 Suhyung Cho1*†
Suhyung Cho1*† Sang-Hyeok Cho1†
Sang-Hyeok Cho1† So-Ra Ko2
So-Ra Ko2 Yujin Jeong1
Yujin Jeong1 Eunju Lee1
Eunju Lee1 Sangrak Jin1Bo-Seong Jeong1Byung-Ha Oh1Hee-Mock Oh2
Sangrak Jin1Bo-Seong Jeong1Byung-Ha Oh1Hee-Mock Oh2 Chi-Yong Ahn2
Chi-Yong Ahn2 Byung-Kwan Cho1*
Byung-Kwan Cho1*- 1Department of Biological Sciences and KI for the BioCentury, Korea Advanced Institute of Science and Technology, Daejeon, South Korea
- 2Biological Resource Center, Korea Research Institute of Bioscience and Biotechnology, Daejeon, South Korea
The marine dinoflagellate Alexandrium is associated with harmful algal blooms (HABs) worldwide, causing paralytic shellfish poisoning (PSP) in humans. We found that the marine bacterium Pseudoruegeria sp. M32A2M exhibits algicidal activity against Alexandrium catenella (Group I), inhibiting its motility and consequently inducing cell disruption after 24 h of co-culture. To understand the communication between the two organisms, we investigated the time-course cellular responses through genome-wide transcriptome analysis. Functional analysis of differentially expressed genes revealed that the core reactions of the photosystem in A. catenella were inhibited within 2 h, eventually downregulating the entire pathways of oxidative phosphorylation and carbon fixation, as well as associated metabolic pathways. Conversely, Pseudoruegeria upregulated its glycolysis, tricarboxylic acid cycle, and oxidative phosphorylation pathways. Also, the transporters for nutrients such as C3/C4 carbohydrates and peptides were highly upregulated, leading to the speculation that nutrients released by disrupted A. catenella cells affect the central metabolism of Pseudoruegeria. In addition, we analyzed the secondary metabolite-synthesizing clusters of Pseudoruegeria that were upregulated by co-culture, suggesting their potential roles in algicidal activity. Our time-course transcriptome analysis elucidates how A. catenella is affected by algicidal bacteria and how these bacteria obtain functional benefits through metabolic pathways.
Introduction
Over the last several decades, harmful algal blooms (HABs) have been arising globally in both marine and freshwater bodies, causing severe ecological and economic damage to aquatic ecosystems and related industries (Asakawa et al., 2005; Peperzak, 2005; Anderson et al., 2008; Furuya et al., 2018). The representative microorganisms causing HABs are dinoflagellates or diatoms (Anderson, 1998; Heil et al., 2001; Shin et al., 2018), and dinoflagellates are known to account for 75% of all harmful algal species that cause HABs (Smayda, 1997).
In particular, the Alexandrium species complex of dinoflagellates, which includes A. tamarense, A. fundyense, and A. catenella, produces toxic molecules such as saxitoxins that cause paralytic shellfish poisoning (PSP) (Anderson et al., 2012). The blooms by Alexandrium species have been reported from coasts worldwide including the United States (Anderson, 1997), Europe (Hakanen et al., 2012), and Asia (Han et al., 1992; Wang and Wu, 2009; Ishikawa et al., 2014). In particular, blooms of A. catenella (Group I) have been observed regularly during the spring season along the Asian coast, and the southeastern coastal areas of Korea have been reported as hotspots for monitoring PSP (Shin et al., 2017).
Marine microorganisms form intricate interaction networks with other neighboring species beyond the predator–prey relationship (Amin et al., 2012). Microorganisms communicate and interact by secreting various molecules within the phycosphere, although they are not bound directly to each other (Seymour et al., 2017). Recent studies are emphasizing the importance of biotic relationships between harmful algae and other microorganisms, such as bacteria (Yang et al., 2014), grazers (Jeong et al., 2008), and parasites (Lu et al., 2016; Kim et al., 2017). In a mutualistic relationship, phytoplankton provide diverse organic matters to bacteria, including carbon, nitrogen, and phosphorus. For example, organic sulfur in the form of dimethylsulfoniopropionate or dimethyl sulfide influences the metabolic activity and proliferation of bacteria (Gonzalez et al., 1999; Azam and Malfatti, 2007; Jiao et al., 2010). The phytoplankton-associated bacterium Roseobacter takes up heme from algal cellular debris for its iron requirements (Hogle et al., 2017). In return, algae acquire vitamins B1, B7, and B12 from bacteria for the regulation of cellular metabolic pathways and cell propagation, as well as signaling molecules such as indole-3-acetic acid, which promote cell division (Croft et al., 2005, 2006; Tang et al., 2010; Amin et al., 2015).
Meanwhile, some bacteria are detrimental to diatoms/dinoflagellates owing to the release of algicidal molecules, proteases, nucleases, biosurfactants, siderophores for chelating Fe3+ ions, and quorum-sensing molecules such as N-acyl homoserine lactones (Imamura et al., 2000; Lee et al., 2002; Ahn et al., 2003; Paul and Pohnert, 2011; Amin et al., 2012; Qi and Han, 2018). Recent studies have suggested that heterotrophic marine bacteria often affect marine phytoplankton blooms or bloom decline, playing a key role in balancing primary productivity in the marine ecosystem (Buchan et al., 2014; Hogle et al., 2016). Alexandrium is reported to be associated with Proteobacteria and Bacteroidetes at the phylum level and with multiple genera of Flavobacteriales at the order level during bloom peaks (Hattenrath-Lehmann and Gobler, 2017). Some bacteria under the Roseobacter lineage are known to interact mutualistically with phytoplankton for the transformation of carbon, nitrogen, phosphorus, and sulfur; however, these bacteria sometimes secrete pathogenic compounds when nutrient resources are limited, terminating algal blooms (Amaro et al., 2005; Wagner-Dobler and Biebl, 2006). Therefore, many marine bacteria showing algicidal effects have been currently recognized as potential tools to control HABs (Kim et al., 1998; Mayali and Azam, 2004; Zhang et al., 2014a).
Understanding the mutual associations of marine organisms between dinoflagellates/diatom and bacteria has becomes important; therefore, community analyses of phytoplankton and bacteria have been intensively performed through metagenome analyses (Hattenrath-Lehmann and Gobler, 2017; Zhou et al., 2018; Cui et al., 2020). Metagenome analyses demonstrate changes in the diversity of bacterial community caused by various biotic and abiotic factors, including HABs. Bacterial community data provide comprehensive information to analyze the characteristics of abundant bacteria during blooms; however, the information remains insufficient to understand specific functional interactions and underlying mechanisms.
To understand the cellular functions of dinoflagellates or cohabiting bacteria, transcriptome analyses of Alexandrium strains have been performed in optimized media or under nutrient deficiency (Zhang et al., 2014b; Meng et al., 2019; Vingiani et al., 2020), which have enabled the identification of functional characteristics or functional changes in different culture conditions. In addition, the transcriptome analysis of the marine bacterium Ruegeria pomeroyi, a member of the Roseobacter clade, has been performed in a co-culture with the dinoflagellate A. tamarense and diatom Thalassiosira pseudonana (Landa et al., 2017). Despite these advances, few studies have systematically elucidated the mutual interaction between Alexandrium and bacteria from the perspective of the metatranscriptome.
In this study, we found that the marine bacterium Pseudoruegeria sp. M32A2M exhibits algicidal activity against A. catenella (Group I). Pseudoruegeria sp. is representative of the marine Roseobacter group, which has been reported to appear frequently in various regions, including seas in East Asia and the North Sea (Park et al., 2014; Pohlner et al., 2017; Zhang et al., 2017). However, its function and characteristics as an algicidal bacterium are unknown. We monitored the sequential changes of the metatranscriptome occurring under the co-culture of A. catenella and Pseudoruegeria sp. Through differentially expressed gene (DEG) analysis, we elucidated how A. catenella is affected by Pseudoruegeria sp. and vice versa, with respect to various functional changes in metabolic pathways.
Materials and Methods
A. catenella Pyrosequencing and BLASTN Analysis
Prior to cell culture, pyrosequencing of the 18S rRNA (3′ end), internal transcribed spacer 1 (ITS1), 5.8S rRNA, ITS2, and large subunit (LSU) rRNA genes was performed to identify the Alexandrium strain with the following paired primers: (i) ITS_F (GAGGAAGGAGAAGTCGTAACAAGG) and R732 (CCTTGGTCCGTGTTTCAAGAC), (ii) F341 (AGCGCA CAAGTACCATGAGG) and R1344 (TTCGGCAGGTGAG TTGTTACACAC), (iii) F918 (AGGTTCGTATCGATACTGACG TGC) and R2242 (CAGAGCACTGGGCAGAAATCAC), (iv) F1976 (GAAAAGGATTGGCTCTGAGG) and R3016 (AAACTAACCTTCTCACGACGGTC), and (v) F2843 (TTAC CACAGGGATAACTGGCTTG) and ETS_R (GCAGGGACA TAATCAGCACAC). Further, BLASTN analysis was performed using the pyrosequencing results (Supplementary Table 1).
Cell Culture
A. catenella (Group I) collected from the South Sea in South Korea, was isolated using the micropipetting method, and maintained in sterilized Guillard’s (F/2) medium (Sigma, St. Louis, MO, United States) with Red sea salt of 31 PSU (Red Sea, Houston, TX, United States), and subcultured with a 1/10 dilution approximately 40 times under a light–dark cycle (12 h each) with 50 μmol⋅m–2⋅s–1 photons at 20°C ± 1°C. Pseudoruegeria sp. M32A2M was isolated from surface water of the South Sea in South Korea, where HABs of Alexandrium are frequently observed (Cho et al., 2020). Pseudoruegeria sp. was primarily cultured on marine agar 2216 plates (BD, Franklin Lakes, NJ, United States) at 25°C for 2 days. The cells scraped from agar plates were resuspended using gentle pipetting in F/2 media with Red sea salt and incubated for 2 days at 20°C ± 1°C for adaptation in liquid media prior to co-culture with A. catenella. Thereafter, the Pseudoruegeria sp. culture medium was filtered through a 20 μm nylon net by gravity to remove some aggregated cells. Then, the OD600nm of filtrated cells was measured before inoculation into A. catenella cell medium. For this study, a total of 10 L of A. catenella (2,000 cells⋅mL–1) and 1 L of Pseudoruegeria sp. (OD 0.3 at 600 nm) were cultured in F/2 medium.
For the co-culture of the two microorganisms, 100 mL of Pseudoruegeria sp. culture (OD 0.3 at 600 nm) estimated to be 6 × 1010 cells in total, were each loaded into nine flasks containing 1 L of A. catenella culture (2,000 cells⋅mL–1) of 2 × 106 cells in total. Three sets of flasks with three biological replicates were exposed to light for 2, 6, and 12 h with 50 μmol⋅m–2⋅s–1 photons at 20°C ± 1°C, respectively. As control groups, 100 mL of fresh medium was added into 1 L of pure-cultured A. catenella (2,000 cells⋅mL–1), and 1 L of fresh medium was added into 100 mL of Pseudoruegeria sp. medium. Then, diluted cells were collected again using centrifugation prior to sequencing preparation.
Photosynthetic Pigment Assay
Chlorophyll-a (Chl-a) and carotenoid were extracted from A. catenella with 90% ethanol overnight at 4°C. These compounds were measured spectrophotometrically as follows: Chl-a (mg⋅L–1) = (12.7 × A665) - (2.69 × A645), and carotenoid (mg⋅L–1) = (1,000 × A470 - 2.05 × CChl–a)/245 (where A665, A645, and A470 indicate the absorbance at wavelengths of 665, 645, and 470 nm, respectively, and CChl–a represents the Chl-a content) (Li et al., 2015a).
Cell Separation
The co-cultured A. catenella and Pseudoruegeria sp. mixtures were separated through 20 μm nylon net filter membranes (Merck Millipore, Burlington, MA, United States) by vacuum to avoid cross contamination of mRNA and to further construct clearer sequencing libraries. Then, the membrane was washed with 200 mL of fresh F/2 medium. The A. catenella collected on a filter membrane was resuspended with 50 mL of F/2 medium and centrifuged at 1,503 × g at 4°C for 10 min. The bacterial cells filtrated through the membrane were centrifuged at a high g-force of 3,381 × g for 10 min to collect the bacterial cells. The centrifugation at a high g-force for bacterial cells also helped disrupt the fragile cell membrane of a few residual A. catenella. A. catenella and bacterial cell pellets were resuspended with RNAlaterTM (Invitrogen, Carlsbad, CA, United States) and incubated at 4°C for 1 h. Each pellet was stored at -80°C until further experiments.
Library Preparation for High-Throughput Sequencing
For total RNA extraction, cells were washed with extraction buffer comprising 200 mM Tris–HCl (pH 7.5), 25 mM EDTA, 250 mM NaCl, and 0.5% SDS. Then, each pellet was instantly frozen using liquid nitrogen and ground to a fine powder. For RNA extraction of A. catenella and Pseudoruegeria sp., TRIzol (Invitrogen) and phenol (Sigma) at 65°C were added to the powdered pellet, and this was immediately followed by ethanol precipitation.
The RNA-seq library was constructed using the TruSeq mRNA Sample Prep Kit (Illumina, San Diego, CA, United States) according to the manufacturer’s instructions. Briefly, the poly-A-tailed mRNA of A. catenella was purified with oligo-dT magnetic beads. High-throughput sequencing with 100 bp single-end reads technically requires at least 100 bp RNA fragments. However, it is important to mention that RNA fragmentation causes a broad length distribution. To account for this, the purified mRNAs were further fragmented for 2 min to obtain RNA fragments with a median value of about 150 bp. cDNAs synthesized from the fragmented RNAs were ligated to adaptors and then amplified using qPCR.
For bacterial mRNA preparation, rRNAs from purified total RNAs (4 μg) were first depleted using the Ribo-zeroTM rRNA Removal Kit for Epidemiology according to the manufacturer’s instructions (Illumina). After ethanol precipitation of mRNAs (∼0.4 μg), the rRNA removal was confirmed using a TapeStation system 2200 (Agilent, Santa Clara, CA, United States). Then, the bacterial mRNAs were also fragmented to lengths of a median value of about 150 bp using a TruSeq mRNA Sample Prep Kit (Illumina), followed by ethanol precipitation.
The total library length for A. catenella and Pseudoruegeria sp. was approximately 250–300 bp by adaptor ligation and overlap extension qPCR for sequencing. The resulting library was sequenced by a HiSeq 2500 instrument with 100 bp single-end reads (Illumina).
De novo Transcriptome Assembly and Differential Expression Analysis
After RNA-seq raw reads of A. catenella and Pseudoruegeria sp. were obtained from Illumina sequencing, the adapter sequences and low-quality sequences were trimmed using IlluQC software included in the NGSQCToolkit (Patel and Jain, 2012) and CLC Genomics Workbench 6.5.1 (CLCbio, Denmark) with the default setting. Then, de novo transcriptome assembly from RNA-seq raw reads of pure-cultured A. catenella was performed using Trinity v 2.0.6, with default settings for strand-specific RNA reads (Grabherr et al., 2011).
The RNA-seq reads from A. catenella were then aligned to the Trinity-assembled transcriptome by Bowtie2 to remove the misassembled sequences and their abundance was calculated using RNA-seq by Expectation Maximization (RSEM) (Li and Dewey, 2011; Langmead and Salzberg, 2012). RSEM results were transformed to fragments per kilobase of transcript per million (FPKM) values, and mis-assembled transcripts (FPKM cut-off = 1) were filtered out (Trapnell et al., 2010). Further, redundant isoform unigenes were removed by clustering with the BLAST-like alignment tool (BLAT) using tileSize = 8 and stepSize = 5, with other parameters set to default (Kent, 2002; Zhang et al., 2014b). Because this process results in a non-redundant set of the longest representative transcripts (i.e., unigenes), the analysis for various RNA isoforms by the alternative splicing or alternative polyadenylation was excluded.
From the unigene reference, the coding regions of the assembled transcript sequences were predicted using TransDecoder1 with the assistance of UniProt, BLASTP, Pfam, and HMMscan (HMMER v.3.2.1) (Haas et al., 2013; El-Gebali et al., 2018). For function annotation from coding sequences, BLASTx alignment was performed against the UniProt database with an E-value threshold of 10–6 (Camacho et al., 2009; UniProt: a hub for protein information, 2014). In addition, Gene Ontology (GO) functional annotation was performed based on the annotated UniProt ID (Gene Ontology Consortium, 2014). Together, Kyoto Encyclopedia of Genes and Genomes (KEGG) Ortholog ID and EuKaryotic Orthologous Groups (KOG) categorization were conducted (Koonin et al., 2004; Kanehisa et al., 2015).
Raw RNA reads from Pseudoruegeria sp. were mapped against the Pseudoruegeria sp. M32A2M reference genome in DDBJ/ENA/GenBank (GenBank assembly accession, GCA_010374725.1) using the CLC Genomics Workbench (version 6.5.1) (similarity score = 0.9 and length fraction = 0.9). GFF files were visualized using SignalMap software v.2.0.0.5 (Roche, Pleasanton, CA). Further, KEGG Ortholog ID and Clusters of Orthologous Groups (COG) categorization was performed based on the genome sequence of Pseudoruegeria sp. (Tatusov et al., 2000; Kanehisa et al., 2015).
For quantitative analysis of RNA expression, the expression levels of the unigenes of A. catenella and transcripts of Pseudoruegeria sp. were normalized by DESeq2 and calculated as reads per million (Love et al., 2014). Differentially expressed genes (DEGs) were classified when the fold change in the normalized RNA expression value of the co-cultured cells against that of the pure-cultured cells was more than twofold (|log2Fold change (FC)| ≥ 1) with a low p-value (DESeq P < 0.05). Transcripts with low expression levels < 50 reads per million in the pure-cultured cells were excluded for DEG analysis to avoid over-interpretation. GO enrichment analysis with DEGs was conducted using the BiNGO Cytoscape application (Maere et al., 2005).
Results
Algicidal Effect of Pseudoruegeria sp. on A. catenella
First, we identified the Alexandrium strain to be used in this study through the pyrosequencing of the LSU rRNA gene and the 18S rRNA fragment (Supplementary Table 1). We found that the Alexandrium strain was highly matched with the top four A. catenella strains screened in South Korea (DQ785887.1, DQ785886.1, AY347308.2, and DQ785885.1) in terms of total score from BLASTN analysis (Supplementary Table 1). In a previous study, we screened the marine bacterium Pseudoruegeria sp. M32A2M from regions where A. catenella was frequently found, and we characterized its genomic information through 16S rRNA sequencing and genome sequencing (Cho et al., 2020).
To investigate the algicidal activity of Pseudoruegeria sp., the Chl-a and carotenoid concentrations in A. catenella were measured according to the time of co-culture with Pseudoruegeria sp. (Figure 1A). The Chl-a and carotenoid levels started to decrease from 2 h of co-culture and decreased significantly at 12 h of co-culture to 38.7 and 28.5% of the corresponding levels in pure-cultured cells, respectively. In pure culture, A. catenella had an integrated membrane with four chains; however, the cells began to remarkably lose membrane integrity following 24 h of co-culture (Figure 1B). A. catenella has highly active flagella, as indicated by the phylum name Dinoflagellata. A. catenella showed highly active motility in pure culture (Supplementary Video 1), but after 24 h of co-culture, its motility was greatly decreased (Supplementary Video 2).
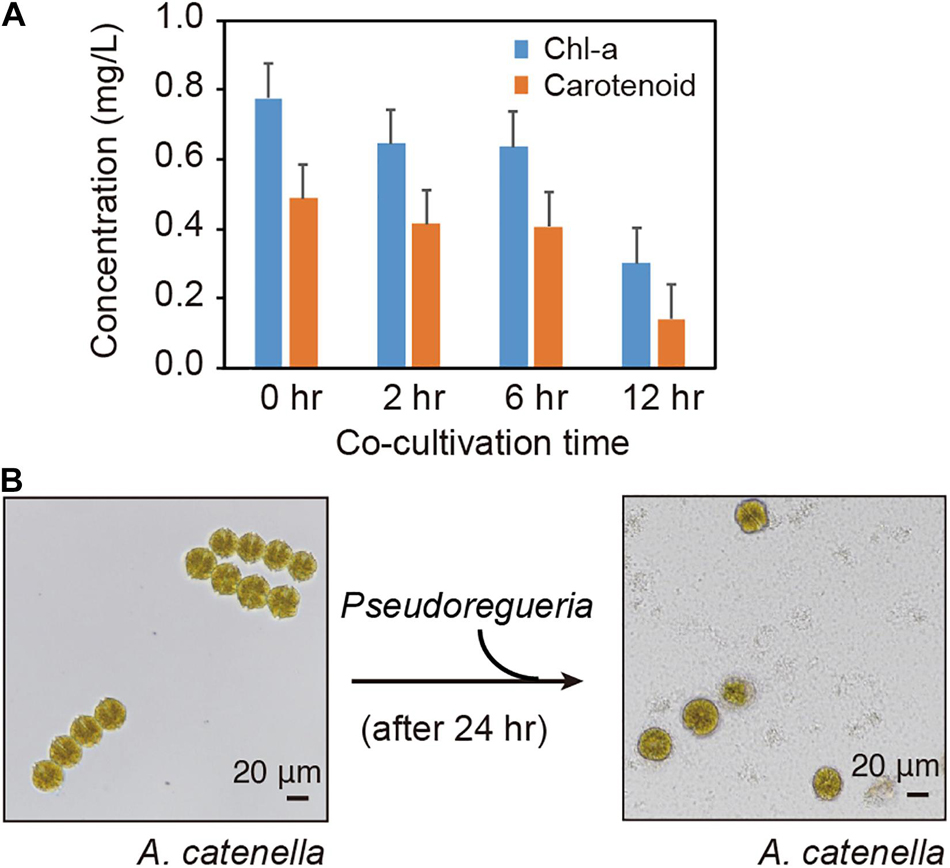
Figure 1. Co-culture of Alexandrium catenella and Pseudoruegeria sp. (A). Changes in Chl-a and carotenoid concentrations in A. catenella were measured at 0 (pure), 2, 6, and 12 h of co-culture. (B) The morphological changes of A. catenella were observed before and after treatment with Pseudoruegeria sp. (after 24 h).
Transcriptome Analysis of A. catenella and Pseudoruegeria sp.
To understand how A. catenella and Pseudoruegeria sp. communicate with each other during co-culture and how their cellular functions have been altered, we investigated the changes in the cellular transcriptomes in both organisms with the passage of co-culture time. As a control, pure-cultured A. catenella and Pseudoruegeria sp. were used for subsequent comparison with co-cultured cells (Figure 2A). For co-culture, the two microorganisms were incubated together for 2, 6, and 12 h with light exposure. Mixed cells with a large difference in size were isolated using a net membrane filter (Figure 2A), because A. catenella and Pseudoruegeria sp. lengths are 20–48 μm and 1–5 μm, respectively (Fukuyo, 1985; Cho et al., 2020). From the two isolated organisms, mRNAs for eukaryotes and bacteria were individually purified and sequenced using high-throughput sequencing (Figure 2A and Supplementary Table 2). Consequently, the total mapped reads from A. catenella in high-throughput sequencing were 252,490,204 (Figure 2B). The genome sequencing of A. catenella, which has a large genome size of more than hundreds of giga base pairs, is challenging. Therefore, to create the reference for A. catenella, the RNA-seq raw reads from pure-cultured A. catenella were de novo assembled using Trinity V 2.0.6. The total number of assembled transcripts was 245,489 (Figure 2B). After the exclusion of isoforms by BLAT, 173,134 unigenes were obtained. Among the unigenes, 39,759 genes were functionally annotated through the functional retrieval procedures based on UniProt, GO, KEGG, and KOG (Supplementary Dataset 1 and Figure 2B).
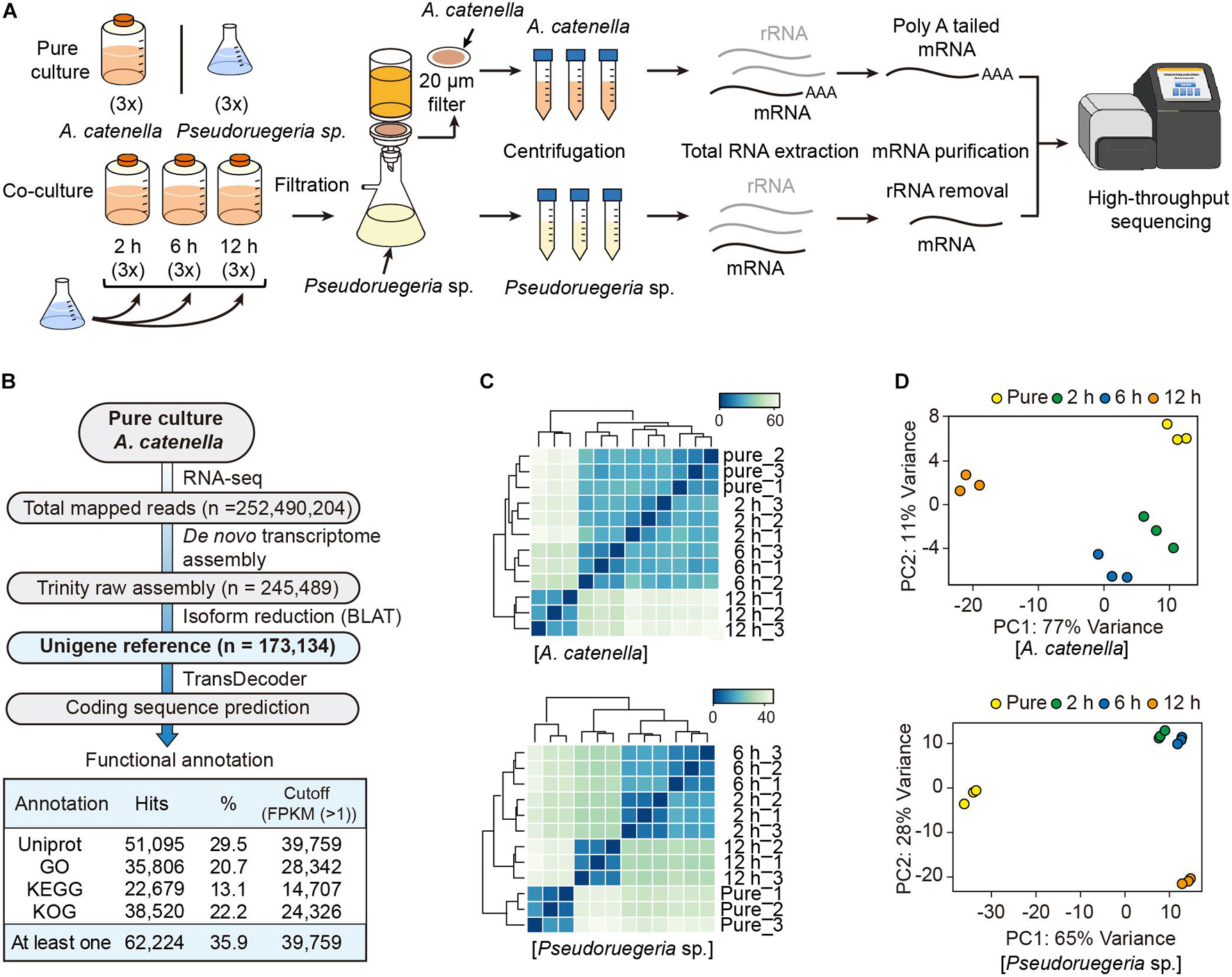
Figure 2. Transcriptome analysis of A. catenella and Pseudoruegeria sp. (A) Scheme of the transcriptome analysis process. A. catenella and Pseudoruegeria sp. were each pure- cultured as the control. For the co-culture of the two organisms, 1 L of A. catenella (2,000 cells⋅mL–1) and 100 mL of Pseudoruegeria sp. (OD600 nm 0.3) were cultured together for 2, 6, and 12 h with light exposure. All cultures had three experimental replicates. After co-culture, the two organisms were again isolated using filtration and then collected separately for RNA extraction. Finally, the purified mRNA was sequenced using RNA-seq. (B) Pipeline for transcriptome analysis of A. catenella. (C,D) Pearson’s correlation analysis (C) and principal component analysis of A. catenella and Pseudoruegeria sp. (D) were performed from the transcriptomes of pure-cultured cells and co-cultured cells for 2, 6, and 12 h.
The transcriptomes for pure-cultured and co-cultured (2, 6, and 12 h) Pseudoruegeria sp. were analyzed for the reference contigs (4,979 genes) (Cho et al., 2020; Supplementary Dataset 2). To verify experimental reproducibility between three biological replicates and to observe transcriptome changes in different culture conditions, we calculated Pearson’s correlation coefficient for the unigene reference genes using the mapped read counts normalized by DESeq2. As a result, both A. catenella and Pseudoruegeria sp. showed high Pearson’s correlation coefficient of over 0.99 between three biological replicates (Figure 2C). When observed between incubation times, A. catenella showed slight transcriptome variations for 2 h and 6 h but significant changes after 12 h (Figure 2C). On the other hand, the transcriptome of Pseudoruegeria sp. started to change significantly from 2 h of co-culture. Similarly, in the principal component analysis, A. catenella and Pseudoruegeria sp. showed distinct transcriptome changes according to the co-culture time with high reproducibility (Figure 2D). Taken together, the cellular transcriptomes of A. catenella and Pseudoruegeria sp. affected each other and showed drastic changes at 12 h of co-culture.
Differentially Expressed Genes During the Co-culture of A. catenella and Pseudoruegeria sp.
To investigate the reciprocal responding genes by co-culture, we selected the DEGs that satisfied the transcriptional changes over twofold with low p-values (DESeq P < 0.05) after co-culture (see volcano plots in Supplementary Figure 1). Consequently, for A. catenella, among 39,759 genes, 1,716 (4.3%) and 2,602 (6.5%) genes were found to be upregulated and downregulated, respectively (Figure 3A). In particular, 120 genes were commonly downregulated at 2, 6, and 12 h. For Pseudoruegeria sp., more than 20% of 4,246 genes were differentially regulated, i.e., 904 (21.3%) and 946 (22.3%) were upregulated and downregulated, respectively (Figure 3B).
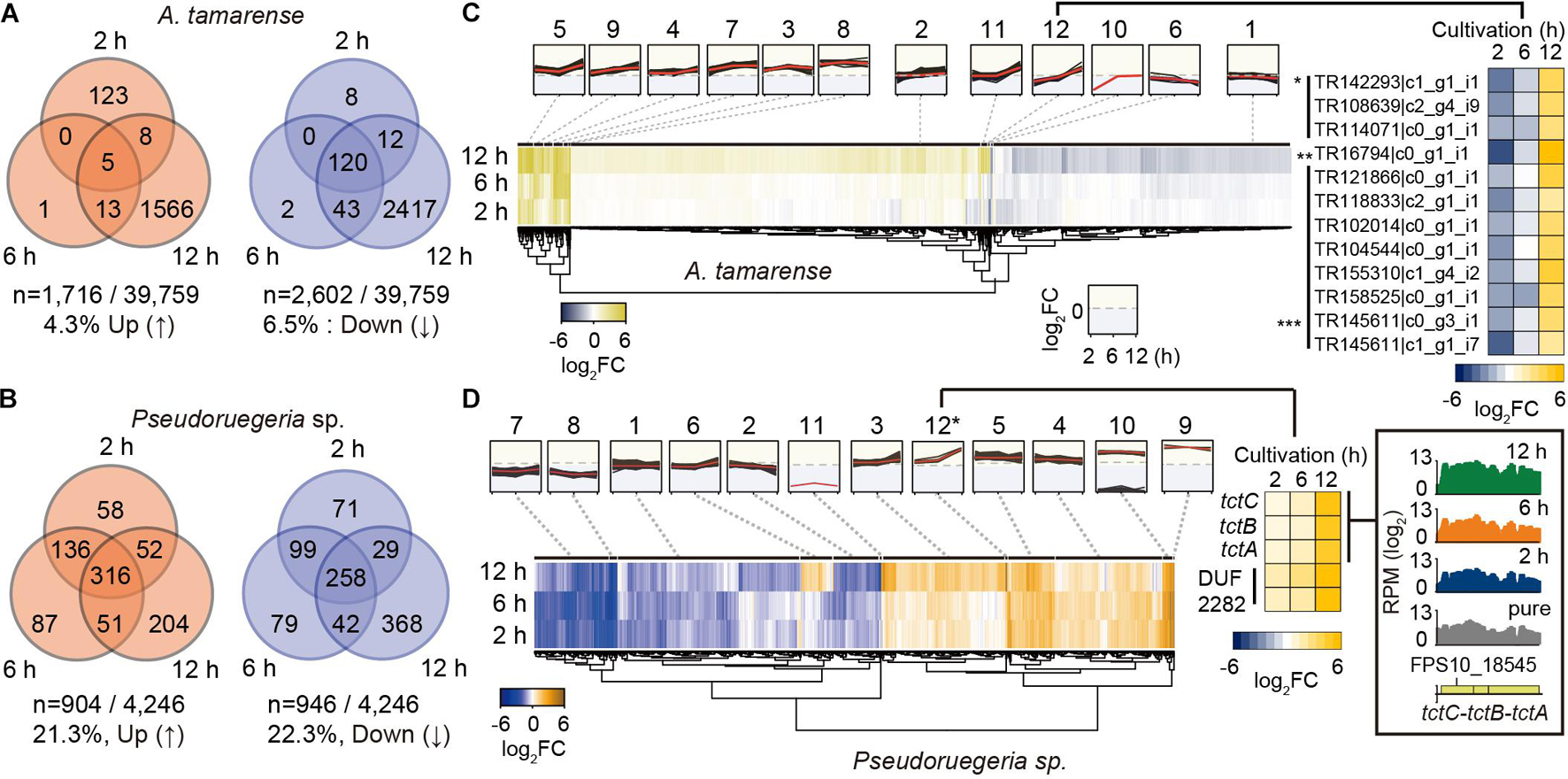
Figure 3. Collection and grouping of differentially expressed genes (DEGs) during the co-culture of A. catenella and Pseudoruegeria sp. (A,B). The number of upregulated and downregulated genes from the gene dataset satisfying the DEG condition (|log2 (Normalized RPMcoculture/Normalized RPMpure–culture)| ≥ 1 and low p-value (DESeq P < 0.05), reads per million > 50 in pure cells) were counted for A. catenella (A) and Pseudoruegeria sp. (B) according to the co-culture time. (C) DEGs of A. catenella were classified into 12 groups according to the RNA expression profile and are displayed using a heatmap. The RPM fold change of the genes in the 12th group are separately shown using a heatmap. An asterisk (*) indicates cell wall-associated hydrolase- coding genes; double asterisks (**) indicate a putative blue light receptor-coding gene; and triple asterisks (***) indicate uncharacterized protein-coding genes. (D) The DEGs of Pseudoruegeria sp. were also classified into 12 groups according to the RNA expression profile. For example, the tripartite tricarboxylate transporter cluster (tctC-B-A) and DUF2282 domain containing membrane protein-coding genes were assigned in the 12th cluster, and their expression fold changes after co-culture are displayed using a heatmap. The expression profiles of the tct gene cluster at the pure-culture and co-cultures for 2, 6, and 12 h are displayed using SignalMap.
The transcriptomes of Pseudoruegeria sp. and A. catenella also remarkably changed at 12 h of co-culture. To examine the patterns of transcriptome changes by co-culture time, all DEGs of both microorganisms were classified into 12 groups according to their fold change and expression pattern (Supplementary Datasets 1, 2) and were visualized using heatmaps (Figures 3C,D). For example, the 12th group of A. catenella, which showed a highly increased expression pattern only at 12 h of co-culture, consisted of transcripts for three cell wall-associated hydrolases (∗), which may cause membrane destruction; a blue light receptor (∗∗); and uncharacterized proteins (∗∗∗). For Pseudoruegeria sp., the tripartite tricarboxylated transporter clusters (tctC-tctB-tctA) and DUF2282 domain containing predicted integral membrane proteins were assigned to the 12th group, which showed rapid increase at 12 h of co-culture, suggesting that the uptake of short carbohydrates can be active at 12 h of co-culture (Figure 3D).
Functional Analysis of DEGs of A. catenella During Co-culture
To systematically investigate the cellular functions of genes changed by the co-culture of the two organisms, the DEGs in A. catenella were classified through GO enrichment analysis (Supplementary Table 3). Interestingly, the genes involved in the photosynthetic electron transport chain and PSII started to be downregulated from 2 h (Cluster 1/1) (Figure 4A). At 12 h, downregulation expanded to genes related to carbohydrate metabolism (Cluster 1/13); phosphorus/phosphate metabolic processes (Cluster 2/13); and the detection of external stimuli, including light- and abiotic factor-related genes (Cluster 3/13), as well as photosynthesis. In addition, the genes related to the movement of microtubules, which are associated with cell motility and division, as a part of the cytoskeleton (Vale, 2003), were jointly downregulated (Supplementary Table 3). These results are consistent with the drastic decrease in Chl-a and carotenoids, which are important for photosynthesis, and reduced motility and membrane disintegration of A. catenella. For upregulated clusters, the transcription of macromolecule biosynthetic process/metabolic pathways, ribosome assembly, and translation-associated genes increased from 2 h (Supplementary Table 3). At 12 h, the transcription levels of RNA synthesis/processing/modification and ribonucleotide/nucleoside-related processes markedly increased.
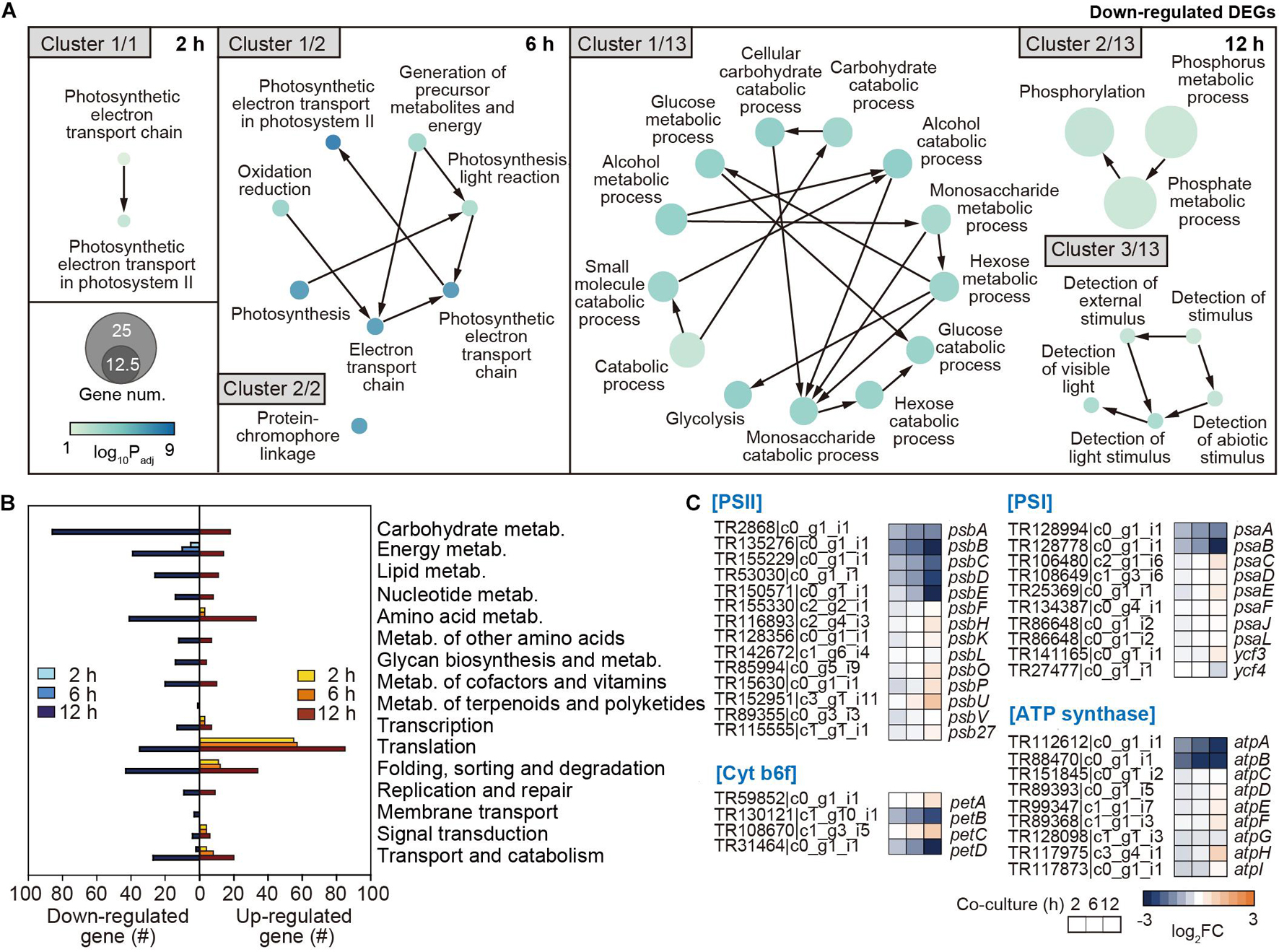
Figure 4. Functional analysis of downregulated genes in A. catenella by co-culture. (A) Functional clustering through GO enrichment analysis was executed by co-culture time (2, 6, and 12 h) from downregulated DEGs in A. catenella, among which core clusters are displayed according to gene abundance. The node size indicates the gene number, and the color key indicates the log10 p-value of DEG. (B) KEGG analysis results from the DEGs in A. catenella are also shown by co-culture time. (C) Transcriptome changes after co-culture in the genes of photosystem II (PSII), cytochrome b6f, PSI, and ATP synthase in the oxidative phosphorylation pathway are displayed using a heatmap, according to the co-culture time. FC indicates the fold change in transcriptome after co-culture.
Furthermore, the functional changes from the perspective of metabolic pathway were investigated through KEGG analysis. As the reference genome for KEGG analysis, the first fully genome-sequenced eukaryotic marine phytoplankton, Thalassiosira pseudonana, was used (Armbrust et al., 2004). Overall, A. catenella expressed a drastically changed transcriptome at 12 h (Figure 4B). However, energy metabolism-associated genes, including photosynthesis and oxidative phosphorylation, uniquely began to be downregulated continuously from 2 h, and the number of downregulated genes gradually increased with co-culture time (Figure 4B and Supplementary Figure 2A). At 12 h, energy metabolism genes associated with carbon fixation, nitrogen metabolism, and sulfur metabolism were additionally downregulated (Figure 4B and Supplementary Figure 2A). Furthermore, carbohydrate metabolism genes associated with glycolysis, the tricarboxylic acid (TCA) cycle, and the pentose phosphate pathway were intensively downregulated at 12 h (Figure 4B and Supplementary Figure 3).
The results of GO and KEGG analysis together show that the transcription of genes associated with photosynthesis and oxidative phosphorylation in A. catenella is primarily deactivated by co-culture with Pseudoruegeria sp., which seems to significantly affect carbon fixation- and carbohydrate-associated metabolism upon long-term exposure. In detail, we investigated the transcriptomic changes in photosynthesis-related oxidative phosphorylation among metabolic pathways at the gene level (Figure 4C). Among photosynthesis pathways, PSII is the first light-dependent oxygenic protein complex (Supplementary Figure 3). It consists of D1 protein (PsbA), which binds chlorophyll P680, and D2 proteins (PsbD) as reaction-center proteins. It is also surrounded by a CP43/CP47 (PsbB/PsbC) core complex of antenna and light-harvesting complexes to employ chlorophyll and pigments to absorb light. It also forms a complex with Cyt b559 subunits (PsbE/PsbF), oxygen-evolving complex (PsbO and PsbP), and other complex proteins. The transcription levels of psbA, psbB, psbC, psbD, and psbE in the PSII complex were very high, ranking as 1st, 2nd, 13th, 22nd, and 30th, respectively, in the overall transcript expression profile (Supplementary Dataset 1); however, they were all significantly downregulated upon short-term exposure to Pseudoruegeria sp. (Figure 4C). The core petB (cyt b6) and petD (cyt b6f-IV) subunits of the cytochrome b6f complex that transfers electrons from PSII to PSI (Kurisu et al., 2003) were gradually downregulated with an increase in co-culture time (Figure 4C and Supplementary Figure 3). Consecutively, similar to PSII, PSI reduced the transition from NADP to NADPH through light-driven reactions of photosynthesis. PsaA (A1) and PsaB (A2), which are photosynthetic reaction center proteins among the PSI complex, were also downregulated upon co-culture (Figure 4C). This is consistent with the downregulation pattern of PSII center proteins (D1 and D2 proteins). The downregulation of the center proteins in the two photosystems can inhibit the assembly of the complex, including the antenna proteins and pigments. In addition, the transcription of atpA and atpB of F1, which is responsible for hydrolyzing ATP in the ATP synthase complex, was highly downregulated (Figure 4C). ATP from ATP synthase and NADPH from PSI are necessary for carbon fixation. As a result, the downregulation of photosynthesis-associated core genes consequently led to the downregulation of carbon fixation metabolic processes, including rubisco, glycolysis, and pentose phosphate pathways (Supplementary Figure 3).
Functional Analysis of the Differentially Expressed Genes of Pseudoruegeria sp. During Co-culture
The DEGs in Pseudoruegeria sp. were functionally classified through GO enrichment analysis (Supplementary Table 4). In particular, translation-associated genes (Cluster 1/3) and electron transport chain genes (Cluster 2/3) were continuously upregulated from 2 h onward (Figure 5A). Unlike the observations in A. catenella, ATP biosynthetic process genes (Cluster 1/7) and energy generation by respiration and TCA genes (Cluster 2/7) were highly upregulated at 12 h. However, genes for biosynthetic processes of vitamins/porphyrins were downregulated at 2 and 6 h of co-culture, and genes for transmembrane transport and some response networks to external stimuli were also downregulated at 12 h of co-culture (Supplementary Table 4).
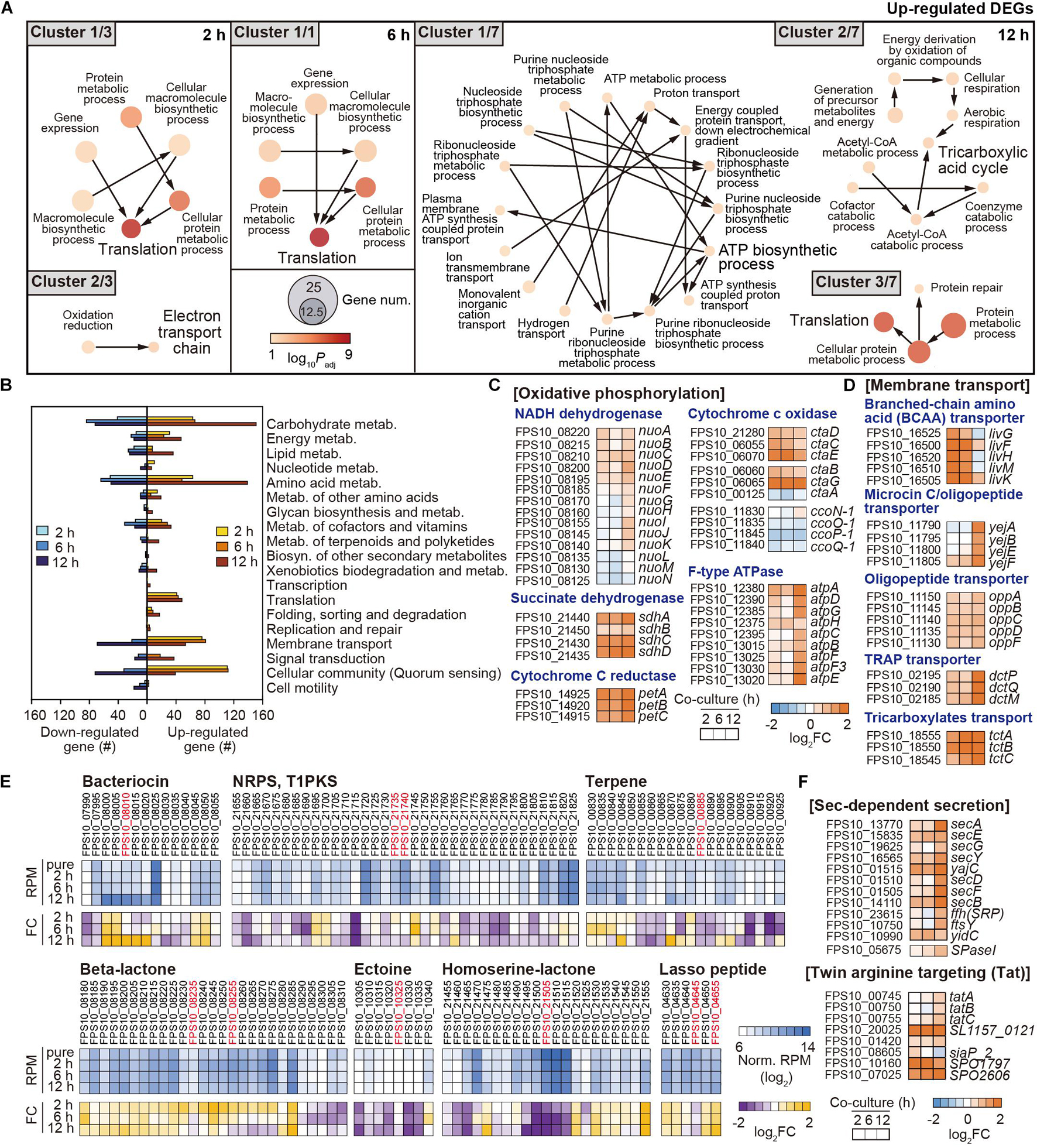
Figure 5. Functional changes in Pseudoruegeria sp. by co-culture. (A) Functional clustering through GO enrichment analysis from upregulated DEGs in Pseudoruegeria sp. are displayed by culture time. The node size indicates the gene number, and the color key indicates the log10 p-value of DEG. (B) KEGG analysis from the DEGs in Pseudoruegeria sp. are also shown during co-culture. (C,D) Transcriptome fold changes (FC) of the genes related to oxidative phosphorylation (C) and membrane transport (D) are displayed by the co-culture time using a heatmap. (E) RNA expression and its changes in secondary metabolite-synthesizing gene clusters in Pseudoruegeria sp. are shown using a heatmap, according to the co-culture time. (F) Transcriptome fold changes of the bacterial secretion systems, the Sec-dependent secretion (Sec) and twin-arginine targeting (Tat) systems, are shown by co-culture time using a heatmap. FC indicates the fold change in transcriptome after co-culture.
Furthermore, the DEGs in Pseudoruegeria sp. were classified using KEGG, with the reference being Ruegeria pomeroyi, which is a member of the marine Roseobacter clade and is phylogenetically close to Pseudoruegeria sp. (Park et al., 2014). Carbohydrate metabolism genes involved in glyoxylate and dicarboxylate metabolism, TCA cycle, and glycolysis were particularly highly upregulated (Figure 5B and Supplementary Figures 2B, 4). Furthermore, energy metabolism, amino acid metabolism, membrane transport, and cellular community-associated genes were together highly upregulated upon co-culture (Figure 5B). Especially, the transcriptomes of the genes associated with oxidative phosphorylation among the category of energy metabolism, such as NADH dehydrogenase (Nuo), succinate dehydrogenase (Sdh), cytochrome C reductase (Pet), cytochrome C oxidase (Cta), and F-type ATPase (Atp), significantly increased after co-culture with A. catenella (Figure 5C).
In addition, the transcription levels of membrane transport genes were observed to be highly upregulated following 2 h of co-culture (Figure 5D). In particular, the genes associated with branched-chain amino acid (BCAA) ABC transporter (livG-F-H-M-K); microcin C transporter (yejA-B-E-F); and oligopeptide transporter (oppA-B-C-D-F), which play a role in transferring dipeptides, branched-chain amino acids, oligopeptides, and proteins into the cytoplasm, were highly upregulated (Figure 5D). In addition, tripartite ATP-independent periplasmic (TRAP) transporter (dctP-Q-M) and tricarboxylate transporter (tctA-B-C) genes used for the uptake of tricarboxylates such as citrate and C4-dicarboxylates as metabolites in the TCA cycle, were also substantially upregulated (Figure 5D).
In both KEGG and GO analyses, genes associated with vitamins and the metabolism of cofactors were downregulated or upregulated according to the type of vitamin. Several genes for porphyrin and chlorophyll metabolism for cobalamin (vitamin B12) biosynthesis and the metabolism of thiamine (vitamin B1), pyridoxine (vitamin B6), and nicotinamide (vitamin B3) were downregulated (Figure 5E and Supplementary Figures 2B, 5).
Transcriptome Analysis of the Clusters for Secondary Metabolite Synthesis/Secretion System
Interestingly, algicidal effect by co-culture with Pseudoruegeria sp. was also observed in the supernatant after the removal of cells from co-culture, indicating that Pseudoruegeria sp. secretes algicidal compounds into the extracellular environment. To predict the metabolites secreted by Pseudoruegeria sp. that could cause this algicidal effect, we searched the secondary metabolite synthesis clusters, which had been predicted using Antismash (Cho et al., 2020). As a result, several secondary metabolite-synthesizing gene clusters encoding bacteriocin, NRPS/T1PKS, terpene, β-lactone, ectoine, L-homoserine lactone, and lasso peptide were found from the Pseudoruegeria sp. genome sequences (Figure 5E and Supplementary Table 5). Further, when the transcriptome changes of these clusters were examined, the core genes of the peptide toxin bacteriocin, which is known to be harmful to some mammals and other bacteria (Cornut et al., 2008; Cotter et al., 2013), and the lasso peptide gene cluster, which encodes a type of bacteriocin, were highly upregulated under co-culture. In addition, the clusters associated with the synthesis of β-lactone, which is known as a natural algicidal molecule, showed high RNA expression levels and significantly increased transcriptomic expression during co-culture (Figure 5E).
If the algicidal molecule secreted from bacteria is a peptide or protein-based molecule, the bacteria may also use their own secretion system. Accordingly, we monitored the transcription of bacterial secretion systems, including the sec-dependent system, e.g., secA, secDF, secYEG, secB, yajC, ftsY, and ffh, and the twin-arginine translocation system, e.g., tatA and tatBC, for the secretion of peptides/proteins such as bacteriocin. The transcription of such genes also started to increase from 2 h of co-culture and indicated the highest change at 12 h of co-culture (Figure 5F).
Discussion
Molecular understanding of the relationship between HABs and algicidal bacteria remains insufficient. In this study, we observed that A. catenella exhibited a noticeable decrease in motility and thereafter lost membrane integrity after 24 h of co-culture with Pseudoruegeria sp. M32A2M. Through interactive transcriptome analysis at different time points of co-culture, we further investigated how A. catenella was affected by co-culture and how Pseudoruegeria would benefit from the association.
Although the phenotype of A. catenella showed no significant changes at 6 h, the cellular transcriptomes of a few genes began to change within 2 h. These were photosynthesis-associated genes for oxidative phosphorylation; photosynthetic reaction center proteins, including psbAB of PSII, psaAB of PSI, petBD of Cyt b6f, and atpAB of ATP synthase. The photosynthesis-related genes started to be uniquely downregulated from 2 h of co-culture and showed the highest down-regulation at 12 h of co-culture (Figure 6). Meanwhile, the down-regulation of carbohydrates metabolism related genes was noticeably detected only at 12 h of co-culture. Time-course transcriptome analysis using GO enrichment and KEGG analysis reflected that the rapid down-regulation of photosynthetic genes affect the deactivation of carbon fixation, glycolysis, and its associated pathways; DNA topological changes; and a decrease in microtubule-based movement at 12 h (Figure 6). These results were in accordance with our observations in Figure 1 that Chl-a and carotenoid concentrations of A. catenella started to be downregulated from 2 h of co-culture and were noticeably the lowest at 12 h of co-culture.
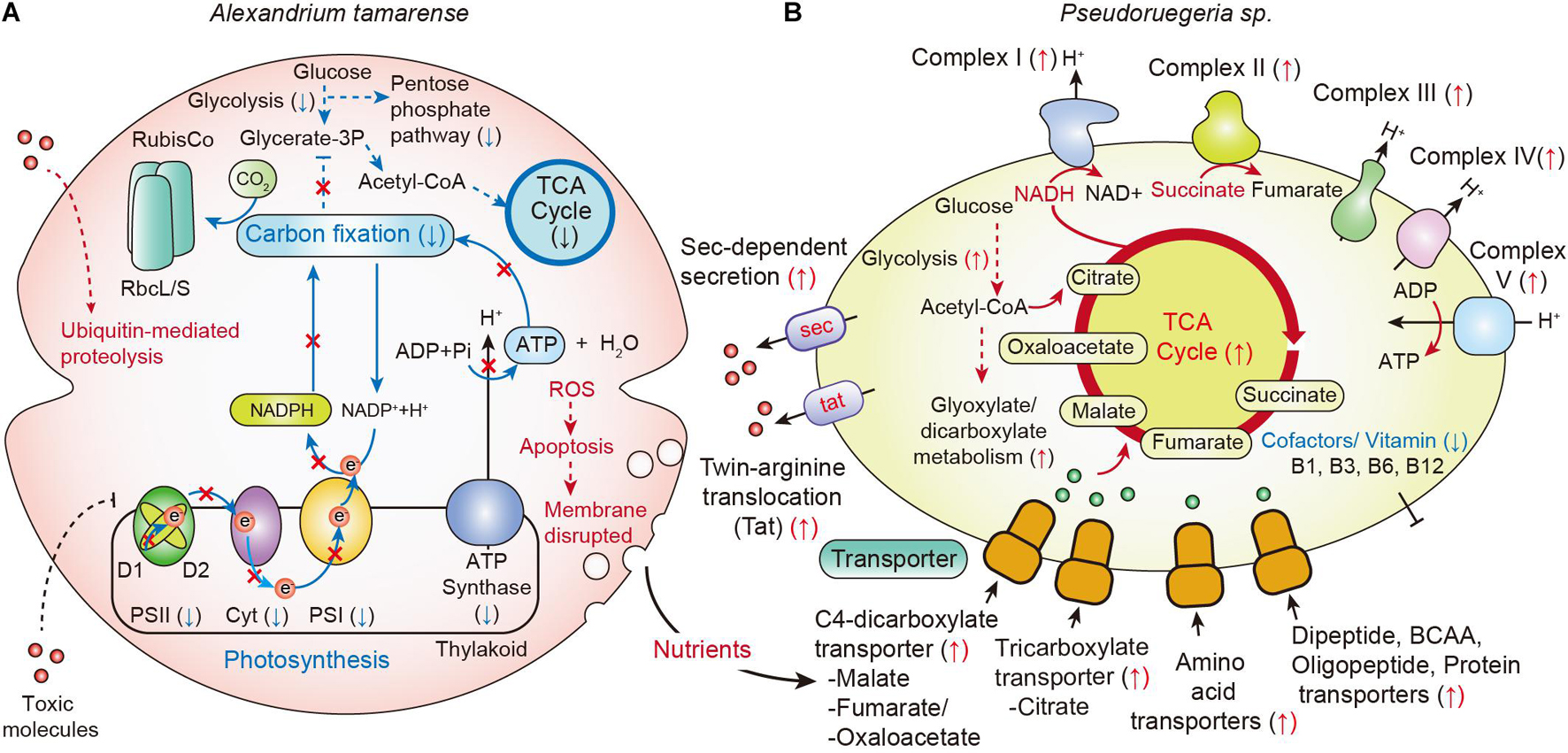
Figure 6. Changes in metabolic pathways by communication between A. catenella and Pseudoruegeria sp. (A,B) A scheme for the communication of A. catenella (A) and Pseudoruegeria sp. (B) during co-culture is displayed based on transcriptome analysis. Blue and red arrows indicate the downregulated (↓) and upregulated (↑) genes, respectively. The dashed line indicates multiple reaction steps.
When cells are disrupted, many cellular nutrients are released into the environment, and the surrounding microorganisms can use these nutrients. Interestingly, in case of Pseudoruegeria sp., the transcription of transporters for the uptake of organic matters, such as C4-dicarboxylate transporter for malate, fumarate, and oxaloacetate and tricarboxylate transporter for citrate, which are core metabolites for the TCA cycle, was substantially increased after co-culture. In addition, the transporters for amino acids, dipeptides and oligopeptides, and proteins were also upregulated from 2 h of co-culture. The upregulation of various transporters for nutrient uptake seems to be closely related with the upregulation of its central metabolism, including the TCA cycle and oxidative phosphorylation pathways, by NADH dehydrogenase (Complex I), fumarate reductase (Complex II), cytochrome bc1 complex (Complex III), cytochrome c oxidase (Complex IV), and ATP synthase (Complex V) on the membrane after co-culture with A. tamarense (Figure 6).
Further, the secondary metabolite biosynthesis gene clusters were found in Pseudoruegeria sp. Among several clusters, the transcriptomes of bacteriocin- and lasso peptide-synthesizing related genes were specifically upregulated. Bacteriocin and lasso peptide are known to be harmful to some mammals and other bacteria (Cornut et al., 2008; Cotter et al., 2013). In addition, the transcription of the β-lactone gene cluster significantly increased during co-culture. Ruegeria pomeroyi, a member of the Roseobacter clade, like Pseudoruegeria sp., produces lactones showing algicidal activity, suggesting the important ecological role of lactone in the interaction between these bacteria and algae (Riclea et al., 2012). Therefore, the increase in the transcription levels of the bacteriocin, the lasso peptide, and the β-lactone cluster implies their potential as algicides. Furthermore, we observed the increase in the transcriptome of the Sec and Tat bacterial secretion systems for unfolded proteins and folded protein secretion, respectively. This suggests that Pseudoruegeria sp. may use these secretion systems for the secretion of extracellular molecules such as peptide toxins during co-culture with A. catenella (Figure 6).
However, the cofactors and vitamins B1, B3, B6, and B12, necessary for the cellular regulation and propagation of dinoflagellates, were extremely downregulated. Dinoflagellates such as A. catenella are known to require vitamins B1, B7, and B12 for their viability (Tandon et al., 2017). Ruegeria supplies these vitamins to vitamin-auxotroph algae under commensal relationships (Croft et al., 2005, 2006; Tandon et al., 2017). However, the synthesis of vitamins B1 and B12 in Pseudoruegeria sp. was downregulated upon co-culture with A. catenella, supporting that it might not have a commensal relationship with A. catenella based on vitamin supply. Overall, our results indicate that Pseudoruegeria sp. exhibits a typical algicidal effect that inactivates the photosystems and disrupts cell membranes, ultimately leading to the death of A. catenella. However, it is not clear yet whether the inactivation of the photosystems is due to direct targeting of the algicides or an indirect result through the inhibition of the photosystem precursor metabolic pathway. Nevertheless, the unique downregulation of photosystems among all metabolic pathways at 2 h of co-culture suggests that the algicides from Pseudoruegeria sp. may closely participate in the initiative process of photosynthesis. In fact, there are known herbicides or algicides that target photosystem II, e.g., triazine, diuron, ametryn, atrazine, tebuthiuron, hexazinone, DCMU (3-(3,4-dichlorophenyl)-1,1-dimethylurea), and terbutryn. These compounds inhibit the photosynthesis by blocking electron transfer from QA to QB, by substituting the plastoquinone binding site on the D1 protein of PSII (Shaw et al., 2010; Broser et al., 2011). Accordingly, we surmise that the algicides secreted by Pseudoruegeria sp., may play a role similar to that of herbicides that block the QB plastoquinone binding site of PSII. Further research on the algicides secreted from Pseudoruegeria sp. is expected to provide a more comprehensive explanation of the relationship between A. catenella and algicidal bacteria.
As we mentioned earlier, the use of algicidal bacteria, with various reported algicidal mechanisms, has been proposed as a biological control method against HABs. Li et al. isolated an algicidal bacterium, Altererythrobacter sp. LY02, which acts against A. tamarense (Li et al., 2016). They suggested that the active substance causes severe morphological and structural damage to the cells and organelles. They also found another algicidal bacterium of A. tamarense, Deinococcus xianganensis, which produces deinoxanthin (Li et al., 2015b). Deinoxanthin induces the production of reactive oxygen species (ROS) in A. tamarense, resulting in serious damage to the structure of organelles, including the chloroplasts and mitochondria. Yang et al. (2014) reported that the algicidal bacterium Joostella sp. DH77-1 can kill A. tamarense. The substance secreted by this bacterium was found to affect membrane integrity and the activity of antioxidant enzymes, such as superoxide dismutase and peroxidase; protein production; and pigment contents. Antioxidant defense enzymes, such as superoxide dismutases (SOD), catalase, glutathione peroxidase, and peroxiredoxin, can be induced by the generation of ROS that promotes the apoptosis by extrinsic and intrinsic signaling pathways (Redza-Dutordoir and Averill-Bates, 2016).
Accordingly, we examined the transcriptome of ROS-related genes in A. catenella when co-cultured with Pseudoruegeria sp. Interestingly, at 12 h of co-culture, we observed the 12 upregulated ROS-related genes (log2FC > 1), including glutathione S-transferase, RAC serin/threonine-protein kinase, arsenite methyltransferase, and soluble epoxide hydrolase/lipid-phosphate phosphatase, when mapped against the total reference genome database in KEGG. To speculate whether the increase of ROS in A. catenella affects the apoptosis, the transcriptional changes of apoptosis-related genes in A. catenella were further investigated after co-culture. In fact, the apoptosis mechanism is well described in mammals but is not well-known in algae. Recently, a marine bacterium, Phaeobacter inhibens, was reported to induce the apoptosis-like programmed cell death through cellular caspase like molecules in Emiliania huxleyi, which forms expansive blooms (Bramucci and Case, 2019). We also found that the apoptosis-related cathepsin or cathepsin like proteases, cathepsin B, cathepsin D, cathepsin F, and cathepsin X, were particularly upregulated at 12 h of co-culture. Especially, lysosomal hydrolases, a cathepsin family of proteases, is known to promote the lysosomal damage and determine the cell fate (Johansson et al., 2010). This suggests that A. catenella may induce its apoptosis through the activation of corresponding proteases promoted by ROS during co-culture with algicidal bacteria, and finally induce the destruction of the membrane after 24 h of long-term co-culture (Figure 6). However, it is still not clear whether A. catenella uses a similar mechanism for apoptosis because cathepsin is a usual protease found in mammalian cells.
Zhang et al. (2018) isolated the marine bacterium Paracoccus sp. strain Y42, which acts against the HAB-causing dinoflagellate Prorocentrum donghaiense. Y42 primarily targets dinoflagellates, showing algicidal activities against A. minutum, Scrippsiella trochoidea, and Skeletonema costatum. The potential algicides from Y42 have been found to cause the loss of photosynthetic pigments and the decline of photosynthesis efficiency and electron transport rate, ultimately leading to death. Although the algicide structure of Y42 has not been identified, the algicidal activity seems to be similar with our changes in that it inhibits the photosynthesis and electron transport. In plant, microorganisms sometimes induce the ubiquitination-mediated protein degradation (Zeng et al., 2006). Ubiquitination system is important for developmental processes and responses to abiotic and biotic stresses. Uniquely at 12 h of co-culture, A catenella showed the upregulation in the transcriptome of the ubiquitin mediated proteolysis related genes. It suggests that A. catenella may utilize a defense system such as ubiquitination system against the algicidal bacterium Pseudoruegeria sp. similarly to plant-microbe interactions.
HABs caused by the dinoflagellate Alexandrium species are a major issue worldwide, and the phenomenon is being accelerated by increasing water temperatures and by the flow of excess nutrients from land into water bodies and oceans. To account for this, many marine bacteria showing algicidal effects have been recognized as a potential tool to control HABs (Kim et al., 1998; Mayali and Azam, 2004; Zhang et al., 2014a). Accordingly, the biotic relationships between HABs and algicidal bacteria are becoming more important. Hence, in this study, the algicidal mechanism through the metatranscriptome analysis was systematically analyzed between A. catenella and its algicidal bacterium Pseudoruegeria sp. Our time-course transcriptome analysis elucidates how A. catenella is affected by algicidal bacteria and how these bacteria obtain functional benefits through metabolic pathways.
Data Availability Statement
The sequencing datasets for this study are available under GenBank BioProject PRJNA604093. Biosample accession numbers for A. catenella and Pseudoruegeria sp. are SAMN13951753 and SAMN15903161, respectively.
Author Contributions
SC, C-YA, and B-KC conceived, designed, and supervised the study. SC, S-RK, YJ, S-HC, EL, SJ, and B-SJ, performed the experiments. SC, YJ, S-HC, B-HO, H-MO, C-YA, and B-KC analyzed the data. SC and B-KC wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Basic Core Technology Development Program for the Oceans and the Polar Regions (2016M1A5A1027455 to SC, 2016M1A5A1027453 to C-YA, and 2016M1A5A1027458 to B-KC) through the National Research Foundation (NRF), funded by the Ministry of Science and ICT (MSIT).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.728890/full#supplementary-material
Footnotes
References
Ahn, C. Y., Joung, S. H., Jeon, J. W., Kim, H. S., Yoon, B. D., and Oh, H. M. (2003). Selective control of cyanobacteria by surfactin-containing culture broth of Bacillus subtilis C1. Biotechnol. Lett. 25, 1137–1142. doi: 10.1023/A:1024508927361
Amaro, A. M., Fuentes, M. S., Ogalde, S. R., Venegas, J. A., and Suarez-Isla, B. A. (2005). Identification and characterization of potentially algal-lytic marine bacteria strongly associated with the toxic dinoflagellate Alexandrium catenella. J. Eukaryot. Microbiol. 52, 191–200. doi: 10.1111/j.1550-7408.2005.00031.x
Amin, S. A., Hmelo, L. R., van Tol, H. M., Durham, B. P., Carlson, L. T., Heal, K. R., et al. (2015). Interaction and signalling between a cosmopolitan phytoplankton and associated bacteria. Nature 522, 98–101. doi: 10.1038/nature14488
Amin, S. A., Parker, M. S., and Armbrust, E. V. (2012). Interactions between diatoms and bacteria. Microbiol. Mol. Biol. Rev. 76, 667–684. doi: 10.1128/Mmbr.00007-12
Anderson, D. M. (1997). Bloom dynamics of toxic Alexandrium species in the northeastern US. Limnol. Oceanogr. 42, 1009–1022. doi: 10.4319/lo.1997.42.5_part_2.1009
Anderson, D. M. (1998). Physiology and bloom dynamics of toxic Alexandrium species, with emphasis on life cycle transitions. Nato Asi. Ser. G Ecol. Sci. 41, 29–48.
Anderson, D. M., Alpermann, T. J., Cembella, A. D., Collos, Y., Masseret, E., and Montresor, M. (2012). The globally distributed genus Alexandrium: multifaceted roles in marine ecosystems and impacts on human health. Harmful Algae 14, 10–35. doi: 10.1016/j.hal.2011.10.012
Anderson, D. M., Burkholder, J. M., Cochlan, W. P., Glibert, P. M., Gobler, C. J., Heil, C. A., et al. (2008). Harmful algal blooms and eutrophication: Examining linkages from selected coastal regions of the United States. Harmful Algae 8, 39–53. doi: 10.1016/j.hal.2008.08.017
Armbrust, E. V., Berges, J. A., Bowler, C., Green, B. R., Martinez, D., Putnam, N. H., et al. (2004). The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306, 79–86. doi: 10.1126/science.1101156
Asakawa, M., Takayama, H., Beppu, R., and Miyazawa, K. (2005). Occurrence of paralytic shellfish poison (PSP)-producing dinoflagellate Alexandrium tamarense in Hiroshima Bay, Hiroshima Prefecture, Japan, during 1993-2004 and its PSP profiles. Shokuhin Eiseigaku Zasshi 46, 246–250.
Azam, F., and Malfatti, F. (2007). Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 5, 782–791. doi: 10.1038/nrmicro1747
Bramucci, A. R., and Case, R. J. (2019). Phaeobacter inhibens induces apoptosis-like programmed cell death in calcifying Emiliania huxleyi. Sci. Rep. 9:5215. doi: 10.1038/s41598-018-36847-6
Broser, M., Glockner, C., Gabdulkhakov, A., Guskov, A., Buchta, J., Kern, J., et al. (2011). Structural basis of cyanobacterial photosystem II Inhibition by the herbicide terbutryn. J. Biol. Chem. 286, 15964–15972. doi: 10.1074/jbc.M110.215970
Buchan, A., LeCleir, G. R., Gulvik, C. A., and Gonzalez, J. M. (2014). Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 12, 686–698. doi: 10.1038/nrmicro3326
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformat. 10:421. doi: 10.1186/1471-2105-10-421
Cho, S. H., Lee, E., Ko, S. R., Jin, S., Song, Y., Ahn, C. Y., et al. (2020). Elucidation of the Biosynthetic Pathway of Vitamin B Groups and Potential Secondary Metabolite Gene Clusters Via Genome Analysis of a Marine Bacterium Pseudoruegeria sp. M32A2M. J. Microbiol. Biotechnol. 30, 505–514. doi: 10.4014/jmb.1911.11006
Cornut, G., Fortin, C., and Soulieres, D. (2008). Antineoplastic properties of bacteriocins: revisiting potential active agents. Am. J. Clin. Oncol. 31, 399–404. doi: 10.1097/COC.0b013e31815e456d
Cotter, P. D., Ross, R. P., and Hill, C. (2013). Bacteriocins - a viable alternative to antibiotics? Nat. Rev. Microbiol. 11, 95–105. doi: 10.1038/nrmicro2937
Croft, M. T., Lawrence, A. D., Raux-Deery, E., Warren, M. J., and Smith, A. G. (2005). Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 438, 90–93. doi: 10.1038/nature04056
Croft, M. T., Warren, M. J., and Smith, A. G. (2006). Algae need their vitamins. Eukaryot. Cell 5, 1175–1183. doi: 10.1128/Ec.00097-06
Cui, Y., Chun, S. J., Baek, S. S., Baek, S. H., Kim, P. J., Son, M., et al. (2020). Unique microbial module regulates the harmful algal bloom (Cochlodinium polykrikoides) and shifts the microbial community along the Southern Coast of Korea. Sci. Total Environ. 721:137725. doi: 10.1016/j.scitotenv.2020.137725
El-Gebali, S., Mistry, J., Bateman, A., Eddy, S. R., Luciani, A., Potter, S. C., et al. (2018). The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. doi: 10.1093/nar/gky995
Fukuyo, Y. (1985). Morphology of Protogonyaulax-Tamarensis (Lebour) Taylor and Protogonyaulax-Catenella (Whedon and Kofoid) Taylor from Japanese Coastal Waters. Bull. Mar. Sci. 37, 529–537.
Furuya, K., Iwataki, M., Lim, P. T., Lu, S., Leaw, C.-P., Azanza, R. V., et al. (2018). “Overview of Harmful Algal Blooms in Asia,” in Global Ecology and Oceanography of Harmful Algal Blooms, eds P. M. Glibert, E. Berdalet, M. A. Burford, G. C. Pitcher, and M. Zhou (Berlin: Springer-Verlag), 289–308.
Gene Ontology Consortium (2014). Gene Ontology Consortium: going forward. Nucleic Acids Res. 43, D1049–D1056. doi: 10.1093/nar/gku1179
Gonzalez, J. M., Kiene, R. P., and Moran, M. A. (1999). Transformation of sulfur compounds by an abundant lineage of marine bacteria in the alpha-subclass of the class Proteobacteria. Appl. Environ. Microbiol. 65, 3810–3819. doi: 10.1128/AEM.65.9.3810-3819.1999
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644. doi: 10.1038/nbt.1883
Haas, B. J., Papanicolaou, A., Yassour, M., Grabherr, M., Blood, P. D., Bowden, J., et al. (2013). De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 8, 1494–1512. doi: 10.1038/nprot.2013.084
Hakanen, P., Suikkanen, S., Franzen, J., Franzen, H., Kankaanpaa, H., and Kremp, A. (2012). Bloom and toxin dynamics of Alexandrium ostenfeldii in a shallow embayment at the SW coast of Finland, northern Baltic Sea. Harmful Algae 15, 91–99. doi: 10.1016/j.hal.2011.12.002
Han, M. S., Jeon, J. K., and Kim, Y. O. (1992). Occurrence of Dinoflagellate Alexandrium-Tamarense, a Causative Organism of Paralytic Shellfish Poisoning in Chinhae Bay, Korea. J. Plankton Res. 14, 1581–1592. doi: 10.1093/plankt/14.11.1581
Hattenrath-Lehmann, T. K., and Gobler, C. J. (2017). Identification of unique microbiomes associated with harmful algal blooms caused by Alexandrium fundyense and Dinophysis acuminata. Harmful Algae 68, 17–30. doi: 10.1016/j.hal.2017.07.003
Heil, C. A., Glibert, P. M., Al-Sarawi, M. A., Faraj, M., Behbehani, M., and Husain, M. (2001). First record of a fish-killing Gymnodinium sp. bloom in Kuwait Bay, Arabian Sea: chronology and potential causes. Mar. Ecol. Prog. Ser. 214, 15–23.
Hogle, S. L., Brahamsha, B., and Barbeau, K. A. (2017). Direct Heme Uptake by Phytoplankton-Associated Roseobacter Bacteria. mSystems 2, 124–116. doi: 10.1128/mSystems.00124-16
Hogle, S. L., Bundy, R. M., Blanton, J. M., Allen, E. E., and Barbeau, K. A. (2016). Copiotrophic marine bacteria are associated with strong iron-binding ligand production during phytoplankton blooms. Limnol. Oceanogr. Lett. 1, 36–43. doi: 10.1002/lol2.10026
Imamura, N., Motoike, I., Noda, M., Adachi, K., Konno, A., and Fukami, H. (2000). Argimicin A, a novel anti-cyanobacterial compound produced by an algae-lysing bacterium. J. Antibiot. 53, 1317–1319. doi: 10.7164/antibiotics.53.1317
Ishikawa, A., Hattori, M., Ishii, K. I., Kulis, D. M., Anderson, D. M., and Imai, I. (2014). In situ dynamics of cyst and vegetative cell populations of the toxic dinoflagellate Alexandrium catenella in Ago Bay, central Japan. J. Plankton Res. 36, 1333–1343. doi: 10.1093/plankt/fbu048
Jeong, H. J., Kim, J. S., Yoo, Y. D., Kim, S. T., Song, J. Y., Kim, T. H., et al. (2008). Control of the harmful alga Cochlodinium polykrikoides by the naked ciliate Strombidinopsis jeokjo in mesocosm enclosures. Harmful Algae 7, 368–377. doi: 10.1016/j.hal.2007.12.004
Jiao, N., Herndl, G. J., Hansell, D. A., Benner, R., Kattner, G., Wilhelm, S. W., et al. (2010). Microbial production of recalcitrant dissolved organic matter: long-term carbon storage in the global ocean. Nat. Rev. Microbiol. 8, 593–599. doi: 10.1038/nrmicro2386
Johansson, A. C., Appelqvist, H., Nilsson, C., Kagedal, K., Roberg, K., and Ollinger, K. (2010). Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 15, 527–540. doi: 10.1007/s10495-009-0452-5
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2015). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kent, W. J. (2002). BLAT–the BLAST-like alignment tool. Genome Res. 12, 656–664. doi: 10.1101/gr.229202
Kim, J. H., Jeong, H. J., Lim, A. S., Kwon, J. E., Lee, K. H., Park, K. H., et al. (2017). Removal of two pathogenic scuticociliates Miamiensis avidus and Miamiensis sp using cells or culture filtrates of the dinoflagellate Alexandrium andersonii. Harmful Algae 63, 133–145. doi: 10.1016/j.hal.2017.02.002
Kim, M. C., Yoshinaga, I., Imai, I., Nagasaki, K., Itakura, S., and Ishida, Y. (1998). A close relationship between algicidal bacteria and termination of Heterosigma akashiwo (Raphidophyceae) blooms in Hiroshima Bay, Japan. Mar. Ecol. Prog. Ser. 170, 25–32. doi: 10.3354/meps170025
Koonin, E. V., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Krylov, D. M., Makarova, K. S., et al. (2004). A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5:R7. doi: 10.1186/gb-2004-5-2-r7
Kurisu, G., Zhang, H., Smith, J. L., and Cramer, W. A. (2003). Structure of the cytochrome b6f complex of oxygenic photosynthesis: tuning the cavity. Science 302, 1009–1014. doi: 10.1126/science.1090165
Landa, M., Burns, A. S., Roth, S. J., and Moran, M. A. (2017). Bacterial transcriptome remodeling during sequential co-culture with a marine dinoflagellate and diatom. ISME J. 11, 2677–2690. doi: 10.1038/ismej.2017.117
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, S. O., Kato, J., Nakashima, K., Kuroda, A., Ikeda, T., Takiguchi, N., et al. (2002). Cloning and characterization of extracellular metal protease gene of the algicidal marine bacterium Pseudoalteromonas sp strain A28. Biosci. Biotechnol. Biochem. 66, 1366–1369. doi: 10.1271/bbb.66.1366
Li, B., and Dewey, C. N. (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323
Li, Y., Liu, L., Xu, Y., Guan, C., Lei, X., Zheng, W., et al. (2016). First evidence of Altererythrobacter sp. LY02 with indirect algicidal activity on the toxic dinoflagellate, Alexandrium tamarense. Curr. Microbiol. 73, 550–560. doi: 10.1007/s00284-016-1093-x
Li, Y., Zhu, H., Lei, X., Zhang, H., Cai, G., Chen, Z., et al. (2015a). The death mechanism of the harmful algal bloom species Alexandrium tamarense induced by algicidal bacterium Deinococcus sp. Y35. Front. Microbiol. 6:992. doi: 10.3389/fmicb.2015.00992
Li, Y., Zhu, H., Lei, X., Zhang, H., Guan, C., Chen, Z., et al. (2015b). The first evidence of deinoxanthin from Deinococcus sp. Y35 with strong algicidal effect on the toxic dinoflagellate Alexandrium tamarense. J. Hazard. Mater. 290, 87–95. doi: 10.1016/j.jhazmat.2015.02.070
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550–550. doi: 10.1186/s13059-014-0550-8
Lu, Y. M., Wohlrab, S., Groth, M., Glockner, G., Guillou, L., and John, U. (2016). Transcriptomic profiling of Alexandrium fundyense during physical interaction with or exposure to chemical signals from the parasite Amoebophrya. Mol. Ecol. 25, 1294–1307. doi: 10.1111/mec.13566
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics 21, 3448–3449. doi: 10.1093/bioinformatics/bti551
Mayali, X., and Azam, F. (2004). Algicidal bacteria in the sea and their impact on algal blooms. J. Eukaryot. Microbiol. 51, 139–144. doi: 10.1111/j.1550-7408.2004.tb00538.x
Meng, F. Q., Song, J. T., Zhou, J., and Cai, Z. H. (2019). Transcriptomic Profile and Sexual Reproduction-Relevant Genes of Alexandrium minutum in Response to Nutritional Deficiency. Front. Microbiol. 10:2629. doi: 10.3389/fmicb.2019.02629
Park, S., Jung, Y. T., Won, S. M., and Yoon, J. H. (2014). Pseudoruegeria sabulilitoris sp. nov., isolated from seashore sand. Int. J. Syst. Evol. Microbiol. 64, 3276–3281. doi: 10.1099/ijs.0.066258-0
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Paul, C., and Pohnert, G. (2011). Interactions of the algicidal bacterium Kordia algicida with diatoms: regulated protease excretion for specific algal lysis. PLoS One 6:e21032. doi: 10.1371/journal.pone.0021032
Peperzak, L. (2005). Future increase in harmful algal blooms in the North Sea due to climate change. Water Sci. Technol. 51, 31–36.
Pohlner, M., Marshall, I., Schreiber, L., Cypionka, H., and Engelen, B. (2017). Draft Genome Sequence of Pseudoruegeria sp. SK021, a Representative of the Marine Roseobacter Group, Isolated from North Sea Sediment. Genome Announc. 5, 541–517. doi: 10.1128/genomeA.00541-17
Qi, B., and Han, M. (2018). Microbial siderophore Enterobactin promotes mitochondrial iron uptake and development of the host via interaction with ATP synthase. Cell 175, 571–582e511. doi: 10.1016/j.cell.2018.07.032
Redza-Dutordoir, M., and Averill-Bates, D. A. (2016). Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 1863, 2977–2992. doi: 10.1016/j.bbamcr.2016.09.012
Riclea, R., Gleitzmann, J., Bruns, H., Junker, C., Schulz, B., and Dickschat, J. S. (2012). Algicidal lactones from the marine Roseobacter clade bacterium Ruegeria pomeroyi. Beilstein J. Org. Chem. 8, 941–950. doi: 10.3762/bjoc.8.106
Seymour, J. R., Amin, S. A., Raina, J. B., and Stocker, R. (2017). Zooming in on the phycosphere: the ecological interface for phytoplankton-bacteria relationships. Nat. Microbiol. 2:17065. doi: 10.1038/nmicrobiol.2017.65
Shaw, M., Furnas, M. J., Fabricius, K., Haynes, D., Carter, S., Eaglesham, G., et al. (2010). Monitoring pesticides in the Great Barrier Reef. Mar. Pollut. Bull. 60, 113–122. doi: 10.1016/j.marpolbul.2009.08.026
Shin, H. H., Li, Z., Kim, E. S., Park, J. W., and Lim, W. A. (2017). Which species, Alexandrium catenella (Group I) or A-pacificum (Group IV), is really responsible for past paralytic shellfish poisoning outbreaks in Jinhae-Masan Bay, Korea? Harmful Algae 68, 31–39. doi: 10.1016/j.hal.2017.07.006
Shin, H., Lee, E., Shin, J., Ko, S.-R., Oh, H.-S., Ahn, C.-Y., et al. (2018). Elucidation of the bacterial communities associated with the harmful microalgae Alexandrium tamarense and Cochlodinium polykrikoides using nanopore sequencing. Sci. Rep. 8, 1–8.
Smayda, T. J. (1997). Harmful algal blooms: Their ecophysiology and general relevance to phytoplankton blooms in the sea. Limnol. Oceanogr. 42, 1137–1153. doi: 10.4319/lo.1997.42.5_part_2.1137
Tandon, P., Jin, Q., and Huang, L. (2017). A promising approach to enhance microalgae productivity by exogenous supply of vitamins. Microb. Cell. Fact. 16:219. doi: 10.1186/s12934-017-0834-2
Tang, Y. Z., Koch, F., and Gobler, C. J. (2010). Most harmful algal bloom species are vitamin B1 and B12 auxotrophs. Proc. Natl. Acad. Sci. U S A. 107, 20756–20761. doi: 10.1073/pnas.1009566107
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36.
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28:511. doi: 10.1038/nbt.1621
UniProt: a hub for protein information (2014). UniProt: a hub for protein information. Nucleic Acids Res. 43, D204–D212. doi: 10.1093/nar/gku989
Vale, R. D. (2003). The molecular motor toolbox for intracellular transport. Cell 112, 467–480. doi: 10.1016/s0092-8674(03)00111-9
Vingiani, G. M., Stalberga, D., De Luca, P., Ianora, A., De Luca, D., and Lauritano, C. (2020). De novo Transcriptome of the Non-saxitoxin Producing Alexandrium tamutum Reveals New Insights on Harmful Dinoflagellates. Mar. Drugs 18:md18080386. doi: 10.3390/md18080386
Wagner-Dobler, I., and Biebl, H. (2006). Environmental biology of the marine Roseobacter lineage. Annu. Rev. Microbiol. 60, 255–280. doi: 10.1146/annurev.micro.60.080805.142115
Wang, J., and Wu, J. (2009). Occurrence and potential risks of harmful algal blooms in the East China Sea. Sci. Total Environ. 407, 4012–4021. doi: 10.1016/j.scitotenv.2009.02.040
Yang, X. R., Li, X. Y., Zhou, Y. Y., Zheng, W., Yu, C. P., and Zheng, T. L. (2014). Novel insights into the algicidal bacterium DH77-1 killing the toxic dinoflagellate Alexandrium tamarense. Sci. Total Environ. 482, 116–124. doi: 10.1016/j.scitotenv.2014.02.125
Zeng, L. R., Vega-Sanchez, M. E., Zhu, T., and Wang, G. L. (2006). Ubiquitination-mediated protein degradation and modification: an emerging theme in plant-microbe interactions. Cell Res. 16, 413–426. doi: 10.1038/sj.cr.7310053
Zhang, B. Z., Cai, G. J., Wang, H. T., Li, D., Yang, X. J., An, X. L., et al. (2014a). Streptomyces alboflavus RPS and Its Novel and High Algicidal Activity against Harmful Algal Bloom Species Phaeocystis globosa. PLoS One 9:0092907. doi: 10.1371/journal.pone.0092907
Zhang, F., Ye, Q., Chen, Q., Yang, K., Zhang, D., Chen, Z., et al. (2018). Algicidal Activity of Novel Marine Bacterium Paracoccus sp. Strain Y42 against a Harmful Algal-Bloom-Causing Dinoflagellate, Prorocentrum donghaiense. Appl. Environ. Microbiol. 84, 1015–1018. doi: 10.1128/AEM.01015-18
Zhang, S., Sui, Z., Chang, L., Kang, K., Ma, J., Kong, F., et al. (2014b). Transcriptome de novo assembly sequencing and analysis of the toxic dinoflagellate Alexandrium catenella using the Illumina platform. Gene 537, 285–293. doi: 10.1016/j.gene.2013.12.041
Zhang, Y., Xu, Y., Fang, W., Wang, X., Fang, Z., and Xiao, Y. (2017). Pseudoruegeria marinistellae sp. nov., isolated from an unidentified starfish in Sanya, China. Antonie Van Leeuwenhoek 110, 187–194. doi: 10.1007/s10482-016-0789-z
Keywords: harmful algae, Alexandrium, transcriptome, algicidal bacteria, symbiosis
Citation: Cho S, Cho S-H, Ko S-R, Jeong Y, Lee E, Jin S, Jeong B-S, Oh B-H, Oh H-M, Ahn C-Y and Cho B-K (2021) Elucidation of the Algicidal Mechanism of the Marine Bacterium Pseudoruegeria sp. M32A2M Against the Harmful Alga Alexandrium catenella Based on Time-Course Transcriptome Analysis. Front. Mar. Sci. 8:728890. doi: 10.3389/fmars.2021.728890
Received: 22 June 2021; Accepted: 08 September 2021;
Published: 29 September 2021.
Edited by:
Irene Wagner-Doebler, Helmholtz Center for Infection Research, Helmholtz Association of German Research Centers (HZ), GermanyReviewed by:
Chih-Ching Chung, National Taiwan Ocean University, TaiwanDavid H. Green, Scottish Association for Marine Science, United Kingdom
Christopher John Stanley Bolch, University of Tasmania, Australia
Copyright © 2021 Cho, Cho, Ko, Jeong, Lee, Jin, Jeong, Oh, Oh, Ahn and Cho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Suhyung Cho, c2hjaG85NUBrYWlzdC5hYy5rcg==; Byung-Kwan Cho, YmNob0BrYWlzdC5hYy5rcg==
†These authors have contributed equally to this work