 Cheryl Lewis Ames1,2,3*
Cheryl Lewis Ames1,2,3* Aki H. Ohdera3,4
Aki H. Ohdera3,4 Sophie M. Colston1
Sophie M. Colston1 Allen G. Collins3,5
Allen G. Collins3,5 William K. Fitt6
William K. Fitt6 André C. Morandini7,8
André C. Morandini7,8 Jeffrey S. Erickson9
Jeffrey S. Erickson9 Gary J. Vora9*
Gary J. Vora9*- 1National Research Council, National Academy of Sciences, U.S. Naval Research Laboratory, Washington, DC, United States
- 2Graduate School of Agricultural Science, Faculty of Agriculture, Tohoku University, Sendai, Japan
- 3Department of Invertebrate Zoology, National Museum of Natural History, Smithsonian Institution, Washington, DC, United States
- 4Division of Biology and Biological Engineering, California Institute of Technology, Pasadena, CA, United States
- 5National Systematics Laboratory of the National Oceanic Atmospheric Administration Fisheries Service, National Museum of Natural History, Smithsonian Institution, Washington, DC, United States
- 6Odum School of Ecology, University of Georgia, Athens, GA, United States
- 7Departamento de Zoologia, Instituto de Biociências, University of São Paulo, São Paulo, Brazil
- 8Centro de Biologia Marinha, University of São Paulo, São Sebastião, Brazil
- 9Center for Bio/Molecular Science and Engineering, U.S. Naval Research Laboratory, Washington, DC, United States
Recent advances in molecular sequencing technology and the increased availability of fieldable laboratory equipment have provided researchers with the opportunity to conduct real-time or near real-time gene-based biodiversity assessments of aquatic ecosystems. In this study, we developed a workflow and portable kit for fieldable environmental DNA sequencing (FeDS) and tested its efficacy by characterizing the breadth of jellyfish (Medusozoa) taxa in the coastal waters of the Upper and Lower Florida Keys. Environmental DNA was isolated from seawater collection events at eight sites and samples were subjected to medusozoan 16S rRNA gene and metazoan mitochondrial cytochrome oxidase 1 gene profiling via metabarcoding onsite. In total, FeDS yielded 175,326 processed sequence reads providing evidence for 53 medusozoan taxa. Our most salient findings revealed eDNA from: (1) two venomous box jellyfish (Cubozoa) species, including taxa whose stings cause the notorious Irukandji envenomation syndrome; (2) two species of potentially introduced stalked jellyfish (Staurozoa); and (3) a likely cryptic species of upside-down jellyfish (Scyphozoa). Taken together, the results of this study highlight the merits of FeDS in conducting biodiversity surveys of endemic and introduced species, and as a potential tool for assessing envenomation and/or conservation-related threats.
Introduction
Over the past decade, the study of environmental DNA (eDNA) coupled with next-generation sequencing (NGS) has emerged as a promising means to assess and monitor the biodiversity of a habitat which may in turn influence conservation action and intervention. These techniques, which have been commonly used in microbial ecology studies for some time, are now being similarly adopted for the study of macroorganismal ecology. Timely and accurate assessments of organisms in their native habitats are critical to understanding the dynamics of population trends with respect to normal conditions and potentially disruptive environmental events. Conservation interests have increasingly shifted from surveys with a narrow scope, such as single species, taxon-targeted approaches, to those that evaluate ecosystems more broadly (i.e., community-targeted approaches), within both healthy and compromised ecosystems (Thomsen et al., 2012; Thomsen and Willerslev, 2015; Goldberg et al., 2016; Port et al., 2016).
As the name indicates, eDNA is genetic material that has been deposited into the environment via numerous sources, such as skin, feces, urine, larvae, and gametes. DNA extracted directly from environmental samples (e.g., filtered seawater, soil, and sand) can be sequenced to identify source organisms occupying the proximate area of the collected sample (Laramie et al., 2015; Minamoto et al., 2016; Hinlo et al., 2017; Dibattista et al., 2020). Accordingly, eDNA-based investigations have proven to be as valuable as traditional survey methods that require capture and subsampling of target organisms. Importantly, eDNA methods are a less-intrusive approach to studying invasive or declining species (Zhou et al., 2013; Bucklin et al., 2016; Holman et al., 2019; Nelson-Chorney et al., 2019), documenting the distribution of difficult to sample taxa (Parsons et al., 2018), and estimating relative seasonal biomass of target species (Takasu et al., 2019; Stoeckle et al., 2020). Marine-derived eDNA has also been likened to a “barometer of disturbance” with respect to its potential to assess anthropogenic effects on ecosystems (Dibattista et al., 2020).
While the depth and resolution of eDNA sequencing make it an attractive approach for community characterization, often there is a significant delay between sample collection in the field and sequence generation and analysis in the laboratory. Marine ecosystems, which are dynamic and subject to fluctuations in tides, reproductive cycles, seasonal disasters and stochastic events, require the generation of actionable data in a timelier manner. For such applications, we developed the fieldable eDNA sequencing (FeDS) kit which utilizes the field-deployable MinION sequencing platform Oxford Nanopore Technologies (ONT) [reviewed in Jain et al. (2016); Pomerantz et al. (2018), Krehenwinkel et al. (2019); Watsa et al. (2020)].
In this study, FeDS was used to assess the biodiversity of jellyfishes (phylum Cnidaria; subphylum Medusozoa) in order to create a more comprehensive survey of medusozoan taxa surrounding the Florida Keys. A critical FeDS capability would be the detection of two resident species for which draft genomes were recently published (Ohdera et al., 2019) – the upside-down jellyfish Cassiopea xamachana (class Scyphozoa) and the venomous box jellyfish Alatina alata (class Cubozoa). C. xamachana is considered a valuable bioindicator species, with potential applications to ecotoxicological assessments (Ohdera et al., 2018). A. alata is known as a notorious stinger during monthly inshore spawning aggregations in tropical and subtropical waters (Lewis et al., 2013; Lewis Ames et al., 2016).
We deployed the FeDS kit at sampling locations in the Upper Keys (Key Largo and Marathon Key) where ongoing studies on viable C. xamachana populations have continued for decades (Fitt and Trench, 1983; Hofmann et al., 1996; Fitt and Costley, 1998). As the life cycle of Cassiopea (like many other jellyfishes) involves an alternation of generations between a sexually reproducing jellyfish (medusa), microscopic swimming larva (planula), and a sessile asexual stage (polyp) (Supplementary Figure 1), we anticipated that there would be sufficient eDNA in the water column for FeDS-based detection of these medusozoans. Sampling sites were also selected in the Lower Keys (Fleming Key) to gauge the potential for FeDS to serve as a tool for assessing envenomation risks due to difficult-to-detect venomous jellyfish. These sites corresponded to locations where unresolved jellyfish stings had been reported (Grady and Burnett, 2003).
Materials and Methods
Collection Sites and Samples
Triplicate seawater samples (1 L) were collected from the following locations: (a) three nearshore sites in Upper Keys (Key Largo, FL, United States) with one artificial site, as a positive control (May 14–16, 2018), and (b) four nearshore sites in Lower Keys (Fleming Key, FL, United States), with one of these sites serving as a process negative control (May 16–18, 2018) (Table 1 and Supplementary Figure 2).

Table 1. Florida Keys eDNA collection sites. “Sheltered” and “Pelagic” are qualitative descriptions of the general connectedness observed between the sites and open ocean.
Collection sites in the Upper Keys, which included Key Largo and Marathon Key (BC01 – BC03) (Table 1 and Supplementary Figures 2A–E), were chosen based on historic reports of large smacks of the upside-down jellyfish C. xamachana (Figures 1a–c), often together with Cassiopea frondosa (Figures 1d–f; Hofmann et al., 1996; Fitt and Costley, 1998; Ohdera et al., 2018). The presence of Cassiopea medusae has been documented at all four locations by participants of the Annual International Cassiopea Workshop, held at the Key Largo Marine Research Laboratory since May 2017. As an ostensible positive control (BC04), an outdoor aquarium was established at the Key Largo Marine Research Station, consisting of a 10 L plastic container filled with locally sourced seawater and live C. frondosa medusae (n = 6) (Figures 1g,h) collected at BC02 (Table 1, Supplementary Figure 2E, and Figures 1g,h).

Figure 1. Cassiopea medusae at Upper Keys collection sites: (a–c) Quarry (BC01). (a) Large C. xamachana medusae in patches of high abundance (n = 5 in view), on the substrate, but not overlapping, approximately 17–30 cm diameter. (b) Large individual C. xamachana medusa. (c) Large individual C. xamachana medusa. (d–f) Rock Harbor (BC02). (d) Medium to large C. xamachana and C. frondosa medusae on the substrate in high abundance (n = 16 in view), overlapping one another, approximately 15–20 cm diameter. (e) Medium individual C. xamachana medusa. (f) Medium individual C. frondosa medusa. (g,h) Aquarium (BC04). (g) Medium C. frondosa medusae in aquarium with water from the local source. (h) Close up on medium C. frondosa medusae from (g), showing mucus being released. No Cassiopea medusae were photographed from BC03, as only a few small medusae were witnessed. Sampling locations mapped in Figure 6.
Collection sites in the Lower Keys – Fleming Key (BC05 – BC08) (Table 1 and Supplementary Figures 2F–H) were primarily chosen based on proximity to the United States Special Forces Underwater Operations School (SFUWOS), where United States military divers previously reported experiencing systemic envenomation syndrome similar to “Irukandji syndrome” (Barnes, 1964), and at least one reported jellyfish-related fatality has been documented (Burnett and Gable, 1989; Grady and Burnett, 2003). Seawater samples were taken from BC08 – an artificial enclosure away from the dive drop sites, to serve as a process negative control (Table 1). Although no jellyfish species were positively identified in the reported sting incidences, the venomous box jellyfish A. alata is a suspected culprit, given its “Irukandji-like” sting and well-documented presence in Caribbean and Florida waters (Lawley et al., 2016; Lewis Ames et al., 2016; Bouyer-Monot et al., 2017).
Filtration of Seawater Samples
Triplicate seawater samples from each site were collected by rinsing the contents of a plankton net (towed by kayaking or snorkeling in shallow coastal waters) into a 1 L bottle (Nalgene), and subjected to coarse filtration with 250 μm sieve (20 cm mouth, Fisherbrand), homogenization by hand-held USB-powered Juicer Cup Blender (380 ml volume, Huatop) for 25–35 s to remove debris and large particles, and then fine filtration through a 47 mm diameter water-testing membrane filter (0.45 μm Gridded Sterile Cellulose Nitrate Membrane, Sartorius) using a magnetic filter funnel (500 ml, PALL) attached to a polypropylene vacuum flask (1 L, #8 stopper, Nalgene) connected to a battery-powered portable vacuum filtration system (Argos Technologies PV000 P-VAC System) (Figure 2A). Filter membranes (containing eDNA filtrate) were transferred to custom-made 2-chamber tubes [0.5 ml PCR tubes with a hole drilled in the bottom, suspended within a 2 ml DNA LoBind tube (Eppendorf)] containing 400 μl of tissue lysis buffer and 40 μl of protease K (ATL Buffer, DNeasy Blood and Tissue Isolation Kit, Qiagen). Tubes containing membranes in buffer were kept at ambient temperature until DNA extraction was conducted (Figure 2B). All equipment was wiped between stations with HYPE-WIPE 3% Bleach Towelettes (7.6 cm, Current Technologies) and rinsed with distilled water and 70% ethanol, before processing subsequent samples.

Figure 2. Overview of FeDS workflow used in this study for collection sites in both Upper Keys (BC01 – BC04) and Lower Keys (BC05 – BC08). (A) Triplicate seawater samples were collected from each sampling location and processed in the field within 1–2 h post-collection. Coarse filtration was conducted, followed by homogenization and fine filtration on a water-testing membrane filter. (B) Filter membranes (containing eDNA filtrate) were kept in buffer at ambient temperature until eDNA was extracted; in the final step, isolated eDNA was concentrated in 70 μl of elution buffer. All equipment was sanitized between stations. (C) Molecular protocols including PCR were carried out using an USB-operated 8-well thermocycler (miniPCR). (D) Nanopore sequencing metabarcoding with MinION commenced in Upper Keys (BC01 – BC04) outside on a bluff overlooking BC03, and in Key West (BC05 – BC08) in a rental car.
Isolation of eDNA From Filtrate
DNA extraction and molecular protocols were carried out using the following battery-operated (12V Lithium battery pack (RAVPower) portable equipment: mini centrifuge (6,000RPM/2,960G RPM, TOMY) (Figure 2B), mini dry bath (MyBlock, Benchmark Scientific) and USB-operated 8-well thermocycler (miniPCR) (Figure 2C), Mini Vortex mixer (NISSIN) (AA alkaline batteries), and USB-operated fluorometer (Quantus Promega) with the QuantiFluor ONE dsDNA System. Environmental DNA was extracted using modifications to the protocol for the DNeasy Blood and Tissue Isolation Kit (Qiagen), as previously published (Yamamoto et al., 2016). Specifically, tubes containing membranes in buffer were incubated for 90 min at 56°C, 200 μl TE 1X buffer was added to membranes to maximize yield. After centrifugation, 600 μl was added to the collected liquid (filter membrane discarded), and in the final step eDNA was eluted in a volume of 70 μl. Extracted eDNA was purified with AMPure XP beads (Agencourt AMPURE XP, Beckman Coulter) on a magnetic bead separation rack (Bel-ArtTM SP Scienceware), at ambient temperature using a 1–1.6X ration (to remove fragments smaller than 100 bp), and quantified using the fluorometer. Of the triplicate samples for each of the eight collection sites, the sample with the highest DNA concentration for each site was selected for downstream analysis, starting with first round PCR in the USB-operated 8-well thermocycler (miniPCR) (Figures 2, 3).
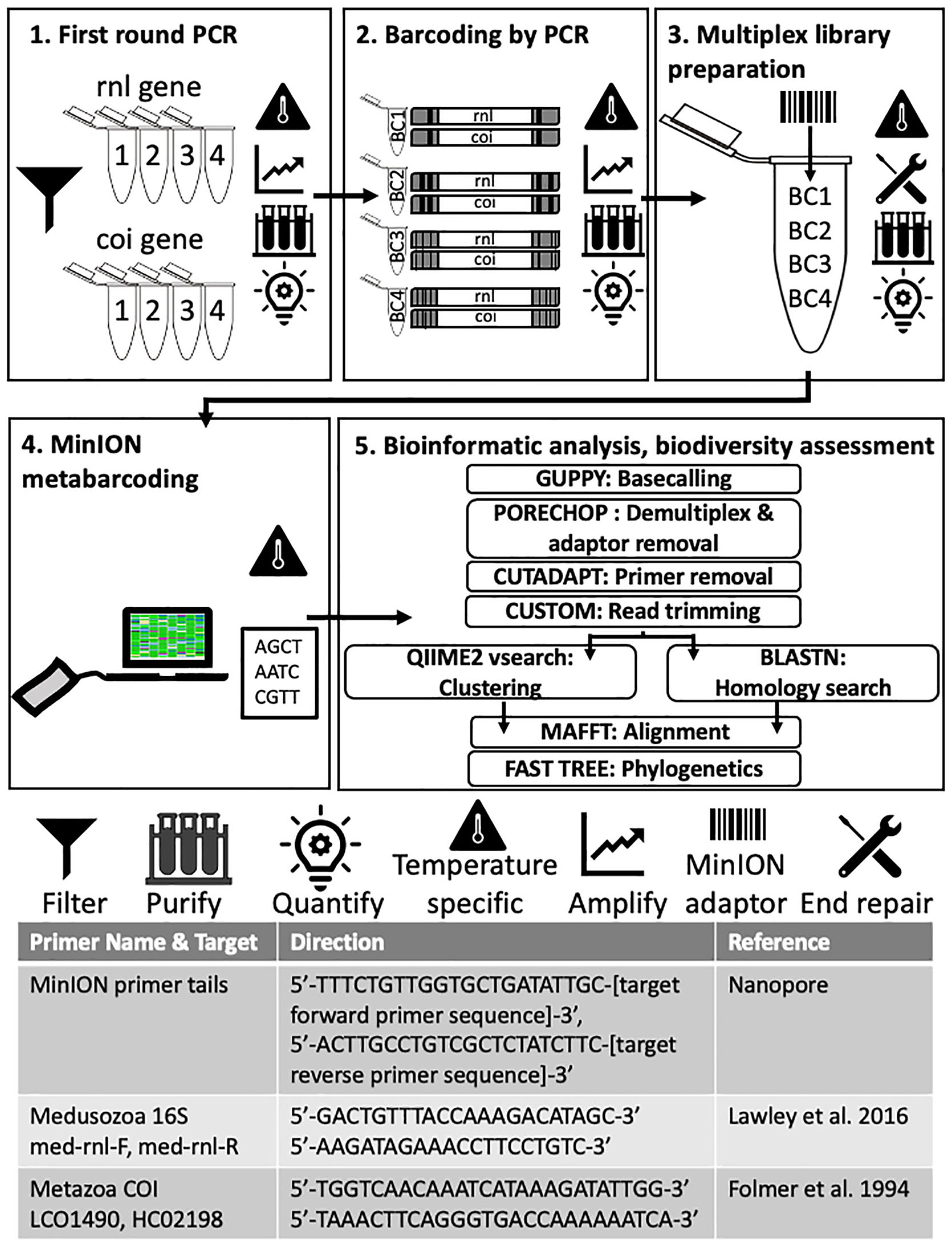
Figure 3. Molecular and bioinformatic components of the FeDS workflow. (1) First round PCR. Two genetic targets were amplified from eDNA samples (16S rRNA gene and COI, 4 miniPCR wells each). Primers and MinION primer tails provided in the table at the bottom. Amplicons pooled by collection site. (2) Barcoding by PCR. In the second round PCR, unique nanopore barcodes were added to each pooled amplicon sample. (3) Multiplexed library preparation. Input DNA was end-repaired and dA-tailed, and adapters were added. (4) MinION metabarcoding. The prepared sequencing library was loaded onto an R9.4 flow cell (FLO-MIN106, ONT) and sequenced using the MinION Mk-1B and MinKNOW v 1.2.8. (offline version), on a laptop. Icons = Filter: Seawater filtered through 0.45 μm Gridded Sterile Cellulose Nitrate Membrane, and eDNA isolated using extraction kit. Purification: Impurities removed with AMPure XP beads; purified DNA eluted in 31 μl nuclease-free water followed by quantification with the QuantiFluor portable fluorometer using QuantiFluor ONE dsDNA HS assay, after every step. Temperature specific: PCR step (amplification), or cold incubation 20°C, or controlling MinION temperature (to = < 35°C) with battery-powered mini-fan. MinION adaptor: unique nanopore barcodes (EXP-PBC001, ONT) added to each amplicon sample. End repair: End-repaired and dA-tailed using NEBNext End Repair/dA-tailing (NEB). (5) Bioinformatic analyses and biodiversity assessments. Software programs in upper case letters, followed by respective application. Refer to code: https://github.com/aohdera/Ames_et_al_2020.
Target eDNA Amplification, Sample Barcoding, and Pooling
Two mitochondrial gene fragments, one from the 16S rRNA gene (large subunit of the ribosome; 565 bp) and the other from cytochrome oxidase subunit 1 (COI) gene (720 bp), were targeted using primers with an additional 5′ sequence which allows for unique ONT barcodes to be added during library preparation (Figure 3). The COI primers were designed to amplify a broad diversity of metazoans (Geller et al., 2013), while the 16S rRNA gene primers specifically target most medusozoans (Lawley et al., 2016). For each 20 μl reaction, 3 μl of template DNA was combined with 1.4 μl dNTPs, 2 μl Advantage 2 DNA buffer, Advantage 2 Polymerase (Advantage 2 Enzyme System Kit, Takara Bio), 1 μl each of forward and reverse primers (10 μM), and 11.2 μl nuclease-free water. Subsequently, amplicons were prepared for sequencing using the 1D ligation kit (LSK-SQK108, ONT) in accordance with the manufacturer’s instructions. All PCR steps were conducted on the miniPCR. First-round PCR (Figure 3, Step 1, Advantage 2 Enzyme System Kit, Takara Bio) conditions were as follows: initial denaturation at 95°C (5 min); three cycles of denaturation at 95°C (30 s), annealing at 54°C (30 s), and extension at 72°C (45 s), followed by a final extension executed at 72°C (5 min). Following first round PCR, amplicons for each gene were pooled by collection site by taking equimolar concentrations of each sample to generate a single 20 μl purified amplicon sample. In the second round PCR (Figure 3, Step 2, Barcoding by PCR), unique ONT barcodes (EXP-PBC001, ONT) were added to each pooled amplicon sample using LongAmpTaq 2X master mix (New England BioLabs) with the following reaction conditions: initial denaturation at 95°C (3 min); 15 cycles of denaturation at 95°C (15 s), annealing at 62°C (15 s) and extension at 65°C (45 s), followed by a final extension at 65°C (5 min). For final multiplex library preparation (Figure 3, Step 3), ∼1 μg of input DNA (equimolar pooled barcoded amplicons) was end-repaired and dA-tailed using NEBNext End Repair/dA-tailing (NEB), then incubated at 20°C (using Medi-Pak Instant Cold Packs) and then at 65°C for 5 min each. Adapters were added in a ligation reaction using the 1D adapter mix (AMX) and Blunt/TA Ligase Master Mix (NEB) and incubated at ambient temperature for 10 min.
MinION Metabarcoding
Separately for the Upper Keys and Lower Keys samples, the prepared library (∼300 ng of NEB end-prepped template) was loaded onto an R9.4 flow cell (FLO-MIN106, ONT) and sequenced using the MinION Mk-1B and MinKNOW v 1.2.8 (offline version), on a MacBook Pro (Sierra Version 10.12.6, 16GB, 17 quad core) connected by an USB to USB-C adapter (Figure 3, Step 4). The resulting average sequence length of 16S rRNA gene and COI reads for the two MinION runs was 700 and 825 bp, respectively. MinION sequencing for BC01-BC04 was initiated at 21:55 (May 15, 2018) outside on a bluff (elevation ∼ 4 m) overlooking Buttonwood Sound (ambient temperature 26.5°C, 81% humidity) (Figure 2D), completed inside the Key Largo Marine Science Station, and lasted 7 hrs and 50 min, generating 578K reads. MinION sequencing for BC05-BC08 was initiated at 12:00 (May 18, 2018) inside a rent-a-car (ambient temperature 27°C, 87% humidity) (Figure 2D), completed inside the Naval Research Laboratory, Key West, and lasted 4 hr and 30 min, generating 521K reads.
Metabarcoding Data Analysis and Interpretation
Bioinformatic analyses (Figure 3, Step 5) were conducted after returning from the field, on the Smithsonian Institution High Performance Cluster (SI/HPC) (Code available at: https://github.com/aohdera/Ames_et_al_2020). Basecalling of raw reads was done using Guppy (ONT) (Balachandran et al., 2017; Wick, 2017; De Coster et al., 2018) with a quality score cutoff of 9. Demultiplexing and barcode filtering were performed using Porechop v0.2.3 (Wick, 2017) with a strict 85% sequence identity for the forward and reverse barcodes. Alignments were conducted to effectively remove end adaptors and sequences with internal adaptors prior to downstream analyses. As an added conservative measure to ensure removal of residual MinION adaptors and barcodes, Nanofilt (De Coster et al., 2018) was used to trim 50 bp from either end of the sequences. Strictly filtered reads were further trimmed using Cutadapt (Martin, 2001) to specifically remove any untrimmed primers and MinION adaptors, and reads shorter than 550 bp were removed using a custom script1.
Reads were classified as either 16S rRNA gene or COI gene sequences using BLASTN against a curated NCBI nucleotide database installed locally from the MIDORI server2 (Machida et al., 2017). Due to our rigorous quality control protocol and limited reference database, the number of reads with homology matches to metazoan taxa diminished the datasets for all eight metabarcoding samples. As expected, the number of reads represented by the process negative control (BC08) amounted to zero and, as such, BC08 was not included in downstream analyses. The seven remaining trimmed datasets (BC01-BC07) were subsequently concatenated into a single file (containing 64K sequences) for downstream biodiversity assessments (trimmed 16S rRNA gene and COI reads available at: https://github.com/aohdera/Ames_et_al_2020).
Medusozoan Biodiversity Assessment
Classification of the eDNA samples was conducted using a modified pipeline for the Qiime2 software (Bolyen et al., 2019; Figure 3, Step 5). In order to capture a suitable number of OTUs that might reflect the true biodiversity at sample collection sites, we conducted closed vsearch trial runs in 5% increments for percent identity values from 70–95% (as de novo “open” vsearch analysis proved unsuitable for our dataset – see Supplementary Figures 3,4). Furthermore, homology search was also conducted with BLASTN on all reads meeting the quality threshold, with BLASTN hits filtered for coverage (80%) and two identity cutoffs of 80 and 90% for comparison (Figure 4). To validate our method, we compared species-richness calculated from the Qiime2 and BLASTN classification methods for both 80% and 90% identity cutoffs (Supplementary Figure 5), and settled on a vsearch threshold of 80% clustering. While separate Qiime2 and BLASTN analyses were conducted on both 16S rRNA gene and COI datasets using the above parameters, we focus mainly on the 16S rRNA gene results since those primers preferentially target medusozoan taxa. The COI primers used in this study broadly target metazoans and were chosen to supplement the 16S rRNA gene libraries to ensure that the starting concentration of total DNA in the multiplex samples was sufficient for optimal sequencing on the MinION, in addition to the potential for recovering COI sequences of medusozoan taxa. To validate the Qiime2 taxonomic assignments, 16S rRNA gene reads showing greater than 80 and 90% similarity to sequences associated with species of the four medusozoan classes – Cubozoa, Hydrozoa, Scyphozoa, and Staurozoa – were aligned using MAFFT (E-INSI algorithm) (Katoh and Standley, 2013). Majority rule consensus sequences were generated in cases where three or more sequences corresponded to a given species (Table 2).
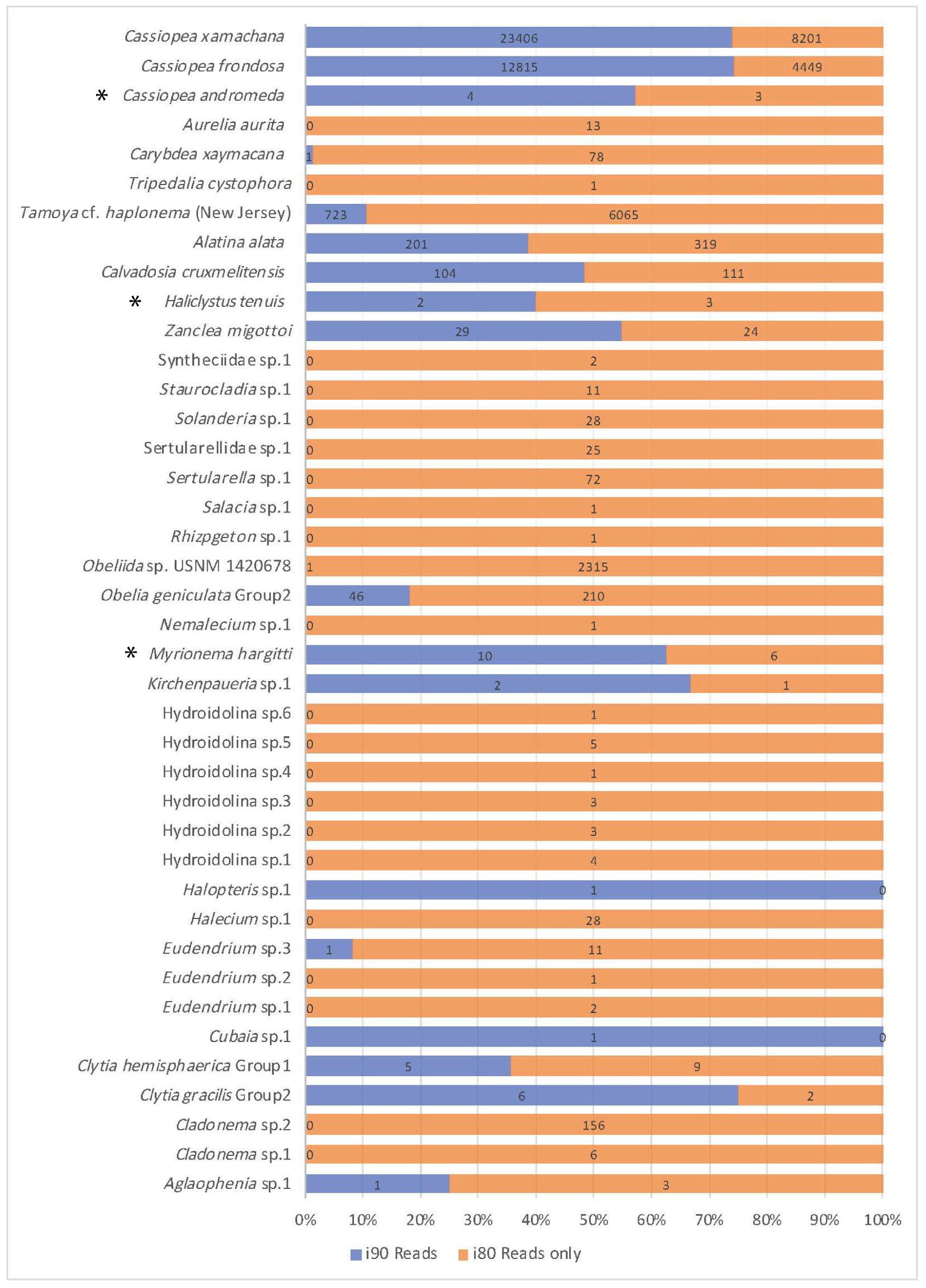
Figure 4. Comparison of read number for jellyfish taxa recovered. Jellyfish taxa comprising 40 OTUs (putative species) identified at 80 and 90% identity cutoffs with BLASTN, recovered from medusozoan 16S rRNA gene eDNA metabarcoding data (shown are matches comprising three or more sequences). Taxonomic identity was determined against the GenBank sequence database (NCBI). Values reflect proportions prior to rarefaction analysis (subsample normalization), which resulted in loss of three minimally represented taxa (∗) across all sites.
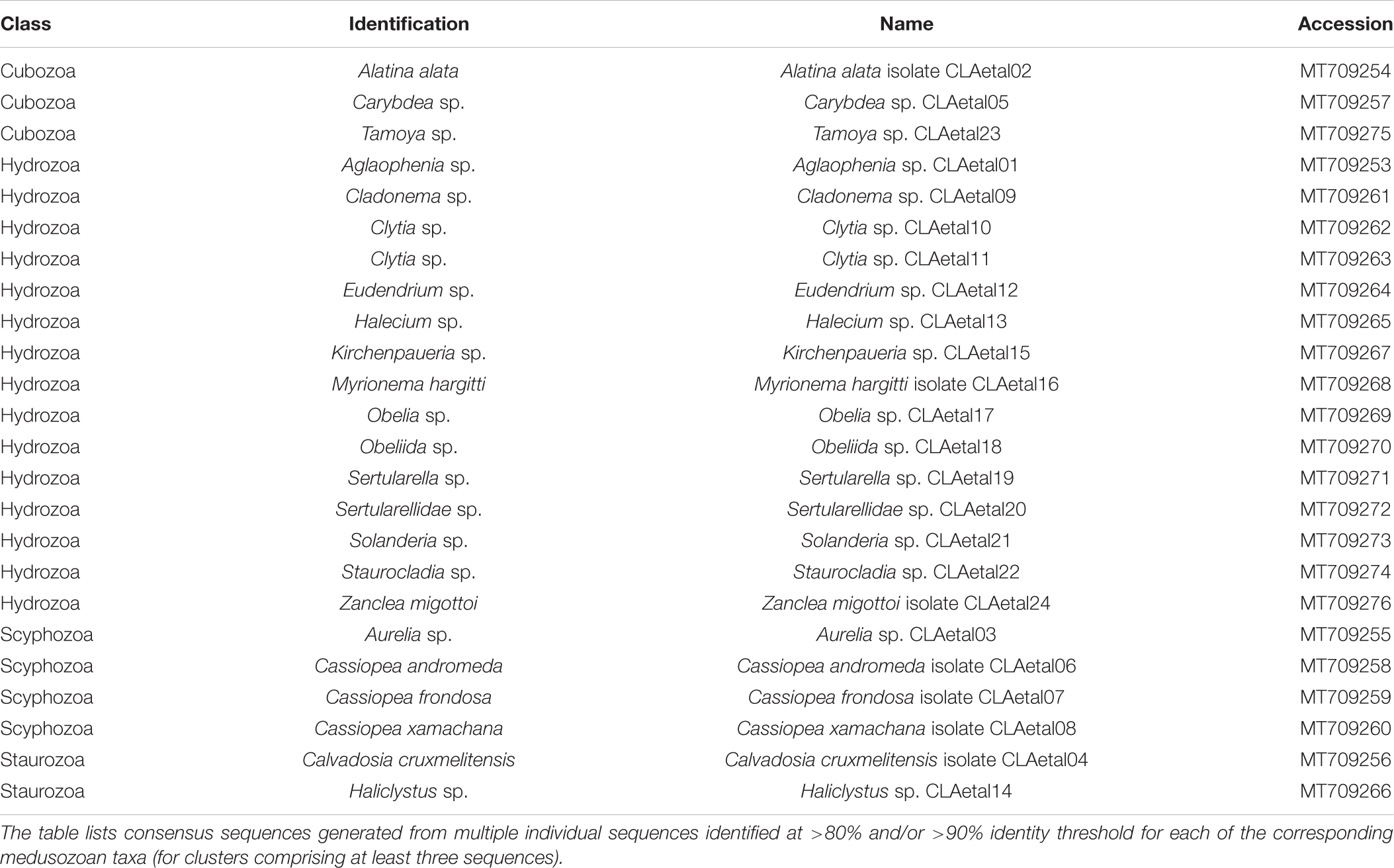
Table 2. GenBank accession numbers corresponding to consensus sequences (16S rRNA gene) for medusozoan taxa detected as eDNA using FeDS.
Phylogenetic Reconstruction
Phylogenetic analyses were conducted using FastTree (Price et al., 2010) (assuming a GTR model of nucleotide evolution, with optimization of the Gamma20 likelihood) as a plugin in Geneious prime version 2019.1.13. In order to assess the overall coherence of our metabarcoding data to the current understanding of medusozoan phylogeny (Zapata et al., 2015), we reconstructed a topology of all sequences (under both identity thresholds, >80% and >90%) including individual or pairs of reads when fewer than three sequences showed similarity to a given medusozoan target (Figure 5A). Separate trees were also constructed for the classes Cubozoa, Scyphozoa and Staurozoa that included sequences for our recovered medusozoan taxa, as well as corresponding 16S rRNA gene sequences from GenBank (Figure 5). Additionally, distance plots for selected taxa of these three classes were reconstructed to validate species-level matches to ascertain whether OTUs represented distinct species or genetic variants of a single species (calculations and distance plots available at https://github.com/aohdera/Ames_et_al_2020). Note that because of the much higher species richness for Hydrozoa (and associated large number of 16S rRNA gene sequences in GenBank), a summary tree was not constructed separately for this class.
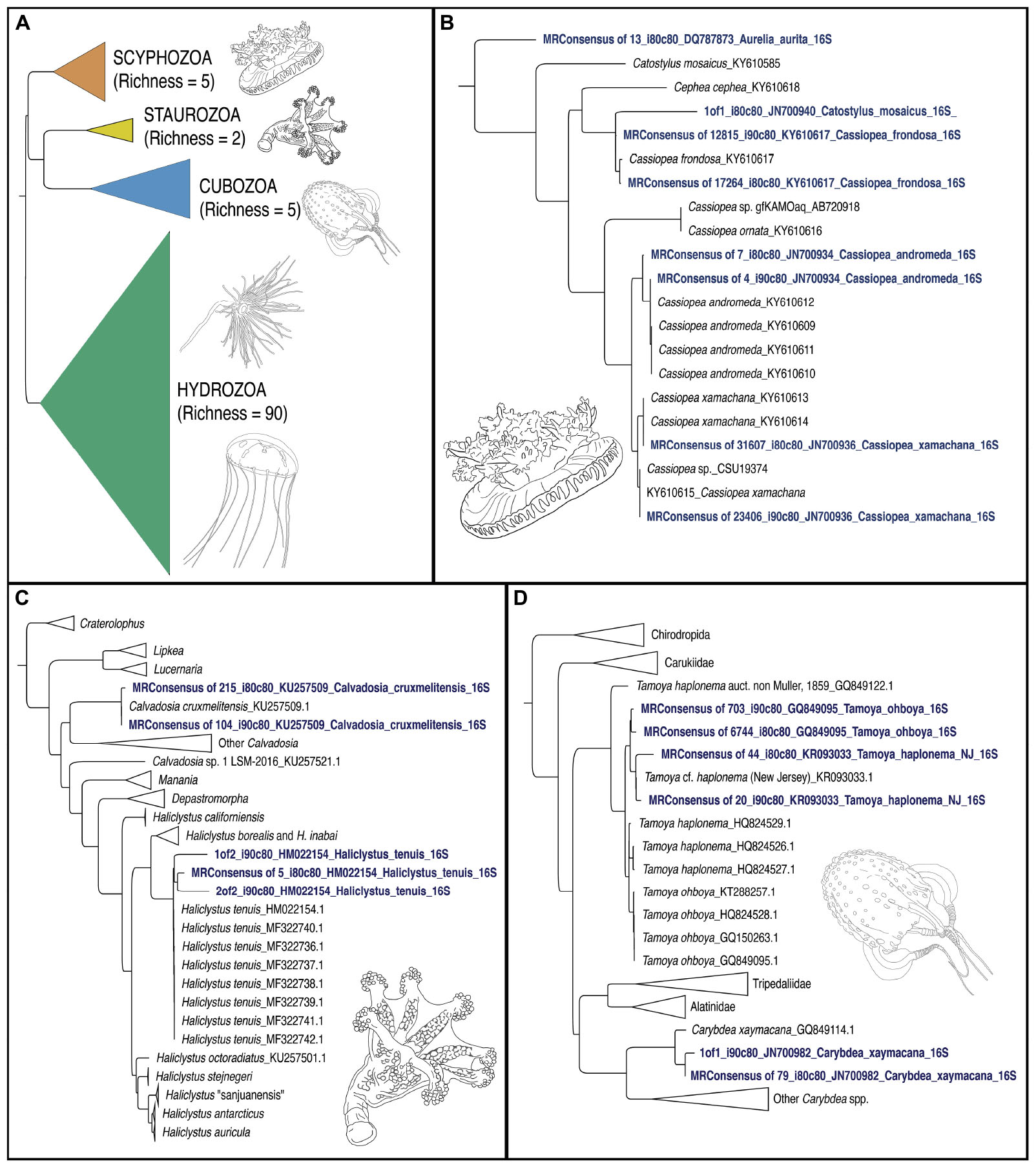
Figure 5. Phylogenetic trees of jellyfish (Medusozoa) based on mitochondrial 16S rRNA gene eDNA profiling from the Florida Keys. (A) Phylogenetic reconstruction of representatives of all four medusozoan classes detected in eDNA samples. Terminal taxa consist of majority-rule consensus sequences of aligned sequence reads that showed BLAST similarity to target sequences at two thresholds (1) Percent identity >90% and percent coverage >80%, and (2) Percent identity >80% and percent coverage >80%) in cases where there were three or more such reads (see Figure 4). In cases where there were less than three such reads, these reads were included as terminals (GenBank accession numbers in Table 2). Richness refers to the number of OTUs for each class. Colors correspond to classes represented in mapped pie charts in Figure 6. Phylogenetic reconstruction of 16S rRNA gene sequences in GenBank (NCBI) in tandem with relevant taxa detected as metabarcodes (denoted in blue letters). (B) Scyphozoa. Note that one read identified as >80% identical to Catostylus mosaicus (an Indo-Pacific species) appears more likely to represent an aberrant read of C. frondosa. (C) Staurozoa. Representing the first record of Staurozoa in the Florida Keys. (D) Cubozoa. Note that two sets of reads showing high identity to Tamoya cf. haplonema from New Jersey (KR093033) and T. ohboya (GQ849095), respectively, both group with Tamoya cf. haplonema from New Jersey (KR093033) suggesting that the two sets of sequences may be associated with a single species rather than two. Hydrozoa phylogenetic tree not provided. Sequence alignments conducted with MAFFT (E-INSI algorithm), and tree reconstructed with FASTTREE on Geneious Prime version 2019.1.1.
Results
We retained 175,326 total reads after stringent quality filtering and trimming, ranging from 3,965 to 50,063 reads across the seven sampling sites. Of those, 59,705 were identified through BLAST as 16S rRNA gene sequences. BC07 retained the lowest number of reads with 506, while 27,629 reads remained for BC02. Qiime2 analysis using closed vsearch with an identity cutoff of 80% identified a total of 53 medusozoan OTUs (Supplementary Figure 4). Despite the low read count for several samples, alpha-diversity rarefaction analysis suggested our method were sufficient to assess progress in recovering representative medusozoan eDNA by sample location, given our sequencing depth (Supplementary Figures 5,6). A BLASTN search of our recovered 16S rRNA gene filtered dataset yielded 59 unique medusozoan taxa, suggesting minor discrepancies can occur with variations in database and algorithm (clustering versus homology) used for read classification. Furthermore, although sequences corresponding to eDNA for the COI gene were largely a secondary target in this study, we were successful in detecting three medusozoan OTUs comprising two hydrozoans and C. frondosa (details provided in Supplementary Figures 5A, B); these data were not incorporated into our medusozoan biodiversity analyses.
Medusozoan Fauna in the Florida Keys
Overall, medusozoan OTU richness was lowest at Rock Harbor-BC02 (24 OTUs), followed by the Aquarium control-BC04 (15 OTUs) (Figures 6C,D). Buttonwood Sound (BC03) and Finger Pier (BC05), both pelagic habitats, had the greatest number of OTUs (42) (Figures 6B,E). On average, we detected 30 OTUs from sheltered sites (BC01 and BC02) and 36 OTUs from pelagic sites (BC03, BC05, BC06, and BC07) (Figure 6). Although Hydrozoa represented the class with the highest number of OTUs detected, on average, 65.2% of reads were assigned to Scyphozoa, 26.7% to Hydrozoa, followed by 18.6% to Cubozoa, with Staurozoa represented by the fewest reads (0.9%). With the exception of Buttonwood Sound, Cassiopea was consistently the most represented taxon (based on total corresponding reads) at each site.
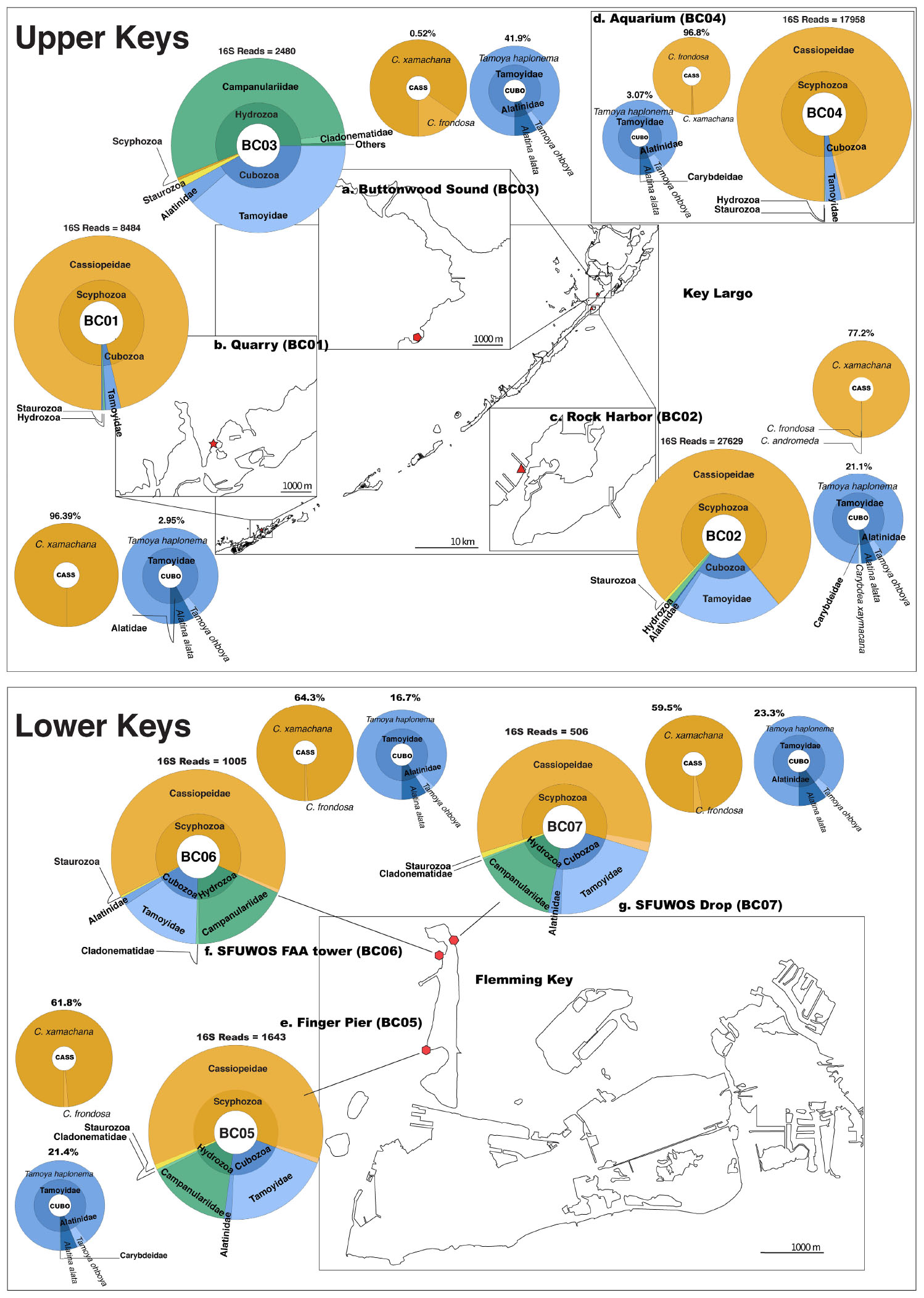
Figure 6. Map depicting distribution of jellyfish species at Florida Keys sampling sites based on 16S rRNA gene. Upper Keys (Key Largo and Marathon Key), water samples (May 14–16, 2018). (a) Buttonwood Sound (BC03). 25.10143, –80.43861. (b) Quarry (BC01). 24.74975, –80.97812. (c) Rock Harbor (BC02). 25.07924, –80.45245. (d) Aquarium (BC04). 25.10135, –80.43861. Lower Keys (Fleming Key), water samples (May 17, 2018). (e) Finger Pier (BC05). 24.57581, –81.79922. (f) SFUWOS FAA Tower (BC06). 24.59015, –81.79704. (g) SFUWOS Drop (BC07). 24.59181, –81.79487; Negative control (BC08) not depicted. Red symbols correspond to sampling locations on the map. Large, multicolored pie charts show medusozoan diversity detected at each site and total number of 16S rRNA gene reads detected per location based on Qiime2 analysis. Percentages above smaller monochromatic pie charts highlight the percent of total reads corresponding to Cassiopea and cubozoan species, respectively. Results from the Qiime2 barplot function were modified and visualized here using Krona (Ondov et al., 2011). Percentages reflect proportions prior to rarefaction analysis (subsample normalization), which resulted in loss of three minimally represented taxa.
Cassiopea reads were recovered from all sampling sites, but most of the reads were from sheltered sites, with C. xamachana reads encompassing 96.3% of Cassiopea reads generated for Rock Harbor (Figure 6 and Supplementary Figure 6). Although Cassiopea medusae were not visually confirmed at Fleming Key, the detection of eDNA for this taxon was expected given that the upside-down jellyfish is the most common jellyfish in the Florida Keys and has been documented in mangroves of neighboring Key West, several kilometers away (NOAA, 2020). Phylogenetic analysis of consensus reads, as determined by BLASTN revealed that three putative Cassiopea species are likely present in the Florida Keys. C. xamachana dominates throughout the Florida Keys with respect to detected eDNA (Figure 7) and visual confirmation (Ohdera et al., 2018), whereas C. frondosa occurs less frequently (Table 1).
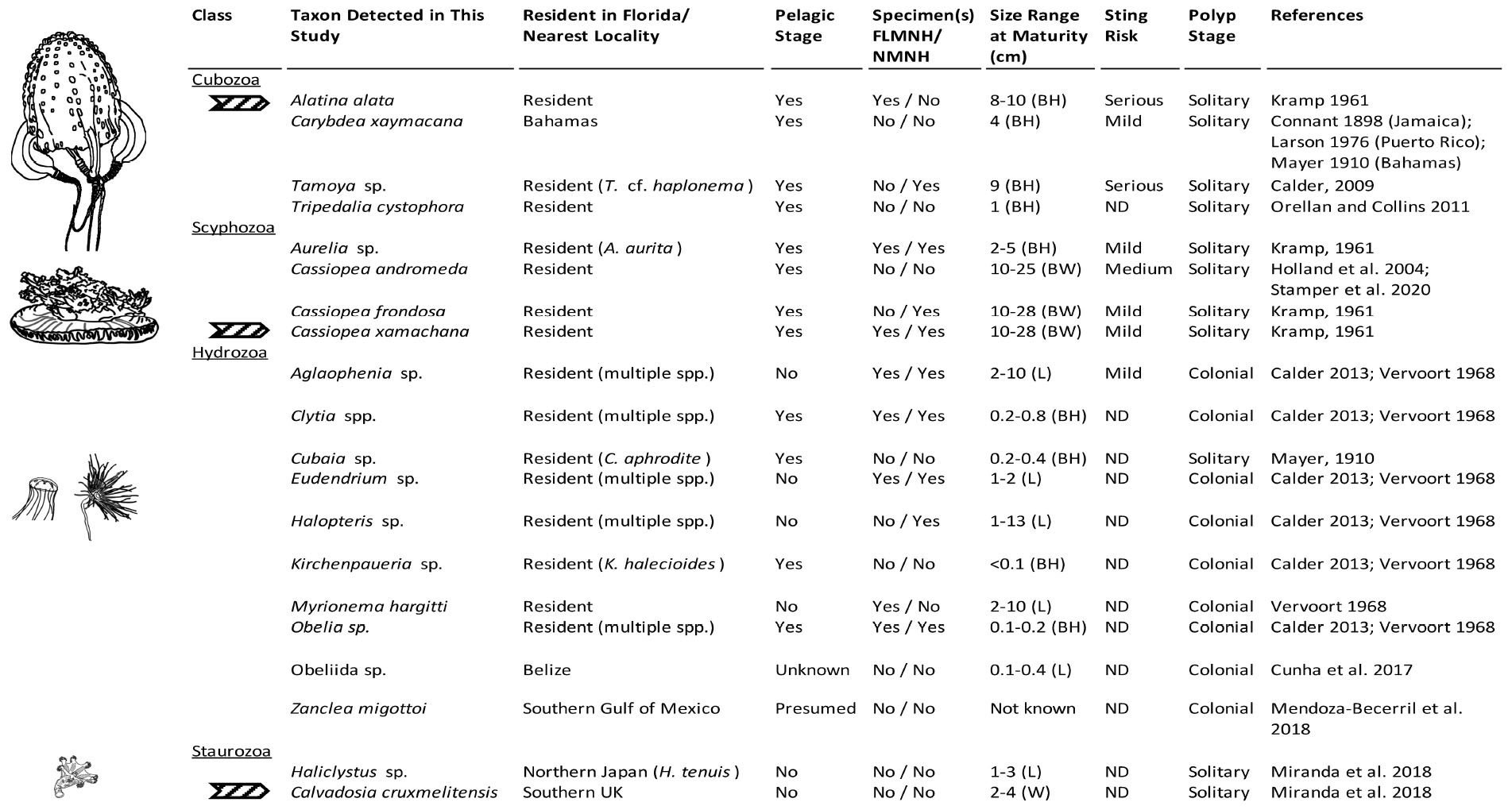
Figure 7. Medusozoa fauna identified from 16S rRNA gene using FeDS. The table highlights morphological characteristics and residency in the Florida Keys sampling areas or closest geographic location. Line drawings to the left depict a representative taxon for each class, drawn in comparative scale. Arrows indicate species for which partially annotated genomes have been published (Ohdera et al., 2019). BH, bell height; BW, bell width; L, length of hydroid; W, width; ND, no data available; FLMNH, Florida Museum of Natural History; NMNH, National Museum of Natural History, Smithsonian Institution.
In addition to Cassiopea, we detected a second scyphozoan genus Aurelia, for which the consensus of reads at the 80% identity threshold (BLASTN) shared ∼86% similarity to Aurelia aurita (GenBank DQ787873) – a broadly distributed species complex of jellyfish known as moon jellyfish (Dawson, 2003), with at least one species known in Florida waters (Figure 7). Using Qiime2, this match was only recovered using percent identity cutoffs below 75%. Interestingly, the consensus of reads at the 80% identity threshold in our BLASTN search showed >98% identity to unpublished sequences of Aurelia sp. samples from southern Brazil (J. Lawley, pers. comm.). Therefore, it is likely that the DNA sequences of the species we detected is yet lacking from GenBank, highlighting the importance of building robust reference databases of genetic barcodes.
Venomous species of box jellyfish (Cubozoa) detected by eDNA metabarcoding include taxa of the cubozoan families Alatinidae and Tamoyidae (Figures 4–7). Though previously reported from the Caribbean and Florida Keys region (Figure 7), no medusa of either family was visually confirmed during this study. As these box jellyfish medusae are rather large and conspicuous (Figure 7), it is conceivable that our sequences corresponded to eDNA from microscopic life stages (planulae or polyps) present at collection sites, in line with recent findings of eDNA signal detected for benthic cnidarians (Sawaya et al., 2019; Bolte et al., 2021). A. alata is the best documented species of the family Alatinidae, but at least one other has been reported in the Gulf of Mexico (Graham, 1998; Lewis et al., 2013; Lasley et al., 2016; Lawley et al., 2016). Our initial results indicated that two species of Tamoyidae had been detected in this study: Tamoya cf. haplonema (matching a New Jersey sample; GenBank KR093033) and a very similar sequence of Tamoya ohboya (matching a Caribbean sample; GenBank GQ150263). Tamoya species have been previously reported in this geographical region (Collins et al., 2011; Figure 7), although further studies are needed to properly delineate species within this genus. While two different exemplar sequences were identified by maximal BLASTN scores, consensus sequences of these reads both had nearly 100% identity to the T. cf. haplonema rRNA gene from New Jersey. Furthermore, the tiny cubozoan, Tripedalia cystophora, was detected via eDNA as well (as a singleton). First described from Jamaica, T. cystophora has only recently been documented in Florida waters (Orellana and Collins, 2011; Lasley et al., 2016). Finally, a set of 17 reads corresponding to an unidentified species of Carybdea (closely matching sequences of the Caribbean species Carybdea xaymacana), suggest the presence of another yet-to-be described species of box jelly in the Florida Keys (Figure 7).
Although no species of the benthic stalked jellyfishes in the class Staurozoa were reported previously from Florida (Miranda et al., 2018), in this study we identified eDNA of the staurozoans Calvadosia cruxmelitensis and Haliclystus cf. tenuis (Figures 4–7). The former is of particular interest as its genome was recently published together with that of C. xamachana and A. alata (Ohdera et al., 2019). Staurozoans are relatively small medusozoans (Figure 7), and often cryptic and difficult to find in the field because they live on and blend in well with macroalgae. These species are exclusively benthic, so the presence of sequences corresponding to these taxa (C. cruxmelitensis being represented at every sampling location in this study) suggests that FeDS was able to detect eDNA of benthic species in addition to that of pelagic jellyfishes. H. tenuis was originally described from Japan (Kishinouye, 1910), but has recently been considered an introduced species in the north Atlantic (Holst and Laakmann, 2019); it has never been reported from the western Atlantic (Figure 7). C. cruxmelitensis is distributed throughout the British Isles, while congeners are the only known staurozoans to be distributed in warm tropical and subtropical waters, including reports of C. hawaiiensis from Hawaii (Edmondson, 1930), an undetermined species from India (Panikkar, 1944), and C. corbini from Brazil (Grohmann et al., 1999), Puerto Rico (Capriles and Martinez, 1970; Larson, 1980), and the western Gulf of Mexico (Lechuga and Fernández-Álamo, 2005). While known geographic distributions of staurozoans (Miranda et al., 2018) would suggest that C. corbini is the most likely species to be encountered in Florida waters, our data unequivocally indicate that C. cruxmelitensis inhabits the Florida Keys (Figure 7). Nevertheless, this assertion should be confirmed through visual inspection of suitable coastal Florida habitats to assess a hypothetical introduction. Overall, these results suggest that extensive eDNA analysis could rapidly advance our understanding of the distribution of cryptic organisms.
We identified 36 Hydrozoa OTUs, despite only 15% of total reads mapping to hydrozoan sequences. Several of these taxa (e.g., species of Aglaophenia, Eudendrium, and Halopteris) lack a medusa stage, providing more evidence that our eDNA sampling captured benthic taxa (Figures 4, 7). Given the extensive hydrozoan diversity of over 3,500 known species, including hundreds described from the Caribbean, the more than 50% representation of all OTUs by hydrozoan taxa was not unexpected (Figures 4, 5).
Discussion
In this study, we have successfully demonstrated the efficacy of our FeDS kit which leverages the portable MinION sequencer to enable rapid, onsite metabarcoding of eDNA corresponding to 53 medusozoan taxa in the Florida Keys. We executed all necessary steps, from collection to data generation, using accessible battery-operated equipment to conduct a multi-site biodiversity analysis of jellyfish in the Florida Keys. Despite the difficulty in visually identifying medusozoans that lack a pelagic swimming stage, our seawater eDNA analysis proved to be effective for detecting several species of hydroids and staurozoans (stalked jellyfish) lacking a swimming medusa stage. Although no single literature source exists that comprehensively documents the jellyfish fauna of Florida and the Caribbean, the results of our comparative evaluation of jellyfish biodiversity in these coastal habitats were generally consistent with the literature on medusozoans present in the region (Conant, 1897; Bigelow, 1900, 1918, 1938; Mayer, 1910; Kramp, 1961; Vervoort, 1967; Larson, 1976; Humann and Deloach, 2002; Holland et al., 2004; Calder, 2009, 2013; Orellana and Collins, 2011; Lasley et al., 2016; Cunha et al., 2017; Mendoza-Becerril et al., 2017; Miglietta et al., 2018; Miranda et al., 2018; Ohdera et al., 2018; NOAA, 2020) (summarized herein in Figure 7). Due to the large amount of sequence data generated, we were able to filter reads stringently and still recover a great deal of medusozoan biodiversity as a proof-of-concept that eDNA metabarcoding with FeDS identified higher overall biodiversity than could be detected with traditional survey methods alone, validating its utility for field applications.
Overall, FeDS-generated 16S rRNA gene sequences primarily corresponded to Cassiopea (up to 96% of sequences in Upper Keys sites and 64% in Lower Keys sites), with the majority of sequences corresponding to C. xamachana. In the case of Cassiopea andromeda, although we detected its eDNA in the Florida Keys some workers have suggested that C. xamachana and C. andromeda may be the same species (Holland et al., 2004). However, in our analysis, consensus sequences of putative C. andromeda and C. xamachana diverge by roughly 4%, raising the hypothesis that a separate species is present, which is corroborated by recent findings (Stampar et al., 2020). The fact that resident Cassiopea species, C. xamachana and C. frondosa, can be readily distinguished based on both morphology and genetics is evidence for two reproductively isolated species. C. andromeda was originally described from the Red Sea, from where it has recently spread throughout the world to warm, coastal waters, possibly introduced at the microscopic life stage in ballast water or as polyps on ship hulls (Holland et al., 2004; Stampar et al., 2020).
Cassiopea xamachana medusae are known to release large amounts of mucus that contains motile clusters of stinging cells called cassiosomes (Ames et al., 2020). Thought to be an important component of healthy mangrove ecosystems, both spawn and mucus likely contributed to our success in preferentially amplifying Cassiopea eDNA (representing 82% of all matched reads). Together with our recent publication of the reference genome of the upside-down jellyfish C. xamachana (Ohdera et al., 2019), recent works have brought attention to this model system as a viable biomonitor species with promising applications for coastal ecosystem management and conservation (Todd et al., 2006; Newkirk et al., 2020). At BC03, however, FeDS detected a diminutive eDNA signature for Cassiopea (Supplementary Figure 2C). This low-level detection at this site may be related to the effects of the devastating Hurricane Irma (Dilling et al., 2017), which in September 2017 depleted the Cassiopea population at this site almost entirely (Supplementary Figure 2D). However, at that same location during the 3rd annual Cassiopea International Workshop (a year after deploying FeDS), coauthors of this study (WF and AO) verified that the Cassiopea medusa numbers had rebounded to pre-Irma abundances. While tentative, it appears that our FeDS kit was able to detect a small eDNA signal during the early stages of a population recovery. Like many jellyfishes, Cassiopea exhibits alternation of generations in its life cycle (Supplementary Figure 1); the advantage conferred by the persistence of the asexual polyp stage may be the key to their resilience, despite anthropogenic or natural pressures.
Our eDNA findings also indicated a significant presence of two species of venomous box jellyfish (Tamoya sp., A. alata) in the Upper Keys (up to 42% of sequences), and in the Lower Keys (up to 23% of sequences); the latter are in the vicinity of SFUWOS SCUBA diving drop-off sites. In the absence of visual confirmation of these relatively large medusae, it is conceivable that unseen minute stages (e.g., juvenile medusae) have been the cause of serious envenomation reports (Guest, 1950; Burnett and Gable, 1989; Grady and Burnett, 2003; Lawley et al., 2016).
Despite the inherent difficulty in visually identifying medusozoans that lack a pelagic swimming stage, FeDS was shown to be effective in detecting several species of hydroids and staurozoans (stalked jellyfish) lacking a swimming medusa stage. Therefore, given the proven applications of eDNA for rapid detection of cryptic, dangerous and/or introduced species in coastal environments (Berry et al., 2019; Holman et al., 2019), our FeDS kit could serve as a powerful tool to test whether jellyfish are resilient to global shifts toward warming oceans, or if changes in climate might drastically alter patterns of jellyfish proliferation [discussed in Purcell et al. (2007); Bayha and Graham (2013), Condon et al. (2013), Olguín-Jacobson et al. (2020)].
The speed of our FeDS protocol (sample-to-sequence) was in stark contrast to the time-lag involved in developing a suitable pipeline to analyze multi-site nanopore metabarcoding data (sequence-to-assessment) due to the limited availability of appropriate bioinformatic tools [for a review of computational challenges of nanopore data and potential software see Santos et al. (2020); Watsa et al. (2020)]. Despite initial data analysis setbacks, we are confident that the final custom pipeline we developed is a suitable, reliable bioinformatics approach for handling the data with ease of reproducibility for any study system. Recently, several third-party freeware options have been developed to analyze long read data generated by nanopore sequencing devices (Chang et al., 2020a; de Koning et al., 2020; Rodríguez-pérez et al., 2020), offering promise for a standardized pipeline in the near future. For our homology search, we used a subset of all metazoan sequences in GenBank with species level identifications, called the MIDORI database (Machida et al., 2017). While this public database is deemed trustworthy (Leray et al., 2019), it is far from an exhaustive repertoire of Florida Keys fauna. Future expansion of the initial medusozoan reference database generated in this study from eDNA sequences will facilitate large-scale biodiversity studies of the region. Furthermore, once FeDS is equipped with the latest nanopore sequencing chemistry (R10.3 barcodes) researchers can achieve fieldable eDNA biodiversity studies with ∼99.9% accuracy when compared with Illumina reference barcodes at an affordable price (Chang et al., 2020a, b). With a number of well-established regional databases becoming available to the public [e.g., Smithsonian Marine Global Earth Observatory (MarineGEO) monitoring site (Nguyen et al., 2020)], FeDS becomes a highly attractive option for timely and accurate eDNA detection in aquatic environments toward discovery of regional biodiversity patterns.
Conclusion
Our FeDS workflow is easily adaptable for biodiversity assessments given its: (1) Low-complexity protocol with a relatively short “sample-to-sequence” timeframe; (2) Fieldable eDNA metabarcoding capabilities for austere environments through portable, manual and/or battery-powered equipment, ensuring a minimal environmental footprint; and (3) Multiplexing capabilities for the simultaneous evaluation of multiple collection sites and genetic markers. FeDS has surmounted the hurdles previously impeding rapid biodiversity assessments in the field, and has demonstrated the ability to perform effective identification assays under resource-limited, offline conditions. Our FeDS kit is poised as a practical molecular tool for both civilian and naval management to regularly and proactively analyze water samples for early detection of declining endemic species, as well as aquatic threats, such as venomous jellyfish. The equipment and other components of the mobile kit assembled for this study are affordable, readily available and can be optimized for any situation, eliminating the need for access to expensive, space- and energy-consuming laboratory equipment. While we suspect that the fieldable eDNA sequencing tools described herein can be deployed to conduct regular, real-time analysis of seawater samples for the purposes of sting mitigation, resource management and even predicting recovering populations following natural disasters, further assessments of that assertion are warranted. Finally, as nanopore chemistry has and will continue to improve, further enhancements to our FeDS kit, such as the integration of a device with remote communication capabilities, could eventually lead to the development of an autonomous environmental DNA sequencing system, important commercially, and for public safety and conservation.
Data Availability Statement
Filtered and trimmed 16S and COI MinION data sets have been made available at: https://github.com/aohdera/Ames_et_al_2020. Nanopore sequences for medusozoans (16S; classes Scyphozoa, Cubozoa, and Staurozoa) have been separately accessioned into GenBank (see Table 2). All code and data sets used in this study are provided as Supplementary Data Sheet 1: https://github.com/aohdera/Ames_et_al_2020.
Author Contributions
CA conceived, designed, and carried out sample collection, and all steps in the FeDS workflow in Key Largo and Fleming Key, conducted bioinformatic analyses and phylogenetic analyses, coauthored the first draft of the manuscript, and prepared figures and tables. AO carried out sample collection and MinION sequencing in Key Largo, conducted bioinformatic analyses, prepared figures and tables, and coauthored the first draft. SC helped to design the experiments, provided training in MinION technology, assisted with MinION read processing, and prepared figures and tables. AC conducted phylogenetic analyses and interpretation of results, historical data assessment and synopsis, prepared figures and tables, and coauthored the first draft. WF assisted with sample collection in Key Largo, provided ecological data related to pre- and post-Irma conditions in Key Largo, and conducted photo-documentation. ACM assisted with sample collection in Key Largo, species identification, and conducted photo-documentation. JE provided technical guidance during experimental design and developing equipment for portability. GV helped to conceive of the experiments, oversaw all aspects of the project from start to finish, assisted with FeDS kit design, and contributed reagents, materials, and analysis tools. All authors provided content for each manuscript draft and approved the final draft.
Funding
This research was supported by the Office of Naval Research via Naval Research Laboratory core funds (work unit 6B84). CA and SC acknowledge postdoctoral fellowships through the National Research Council’s Research Associateship Program. ACM was supported by FAPESP 2015/21007-9 and CNPq 309440/2019-0.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Mónica Medina, whose assistance in establishing the Cassiopea model meeting in Key Largo (FL, United States) allowed us to conduct this research, and the meeting attendees for their assistance with collecting. Additionally, we acknowledge technical support received from Vanessa Molina, NRL Key West, and CPT Brett Ambroson, Diving Medical Officer, Special Forces Underwater Operations School, Key West, for logistical assistance in Fleming Key (FL, United States). Finally, we are grateful to two reviewers and the Editor for their constructive comments. This is a contribution of NP-BioMar USP. The opinions and assertions contained herein are those of the authors and are not to be construed as those of the U.S. Navy, military service at large or U.S. Government.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.640527/full#supplementary-material
Footnotes
- ^ https://github.com/aohdera/Ames_et_al_2020
- ^ http://reference-midori.info/download.php#
- ^ https://www.geneious.com
References
Ames, C. L., Klompen, A. M. L., Badhiwala, K., Muffett, K., Reft, A. J., Kumar, M., et al. (2020). Cassiosomes are stinging-cell structures in the mucus of the upside-down jellyfish Cassiopea xamachana. Commun. Biol. 3:67.
Balachandran, P., Trevino, R., Wang, X., Standage-Beier, K., and Faucon, P. (2017). “High accuracy base calls in Nanopore sequencing,” in Proceedings of the 2017 6th International Conference on Bioinformatics and Biomedical Science, ICBBS 2017 Association for Computing Machinery, New York, NY, 12–16.
Barnes, J. H. (1964). Cause and efects of Irukandji stingings. Med. J. Aust. 1, 897–904. doi: 10.5694/j.1326-5377.1964.tb114424.x
Bayha, K. M., and Graham, W. M. (2013). “Nonindigenous marine jellyfish: invasiveness, invasibility and impacts,” in Jellyfish Blooms, eds K. A. Pitt and C. H. Lucas (Dordrech: Springer).
Berry, T. E., Saunders, B. J., Coghlan, M. L., Stat, M., Jarman, S., Richardson, A. J., et al. (2019). Marine environmental DNA biomonitoring reveals seasonal patterns in biodiversity and identifies ecosystem responses to anomalous climatic events. PLoS Genet 15:e1007943. doi: 10.1371/journal.pgen.1007943
Bigelow, H. B. (1918). Some Medusae and Siphonophora from the western Atlantic. (Intergovernmental Panel on Climate Change, Ed). Bull. Museum Comp. Zool. Harvard Coll. 62, 363–442.
Bigelow, H. B. (1938). Plankton of Bermuda oceanographic expeditions. VIII. Medusae taken during the Years 1929 and 1930. Zool. Sci. Contrib. N. Y. Zool. Soc. 23, 99–189.
Bigelow, R. P. (1900). The Anatomy and Development of Cassiopea xamachana. Boston, MA: Boston Society of Natural History.
Bolte, B., Goldsbury, J., Jerry, D., and Kingsford, M. (2021). Validation of eDNA as a viable method of detection for dangerous cubozoan jellyfish. Environmental DNA 1–11. doi: 10.1002/edn3.181
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857.
Bouyer-Monot, D., Pelczar, S., Ferracci, S., and Boucaud-Maitre, D. (2017). Retrospective study of jellyfish envenomation in emergency wards in Guadeloupe between 2010 and 2016: when to diagnose Irukandji syndrome? Toxicon 137, 73–77. doi: 10.1016/j.toxicon.2017.07.011
Bucklin, A., Lindeque, P. K., Rodriguez-Ezpeleta, N., Albaina, A., and Lehtiniemi, M. (2016). Metabarcoding of marine zooplankton: prospects, progress and pitfalls. J. Plankton Res. 38, 393–400. doi: 10.1093/plankt/fbw023
Burnett, J. W., and Gable, W. D. (1989). A fatal jellyfish envenomation by the Portuguese man-o’war. Toxicon 27, 823–824. doi: 10.1016/0041-0101(89)90050-0
Calder, D. R. (2009). Cubozoan and scyphozoan jellyfishes of the Carolinian biogeographic province, southeastern USA. R. Ont. Mus. Contrib. Sci. 3, 1–58. doi: 10.1007/978-94-007-6288-6_67-1
Calder, D. R. (2013). Some shallow-water hydroids (Cnidaria: Hydrozoa) from the central east coast of Florida, USA. Zootaxa 3648:1. doi: 10.11646/zootaxa.3648.1.1
Capriles, V. A., and Martinez, H. (1970). First report of a Stauromedusae from Puerto Rico. Caribb. J. Sci. 10:106.
Chang, J. J. M., Ip, Y. C. A., Bauman, A. G., and Huang, D. (2020a). MinION-in-ARMS: nanopore sequencing to expedite barcoding of specimen-rich macrofaunal samples from autonomous reef monitoring structures. Front. Mar. Sci. 7:448. doi: 10.3389/fmars.2020.00448
Chang, J. J. M., Ip, Y. C. A., Ng, C. S. L., and Huang, D. (2020b). Takeaways from mobile DNA barcoding with BentoLab and MinION. Genes (Basel) 11:1121. doi: 10.3390/genes11101121
Collins, A. G., Bentlage, B., Gillan, W., Lynn, T. H., Morandini, A. C., and Marques, A. C. (2011). Naming the Bonaire banded box jelly, Tamoya ohboya, n. sp. (Cnidaria: Cubozoa: Carybdeida: Tamoyidae). Zootaxa 68, 53–68. doi: 10.11646/zootaxa.2753.1.3
Condon, R. H., Duarte, C. M., Pitt, K. A., Robinson, K. L., Lucas, C. H., Sutherland, K. R., et al. (2013). Recurrent jellyfish blooms are a consequence of global oscillations. Proc. Natl. Acad. Sci. U.S.A. 110, 1000–1005.
Cunha, A. F., Collins, A. G., and Marques, A. C. (2017). Phylogenetic relationships of Proboscoida Broch, 1910 (Cnidaria, Hydrozoa): are traditional morphological diagnostic characters relevant for the delimitation of lineages at the species, genus, and family levels? Mol. Phylogenet. Evol. 106, 118–135. doi: 10.1016/j.ympev.2016.09.012
Dawson, M. N. (2003). Macro-morphological variation among cryptic species of the moon jellyfish, Aurelia (Cnidaria: Scyphozoa). Mar. Biol. 143, 369–379. doi: 10.1007/s00227-003-1070-3
De Coster, W., D’Hert, S., Schultz, D. T., Cruts, M., and Van Broeckhoven, C. (2018). NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 34, 2666–2669. doi: 10.1093/bioinformatics/bty149
de Koning, W., Miladi, M., Hiltemann, S., Heikema, A., Hays, J. P., Flemming, S., et al. (2020). NanoGalaxy: nanopore long-read sequencing data analysis in Galaxy. Gigascience 9:giaa105.
Dibattista, J. D., Reimer, J. D., Stat, M., Masucci, G. D., Biondi, P., De Brauwer, M., et al. (2020). Environmental DNA can act as a biodiversity barometer of anthropogenic pressures in coastal ecosystems. Sci. Rep. 10:8365.
Dilling, L., Morss, R., and Wilhelmi, O. (2017). Learning to expect surprise: hurricanes Harvey, Irma, Maria, and beyond. J. Extrem. Events 04:1771001. doi: 10.1142/s2345737617710014
Fitt, W. K., and Costley, K. (1998). The role of temperature in survival of the polyp stage of the tropical rhizostome jellyfish Cassiopea xamachana. J. Exp. Mar. Biol. Ecol. 222, 79–91. doi: 10.1016/s0022-0981(97)00139-1
Fitt, W. K., and Trench, R. K. (1983). Endocytosis of the symbiotic dinoflagellate Symbiodinium microadriaticum Freudenthal by endodermal cells of the scyphistomae of Cassiopea xamachana and resistance of algae to host digestion. J. Cell Sci. 64, 195–212.
Geller, J., Meyer, C., Parker, M., and Hawk, H. (2013). Redesign of PCR primers for mitochondrial cytochrome c oxidase subunit I for marine invertebrates and application in all-taxa biotic surveys. Mol. Ecol. Resour. 13, 851–861. doi: 10.1111/1755-0998.12138
Goldberg, C. S., Turner, C. R., Deiner, K., Klymus, K. E., Thomsen, P. F., Murphy, M. A., et al. (2016). Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol. Evol. 7, 1299–1307. doi: 10.1111/2041-210x.12595
Grady, J. D., and Burnett, J. W. (2003). Irukandji-like syndrome in South Florida divers. Ann. Emerg. Med. 42, 763–766. doi: 10.1016/s0196-0644(03)00513-4
Graham, M. (1998). Short papers and notes: first report of Carybdea alata var. grandis (Reynaud 1830) (Cnidaria: Cubozoa) from the Gulf of Mexico. Gulf Mex. Sci. 1, 28–30.
Grohmann, P. A., Magalhaes, M. P., and Hirano, Y. M. (1999). First record of the order stauromedusae (Cnidaria, Scyphozoa) from the Tropical Southwestern Atlantic, with a review of the distribution of Stauromedusae in the Southern hemisphere. Species Divers 4, 381–388. doi: 10.12782/specdiv.4.381
Guest, W. C. (1950). The occurrence of the jellyfish Chiropsalmus quadrumanus in Matagorda Bay, Texas. Texas Game Fish Comm. Mar. Lab. 9, 79–83.
Hinlo, R., Gleeson, D., Lintermans, M., and Furlan, E. (2017). Methods to maximise recovery of environmental DNA from water samples (H Doi, Ed). PLoS One 12:e0179251. doi: 10.1371/journal.pone.0179251
Hofmann, D. K., Fitt, W. K., and Fleck, J. (1996). Checkpoints in the life-cycle of Cassiopea spp.: control of metagenesis and metamorphosis in a tropical jellyfish. Int. J. Dev. Biol. 40, 331–338.
Holland, B. S., Dawson, M. N., Crow, G. L., and Hofmann, D. K. (2004). Global phylogeography of Cassiopea (Scyphozoa: Rhizostomeae): molecular evidence for cryptic species and multiple invasions of the Hawaiian Islands. Mar. Biol. 145, 1119–1128. doi: 10.1007/s00227-004-1409-4
Holman, L. E., de Bruyn, M., Creer, S., Carvalho, G., Robidart, J., and Rius, M. (2019). Detection of introduced and resident marine species using environmental DNA metabarcoding of sediment and water. Sci. Rep. 9:11559.
Holst, S., and Laakmann, S. (2019). First record of the stalked jellyfish Haliclystus tenuis Kishinouye, 1910 (Cnidaria: Staurozoa) in Atlantic waters. Mar. Biodivers. 49, 1061–1066. doi: 10.1007/s12526-018-0888-3
Humann, P., and Deloach, N. (2002). Reef Creature Identification: Florida, Caribbean, Bahamas. Jacksonville, FL: New World Publications Inc.
Jain, M., Olsen, H. E., Paten, B., and Akeson, M. (2016). Erratum to: the Oxford nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol. 17:256.
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Krehenwinkel, H., Pomerantz, A., and Prost, S. (2019). Genetic biomonitoring and biodiversity assessment using portable sequencing technologies: Current uses and future directions. Genes (Basel) 10:858. doi: 10.3390/genes10110858
Laramie, M. B., Pilliod, D. S., Goldberg, C. S., and Strickler, K. M. (2015). Environmental DNA sampling protocol - filtering water to capture DNA from aquatic organisms. US Geol. Surv. Tech. Methods Book 2:15.
Larson, R. J. (1976). Marine Flora and Fauna of the Northeastern United States. Cnidaria: Scyphozoa. Washington, DC: United States Government Printing Office.
Larson, R. J. (1980). A new stauromedusa, Kishinouyea corbini (Scyphozoa, Stauromedusae) from the tropical western Atlantic. Bull. Mar. Sci. 30, 102–107.
Lasley, R. M., Ames, C. L., Erdman, R., Parks, S., and Collins, A. G. (2016). First record of the box jellyfish Tripedalia cystophora (Cnidaria: Cubozoa: Tripedaliidae) in the Gulf of Mexico. Proc. Biol. Soc. Washingt. 129, 164–172. doi: 10.2988/0006-324x-129.q2.164
Lawley, J. W., Ames, C. L., Bentlage, B., Yanagihara, A., Goodwill, R., Kayal, E., et al. (2016). Box jellyfish Alatina alata has a circumtropical distribution. Biol. Bull. 231, 152–169.
Lechuga, G. R., and Fernández-Álamo, M. A. (2005). Primer registro de Kishinouyea corbini Larson, 1980 (Cnidaria: Scyphozoa, Stauromedusae) para México. Rev. Soc. Mex. Hist. Nat. 7, 107–110.
Leray, M., Knowlton, N., Ho, S. L., Nguyen, B. N., and Machida, R. J. (2019). GenBank is a reliable resource for 21st century biodiversity research. Proc Natl Acad Sci U.S.A. 116, 22651–22656. doi: 10.1073/pnas.1911714116
Lewis Ames, C., Ryan, J. F., Bely, A. E., Cartwright, P., and Collins, A. G. (2016). A new transcriptome and transcriptome profiling of adult and larval tissue in the box jellyfish Alatina alata, an emerging model for studying venom, vision and sex. BMC Genomics 17:650. doi: 10.1186/s12864-016-2944-3
Lewis, C., Bentlage, B., Yanagihara, A. A., Gillan, W., van Blerk, J., Keil, D. P., et al. (2013). Redescription of Alatina alata (Reynaud, 1830) (Cnidaria: Cubozoa) from Bonaire, Dutch Caribbean. Zootaxa 3737, 473–487. doi: 10.11646/zootaxa.3737.4.8
Machida, R. J., Leray, M., Ho, S.-L., and Knowlton, N. (2017). Metazoan mitochondrial gene sequence reference datasets for taxonomic assignment of environmental samples. Sci. Data 4:170027.
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 17, 10–12. doi: 10.14806/ej.17.1.200 (accessed March 29, 2021).
Mayer, A. G. (1910). Medusae of the World Volume III the Scyphomedusae. Washington, DC: Carnegie Institution of Washington.
Mendoza-Becerril, M. A., Simoes, N., and Genzano, G. (2017). Benthic hydroids (Cnidaria, Hydrozoa) from Alacranes. Bull. Mar. Sci. 94, 125–142. doi: 10.5343/bms.2017.1072
Miglietta, M. P., Piraino, S., Pruski, S., Gonzalez, M. A., Iglesias, S. C., Jerónimo-Aguilar, S., et al. (2018). An integrative identification guide to the Hydrozoa (Cnidaria) of Bocas del Toro. Panama. Neotrop. Biodivers. 4, 103–113. doi: 10.1080/23766808.2018.1488656
Minamoto, T., Naka, T., Moji, K., and Maruyama, A. (2016). Techniques for the practical collection of environmental DNA: filter selection, preservation, and extraction. Limnology 17, 23–32. doi: 10.1007/s10201-015-0457-4
Miranda, L. S., Mills, C. E., Hirano, Y. M., Collins, A. G., and Marques, A. C. (2018). A review of the global diversity and natural history of stalked jellyfishes (Cnidaria, Staurozoa). Mar. Biodivers. 48, 1695–1714. doi: 10.1007/s12526-017-0721-4
Nelson-Chorney, H. T., Davis, C. S., Poesch, M. S., Vinebrooke, R. D., Carli, C. M., and Taylor, M. K. (2019). Environmental DNA in lake sediment reveals biogeography of native genetic diversity. Front. Ecol. Environ. 17:313–318. doi: 10.1002/fee.2073
Newkirk, C. R., Frazer, T. K., Martindale, M. Q., and Schnitzler, C. E. (2020). Adaptation to bleaching: are thermotolerant Symbiodiniaceae strains more successful than other strains under elevated temperatures in a model symbiotic cnidarian? Front. Microbiol. 11:822. doi: 10.3389/fmicb.2020.00822
Nguyen, B. N., Shen, E. W., Seemann, J., Correa, A. M. S., O’Donnell, J. L., Altieri, A. H., et al. (2020). Environmental DNA survey captures patterns of fish and invertebrate diversity across a tropical seascape. Sci. Rep. 10:6729.
NOAA (2020). What are the Most Common Jellies in the Keys?. Available online at: https://floridakeys.noaa.gov/animals/commonkeysjellies.html (accessed Aug 11, 2019).
Ohdera, A. H., Abrams, M. J., Ames, C. L., Baker, D. M., Suescún-Bolívar, L. P., Collins, A. G., et al. (2018). Upside-down but headed in the right direction: Review of the highly versatile Cassiopea xamachana system. Front. Ecol. Evol. 6:35. doi: 10.3389/fevo.2018.00035
Ohdera, A., Ames, C. L., Dikow, R. B., Kayal, E., Chiodin, M., Busby, B., et al. (2019). Box, stalked, and upside-down? Draft genomes from diverse jellyfish (Cnidaria, Acraspeda) lineages: Alatina alata (Cubozoa), Calvadosia cruxmelitensis (Staurozoa), and Cassiopea xamachana (Scyphozoa). Gigascience 8:giz069.
Olguín-Jacobson, C., Pitt, K. A., Carroll, A. R., and Melvin, S. D. (2020). Polyps of the Jellyfish Aurelia aurita are unaffected by chronic exposure to a combination of pesticides. Environ. Toxicol. Chem. 39, 1685–1692. doi: 10.1002/etc.4750
Ondov, B. D., Bergman, N. H., and Phillippy, A. M. (2011). Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 12:385. doi: 10.1186/1471-2105-12-385
Orellana, E. R., and Collins, A. G. (2011). First report of the box jellyfish Tripedalia cystophora (Cubozoa: Tripedaliidae) in the continental USA, from Lake Wyman, Boca Raton, Florida. Mar. Biodivers. Rec. 4:e54.
Parsons, K. M., Everett, M., Dahlheim, M., and Park, L. (2018). Water, water everywhere: environmental DNA can unlock population structure in elusive marine species. R. Soc. Open Sci. 5:180537. doi: 10.1098/rsos.180537
Pomerantz, A., Peñafiel, N., Arteaga, A., Bustamante, L., Pichardo, F., Coloma, L. A., et al. (2018). Real-time DNA barcoding in a rainforest using nanopore sequencing: opportunities for rapid biodiversity assessments and local capacity building. Gigascience 7:giy033.
Port, J. A., O’Donnell, J. L., Romero-Maraccini, O. C., Leary, P. R., Litvin, S. Y., Nickols, K. J., et al. (2016). Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol. Ecol. 25, 527–541. doi: 10.1111/mec.13481
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 Approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Purcell, J. E., Uye, S. I., and Lo, W. T. (2007). Anthropogenic causes of jellyfish blooms and their direct consequences for humans: a review. Mar. Ecol. Prog. Ser. 350, 153–174. doi: 10.3354/meps07093
Rodríguez-pérez, H., Ciuffreda, L., and Flores, C. (2020). NanoCLUST: a species-level analysis of 16S rRNA nanopore sequencing data. bioRxiv [Preprint]. doi: 10.1101/2020.05.14.087353
Santos, A., van Aerle, R., Barrientos, L., and Martinez-Urtaza, J. (2020). Computational methods for 16S metabarcoding studies using Nanopore sequencing data. Comput. Struct. Biotechnol. J. 18, 296–305. doi: 10.1016/j.csbj.2020.01.005
Sawaya, N. A., Djurhuus, A., Closek, C. J., Hepner, M., Olesin, E., Visser, L., et al. (2019). Assessing eukaryotic biodiversity in the Florida Keys National Marine Sanctuary through environmental DNA metabarcoding. Ecol. Evol. 9, 1029–1040. doi: 10.1002/ece3.4742
Stampar, S. N., Gamero-Mora, E., Maronna, M. M., Fritscher, J. M., Oliveira, B. S. P., Sampaio, C. L. S., et al. (2020). The puzzling occurrence of the upside-down jellyfish cassiopea (Cnidaria: Scyphozoa) along the Brazilian coast: a result of several invasion events? Zoologia 37, 1–10. doi: 10.3897/zoologia.37.e50834
Stoeckle, M. Y., Adolf, J., Charlop-powers, Z., Dunton, K. J., Hinks, G., and VanMorter, S. M. (2020). Trawl and eDNA assessment of marine fish diversity, seasonality, and relative abundance in coastal New Jersey, USA. ICES J. Mar. Sci. 78, 293–304. doi: 10.1093/icesjms/fsaa225
Takasu, H., Inomata, H., Uchino, K., Tahara, S., Mori, K., Hirano, Y., et al. (2019). Spatio-temporal distribution of environmental DNA derived from Japanese sea nettle jellyfish Chrysaora pacifica in Omura Bay, Kyushu, Japan. Plankt. Benthos Res. 14, 320–323. doi: 10.3800/pbr.14.320
Thomsen, P. F., and Willerslev, E. (2015). Environmental DNA - An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18. doi: 10.1016/j.biocon.2014.11.019
Thomsen, P. F., Kielgast, J., Iversen, L. L., Møller, P. R., Rasmussen, M., and Willerslev, E. (2012). Detection of a diverse marine fish fauna using environmental DNA from seawater samples (S Lin, Ed). PLoS One 7:e41732. doi: 10.1371/journal.pone.0041732
Todd, B. D., Thornhill, D. J., and Fitt, W. K. (2006). Patterns of inorganic phosphate uptake in Cassiopea xamachana: a bioindicator species. Mar. Pollut. Bull. 52, 515–521. doi: 10.1016/j.marpolbul.2005.09.044
Vervoort, W. (1967). Report on a collection of hydroida from the Caribbean Region, including an annotated checklist of caribbean hydroids. Zool. Verh. 92, 1–124. doi: 10.11646/zootaxa.3171.1.1
Watsa, M., Erkenswick, G. A., Pomerantz, A., and Prost, S. (2020). Portable sequencing as a teaching tool in conservation and biodiversity research. PLoS Biol. 18:e3000667. doi: 10.1371/journal.pbio.3000667
Wick, R. (2017). Porechop. Available online at: https://github.com/rrwick/Porechop (accessed March 29, 2021).
Yamamoto, S., Minami, K., Fukaya, K., Takahashi, K., Sawada, H., Murakami, H., et al. (2016). Environmental DNA as a ‘snapshot’ of fish distribution: a case study of Japanese Jack Mackerel in Maizuru Bay, Sea of Japan. PLoS One 11:e0149786. doi: 10.1371/journal.pone.0149786
Zapata, F., Goetz, F. E., Smith, S. A., Howison, M., Siebert, S., Church, S. H., et al. (2015). Phylogenomic analyses support traditional relationships within Cnidaria. PLoS One 10:e0139068. doi: 10.1371/journal.pone.0139068
Keywords: eDNA, envenomation, upside-down jellyfish, conservation, biodiversity, portable lab kit
Citation: Ames CL, Ohdera AH, Colston SM, Collins AG, Fitt WK, Morandini AC, Erickson JS and Vora GJ (2021) Fieldable Environmental DNA Sequencing to Assess Jellyfish Biodiversity in Nearshore Waters of the Florida Keys, United States. Front. Mar. Sci. 8:640527. doi: 10.3389/fmars.2021.640527
Received: 11 December 2020; Accepted: 22 March 2021;
Published: 13 April 2021.
Edited by:
Frank Edgar Muller-Karger, University of South Florida, United StatesReviewed by:
Chih-Ching Chung, National Taiwan Ocean University, TaiwanYu Zhang, Shanghai Jiao Tong University, China
Copyright © 2021 Ames, Ohdera, Colston, Collins, Fitt, Morandini, Erickson and Vora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cheryl Lewis Ames, YW1lcy5jaGVyeWwubHlubi5hMUB0b2hva3UuYWMuanA=; Gary J. Vora, Z2FyeS52b3JhQG5ybC5uYXZ5Lm1pbA==