 Wei Zhang1,2,3
Wei Zhang1,2,3 Beiping Tan1,2,3*
Beiping Tan1,2,3* Junming Deng1,2,3
Junming Deng1,2,3 Xiaohui Dong1,2,3Qihui Yang1,2,3
Xiaohui Dong1,2,3Qihui Yang1,2,3 Shuyan Chi1,2,3
Shuyan Chi1,2,3 Hongyu Liu1,2,3
Hongyu Liu1,2,3 Shuang Zhang1,2,3
Shuang Zhang1,2,3 Shiwei Xie1,2,3Haitao Zhang3
Shiwei Xie1,2,3Haitao Zhang3- 1Laboratory of Aquatic Animal Nutrition and Feed, College of Fisheries, Guangdong Ocean University, Zhanjiang, China
- 2Aquatic Animals Precision Nutrition and High Efficiency Feed Engineering Research Center of Guangdong Province, Zhanjiang, China
- 3Key Laboratory of Aquatic, Livestock and Poultry Feed Science and Technology in South China, Ministry of Agriculture, Zhanjiang, China
Intestinal inflammatory disease induced by excessive soy protein substitutions for fish meal (FM) protein is a common phenomenon. The pearl gentian grouper, Epinephelus fuscoguttatus♀ × Epinephelus lanceolatus♂, a marine fish with important economical and nutritional values, exhibited a similar problem. As far as we know, there are no reports on the full-length transcriptome of the pearl gentian grouper. In the present study, seven isonitrogenous and isolipidic (10% lipid) diets were prepared and fed to fish for 10 weeks. The water volume in each barrel was about 1 m3, using natural light and temperature. The results showed that 40% dietary soy proteins significantly negatively affected the growth performance of the pearl gentian grouper. Compared to the FM control, the content of immunoglobulin M and the enzyme activities of glutathione reductase, glutathione peroxidase, and total superoxide dismutase in the intestine significantly increased; the content of malondialdehyde in the intestine significantly decreased; and the enzyme activities of alanine transaminase and aspartate transaminase in the liver significantly increased. A library composed of seven different treated distal intestine tissues, including the FM control group, 20% soybean meal substitute for FM (SBM20), SBM40, 20% soybean protein concentrate (SPC20), SPC40, 20% fermented soybean meal substitute for FM (FSBM20), and FSBM40, was constructed and sequenced using PacBio single-molecule real-time (SMRT) and the RNA-Seq technology. As a whole, this study obtained 420,006 full-length non-chimeric (FLNC) reads. After error correction, sequence clustering, and redundancy, 82,351 transcripts with high quality were obtained. In addition, a total of 77,815 transcripts were annotated in seven databases (non-redundant protein sequences, non-redundant nucleotide sequences, Protein family, Clusters of Orthologous Groups of proteins, Gene Ontology, Swiss-Prot, and KEGG Orthology). Also, 49,093 long non-coding RNAs (lncRNAs) and 141,702 simple sequence repeats were identified. Based on full-length transcriptome sequencing, the present study found that the Toll-like receptor/nuclear factor kappa-B signaling pathway plays an important role in the development of SBM- and FSBM-induced enteritis. SPC-induced enteritis is mainly accompanied by a general imbalance of the nutrition absorption-related signaling pathways, which only affects a small part of the immune-related signaling pathways. This study supplies new and valuable reference transcripts, which would better facilitate further research on the pearl gentian grouper.
Introduction
Fish meal (FM) is an important source of protein in aquatic feed, especially for carnivorous species. Most of the FM originates from wild oil-rich fishes, and its supply is unsustainable due to the limited abundance of these populations (Booman et al., 2018; Cai et al., 2020). The total amount of global aquaculture continues to increase, while FM production is relatively constant (Guardiola et al., 2016). Therefore, it is very important to find new protein sources with good potential to replace FM (Teves and Ragaza, 2014). At present, soy products are the first choice to replace FM protein, such as soybean meal (SBM), soybean protein concentrate (SPC), and fermented soybean meal (FSBM) (Xiang, 2017). However, intestinal inflammation disease induced by high levels of soy protein, especially SBM as a substitute for FM, is a common phenomenon in aquaculture, which would affect fish growth and feed utilization.
Up to now, the exact reason and mechanism of fish enteritis induced by soy proteins are not very clear, and the molecular mechanism of enteritis formation is still lacking of a systematic research. In order to further understand the effect of aquatic feed on fish physiology, it is necessary to apply different research methods to investigate the relationship between diet and intestinal health from different perspectives. New omics technologies such as transcriptomics can provide great potential for studying the complex relationship between nutrition and the immunity of fish in health and disease (Martin and Król, 2017). Despite the reduced cost of deep sequencing, only a few partially completed genome information are available of the key aquaculture fish species, such as the Atlantic cod (Gadus morhua) (Star et al., 2011), common carp (Cyprinus carpio) (Xu et al., 2014), European sea bass (Dicentrarchus labrax) (Tine et al., 2014), tilapia (Oreochromis niloticus) (Brawand et al., 2014), grass carp (Ctenopharyngodon idellus) (Wang et al., 2015), and the channel catfish (Ictalurus punctatus) (Liu et al., 2016).
The pearl gentian grouper (Epinephelus fuscoguttatus♀ × E. lanceolatus♂) is a carnivorous fish species that has the advantages of fast growth, high market value, and good disease resistance (Zhou et al., 2020). As far as we know, the pearl gentian grouper is a non-model species that has not been reported in any published genomic library. Previously, our lab carried out grouper transcriptome sequencing using Illumina, such as the liver, blood, and intestine. However, the length of the sequencing reads was short (usually 100–250 bp) and the full-length transcripts obtained by splicing were not complete, which would hinder further study of the molecular mechanism.
The third-generation sequencing technology is also called de novo sequencing technology, namely, the single-molecule real-time (SMRT) DNA sequencing. The third-generation sequencing technology is the development trend in the future, which is mainly used in genome sequencing, methylation research, and mutation identification (SNP detection) (Jia et al., 2020). Compared with the next-generation sequencing technologies, the SMRT sequencing technology shows many superiorities, such as (1) obtaining full-length transcripts directly without transcript splicing; (2) a longer sequencing length and an ultrahigh sequencing flux; (3) discovering new functional genes and supplementing genome annotation; and (4) the analysis of alternative splicing (Roberts et al., 2013). Full-length sequencing is crucial for fully characterizing the transcriptomes of lesser studied and non-model organisms (Workman et al., 2018), but up to now, the application of which in aquaculture is scare.
Previously, our lab found that different levels of soybean diets, including 20% SBM protein as a substitute for FM protein (SBM20), SBM40, 20% soybean protein concentrate (SPC20), SPC40, 20% fermented soybean meal as substitute for FM (FSBM20), or FSBM40, can induce enteritis of the pearl gentian grouper. In this study, sequencing was carried out to generate the full-length transcriptome of the pearl gentian grouper intestine using the Pacific Biosciences (PacBio) SMRT sequencing technology (PacBio, Menlo Park, CA, United States) for the first time. Based on the obtained transcriptome data, the present study performed transcript functional annotation, coding sequence prediction, long non-coding RNA (LncRNA) prediction, and simple sequence repeat (SSR) analysis. In addition, the differential mechanisms of enteritis in the pearl gentian grouper induced by three soy proteins were preliminarily investigated. This study would be a valuable genome resource for further research of the pearl gentian grouper and also provides more reference results for the study of soy meal-induced enteritis in fish.
Materials and Methods
Experimental Diets
The composition and chemical analysis of the experimental diets are presented in Supplementary Table 1. The red FM used in this study containing 72.53% crude protein and 8.82% total lipid was provided by Corporación Pesquera Inca S.A.C., Bayovar Plant, Peru. The SBM and SPC used in this study contained 48.92 and 70.72% crude protein, respectively, which were provided by Zhanjiang Haibao Feed Co., Ltd. (Zhanjiang, China). The FSBM used in this study that contained 60.75% crude protein was provided by Xijie Foshan Co., Ltd. (Foshan, China). The fermentation strain is Bacillus subtilis. Seven isonitrogenous (approximately 50% crude protein) and isolipidic (10% total lipid) experimental diets were formulated to replace 0, 20, and 40% of FM protein with SBM, SPC, and FSBM protein, which were named FM (control), SBM20, SBM40, SPC20, SPC40, FSBM20, and FSBM40, respectively. Lysine and methionine were used to compensate for the amino acid imbalance of the diets (Miao et al., 2018). The detailed preparation process and the storage conditions of the experimental diets are described in our previously published literature (Zhang et al., 2019). Briefly, the raw materials were ground into a fine powder, crushed through a 60-mesh sieve, and weighed accurately according to the formula. The micro-constituents were mixed homogenously using the sequential expansion method. Then, deionized water and lipids were added and stirred evenly to obtain a homogenous mixture. After that, the mixture was passed through a pelletizer with 2.0 and 3.00 mm diameter. The pellets were air-dried to 10% moisture, sealed in plastic bags, and stored at –20°C until use. The essential amino acid contents of the diets are shown in Supplementary Table 2.
Feeding Trial and Experimental Condition
The detailed feeding trial and experimental conditions are described in our previously published literature (Zhang et al., 2019). Briefly, when juvenile groupers have adapted to the experimental environment, fish of similar size were randomly distributed into 1,000-L cylindrical fiberglass tanks. The fish had initial weight of about 12.55 g and length of about 7.66 cm. Each tank had 60 fish. Each diet group was fed to four replicates twice daily at 0800 and 1600 h, respectively, until apparent satiation for 10 weeks. The experiment was carried out at the indoor farming systems of the Marine Biological Research Base, Zhanjiang, China. All tanks were provided with continuous aeration by air stones. The light cycle used natural conditions, and the temperature was 29 ± 1°C. Ammonia and nitrate were no more than 0.03 mg L–1, and dissolved oxygen was not less than 7 mg L–1. In the first 2 weeks, 60% of the water in each tank was changed every day; all of the water was changed every day thereafter.
Sampling
Samples were collected at the end of the experiment. Before the experiment, the fish were starved for 24 h and then anesthetized with eugenol (1:10,000) for sampling. After cutting the abdomen along the midline, the intestine was gently pulled out and the mesenteric adipose tissue cleared up, and then the external residue was washed off with deionized water. Subsequently, some distal intestine (DI) and liver samples were quickly put into liquid nitrogen immediately after being placed in a cryopreservation tube. After sampling, the samples were stored at –80°C for transcriptome sequencing and enzyme activity analysis. Some DI samples were cut into pieces and placed into a tube containing RNAlater. After storing overnight at 4°C, the samples were then stored at –80°C for gene expression determination. Some DI tissues were stored in 4% paraformaldehyde general tissue fixative (Wuhan Servieobio Technology Co., Ltd., Wuhan, China) for 24 h for histology observation. In addition, the present study mainly analyzed the physiological changes in the 40% substitution groups. The weight gain rate (WGR), specific growth rate (SGR), feed conversion ratio (FCR), hepatosomatic index (HSI), and the survival rate (SR) were evaluated.
Histology
Intestinal histological observation (hematoxylin and eosin staining) was done according to Zhang et al. (2019). Briefly, the height-and-width ratio of the plica, the width of the lamina propria, and the length of the microvilli were determined. Each index was determined through 10 different scans. The stained sections were observed and photographed with an optical microscope (Olympus CKX41 microscope, Tokyo, Japan).
Analysis of Biochemical Indicators
The enzyme activities of glutathione reductase (GR), glutathione peroxidase (GPx), and total superoxide dismutase (T-SOD) in DI tissues and alanine aminotransferase (ALT) and aspartate transaminase (AST) in liver tissues were detected by fish enzyme-linked immunosorbent assay (ELISA) kits. The immunoglobulin M (IgM) and malondialdehyde (MDA) contents in DI tissues were determined by the fish ELISA kit. All the kits were purchased from Shanghai Jianglai Biotechnology Co., Ltd. (Shanghai, China). The detailed test steps were according to the instruction manual.
RNA Extraction
Total RNA was extracted separately from each group of distal intestinal tissues of the pearl gentian grouper using the RNeasy Plus Mini Kit (QIAGEN, Valencia, CA, United States). The quality of RNA is usually measured by 1% agarose gels and its purity and concentration usually measured by a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, United States) with an OD260/OD280 reading value. The integrity of RNA was assessed by the RNA Nano 6000 Assay Kit of Agilent Bioanalyzer 2100 system (Agilent, Santa Clara, CA, United States). For PacBio isoform sequencing (Iso-Seq), only the total RNA samples from seven groups with an RNA integrity number (RIN) >7 were mixed together for sequencing. For Illumina RNA sequencing (RNA-Seq), equal amounts of total RNA from three fish were pooled for each group. Indexed complementary DNA (cDNA) libraries were then prepared for each group.
SMRT Library Construction and Sequencing
The Iso-Seq library was prepared according to the isoform sequencing protocol using the (Clontech, Japan) SMARTer PCR cDNA Synthesis Kit and the BluePippin Size Selection System protocol as described by PacBio (PN 100-092-800-03). Briefly, after enrichment by Oligo(dT) magnetic beads, the messenger RNA (mRNA) was reverse transcribed into cDNA using the SMARTer PCR cDNA Synthesis Kit. PCR was used to amplify and enrich the synthesized cDNA, and the optimal conditions for PCR were determined by cycle optimization. Part of the cDNA was screened by BluePippin, and the >4-kb fragments were enriched; then, the screened fragments were subjected to large-scale PCR to obtain enough cDNA quantity. The full-length cDNA was used for damage repair, end repair, and connection of the SMRT dumbbell-shaped connector. The equimolar library of the non-screened fragments and fragments larger than 4 kb was constructed. Exonuclease digestion was used to remove the sequence of the unconnected junctions at both ends of cDNA. Finally, a complete SMRT bell library was constructed by binding the primers and DNA polymerase. After passing library inspection, the library was sequenced by the PacBio Sequel platform according to the effective concentration of the library and data output requirements.
Illumina Library Construction and Sequencing
After the RNA samples of each individual were mixed equally, the cDNA library was constructed according to Li et al. (2013). Briefly, polyadenylated (polyA) mRNA was enriched using magnetic beads containing Oligo(dT), and the fragment buffer was added to make it into short fragments. The short mRNA was used as a template to synthesize cDNA. Terminal repair, polyA addition, and sequencing adaptor were performed. Then, the target fragments were recovered for PCR amplification to complete the preparation of the whole library. Finally, the libraries were sequenced using the Illumina HiSeqTM 4000 by Gene Denovo Co., Ltd. (Guangzhou, China).
PacBio SMRT Data Processing and Error Correction
After sequence completion, the off-line raw data are despliced and read with low quality. The output was filtered and processed by the software SMRTlink V5.1. The parameters are –minlength = 200 and –minreadscore = 0.65; then, the final data are the valid data. In order to obtain the full-length transcripts, first, the subread sequence was self-corrected to form circular consensus sequencing (CCS; parameters: –minpasses = 2, minpredicted accuracy = 0.8), and a high-quality consistent transcript sequence was obtained. A non-chimeric sequence with a 5′-end primer, a 3′-end primer, and a polyA tail is called a full-length non-chimeric (FLNC) sequence. An iterative isoform clustering (IEC) algorithm was used to cluster the FLNC sequences of the same transcript to obtain CCS, and then non-full-length sequences were used to correct the CCS. Then, the fused consensus sequences (CS) were obtained for subsequent analysis. After that, the Illumina data were used to correct the polished consensus sequence with the LoRDEC software (parameters: –k21 and –s3) to further improve the accuracy of sequencing. Finally, CD-HIT v4.6.7 (–c0.95 –T6 –G0 –aL0.00 –aS0.99) software was used to cluster and compare the protein or nucleic acid sequences by sequence alignment and to remove redundant and similar sequences.
Functional Annotation
Full-length (FL) transcripts were searched against the NCBI non-redundant protein sequences (Nr), NCBI non-redundant nucleotide sequences (Nt), Protein family (Pfam), Clusters of Orthologous Groups of proteins (KOG/COG), Swiss-Prot, Kyoto Encyclopedia of Genes and Genomes Orthology database (KEGG Orthology), and Gene Ontology (GO). Diamond BLASTX software was used for functional annotation with an e-value of 1e–10 in the Nr, KOG, Swiss-Prot, and KEGG database analysis. BLAST software with the e-value of 1e–10 was used in the Nt database analysis. The Hmmscan software was used in the Pfam database analysis. GO annotation was analyzed using the Blast2GO software (Conesa et al., 2005) with the Nr annotation results of transcripts.
Coding Sequencing and LncRNA Prediction
The ANGEL pipeline, a long-read implementation of ANGLE, was used in order to determine the protein coding sequences (CDS) from the cDNAs. We used this species’ or the confident protein sequences of closely related species for ANGEL training and then ran the ANGEL prediction for the given sequences (Shimizu et al., 2006).
Long non-coding RNAs (lncRNAs) are RNA molecules with transcripts longer than 200 nt and do not encode proteins. Due to the limitation of the principle of library construction, we can only obtain lncRNAs with a polyA tail. Usually, four software—CNCI (Sun et al., 2013), CPC (Kong et al., 2007), Pfam (Finn et al., 2016), and PLEK (Aimin et al., 2014)—are used to predict the coding potential of genes obtained from CD-HIT de-redundancy.
Simple Sequence Repeats Analysis
Simple sequence repeats (SSRs) are also known as short tandem repeats or microsatellites. They are a class of repeats consisting of several nucleotides (one to six) as repeat units, which are short in length and widely distributed in eukaryotic genomes. In our analysis, MISA software (version 1.0, default parameters) was used. The minimum repetition times of each unit size were 1–10, 2–6, 3–5, 4–5, 5–5, and 6–5 to detect genes by (Simple Sequence Repeats Analysis) SSRs (Thiel, 2003; Gulcher, 2012).
Analysis of the Differentially Expressed Genes
The present study compared the effects of SBM40, SPC40, and FSBM40 on the transcriptome level of the distal intestine in the pearl gentian grouper. Firstly, the differentially expressed genes (DEGs) in the SBM40, SPC40, and FSBM40 groups were screened. The screened thresholds were | Log2FC| > 1 and P < 0.05. The genes meeting the above conditions were identified as DEGs. Then, the DEGs in the SBM40, SPC40, and FSBM40 groups were analyzed by a Venn diagram for common and unique DEGs. Finally, GO annotation and KEGG enrichment analyses were conducted for the common and unique DEGs, respectively, and the signaling pathways related to immune diseases/system, infectious diseases, and signal transduction that were significantly affected in the KEGG enrichment results were analyzed (P < 0.05).
Validation of Real-Time Quantitative PCR
In order to test the accuracy of the full-length PacBio SMRT sequencing results, samples stored at –80°C were selected for quantitative reverse transcription PCR (RT-qPCR). In this study, 18 genes related to immune and inflammatory development were selected, which included TLR1, TLR2, TLR3, TLR5, TLR8, TLR9, TLR13, TLR21, TLR22, IgA, pIgR, IL4, IL5, IL10, MyD88, IκBα, and p65. The primers were designed by the Primer Premier 5.0 software and the primer sequence templates were from the full-length PacBio SMRT transcriptome sequencing database. The primers were synthesized by Shenggong Bioengineering Co., Ltd. (Shanghai, China). The internal reference gene is β-actin. The primers are displayed in Supplementary Table 3. The PCR reaction conditions were: 95°C for 2 min, 1 cycle; 95°C for 15 s, 60°C for 10 s, 72°C for 20 s, 40 cycles. The expressions of the target genes were determined by 2–ΔΔCt (Livak and Schmittgen, 2001).
Statistics
Analysis of the omics data refers to the above mentioned. The rest of the data were analyzed using SPSS 22.0 software (SPSS Inc., Chicago, IL, United States). The results were presented as the mean ± standard deviation (x ± SD). In order to test differences among groups, one-way ANOVA was used after the homogeneity variance test. The significance threshold was P < 0.05. Growth performance was calculated using the following formulas:
Results
Growth Performance
Figure 1 shows that, compared to the FM control group, there was a significant decrease in the WGR and SGR in the experimental groups (P < 0.05), and there was no significant difference among the experimental groups (P > 0.05). There was a significant increase in the FCR in the experimental groups (P < 0.05), which indicated that fish fed diets containing different soy proteins had worse FCR values; there was no significant difference among the experimental groups (P > 0.05). The HSI and SR had no significant differences among the groups (P > 0.05) (Supplementary Figure 1).
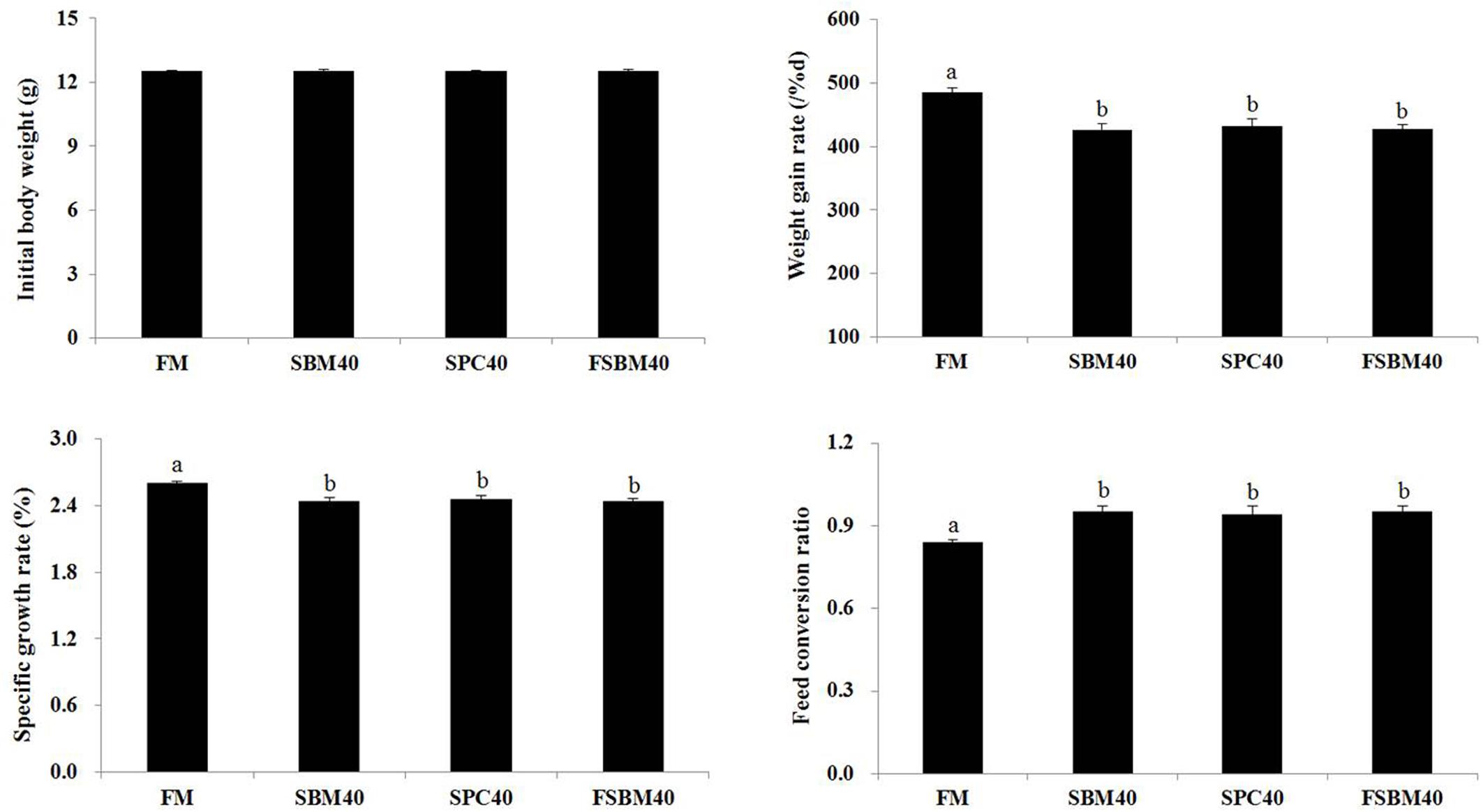
Figure 1. Effect of the different soy proteins at 40% substitution levels for fish meal on the growth of the pearl gentian grouper (n = 3).
Histological Observation of Enteritis
The results illustrated that the plica height/width and the microvilli length significantly decreased in each experimental group compared to the FM control group (P < 0.05). On the contrary, the lamina propria width significantly increased in each experimental group (P < 0.05) (Supplementary Figure 2 and Table 1).

Table 1. Effect of different soy protein substitutions for fish meal protein on the distal intestine morphology of the pearl gentian grouper (n = 10).
Biochemical Indicators
Table 2 exhibits that, compared to the FM control group, the enzyme activities of T-SOD, GR, and GPX significantly increased in the experimental groups (P < 0.05), and the highest value appeared in SPC40, followed by the SBM40 and FSBM40 groups. The MDA content also significantly increased in the experimental groups (P < 0.05), and the highest value appeared in SBM40, followed by the FSBM40 and SPC40 groups. The content of IgM significantly decreased in the experimental groups (P < 0.05), and the highest value appeared in SBM40, followed by the FSBM40 and SPC40 groups. The enzyme activities of ALT and AST in the liver were significantly increased in the experiment groups (P < 0.05), and the highest value appeared in SPC40, followed by the FSBM40 and SPC40 groups.
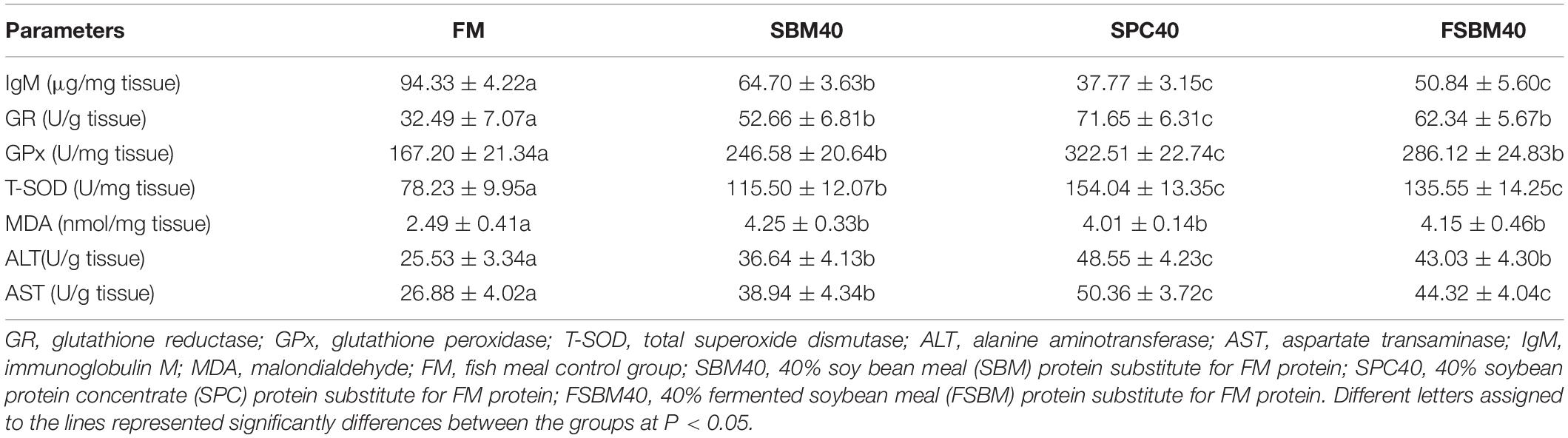
Table 2. Effect of the different soy protein substitutions for fish meal protein on the enzyme activities of the pearl gentian grouper (n = 3).
SMRT Sequencing of the Intestine
The flow process diagram of the transcriptome of the pearl gentian grouper by SMRT and Illumina sequencing is shown in Supplementary Figure 3A. In total, there were 487,152 CCS reads with an average length of 3,013 bp isolated from the PacBio SMRT raw data (30.31 G of subreads) in the mixed library (Supplementary Table 4), among which FLNC made up 86.22%, while the ratios of the non-full-length (NFL), full-length chimeric (FLC), and short reads were 11.39, 0.89, and 1.50%, respectively (Supplementary Figure 3B).
After ICE correction (iterative correction and eigenvector decomposition), 225,854 CS were obtained. Then, the Illumina sequencing data were used to further correct the CS using LoRDCE software. Thereafter, the sequences were removed by CD-HIT software; 225,854 non-redundant transcripts (2,998 bp on average) and 82,351 unigenes (3,486 bp on average, N50 = 4,131 and N90 = 2,173) in all were obtained. The length distribution of the unigenes and the number of transcripts corresponding to genes are shown in Supplementary Figure 3C and Figure 2D. Transcripts with lengths ranging from 1,700 to 5,100 bp make up 73.29% of the unigenes.
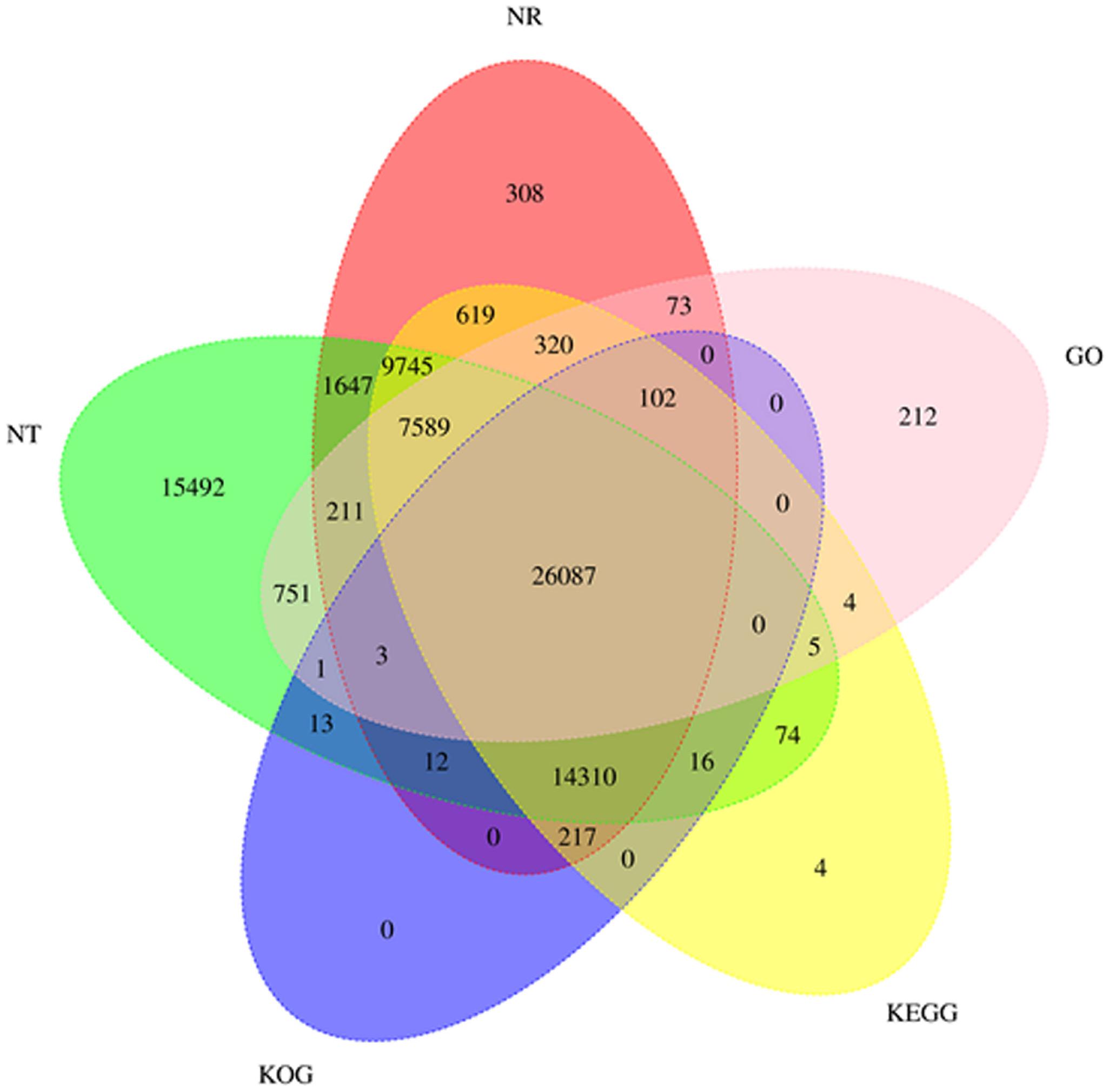
Figure 2. Venn diagram of the functional annotation of the long-read transcriptomes in Nr, Nt, GO, KOG, and KEGG databases. Nr, non-redundant protein database; Nt, non-redundant nucleotide sequences; GO, Gene Ontology; KOG, Clusters of Orthologous Eukaryotic Genes; KEGG, Kyoto Encyclopedia of Genes and Genomes.
The unigene length sequenced by third-generation technology in this study is much longer than that sequenced using Illumina, in which the N50 of the non-assembled unigenes by PacBio sequencing is 4,131 bp; however, the N50 values of the unigenes sequenced in our previous unpublished transcriptomes using Illumina for the pearl gentian grouper intestine and liver were 1,886 and 1,921 bp, respectively (Table 3).

Table 3. Comparison of unigenes from the PacBio and Illumina sequencing platforms of the pearl gentian grouper.
Functional Annotation of the Full-Length Transcripts
All of the full-length transcripts were blasted against seven databases, including the Nr, Swiss-Prot, KEGG, KOG, GO, Nt, and Pfam databases (Table 4). Based on the annotation results of the seven databases, five databases were selected to draw the Venn diagram (Figure 2). In total, 77,815 (94.5%) FL transcripts were annotated in at least one of the databases. The Nt database annotated the highest gene number (75,956), followed by Nr (61,243), KEGG (59,092), Swiss-Prot (52,750), KOG (40,761), and GO (35,358) (Figure 3). The species distribution annotation in the Nr database showed that the top 10 species were Lates calcarifer (27.03%), Larimichthys crocea (17.12%), Stegastes partitus (6.93%), Notothenia coriiceps (3.43%), Paralichthys olivaceus (2.44%), O. niloticus (2.15%), Epinephelus coioides (1.77%), Nothobranchius furzeri (0.93%), D. labrax (0.92%), and Epinephelus lanceolatus (0.76%) (Figure 4). However, the remaining 36.52% of the matched FL transcripts showed similarities to other species due to limited genome information. This suggested that the FL transcripts of the pearl gentian grouper should be further annotated with updated published fish genes and related gene background information.

Table 4. Annotation results of seven databases.
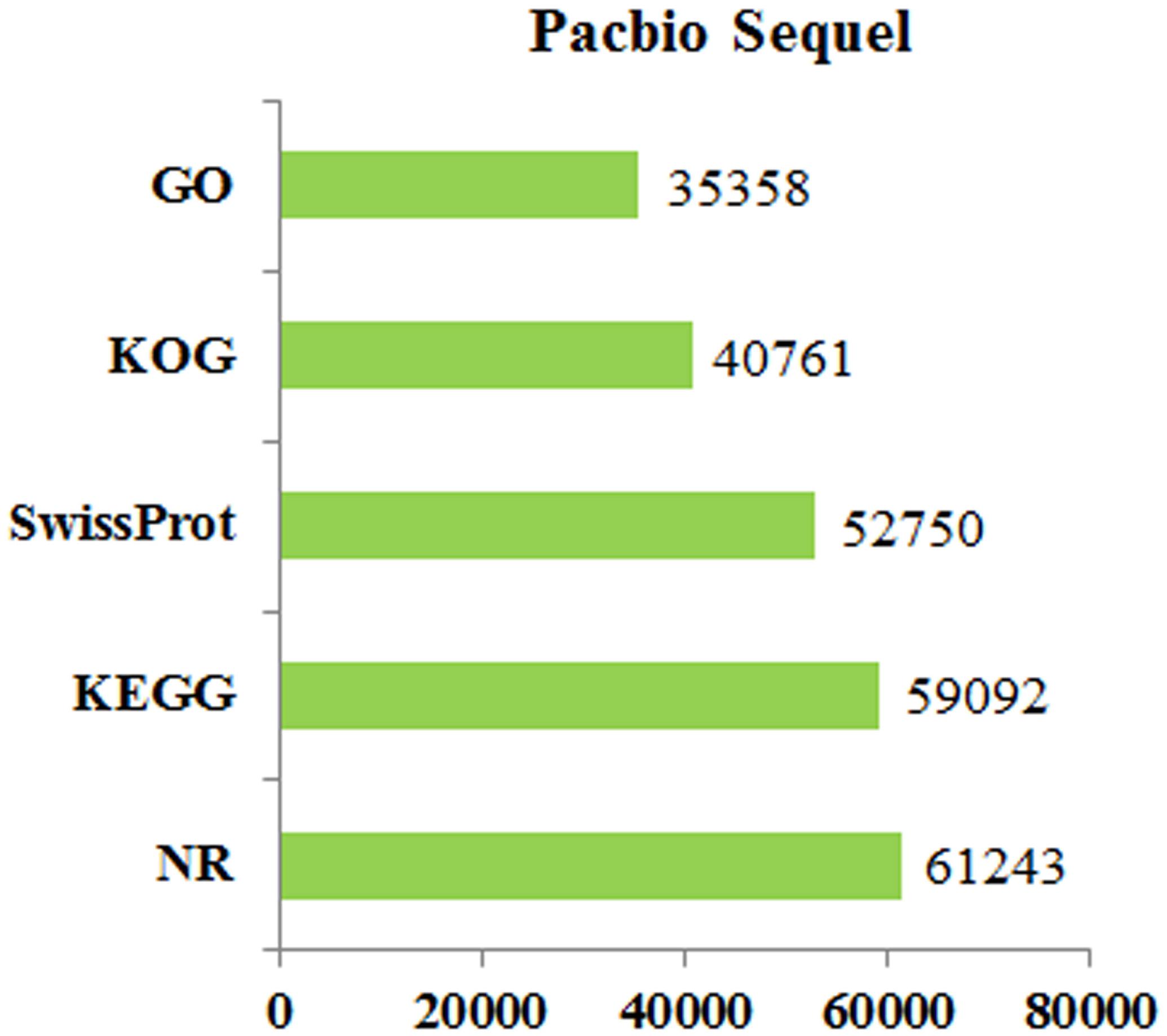
Figure 3. Annotated gene number of single-molecule real-time (SMRT) sequencing.
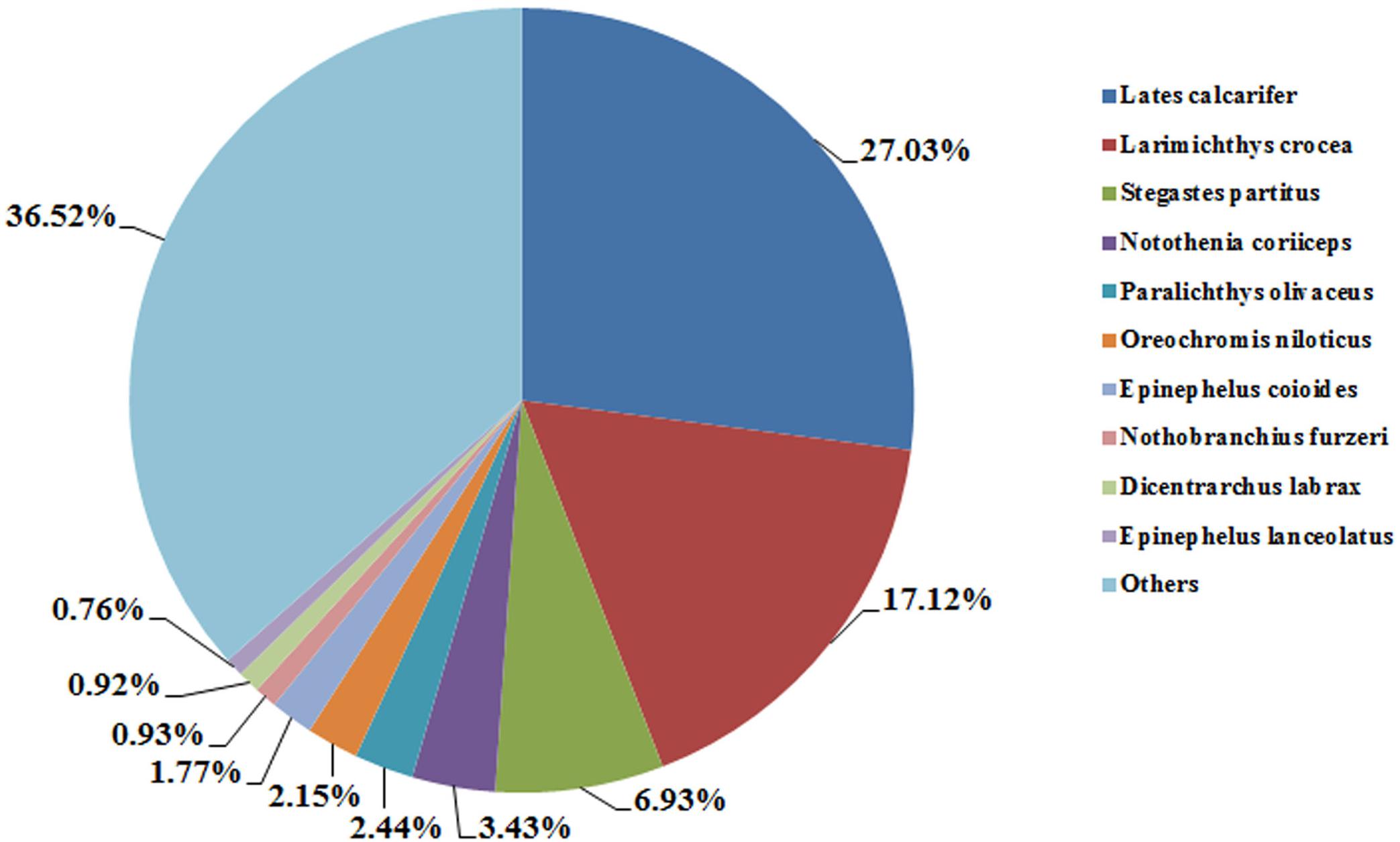
Figure 4. Species distribution of the Nr annotation of the long-read transcripts.
In the KOG database, the 77,815 annotated FL transcripts were categorized into 26 KOG classifications (Figure 5A). The largest cluster was “general function prediction only (R)” (7,805 isoforms), which indicates that the functions of most genes still need experimental confirmation. This was followed by “signal transduction mechanisms (T)” (7,797 isoforms), “posttranslational modification, protein turnover, chaperones (O)” (4,178 isoforms), and “function unknown (S)” (2,740 isoforms).
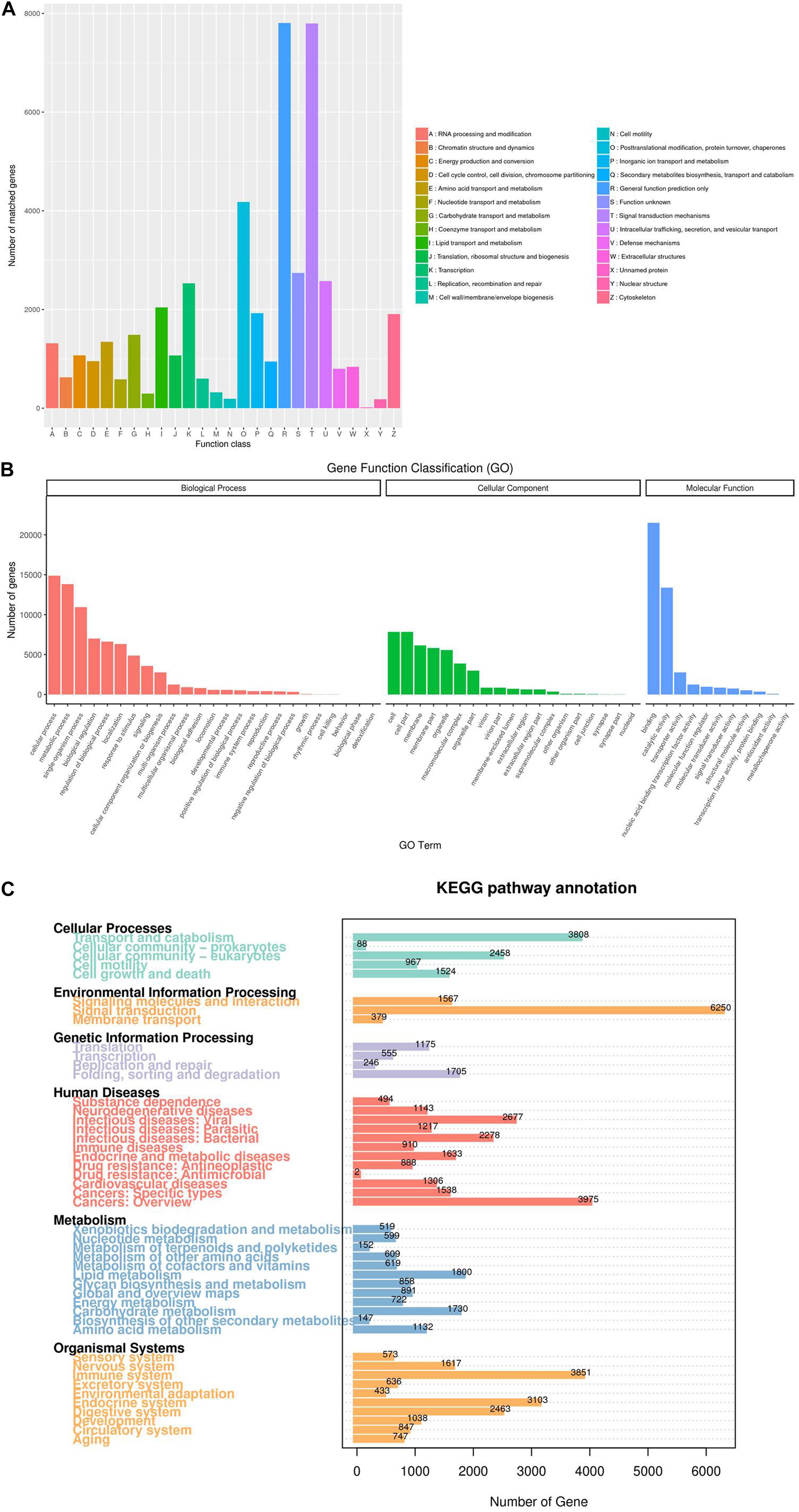
Figure 5. Functional classification of the long-read transcripts. (A) Clusters of Orthologous Eukaryotic Genes (KOG) classification. (B) Gene Ontology (GO) classification. (C) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway classification.
Functional annotation using GO exhibits the properties of the gene products by classifying them into the “biological process,” “cellular component,” and “molecular function” categories. Figure 5B showed that there were 77,139, 44,427, and 42,465 transcripts assigned to the “molecular function,” “cellular component,” and “biological process” categories, respectively. In detail, in the “molecular function” annotation, “binding” (21,517 isoforms) and “catalytic activity” (13,382 isoforms) were the top two abundant terms; in the “cellular component” annotation, “cell” (7,847 isoforms), “cell part” (7,847 unigenes), and “membrane” (6,164 isoforms) were the majority of level 2 terms. In the “biological process” annotation, “cellular process” (14,884 isoforms), “metabolic process” (13,824 isoforms), and “single-organism process” (10,951 isoforms) comprised the largest proportions.
KEGG annotation found that 23,082 genes were assigned to five pathways (level 1), including “cellular processes,” “environmental information processing,” “genetic information processing,” “metabolism,” and “organismal systems.” At level 2 of the KEGG pathways, “signal transduction,” (6,250 isoforms), “cancers: overview” (3,975 isoforms), “immune system” (3,851 isoforms), “transport and catabolism” (3,808 isoforms), and “endocrine system” (3,103 isoforms) were the most abundant terms (Figure 5C).
CDS and LncRNA Prediction
Coding sequences (CDS) is the sequence that encodes a protein product, which completely corresponds to the codon of the protein. After BLAST comparison of the obtained polished consensus in the protein database, 8,243 CDS were found. The lengths of the sequences ranged from 0 to 5,000 bp, mainly concentrated in 0–2,500 bp (Figure 6), indicating that the unigenes had good sequence quality.
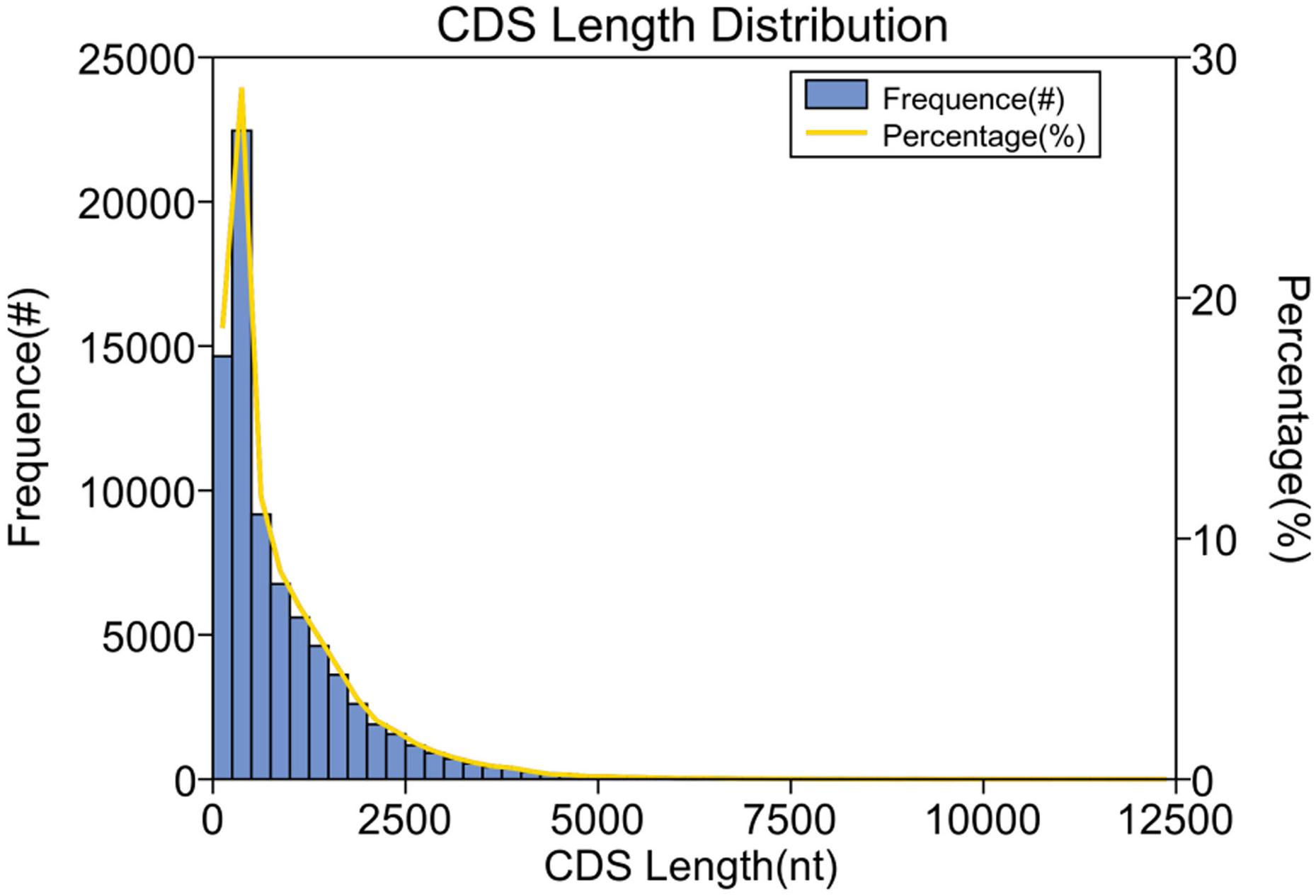
Figure 6. Length distribution of the coding sequences (n = 4). The x-axis represents the coding sequence length and the y-axis represents the number of predicted coding sequences.
The results showed that 38,219 lncRNAs were identified using the CNCI software, followed by 24,640 lncRNAs identified using the CPC software, 16,655 lncRNAs using the PLEK software, and 44,249 lncRNAs using the Pfam software, among which 8,874 common lncRNAs were identified by four different bioinformatics software (Figure 7A). From the length distribution of the lncRNAs and mRNAs, it can be seen that the peak value of lncRNA length is about 2,000 bp and that of mRNA length is about 2,500 bp (Figure 7B).
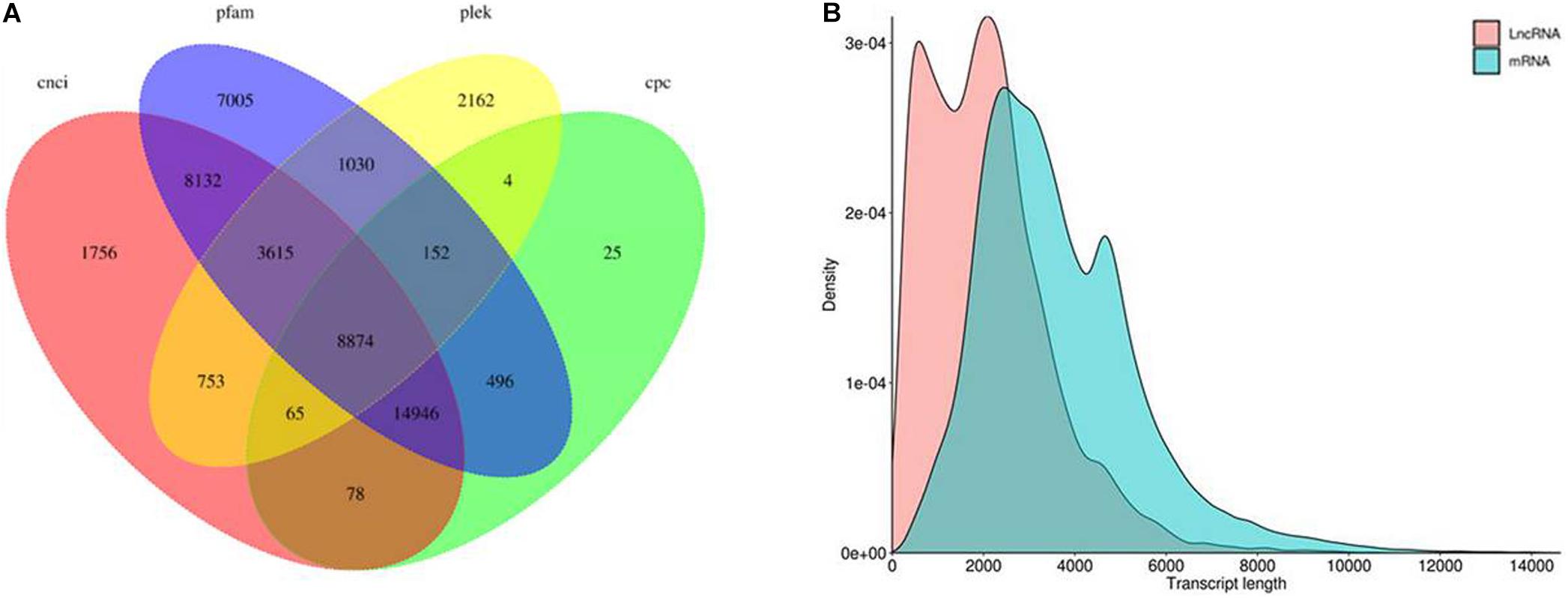
Figure 7. Long non-coding RNA (lncRNA) prediction. (A) Venn diagram of the lncRNA prediction results by four software. (B) Length distribution of lncRNAs and mRNAs.
Analysis of SSRs
In aggregate, 63,118 SSRs were obtained from 53,759 unigenes, among which had at least one SSR. Most of the SSRs were mononucleotide repeats, accounting for 59.86%, followed by the dinucleotide repeats accounting for 25.97%, trinucleotide repeats accounting for 11.01%, tetranucleotide repeats accounting for 2.53%, pentanucleotide repeats accounting for 0.56%, and the hexanucleotide repeats accounting for 0.08% (Supplementary Figure 4 and Supplementary Table 5).
Statistics of the DEGs
Table 5 shows that the SBM40 group had 2,305 significant DEGs (P < 0.05), of which 1,256 were significantly upregulated and 1,049 were significantly downregulated. The SPC40 group had 4,076 significant DEGs (P < 0.05), of which 2,328 were significantly upregulated and 1,748 were significantly downregulated. The FSBM40 group had 3,462 significant DEGs (P < 0.05), of which 2,005 were significantly upregulated and 1,457 were significantly downregulated.
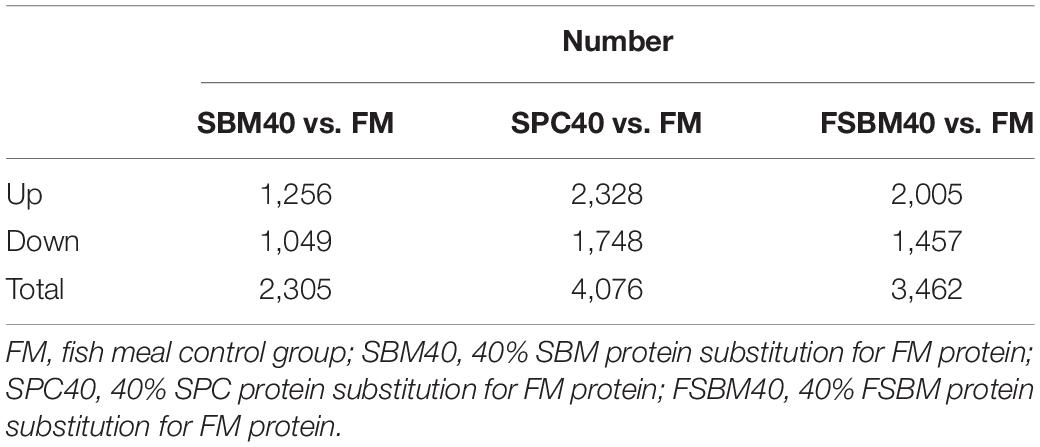
Table 5. Comparison of the significantly differential expressed genes of the three soy protein substitutions for fish meal in the distal intestine of the pearl gentian grouper (n = 4).
The Venn diagram of the DEGs displayed that, compared to the FM control group, the common DEGs in SBM40, SPC40, and FSBM40 were 554, named Profile G; 1,003 unique DEGs in the SBM40 group, named profile H; 2,254 unique DEGs in the SPC40 group, name Profile I; and 1,656 unique DEGs in the FSBM40 group, name Profile J (Figure 8). Only 7.80% (554/7,101) of the DEGs in the three groups have similar expression patterns, indicating that the three soy proteins have different metabolic strategies.
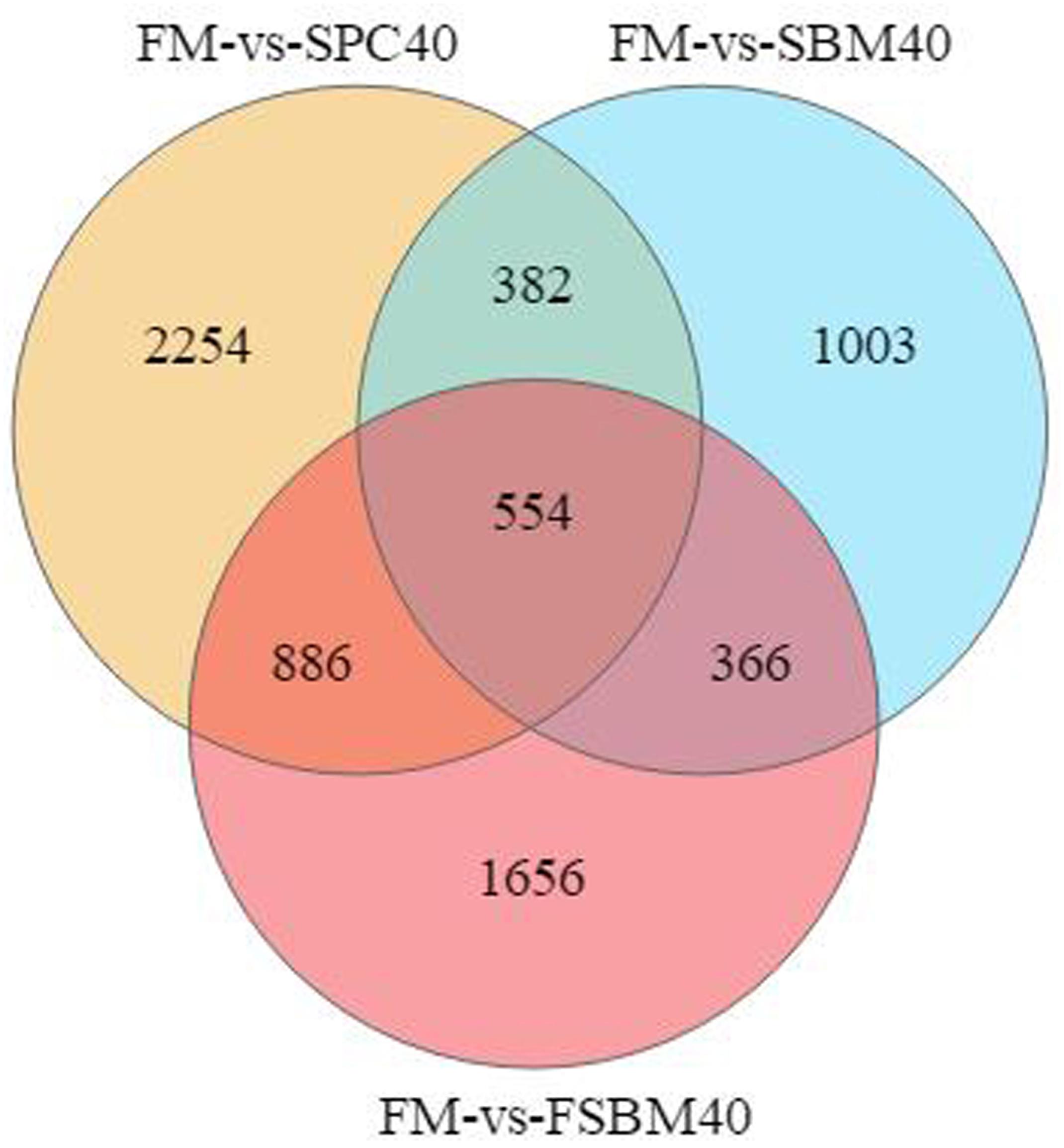
Figure 8. Venn diagram analysis of the significantly differentially expressed genes (DEGs) of the soy protein substitutes for fish meal in the distal intestine of the pearl gentian grouper (n = 4).
KEGG Enrichment Analysis of the DEGs
KEGG enrichment analysis was performed on profiles G, H, I, and J. The enrichment of Profile G showed that 238 pathways were enriched and 30 pathways were significant (P < 0.05). Among all the pathways, 73 were related to immune disease/system, infectious diseases, and signal transduction, nine of which were significantly enriched (P < 0.05). That is to say, 30% (9/30) of all the pathways were related to immune diseases/system, infectious diseases, and signal transduction (Figure 9A). Profile H enrichment results found that 297 pathways were enriched and 51 pathways were significant (P < 0.05). Among all the pathways, 79 were related to immune disease/system, infectious diseases, and signal transduction, 23 of which were significantly enriched (P < 0.05). That is to say, 45.10% (23/51) of all the pathways were related to immune diseases/system, infectious diseases, and signal transduction (Figure 9B). Profile I enrichment results found that 320 pathways were enriched and 35 pathways were significant (P < 0.05). Among all the pathways, 80 were related to immune disease/system, infectious diseases, and signal transduction, one of which was significantly enriched (P < 0.05). That is to say, 2.86% (1/35) of all the pathways were related to immune diseases/system, infectious diseases, and signal transduction. Most of the significant pathways were related to fat digestion and absorption, alpha-linolenic acid metabolism, glycerophospholipid metabolism, fatty acid metabolism, linoleic acid metabolism, biosynthesis of unsaturated fatty acid and protein digestion and absorption, etc., accounting for 85.71% (30/35) (Figure 9C). Profile J enrichment results found that 305 pathways were enriched and 38 pathways were significant (P < 0.05). Among all the pathways, 81 were related to immune disease/system, infectious diseases, and signal transduction, 23 of which were significantly enriched (P < 0.05). That is to say, 60.53% (23/38) of all the pathways were related to immune diseases/system, infectious diseases, and signal transduction (Figure 9D).
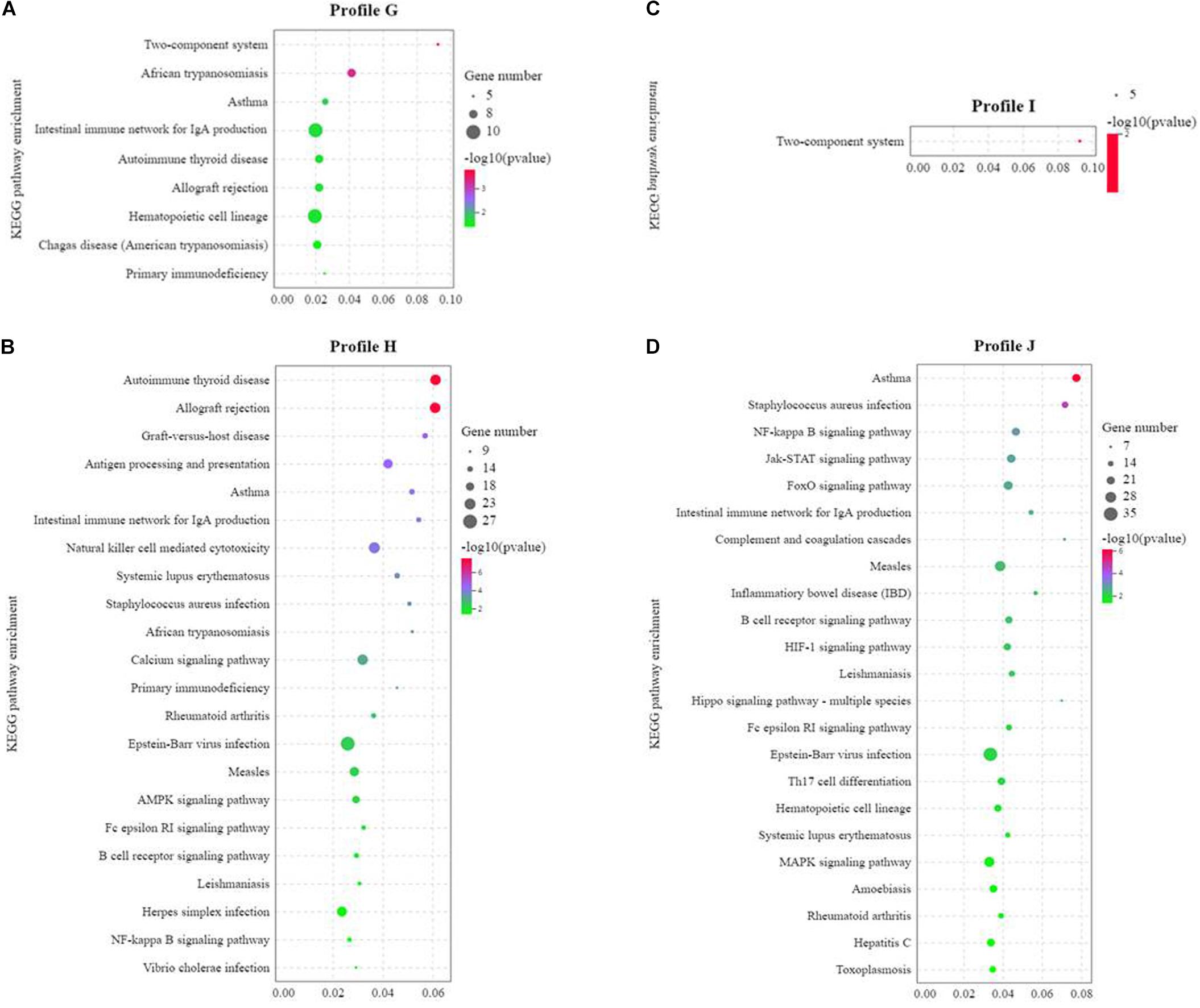
Figure 9. Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of the significantly differentially expressed genes (DEGs) of the three soy protein substitutes for fish meal in the distal intestine of the pearl gentian grouper (n = 4). (A) The KEGG enrichment analysis of the common differential genes in SBM40, SPC40, and FSBM40 groups. (B) The KEGG enrichment analysis of the unique differential genes in SBM40 group. (C) The KEGG enrichment analysis of the unique differential genes in SPC40 group. (D) The KEGG enrichment analysis of the unique differential genes in FSBM40 group.
Validation of the RNA-Seq Data by RT-qPCR
Generally speaking, the trend of the RT-qPCR results was consistent with that of the transcriptome sequencing data, indicating that the RNA-Seq results were relatively accurate (Figure 10). These results further confirmed the reliability of the “3 + 2” transcriptome sequencing strategy.
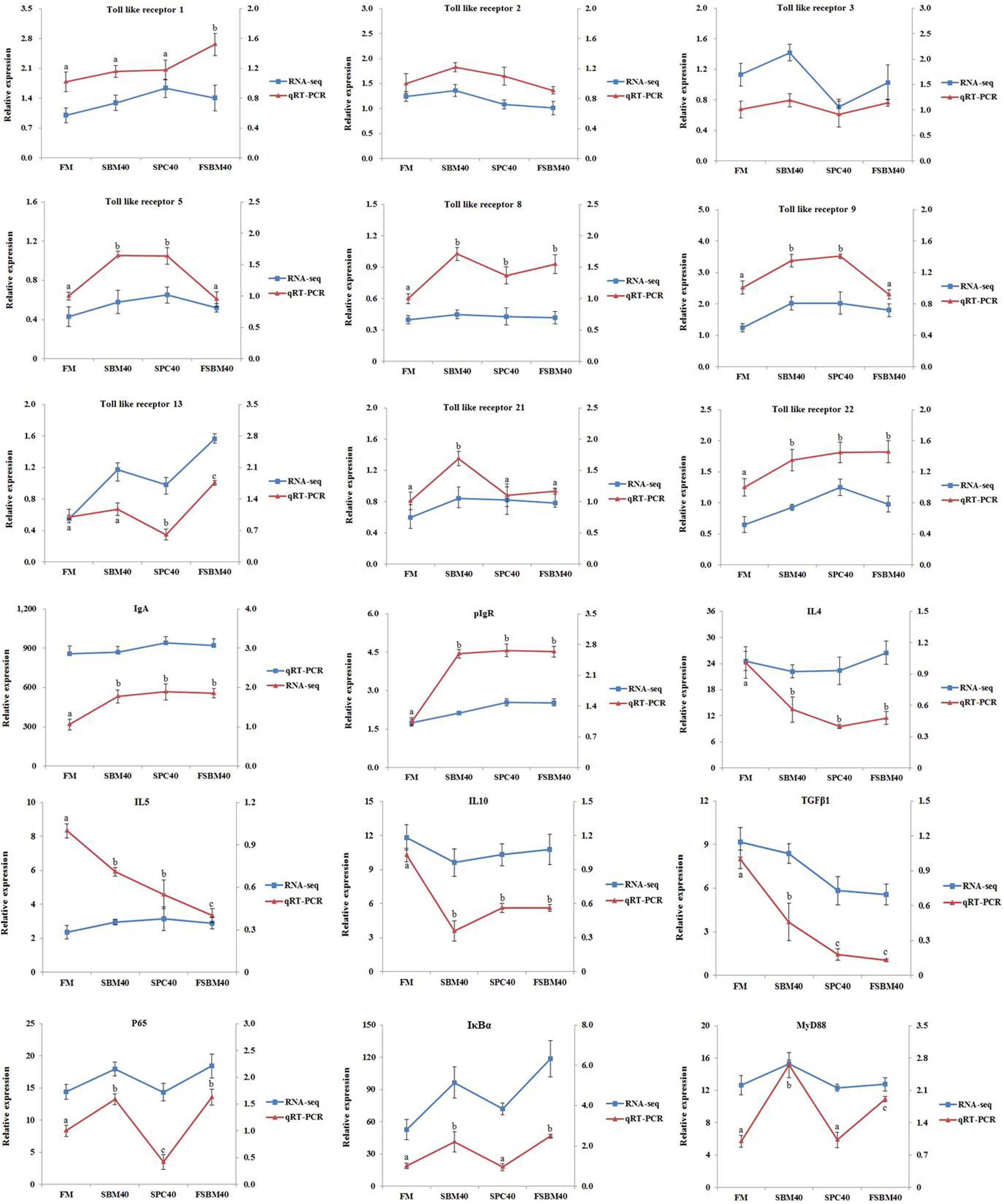
Figure 10. Comparison of the RNA sequencing (RNA-Seq) and the quantitative reverse transcription PCR (RT-qPCR) results. In order to validate the RNA-Seq results, RT-qPCR was used to detect the gene expression levels of the TLR/myD88/NF-κB pathway and the intestinal immune network for IgA production pathway in the distal intestine (DI) tissues of the pearl gentian grouper. The mRNA expression level in RT-qPCR was normalized by β-actin. The relative expression level in the RNA-Seq analysis was calculated by the FPKM (fragments per kilobase of transcript per million mapped reads) value. The statistical results were expressed as the mean ± SD. Different letters assigned to the lines represent significant differences among the groups at P < 0.05. FM, fish meal control group; SBM40, 40% soybean meal (SBM) protein replacement level for FM protein; SPC40, 40% soybean protein concentrate (SPC) protein replacement level for FM protein; FSBM40, 40% fermented soybean meal (FSBM) protein replacement level for FM protein.
Discussion
The present study showed that experimental levels of soy proteins from SBM, SPC, and FSBM as substitutes for FM presented significantly negative effects on the growth performance and intestinal health of the pearl gentian grouper. A related study on E. coioides (initial weight = 84 ± 2.5 g) found that fish had the best growth performance at the level of 20% SBM substitution for FM (basal FM = 60%) (An et al., 2018). The unpublished research in our lab also found that the optimal substitution level of SBM for the pearl gentian grouper (initial weight = 17.01 ± 0.01 g) was 12.05% (basal FM = 50%). Research has found that 20–50% SPC substitution for FM (50% basal FM) had no significant effects on growth performance in the brown-marbled grouper (initial weight = 6.1 ± 0.7 g), while the growth performance significantly decreased at 60% SPC substitution level (Faudzi et al., 2018). However, it was found that the replacement of FM with SPC less than 40% had significantly negative effects on the WGR, SGR, and feed efficiency of Scophthalmus maximus and P. olivaceus (Deng et al., 2006; Liu et al., 2014). There were no significant effects on the final body weight, SGR, and FCR by replacing 30% of FM with FSBM (50% basal FM) in the diet of Acanthopagrus latus, while in the diet of Micropterus salmoides, fish had better growth, physiology, and apparent digestibility when the FSBM substitution for FM was no more than 10% (Wang, 2009; Ehsani et al., 2014). A previous study on E. coioides showed that a 14% dietary FSBM did not significantly affect the WGR and SGR values; however, at higher levels, the WGR and SGR values decreased significantly. The optimal level of FSBM substitution was 10% (52% basal FM) (Luo et al., 2004). In general, the present study obtained consistent results. The difference may be caused by the different varieties of breeding animals.
Previous studies have shown that the characteristics of soy meal-induced enteritis include a reduced mucosal fold height, swelling of the lamina propria and subepithelial mucosa, loss of normal enterocyte supranuclear absorptive vacuolization, and profound infiltration of various inflammatory cells, which decreased the capacity of the DI to digest and absorb nutrients (Gu et al., 2018). The present study found a similar phenomenon. The intestinal structure of fish is very sensitive to oxidative damage. Fish can resist oxidative damage through antioxidant enzymes, such as total GR, GPx, and T-SOD (Zhao et al., 2014). The significant increases of the GR, GPx, and T-SOD enzyme activities in this study indicated that soy proteins induced intestinal stress. IgM is an important component of specific immunity. The present study found that the content of IgM significantly decreased, indicating an impairment of the intestinal immune function of the pearl gentian grouper. MDA is one of the final products of oxidative stress, the concentrations of which indicate the rate or intensity of lipid peroxidation in tissues and cells (Wen et al., 2015). In the present study, the concentration of MDA in the DI tissues significantly increased in the experimental groups, indicating that soy proteins caused intestinal injury in the pearl gentian grouper. ALT and AST are two important and sensitive indicators of hepatocyte injury when liver lesions occur (Kalhoro et al., 2018; Zhou et al., 2018). Previous research also revealed that excessive dietary SBM for the Atlantic salmon and grass carp induced liver lesions (De Santis et al., 2015; Wu et al., 2018). The present study found similar results. Based on the above analysis, the pearl gentian grouper showed the typical characteristics of fish intestinal health issues caused by soy proteins. In order to conduct a systematic and in-depth study, the present study constructed and analyzed the characteristics of the full-length transcriptome database of the pearl gentian grouper by using omics technology.
Genome and transcriptome sequencing is a fundamental work in the field of life science. Due to the lack the genomic data for most non-model organisms, full-length transcriptome sequencing becomes particularly important. Full-length transcripts can greatly improve the basic and applied research on gene function, gene expression regulation, and the evolutionary relationships of these species (Ren et al., 2006). Previously, obtainment of a full-length gene on a large scale is almost impossible, which is also time-consuming and expensive through RACE and Illumina technology in general (Wan et al., 2019). Currently, most of the transcriptome data are obtained based on next-generation sequencing technologies, such as Illumine, Heliscope, Roche 454, Solexa, and SOLID (Lobato et al., 2017). The length of the sequences obtained by second-generation sequencing technologies is short, and the splicing of short sequences cannot provide a large number of long transcripts and lose important information, such as alternative splicing (Sun, 2016). Therefore, the third-generation sequencing technology of PacBio SMRT is usually used for transcriptome de novo sequencing.
Full-length transcripts are very important for the research of genome assembly and gene function, and the PacBio SMRT sequencing technology can obtain full-length transcripts on a large scale (Wong et al., 2019). This study obtained 82,351 high-quality unique transcripts, and 86.22% were full-length transcripts. This result showed that the third-generation sequencing technology is more efficient than the next-generation sequencing technology (Yang et al., 2017). According to the published literature, only a small number of species had their transcriptomes obtained based on PacBio platform, including the transcriptome data of the hybrid splicing of second- and third-generation sequencing or the corrected third generation through second-generation sequencing technology. Most of the transcriptome data obtained completely based on the PacBio platform are from human beings (Au et al., 2013), and there are also data on HIV virus (Ocwieja et al., 2012), bovine (Larsen and Smith, 2012), Mus musculus (Treutlein et al., 2014), Propithecus coquereli (Larsen et al., 2014), etc. However, research on full-length transcriptome sequencing based on the PacBio platform was just carried out in recent years, and it was not until 2015 that the sequencing of fungi (Gordon et al., 2015), Gossypium hirsutum (van Eijk, 2015), and Sepia officinalis (Worley, 2015) was carried out. The full-length transcripts obtained in this research would improve further investigation of the pearl gentian grouper.
The longest transcript obtained in this study is 14,637 bp and the N50 is 4,131 bp, which is much longer than that in the pearl gentian grouper used in the Illumina sequencing. For example, our unpublished research found that the N50 values of the assembled unigenes were only 1,886 and 1,921 bp in the intestine and liver transcriptomes of the pearl gentian grouper, respectively. The results indicate that the PacBio SMRT sequencing technology has more advantage in terms of reading the sequence length.
Previous studies have pointed out that the annotation rate of the third-generation sequencing data is higher than that of the second-generation sequencing data (Zeng et al., 2018). In our unpublished research on the pearl gentian grouper transcriptomes of the intestine and liver tissues, the transcript annotation rates were 32.64 and 36.58%, respectively. Also, in our published articles on M. salmoides and Trachinotus ovatus, in which the transcriptomes were sequenced by the Illumina 2000 platform, the annotation rates of the transcripts were 52.98% (26,886/50,743) (Zhang et al., 2019) and 43.30% (27,366/62,377) (Liu et al., 2019), respectively. Although the raw data obtained from third-generation sequencing had relatively more error, it can be corrected through the data obtained from next-generation sequencing (Hackl et al., 2014). The raw data in this study had been corrected using the transcriptome data sequenced by the Illumina 4000 platform, which would ensure the accuracy of the PacBio SMRT results. Finally, the annotation rate of the transcripts in this study is 94.5%, which is much higher than that previously obtained using the Illumina sequencing technology.
Public databases such as the Nr, Nt, Pfam, KOG, Swiss-Prot, KEEG, and GO have been widely applied for functional annotation of transcriptome sequences. Nr and Nt are the official protein and nucleic acid sequence databases in NCBI (Feng et al., 2019). In this study, 78.70 and 97.61% of the FL transcripts were annotated in Nr and Nt, respectively, which indicated that most of the transcripts were annotated and only contained few non-coding sequences, such as lncRNAs. For the rest of the databases, the highest ratio of the transcripts was in KEGG (75.94%), followed by Swiss-Prot (67.79%), KOG (52.38%), GO (45.44%), and Pfam (45.44%). In our previous second-generation transcriptome sequencing data of pearl gentian grouper intestinal tissues, the annotation rate in Nr was 30.78%, followed by KOG (18.11%), KEGG (17.18%), and Swiss-Prot (15.35%, unpublished). The percentage of the annotated transcripts in this study was higher than those reported by RNA-Seq, which also showed advantages of the third-generation sequencing technology.
lncRNAs are rapidly evolving and are often species-specific, which play vital roles in many physiological processes such as translation, transcription, differentiation, splicing, immune responses, epigenetic regulation, and cell cycle control (Chen and Yan, 2013). Previous research reported that the function and mechanism of lncRNAs are complex and may have competitive relationship with miRNAs when interacting with lncRNAs (Yoon et al., 2014). However, the identification of lncRNAs in the pearl gentian grouper using full-length sequencing technology has not been reported yet. There are 8,874 common lncRNAs that were predicted by the four software in this study, which would be useful for further research on the pearl gentian grouper, including epigenetics, immunology, and phylogenomics (Zeng et al., 2018).
Based on PacBio SMRT full-length transcriptome sequencing, the present study preliminarily investigated the differential mechanisms of enteritis in the pearl gentian grouper induced by different soy proteins. Similar to previous studies on plant protein-induced fish enteritis, some conserved signaling pathways, such as the nuclear factor kappa B (NF-κB) signaling pathway, were found in the intestine transcriptome of pearl gentian fed with the SBM40 and FSBM40 diets. Previous studies indicated that Atlantic salmon fed with SPC did not show changes in the transcriptome levels similar to SBM-induced enteritis (Król et al., 2016). The present study also found that the intestinal transcription profile of pearl gentian grouper fed the SPC40 diet was significantly different from those of the SBM40 and FSBM40 diets. Only 2.86% of the signaling pathways related to immune diseases/system, infectious diseases, and signal transduction were significantly affected, while 85.71% of the signaling pathways related to nutrition digestion and absorption were significantly affected. However, in the common Profile G, some signaling pathways closely related to intestinal immunity were also enriched, such as intestinal immune network for immunoglobulin A (IgA) production.
The intestinal tract is the largest lymphoid tissue in the human body. A remarkable feature of intestinal immunity is that it can produce a large number of IgA antibodies as the first line of defense against microorganisms (Mestecky et al., 1999). There are a few studies on the signaling pathway of the intestinal immune network for IgA production in fish. In mammalian studies, it has been found that IgA production is induced by the interaction of specific antigen and innate immune receptors, such as Toll-like receptor 2 (TLR2), TLR4, and TLR9 (Suzuki and Fagarasan, 2008). Related studies also revealed that the TLR/NF-κB signaling pathway is the main component of inflammation and immune response (Tan et al., 2016). Based on the above analysis, this study focused on the role of the TLR-mediated NF-κB signaling pathway and the intestinal immune network for IgA production pathway in the development of SBM-, SPC-, and FSBM-induced enteritis in the pearl gentian grouper.
A total of nine TLR members were found in the intestinal tissues of the pearl gentian grouper, including TLR1, TLR2, TLR3, TLR5, TLR8, TLR9, TLR13, TLR21, and TLR22. At present, there are 20 TLRs found in fish, at least. Among the TLRs found in this experiment, TLR1, TLR2, TLR3, TLR5, TLR9, TLR21, and TLR22 have been reported as sensors for bacterial ligands in fish (Wei et al., 2011; Yeh et al., 2013; Byadgi et al., 2014). The present study showed that the expression levels of TLR5, TLR8, TLR9, TLR21, and TLR22 were significantly increased with the addition of the SBM40 diet; TLR5, TLR8, TLR9, and TLR22 were significantly increased with the SPC40 diet addition; and TLR1, TLR8, TLR13, and TLR22 were significantly increased with the addition of the FSBM40 diet, indicating that the signal transduction of the TLRs was activated by various bacterial components/products after different soy protein substitutions for FM protein.
In addition, the present study found that, compared with the control group, the addition of SBM, SPC, and FSBM resulted in the significant downregulation of IL4, IL5, IL10, and TGF-β expressions, but the expression of IgA increased significantly. Relevant studies pointed out that IL4, IL5, IL6, and IL10 secreted by helper T cells may play important roles in promoting IgA secretion (Barnes et al., 2011). In this experiment, the three soy protein diets all caused the high expression of IgA, which may be the manifestation of intestinal immune imbalance in the pearl gentian grouper. The specific reasons need to be further studied.
Taken together, this study analyzed the full-length transcriptome of the pearl gentian grouper intestine using the PacBio SMRT sequencing technology, which represents the first third-generation long-read transcriptome sequencing of the pearl gentian grouper. The obtained transcriptome data may improve further studies on the pearl gentian grouper.
Data Availability Statement
The original contributions presented in the study are publicly available. The Pacbio SMRT sequencing raw reads and Illumina sequencing raw reads are deposited in NCBI Sequence Read Archive (SRA) and the accession numbers are PRJNA664623 and PRJNA664416, respectively.
Ethics Statement
The animal protocol was approved by the Ethics Review Board of the Guangdong Ocean University. All procedures were performed in accordance with the standards of the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 8023, revised 1978) and relevant Chinese policies.
Author Contributions
WZ designed and took part in the whole process of the experiment and wrote the draft of this manuscript. BT and JD co-conceived the experiment and revised the draft critically for important intellectual content. XD and QY participated in the experiments. SC revised the first draft. HL and SZ analyzed the data. SX and HZ approved the final version. All authors contributed to the article and approved the submitted version.
Funding
This research was supported by the National Key R&D Program of China (2019YFD0900200).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the Key Laboratory of Aquatic, Livestock and Poultry Feed Science and Technology in South China, Ministry of Agriculture, for providing technical assistances.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.688601/full#supplementary-material
Supplementary Figure 1 | Effect of different soy proteins at 40% substitution levels for fish meal protein on the hepatosomatic index and survival rate of pearl gentian grouper (n = 3).
Supplementary Figure 2 | Hematoxylin-eosin staining in the distal intestine of pearl gentian grouper. Representative images of increased width and cellular (leucocyte) infiltration (asterisk) of the lamina propria (arrow) in distal intestine depicting the inflammatory changes of grouper fed the FM (A), SBM40 (B), SPC40 (C), and FSBM40 (D) diets. FM, fish meal control group; SBM40, 40% SBM protein substitution for FM protein; SPC40, 40% SPC protein substitution for FM protein; FSBM40, 40% FSBM protein substitution for FM protein.
Supplementary Figure 3 | Analysis of long read transcriptome of pearl gentian grouper by SMRT sequencing. (A) The flow chart of transcriptome analysis of hybrid grouper in this study; (B) classification of total consensus sequence reads; (C) length distribution of the unigenes; (D) number of transcripts corresponding to unigenes.
Supplementary Figure 4 | Predicted simple sequence repeats (SSRs) of the long read transcripts (n = 4).
References
Aimin, L., Zhang, J. Y., and Zhou, Z. Y. (2014). Plek: a tool for predicting long non-coding RNAs and messenger RNAs based on an improved k-mer scheme. BMC Bioinformatics 15:311. doi: 10.1186/1471-2105-15-311
An, M. L., Fan, Z., Wang, Q. K., Sun, J. H., Xu, D. W., Guo, Y. J., et al. (2018). Effect of replacing fish meal with soybean meal on growth, digestion and antioxidant ability of Epinephelus coioides. Jiangsu Agric. Sci. 46, 128–132. doi: 10.15889/j.issn.1002-1302.2018.16.032
Au, K. F., Sebastiano, V., Afshar, P. T., Durruthy, J. D., Lee, L., Williams, B. A., et al. (2013). Characterization of the human ESC transcriptome by hybrid sequencing. Proc. Natl. Acad. Sci. U.S.A. 110, E4821–E4830. doi: 10.1073/pnas.1320101110
Barnes, S., Prasain, J., D’Alessandro, T., Arabshahi, A., Botting, N., Lila, M., et al. (2011). The metabolism and analysis of isoflavones and other dietary polyphenols in foods and biological systems. Food Funct. 2, 235–244. doi: 10.1039/c1fo10025d
Booman, M., Forster, I., Vederas, J. C., Groman, D. B., and Jones, S. R. M. (2018). Soybean meal-induced enteritis in Atlantic salmon (Salmo salar) and Chinook salmon (Oncorhynchus tshawytscha) but not in pink salmon (O. gorbuscha). Aquaculture 483, 238–243. doi: 10.1016/j.aquaculture.2017.10.025
Brawand, D., Wagner, C. E., Li, Y. I, Malinsky, M., Keller, I., Fan, S., et al. (2014). The genomic substrate for adaptive radiation in African cichlid fish. Nature 513, 375–381. doi: 10.1038/nature13726
Byadgi, O., Puteri, D., Lee, Y. H., Lee, J. W., and Cheng, T. C. (2014). Identification and expression analysis of cobia (Rachycentron canadum) Toll-like receptor 9 gene. Fish Shellfish Immun. 36, 417–427. doi: 10.1016/j.fsi.2013.12.017
Cai, L. S., Wang, L., Song, K., Lu, K. L., Zhang, C. X., and Rahimnejad, S. (2020). Evaluation of protein requirement of spotted seabass (Lateolabrax maculatus) under two temperatures, and the liver transcriptome response to thermal stress. Aquaculture 516:734615. doi: 10.1016/j.aquaculture.2019.734615
Chen, X., and Yan, G. Y. (2013). Novel human lncRNA-disease association inference based on lncRNA expression profles. Bioinformatics 29, 2617–2624. doi: 10.1093/bioinformatics/btt426
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
De Santis, C., Ruohonen, K., Tocher, D. R., Martin, S. A. M., Król, E., Secombes, C. J., et al. (2015). Atlantic salmon (Salmo salar) parr as a model to predict the optimum inclusion of air classified faba bean protein concentrate in feeds for seawater salmon. Aquaculture 444, 70–78. doi: 10.1016/j.aquaculture.2015.03.024
Deng, J. M., Mai, K. S., Ai, Q. H., Zhang, W. B., Wang, X. J., Xu, W., et al. (2006). Effects of replacing fish meal with soy protein concentrate on feed intake and growth of juvenile Japanese flounder, Paralichthys olivaceus. Aquaculture 258, 503–513. doi: 10.1016/j.aquaculture.2006.04.004
Ehsani, J., Maniat, M., Azarm, H. M., and Ghabtani, A. (2014). Effects of partial substitution of dietary fish meal by fermented soybean meal on growth performance, body composition and activity of digestive enzymes of juvenile yellowfin sea bream (Acanthopagrus latus). Int. J. Biosci. 5, 99–107. doi: 10.12692/ijb/5.4.99-107
Faudzi, N. M., Yong, A., Shapawi, R., Senoo, S., and Takii, K. (2018). Soy protein concentrate as an alternative in replacement of fish meal in the feeds of hybrid grouper, brown-marbled grouper (Epinephelus fuscoguttatus) × giant grouper (E. lanceolatus) juvenile. Aquac. Res. 49, 431–441. doi: 10.1111/are.13474
Feng, X., Jia, Y. T., Zhu, R., Chen, K., and Chen, Y. F. (2019). Characterization and analysis of the transcriptome in Gymnocypris selincuoensis on the Qinghai-Tibetan Plateau using single-molecule long-read sequencing and RNA-seq. DNA Res. 26, 353–363. doi: 10.1093/dnares/dsz014
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Gordon, S. P., Tseng, E., Salamov, A., Zhang, J., Meng, X., Zhao, Z., et al. (2015). Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One 10:e0132628. doi: 10.1371/journal.pone.0132628
Gu, M., Jia, Q., Zhang, Z. Y., Bai, N., Xu, X. J., and Xu, B. (2018). Soya-saponins induce intestinal inflammation and barrier dysfunction in juvenile turbot (Scophthalmus maximus). Fish Shellfish Immun. 77, 264–272. doi: 10.1016/j.fsi.2018.04.004
Guardiola, F. A., Porcino, C., Cerezuela, R., Cuesta, A., Faggio, C., and Esteban, M. A. (2016). Impact of date palm fruits extracts and probiotic enriched diet on antioxidant status, innate immune response and immune-related gene expression of European seabass (Dicentrarchus labrax). Fish Shellfish Immun. 52, 298–308. doi: 10.1016/j.fsi.2016.03.152
Gulcher, J. (2012). Microsatellite markers for linkage and association studies. Cold Spring Harb. Protoc. 4:425. doi: 10.1101/pdb.top068510
Hackl, T., Hedrich, R., Schultz, J., and Forster, F. (2014). Proovread: large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30, 3004–3011. doi: 10.1093/bioinformatics/btu392
Jia, X., Tang, L., Mei, X., Liu, H., and Su, J. (2020). Single-molecule long-read sequencing of the full-length transcriptome of Rhododendron lapponicum L. Sci. Rep. 10:6755. doi: 10.1038/s41598-020-63814-x
Kalhoro, H., Zhou, J., Hua, Y., Ng, W. K., Ye, L., Zhang, J., et al. (2018). Soy protein concentrate as a substitute for fish meal in diets for juvenile Acanthopagrus schlegelii: effects on growth, phosphorus discharge and digestive enzyme activity. Aquac. Res. 49, 1896–1906. doi: 10.1111/are.13645
Kong, L., Zhang, Y., Ye, Z. Q., Liu, X. Q., Zhao, S. Q., Wei, L., et al. (2007). CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349. doi: 10.1093/nar/gkm391
Król, E., Douglas, A., Tocher, D. R., Crampton, V. O., Speakman, J. R., Secombes, C. J., et al. (2016). Differential responses of the gut transcriptome to plant protein diets in farmed Atlantic salmon. BMC Genomics 17:156–172. doi: 10.1186/s12864-016-2473-0
Larsen, P. A., Campbell, C. R., and Yoder, A. D. (2014). Next-generation approaches to advancing eco-immunogenomic research in critically endangered primates. Mol. Ecol. Resour. 14, 1198–1209. doi: 10.1111/1755-0998.12274
Larsen, P. A., and Smith, T. P. (2012). Application of circular consensus sequencing and network analysis to characterize the bovine IgG repertoire. BMC Immunol. 13:52. doi: 10.1186/1471-2172-13-52
Li, P., Deng, W., Li, T., Song, B., and Shen, Y. (2013). Illumina-based de novo transcriptome sequencing and analysis of Amanita exitialis basidiocarps. Gene 532, 63–71. doi: 10.1016/j.gene.2013.09.014
Liu, K., Tan, B. P., Zhang, W., Liu, H. Y., Dong, X. H., Yang, Q. H., et al. (2019). Transcriptome, enzyme activity and histopathology analysis reveal the effects of high level of dietary carbohydrate on glycometabolism in juvenile golden pompano, Trachinotus ovatus. Aquac. Res. 1, 1–15. doi: 10.1111/are.14096
Liu, X. W., Ai, Q. H., Mai, K. S., Liu, F. Z. G., and Xu, W. (2014). Effects of replacing fish meal with soy protein concentrate on feed intake and growth of turbot (Scophthalmus maximus). J. Fish. China 38, 91–98. doi: 10.3724/SP.J.1231.2014.48852
Liu, Z., Liu, S., Yao, J., Bao, L., Zhang, J., Li, Y., et al. (2016). The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat. Commun. 7:11757. doi: 10.1038/ncomms11757
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2–ΔΔ method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lobato, F. M. F., Damasceno, C. D., Leite, D. S., dos Santos, ÂK. R., and De Santana, ÁL. (2017). Data analysis of multiplex sequencing at solid platform: a probabilistic approach to characterization and reliability increase. Am. J. Resp. Cell. Mol. 8, 26–38. doi: 10.4236/ajmb.2018.81003
Luo, Z. Y., Liu, Y. J., Mai, K. S., Tian, L. X., Liu, D. H., and Tan, X. Y. (2004). Partial replacement of fish meal by soybean protein in diets for grouper Epinephelus coioides juveniles. J. Fisheries China 2, 175–181.
Martin, S. A. M., and Król, E. (2017). Nutrigenomics and immune function in fish: new insights from omics technologies. Dev. Comp. Immunol. 75, 86–98. doi: 10.1016/j.dci.2017.02.024
Mestecky, J., Moro, I., and Underdown, B. J. (1999). Mucosal Immunology, eds P. Ogra et al. (San Diego, CA: Academic Press), 133–152.
Miao, S., Zhao, C., Zhu, J., Hu, J., Dong, X., and Sun, L. (2018). Dietary soybean meal affects intestinal homoeostasis by altering the microbiota, morphology and inflammatory cytokine gene expression in northern snakehead. Sci. Rep. 8, 113–123. doi: 10.1038/s41598-017-18430-7
Ocwieja, K. E., Scott, S. M., Rithun, M., Rebecca, C. A., Patricia, D., Michael, B., et al. (2012). Dynamic regulation of HIV-1 mRNA populations analyzed by single-molecule enrichment and long-read sequencing. Nucleic Acids Res. 40, 10345–10355. doi: 10.1093/nar/gks753
Ren, Y. P., Zhang, J. Q., Sun, Y., Wu, Z. F., Ruan, J. S., He, B. J., et al. (2006). Full-length transcriptome sequencing on PacBio platform. Sci. Bull. 61, 1250–1254. doi: 10.1360/N972015-01384
Roberts, R. J., Carneiro, M. O., and Schatz, M. C. (2013). The advantages of SMRT sequencing. Genome Biol. 14:405. doi: 10.1186/gb-2013-14-7-405
Shimizu, K., Adachi, J., and Muraoka, Y. (2006). ANGLE: a sequencing errors resistant program for predicting protein coding regions in unfinished cDNA. J. Bioinf. Comput. Biol. 4, 649–664. doi: 10.1142/S0219720006002260
Star, B., Nederbragt, A. J., Jentoft, S., Grimholt, U., Malmstrøm, M., Gregers, T. F., et al. (2011). The genome sequence of Atlantic cod reveals a unique immune system. Nature 477, 207–210. doi: 10.1038/nature10342
Sun, L., Luo, H. T., Bu, D. C., Zhao, G. G., Yu, K. T., Zhang, C. H., et al. (2013). Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 41:e166. doi: 10.1093/nar/gkt646
Sun, L. R. (2016). Design and Evaluation of Two Hybrid Genome Assembly Approaches Using Illumina, Roche 454, and PacBio Datasets. Fargo, ND: North Dakota State University.
Suzuki, K., and Fagarasan, S. (2008). How host-bacterial interactions lead to IgA synthesis in the gut. Trends Immunol. 29, 523–531. doi: 10.1016/j.it.2008.08.001
Tan, P., Dong, X. J., Mai, K. S., Xu, W., and Ai, Q. H. (2016). Vegetable oil induced inflammatory response by altering TLR-NF-κB signalling, macrophages infiltration and polarization in adipose tissue of large yellow croaker (Larimichthys crocea). Fish Shellfish Immun. 59, 398–405. doi: 10.1016/j.fsi.2016.11.009
Teves, J. F. C., and Ragaza, J. A. (2014). The quest for indigenous aquafeed ingredients: a review. Rev. Aquac. 8, 154–171. doi: 10.1111/raq.12089
Thiel, T. (2003). MISA-Microsatellite Identifcation Tool. Available online at: http://pgrc.ipk-gatersleben.de/misa/ (accessed June 17, 2016).
Tine, M., Kuhl, H., Gagnaire, P. A., Louro, B., Desmarais, E., Martins, R. S. T., et al. (2014). European sea bass genome and its variation provide insights into adaptation to euryhalinity and speciation. Nat. Commun. 5:5770. doi: 10.1038/ncomms6770
Treutlein, B., Gokce, O., Quake, S. R., and Südhof, T. C. (2014). Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc. Natl. Acad. Sci. U.S.A. 111, E1291–E1299. doi: 10.1073/pnas.1403244111
van Eijk, M. (2015). “Genome assembly and Iso-Seq transcriptome sequencing of tetraploid cotton,” in Proceedings of the Plant and Animal Genome XXIII Conference. Plant and Animal Genome, San Diego, CA.
Wan, H. F., Jia, X. W., Zou, P. F., Zhang, Z. P., and Wang, Y. L. (2019). The Single-molecule long-read sequencing of Scylla paramamosain. Sci. Rep. 9:12401. doi: 10.1038/s41598-019-48824-8
Wang, X. X. (2009). Study of fermented soybean meal as a substitute for fish meal in California perch feed. Aquatic. Feed Techn. 1, 58–61.
Wang, Y. P., Lu, Y., Zhang, Y., Ning, Z. M., Li, Y., Zhao, Q., et al. (2015). The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nat. Genet. 47, 625–631. doi: 10.1038/ng.3280
Wei, Y. C., Pan, T. S., Ming, X. C., Bei, H., Zhen, X., Luo, T. R., et al. (2011). Cloning and expression of toll-like receptors 1 and 2 from a teleost fish, the orange-spotted grouper epinephelus coioides. Vet. Immunol. Immunop. 141, 173–182. doi: 10.1016/j.vetimm.2011.02.016
Wen, L. M., Jiang, W. D., Liu, Y., Wu, P., Zhao, J., Jiang, J., et al. (2015). Evaluation the effect of thiamin deficiency on intestinal immunity of young grass carp (Ctenopharyngodon idella). Fish Shellfish Immun. 46, 501–515. doi: 10.1016/j.fsi.2015.07.001
Wong, K. C., Zhang, J., Yan, S., Li, X., Lin, Q., Kwong, S., et al. (2019). DNA sequencing technologies: sequencing data protocols and bioinformatics tools. ACM Comput. Surv. 52, 1–30. doi: 10.1145/3340286
Workman, R. E., Myrka, A. M., William, W. G., Elizabeth, T., Welch, K. C., and Winston, T. (2018). Single molecule, full-length transcript sequencing provides insight into the extreme metabolism of ruby-throated hummingbird Archilochus colubris. GigaScience 7, 1–12. doi: 10.1093/gigascience/giy009
Worley, K. C. (2015). “European cuttlefish whole transcriptome sequencing: a single-molecule full length transcript survey with Iso-Seq method,” in Proceedings of the Lant and Animal Genome XXIII Conference. Plant and Animal Genome, San Diego, CA.
Wu, N., Wang, B., Cui, Z. W., Zhang, X. Y., Cheng, Y. Y., Xu, X., et al. (2018). Integrative transcriptomic and microRNAomic profiling reveals immune mechanism for the resilience to soybean meal stress in fish gut and liver. Front. Physiol. 9:1154. doi: 10.3389/fphys.2018.01154
Xiang, F. Q. (2017). Effects of Sea Bass on Growth, Immune and Intestinal Flora by Soy Protein Concentrate. Changsha: Hunan Agricultural University.
Xu, P., Zhang, X., Wang, X., Li, J., Liu, G., Kuang, Y., et al. (2014). Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 46, 1212–1219. doi: 10.1038/ng.3098
Yang, X., Ikhwanuddin, M., Li, X., Lin, F., Wu, Q., Zhang, Y., et al. (2017). Comparative transcriptome analysis provides insights into differentially expressed genes and long non-coding RNAs between ovary and testis of the Mud crab (Scylla paramamosain). Mar. Biotechnol. 20, 20–34. doi: 10.1007/s10126-017-9784-2
Yeh, D. W., Liu, Y. L., Lo, Y. C., Yuh, C. H., Yu, G. Y., Lo, J. F., et al. (2013). Toll-like receptor 9 and 21 have different ligand recognition profiles and cooperatively mediate activity of CpG-oligodeoxynucleotides in zebrafish. Proc. Natl. Acad. Sci. U.S.A. 110, 20711–20716. doi: 10.1073/pnas.1305273110
Yoon, J. H., Abdelmohsen, K., and Gorospe, M. (2014). Functional interactions among microRNAs and long noncoding RNAs. Semin. Cell Dev. Biol. 34, 9–14. doi: 10.1016/j.semcdb.2014.05.015
Zeng, D. G., Chen, X. L., Peng, J. X., Yang, C. L., Peng, M., Zhu, W. L., et al. (2018). Single-molecule long-read sequencing facilitates shrimp transcriptome research. Sci. Rep. 8:16920. doi: 10.1038/s41598-018-35066-3
Zhang, W., Liu, K., Tan, B. P., Liu, H. Y., Dong, X. H., Yang, Q. H., et al. (2019). Transcriptome, enzyme activity and histopathology analysis reveal the effects of dietary carbohydrate on glycometabolism in juvenile largemouth bass, Micropterus salmoides. Aquaculture 504, 39–51. doi: 10.1016/j.aquaculture.2019.01.030
Zhao, J., Feng, L., Liu, Y., Jiang, W., Wu, P., Jiang, J., et al. (2014). Effect of dietary isoleucine on the immunity, antioxidant status, tight junctions and microflora in the intestine of juvenile Jian carp (Cyprinus carpio var. Jian). Fish Shellfish Immun. 41, 663–673. doi: 10.1016/j.fsi.2014.10.002
Zhou, W., Samad, R., Lu, K., Wang, L., and Liu, W. (2018). Effects of berberine on growth, liver histology, and expression of lipid-related genes in blunt snout bream (Megalobrama amblycephala) fed high-fat diets. Fish Physiol. Biochem. 45, 1–9. doi: 10.1007/s10695-018-0536-7
Keywords: Epinephelus fuscoguttatus♀ × Epinephelus lanceolatus♂, full-length transcriptome sequencing of groupe, Illumina sequencing, intestinal health, soy protein, third-generation sequencing
Citation: Zhang W, Tan B, Deng J, Dong X, Yang Q, Chi S, Liu H, Zhang S, Xie S and Zhang H (2021) The Single-Molecule Long-Read Sequencing of Intestine After Soy Meal-Induced Enteritis in Juvenile Pearl Gentian Grouper, Epinephelus fuscoguttatus♀ × Epinephelus lanceolatus♂. Front. Mar. Sci. 8:688601. doi: 10.3389/fmars.2021.688601
Received: 31 March 2021; Accepted: 21 May 2021;
Published: 02 July 2021.
Edited by:
Menghong Hu, Shanghai Ocean University, ChinaReviewed by:
Kang-le Lu, Jimei University, ChinaMansour Torfi Mozanzadeh, South Iran Aquaculture Research Center, Iran
Min Jin, Ningbo University, China
Copyright © 2021 Zhang, Tan, Deng, Dong, Yang, Chi, Liu, Zhang, Xie and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beiping Tan, YnB0YW5AMTI2LmNvbQ==