 Zoe A. Pratte1
Zoe A. Pratte1 Christina A. Kellogg2*
Christina A. Kellogg2*- 1Department of Microbiology and Immunology, Montana State University, Bozeman, MT, United States
- 2St. Petersburg Coastal and Marine Science Center, United States Geological Survey, St. Petersburg, FL, United States
All animals are host to a multitude of microorganisms that are essential to the animal’s health. Host-associated microbes have been shown to defend against potential pathogens, provide essential nutrients, interact with the host’s immune system, and even regulate mood. However, it can be difficult to preserve and obtain nucleic acids from some host-associated microbiomes, making studying their microbial communities challenging. Corals are an example of this, in part due to their potentially remote, underwater locations, their thick surface mucopolysaccharide layer, and various inherent molecular inhibitors. This study examined three different preservatives (RNAlater, DNA/RNA Shield, and liquid nitrogen) and two extraction methods (the Qiagen PowerBiofilm kit and the Promega Maxwell RBC kit with modifications) to determine if there was an optimum combination for examining the coral microbiome. These methods were employed across taxonomically diverse coral species, including deep-sea/shallow, stony/soft, and zooxanthellate/azooxanthellate: Lophelia pertusa, Paragorgia johnsoni, Montastraea cavernosa, Porites astreoides, and Stephanocoenia intersepta. Although significant differences were found between preservative types and extraction methods, these differences were subtle, and varied in nature from coral species to coral species. Significant differences between coral species were far more profound than those detected between preservative or extraction method. We suggest that the preservative types presented here and extraction methods using a bead-beating step provide enough consistency to compare coral microbiomes across various studies, as long as subtle differences in microbial communities are attributed to dissimilar methodologies. Additionally, the inclusion of internal controls such as a mock community and extraction blanks can help provide context regarding data quality, improving downstream analyses.
Introduction
In recent decades researchers have come to realize the importance and diversity of the host-associated microbiome. Microbes are attributed with beneficial contributions such as the production of essential metabolites, priming a host’s immune system, and directly affecting disease risk (Holmes et al., 2011; Nicholson et al., 2012), while dysbiosis, or an altered state of the microbiome, has been associated with disease and stress (Hamdi et al., 2011; Nicholson et al., 2012; Apprill, 2017; Sweet and Bulling, 2017). The bulk of this knowledge has come from studying the human and mouse microbiomes. However, some host-associated microbiomes are far more difficult to study than others. For example, much of the coral microbial population resides in the mucopolysaccharide layer above the coral tissue, making nucleotide extraction difficult (Ducklow and Mitchell, 1979; Sweet et al., 2011; Apprill et al., 2016). In addition to the mucus, the presence of mesoglea, with its combination of amorphous gel and collagen fiber matrix has been postulated to impede high quality DNA extraction from coral tissues (Weber et al., 2017). Coral tissues and mucus also contain an abundance of polymerase chain reaction (PCR) inhibitors, including calcium, polysaccharides, salts, and possibly melanin (Schrader et al., 2012; Baker and Kellogg, 2014; Weber et al., 2017). Physically accessing some hosts, such as deep-sea corals or remote tropical reefs, is also quite challenging, and requires preservation methods that can survive long transportation or shipment, independent of electricity. The high host-DNA to microbial-DNA ratio is problematic even for bacterial-specific primers (Galkiewicz and Kellogg, 2008; Baker and Kellogg, 2014; Weber et al., 2017; Reigel et al., 2020), providing additional difficulties once samples are back in the laboratory. There is little consensus across the field when it comes to methodologies, perhaps because of the variability of technique efficacy across different coral species. In fact, it would seem that these challenges have resulted in a broader spread of methodologies, resulting in a variety of preservation and DNA extraction combinations (Sweet and Bulling, 2017).
The vast majority of studies on preservation and extraction practices are focused upon the human microbiome (Yuan et al., 2012; Abusleme et al., 2014; Dominianni et al., 2014; Choo et al., 2015; Lim et al., 2018; Chen et al., 2019), and therefore don’t address the challenges of salt, PCR inhibitors, gelatinous matrix, and remote field operations. For example, Dominianni et al. (2014) found that human fecal samples were sufficiently preserved on fecal occult blood test cards for microbiome analyses, while Gray et al. (2013) found similar products, the FTA and FTA Elute cards, were not effective solutions for coral-based microbial community preservation and subsequent PCR amplification. Three coral-specific studies examined different extraction kits for nucleic acid isolation, but did not address preservative type, and examined a limited number of coral species (Santos et al., 2012; Baker and Kellogg, 2014; Weber et al., 2017). One recent study compared three preservation methods (DMSO, liquid nitrogen, and 4% paraformaldehyde solution) and two extraction methods (bead-beating versus crushing) in only two shallow-water corals (Hernandez-Agreda et al., 2018). Thus, there is a need for further examination of the effectiveness and biases connected with different preservatives and extraction methods when studying challenging host-associated microbiomes.
The purpose of this study was to compare preservation and extraction methods across various species of deep-water (Lophelia pertusa and Paragorgia johnsoni) and shallow-water corals (Montastraea cavernosa, Porites astreoides, and Stephanocoenia intersepta) with disparate life-history traits, including soft and stony corals, and zooxanthellate and azooxanthellate species (see Table 1). We tested three preservative types commonly used for environmental microbiome preservation (RNAlater, DNA/RNA Shield, and liquid nitrogen), combined with two different extraction kits (the Qiagen PowerBiofilm kit and the Promega Maxwell RBC kit with modifications), and identified the effectiveness and associated biases.
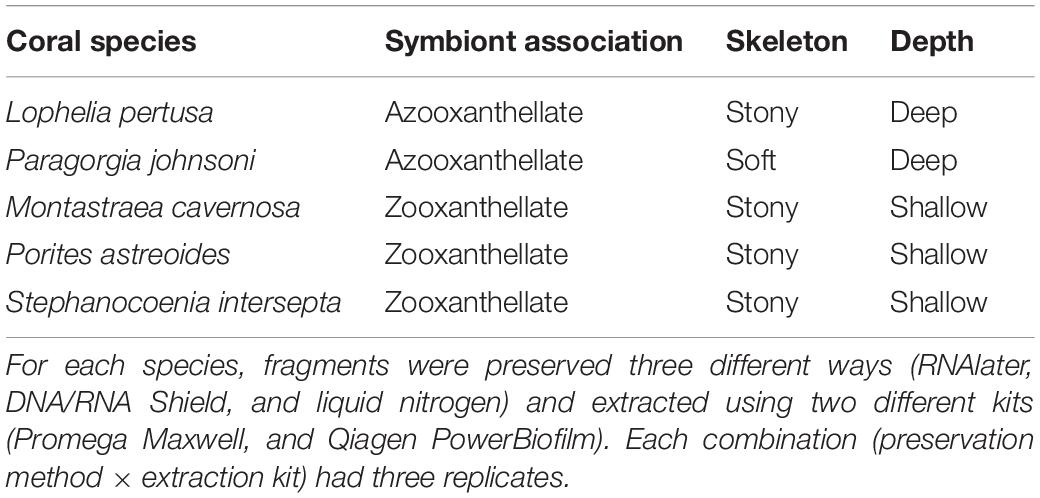
Table 1. Coral species sampled and their associated life-history traits.
Materials and Methods
Sample Collection
Five coral species, three shallow-tropical and two deep-water, were collected for this experiment. Details of life-history traits and characteristics of each coral are given in Table 1. Tropical, zooxanthellate corals were collected in March 2018 from the Florida Keys National Marine Sanctuary coral nursery, which was temporarily housed at the Mote Marine Laboratory at that time. Note that these corals were being maintained in water tables and therefore the microbiomes are expected to reflect some level of tank effect and may not represent the same bacterial diversity that would be observed on the reef. These corals were sampled under permit #FKNMS-2017-064. Species included Montastraea cavernosa, Porites astreoides, and Stephanocoenia intersepta. A single colony (approximately 10 cm2) of each species was placed into a sterile aluminum weigh boat and fragmented using a sterile hammer and chisel. Small fragments were rinsed with sterile 1× PBS (phosphate-buffered saline) to remove any loosely adhered microbes prior to being placed into preservative.
Deep-sea, azooxanthellate corals Lophelia pertusa and Paragorgia johnsoni were collected during the DeepSEARCH research cruise on the R/V Atlantis using the deep submergence vessel Alvin in August, 2018. Permits are not required for deep-sea coral collections, however, these sample collections were made under Scientific Research Letters of Acknowledgment (LOA) from the National Oceanic and Atmospheric Administration’s National Marine Fisheries Service, both the Greater Atlantic Regional Fisheries Office and the Southeast Regional Office. Individual polyvinyl chloride (PVC) sample quivers were cleaned with ethanol prior to each dive, filled with freshwater, and sealed with rubber stoppers. Coral branches were collected and placed into the quiver (one coral per container) after ambient seawater had evacuated the freshwater and then the quivers were resealed at depth. In the ship’s laboratory, coral samples were removed from the quiver using sterile forceps and rinsed with sterile 1× PBS to remove any loosely adhered microbes. The coral piece was placed into a sterile aluminum weigh boat and either flame-sterilized shears (P. johnsoni) or a flame-sterilized hammer was used to fragment pieces. With the exception of L. pertusa, all coral samples (18 per species) were generated from a single colony per species, in order to eliminate genetic variation as a confounding factor to the study. Three colonies (potential genets) of L. pertusa were collected from the same geographical region on the same dive and distributed evenly across all treatments since there was not sufficient biomass from a single collection.
For each coral species, the fragments generated were preserved in one of three ways. One third were transferred to sterile cryovials containing RNAlater® RNA Stabilization Solution (AmbionTM), one third were transferred to sterile cryovials containing DNA/RNA ShieldTM (Zymo Research), and the last third were double-bagged in two ounce sterile Whirlpak® bags and snap frozen in liquid nitrogen. Fragments were evenly distributed across each preservation method, for a total of six fragments per species per preservation method. Samples preserved with RNAlater or DNA/RNA Shield were kept at 4°C for approximately 24 h until they were transferred to a −20°C freezer, where they remained until further processing. Samples preserved in liquid nitrogen remained in liquid nitrogen until they were transferred to a −80°C freezer, where they remained until further processing.
DNA Extraction
Fragments that were preserved in RNAlater or DNA/RNA Shield were thawed and rinsed with sterile 1× PBS to remove excess preservative. Coral fragments were then crushed using a sterile hammer and placed into the appropriate DNA extraction tube according to each extraction method. Two different extraction methods were tested, the Promega Maxwell® RSC Blood DNA kit and the Qiagen DNeasy® PowerBiofilm kit. Each unique treatment combination (species × preservative × extraction kit) had three replicates.
DNA extracted using the Maxwell RSC kit followed the procedure used by Galand et al. (2018). Briefly: the large ceramic bead was removed from Lysing Matrix A tubes (mpBio) using forceps that had been cleaned with DNA-Away (Thermo Scientific) and then flame-sterilized. Coral samples were added to the tubes with 300 μL of sterile nuclease-free water and placed in a cold-block to keep them chilled. For the bead-beating step, the tubes were then agitated in a FastPrep machine for 20 s at setting 6.5 and then returned to the cold-block to cool. Each tube then received 300 μL of the Promega Maxwell kit’s DNA lysis buffer and 25 μL Proteinase K. Tubes were mixed by inverting three times and then incubated for 1 h in a 56°C water bath. After incubation, the tubes were again inverted 3 times to mix, followed by centrifugation at 2,500 rpm for 30 s. The supernatants were transferred to the front wells of the Promega Maxwell cartridge. The automated cartridge was set up and run following the manufacturer’s instructions. Samples were processed by coral species (i.e., all nine P. astreoides samples were run using a single cartridge) and a 15 min UV-sanitize program was run between cartridges to sterilize the Promega Maxwell instrument.
DNA extracted with the PowerBiofilm kit was extracted according to the manufacturer’s protocol with the substitution of bead-beating in a FastPrep machine running at setting 5 in place of the Powerlyzer 24 at 3200 rpm. To evaluate potential kit contamination, two kit blank samples were processed for each extraction method; for the Maxwell kit, one blank was run with 300 μL of sterile nuclease-free water (KB1PM) and one blank was run with 300 μL of sterile 1× PBS (KB2PM); for the PowerBiofilm kit, one blank was run with 200 μL of sterile nuclease-free water (KB1MB) and one blank was run with 200 μL of sterile 1× PBS (KB2MB).
Amplification and Sequencing
Genomic DNA from the 90 coral samples and 4 kit extraction blanks was sent to Glomics Inc. (Norman, OK, United States) for all PCR amplification and sequencing. All samples were diluted to 2 ng/μL and bacterial and archaeal V4 small subunit 16S rRNA gene fragments were first amplified in triplicate from genomic DNA using the primer set 515F: GTGCCAGCMGCCGCGGTAA and 806RB: GGACTACNVGGGTWTCTAAT (Caporaso et al., 2012; Apprill et al., 2015). A second reaction appended unique forward and reverse barcodes to pooled triplicate reactions. All reactions were 25 μL and comprised of an in-house buffer and taq polymerase, 1 μL each forward and reverse primer (10 μM), and 5 or 15 μL template DNA for the 515F/806RB and barcoding reactions, respectively. PCR conditions for both reactions were an initial denaturation at 94°C for 1 min, followed by 11 (515F/806RB) or 22 (barcoding) cycles of 94°C for 20 s, 53°C for 25 s, 68°C for 45 s, and a final extension at 68°C. Barcoded amplicons were pooled in equal-molar concentration for sequencing and purified using Ampure XP beads. PCR products were run out on a 1% agarose gel, and the target band purified with the QIAGEN Gel extraction kit. Amplicons were sequenced using Illumina MiSeq technology with v2 chemistry and 2 × 250 cycles, with the final library spiked with 12.5% PhiX. To assess amplification bias and sequencing error rate, a Glomics-supplied mock community positive control was amplified and sequenced as described above on the same sequencing run (Supplementary Table 1). The number of 16S rRNA gene copies per mock community member were identified using rrnDB version 5.6 (Stoddard et al., 2015). All raw fastq files are publicly available via USGS data release (Kellogg et al., 2021) and from NCBI under Bioproject PRJNA544686.
Sequence Analysis
Sequences were imported into QIIME2 (Bolyen et al., 2019) and trimmed, merged, and sorted into amplicon sequence variants (ASVs) using DADA2 (Callahan et al., 2016) and the parameters ‘–p-trim-left-f 25 –p-trim-left-r 25 –p-trunc-len-f 200 –p-trunc-len-r 200.’ Taxonomy was assigned using the Silva database release silva-132-99-515-806. All ASVs assigned as “mitochondria” or “chloroplast” were removed. Sequences identified only to the domain “Bacteria” were searched against NCBI’s nr database using the Basic Local Alignment Search Tool (BLAST; Altschul et al., 1990) and any sequences identified as eukaryotic in origin were subsequently removed from the dataset. “Bacteria” ASVs identified as coral mitochondria were further analyzed as a proxy for contamination and PCR/sequencing error. Samples comprising more than 1% mock community ASVs were removed from the dataset, including the mock community and three kit extraction blanks (Supplementary Figures 1, 2). One of the kit extraction blanks (KB1MB) only had three sequence reads and therefore was removed. Of the original 90 coral samples, 72 passed quality controls and rarefaction (Table 2). The final sample ASV table was then rarefied to 5,203 sequences (Supplementary Table 2).
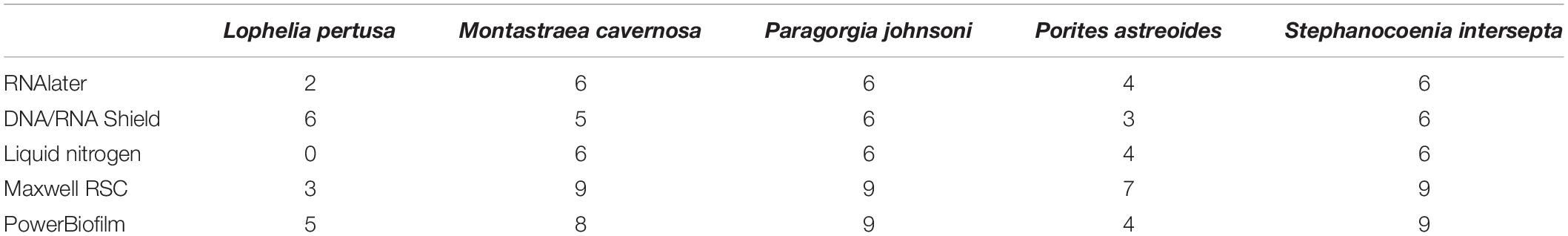
Table 2. Number of samples that passed quality control and rarefaction depth for each combination of coral species, preservative (RNAlater, DNA/RNA Shield, and liquid nitrogen), and extraction method (Maxwell RSC and PowerBiofilm).
Diversity Analysis
Alpha and beta diversity metrics were calculated for all samples that passed through all quality controls (described above) using the QIIME2 diversity ‘core-metrics-phylogenetic’ wrapper. For alpha diversity, this included observed ASVs, Shannon diversity index, Pielou’s evenness, and Faith’s Phylogenetic Diversity. For beta diversity, this included Bray–Curtis (Bray and Curtis, 1957), Jaccard (Jaccard, 1908), weighted and unweighted UniFrac distance matrices (Lozupone and Knight, 2005). A permutational multivariate analysis of variance (PERMANOVA; Anderson, 2001) was conducted for each alpha diversity metric using the ‘diversity alpha-group-significance’ plug-in, and an analysis of similarities (ANOSIM; Clarke, 1993) was conducted for each beta diversity metric using the ‘diversity beta-group-significance’ plug-in. Diversity data were imported into R using the qiime2R package for figure generation. Finally, an unrarefied table was imported into R using the package qiime2R, converted to a phyloseq object (McMurdie and Holmes, 2013), and then significantly different ASVs detected for each combination of coral species, preservative, and extraction method using DESeq2 (Love et al., 2014). All analyses were conducted for all coral species pooled, and then individually for each species.
Results
Five species of corals (Table 1) were preserved using three treatments (n = 6 per species, per treatment): RNAlater, DNA/RNA Shield, and liquid nitrogen. Three replicates per preservative were then processed via each of two extraction methods: Qiagen’s PowerBiofilm kit and Promega’s Maxwell RBC kit with modifications.
Diversity Metrics-Coral Species
Not surprisingly, significant differences in alpha diversity metrics were found between coral species, particularly between deep- and shallow-water corals (Figure 1). Deep-water corals had a significantly lower number of observed ASVs (Figure 1 and Supplementary Figure 3) and Faith’s phylogenetic diversity (p < 0.001). The average number of observed ASVs in L. pertusa (53) and P. johnsoni (50) was an order of magnitude below the zooxanthellate corals: M. cavernosa (532), S. intersepta (386), and P. astreoides (376).
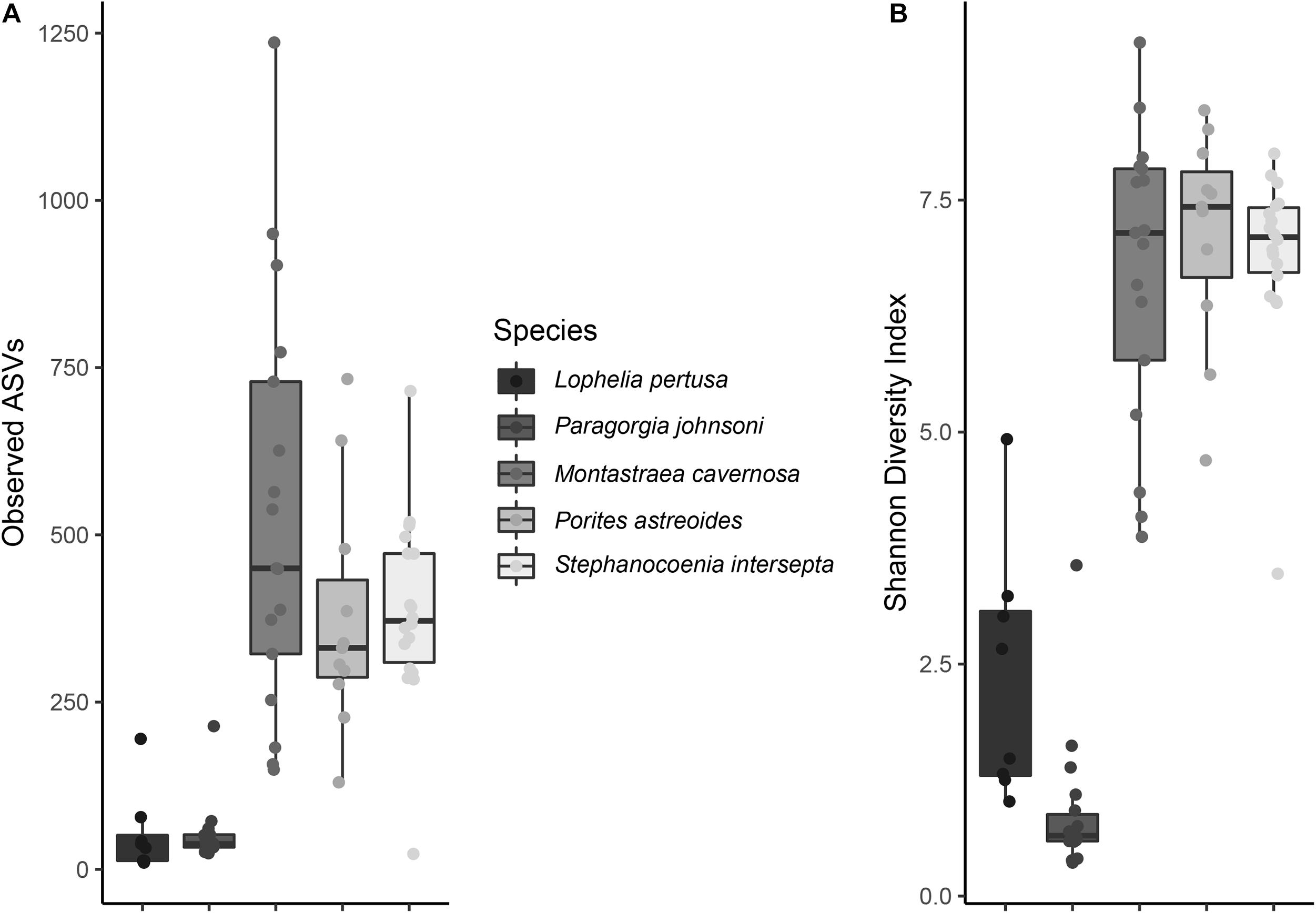
Figure 1. (A) Observed amplicon sequence variants (ASVs) and (B) Shannon diversity index values for all five coral species. All preservatives (RNAlater, DNA/RNA Shield, and liquid nitrogen), and extraction methods (PowerBiofilm and Maxwell RSC) are pooled.
No significant differences existed among deep-water groups or shallow-water groups for observed ASVs or Faith’s phylogenetic diversity. Evenness was significantly different between all corals (p < 0.001), with the exception of M. cavernosa and P. astreoides (p = 0.057), M. cavernosa and S. intersepta (p = 0.156), and P. astreoides and S. intersepta (p = 0.066).
Shannon diversity index was significantly different between all corals (p < 0.01), with the exception of M. cavernosa and P. astreoides (p = 0.690), M. cavernosa and S. intersepta (p = 0.869), and P. astreoides and S. intersepta (p = 0.403). Shannon diversity indices are shown in Figure 1.
When all species were analyzed together, samples clustered strongly according to coral species rather than extraction method or preservative type, independent of the beta diversity metric used (Supplementary Figure 4). Large differences were also seen between coral species and community composition (Figure 2), with the shallow-water corals possessing more Alphaproteobacteria, while P. johnsoni was completely dominated by Mollicutes and L. pertusa being primarily composed of Gammaproteobacterial and unknown bacteria sequences.
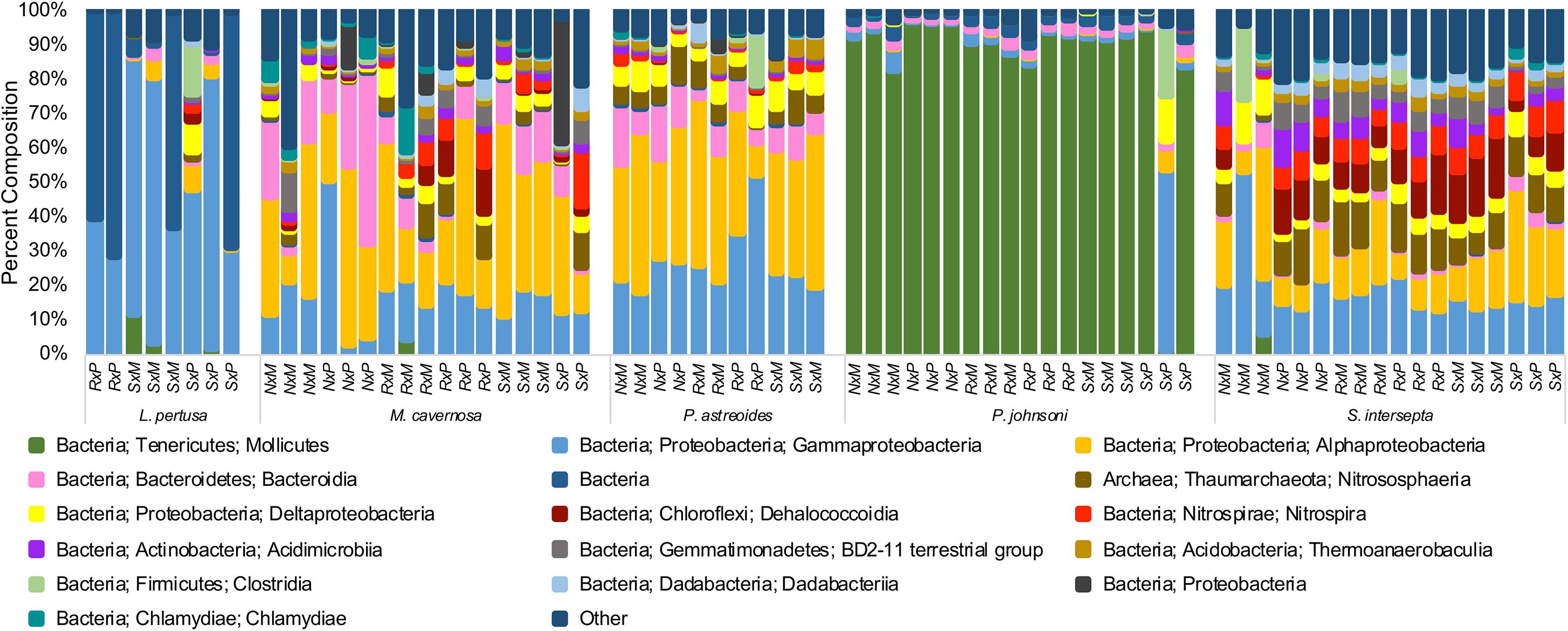
Figure 2. Taxonomic composition of bacterial communities from five coral species, Lophelia pertusa, Montastraea cavernosa, Porites astreoides, Paragorgia johnsoni, and Stephanocoenia intersepta. Preservative abbreviations: R, RNAlater; S, DNA/RNA Shield; N, liquid nitrogen. Extraction method abbreviations: M, Maxwell RSC; P, PowerBiofilm. The single P. johnsoni sample not primarily composed of Mollicutes was removed as an extreme outlier in other analyses. Several groups could not be resolved to the class level.
Quality Control
All coral samples were found to contain from one to four mitochondrial sequences that matched to a different coral than the sample host, with negative controls (extraction kit blanks for each method) also containing coral mitochondrial sequences from up to five different species (Supplementary Figure 1). A mock community of known bacteria was amplified and sequenced in the same manner as the coral samples and found to contain mitochondrial sequences representing two coral species. There is sequence homology between bacterial and mitochondrial 16S ribosomal genes, which sometimes causes coral mitochondrial DNA to be amplified by “bacterial-specific” primers (Galkiewicz and Kellogg, 2008). This, combined with cross-talk or sample bleeding during amplification and sequencing (Mitra et al., 2015; Wright and Vetsigian, 2016), resulted in the detection of coral mitochondrial ASVs in the mock community and other samples.
Amplicon sequence variants (ASVs) for all 16 mock community members were correctly identified, although resolution was not precise for many (Supplementary Table 1). It was confirmed that mock community ASVs that classified to the genus or species level only produced one sequence variant. All samples were assessed for mock community contamination and the lower biomass samples such as the extraction kit blanks were particularly dominated (96.2–99.8% relative abundance; Supplementary Figure 2 and Supplementary Table 3). Additionally, the mock community and kit extraction blanks had 0.19–3.6% relative abundance of what are likely cross-talk microbial ASVs from coral samples; these ASVs were also present in two or more coral samples and had read numbers of one to two orders of magnitude higher than the control reads (Supplementary Table 3). The relative abundance of likely kit contaminants was extremely small (less than 0.2% relative abundance; Supplementary Table 3). For quality control, any samples containing over 1% of mock community members (combined) were removed from the data set. All negative control (extraction blank) samples did not pass the quality control parameters and were removed, along with 18 of the 90 coral samples (Table 2). Lophelia pertusa had the most samples removed (10), followed by P. astreoides (7) and M. cavernosa (1). Zero samples were removed for P. johnsoni and S. intersepta. The total number of observed ASVs per sample after quality control and rarefaction (Supplementary Table 2) ranged from 10 to 1,236, with an average of 298 (standard deviation ± 264).
Diversity Metrics-Preservatives
When all five coral species (Table 1) were pooled together for statistical analysis, no significant differences were found between preservative type (RNAlater, DNA/RNA Shield, or liquid nitrogen) for any alpha diversity metric (Table 3). When examined by individual coral species, differences between preservation method varied greatly between coral species and the alpha diversity metric used (Table 3), although the only significant difference detected was when comparing evenness between RNAlater and DNA/RNA Shield in S. intersepta. However, in all significant or lower p-value cases, DNA/RNA Shield had the higher alpha diversity metric compared to both RNAlater and liquid nitrogen (lower p-values are reported here in consideration for the small sample sizes per treatment). Comparisons between liquid nitrogen and RNAlater were less consistent and different between species, although none of these comparisons were significantly different. Similarly, the number of significantly different ASVs (as detected by DESeq2) varied between coral species and preservative type (Table 4 and Supplementary Table 4). For example, when comparing DNA/RNA Shield to liquid nitrogen, S. intersepta had 87 significantly different ASVs between the two preservation methods, while P. johnsoni had none. In general, there were more significantly different ASVs when comparing RNAlater and DNA/RNA Shield to liquid nitrogen. There was no clear pattern as to specific taxa that were significantly more abundant among all coral species and treatments, although some taxa such as SAR202 appear to be less abundant in liquid nitrogen preserved samples compared to the other two preservation methods (Supplementary Table 4). For all beta diversity metrics, no significant differences were detected between preservation methods (Table 5). This was also reflected in the principal coordinate plots of individual species, which showed little clustering by way of preservation (Figure 3).
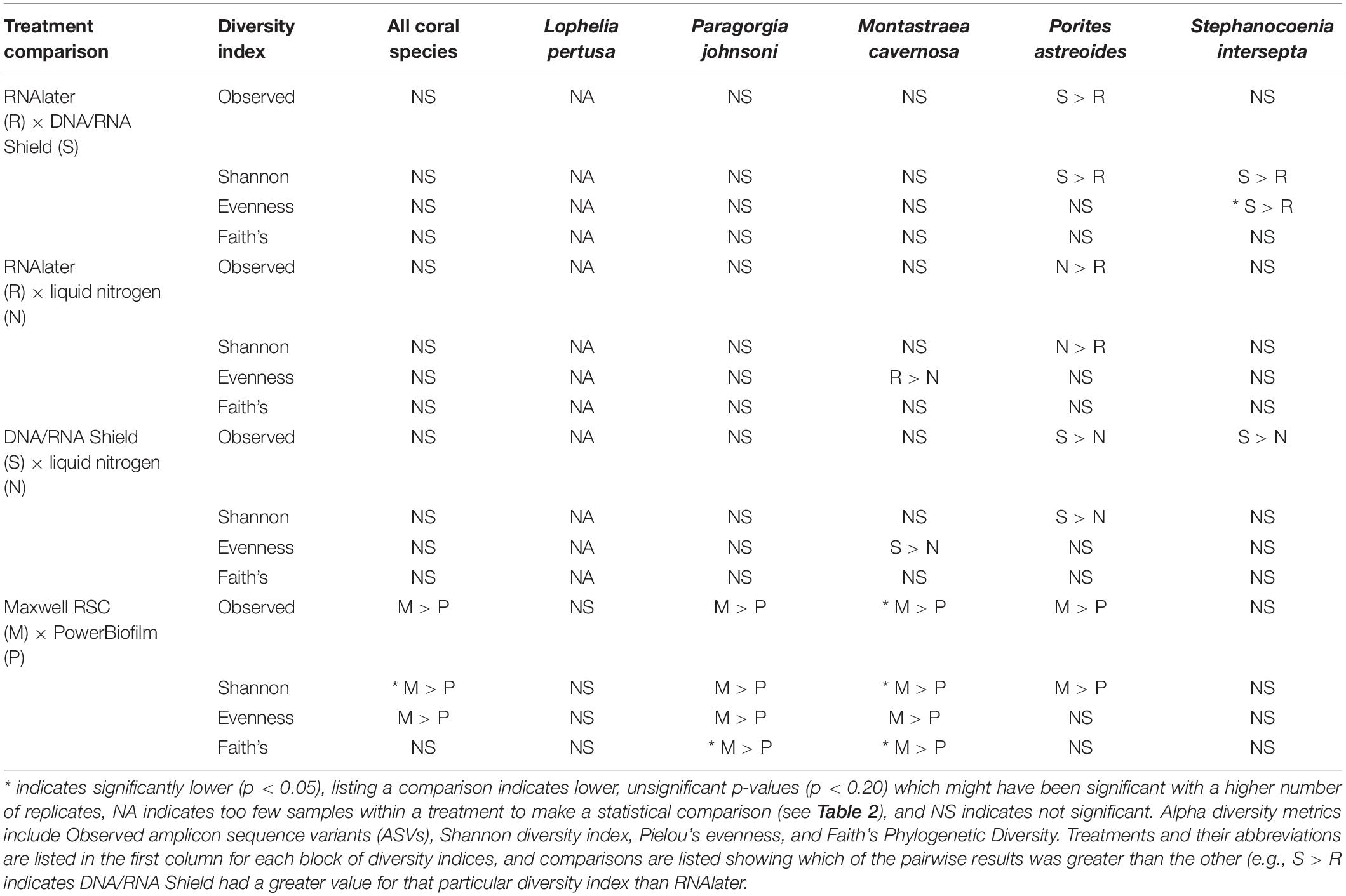
Table 3. Pairwise results for each permutational multivariate analysis of variance (PERMANOVA) for all alpha diversity metrics, coral species, extraction methods (PowerBiofilm, Maxwell RSC), and preservatives (RNAlater, DNA/RNA Shield, liquid nitrogen).
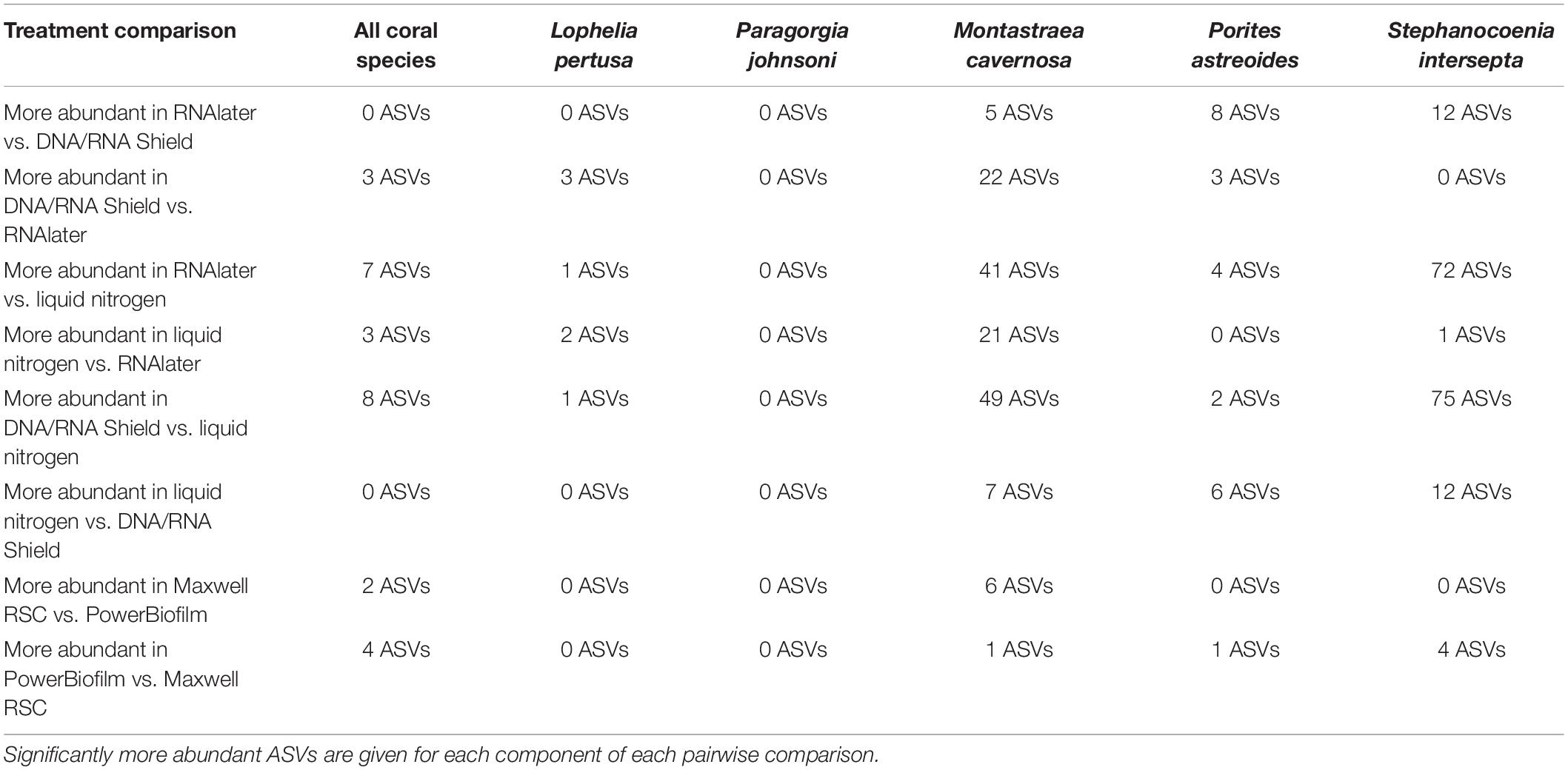
Table 4. Number of significantly different amplicon sequence variants (ASVs) as determined by DESeq2 using an unrarefied ASV table.
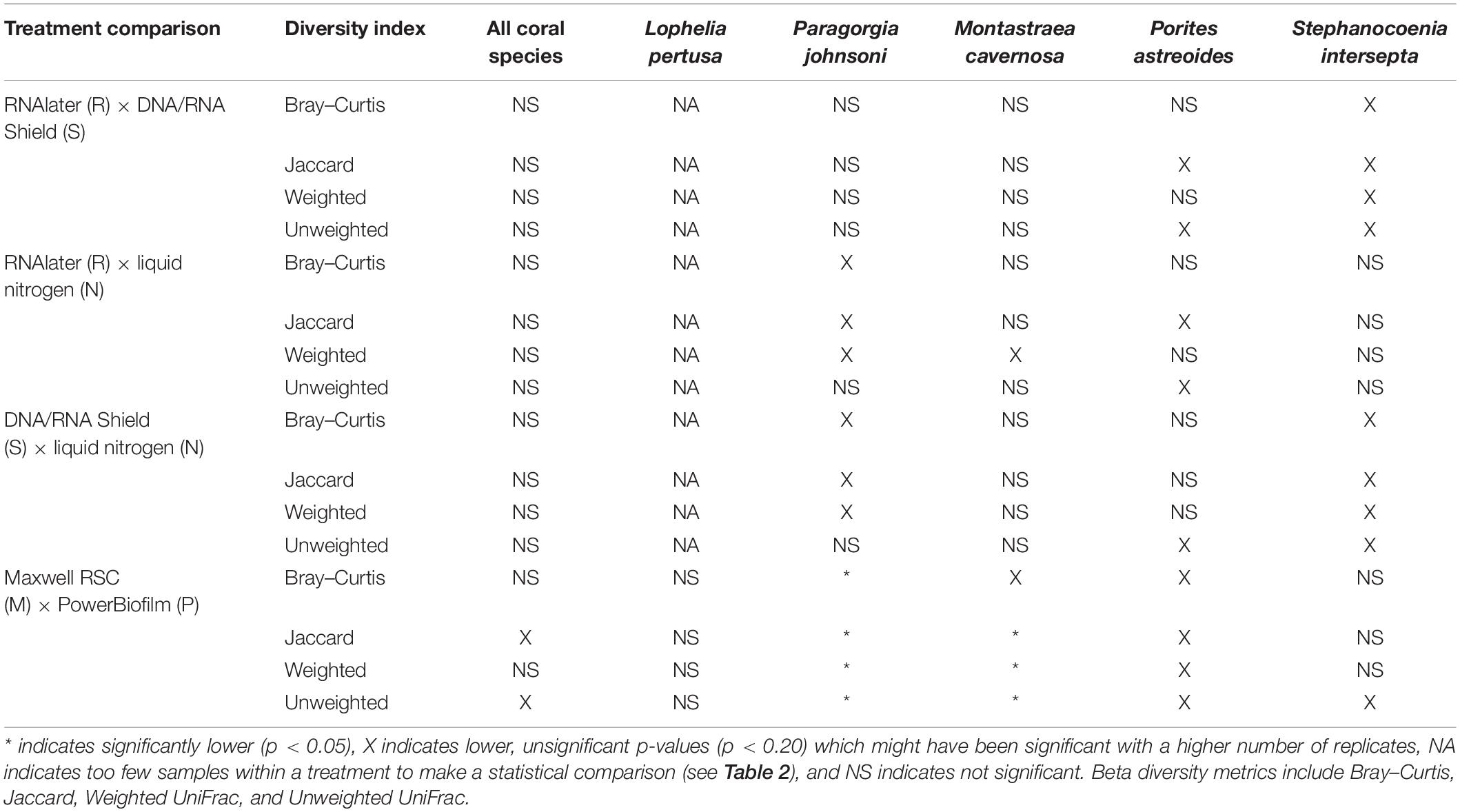
Table 5. Pairwise results for all analysis of similarities (ANOSIM) for all beta diversity metrics, coral species, extraction methods (PowerBiofilm, Maxwell RSC), and preservatives (RNAlater, DNA/RNA Shield, liquid nitrogen).
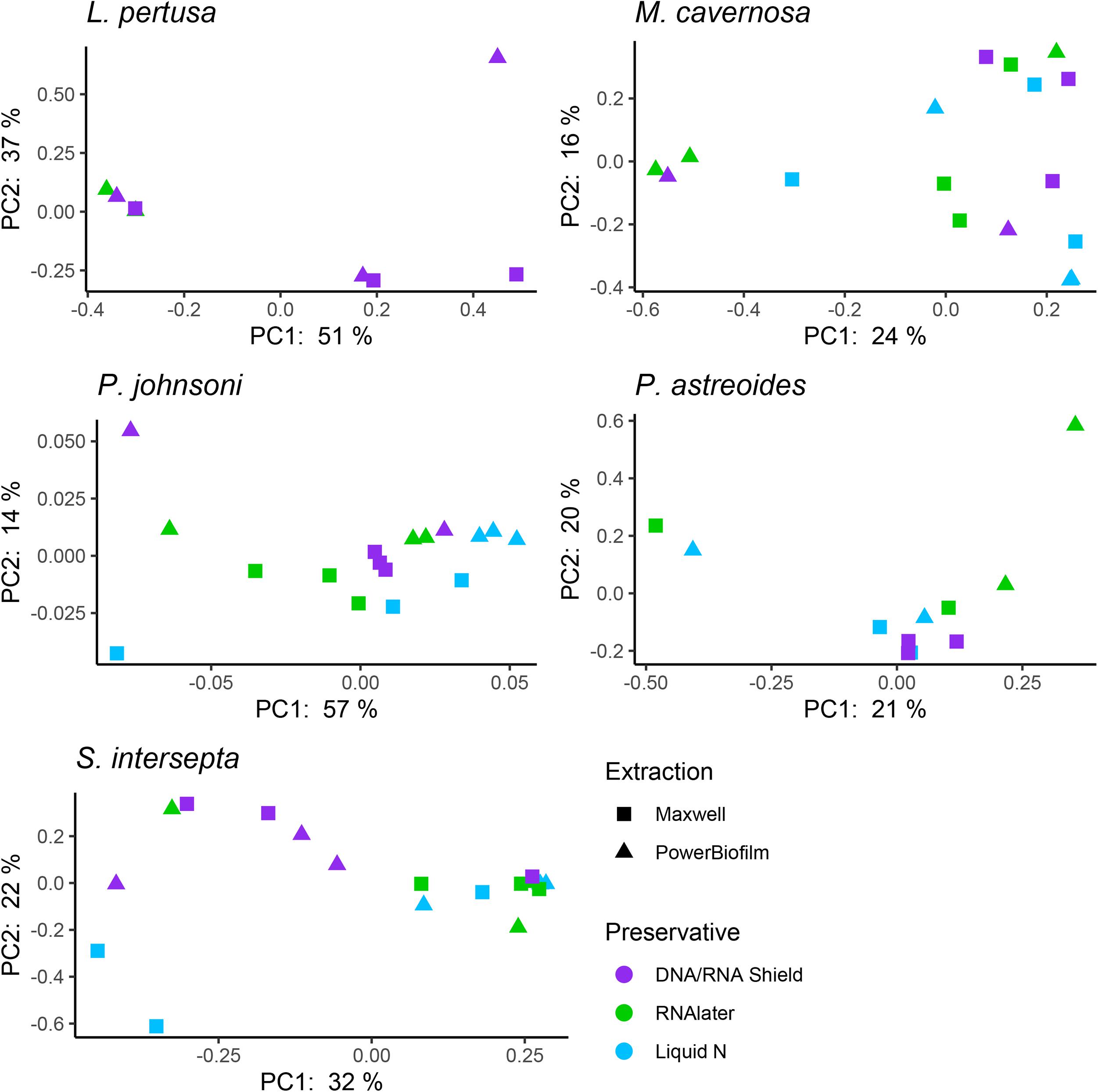
Figure 3. Principal coordinate plots based upon a Bray–Curtis dissimilarity matrix of individual coral species Lophelia pertusa, Paragorgia johnsoni, Montastraea cavernosa, Porites astreoides, and Stephanocoenia intersepta. Strong clustering is not observed for preservative or extraction, but some significant differences were detected through an analysis of similarities (ANOSIM; Table 5).
Diversity Metrics-Extraction Method
Compared to the preservative used, more significant differences were detected according to extraction method, although there was substantial variation in differences depending upon coral species and the alpha diversity metric used (Table 3). In three of the five corals, as well as for all coral species pooled, the Maxwell RBC extraction method exhibited higher alpha diversity metrics than the PowerBiofilm method (Table 3). However, the extraction method used also demonstrated far fewer significantly different ASVs when compared to preservative type (Table 4). Beta diversity metrics were also significantly different or had lower p-values for most coral species (Table 5), although principal coordinate plots for individual species of corals did not demonstrate any strong clustering according to extraction method (Figure 3). Paragorgia johnsoni was the only coral to have all four beta diversity metrics show significant differences and there is some indication of the extraction methods being separated on the PC2 axis of Figure 3. It should be noted that for all comparisons, the lack of significant differences detected in the L. pertusa samples is likely due to the small number of L. pertusa samples passing quality control (0 for liquid nitrogen treatment and 2 for RNAlater; Table 2).
Discussion
The Shannon diversity index values for L. pertusa are consistent with findings from other studies (Meistertzheim et al., 2016; Kellogg et al., 2017; Jensen et al., 2019; Chapron et al., 2020) and the value for P. johnsoni is consistent with that calculated for congener P. arborea (Jensen et al., 2019). The shallow-water coral values were markedly higher than values recorded in the literature (Morrow et al., 2012; Glasl et al., 2019). This may be a result of their maintenance in a circulating water table since it is known that captivity affects coral microbiomes; however, it is typically the wild corals that have higher diversity rather than the tank corals (Kooperman et al., 2007; Röthig et al., 2017).
Results indicate that there is no single combination of preservative (RNAlater, DNA/RNA Shield, or liquid nitrogen) and extraction method (PowerBiofilm or Maxwell RBC kits) that was superior to the others across all coral species. Subtle yet significant differences were detected for both preservation types and extraction methods but were dependent upon statistical analysis used and were different for each coral species. For example, the combination of DNA/RNA Shield and PowerBiofilm performed best for Lophelia samples in terms of samples that sequenced well enough to pass quality controls and rarefaction (Table 2). Further, DNA/RNA Shield resulted in a slightly higher alpha diversity for Lophelia and Stephanocoenia, indicating that it preserved more bacterial taxa. However, for Porites astreoides, the Maxwell RSC method resulted in a higher number of samples passing quality controls and rarefaction, suggesting that regardless of preservation, this extraction method might be more effective for this particular coral. In Stephanocoenia, the combination of preservation in DNA/RNA Shield with extraction via Maxwell resulted in significantly lower detection of Neisseriaceae, Rhodospirillum rubrum, and Enterobacteriaceae (Supplementary Table 4). For P. astreoides, the combination of DNA/RNA Shield with Maxwell resulted in a significantly lower detection of Gammaproteobacteria JTB23 (Supplementary Table 4). For the rest of the coral species, no clear trends were apparent (Table 2 and Supplementary Table 4). Additionally, statistical comparison of alpha and beta diversity metrics detected more significant differences between the extraction methods, while the majority of significantly different ASVs were identified between preservative types.
No significant differences were detected between any alpha or beta diversity metrics and preservative, although some combinations produced a lower p-value that may be significant with more statistical power. In similar studies that examined the effect of preservation type on the human fecal microbiome, RNAlater exhibited the largest shift from the true microbial community among various preservative types, some including DNA/RNA Shield (Dominianni et al., 2014; Choo et al., 2015; Chen et al., 2019). However, these studies all stored RNAlater-preserved samples at room temperature, and samples preserved in RNAlater and kept under cold conditions (4 or −20°C) do not exhibit this change (Hallmaier-Wacker et al., 2018). This may also be due to differences in relative penetration of RNAlater into the sample, with fecal samples making it difficult for RNAlater to rapidly penetrate the bacterial cells for preservation (Chen et al., 2019). Although we cannot know the true composition of the microbial communities in our study, samples preserved in RNAlater were kept under cold conditions and were most similar to samples preserved in DNA/RNA Shield, indicating minimal shifts in the detected microbial community. The majority of significantly different ASVs were higher in either RNAlater or DNA/RNA Shield compared to liquid nitrogen, suggesting a loss of diversity in the liquid nitrogen preserved samples (Supplementary Table 4). Notable across all coral species, liquid nitrogen preservation resulted in significantly lower detection of SAR202 clade, Nitrospinae, and uncultured Syntrophobacterales; however, individual coral species had distinct patterns (Supplementary Table 4). Also across all coral species, RNAlater preservation resulted in significantly lower detection of Rhodobacteraceae and Chlamydiales (Supplementary Table 4). However, no clear patterns could be determined in the significantly different ASVs. Regardless, we suggest that coral samples be placed in a preservative such as RNAlater or DNA/RNA Shield and stored under cold conditions (4 or −20°C) whenever possible.
In agreement with prior coral methodology studies (Baker and Kellogg, 2014; Weber et al., 2017), we did not detect any drastic difference between extraction kits when applied to five different coral species, although more subtle differences were detected. This is likely because each of the extraction kits in these studies implemented a bead-beating step. Across the extraction methodology literature, bead-beating protocols consistently result in higher alpha-diversity metrics than non-bead-beating methods, indicating that bead-beating is the best uniform method for cell lysis (de Lipthay et al., 2004; Willner et al., 2012; Yuan et al., 2012; Djurhuus et al., 2017; Lim et al., 2018; Pollock J. et al., 2018), particularly in biofilm communities such as the oral microbiome (Abusleme et al., 2014). Both extraction methods analyzed in our study had a bead-beating step, which we hypothesize to be why there were very few significant differences between extraction methods for all corals and diversity metrics analyzed. Both extraction methods pair chemical lysis with mechanical lysis (bead-beating), with the proprietary differences being their purification steps: The Maxwell RBC kit uses a paramagnetic particle and cellulose-based binding of nucleic acids during washing steps to purify DNA, while the Qiagen PowerBiofilm kit employs “Inhibitor Removal Technology (IRT)” to remove inhibitors such as humic acid, metals, and salts during purification. In all significant cases, the Maxwell RBC kit and its modified extraction method resulted in a higher alpha diversity metric than the Qiagen PowerBiofilm kit. It is therefore recommended that all extraction protocols for coral or similar samples contain a bead-beating step to increase the level of uniformity among methodologies (Pollock J. et al., 2018).
The significant differences between preservation type and extraction method were a far second to the differences detected between coral species, suggesting that comparisons between coral species will be less impacted with differing preservation and extraction techniques than comparisons within the same coral species. Similarly, differences between individual human fecal samples were far more significant than those found between preservative or extraction methods (Dominianni et al., 2014; Choo et al., 2015; Wagner Mackenzie et al., 2015; Gill et al., 2016; Chen et al., 2019). Cruaud et al. (2014) also found that geographical differences between deep-sea sediment sites explained the majority of inter-sample variation compared to extraction method. The sum of these studies suggests that different preservation types and extraction methods may affect comparisons when looking for subtle differences between microbial communities. However, those studies that are between species, geographic locations, and even individuals are less affected by small variations in preservative and extraction method.
Differences in apparent microbial composition of mock communities can occur depending upon the 16S region targeted (Sipos et al., 2007; Klindworth et al., 2012; Chen et al., 2019). These apparent differences might be even more profound in deep-sea corals because the 515/806 V4 primers (Caporaso et al., 2012; Apprill et al., 2015) have been shown to be more sensitive to Fusobacteria and Mollicutes in the fecal microbiome, and P. johnsoni in this study and L. pertusa in other studies (Kellogg et al., 2009) are almost entirely composed of Mollicutes. Indeed Cruaud et al. (2014) and Fouhy et al. (2016) show that primer choice has a far deeper influence on microbial community than extraction method, although they did compare two bead-beating methods so the differences would be minimized. Primer choice is also particularly important, as some bacterial 16S primers are known to amplify coral mitochondrial DNA (Galkiewicz and Kellogg, 2008; Weber et al., 2017; Pollock F. J. et al., 2018; Meyer et al., 2019). As shown in our study, using the 515/806 V4 primers, L. pertusa may be particularly prone to mitochondrial amplification compared to the other coral species, resulting in fewer samples making it through quality control. This has also been reported for other coral species such as Porites lobata and Pocillopora verrucosa (Weber et al., 2017; Sonett et al., 2021). Thus far, the coral microbiology community has employed a variety of solutions to combat this molecular hurdle: (1) deeper sequencing, to allow for discarding the mitochondrial bycatch (Rosales et al., 2019); (2) agarose gel size separation of the 16S rRNA band from the coral mitochondrial band (Apprill et al., 2016); (3) alternative primer sets (Galkiewicz and Kellogg, 2008; Bayer et al., 2013; Liang et al., 2017); and (4) PNA clamps (Reigel et al., 2020). Further, the magnitude of under-annotation of coral mitochondria by existing bioinformatic taxonomies has recently been quantified and in silico improvements offered (Sonett et al., 2021).
No studies that compared preservation and extraction methods with known community compositions (either by immediate freezing of samples at −80°C or known mock communities) were able to identify methodologies that returned the expected microbial communities (Willner et al., 2012; Yuan et al., 2012; Gray et al., 2013; Dominianni et al., 2014; Brooks et al., 2015; Choo et al., 2015; Fouhy et al., 2016; Hallmaier-Wacker et al., 2018; Chen et al., 2019), confirming that no methodology is perfect. Yet, the addition of a mock community allowed us to improve our data quality control (Pollock J. et al., 2018). The three extraction blanks that passed rarefaction demonstrated the highest numbers of mock community ASV contamination, suggesting that samples with low (or no) DNA yields are more prone to cross-talk contamination (Supplementary Table 3), a concept that has been previously reported (Willner et al., 2012; Fouhy et al., 2016; Vohsen et al., 2020). This could also apply to samples that have high coral-to-microbe DNA ratios (i.e., low microbial DNA, yet high DNA yields from coral and zooxanthellae algal symbionts). Additionally, the lack of consistently present ASVs according to preservation type or extraction kit across all coral species and the unexpected presence of multiple coral mitochondrial sequences indicates that contamination is a result of cross-talk during amplification and sequencing, rather than introduction by reagent contamination (Salter et al., 2014; Glassing et al., 2016). Similar cross-contamination revealed by the inclusion of mock community analysis have also been shown by Willner et al. (2012). The high number of coral mitochondrial sequences detected in this study, as well as cross-talk between primers, emphasize the importance of assessing each dataset by hand, in order to produce the highest quality dataset possible, and the sequencing of technical replicates with independent barcodes could allow researchers to assess the level of cross-talk among samples and improve quality control.
Our study suggests that methodology is important in assessing an environmental microbial community, but components such as preservation method and extraction method may be less important than components such as 16S rRNA gene region and the inclusion of a mock community to allow for the assessment of bias and contamination (Brooks et al., 2015). Every combination of preservative and extraction method is going to introduce biases. However, molecular methods are rapidly evolving. Testing and comparisons of continually evolving preservation and extraction methods should be ongoing, as no combination of methods is ever going to be without bias. However, the sum of these biases does not exceed the differences between individuals or species and should not prevent comparisons across extraction methods and preservatives, with careful acknowledgment of the biases in each study.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA544686 and http://dx.doi.org/10.5066/P96GBWDM, USGS Data Release.
Author Contributions
CK designed the experiment, collected and processed the samples, and wrote sections of the manuscript. ZP performed the data analysis and wrote the first draft of the manuscript. Both authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the U.S. Geological Survey’s Environments Program.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank chief scientist, Dr. Erik Cordes of Temple University, as well as the science party, master, and crew of the R/V Atlantis and DSV Alvin during the DeepSEARCH (AT-41) cruise. We would also like to thank Dr. Leila Meistertzheim for encouragement to try the Promega Maxwell extraction system and discussions about the experiment; Sara Snader for assistance in the lab; and Dr. Dawn Goldsmith for compiling the associated USGS data release and submission of sequencing data and metadata to NCBI. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the U.S. Government.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.684161/full#supplementary-material
References
Abusleme, L., Hong, B.-Y., Dupuy, A. K., Strausbaugh, L. D., and Diaz, P. I. (2014). Influence of DNA extraction on oral microbial profiles obtained via 16S rRNA gene sequencing. J. Oral Microbiol. 6:23990. doi: 10.3402/jom.v6.23990
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410.
Anderson, M. J. (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26, 32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x
Apprill, A. (2017). Marine animal microbiomes: toward understanding host-microbiome interactions in a changing ocean. Front. Mar. Sci. 4:222. doi: 10.3389/fmars.2017.00222
Apprill, A., McNally, S., Parsons, R., and Weber, L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquatic Microb. Ecol. 75, 129–137. doi: 10.3354/ame01753
Apprill, A., Weber, L. G., and Santoro, A. E. (2016). Distinguishing between microbial habitats unravels ecological complexity in coral microbiomes. mSystems 1, e143–e116. doi: 10.1128/mSystems.00143-16
Baker, E. J., and Kellogg, C. A. (2014). Composition of three DNA extraction kits to establish maximum yield and quality of coral-associated microbial DNA: U.S. Geological Survey Open-File Report 2014–1066. doi: 10.3133/ofr20141066 (accessed June 29, 2021).
Bayer, T., Neave, M. J., Alsheikh-Hussain, A., Aranda, M., Yum, L. K., Mincer, T., et al. (2013). The microbiome of the Red Sea coral Stylophora pistillata is dominated by tissue-associated Endozoicomonas bacteria. Appl. Environ. Microbiol. 79, 4759–4762. doi: 10.1128/AEM.00695-13
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bray, J. R., and Curtis, J. T. (1957). An ordination of the upland forest communities of southern wisconsin. Ecol. Monogr. 27, 326–349. doi: 10.2307/1942268
Brooks, J. P., Edwards, D. J., Harwich, M. D. Jr., Rivera, M. C., Fettweis, J. M., Serrano, M. G., et al. (2015). The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol. 15:66. doi: 10.1186/s12866-015-0351-6
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Huntley, J., Fierer, N., et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. doi: 10.1038/ismej.2012.8
Chapron, L., Lartaud, F., Le Bris, N., Peru, E., and Galand, P. E. (2020). Local variability in microbiome composition and growth suggests habitat preferences for two reef-building cold-water coral species. Front. Microbiol. 11:275. doi: 10.3389/fmicb.2020.00275
Chen, Z., Hui, P. C., Hui, M., Yeoh, Y. K., Wong, P. Y., Chan, M. C. W., et al. (2019). Impact of preservation method and 16S rRNA hypervariable region on gut microbiota profiling. mSystems 4, e271–e218. doi: 10.1128/mSystems.00271-18
Choo, J. M., Leong, L. E. X., and Rogers, G. B. (2015). Sample storage conditions significantly influence faecal microbiome profiles. Sci. Rep. 5:16350. doi: 10.1038/srep16350
Clarke, K. R. (1993). Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x
Cruaud, P., Vigneron, A., Lucchetti-Miganeh, C., Ciron, P. E., Godfroy, A., and Cambon-Bonavita, M.-A. (2014). Influence of DNA extraction method, 16S rRNA targeted hypervariable regions, and sample origin on microbial diversity detected by 454 pyrosequencing in marine chemosynthetic ecosystems. Appl. Environ. Microbiol. 80, 4626–4639. doi: 10.1128/AEM.00592-14
de Lipthay, J. R., Enzinger, C., Johnsen, K., Aamand, J., and Sørensen, S. J. (2004). Impact of DNA extraction method on bacterial community composition measured by denaturing gradient gel electrophoresis. Soil Biol. Biochem. 36, 1607–1614. doi: 10.1016/j.soilbio.2004.03.011
Djurhuus, A., Port, J., Closek, C. J., Yamahara, K. M., Romero-Maraccini, O., Walz, K. R., et al. (2017). Evaluation of filtration and DNA extraction methods for environmental DNA biodiversity assessments across multiple trophic levels. Front. Mar. Sci. 4:314. doi: 10.3389/fmars.2017.00314
Dominianni, C., Wu, J., Hayes, R. B., and Ahn, J. (2014). Comparison of methods for fecal microbiome biospecimen collection. BMC Microbiol. 14:103. doi: 10.1186/1471-2180-14-103
Ducklow, H. W., and Mitchell, R. (1979). Bacterial populations and adaptations in the mucus layers on living corals. Limnol. Oceanogr. 24, 715–725. doi: 10.4319/lo.1979.24.4.0715
Fouhy, F., Clooney, A. G., Stanton, C., Claesson, M. J., and Cotter, P. D. (2016). 16S rRNA gene sequencing of mock microbial populations–impact of DNA extraction method, primer choice and sequencing platform. BMC Microbiol. 16:123. doi: 10.1186/s12866-016-0738-z
Galand, P. E., Chapron, L., Meistertzheim, A.-L., Peru, E., and Lartaud, F. (2018). The effect of captivity on the dynamics of active bacterial communities differs between two deep-sea coral species. Front. Microbiol. 9:2565. doi: 10.3389/fmicb.2018.02565
Galkiewicz, J. P., and Kellogg, C. A. (2008). Cross-kingdom amplification using Bacteria-specific primers: complications for studies of coral microbial ecology. Appl. Environ. Microbiol. 74, 7828–7831. doi: 10.1128/AEM.01303-08
Gill, C., van de Wijgert, J. H. H. M., Blow, F., and Darby, A. C. (2016). Evaluation of lysis methods for the extraction of bacterial DNA for analysis of the vaginal microbiota. PLoS One 11:e0163148. doi: 10.1371/journal.pone.0163148
Glasl, B., Bourne, D. G., Frade, P. R., Thomas, T., Schaffelke, B., and Webster, N. S. (2019). Microbial indicators of environmental perturbations in coral reef ecosystems. Microbiome 7:94. doi: 10.1186/s40168-019-0705-7
Glassing, A., Dowd, S. E., Galandiuk, S., Davis, B., and Chiodini, R. J. (2016). Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathogens 8:24. doi: 10.1186/s13099-016-0103-7
Gray, M. A., Pratte, Z. A., and Kellogg, C. A. (2013). Comparison of DNA preservation methods for environmental bacterial community samples. FEMS Microbiol. Ecol. 83, 468–477. doi: 10.1111/1574-6941.12008
Hallmaier-Wacker, L. K., Lueert, S., Roos, C., and Knauf, S. (2018). The impact of storage buffer, DNA extraction method, and polymerase on microbial analysis. Sci. Rep. 8:6292. doi: 10.1038/s41598-018-24573-y
Hamdi, C., Balloi, A., Essanaa, J., Crotti, E., Gonella, E., Raddadi, N., et al. (2011). Gut microbiome dysbiosis and honeybee health. J. Appl. Entomol. 135, 524–533. doi: 10.1111/j.1439-0418.2010.01609.x
Hernandez-Agreda, A., Leggat, W., and Ainsworth, T. D. (2018). A comparative analysis of microbial DNA preparation methods for use with massive and branching coral growth forms. Front. Microbiol. 9:2146. doi: 10.3389/fmicb.2018.02146
Holmes, E., Li, J. V., Athanasiou, T., Ashrafian, H., and Nicholson, J. K. (2011). Understanding the role of gut microbiome–host metabolic signal disruption in health and disease. Trends Microbiol. 19, 349–359. doi: 10.1016/j.tim.2011.05.006
Jaccard, P. (1908). Nouvelles recherches sur la distribution florale. Bull. Soc. Vaud. Sci. Nat. 44, 223–270.
Jensen, S., Hovland, M., Lynch, M. D. J., and Bourne, D. G. (2019). Diversity of deep-water coral associated bacteria and comparison across depth gradients. FEMS Microbiol. Ecol. 95:fiz091. doi: 10.1093/femsec/fiz091
Kellogg, C. A., Goldsmith, D. B., and Gray, M. A. (2017). Biogeographic comparison of Lophelia-associated bacterial communities in the western Atlantic reveals conserved core microbiome. Front. Microbiol. 8:796. doi: 10.3389/fmicb.2017.00796
Kellogg, C. A., Goldsmith, D. B., and Voleschow, J. J. (2021). Coral Microbiome Preservation and Extraction Method Comparison—Raw Data: U.S. Geological Survey data Release. doi: 10.5066/P96GBWDM (accessed June 29, 2021).
Kellogg, C. A., Lisle, J. T., and Galkiewicz, J. P. (2009). Culture-independent characterization of bacterial communities associated with the cold-water coral Lophelia pertusa in the northeastern Gulf of Mexico. Appl. Environ. Microbiol. 75, 2294–2303. doi: 10.1128/AEM.02357-08
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2012). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Kooperman, N., Ben-Dov, E., Kramarsky-Winter, E., Barak, Z. E., and Kushmaro, A. (2007). Coral mucus-associated bacterial communities from natural and aquarium environments. FEMS Microbiol. Lett. 276, 106–113. doi: 10.1111/j.1574-6968.2007.00921.x
Liang, J., Yu, K., Wang, Y., Huang, X., Huang, W., Qin, Z., et al. (2017). Distinct bacterial communities associated with massive and branching scleractinian corals and potential linkages to coral susceptibility to thermal or cold stress. Front. Microbiol. 8:979. doi: 10.3389/fmicb.2017.00979
Lim, M. Y., Song, E.-J., Kim, S. H., Lee, J., and Nam, Y.-D. (2018). Comparison of DNA extraction methods for human gut microbial community profiling. Systematic Appl. Microbiol. 41, 151–157. doi: 10.1016/j.syapm.2017.11.008
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Lozupone, C., and Knight, R. (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71, 8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meistertzheim, A.-L., Lartaud, F., Arnaud-Haond, S., Kalenitchenko, D., Bessalam, M., Le Bris, N., et al. (2016). Patterns of bacteria-host associations suggest different ecological strategies between two reef building cold-water coral species. Deep-Sea Res. Part I-Oceanogr. Res. Papers 114, 12–22. doi: 10.1016/j.dsr.2016.04.013
Meyer, J. L., Castellanos-Gell, J., Aeby, G. S., Häse, C. C., Ushijima, B., and Paul, V. J. (2019). Microbial community shifts associated with the ongoing stony coral tissue loss disease outbreak on the Florida Reef Tract. Front. Microbiol. 10:2244. doi: 10.3389/fmicb.2019.02244
Mitra, A., Skrzypczak, M., Ginalski, K., and Rowicka, M. (2015). Strategies for achieving high sequencing accuracy for low diversity samples and avoiding sample bleeding using Illumina platform. PLoS One 10:e0120520. doi: 10.1371/journal.pone.0120520
Morrow, K. M., Moss, A. G., Chadwick, N. E., and Liles, M. R. (2012). Bacterial associates of two Caribbean coral species reveal species-specific distribution and geographic variability. Appl. Environ. Microbiol. 78, 6438–6449. doi: 10.1128/AEM.01162-12
Nicholson, J. K., Holmes, E., Kinross, J., Burcelin, R., Gibson, G., Jia, W., et al. (2012). Host-Gut microbiota metabolic interactions. Science 336, 1262–1267. doi: 10.1126/science.1223813
Pollock, F. J., McMinds, R., Smith, S., Bourne, D. G., Willis, B. L., Medina, M., et al. (2018). Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat. Commun. 9:4921. doi: 10.1038/s41467-018-07275-x
Pollock, J., Glendinning, L., Wisedchanwet, T., and Watson, M. (2018). The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 84, e2627–e2617. doi: 10.1128/AEM.02627-17
Reigel, A. M., Owens, S. M., and Hellberg, M. E. (2020). Reducing host DNA contamination in 16S rRNA gene surveys of anothozoan microbiomes using PNA clamps. Coral Reefs 39, 1817–1827. doi: 10.1007/s00338-020-02006-5
Rosales, S. M., Miller, M. W., Williams, D. E., Traylor-Knowles, N., Young, B., and Serrano, X. M. (2019). Microbiome differences in disease-resistant vs. susceptible Acropora corals subjected to disease challenge assays. Sci. Rep. 9:18279. doi: 10.1038/s41598-019-54855-y
Röthig, T., Roik, A., Yum, L. K., and Voolstra, C. R. (2017). Distinct bacterial microbiomes associate with the deep-sea coral Eguchipsammia fistula from Red Sea and from aquaria settings. Front. Mar. Sci. 4:259. doi: 10.3389/fmars.2017.00259
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F., et al. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12:87. doi: 10.1186/s12915-014-0087-z
Santos, H. F., Carmo, F. L., Leite, D. C. A., Jesus, H. E., De Carvalho Maalouf, P., Almeida, C., et al. (2012). Comparison of different protocols for the extraction of microbial DNA from reef corals. Brazilian J. Microbiol. 43, 517–527. doi: 10.1590/s1517-83822012000200012
Schrader, C., Schielke, A., Ellerbroek, L., and Johne, R. (2012). PCR inhibitors – occurrence, properties and removal. J. Appl. Microbiol. 113, 1014–1026. doi: 10.1111/j.1365-2672.2012.05384.x
Sipos, R., Székely, A. J., Palatinszky, M., Révész, S., Márialigeti, K., and Nikolausz, M. (2007). Effect of primer mismatch, annealing temperature and PCR cycle number on 16S rRNA gene-targetting bacterial community analysis. FEMS Microbiol. Ecol. 60, 341–350. doi: 10.1111/j.1574-6941.2007.00283.x
Sonett, D., Brown, T., Bengtsson-Palme, J., Padilla-Gamiño, J. L., and Zaneveld, J. R. (2021). The organelle in the room: under-annotated mitochondrial reads bias coral microbiome analysis. bioRxiv [preprint] doi: 10.1101/2021.02.23.431501
Stoddard, S. F., Smith, B. J., Hein, R., Roller, B. R., and Schmidt, T. M. (2015). rrnDB: improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 43, D593–D598. doi: 10.1093/nar/gku1201
Sweet, M. J., and Bulling, M. T. (2017). On the importance of the microbiome and pathobiome in coral health and disease. Front. Mar. Sci. 4:9. doi: 10.3389/fmars.2017.00009
Sweet, M. J., Croquer, A., and Bythell, J. C. (2011). Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs 30, 39–52. doi: 10.1007/s00338-010-0695-1
Vohsen, S. A., Anderson, K. E., Gade, A. M., Gruber-Vodicka, H. R., Dannenberg, R. P., Osman, E. O., et al. (2020). Deep-sea corals provide new insight into the ecology, evolution, and the role of plastids in widespread apicomplexan symbionts of anthozoans. Microbiome 8:34. doi: 10.1186/s40168-020-00798-w
Wagner Mackenzie, B., Waite, D. W., and Taylor, M. W. (2015). Evaluating variation in human gut microbiota profiles due to DNA extraction method and inter-subject differences. Front. Microbiol. 6:130. doi: 10.3389/fmicb.2015.00130
Weber, L., DeForce, E., and Apprill, A. (2017). Optimization of DNA extraction for advancing coral microbiota investigations. Microbiome 5:18. doi: 10.1186/s40168-017-0229-y
Willner, D., Daly, J., Whiley, D., Grimwood, K., Wainwright, C. E., and Hugenholtz, P. (2012). Comparison of DNA extraction methods for microbial community profiling with an application to pediatric bronchoalveolar lavage samples. PLoS One 7:e34605. doi: 10.1371/journal.pone.0034605
Wright, E. S., and Vetsigian, K. H. (2016). Quality filtering of Illumina index reads mitigates sample cross-talk. BMC Genomics 17:876. doi: 10.1186/s12864-016-3217-x
Keywords: preservation, extraction, coral, microbiome, bacteria, RNAlater, liquid nitrogen, PowerBiofilm
Citation: Pratte ZA and Kellogg CA (2021) Comparison of Preservation and Extraction Methods on Five Taxonomically Disparate Coral Microbiomes. Front. Mar. Sci. 8:684161. doi: 10.3389/fmars.2021.684161
Received: 22 March 2021; Accepted: 24 June 2021;
Published: 14 July 2021.
Edited by:
Oren Levy, Bar-Ilan University, IsraelReviewed by:
Maren Ziegler, University of Giessen, GermanyRebecca Lisette Vega Thurber, Oregon State University, United States
Copyright © 2021 Pratte and Kellogg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina A. Kellogg, Y2tlbGxvZ2dAdXNncy5nb3Y=