 Erika García-Bonilla1,2†
Erika García-Bonilla1,2† Diego Chaves-Moreno3†
Diego Chaves-Moreno3† Diego Riaño-Pachón4
Diego Riaño-Pachón4 Wilson Terán5
Wilson Terán5 Alberto Acosta1
Alberto Acosta1 Howard Junca2*
Howard Junca2*- 1Laboratorio Ecosistemas Marinos Estratégicos, Unidad de Ecología y Sistemática - UNESIS, Facultad de Ciencias - Departamento de Biología, Pontificia Universidad Javeriana, Bogotá, Colombia
- 2RG Microbial Ecology: Metabolism, Genomics & Evolution, Division of Ecogenomics & Holobionts, Microbiomas Foundation, Chía, Colombia
- 3Microbial Interactions and Processes Research Group, Helmholtz Centre for Infection Research, Braunschweig, Germany
- 4Laboratório de Biologia Computacional, Evolutiva e de Sistemas, Centro de Energía Nuclear na Agricultura, Universidade de São Paulo, Piracicaba, Brazil
- 5Biología de Plantas y Sistemas Productivos, Departamento de Biología, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá, Colombia
Viruses are among the most abundant biological entities in the ocean, largely responsible of modulating nutrients fluxes and influencing microbial composition and functioning. In marine invertebrate holobionts like sponges and their associated microbiomes, little is known about virome composition. Here, we characterized the Haliclona fulva hologenome, an encrusting low-microbial abundance sponge found across the Western Mediterranean Sea (35–40 m of depth) producer of a large metabolic repertoire of bioactive compounds and harboring a distinct and stable associated microbiome. Assembled contigs from shotgun metagenome sequences obtained from H. fulva specimens were comprehensively analyzed regarding taxonomic and functional content revealing its remarkable and abundant viral community dominated by single-stranded DNA (ssDNA) virus. Viral families consistently detected in contigs are Circoviridae, Phycodnaviridae, Poxviridae, Herelleviridae, Mimiviridae, Microviridae, and notably the first reported encounter of Nanoviridae and Genomoviridae in Porifera, expanding their known host range. The relative abundance of inferred bacteriophages/prophages was low, suggesting that the prokaryotic community in this sponge has a limited host range and susceptibility. H. fulva showed a distinct viral composition supporting the general proposition of specific and coevolving viromes in marine holobionts.
Introduction
Marine sponges represent one of the oldest animal holobiont associations on Earth resulting from the coevolved partnership of the multicellular host with microbial communities, allowing them to colonize different environments, to gain physical and chemical protection against depredators, and to perform functions essential for nutrient acquisition, storage and disposal (Bell, 2008; Webster and Thomas, 2016). Marine sponges are classified according to abundance of their associated microbial community as high microbial-abundance (HMA) and low microbial-abundance (LMA) (Vacelet and Donadey, 1977). HMA sponges are commonly harboring highly diverse microbial communities which contain bacterial and archaeal types that are found in Porifera; these assemblages of microorganisms are known “sponges microbial signatures,” while LMA sponges have a lower and variable diversity, and it can dominated by fewer microorganisms (Thomas et al., 2016). Bacterial and archaeal components of microbial communities have been extensively researched in a wide variety of sponge species over the last years (Hentschel et al., 2003; Giles et al., 2013; Bayer et al., 2014; Thomas et al., 2016), however, little is known about the ecology and functional role of other members such as the viruses present in the holobiont.
The abundance of viral particles in marine ecosystems can reach an average of 107 viruses-like particles/ml of surface seawater and a total estimated of 1030 in the oceans (Wommack and Colwell, 2000). Viruses can control bacterial populations, increasing their diversity and shaping species composition (Rosenwasser et al., 2016). Furthermore, they can help to improve the host’s defense mechanisms, i.e., they are considered part of their immune system, for example, in corals (Bettarel et al., 2015). Others functions related to viruses are associated with biogeochemical cycles, as they can contribute with the releasing organic matter to the environment via cell lysis, providing a carbon resource able to support the growth of several microbial groups (Guidi et al., 2016). Viruses are more likely to infect and lyse fast-growing cells to ensure a faster viral replication, a pattern named “killing the winner” (Thingstad and Lignell, 1997). Alternatively, instead of killing the winner they could integrate into their host genomes, replicating with them as a prophage. This mechanism, named “piggyback the winner,” can protect the bacteria from infections caused by others related phages and improve the host fitness (Weinbauer and Rassoulzadegan, 2004; Silveira and Rohwer, 2016). This scenario supports the exchange of genetic material between the host and the virus by genetic transduction (Coutinho et al., 2017). However, the switch between lytic and lysogenic cycles, as well as the factors driving both processes, are still unknown.
Virus can also reprogram the host’s metabolism increasing their metabolic potential by the introduction of virus-encoded auxiliary metabolic genes (vAMGs) (Breitbart et al., 2007), some examples include the presence of genes related to collagen production, genes coding for ankyrin repeat proteins (ARPs), resistance genes to heavy metals and antimicrobial activity (Laffy et al., 2018; Pascelli et al., 2020).
Although some studies have described marine viral communities, their roles in animals and sponges in these environments is still poorly understood. The presence of virus like particles (VLPs) in marine sponges was observed and reported by Vacelet and Donadey (1977) by transmission electron microscopy (TEM). Recently, the generation of novel viral isolation methods, together with next generation sequencing technologies, have become powerful tools for studying viral communities. So far, only a few recent studies have characterized sponge-associated viral communities (Laffy et al., 2016, 2018; Jahn et al., 2019, 2021; Butina et al., 2020; Pascelli et al., 2020; Urayama et al., 2020), finding members belonging mainly to Caudovirales double-strand DNA (dsDNA) (families: Podoviridae, Siphoviridae, and Myoviridae), Microviridae, and Poxviridae; others representatives found in a lower relative abundance were Megavirales, Parvoviridae, Circoviridae, and Phycodnaviridae. Overall, these recent analysis show that sponges represent niches of viral diversity with a low variability in their assemblages, which is consistent with the microbial status assigned for the sponge. Interestingly, a study found a high degree of individuality in sponge viromes, which suggests unique individual fingerprints (Laffy et al., 2018).
However, research works about marine sponge viromes and their relationship with the microbial status of the holobiont are still scarce. In the present study, we are reporting the characterization by shotgun metagenomics of the functional traits encoded in the virome of Haliclona fulva (Demospongiae: Haplosclerida), selected as a suitable holobiont model given its particular features of: (1) low microbial abundance, largely dominated by two bacterial and archaeal types (García-Bonilla et al., 2019), and (2) remarkable producer of a large metabolic repertoire of bioactive compounds such as fulvynes, alkaloids, peptides, and polyacetylenes (Nuzzo et al., 2012; Tribalat et al., 2016).
Materials and Methods
Sample Collection
Three specimens of H. fulva (HF1, HF2, and HF3) were collected by SCUBA diving in the Mediterranean Sea at the Grotte du Lido, in the bay of Villefranche-sur-Mer, France (latitude: 43° 41′ 31.49″ N; longitude: 7° 19′ 12.19″ E) at 35 m depth. After collection, all individual samples were placed in independent plastic bags. They were preserved in ethanol 70% (v/v) and stored at −20°C until further analysis. The samples were selected based on the findings of our previous study (García-Bonilla et al., 2019) and are coming from exactly the same spots and colonies/specimens analyzed regarding microbiome content by 16S amplicons, showing a stable and consistent microbial composition of low microbial abundance and diversity.
DNA Extraction and Sequencing
Metagenomic DNA was extracted from 15 g sponge wet weight using a MagAttract® HMW DNA kit (Qiagen, Germany) following the manufacturer’s instructions. Tissue lysis was made for 16 h according to protocol. Extracted DNA was eluted with 50 μl water, and its concentration determined by fluorescence using the Qubit® dsDNA BR assay kit (Thermo Fisher Scientific). Nucleic acid integrity was verified by 0.8% agarose gel electrophoresis. Sponge DNA extracts were used as template to perform isothermal multiple displacement hologenome amplification with a phi29 polymerase of high processivity and fidelity in order to increase total DNA concentrations (Lasken, 2007) and to avoid the effect of coextracted enzymatic inhibitors detected in H. fulva, improving further downstream processes (library construction and sequencing). The amplification process was made using REPLI-g Mini kit (Qiagen, Germany), according to manufacturer’s recommendations. Briefly, samples were incubated at 30°C for 11 h followed by 3 min at 65°C for polymerase inactivation. For all assays, a negative control was run to evaluate the presence of contaminants during amplification. Quantity and quality of DNA was measured as described previously.
Genomic libraries were constructed using TruSeq DNA PCR free kit (Illumina, United States). Shotgun hologenome sequencing was performed using paired-end Illumina technology (Macrogen, South Korea). The complete raw sequencing data obtained is publicly accessible at NCBI GenBank SRA under Bioproject PRJNA741981.
Raw Data Processing and Assembly
An initial filtering of the sequencing reads was done after visual evaluation using FastQC (v.0.11.8) consisting of a quality and length-based filtering performed with Trimmomatic (v.0.31). Filtered and processed reads were assembled using Megahit (v.1.0) (Li et al., 2014) and IDBA-UD (Peng et al., 2012) with default parameters. To compare, identify and join overlaps between reads from each assembly, we used minimus2 from the AMOS suite with default parameters (Treangen et al., 2011). Obtained contigs were filtered and those <500 bp were removed. Protein-coding gene prediction was performed using Prodigal (v.2.6.3) (flag –p, metagenomics mode) (Hyatt et al., 2010).
Taxonomical Metagenomic Annotation
In order to allocate contigs across the general taxonomic groups (bacteria, viruses, metazoa, archaea, and fungi), assembled reads from minimus2 were queried against the NCBI nucleotide collection database (nt downloaded December 2019) using blastn (v. 2.10.1), all hits with e-values ≤ 10–5 were processed. The number of hits linked to the distinct taxonomical groups (domains), were computed per contig (Query) and normalized by the number of all hits (per Query). Final assignment of the contig was given to the taxonomical group with the major number of hits, a criterion we are referring as supermajority-rule. Following the same rationale, we used such supermajority-rule to assign contigs to the different microbial orders.
Viral Community Analysis
To enhance the taxonomical classification within the viral contigs, larger putative-viral-contigs than 500 nt were blasted (blastn) against three databases: ViruSite (Stano et al., 2016), Viral RefSeq and viral genomes from NCBI (filtered by host such as bacteria, archaea, and invertebrates). An additional metric, identity ratio (alignment length × ID percentage/contig Length) (Sierra et al., 2010), was computed per blast result, then best hits with e-value ≤ 10–3 and ratio ≤5 were selected as representative per database. Comparison across database searches (per contig) was computed, and taxonomical affiliation per contig was derived from the best scored hit (ratio and e-value) crosswise database.
Viral large contigs (>4,000 pb) obtained from H. fulva metagenomes were blasted against Global Oceans Viromes (GOV) datasets and also against contigs from and ocean viral communities previously reported (Roux et al., 2016); eligible matches were selected following the same criteria mentioned above.
To refine the taxonomical classification, a phylogenetic approach was designed, using the information acquired by the taxonomical assignation cross-linked with the functional annotation of the predicted CDSs (prodigal). Briefly, CDSs were retrieved from presumptive viral contigs (length ≥500 nt) and blasted (blastp) against the NR database from the NCBI. CDSs whose best hits were linked with the Rep-associated-protein (e-values ≤10–3) and were fully contained within the contig, were chosen as representative per contig. Each CDS was related to a contig name that included its taxonomic assignation, this information was added if existing, and that group was named the “rep-group.” In parallel, reference sequences distinctive of known viral families from aquatic environments or animals were retrieved from GenBank (Supplementary Table 1) and named as “seed-group.” These reference sequences were used as guides for the unclassified virome sequences.
Reference sequences (seed-group) either independently or joined with the unclassified Rep proteins (rep-group), were aligned using Muscle (v.3.8.31), followed by phylogenetic inference using RAxML (-m PROTGAMMAGTR -T 20 -p 12345 -x 12345). The best tree per reconstruction (seed-group and combined seed + rep group) were selected and transformed into a distance matrix, using the nwk2mat.py script.1 Similarity matrix was parsed using an in-house script that retrieves, for every unclassified Rep, the closest reference (shortest phylogenetic distance) sequence plus the top five subsequent sequences (Supplementary Table 2). Taxonomical associations were resolved using the closest reference as well as the reference tree, overall distances were computed and used as cut-off, for final identification.
Functional Annotation
To assess the metabolic potential of the model holobiont, the predicted protein sequences were searched against the EggNOG (v. 5.0.0) database of orthologous groups and functional annotation (Huerta-Cepas et al., 2016). Additionally, protein sequences were queried against NCBI-nr database, and conserved patterns or domains were searched against PFAM and TIGRFAM databases using InterProScan (v. 5.46-81) (Jones et al., 2014). The best hit for each protein with an e-value < 10–3 and identity >30% was considered for annotation.
Results
Sequence Data Description
Three H. fulva hologenome samples harboring a well-defined, stable and particular microbial composition (García-Bonilla et al., 2019) were sequenced by Illumina technology. A total of 8,128,740, 5,458,396, and 2,705,172 raw reads were obtained for HF1, HF2, and HF3 samples. After data filtering and trimming, a total of 7,609,456, 4,718,775, and 2,468,369 reads remained for each sample. As different sequencing depths were obtained per sample and all specimens came from the same sampling area and time, and showed a very similar microbial composition (García-Bonilla et al., 2019), therefore, all reads were merged for downstream analyses in order to increase the chances to obtain larger contig assemblies. This larger hologenome assembly generated a total of 11,812 and 14,762 contigs from IDBA-UD and megahit, respectively. The comparison between both assemblers using minimus2 revealed that 9,005 contigs had overlaps, and 6,764 contigs were obtained as unique sequences (singleton). Assembly statistics showed that largest contig had 9,591 bp, N50 and N70 were 860 (n = 3,724) and 594 (n = 6,986), respectively. In total, 2,897 contigs larger than 1 kb were obtained. From this assembly, a total of 17,264 ORFs were predicted by Prodigal.
Haliclona fulva Hologenome Composition
A total of 3,718 contigs, out of 15,769, could be assigned to a taxonomical group, according to the supermajority rule. Of those, 2,164 (58%) contigs were classified as metazoa, 893 (24%) as viruses, 295 (8%) as bacteria, 51 (1%) as fungi, and 22 (0.5%) as archaea (Supplementary Figure 1), each group with its respective GC content (Figure 1).
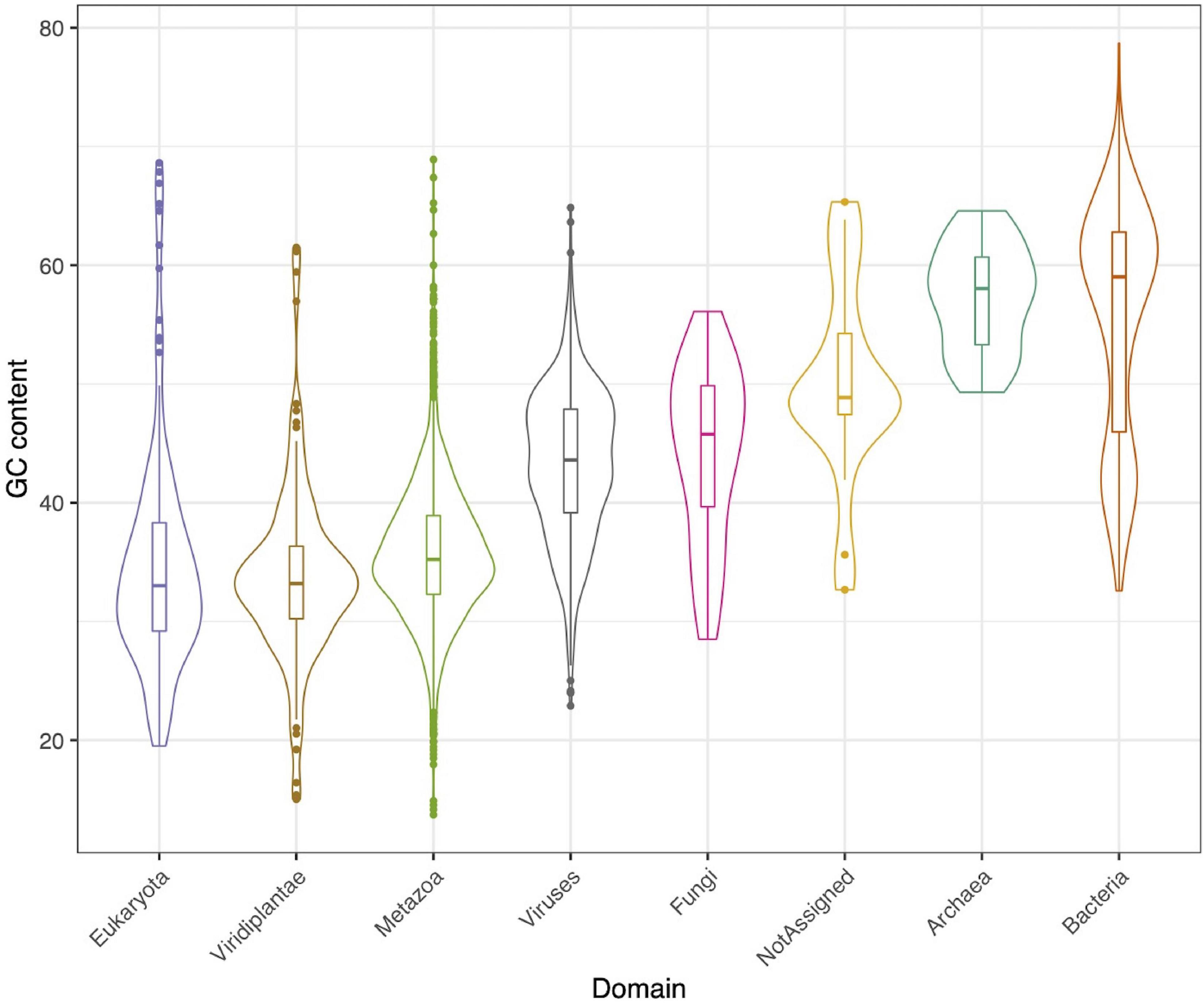
Figure 1. GC content for each identified taxonomical group.
Overall, results showed that most eukaryotic contigs were assigned to metazoan, of them, one quarter of contigs did match with the order Haplosclerida, where H. fulva is classified, thus, this study contributes with the largest genomic information known and publicly available for this sponge species, currently having only a single 28S gene fragment reported (GenBank accession number AJ225829). For fungi, Aspergillaceae family was the most abundant, followed by Saccharomycetaceae (Figure 2A).
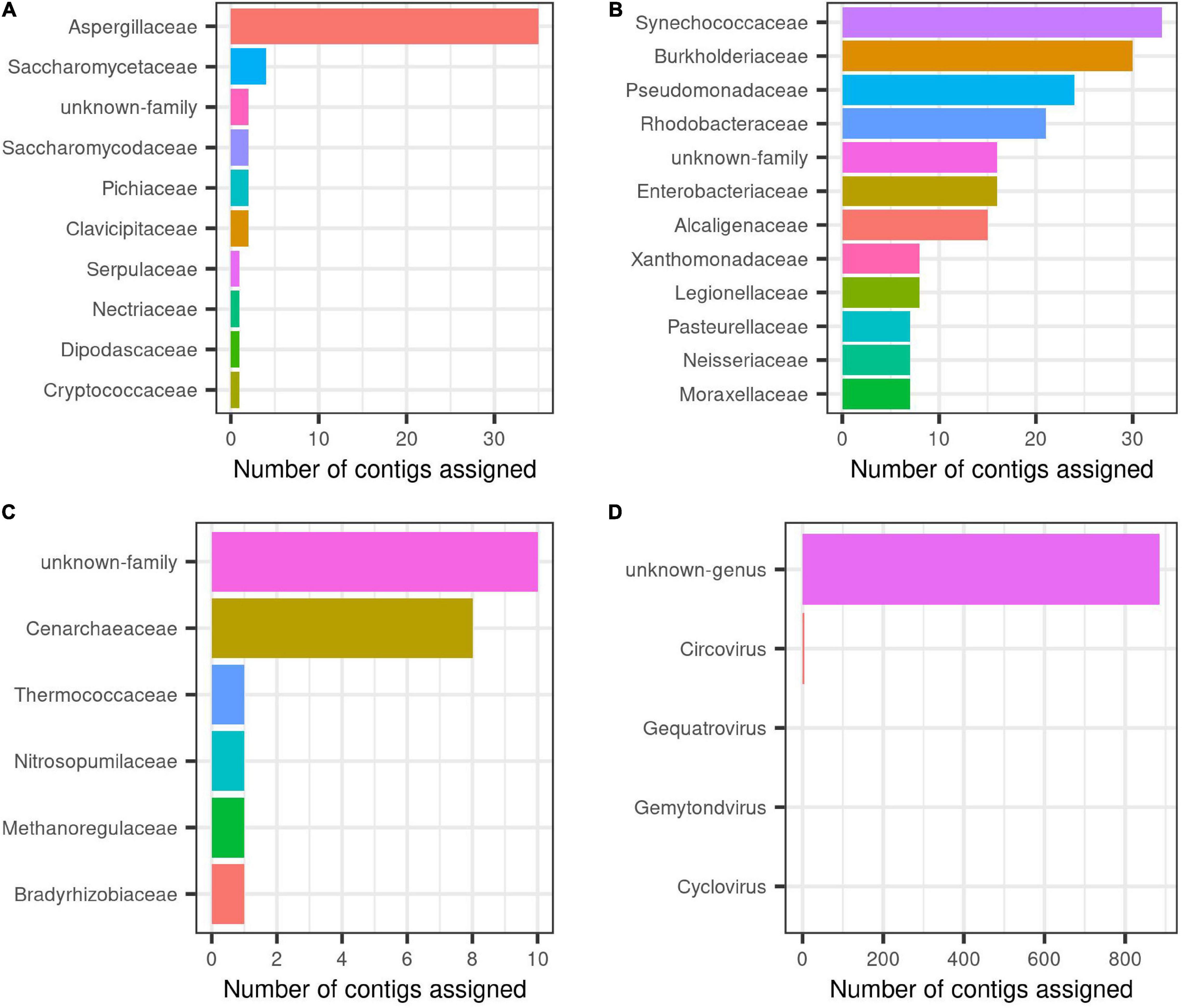
Figure 2. Taxonomic description at family level for each group inside H. fulva-associated microbiome. (A) Fungi, (B) bacteria, (C) archaea, and (D) viruses.
In relation to prokaryotes, bacterial contigs were dominated by high GC content organism (Figure 1). The predominant families were Synechococcaceae and Burkholderiaceae, followed by Pseudomonadaceae and Rhodobacteraceae. Others identified orders in a lower proportion were Enterobacteriaceae, Alcaligenaceae, among others (Figure 2B). In this same trend, for archaea, the majority of contigs could not be classified to a known order, while Cenarchaeaceae was the family most abundant (Figure 2C). Interestingly, in the case of virus, when nt database was used, the majority of contigs could not be taxonomically classified (Figure 2D).
In order to assess the structure of the virome associated to the H. fulva microbiome accounting for 24% of the total contigs found, we developed an approach called supermajority rule to assign viral contigs (details in section “Materials and Methods”), and three reference databases were used for their taxonomic classification of this fraction. We were able to determine that out of 594 contigs that passed the filters and thresholds, 10% could be assigned to a described virus family. Among the used databases, we found a congruent (58%) identification between ViruSite and Viral genomes for viral contigs with assigned family classification. However, the amount of contigs classified varied greatly depending of the database used, ViruSite allowing us to classify 62% of the viral contigs, followed by Viral genomes (28%) (Supplementary Figure 2). Results for RefSeq database showed that it was the less prone to identify sequences because the obtained scores were below the fixed threshold, especially for the calculated ratio parameter. Using this database, only 9% contigs could be classified (Supplementary Figure 2). Analysis of viral community composition showed that single-stranded DNA (ssDNA) viruses were dominant, specifically, those belonging to Circoviridae (26 of identified contigs), unclassified viruses (16), and in a lower proportion Phycodnaviridae, Poxviridae, among others (Figure 3).
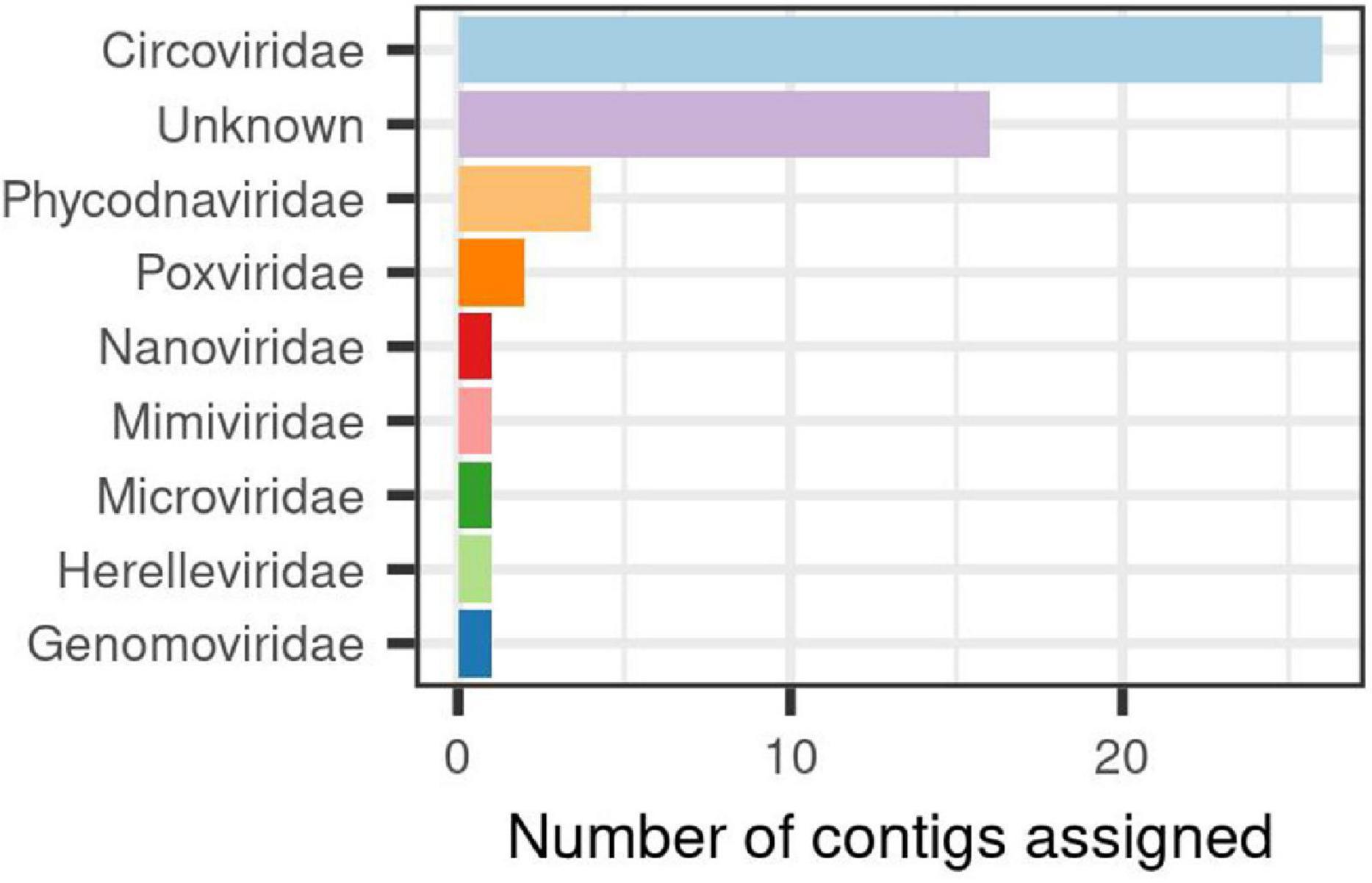
Figure 3. Viral families identified in the associated-virome to Haliclona fulva.
Interestingly, the longest contigs obtained in this large H. fulva hologenome dataset were viral (Supplementary Figure 1). From those large contigs, 49 had a length more than 4 kb and only three were readily classified to a known family, while the remaining were grouped as unknown sequences. In order to mine deeper in these latter sequences, they were compared against GOV dataset. Blast results showed that 23 contigs passed the established criteria and the viral cluster that grouped more contigs was GOV_bin_5855_contig-100_112. However, neither information about taxonomy nor associated host has been linked/reported.
Because only a small fraction of the viruses detected could be identified using the described approaches, we decided to carry out a phylogenetic analysis using the replication associated (Rep) proteins identified in the viral contigs. Analysis showed that 127 ORFs corresponded to the complete protein, derived from 101 contigs. Overall, obtained results from calculated distances between viral sequences and reference sequences (Supplementary Table 2) showed the relationships of the proteins encoded in 54 unique contigs. The biggest cluster, represented by unclassified DNA viruses, contained 21 contigs, followed by the Phycodnaviridae cluster (12 contigs) and a cluster of unclassified ssDNA viruses (10 contigs). In a lower proportion, sequences belonging to Circoviridae, Cressdnaviricota_Circoviridae, and Cressdnaviricota were identified with 5, 4, and 2 clustered contigs, respectively (Figure 4). These branches showed a close relationship, suggesting that REP protein phylogeny is a useful tool for clustering members inside a virome and increase/improve their identification (Supplementary Figure 3). In this study, using both supermajority rule and phylogeny approaches, we could assign a taxonomy to 20% contigs classified primary as viral.
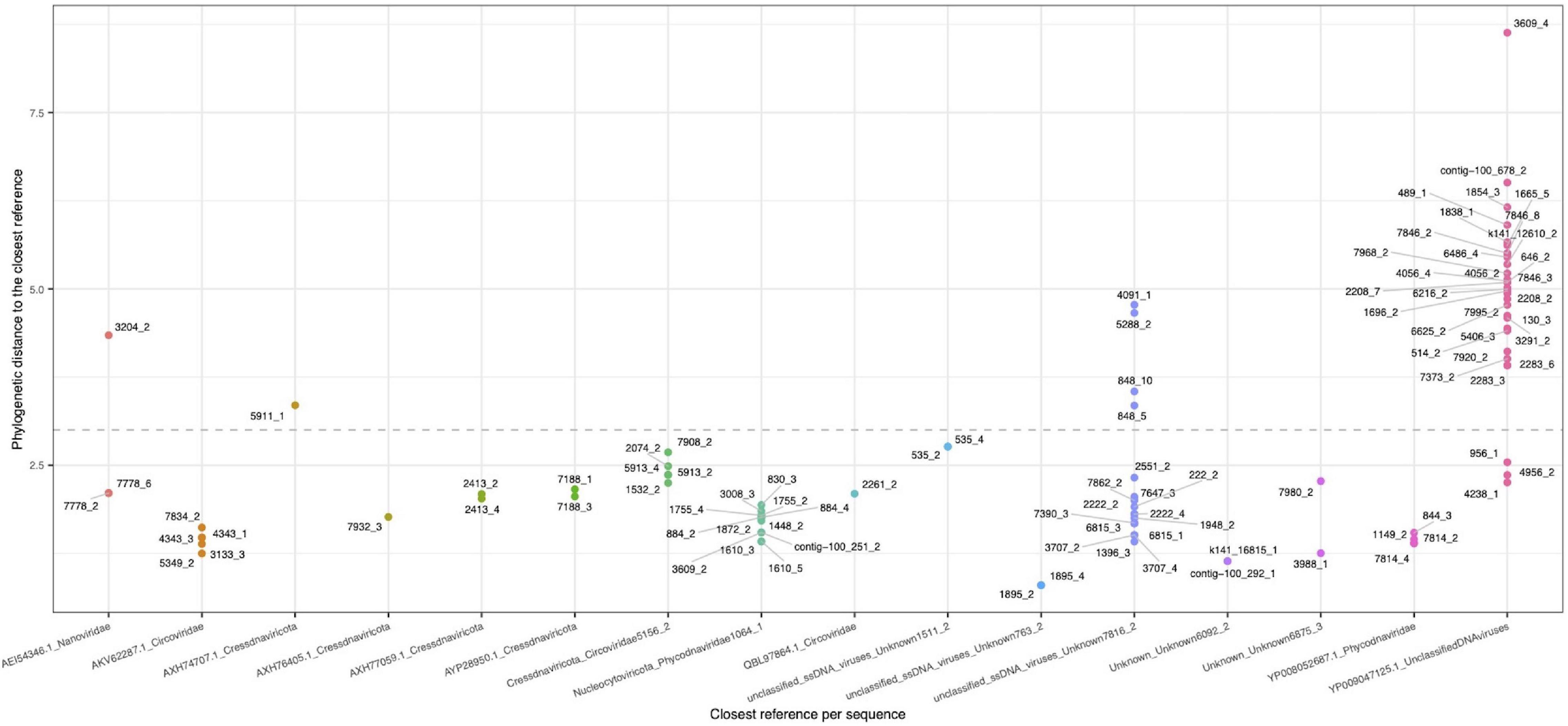
Figure 4. Phylogenetic distance between the retrieved rep-like CDS (dots) representing a contig (rep-group), and the closest reference (seed-group) in the x-axis. A value of 3.0 was used as assignation threshold (dotted line).
Functional Annotation
A total of 2,895 protein coding genes were inferred from the H. fulva hologenome dataset. In summary, 888 proteins were associated to metazoa, 31 to fungi, 206 to bacteria, 32 to archaea, and 1,738 to viruses, which again account for the abundance and functional representativeness of this taxonomic group.
Concerning the eukaryotic host proteins detected, the majority could not be assigned to a specific function, indicating the largely unexplored genomic-encoded functionality and metabolic capabilities of marine sponges in general, and possibly related to the unique and huge chemodiversity reported for H. fulva. Among the identified functions, we observed housekeeping cellular processes such as replication, recombination, and repair; post-translational modifications, protein turnover, chaperones, signal transduction mechanisms, and carbohydrate transport and metabolism (Figure 5). Conserved domain and protein family search using interproscan additionally revealed the presence of proteins classified as ATP-dependent DNA helicase PIF1-like (DNA replication), E3 ubiquitin-protein ligase TRIM71-like (Protein turnover), and ankyrin repeat domain, one of the most common protein domains in eukaryotes involved in protein/protein interaction.
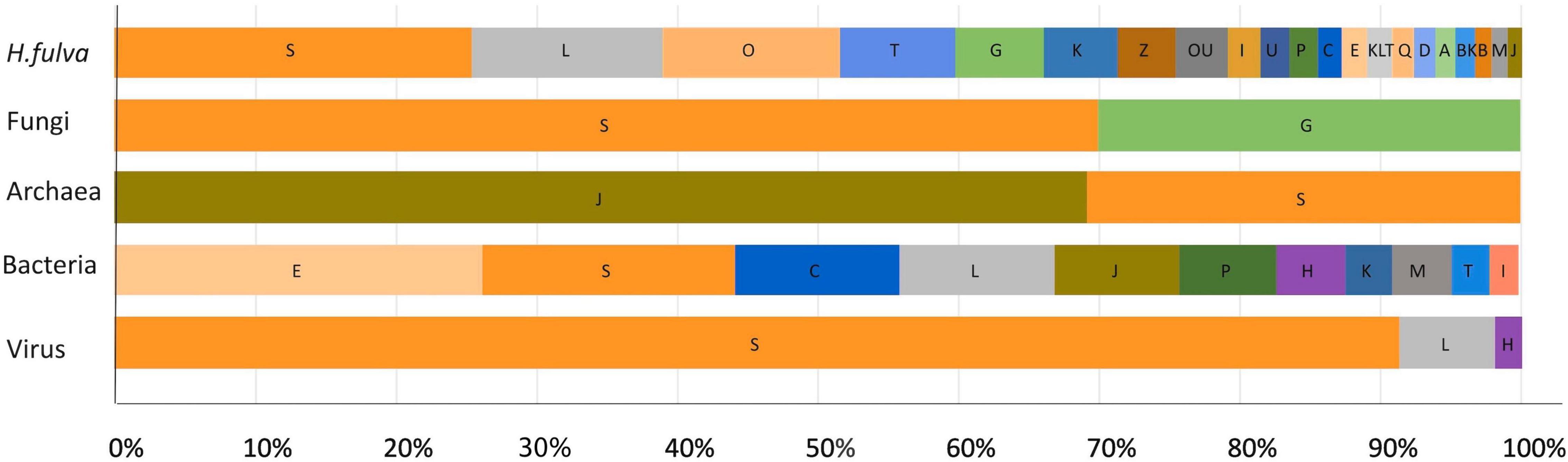
Figure 5. Relative proportion of each known COG functional category predicted from the H. fulva hologenome. Assignment was assigned according to best EggNOG v5.0 match. Functional categories: S, function unknown; L, replication, recombination, and repair; O, post-translational modification, protein turnover, chaperones; T, signal transduction mechanisms; G, carbohydrate transport and metabolism; K, transcription; Z, cytoskeleton; U, intracellular trafficking, secretion, and vesicular transport; I, lipid transport and metabolism; P, inorganic ion transport and metabolism; C, energy production and conversion; E, amino acid transport and metabolism; Q, secondary metabolism biosynthesis, transport, and catabolism; D, cell cycle control, cell division, and chromosome partitioning; A, RNA processing and modification; B, chromatin structure and dynamics; M, cell wall/membrane/envelope biogenesis; J, translation, ribosomal structure, and biogenesis; H, coenzyme transport and metabolism.
Regarding fungal proteins, the same trend was observed, finding associations with housekeeping COG categories such as carbohydrate transport and metabolism, translation, ribosomal structure, and biogenesis; coenzyme transport and metabolism and transcription. Similar results were obtained from nr database and the protein domain search (InterProScan), where the majority of proteins were classified as hypothetical, and the identified domains were related with specific transcription factors and flavin-containing amine oxidoreductases.
With respect to archaea, the most abundant COG category was related to translation, ribosomal structure, and biogenesis (Figure 5), and the most common domains found were associated with deoxyhypusine synthase, carbohydrate kinase, and glutaminase/asparaginase C-terminal domain.
For bacteria, COG analysis using EggNOG database showed that the most abundant functional categories assigned to proteins were amino acid transport and metabolism, followed by function unknown, energy production and conversion, and replication, recombination, and repair (Figure 5). The analysis using nr database indicated the presence of proteins associated with transporters, especially for sugar, iron, amino acid, or ammonium; dehydrogenases, kinases, transferases or lyases, and many hypothetical proteins. Interproscan analysis showed that ABC-transporter-like, phosphoribulokinase/uridine kinase, serine dehydratase-like, and tetrahydrodipicolinate N-succinyltransferase were the most abundant domains.
In relation to viral-specific functions, the principal categories identified were unknown function, followed by replication, recombination, and repair, and coenzyme transport and metabolism (Figure 5). The protein domain analyses showed also that the majority of proteins were assigned as hypothetical belonging to unclassified virus, followed by capsid formation, replication (rep), coat and tail tape measure associated proteins.
Discussion
Community Structure and Functional Analysis
Hologenome analysis showed a diverse composition of the microbiome associated, which includes microbial members reported in other marine sponges. In the case of H. fulva, and to the best of our knowledge, this study would be the first report describing this holobiont functional traits. Sequence analysis of the sponge genomic counterpart remarkably showed little homology with known or functionally characterized sequences in public databases and also reflected the absence of previously reported representative sequences of H. fulva, which limited generating knowledge related with the particular biology of this model. Overall, functional categories associated with molecular processes (e.g., replication, recombination, and repair) and carbohydrate metabolism were dominant. Interestingly, the presence of diverse transposable elements were detected, a particular feature which have been widely reported in sponges (Thomas et al., 2010; Slaby et al., 2017), and seemingly implicated in the evolutionary processes of symbionts within its host, by means of producing rearrangements, deletions of genes, and alteration of metabolic pathways (Moran and Plague, 2004). Other important proteins were identified, among them, E3 ubiquitin-protein ligase TRIM71-like, which has been reported in the sponge Isodictya sp., playing a possible protective function against thermal stress (González-Aravena et al., 2019). Several H. fulva proteins were associated with responses to stress, HSP70 was detected, suggesting that the sponge could use this mechanism to withstand adverse/changing marine environment. The presence of ankyrin domains arises as involved in a probable transduction response to a so called microbial-associated molecular patterns (MAMPs) a role that has been also been identified in Mediterranean sponges as Aplysina aerophoba and Dysidea avara (Pita et al., 2018).
Analysis of the sponge-associated microbial community shows that, at eukaryotic level, the fungal population was shaped mainly by Eurotiales and Saccharomycetales orders, whose dominance has been previously reported in sponges (HMA and LMA) (Zhou et al., 2011; He et al., 2014). The functional role of fungal sequences in our results was associated to carbohydrate transport and metabolism, which suggest that they can participate in substrate degradation synthesized and stored by primary producers or the sponge. These genes were significantly abundant in the hologenome, and thus, seem important on providing and regulating nutrients access for the sponge host (Menezes et al., 2010). Additionally, fungi are also responsible of secondary metabolite production (Höller et al., 2000; Nikolouli and Mossialos, 2012), and they might be key players in the chemical defense of the holobiont.
Regarding prokaryotic members, the archaeal component shows that even though a great portion is unknown, some contigs could be classified inside the Cenarchaeaceae family. These results are in agreement with the described microbial diversity of H. fulva (García-Bonilla et al., 2019), where we found that Cenarchaeum symbiosum was a dominant symbiont in the sponge, representing abundances up to 60% of its microbiome. Cenarchaeaceae group has two main clades, Cenarchaeum and Nitrosopumilus. These genera have dominated microbiomes in sponges thriving across different latitudes (Jackson et al., 2013; Hardoim and Costa, 2014; Turon and Uriz, 2020). At functional level, their presence is related with the nitrogen cycle, specifically, as ammonia-oxidizing (Zhang et al., 2014). In our study, the most common domains were deoxyhypusine synthase, carbohydrate kinase, and glutaminase/asparaginase C-terminal. The first one has been previously detected in various archaeal genomes, specifically in Crenarchaeota, and participate in polyamine metabolism (Prunetti et al., 2016). This metabolic pathway is still poorly understood but is considered being essential for cellular functions as growth translation, and secondary metabolites biosynthesis (Michael, 2016). The second one belongs to the reversible protein phosphorylation group, which is the main used strategy to transduce or respond to environmental stress (e.g., nutrients availability and temperature). In archaea, phosphorylated proteins also participate in functions such as DNA helicase, primase, ATPase, and transcription factors (Gong et al., 2020). Finally, the third one, glutaminase/asparaginase C-terminal domain plays an important role in the holobiont metabolism as: (i) asparaginases catalyze the hydrolysis of asparagine to aspartic acid and ammonia; (ii) L-asparaginase is involved in the biosynthesis of amino acids such as lysine, methionine and threonine; and (iii) glutaminase catalyze the deamidation of glutamine to glutamate with the concomitant releasing of ammonia (Guo et al., 2017). These are key processes since some metabolites can enter as input into the citric acid cycle, providing a carbon source and energy, or they can alternatively provide nitrogen.
Regarding bacteria, analysis showed that the majority of contigs were assigned to families such as Synechococcaceae, Burkholderiaceae, Pseudomonadaceae, and Rhodobacteraceae, which is in agreement with studies reporting them as important relatively abundant members of the sponge-microbiome association (Hentschel et al., 2012; Thomas et al., 2016). Overall, a low bacterial diversity was observed, which is in agreement with its classification as LMA sponge (Moitinho-Silva et al., 2017; García-Bonilla et al., 2019). Curiously, members belonging to Actinobacteria and Bacteroidetes were not detected, despite being reported as core microbium members of this sponge species, suggesting abundant taxa were more likely sequenced than lineages represented in low abundances in this enriched hologenome sequenced.
Our analysis showed that the most enriched functional category for this group was related with the metabolism and amino acids transport. It indicates that these functions collectively play a vital role for the sponge in processes such as nutrients uptake and excretion of waste products. For instance, the presence of different types of transporters and enzymes confirm its functional potential to degrade different substrates. Similar results were reported by Slaby et al. (2017), who found that metabolism and energy production was enriched in a symbiont group identified in the sponge A. aerophoba (Slaby et al., 2017). In this context, our data suggest that for the carbon metabolism and energy production, one of the active pathways could be the autotrophic carbon fixation, which is consistent with the identified phosphoribulokinase domain (Taylor et al., 2007) and related to the lower light access in the microhabitats and depths at which the sponge is adapted (35–40 m). Likewise, we also found serine dehydratase-like domain, which convert serine into pyruvate with the release of ammonia, a key intermediate for the citric acid cycle or gluconeogenesis.
Regarding amino acids metabolism, we found the tetrahydrodipicolinate N-succinyltransferase domain, which participates in lysine biosynthesis, pathway present in most Gram-negative bacteria and mycobacteria (Schnell et al., 2012). However, related genes to this pathway as dipicolinate synthase operon has been detected in Prochlorococcus cyanobacteria (Partensky et al., 1999), one of the most abundant bacterial types reported for H. fulva (García-Bonilla et al., 2019).
Viral Community
Analysis showed that on average 80% of the viral contigs could not be taxonomically assigned, suggesting that available information about this sequence diversity and identity in public databases is still extremely scarce for marine viral genomes. In consequence, the lack of data limits the extent and precision of the diversity description, overlooking its true taxonomical and functional potential (Cassman et al., 2012; Roux et al., 2016).
Analysis of H. fulva virome shows a high predominance of sequences probably originated from ssDNA viruses and specifically hosted in this sponge. While the sequence analyses confirms the outstanding diversity present in this hologenome dataset, the precise abundance should be taken with caution, as phi29-based MDA enrich ssDNA molecules and there is the possibility that naturally occurring ssDNA could be preferentially amplified over the ssDNA of denatured dsDNA (Kim and Bae, 2011). Also, the DNA extraction protocol used, clearly discarded the possibility to detect retro virus as RNA-based viral genomes could not be captured. The identified virus families detected in this H. fulva hologenome study have been widely reported as members of the viromes in invertebrate marine animals (Laffy et al., 2016, 2018; Pascelli et al., 2020), and additionally, it has been suggested that around 50% of marine viruses have RNA or ssDNA genomes, and while it could not be confirmed by conventional count methods (Steward et al., 2013), the sequence frequencies detected are supporting this estimate.
The two analytical approaches that have been undertaken in this study to catch viral contigs and identify them was successful in the first described virome content associated to H. fulva. These approaches revealed that it is composed by eight families, which could be present in mammals, algae, birds, fish, insects, humans, and plants (Delwart and Li, 2012; Krupovic, 2013), and include members belonging to Circoviridae, Phycodnaviridae, Poxviridae, Nanoviridae, Mimiviridae, Microviridae, Herelleviridae, and Genomoviridae. Some of them has been previously reported as part of virome in both LMA and HMA sponges (Pascelli et al., 2020).
Overall, inside the group of ssDNA viruses, we found families belonging to the group of CRESS DNA viruses. They represent the smallest viral genomes known (Krupovic, 2013), having high substitution rates and are highly recombinogenic, which would explain its capacity to infect different hosts and new environments (Kazlauskas et al., 2018). In the marine environment the differential presence of some types of these viruses has been proposed as indicator of coral reef health status (Bettarel et al., 2018). Its detection in sponges was registered in 2015 (Rosario et al., 2015), and only a limited number of studies have described their distribution, which make difficult to determine their prevalence, function and classification by traditional methods (Rosario et al., 2015). As consequence of the limited knowledge and intragenic recombination, REPs phylogeny did not show a clear separation between clades as Circoviridae, Cressdnaviricota_Circoviridae, and Cressdnaviricota, since a diffuse evolutionary relationships between different CRESS DNA virus groups was observed (Roux et al., 2013; Rosario et al., 2015; Kazlauskas et al., 2018).
Other identified (ssDNA) families, represented by a low number of contigs, were Nanoviridae and Genomoviridae. The first one has been reported in aquatic and marine environments (Labonté and Suttle, 2013), while the second, as pathogens of humans and other vertebrates (e.g., domestic animals) (Varsani and Krupovic, 2017). Finding these viral families in hologenomic sequence datasets from Porifera, indicates they are actually replicating in marines sponge cells or on its associated eukaryotic microbiome, possibly the phototrophic fraction. To date, the knowledge about their biological or ecological role is scarce, for instance, Genomoviridae is a viral family recently established (Varsani and Krupovic, 2017), which includes a single classified virus and many sequences without information about their host and biology (Simmonds et al., 2017). Similarly, Nanoviridae has species infecting plants as host and has been poorly studied in marine environments, only its capacity to inhabit in low oxygen zones in the ocean has been reported (Cassman et al., 2012).
Members of the Microviridae family were also found and they were represented by the largest contigs found. They have been widely reported across Porifera (Laffy et al., 2016, 2018; Pascelli et al., 2020), highlighting its abundance and importance in marine invertebrate and environments.
Regarding dsDNA virus, we found eukaryote-infecting families as Phycodnaviridae, Mimiviridae, and Poxviridae, all of them belonging to Megavirales order. They are nucleocytoplasmic large DNA virus (NCLDV) (Colson et al., 2013) shaping reef invertebrate virome (Laffy et al., 2018), and being reported across different species of Porifera (Pascelli et al., 2020). Phycodnaviridae infect algae (Larsen et al., 2008), but in sponges, they might be associated to photosynthetic symbionts. In general, these megaviral families provide a great functional potential for the holobiont, where they are responsible of coding metabolic genes that could integrate in the host genome and related to collagen, ankyrin, or nitrogen metabolism (Pascelli et al., 2020). However, in our results, contigs associated to those families had short length, which could have limited their functional analysis.
Finally, contigs belonging to Herelleviridae (dsDNA) of the Caudovirales order were identified. They were the only class of bacteriophage found in H. fulva virome, and its hosts belong to the phylum Firmicutes. In Porifera, several families have been described such as Myoviridae, Siphoviridae, and Podoviridae (Laffy et al., 2018; Pascelli et al., 2020), which are also found in aquatic environments (Potapov et al., 2019).
Regarding inferred functional traits in H. fulva virome, we mainly identified proteins associated with viral capsid and REP. These results indicate that viromes in this sponge harbor an interesting untapped diversity, and also pointed out about the need for extensive exploration of non-dsDNA viruses that are not having sufficient representation in public databases and can be of importance for sponge health.
It must be noted that inferring the respective hosts of the identified viral largely depends on the known taxonomy of the reported proven host, but the actual host range for a certain family could be indeed wider. On the other hand, many viral contigs could not be classified to any known family. Thus, this important clue remains an open question to understand the biological role of virome components and their ecological function in this holobiont association. It can be hypothesized that a possible role played by these viruses is to contribute on maintaining the stable characteristic composition and LMA status in H. fulva microbiome. This could be exerted either by direct deleterious/antagonistic effects against susceptible commensal microbes hampering their fitness for growth and sponge colonization, or by acting in sponge or microbiome cells modulating metabolome production and composition modulating the interplay of chemical defenses production and nutrients access and disposal. Given the general interest for understanding marine holobiont interactions, this could be an interesting path of research in future endeavors.
In this study, we characterized the virome associated to a LMA sponge as H. fulva, which showed a high predominance of eukaryote-infecting ssDNA viruses. Curiously, a low number of viral contigs related to prokaryotes (bacteriophages) was identified, which can be attributed to its particular and stable microbiome dominated up to 75% by two symbionts: Betaproteobacteria HF1 and C. symbiosum (García-Bonilla et al., 2019). However, as the main bacterial components do not have a phylogenetic closer model, it is difficult to predict possible relationships or interactions with other members inside the microbiome. For Cenarchaeum sp. molecular mechanisms used to inhibit or evade host consumption and defend against viral predation have been described (Hallam et al., 2006). In this context, it is plausible to think that the prokaryotic community in this sponge has a limited host range and susceptibility for prophages, which is coherent with the obtained results. According to that, H. fulva showed a particular viral composition, which is in agreement with results reported by Laffy et al. (2018), who found that viruses infecting reef holobionts are host species specific and they are adapted to their individual host habitats. Probably, as in LMA sponges, the associated microbiome is also host specific, this may also contribute further in the selection of specific viral population strongly associated to the diversity of cell hosts conforming the holobiont.
We hypothesize that ssDNA viruses constitute an important component inside sponge viromes. Knowledge about its biology, diversity, and functional role remains to be explored. Marine viromics is a field relatively new, and Porifera species models can provide unique viral niches for its exploration and understanding.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/bioproject, Bioproject PRJNA741981.
Author Contributions
HJ and EG-B designed the study. EG-B and DC-M wrote the initial draft of the manuscript and performed bioinformatics analyses and figures. HJ, WT, DR-P, and AA checked and corrected the manuscript. All authors approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We acknowledge the financial support given by Pontificia Universidad Javeriana through Postdoctoral fellowship to EG-B (Project No. 8544), project code 9901. We thank Dr. Thierry Pérez (Institut Méditerranéen de Biodiversité et d’Ecologie Marine et Continentale, Marseille, France) for his help in sample collection and sponge taxonomical identification, and we thank Dr. César Pedraza (Universidad Nacional de Colombia, Bogotá) for his excellent support in cloud systems engineering.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.736817/full#supplementary-material
Footnotes
References
Bayer, K., Kamke, J., and Hentschel, U. (2014). Quantification of bacterial and archaeal symbionts in high and low microbial abundance sponges using real-time PCR. FEMS Microbiol. Ecol. 89, 679–690. doi: 10.1111/1574-6941.12369
Bell, J. J. (2008). The functional roles of marine sponges. Estuar. Coast. Shelf Sci. 79, 341–353. doi: 10.1016/j.ecss.2008.05.002
Bettarel, Y., Bouvier, T., Nguyen, H. K. I., and Thu, P. T. H. (2015). The versatile nature of coral-associated viruses. Environ. Microbiol. 17, 3433–3439. doi: 10.1111/1462-2920.12579
Bettarel, Y., Halary, S., Auguet, J. C., Mai, T. C., Van Bui, N., Bouvier, T., et al. (2018). Corallivory and the microbial debacle in two branching scleractinians. ISME J. 12, 1109–1126. doi: 10.1038/s41396-017-0033-5
Breitbart, M., Thompson, L. R., Suttle, C. A., and Sullivan, M. B. (2007). Exploring the vast diversity of marine viruses. Oceanography 20, 135–139.
Butina, T. V., Khanaev, I. V., Kravtsova, L. S., Maikova, O. O., and Bukin, Y. S. (2020). Metavirome datasets from two endemic Baikal sponges Baikalospongia bacillifera. Data Brief. 29:105260. doi: 10.1016/j.dib.2020.105260
Cassman, N., Prieto-Davó, A., Walsh, K., Silva, G. G. Z., Angly, F., Akhter, S., et al. (2012). Oxygen minimum zones harbour novel viral communities with low diversity. Environ. Microbiol. 14, 3043–3065. doi: 10.1111/j.1462-2920.2012.02891.x
Colson, P., De Lamballerie, X., Yutin, N., Asgari, S., Bigot, Y., Bideshi, D. K., et al. (2013). “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 158, 2517–2521. doi: 10.1007/s00705-013-1768-6
Coutinho, F. H., Silveira, C. B., Gregoracci, G. B., Edwards, R. A., Brussaard, C. P. D., Dutilh, B. E., et al. (2017). Marine viruses discovered through metagenomics shed light on viral strategies throughout the oceans. Nat. Commun. 8:15955. doi: 10.1038/ncomms15955
Delwart, E., and Li, L. (2012). Rapidly expanding genetic diversity and host range of the circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res. 164, 114–121. doi: 10.1016/j.virusres.2011.11.021
García-Bonilla, E., Brandão, P. F. B., Pérez, T., and Junca, H. (2019). Stable and enriched Cenarchaeum symbiosum and uncultured betaproteobacteria HF1 in the microbiome of the mediterranean sponge haliclona fulva (Demospongiae: Haplosclerida). Microb. Ecol. 77, 25–36. doi: 10.1007/s00248-018-1201-5
Giles, E. C., Kamke, J., Moitinho-Silva, L., Taylor, M. W., Hentschel, U., Ravasi, T., et al. (2013). Bacterial community profiles in low microbial abundance sponges. FEMS Microbiol. Ecol. 83, 232–241. doi: 10.1111/j.1574-6941.2012.01467.x
Gong, P., Lei, P., Wang, S., Zeng, A., and Lou, H. (2020). Post-translational modifications aid archaeal survival. Biomolecules 10:584. doi: 10.3390/biom10040584
González-Aravena, M., Kenny, N. J., Osorio, M., Font, A., Riesgo, A., and Cárdenas, C. A. (2019). Warm temperatures, cool sponges: the effect of increased temperatures on the Antarctic sponge Isodictya sp. PeerJ 7:e8088. doi: 10.7717/peerj.8088
Guidi, L., Chaffron, S., Bittner, L., Eveillard, D., Larhlimi, A., Roux, S., et al. (2016). Plankton networks driving carbon export in the oligotrophic ocean. Nature 532, 465–470. doi: 10.1038/nature16942.Plankton
Guo, J., Coker, A. R., Wood, S. P., Cooper, J. B., Chohan, S. M., Rashid, N., et al. (2017). Structure and function of the thermostable l -asparaginase from Thermococcus kodakarensis. Acta Crystallogr. Sect. D Struct. Biol. D 73, 889–895. doi: 10.1107/S2059798317014711
Hallam, S. J., Konstantinidis, K. T., Putnam, N., Schleper, C., Watanabe, Y. I., Sugahara, J., et al. (2006). Genomic analysis of the uncultivated marine crenarchaeote Cenarchaeum symbiosum. Proc. Natl. Acad. Sci. U.S.A. 103, 18296–18301. doi: 10.1073/pnas.0608549103
Hardoim, C. C. P., and Costa, R. (2014). Temporal dynamics of prokaryotic communities in the marine sponge sarcotragus spinosulus. Mol. Ecol. 23, 3097–3112. doi: 10.1111/mec.12789
He, L., Liu, F., Karuppiah, V., Ren, Y., and Li, Z. (2014). Comparisons of the fungal and protistan communities among different marine sponge holobionts by pyrosequencing. Microb. Ecol. 67, 951–961. doi: 10.1007/s00248-014-0393-6
Hentschel, U., Fieseler, L., Wehrl, M., Gernert, C., Steinert, M., Hacker, J., et al. (2003). Microbial diversity of marine sponges. Prog. Mol. Subcell. Biol. 37, 59–88.
Hentschel, U., Piel, J., Degnan, S. M., and Taylor, M. W. (2012). Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 10, 641–654. doi: 10.1038/nrmicro2839
Höller, U., Wright, A., and Matthee, G. (2000). Fungi from marine sponges: diversity, biological activity and secondary metabolites. Mycol. Res. 104, 1354–1365.
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). EGGNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Hyatt, D., Chen, G. L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jackson, S. A., Flemer, B., McCann, A., Kennedy, J., Morrissey, J. P., O’Gara, F., et al. (2013). Archaea appear to dominate the microbiome of Inflatella pellicula deep sea sponges. PLoS One 8:e84438. doi: 10.1371/journal.pone.0084438
Jahn, M. T., Arkhipova, K., Markert, S. M., Stigloher, C., Lachnit, T., Pita, L., et al. (2019). A symbiont phage protein aids in eukaryote immune evasion∗. Cell Host Microbe. doi: 10.1101/608950
Jahn, M. T., Lachnit, T., Markert, S. M., Stigloher, C., Pita, L., Ribes, M., et al. (2021). Lifestyle of sponge symbiont phages by host prediction and correlative microscopy. ISME J. 15, 2001–2011. doi: 10.1038/s41396-021-00900-6
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kazlauskas, D., Varsani, A., and Krupovic, M. (2018). Pervasive chimerism in the replication-associated proteins of uncultured single-stranded DNA viruses. Viruses 10:187. doi: 10.3390/v10040187
Kim, K. H., and Bae, J. W. (2011). Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 77, 7663–7668. doi: 10.1128/AEM.00289-11
Krupovic, M. (2013). Networks of evolutionary interactions underlying the polyphyletic origin of ssDNA viruses. Curr. Opin. Virol. 3, 578–586. doi: 10.1016/j.coviro.2013.06.010
Labonté, J. M., and Suttle, C. A. (2013). Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 7, 2169–2177. doi: 10.1038/ismej.2013.110
Laffy, P. W., Wood-Charlson, E. M., Turaev, D., Jutz, S., Pascelli, C., Botté, E. S., et al. (2018). Reef invertebrate viromics: diversity, host specificity and functional capacity. Environ. Microbiol. 20, 2125–2141. doi: 10.1111/1462-2920.14110
Laffy, P. W., Wood-Charlson, E. M., Turaev, D., Weynberg, K. D., Botté, E. S., Van Oppen, M. J. H., et al. (2016). HoloVir: a workflow for investigating the diversity and function of viruses in invertebrate holobionts. Front. Microbiol. 7:822. doi: 10.3389/fmicb.2016.00822
Larsen, J. B., Larsen, A., Bratbak, G., and Sandaa, R. A. (2008). Phylogenetic analysis of members of the Phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl. Environ. Microbiol. 74, 3048–3057. doi: 10.1128/AEM.02548-07
Lasken, R. S. (2007). Single-cell genomic sequencing using multiple displacement amplification. Curr. Opin. Microbiol. 10, 510–516. doi: 10.1016/j.mib.2007.08.005
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2014). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Menezes, C. B. A., Bonugli-Santos, R. C., Miqueletto, P. B., Passarini, M. R. Z., Silva, C. H. D., Justo, M. R., et al. (2010). Microbial diversity associated with algae, ascidians and sponges from the north coast of São Paulo state. Brazil. Microbiol. Res. 165, 466–482. doi: 10.1016/j.micres.2009.09.005
Michael, A. J. (2016). Polyamines in eukaryotes, bacteria, and archaea. J. Biol. Chem. 291, 14896–14903. doi: 10.1074/jbc.R116.734780
Moitinho-Silva, L., Steinert, G., Nielsen, S., Hardoim, C. C. P., Wu, Y. C., McCormack, G. P., et al. (2017). Predicting the HMA-LMA status in marine sponges by machine learning. Front. Microbiol. 8:752. doi: 10.3389/fmicb.2017.00752
Moran, N. A., and Plague, G. R. (2004). Genomic changes following host restriction in bacteria. Curr. Opin. Genet. Dev. 14, 627–633. doi: 10.1016/j.gde.2004.09.003
Nikolouli, K., and Mossialos, D. (2012). Bioactive compounds synthesized by non-ribosomal peptide synthetases and type-I polyketide synthases discovered through genome-mining and metagenomics. Biotechnol. Lett. 34, 1393–1403. doi: 10.1007/s10529-012-0919-2
Nuzzo, G., Ciavatta, M. L., Villani, G., Manzo, E., Zanfardino, A., Varcamonti, M., et al. (2012). Fulvynes, antimicrobial polyoxygenated acetylenes from the mediterranean sponge haliclona fulva. Tetrahedron 68, 754–760. doi: 10.1016/j.tet.2011.10.068
Partensky, F., Hess, W. R., and Vaulot, D. (1999). Prochlorococcus, a marine photosynthetic prokaryote of global significance. Microbiol. Mol. Biol. Rev. 63, 106–127.
Pascelli, C., Laffy, P. W., Botté, E., Kupresanin, M., Rattei, T., Lurgi, M., et al. (2020). Viral ecogenomics across the Porifera. Microbiome 8:144. doi: 10.1186/s40168-020-00919-5
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Pita, L., Hoeppner, M. P., Ribes, M., and Hentschel, U. (2018). Differential expression of immune receptors in two marine sponges upon exposure to microbial-associated molecular patterns. Sci. Rep. 8:16081. doi: 10.1038/s41598-018-34330-w
Potapov, S. A., Tikhonova, I. V., Krasnopeev, A. Y., Kabilov, M. R., Tupikin, A. E., Chebunina, N. S., et al. (2019). Metagenomic analysis of virioplankton from the pelagic zone of lake baikal. Viruses 11:991. doi: 10.3390/v11110991
Prunetti, L., Graf, M., Blaby, I. K., Peil, L., Makkay, A. M., Starosta, A. L., et al. (2016). Deciphering the translation initiation factor 5A modification pathway in halophilic archaea. Archaea 2016:7316725. doi: 10.1155/2016/7316725
Rosario, K., Schenck, R. O., Harbeitner, R. C., Lawler, S. N., and Breitbart, M. (2015). Novel circular single-stranded DNA viruses identified in marine invertebrates reveal high sequence diversity and consistent predicted intrinsic disorder patterns within putative structural proteins. Front. Microbiol. 6:696. doi: 10.3389/fmicb.2015.00696
Rosenwasser, S., Ziv, C., van Creveld, S. G., and Vardi, A. (2016). Virocell metabolism: metabolic innovations during host–virus interactions in the ocean. Trends Microbiol. 24, 821–832. doi: 10.1016/j.tim.2016.06.006
Roux, S., Brum, J. R., Dutilh, B. E., Sunagawa, S., Duhaime, M. B., Loy, A., et al. (2016). Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 537, 689–693. doi: 10.1038/nature19366
Roux, S., Enault, F., Bronner, G., Vaulot, D., Forterre, P., and Krupovic, M. (2013). Chimeric viruses blur the borders between the major groups of eukaryotic single-stranded DNA viruses. Nat. Commun. 4:2700. doi: 10.1038/ncomms3700
Schnell, R., Oehlmann, W., Sandalova, T., Braun, Y., Huck, C., Maringer, M., et al. (2012). Tetrahydrodipicolinate N-Succinyltransferase and dihydrodipicolinate synthase from Pseudomonas aeruginosa: structure analysis and gene deletion. PLoS One 7:e31133. doi: 10.1371/journal.pone.0031133
Sierra, R., Rodríguez-R, L. M., Chaves, D., Pinzón, A., Grajales, A., Rojas, A., et al. (2010). Discovery of Phytophthora infestans genes expressed in planta through mining of cDNA libraries. PLoS One 5:e9847. doi: 10.1371/journal.pone.0009847
Silveira, C. B., and Rohwer, F. (2016). Piggback-the-winner in host. NPJ Biofilms Microbiomes 57, 10–13. doi: 10.1038/npjbio
Simmonds, P., Adams, M. J., Benkő, M., Breitbart, M., Brister, J. R., Carstens, E. B., et al. (2017). Consensus statement: virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 15, 161–168. doi: 10.1038/nrmicro.2016.177
Slaby, B. M., Hackl, T., Horn, H., Bayer, K., and Hentschel, U. (2017). Metagenomic binning of a marine sponge microbiome reveals unity in defense but metabolic specialization. ISME J. 11, 2465–2478. doi: 10.1038/ismej.2017.101
Stano, M., Beke, G., and Klucar, L. (2016). viruSITE—integrated database for viral genomics. Database 2016:baw162. doi: 10.1093/database/baw162
Steward, G. F., Culley, A. I., Mueller, J. A., Wood-Charlson, E. M., Belcaid, M., and Poisson, G. (2013). Are we missing half of the viruses in the ocean? ISME J. 7, 672–679. doi: 10.1038/ismej.2012.121
Taylor, M. W., Radax, R., Steger, D., and Wagner, M. (2007). Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiol. Mol. Biol. Rev. 71, 295–347. doi: 10.1128/MMBR.00040-06
Thingstad, T. F., and Lignell, R. (1997). Theoretical models for the control of bacterial growth rate, abundance, diversity and carbon demand. Aquat. Microb. Ecol. 13, 19–27. doi: 10.3354/ame013019
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7:11870. doi: 10.1038/ncomms11870
Thomas, T., Rusch, D., DeMaere, M. Z., Yung, P. Y., Lewis, M., Halpern, A., et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4, 1557–1567. doi: 10.1038/ismej.2010.74
Treangen, T., Sommer, D., Angly, F., Koren, S., and Pop, M. (2011). Next generation sequence assembly with AMOS∗. Curr. Protoc. Bioinform.
Tribalat, M., Marra, M. V., Mccormack, G. P., and Thomas, O. P. (2016). Does the chemical diversity of the order haplosclerida (Phylum Porifera: Class Demospongia) fit with current taxonomic classification? Planta Med. 82, 843–856. doi: 10.1055/s-0042-105879
Turon, M., and Uriz, M. J. (2020). New insights into the archaeal consortium of tropical sponges. Front. Mar. Sci. 6:789. doi: 10.3389/fmars.2019.00789
Urayama, S., Takaki, Y., Hagiwara, D., and Nunoura, T. (2020). dsRNA-seq reveals novel RNA virus and virus-like putative complete genome sequences from hymeniacidon sp. sponge. Microbes Environ. 35:ME19132. doi: 10.1264/jsme2.ME19132
Vacelet, J., and Donadey, C. (1977). Electron microscope study of the association between some sponges and bacteria. J. Exp. Mar. Biol. Ecol. 30, 301–314.
Varsani, A., and Krupovic, M. (2017). Sequence-based taxonomic framework for the classification of uncultured single-stranded DNA viruses of the family Genomoviridae. Virus Evol. 3:vew037. doi: 10.1093/ve/vew037
Webster, N. S., and Thomas, T. (2016). The sponge hologenome. Am. Soc. 7:e13516. doi: 10.1128/mBio.00135-16
Weinbauer, M. G., and Rassoulzadegan, F. (2004). Are viruses driving microbial diversification and diversity? Environ. Microbiol. 6, 1–11. doi: 10.1046/j.1462-2920.2003.00539.x
Wommack, K. E., and Colwell, R. R. (2000). Virioplankton: viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 64, 69–114. doi: 10.1128/MMBR.64.1.69-114.2000
Zhang, F., Pita, L., Erwin, P. M., Abaid, S., López-Legentil, S., and Hill, R. T. (2014). Symbiotic archaea in marine sponges show stability and host specificity in community structure and ammonia oxidation functionality. FEMS Microbiol. Ecol. 90, 699–707. doi: 10.1111/1574-6941.12427
Keywords: holobiont, Porifera, ssDNA virus, Rep protein, shotgun hologenome, viral ecology
Citation: García-Bonilla E, Chaves-Moreno D, Riaño-Pachón D, Terán W, Acosta A and Junca H (2021) The Hologenome of Haliclona fulva (Porifera, Demospongiae) Reveals an Abundant and Diverse Viral Community. Front. Mar. Sci. 8:736817. doi: 10.3389/fmars.2021.736817
Received: 05 July 2021; Accepted: 13 October 2021;
Published: 02 November 2021.
Edited by:
Zhiyong Li, Shanghai Jiao Tong University, ChinaReviewed by:
Xiubao Li, Hainan University, ChinaFrank O’Neill Aylward, Virginia Tech, United States
Copyright © 2021 García-Bonilla, Chaves-Moreno, Riaño-Pachón, Terán, Acosta and Junca. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Howard Junca, aW5mb0Bob3dhcmRqdW5jYS5jb20=
†These authors have contributed equally to this work and share first authorship