smith, J. M. (2019). Taxonomic Diversity of Bacteria and Archaea in the Clarion-Clipperton Zone of the North Pacific Ocean. Kingston, JM: International Seabed Authority. DeepCCZ Biodiversity Synthesis Workshop Report.
Danovaro, R., Corinaldesi, C., Rastelli, E., and Dell’Anno, A. (2015). Towards a better quantitative assessment of the relevance of deep-sea viruses, Bacteria and Archaea in the functioning of the ocean seafloor. Aquat. Microb. Ecol. 75, 81–90. doi: 10.3354/ame01747
Drazen, J. C., Smith, C. R., Gjerde, K. M., Haddock, S. H. D., Carter, G. S., Choy, C. A., et al. (2020). Midwater ecosystems must be considered when evaluating environmental risks of deep-sea mining. Proc. Natl. Acad. Sci. U.S.A. 117, 17455–17460. doi: 10.1073/pnas.2011914117
Gooday, A. J., Durden, J. M., Holzmann, M., Pawlowski, J., and Smith, C. R. (2020). Xenophyophores (Rhizaria, Foraminifera), including four new species and two new genera, from the western Clarion-Clipperton Zone (abyssal equatorial Pacific). Eur. J. Protistol. 75:125715. doi: 10.1016/j.ejop.2020.125715
Hein, J. R., Koschinsky, A., and Kuhn, T. (2020). Deep-ocean polymetallic nodules as a resource for critical materials. Nat. Rev. Earth Environ. 1, 158–169. doi: 10.1038/s43017-020-0027-0
Hoffmann, K., Bienhold, C., Buttigieg, P. L., Knittel, K., Laso-Pérez, R., Rapp, J. Z., et al. (2020). Diversity and metabolism of Woeseiales bacteria, global members of marine sediment communities. ISME J. 14, 1042–1056. doi: 10.1038/s41396-020-0588-4
International Seabed Authority (2010). Technical Study No. 6: A Geological Model of Polymetallic Nodule Deposits in the Clarion-Clipperton Fracture Zone. Kingston, JM: ISA.
International Seabed Authority (2019). Recommendations for the Guidance of Contractors for the Assessment of the Possibl Environmental Impacts Arising from Exploration for Marine Minerals in the Area. Kingston, JM: ISA. Report ISBA/25/LTC/6/Rev.1.
James, A. K., English, C. J., Nidzieko, N. J., Carlson, C. A., and Wilbanks, E. G. (2020). Giant kelp microbiome altered in the presence of epiphytes. Limnol. Oceanogr. Lett. 5, 354–362. doi: 10.1002/lol2.10157
Jiang, X.-D., Gong, J.-L., Ren, J.-B., Liu, Q.-S., Zhang, J., and Chou, Y. M. (2020). An interdependent relationship between microbial ecosystems and ferromanganese nodules from the Western Pacific Ocean. Sediment. Geol. 398:105588. doi: 10.1016/j.sedgeo.2019.105588
Jones, D. O. B., Ardron, J. A., Colaço, A., and Durden, J. M. (2020). Environmental considerations for impact and preservation reference zones for deep-sea polymetallic nodule mining. Mar. Policy 118:103312. doi: 10.1016/j.marpol.2018.10.025
Kallmeyer, J., Pockalny, R., Adhikari, R. R., Smith, D. C., and D’Hondt, S. (2012). Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. U.S.A. 109, 16213–16216. doi: 10.1073/pnas.1203849109
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1039/nar/gks808
Ku, T. L., and Broecker, W. S. (1969). Radiochemical studies on manganese nodules of deep-sea origin. Deep Sea Res. 16, 625–637. doi: 10.1016/0011-7471(69)90063-1
Laureillard, J., Méjanelle, L., and Sibuet, M. (2004). Use of lipids to study the trophic ecology of deep-sea xenophyophores. Mar. Ecol. Prog. Ser. 270, 129–140. doi: 10.3354/meps270129
Lindh, M. V., Maillot, B. M., Shulse, C. N., Gooday, A. J., Amon, D. J., Smith, C. R., et al. (2017). From the surface to the deep-sea: bacterial distributions across polymetallic nodule fields in the Clarion-Clipperton Zone of the Pacific Ocean. Front. Microbiol. 8:1696. doi: 10.3389/fmicb.2017.01696
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
McQuaid, K. A., Attrill, M. J., Clark, M. R., Cobley, A., Glover, A. G., Smith, C. R., et al. (2020). Using habitat classification to assess representativity of a protected area network in a large, data-poor area targeted for deep-sea mining. Front. Mar. Sci. 7:558860. doi: 10.3389/fmars.2020.558860
Molari, M., Janssen, F., Vonnahme, T. R., Wenzhöfer, F., and Boetius, A. (2020). The contribution of microbial communities in polymetallic nodules to the diversity of the deep-sea microbiome of the Peru Basin (4130-4198 m depth). Biogeosciences 17, 3203–3222. doi: 10.5194/bg-17-3203-2020
Oksanen, J., Blanchet, F. G., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2019). Vegan: Community Ecology Package. R Package Version 2.5-6. Available online at: https://CRAN.R-project.org/package=vegan
Orcutt, B. N., Bradley, J. A., Brazelton, W. J., Estes, E. R., Goordial, J. M., Huber, J. A., et al. (2020). Impacts of deep-sea mining on microbial ecosystem services. Limnol. Oceanogr. 65, 1489–1510. doi: 10.1002/lno.11403
Orcutt, B. N., Sylvan, J. B., Knab, N. J., and Edwards, K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. R. 75, 361–422. doi: 10.1128/MMBR.00039-10
Osenberg, C. W., and Schmitt, R. J. (1996). “Detecting ecological impacts caused by human activities,” in Detecting Ecological Impacts: Concepts and Applications in Coastal Habitats, eds R. J. Schmitt and C. W. Osenberg (Cambridge, MA: Academic Press), 3–16.
Parada, A. E., Needham, D. M., and Fuhrman, J. A. (2016). Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414. doi: 10.1111/1462-2920.13023
Peoples, L. M., Kyaw, T. S., Ugalde, J. A., Mullane, K. K., Chastain, R. A., Yayanos, A. A., et al. (2020). Distinctive gene and protein characteristics of extremely piezophilic Colwellia. BMC Genom. 21:692. doi: 10.1186/s12864-020-07102-y
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ruhl, H. A., Ellena, J. A., and Smith, K. L. Jr. (2008). Connections between climate, food limitation, and carbon cycling in abyssal sediment communities. Proc. Natl. Acad. Sci. U.S.A. 105, 17006–17011. doi: 10.1073/pnas.0803898105
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Shiraishi, F., Mitsunobu, S., Suzuki, K., Hoshino, T., Morono, Y., and Inagaki, F. (2016). Dense microbial community on a ferromanganese nodule from the ultra-oligotrophic South Pacific Gyre: implications for biogeochemical cycling. Earth Planet. Sci. Lett. 447, 10–20. doi: 10.1016/j.epsl.2016.04.021
Shulse, C. N., Maillot, B., Smith, C. R., and Church, M. J. (2017). Polymetallic nodules, sediments, and deep waters in the equatorial North Pacific exhibit highly diverse and distinct bacterial, archaeal, and microeukaryotic communities. Microbiol. Open 6:e428. doi: 10.1002/mbo3.428
Simon-Lledó, E., Bett, B. J., Huvenne, V. A., Schoening, T., Benoist, N. M. A., and Jones, D. O. B. (2019). Ecology of a polymetallic nodule occurrence gradient: implications for deep-sea mining. Limnol. Oceanogr. 64, 1883–1894. doi: 10.1002/lno.11157
Smith, C. R., and Demopoulos, A. W. J. (2003). “The deep Pacific Ocean floor,” in Ecosystems of the World: Ecosystems of the Deep Ocean, Vol. 28, ed. P. A. Tyler (Amsterdam: Elsevier), 179–218.
Smith, C. R., Tunnicliffe, V., Colaço, A., Drazen, J. C., Gollner, S., Levin, L. A., et al. (2020). Deep-sea misconceptions cause underestimation of seabed-mining impacts. Trends Ecol. Evol. 35, 853–857. doi: 10.1016/j.tree.2020.07.002
Sogin, M. L., Morrison, H. G., Huber, J. A., Welch, D. M., Huse, S. M., Neal, P. R., et al. (2006). Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. U.S.A. 103, 12115–12120. doi: 10.1073/pnas.0605127103
Steinert, G., Busch, K., Bayer, K., Kodami, S., Martinez Arbizu, P., Kelly, M., et al. (2020). Compositional and quantitative insights into bacterial and archaeal communities of South Pacific deep-sea sponges (Demospongiae and Hexactinellida). Front. Microbiol. 11:716. doi: 10.3389/fmicb.2020.00716
Sweetman, A. K., Smith, C. R., Shulse, C. N., Maillot, B., Lindh, M., Church, M. J., et al. (2019). Key role of bacteria in the short-term cycling of carbon at the abyssal seafloor in a low particulate organic carbon flux region of the eastern Pacific Ocean. Limnol Oceanogr. 64, 694–713. doi: 10.1002/lno.11069
Talley, L. D. (2007). “Hydrographic atlas of the World Ocean Circulation Experiment (WOCE),” in Pacific Ocean, Vol. 2, eds M. Sparrow, R. Chapman, and J. Gould (Southampton: International WOCE Project Office).
Thurber, A. R., Sweetman, A. K., Narayanaswamy, B. E., Jones, D. O. B., Ingels, J., and Hansman, R. L. (2014). Ecosystem function and services provided by the deep sea. Biogeosciences 11, 3941–3963. doi: 10.5194/bg-11-3941-2014
Tully, B. J., and Heidelberg, J. F. (2013). Microbial communities associated with ferromanganese nodules and the surrounding sediments. Front. Microbiol. 4:161. doi: 10.3389/fmicb.2013.00161
Veillette, J., Sarrazin, J., Gooday, A. J., Galéron, J., Caprais, J.-C., Vangriesheim, A., et al. (2007). Ferromanganese nodule fauna in the Tropical North Pacific Ocean: species richness, faunal cover, and spatial distribution. Deep Sea Res. I 54, 1912–1935. doi: 10.1016/j.dsr.2007.06.011
Vonnahme, T. R., Molari, M., Janssen, F., Wenzhöfer, F., Haeckel, M., Titschack, J., et al. (2020). Effects of a deep-sea mining experiment on seafloor microbial communities and functions after 26 years. Sci. Adv. 6:eaaz5922. doi: 10.1126/sciadv.aaz5922
Walters, W., Hyde, E. R., Berg-Lyons, D., Ackermann, G., Humphrey, G., Parada, A., et al. (2015). Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal Internal Transcribed Spacer marker gene primers for microbial community surveys. mSystems 1:e00009-15. doi: 10.1128/mSystems.00009-15
Wang, C.-S., Liao, L., Xu, H. X., Xu, X. W., Wu, M., and Zhu, L. Z. (2010). Bacterial diversity in the sediment from polymetallic nodule fields of the Clarion-Clipperton Fracture Zone. J. Microbiol. 48, 573–585. doi: 10.1007/s12275-010-0151-5
Washburn, T. W., Jones, D. O. B., Wei, C.-L., and Smith, C. R. (2021). Environmental heterogeneity throughout the Clarion-Clipperton Zone and the representivity of the APEI network. Front. Mar. Sci. 8:661685. doi: 10.3389/fmars.2021.661685
Wear, E. K., Wilbanks, E. G., Nelson, C. E., and Carlson, C. A. (2018). Primer selection impacts specific population abundances but not community dynamics in a monthly time-series 16S rRNA gene amplicon analysis of coastal marine bacterioplankton. Environ. Microbiol. 20, 2709–2726. doi: 10.1111/1462-2920.14091
Wedding, L. M., Friedlander, A. M., Kittinger, J. N., Watling, L., Gaines, S. D., Bennett, M., et al. (2013). From principles to practice: a spatial approach to systematic conservation planning in the deep sea. Proc. R. Soc. B. 280:20131684. doi: 10.1098/rspb.2013.1684
Wei, C.-L., Rowe, G. T., Escobar-Briones, E., Boetius, A., Soltwedel, T., Caley, M. J., et al. (2010). Global patterns and predictions of seafloor biomass using random forests. PLoS One 5:e15323. doi: 10.1371/journal.pone.0015323
Whitman, W. B., Coleman, D. C., and Wiebe, W. J. (1998). Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. U.S.A. 95, 6578–6583. doi: 10.1073/pnas.95.12.6578
Wu, Y. H., Liao, L., Wang, C. S., Ma, W. L., Meng, F. X., Wu, M., et al. (2013). A comparison of microbial communities in deep-sea polymetallic nodules and the surrounding sediments in the Pacific Ocean. Deep Sea Res. Pt. I 79, 40–49. doi: 10.1016/j.dsr.2013.05.004
Xu, M., Wang, P., Wang, F., and Xiao, X. (2005). Microbial diversity at a deep-sea station of the Pacific nodule province. Biodivers. Conserv. 14, 3363–3380. doi: 10.1007/s10531-004-0544-z
Keywords: bacteria, archaea, 16S rRNA gene amplicons, Clarion-Clipperton Zone, polymetallic nodule, sediment, abyssal plain, biodiversity
Citation: Wear EK, Church MJ, Orcutt BN, Shulse CN, Lindh MV and Smith CR (2021) Bacterial and Archaeal Communities in Polymetallic Nodules, Sediments, and Bottom Waters of the Abyssal Clarion-Clipperton Zone: Emerging Patterns and Future Monitoring Considerations. Front. Mar. Sci. 8:634803. doi: 10.3389/fmars.2021.634803
Received: 28 November 2020; Accepted: 12 April 2021;
Published: 13 May 2021.
Edited by:
Cinzia Corinaldesi, Marche Polytechnic University, ItalyReviewed by:
Eugenio Rastelli, University of Naples Federico II, ItalyJulia M. Otte, Aarhus University, Denmark
Copyright © 2021 Wear, Church, Orcutt, Shulse, Lindh and Smith. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emma K. Wear, ZWt3ZWFyLm9jZWFuc0BnbWFpbC5jb20=