 Yuan Liu
Yuan Liu Zhimei Zhu1
Zhimei Zhu1 Zhenghong Sui
Zhenghong Sui- 1Key Laboratory of Marine Genetics and Breeding (Ocean University of China), Ministry of Education, Qingdao, China
- 2College of Agronomy, Qingdao Agricultural University, Qingdao, China
- 3Department of Microbiology, Hazara University Mansehra, Dhodial, Pakistan
Alexandrium pacificum is an organism that has an important impact on the aquaculture industry and human health. In this study, the digital gene expression approach was used to conduct a comparative analysis of differentially expressed genes (DEGs) that influence the explosive growth of A. pacificum following five treatment conditions: normal culture (C), high phosphorus and manganese (M), high irradiance (G), low phosphorus (P), and low nitrogen (N). Compared with the C conditions, a total of 265, 320, 185, and 150 DEGs were detected in the M, G, P, and N treatment groups, respectively. Clustering analysis suggested that A. pacificum acclimated to explosive growth using similar mechanisms in the M and G conditions. Analysis of DEGs showed that upregulation of genes associated with the pentose phosphate pathway and photosynthesis may contribute to explosive growth. Unigenes involved in the cell cycle were also found to be upregulated to promote cell division. The DEGs identified in this study may allow for the elucidation of molecular mechanisms responsible for the explosive growth of A. pacificum.
Introduction
Dinoflagellates, a large group of unicellular algae, are very important marine primary producers and grazers. Importantly, most harmful algae blooms (HAB) are caused by dinoflagellates, such as Karenia spp. and Alexandrium spp. (Zhang et al., 2013). In recent decades, the intensity and impact of HABs have increased drastically resulting in a negative impact on marine resources, water quality and human health (Genovesi et al., 2009). Dinoflagellates are also known for their unique features among eukaryotes. They contain vast amounts of DNA (from 3–250 pg ⋅ cell–1), and have an extremely large genome (up to 80 times of the human genome). Further, their chromosomes are permanently condensed during the cell cycle (Lin, 2006). To date, it remains unclear whether these fascinating biological characteristics are associated with HABs and explosive growth.
Alexandrium pacificum, one of the most well studied and widely distributed dinoflagellates, is known to form toxin blooms and cause paralytic shellfish poisoning (PSP). Studies have shown that not only shellfish, but also humans might be affected by PSP through the consumption of shellfish that have been contaminated by toxins (Hallegraeff et al., 2004). Thus, the development of a treatment strategy for HABs is critical.
Blooms of harmful dinoflagellates are affected by multiple environmental factors. Anthropogenic enrichment is considered to be one of the most pervasive changes altering the environment of offshore areas, especially the input of macronutrients and micronutrients, such as nitrogen and phosphorus (Li et al., 2014). Nitrogenous nutrients are thought to be important in triggering flagellate blooms (Glibert and Terlizzi, 1999). Studies have shown that the main cause of the increasing frequency of HABs is eutrophication. From the 1980s to 2000s, the annual frequency of red tide events in China had increased rapidly and the concentration of dissolved inorganic nitrogen (DIN) was increased from 2 to 5 μM (Wang et al., 2018). With respect to phosphates, the growth rate of Alexandrium tamarense increased with an increase in starting phosphate concentration (ranging from 0 to 10 μM) (Wang and Hsieh, 2005). Here, under laboratory conditions, we cultured algae using F/2 medium containing nitrogen and phosphate at concentrations of 882 and 36 μM, respectively, which are considerably higher than field conditions. Interestingly, some studies have shown that the biomass of A. tamarense observed under high concentrations of nitrogen (2,646 μM) was higher than that observed under low nitrogen concentrations when F/2 medium was used to culture the algae (Zhang et al., 2006). Likewise, when different concentrations of phosphate (3.6, 36, and 108 μM) were used to culture A. tamarense, the maximum biomass and growth rate occurred at the highest phosphate concentration (108 μM), while the lowest phosphate concentration (3.6 μM) limited growth (Yanjun et al., 2003).
Previous studies have shown that manganese could affect the photosynthetic activity and growth of dinoflagellates (Wang et al., 2019). For example, the growth rate of Alexandrium sp. was found to increase with an increasing manganese concentration (ranging from 0.9 to 2.7 μM), while in Coolia tropicalis, manganese was required to maintain the photosynthetic activity and growth rate (Paul and Hauck, 2006). Irradiance is another environmental factor that plays a vital role in stimulating growth. Laabir (2011) reported that the growth and cell yield of Alexandrium catenella increased significantly as the range of irradiance increased from 10 to 90 μmol photons m–2 s–1. Thus, multiple environmental factors impact dinoflagellate growth and may influence HABs.
Many studies have focused on the influence of environmental factors on changes in population structure and size (Toulza et al., 2010). However, the molecular mechanisms underlying these events have been poorly studied and remain unclear. Uribe et al. (2008) developed an expressed sequence tag (EST) library and identified 6,496 unigenes from the EST database. Interestingly, genes associated with photosynthesis were highly expressed during the exponential growth phase. Similarly, an EST database developed by Toulza et al. (2010) found that the largest group of genes identified by this database were also related to photosynthesis. The large size of the dinoflagellate genome makes it difficult to sequence the whole genome. Thus, transcriptome analysis could be an efficient way to identify the underlying mechanisms of HAB development. It is required to understand the functional elements of the genome and identify the molecular constituents of cells and tissues (Laabir, 2011).
In this study, digital gene expression profiling (DGE) was used to comparatively analyze the gene expression and regulation of A. pacificum under the stage of explosive growth through high-throughput transcriptomic sequencing. Based on the environmental conditions under which HAB occur, five treatments were simulated including normal culture (C), high concentrations of phosphorus and manganese (M), high irradiance (G), low phosphorus (P), and low nitrogen (N). The aim of the study was to analyze the gene expression and regulation patterns that affect physiological and biochemical processes when A. pacificum are exposed to different environmental conditions. This database may serve as a valuable source to identify the key genes and pathways, which are activated under the stage of explosive growth of A. pacificum, as well as examine the mechanisms of HAB caused by A. pacificum.
Materials and Methods
Sample Preparation and Collection
Alexandrium pacificum was obtained from the Key Laboratory of Marine Genetics and Breeding, Ministry of Education of China, College of Marine Life Sciences. Stock cultures of A. pacificum were grown at 20 ± 1°C at a photon flux density of 30 μmol m–2 s–l on a light:dark photocycle of 12:12 h and cultures were maintained in F/2 medium (without silicate).
For constructing of a reference transcriptome dataset of A. pacificum, four kinds of cultivating conditions were used including lag phase (Natural seawater with F/2 medium and the cells were collected at lag phase), log phase (Natural seawater with F/2 medium and the cells were collected at log phase) and induced log phase which contains two cultivating conditions (NaH2PO4 ⋅ 2H2O and MnCl2 ⋅ 4H2O were supplied to the final concentration of 0.144 mmol/L and 2.730 μmol/L, respectively, in the f/2 medium. The cell was collected at 7 and 12 days, then mixed for transcriptome analysis). Before treating, the algae which has already grown into logarithmic phase was placed in the dark for 48h to synchronize the cell cycle. The synchronized cells then were inoculated into three cultivating conditions at the initial concentration of 2 × 106 cells/L (Zhang et al., 2014).
For preparing the growth curve, we cultured the algae for 21 days and count the algae number every 2 days using Sedgwick Counting Chamber to observe the growth trend (Figure 1) (Liu et al., 2019). For DGE analysis, five treatments were set up: M, G, C, P, and N (Table 1). In sample M, the concentration of phosphate and manganese was 3 times than normal. In sample P and N, the concentration of nitrogen and phosphate was 10 times lower than normal. Three biological replicates were used in each treatment. Before treatment, the algae which has already grown into logarithmic phase were placed in the dark for 48 h to be synchronized of the growth. The synchronized cells then were inoculated into five treatment conditions at the initial concentration of 2 × 106 cells/L. All the 15 samples (5 treatments and each treatment including 3 biological replicates) were cultured for 12 days when they grow into logarithmic phase and were collected by centrifugation for 6 min at 4,000 g.
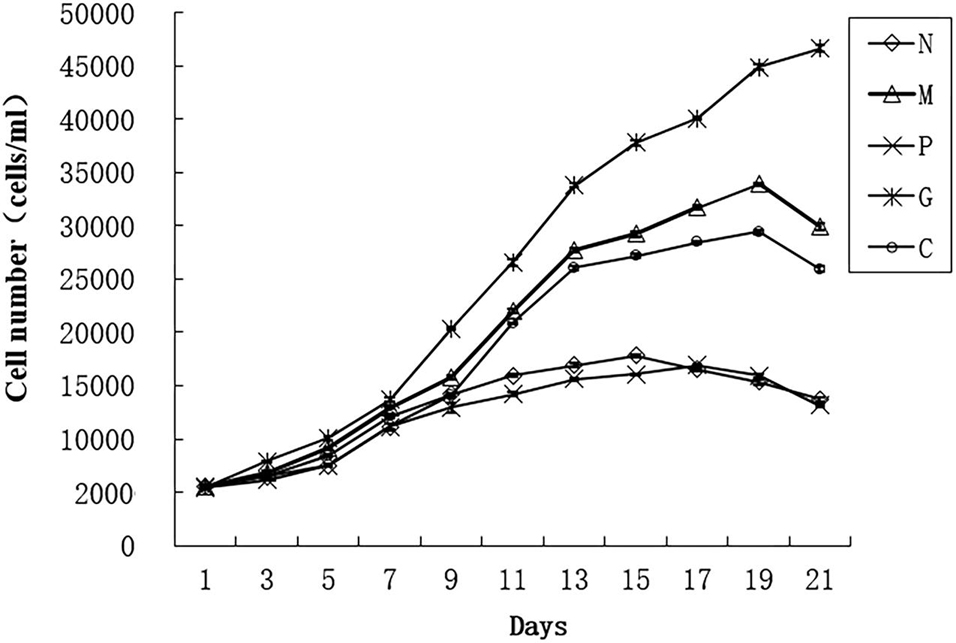
Figure 1. Growth curve of Alexandrium pacificum under five treatments (Liu et al., 2019). Normal culture (C), high concentrations of phosphorus and manganese (M), high irradiance (G), low phosphorus (P), and low nitrogen (N) were the conditions used in the study.

Table 1. Samples used for digital gene expression (DGE) analysis.
To reduce bacterial contamination during collection of cells, every 100 mL of the cultures was filtered through a 10 μm pore size bolting-silk, rinsed with 300 mL sterile F/2 medium and subjected to the following treatments: the washed cells were suspended in 50 mL sterile F/2 medium containing 0.05% Tween-80 and 0.01 M EDTA (at 20°C for 30 min), followed by ultrasonication for 1 min (50 W; ultrasonic treatment time of 5 s with an interval of 5 s). Then lysozyme (0.5 mg/mL, 20°C for 10 min) and SDS (0.25%, 20°C for 10 min) were added. The cells were washed three times with sterile F/2 medium to remove these reagents. The final cultures were stained with acridine orange (0.01%) for 1 or 2 min, then observed using epifluorescence microscopy (Nikon ECLIPSE 80i, Japan), only the axenic samples were harvested and used for the following experiments. Each sample contains 5 × 106 cells which were collected by centrifugation for 6 min at 4 000 g and then the cell pellets in 1.5 mL eppendorf tubes were frozen immediately and stored in liquid nitrogen for RNA extraction.
RNA Extraction and Library Construction for Sequencing
Total RNA was extracted using Trizol reagent (TaKaRa, Japan) following the manufacturer’s instructions, followed by DNA digestion with DNase I (TaKaRa, Japan). The quantity and quality of the RNA samples were measured by gel electrophoresis and NanoDrop 2000 (Thermo, United States). High-quality RNA of the same quantities was used in subsequent experiments.
Fifteen libraries were constructed following the manufacturer’s instructions of the Illumina Gene Expression Sample Prep Kit. The mRNA-seq libraries were sequenced 100 bp paired-end reads on the Illumina Genome Analyzer for DGE analysis. Base calling was performed by the Illumina instrument software.
Quality Control
The original data from HiSeqTM 2000 high-throughput sequencing is converted into the original sequenced sequence (Raw Data) by base calling analysis. FastQC was used for quality control. Raw data (raw reads) were cleaned by removing reads containing adapter, reads in which more than 10% of the bases were unknown, and low quality reads (where more than 50% of bases in a read had a quality value Q ≤ 5). The Q20, Q30, and GC content and sequence duplication level of the clean data were calculated. All the downstream analyses were based on clean, high-quality data.
Mapping of Reads
Transcriptome data of A. pacificum (SRX368254) were selected as the reference sequence to map of the date. Bowtie was used to map single-end clean reads to the unigene sequences. Clean reads were obtained by removing reads containing adapter, reads containing ploy-N and low quality reads from raw data. At the same time, Q20, Q30, and GC content of the clean data were calculated. All the downstream analyses were based on the clean data with high quality.
Quantification of Gene Expression Level and Differential Expression Analysis
Transcript abundance and differential gene expression were calculated with the program Cufflinks1. Gene expression levels were normalized with fragments per kilobase of exon per million mapped reads (FPKM) values to obtain the relative levels of expression. The false discovery rate (FDR) was used to determine the threshold of P-value in multiple test and analysis. Genes were considered to be differentially expressed only when their absolute value of log2 fold change was >1 and FDR was <0.05 between two treatments. The correlation of the detected number of counts between parallel libraries was assessed statistically by calculating the Pearson’s correlation.
Variance-stabilized data obtained using DESeq was used to generate the heatmaps of differentially expressed genes. Clustering analysis was performed using the command hclust in R language which used K-means and SOM methods to calculate distance between clusters. The heatmap was drown using the R packages of ggplot2 and pheatmap. The heatmap and cluster analysis were performed based on the expression level of differential gene RPKM under different experimental conditions include all the 15 samples among the five treatments. Based on researches about RNA-seq analysis (Robinson and Oshlack, 2010) and the Pearson’s correlation tests, we use the average value of RPKM of each treatment to complete the heatmap. Gene ontology (GO) and KEGG enrichment of differentially expressed genes were analyzed after the quantification of gene expression level. GO and KEGG enrichment was based on GOseq and KEGG database. The enrichment analysis was performed using the OmicShare tools, a free online platform for data analysis.
Quantitative Real-Time PCR Validation
Total RNA was extracted as described for DGE library preparation and sequencing. For the first-strand cDNA synthesis experiment, a Transcriptor First Stand cDNA Synthesis Kit (Roche) was used following the manufacturer’s instructions. Quantitative real-time PCR was conducted using SYBR Green dye (LightCycle® 480 SYBR Green I Master). The selected genes including transketolase, phosphoglycerate kinase (PGK), phosphoribulokinase (PRK) and serine-threonine kinase (STK) were verified using the LightCycle® 480 Real-Time PCR System with the following cycling conditions: 95°C for 5 min, followed by 45 cycles of 95°C for 10 s, 60°C for 10 s and 72°C for 20 s. Specificity of the qPCR product was analyzed by melting curve analysis. The sequences of the primers used are given in Supplementary Materials. Actin and GAPDH served as internal controls. The 2–ΔΔCt method was used to calculate relative gene expression values. The mean of fold changes was calculated for each experiment and mean >2 or <0.5 was considered as significant.
Results
Sequencing of 15 Digital Gene Expression Libraries
All fifteen RNA samples of A. pacificum under the five treatments (each treatment contains three biological replicates) were subjected to RNA-seq. Each sample yields 11–12.6 million raw reads. After quality assessment and filtering low quality data, approximately 10.7–12.1 million clean reads were obtained in each library (Table 2). Approximately 69.14 ∼ 80.57% reads were mapped to the transcriptome of A. pacificum (SRX368254), which also proved that the transcriptome data was a reliable reference. The mean Q20 and Q30% of five treatments were ranging from 93.92 to 95.1% and from 83.98 to 86.37% average, respectively, suggesting that the high quality sequencing data made the results more reliable. The mean GC content (%) is 63.5% average. The reproducibility of the DGE was tested by Pearson correlation analysis for every three replicates (Figure 2). Sample C was representative, other results was provided in Supplementary Materials. The rooted squares of the Pearson’s correlation coefficient (R2) were greater than 0.92, mostly ranging from 0.92 to 0.953 (Table 3). The rooted squares of the Pearson’s correlation coefficient (R2) were close to 1, indicating both the better parallelism between samples and the reliability of the experimental results.
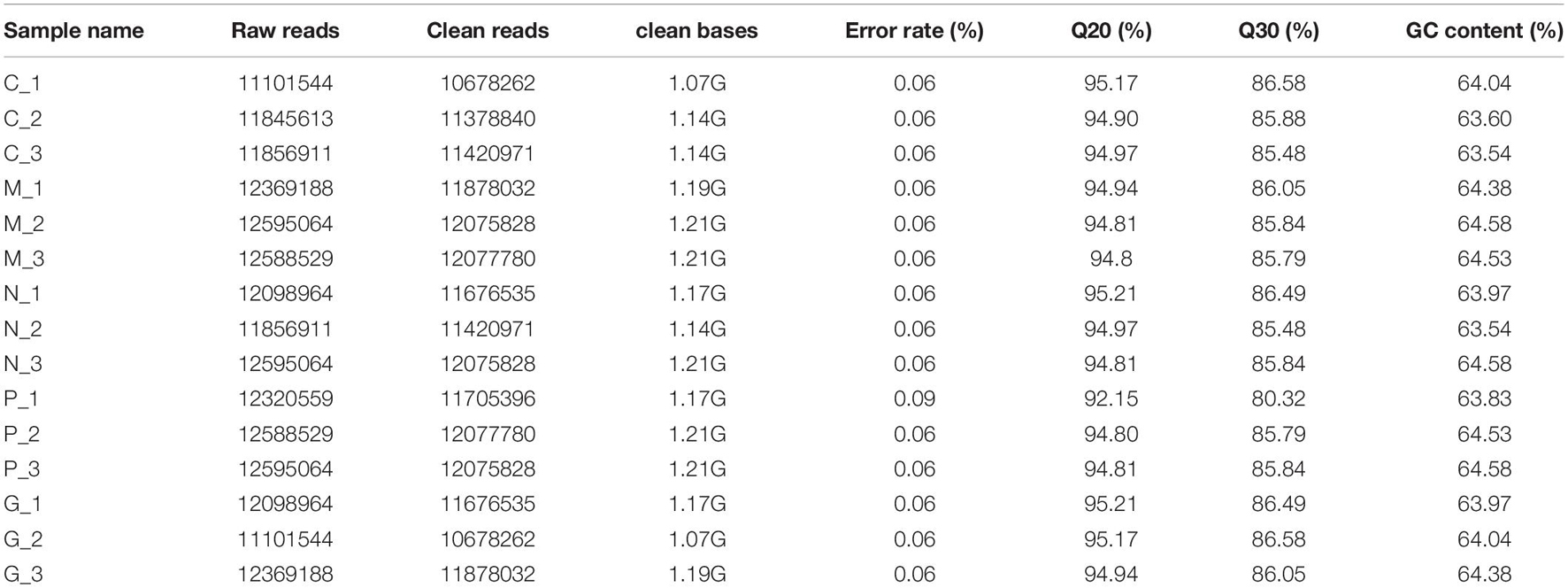
Table 2. Summary of sequencing results for 15 DGE libraries.
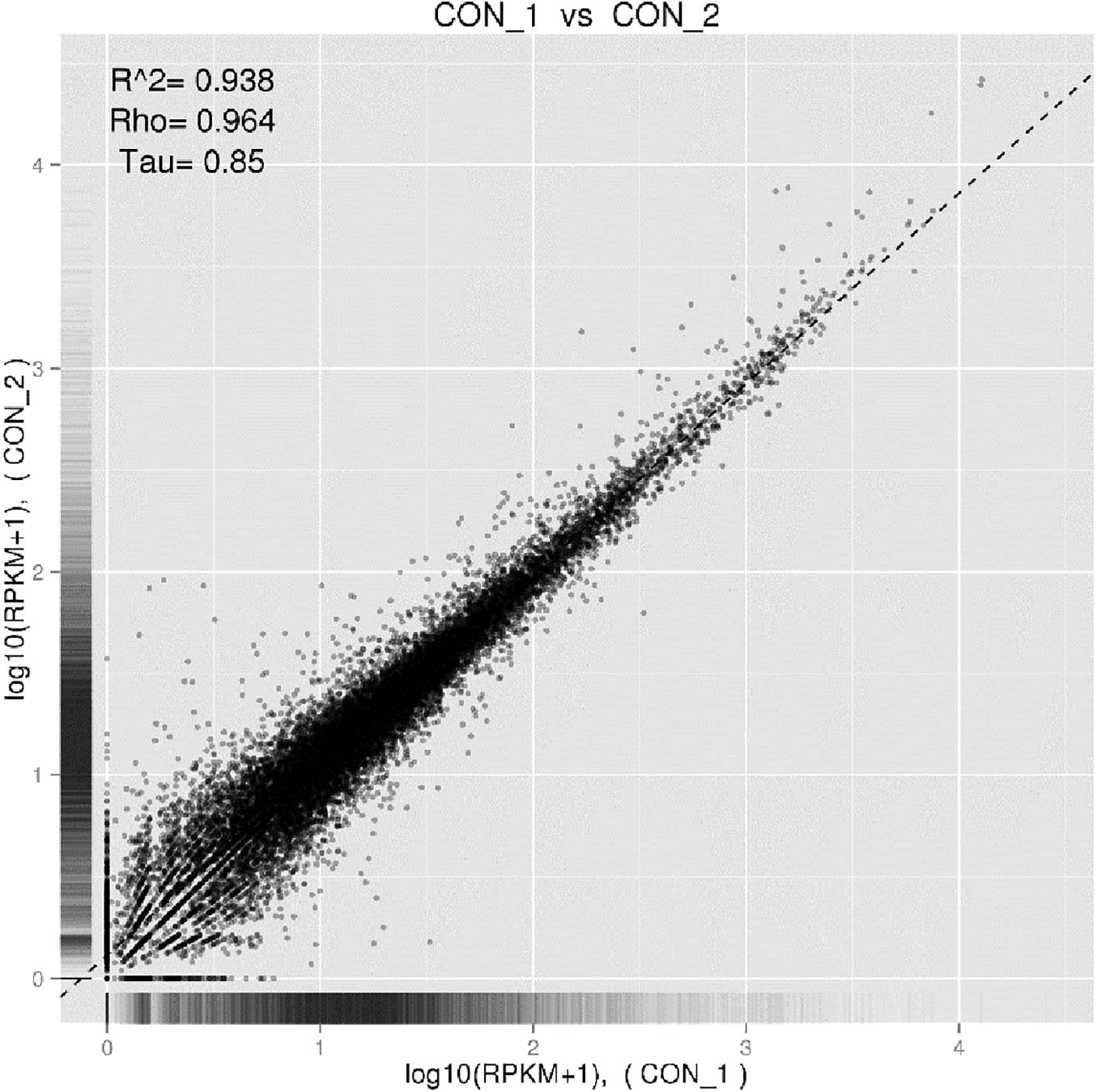
Figure 2. Pearson correlation tests for C1 and C2. The abscissa represents the value log10 (RPKM + 1) of one duplicate; the ordinate represents the value log10 (RPKM + 1) of the other duplicate. R2 is the square of Pearson.
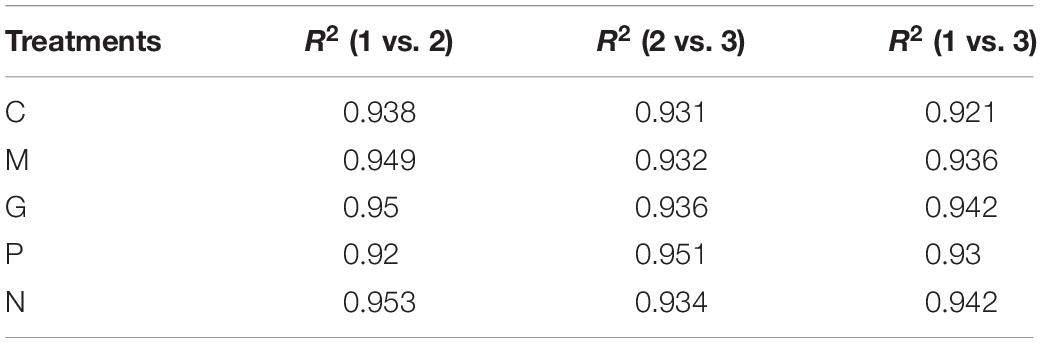
Table 3. The rooted square of the Pearson’s correlation coefficient (R2) between three replicates of 5 different cultivation conditions.
Number of Differential Expression Unigenes
The expression level of mapped unigenes was normalized with a value of FPKM (fragments per kilobase of exon per million fragments mapped). An absolute value of log2 fold change > 1 and false discovery rate < 0.05 was set to declare the differential expressed genes (DEG).
The fold change of the expression levels in the different treatments were compared in pairs (Table 4). Eutrophication conditions (M) and high irradiance conditions (G) compared with C and nutrient limitation conditions (N and P) to find out the DEGs functioned in promoting the growth of the algae. The treatment N compared with P to find out the different mechanisms when the algae cope with the nutrient limitation conditions. The treatment M compared with G to figure out the different mechanisms in promoting the growth of the algae.
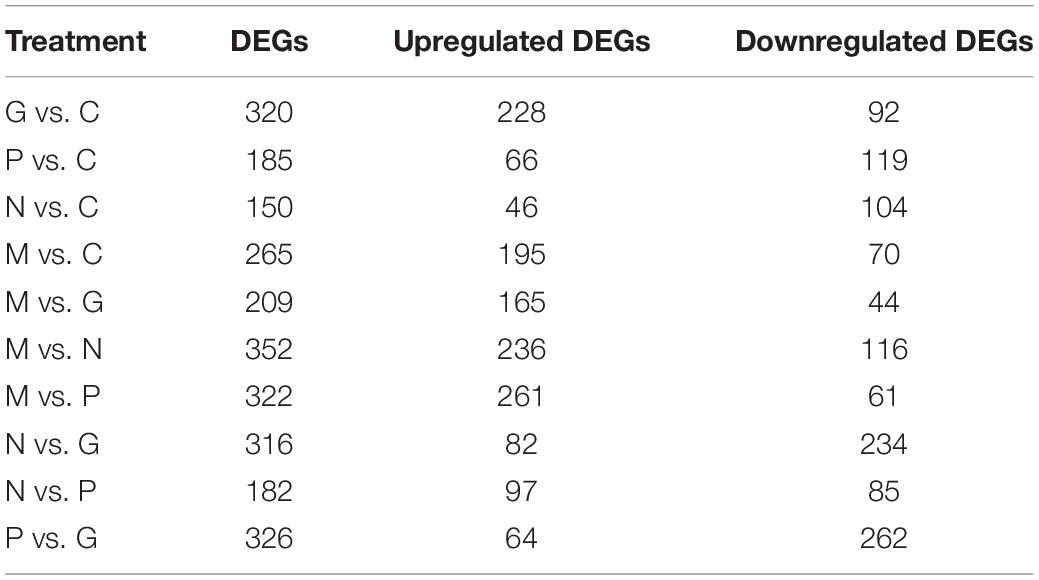
Table 4. The DEGs between treatments.
A hierarchical cluster was used to determine the profiles of the differentially expressed unigenes among the five treatments (Figure 3). Unigenes with the same or similar expression patterns were gathered in the same subcluster. For example, the unigenes from sub-cluster 1 show similar expression trend. The pattern under the M and G conditions were similar which indicates the similar mechanisms under the stage of explosive growth. In subcluster 1, the unigenes were found to relate to the carbohydrate metabolism and energy metabolism which enriched in the pentose phosphate pathway and carbon fixation in photosynthetic organisms. In subcluster 2, the unigenes were associated with the translation of genetic information including genes in the pathway of ribosome biogenesis. In subcluster 3, most of the unigenes were related to the amino acid metabolism, except that there were less unigenes related to the synthesis of carbohydrates (Figure 4).
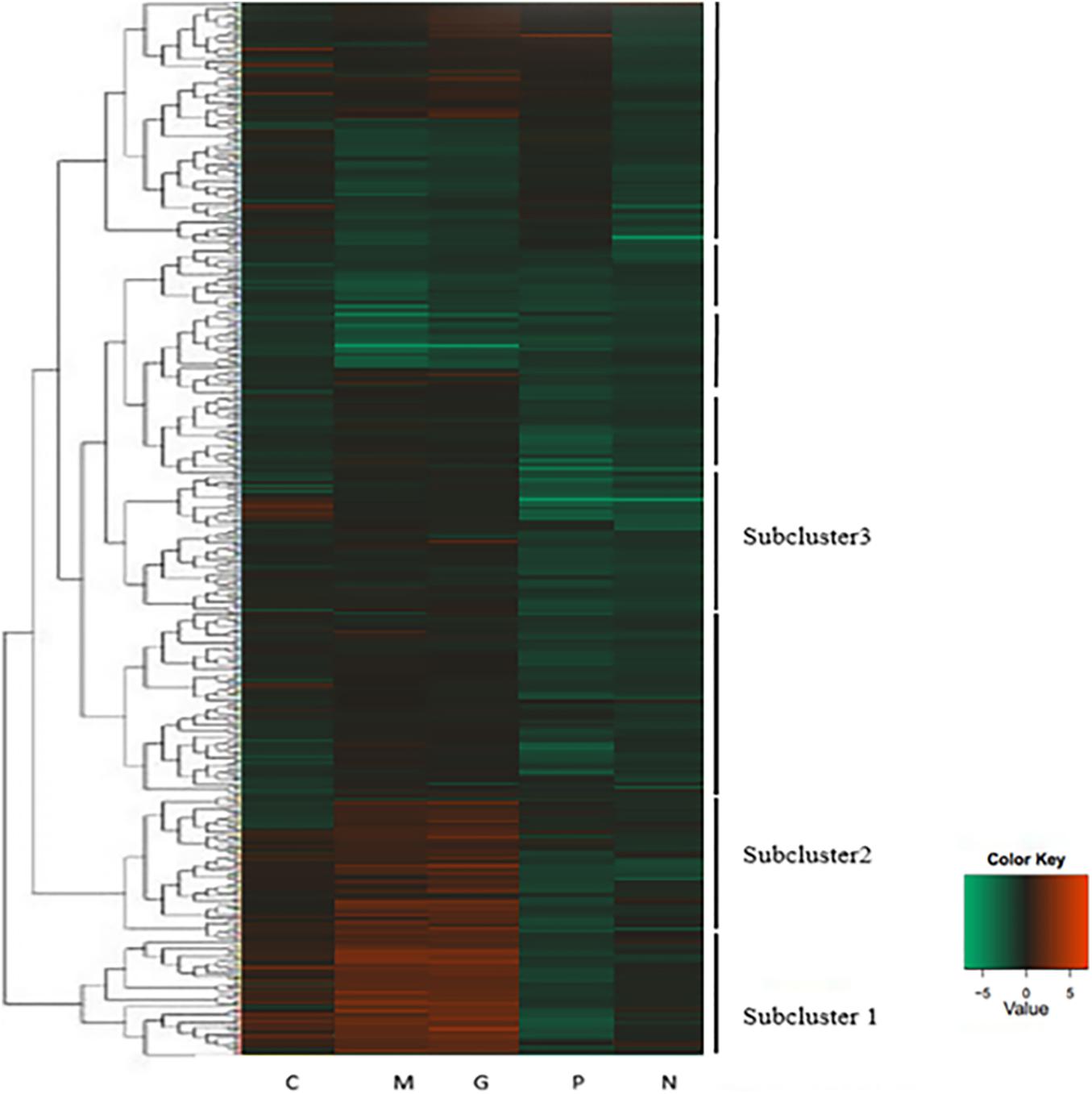
Figure 3. Heatmap of hierarchical clustering. Each column represents a treatment, and each row represents a unigene. Differences in expression were shown in different colors. Negative numbers indicate down-regulated and positive number means up-regulated. Normal culture (C), high concentrations of phosphorus and manganese (M), high irradiance (G), low phosphorus (P) and low nitrogen (N) were the conditions used in the study.
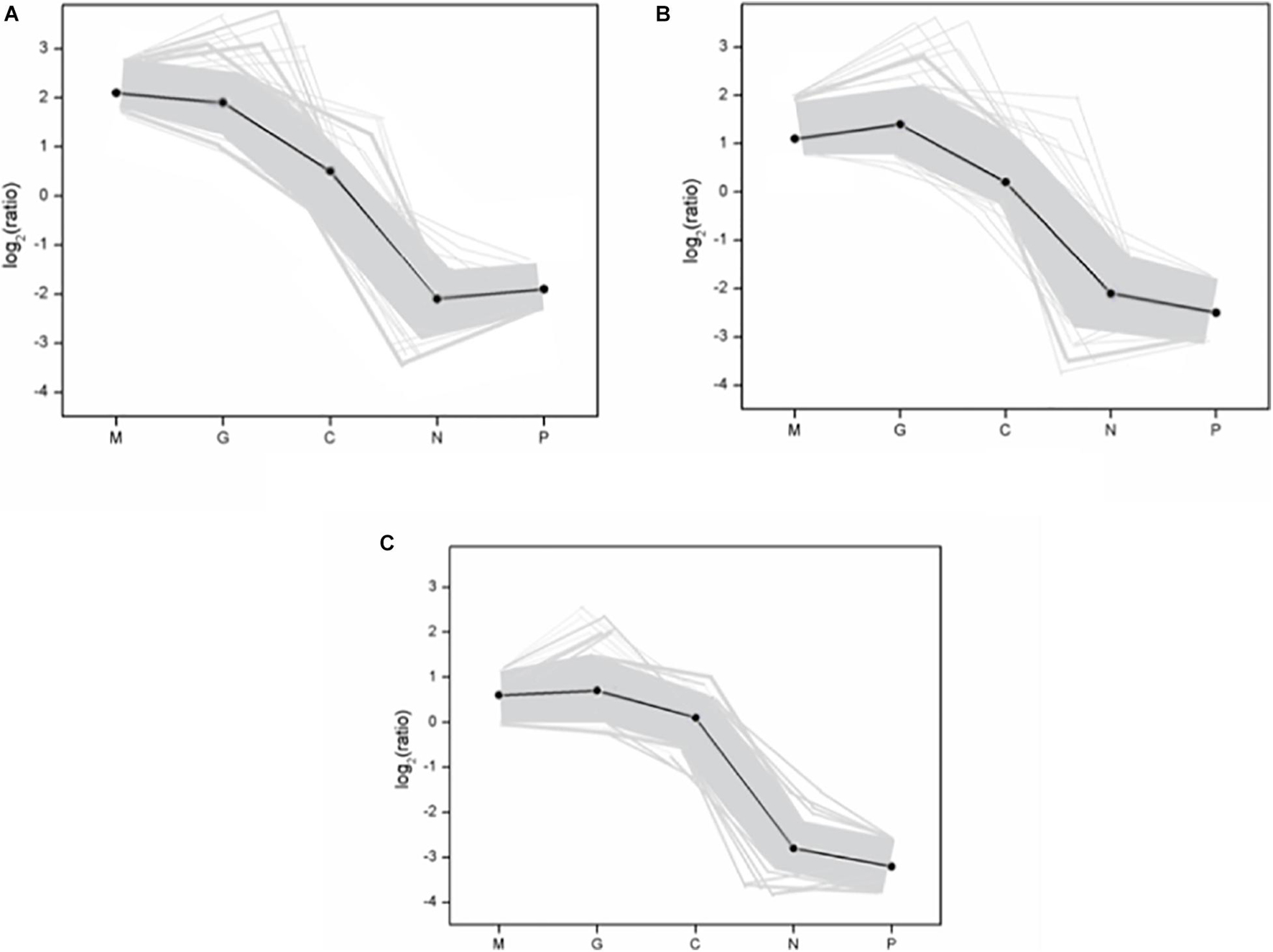
Figure 4. The gene expression pattern of three subclusters. (A) Subcluster 1; (B) subcluster 2; (C) subcluster 3. The “x-” axis represents the treatments. The “y-” axis represents the value of the relative expression level [log2 (ratio)]. In the subcluster, the gray line represents the value of the relative expression levels, the black line represents the mean value of the relative expression level. The subclusters in figure represent the subclusters in Figure 3. Normal culture (C), high concentrations of phosphorus and manganese (M), high irradiance (G), low phosphorus (P), and low nitrogen (N) were the conditions used in the study.
Gene Ontology Functional Analysis of Differentially Expressed Genes
Gene ontology (GO) was carried out to unify the representation of gene and gene product attributed in A. pacificum under different treatment conditions. For the biological process category, the mostly highly represented terms were cellular process and metabolic process. In metabolic process, the number of DEGs under M and G were much more than under N and P indicating the metabolism of the cells were increased to deal with the rapid growth of the algae. For the cellular component category, the mostly highly represented terms were cell and cell parts. For the molecular function category, the two mostly highly represented terms were binding and catalytic activity. And the number of DEGs under M and G were much more compared with those under N and P in these two terms suggesting the cells need more carriers and enzymes to complete the biological process under M and G and the inhibition of parts of the biological process when exposed to nutrient deficiency conditions (Figure 5).
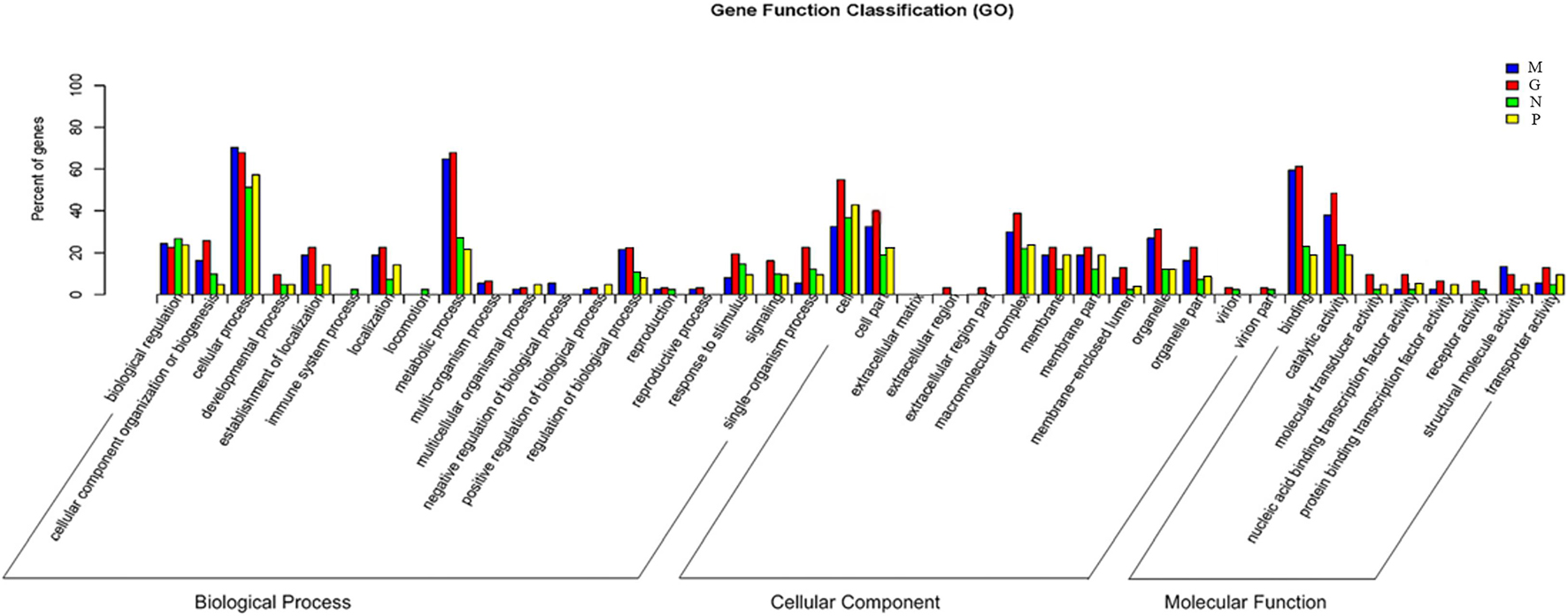
Figure 5. Comparative distribution of the GO terms of DGE in the four treatments relative to C. Normal culture (C), high concentrations of phosphorus and manganese (M), high irradiance (G), low phosphorus (P) and low nitrogen (N) were the conditions used in the study.
Differentially Expressed Genes in Response to Eutrophic Conditions
Phosphorus and manganese are the two main nutritional factors that promote the growth of A. pacificum. A total of 265 DEGs were identified under M conditions relative to C conditions, including 195 upregulated unigenes and 70 downregulated unigenes (Table 4). Based on KEGG analysis, the upregulated unigenes were highly enriched in ribosome biogenesis and the pentose phosphate pathway (PPP). GO enrichment analysis revealed that of these 265 DEGs, 172 upregulated unigenes and 42 downregulated unigenes were significantly enriched in GO terms. Of interest, 75 of the 172 upregulated unigenes were related to the structure of the ribosome. These findings were consistent with the KEGG analysis data.
In higher plants, the PPP is associated with plant development and environmental stress responses. Here, DGE analysis revealed that 12 unigenes involved in the PPP were upregulated under M conditions including 6-phosphogluconate dehydrogenase (6PGDH), glucose-6-phosphate dehydrogenase (G6PDH), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), transketolase and transaldolase.
Upregulation of unigenes involved in carbon assimilation such as ribulose-3-phosphate isomerase, phosphoglycerate kinase and fructose-1, 6-bisphosphate aldolase was also observed under M conditions. Upregulation of such genes would be associated with an increased rate of photosynthesis.
Finally, M treatment conditions led to an upregulation of genes that can affect the development of organisms, including PCNA, calmodulin (CaM), glycine-rich protein and ubiquitin. Nine transcription factors were also found to be differentially regulated included basic leucine zipper (bZIP), basic transcription factor 3 (BTF3), and myeloblastosis oncogene homolog (MYB).
Taken together, these findings indicate that eutrophic conditions promote expression of genes associated with nucleotide synthesis, cell growth and photosynthesis.
Differentially Expressed Genes in Response to High Irradiance Conditions
Under G conditions, the growth rate of A. pacificum was significantly increased and the number of algae in the log phase was significantly higher compared with normal conditions (Table 4). A total of 320 DEGs were identified under G conditions relative to C conditions (Table 4) including 228 upregulated unigenes and 92 downregulated unigenes. Based on GO enrichment analysis, there were 197 and 57 genes associated with the enriched GO terms in the up- and downregulated unigenes, respectively. KEGG analysis revealed that the upregulated unigenes were highly enriched in ribosome biogenesis and carbon fixation in photosynthetic organisms.
Digital gene expression analysis revealed that unigenes associated with electron transfer reactions in photosynthesis were upregulated including PsbO, PsbU, cytochrome b6f complex, ferredoxin and thioredoxin. Among the 320 DEGs, unigenes related to carbon fixation including ribulose-3-phosphate isomerase, phosphoglycerate kinase, ribulose-1,5-biphosphate carboxylase and fructose-bisphosphate aldolase were upregulated under G conditions.
Similar to M conditions, unigenes encoding PCNA, CaM and ubiquitin, which are related to the development of organisms, were upregulated. In addition, three unigenes encoding transcription factors were identified including MYB. Thus, high irradiance promotes expression of genes associated with growth and photosynthesis.
Differentially Expressed Genes in Response to Nutrient Limiting Conditions
A total of 185 DEGs were identified after P treatment compared with C conditions (Table 4). Under low phosphorus conditions, 66 unigenes were upregulated and 119 unigenes were downregulated. Based on GO enrichment analysis, 42 upregulated unigenes and 92 downregulated unigenes were enriched in GO terms. DEGs encoding phosphoglycerate kinase, ribulose-1, 5-bisphosphate carboxylase, GAPDH, transaldolase, ribulose-3-phosphate isomerase, phosphoglycerate kinase and fructose-1, 6-bisphosphate aldolase were downregulated under P conditions.
A total of 150 DEGs were identified under N conditions compared compared with C conditions (Table 4). After treatment with low nitrogen, 46 unigenes were upregulated and 104 unigenes were downregulated. KEGG analysis revealed that the DEGs were highly enriched in nitrogen metabolism. Based on GO enrichment analysis, 21 upregulated unigenes and 84 downregulated unigenes were enriched in GO terms. The downregulation of six unigenes involved in nitrogen metabolism including nitrate reductase, which plays a vital role in nitrogen assimilation, was observed. In addition, that amino acid metabolism is disrupted. Thus, in contrast to other conditions, nutrient limitations promote expression of genes associated with metabolism and stress.
Validation of Gene Expression
To validate the expression profiles obtained by RNA-Seq, RT-PCR was performed on four genes. Normalized gene expression according to 2–ΔΔCT method was shown in Figure 6. Expression of the four genes under M and G were all higher than those under N and P (p < 0.05). The trend in RT-PCR expression was in agreement with the DGE data (Figure 7).
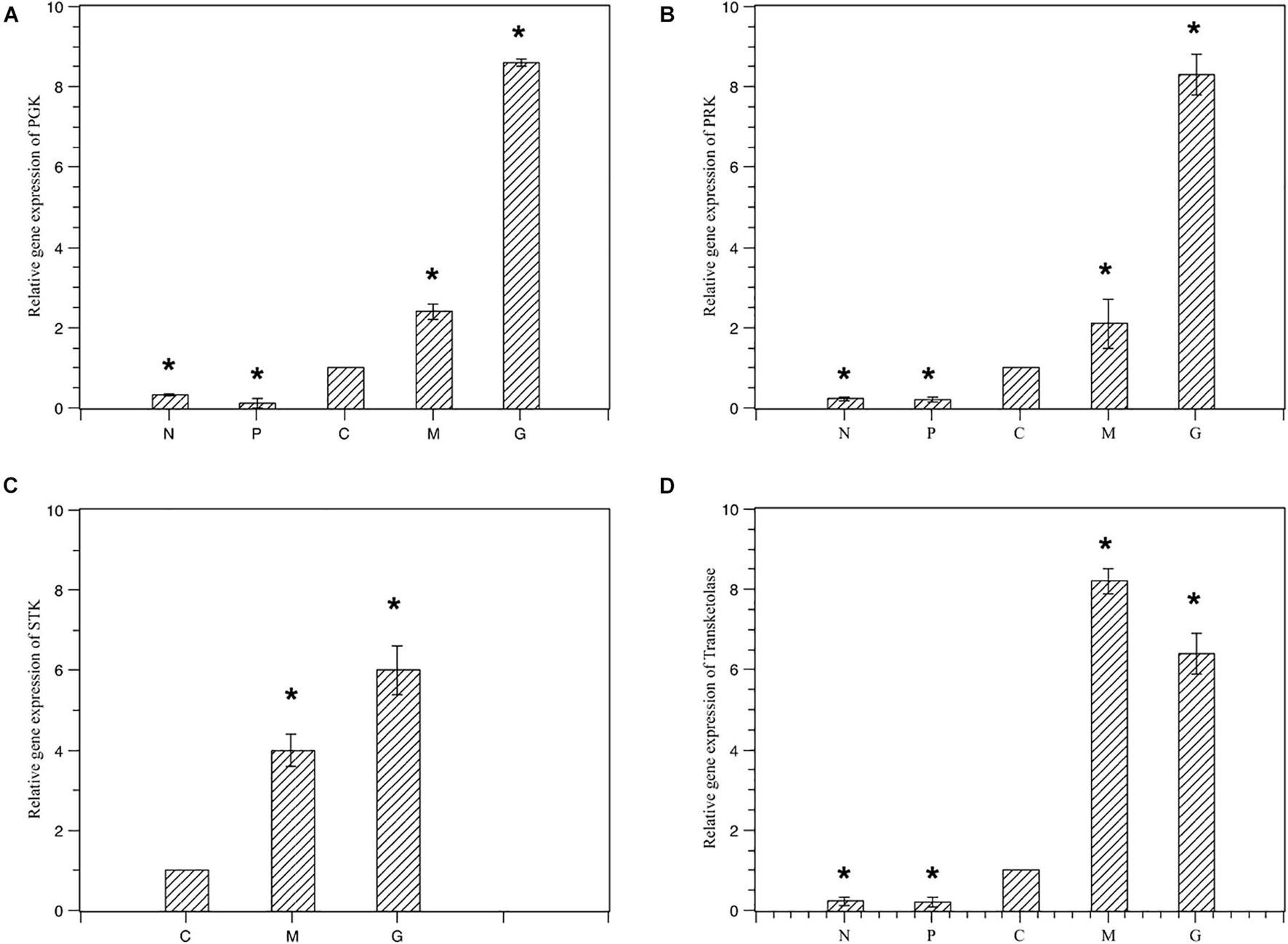
Figure 6. Relative gene expression of the four selected genes under different treatments. Normal culture (C), high concentrations of phosphorus and manganese (M), high irradiance (G), low phosphorus (P) and low nitrogen (N) were the conditions used in the study. (A) PGK; (B) PRK; (C) STK; (D) transketolase. The symbol * represents a significant difference.
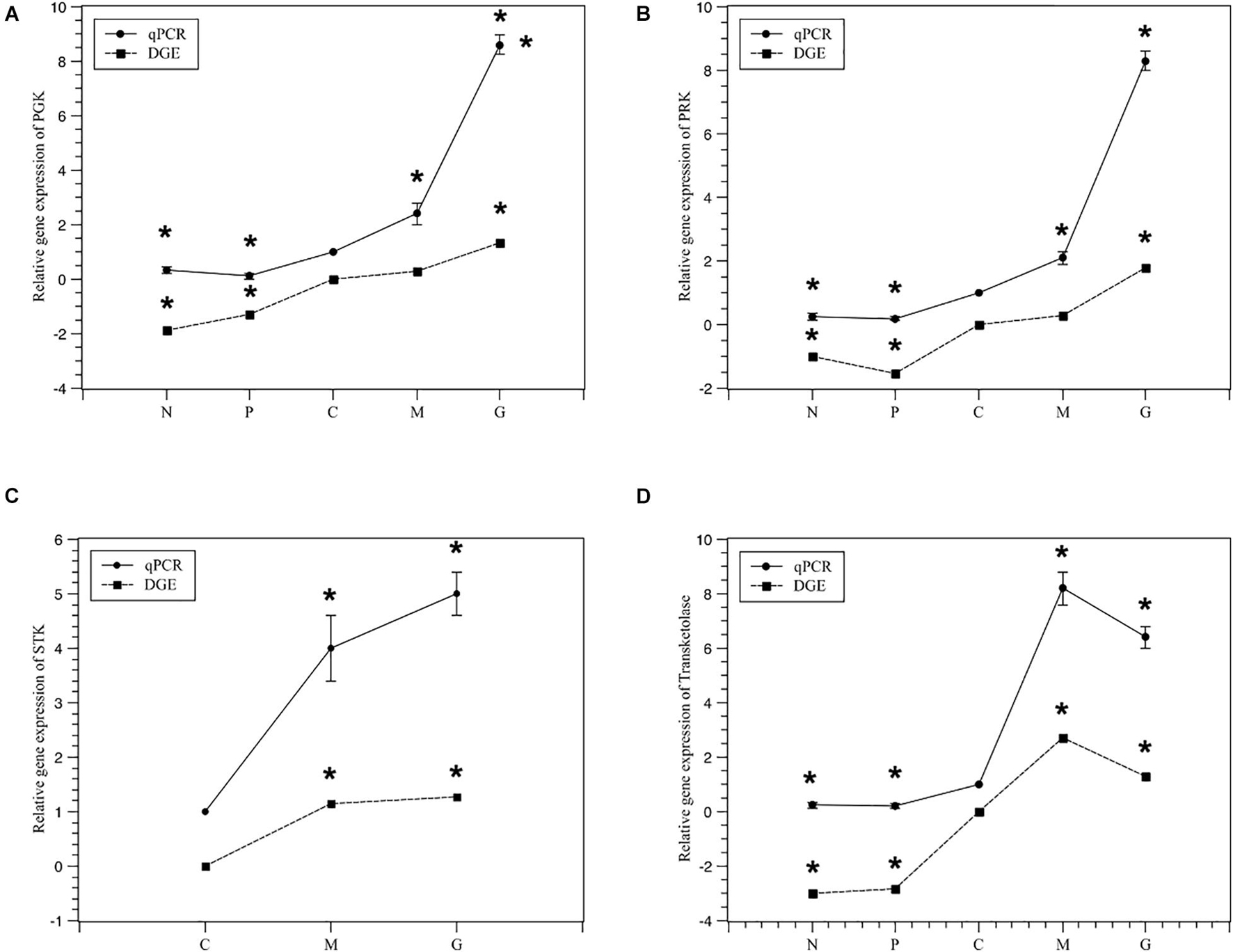
Figure 7. Relativey expression of the five selected genes under different treatments from DGE and qPCR. Normal culture (C), high concentrations of phosphorus and manganese (M), high irradiance (G), low phosphorus (P) and low nitrogen (N) were the conditions used in the study. (A) PGK; (B) PRK; (C) STK; (D) transketolase. The symbol * represents a significant difference.
Discussion
The Role of the Pentose Phosphate Pathway Under the Stage of Explosive Growth
Cell growth requires biosynthesis of critical intermediates such as nucleotides and amino acids. During cellular proliferation, a key feature of the metabolic transformation is the enhancement of biosynthetic capacity (Levine and Puzio-Kuter, 2010). The PPP, a fundamental component of cellular metabolism, can be subdivided into oxidative and non-oxidative PPPs. In this study, 35 unigenes associated with the PPP were found in the transcriptome data (SRX368254). Furthermore, a total of 14 DEGs involved in the PPP were identified under different treatments including G6PDH, 6PGDH, transketolase, transaldolase and ribose 5-phosphate isomerase (Table 5). The upregulation of transketolase and transaldolase under M conditions was consistent with the comparative transcriptome data of A. pacificum (SRX368254). The oxidative branch converts glucose 6-phosphate into ribulose 5-phosphate through the consecutive reactions of G6PDH and 6GPDH (Miclet et al., 2001). In the non-oxidative branch, transketolase and transaldolase are the key enzymes of the non-oxidative PPP, which connect glycolysis and the PPP. They catalyze the conversion of ribose 5-phosphate and sedoheptulose 7-phosphate to complete the PPP (Kochetov and Sevostyanova, 2005; Samland and Sprenger, 2009).
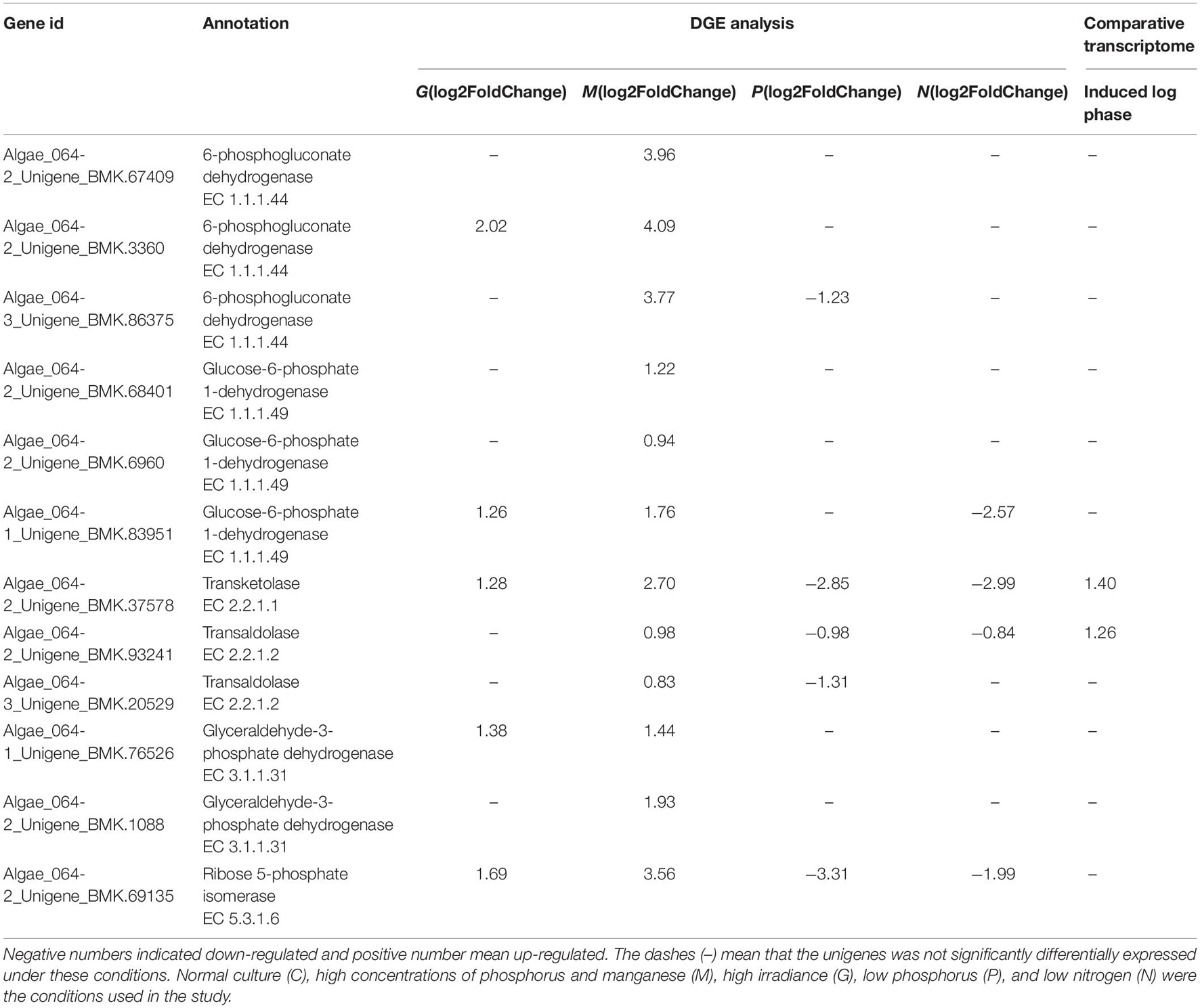
Table 5. Differentially expressed unigenes enriched in PPP in G, M, P, and N relative to C by DGE analysis and in induced log phase compared with lag phase based on comparative transcriptome dataset (SRX368254).
Several studies have demonstrated that the expression levels of G6PDH, transketolase and transaldolase were correlated with the progression of cancer, suggesting that enhanced PPP activity is associated with cell proliferation (Deberardinis et al., 2008). In our study, under M conditions, the unigenes encoding G6PDH, 6PGDH, transketolase, transaldolase, ribose 5-phosphate isomerase and pyruvate kinase were upregulated (Table 5), suggesting increased PPP activity, which may enhance the cell biosynthetic capacity to adapt to rapid proliferation. In addition, transketolase and transaldolase were also found to be upregulated in the induced log phase in the A. pacificum comparative transcriptome dataset (SRX368254).
Under nutrient limiting conditions (P, N), transketolase, transaldolase and ribose 5-phosphate isomerase were downregulated, which reduced the conversion of ribose 5-phosphate and resulted in the inhibition of nucleic acid synthesis and potentially growth arrest. Interestingly, normal expression of the unigenes encoding G6PDH and 6GPDH was observed under N and P conditions compared with C conditions (Table 5). G6PDH and 6GPDH, key enzymes in the oxidative PPP, yield NADPH, which maintains the redox balance under stress conditions (Slekar et al., 1996). Hence, under the nutrient limiting-induced stress, algae still need to express G6PDH and 6GPDH to yield NADPH in order to maintain the redox balance for survival.
Photosynthesis Under the Stage of Explosive Growth
Photosystems are functional and structural units of protein complexes involved in photosynthesis that carry out the primary photochemistry of photosynthesis: the absorption of light and the transfer of energy and electrons. Of the two photosystems [photosystem I (PS I) and PS II], the cytochrome b6f complex mediates electron transfer by oxidizing lipophilic plastoquinone and reducing plastocyanin (Kurisu et al., 2003). In this study, unigenes encoding PsbO, PsbU and the cytochrome b6f complex were found to be upregulated after G treatment (Table 5). The PsbO protein is an extrinsic subunit of PS II and is thought to play a central role in stabilization of the catalytic manganese cluster in which water is split into four protons and O2 via a series of four redox steps (Murakami et al., 2002). PsbU, a 12 kDa protein of PS II, is thought to regulate PS II function in cyanobacteria (Shen et al., 1998).
The ferredoxin/thioredoxin system (FTS), the major redox system responsible for photosynthesis in plant chloroplasts, is composed of three proteins, thioredoxin (Trx), ferredoxin thioredoxin reductase (FTR) and ferredoxin (Fd). In the FTS, Fd transfers electrons received from PS I to FTR, which then reduces Trx, providing a biochemical link between light reactions and the regulation of metabolism (Holmgren, 1995; Schürmann, 2003). In chloroplasts, most thioredoxin-regulated enzymes are associated with carbon metabolism including phosphoribulokinase (PRK), fructose-1,6-bisphosphatase and G6PDH (Ruelland and Miginiac-Maslow, 1999).
Among the DEGs, the unigenes encoding Trx and Fd were upregulated under G conditions (Table 6), suggesting that the FTS was active. Furthermore, together with PsbO, PsbU and the cytochrome b6f complex, Trx and Fd promote the transport of photosynthetic electrons and provide ATP and NADPH for carbon assimilation and the PPP.
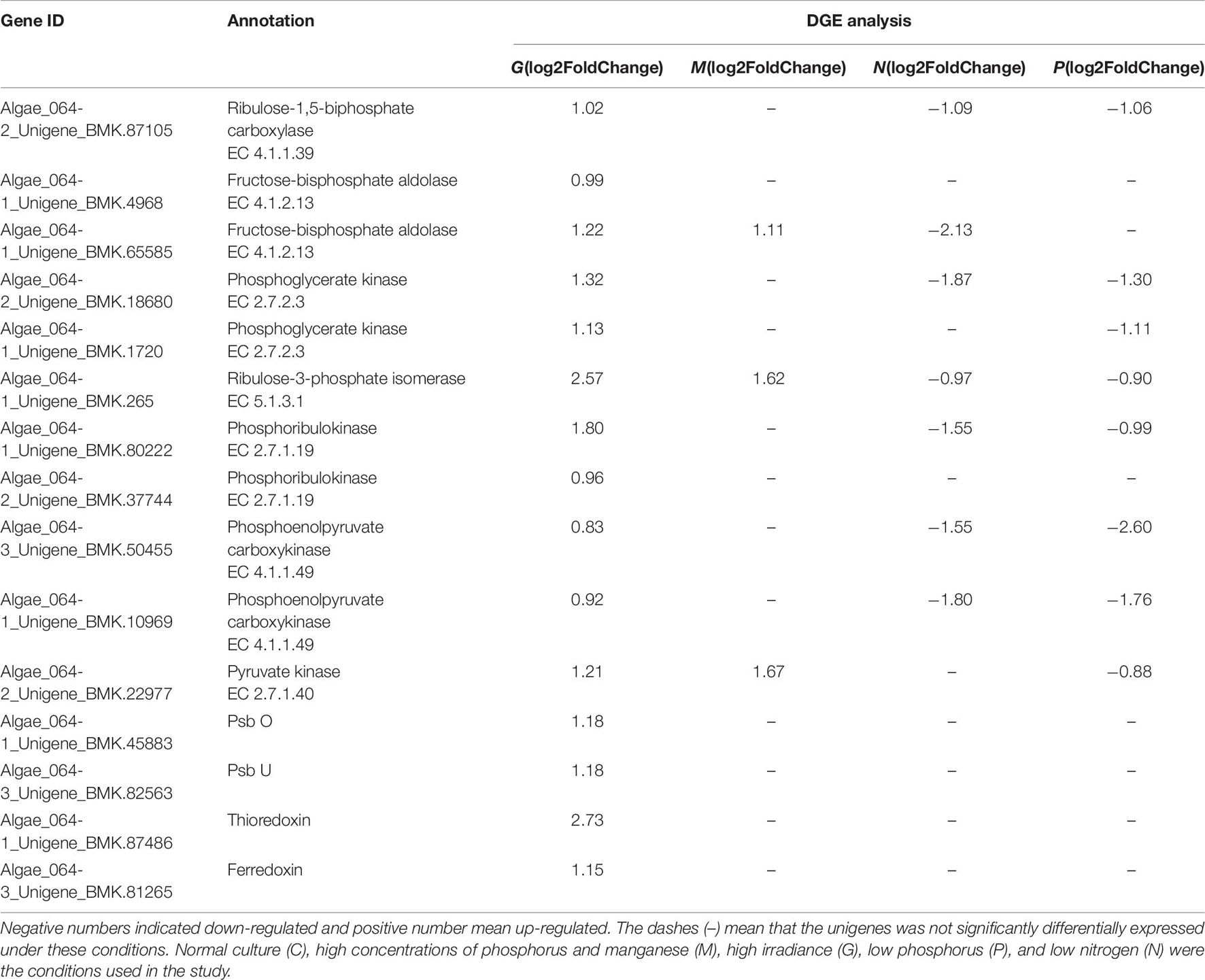
Table 6. Differentially expressed unigenes related to photosynthesis in G, M, P, and N relative to C by DGE analysis.
For algae, many studies have suggested that the Calvin–Benson cycle (C3 pathway) was predominant compared with the C4 and crassulacean acid metabolism (CAM) pathways (Tsuji et al., 2009). However, a recent study suggested that C4 pathways may also be used in algae. Consistent with this, 72 and 52 unigenes associated with the C3 and C4 pathways, respectively, were found in the A. pacificum transcriptome dataset (SRX368254). Among these unigenes, 16 and 9 unigenes associated with the C3 and C4 pathways, respectively, were differentially expressed under different treatment conditions (Table 6). In the C3 pathway, three DEGs encoding components of the ribulose 1, 5-bisphosphate carboxylase (Rubisco) complex, which is the central carboxylation enzyme for CO2 fixation, were upregulated under G and P treatment conditions. The C3 pathway of carbon fixation is catalyzed by Rubisco, producing two three-carbon molecules of 3-phosphoglyceric acid, through the carboxylation of the five-carbon ribulose-1,5-biphosphate (Wang et al., 2011). In addition, G treatment resulted in the upregulation of DEGs associated with the C3 pathway including fructose-bisphosphate aldolase, phosphoglycerate kinase, ribulose-3-phosphate isomerase and PRK. All of the five afore-mentioned genes were involved in the rate-limiting step of the C3 pathway, suggesting that the C3 pathway may be involved in the carbon assimilation process under these conditions.
Unigenes invon the A. pacificum transcriptome dataset (SRX368254). Among these unigenes, the differential expression of enzymes that catalyze the C4lved in the C4 pathway were also found i pathway was observed including phosphoenolpyruvate carboxykinase (PEPCK), pyruvate orthophosphate kinase (PPDK) and pyruvate kinase (PK) (Table 6). In the C4 pathway, PEPCK is a key enzyme that catalyzes the release of CO2 from oxaloacetate to produce phosphoenolpyruvate (PEP) to complete the C4 pathway (Hibberd and Covshoff, 2010). Since PPDK catalyzes the conversion of pyruvate to PEP (Parsley and Hibberd, 2006), upregulated PPDK expression under G treatment conditions could promote the C4 pathway. The function of PK is similar to that of PPDK. However, PK catalyzes an irreversible reaction, and does not consume ATP (Baud et al., 2007). Under high temperature, high light, and the current CO2 concentration in the atmosphere, the C4 pathway is more efficient than C3 photosynthesis because it increases the CO2 concentration around the major CO2 fixating enzyme Rubisco (Bräutigam and Gowik, 2016). When HAB occurs, a rapid increase in the size of the population requires efficient photosynthesis. Hence, the C4 pathway may exist in A. pacificum to promote carbon assimilation and support increased growth.
There is a significant amount of transcriptomic information about C4-related enzyme variations among various algae (Zhao and Su, 2014). In Bacillariophyta, Phaeodactylum tricornutum and Thalassiosira pseudonana have developed a C4-like photosynthesis pathway in addition to the carbon-concentrating mechanism (Armbrust et al., 2004; Kroth et al., 2008), while in Chlorophyta, Ulva linza and Ulva prolifera have been shown to express the C4 pathway (Xu et al., 2012). Taken together, these findings suggest that the C4 pathway may be more complex in A. pacificum. Furthermore, the transcriptome data provides the foundation to elucidate the mechanisms underlying the complete carbon fixation system.
Cell Division
Among the DEGs, 92 unigenes were enriched in the ribosome biogenesis pathway. Ribosome biogenesis underlies a cell’s capacity to grow as cell growth requires a large numbers of ribosomes, the molecular factories that carry out protein synthesis (Lempiäinen and Shore, 2009; Donati et al., 2012). Based on the A. pacificum transcriptome data (SRX368254), 16 unigenes related to DNA replication including PCNA were upregulated, especially in the induced log phase. PCNA was also upregulated under M and G treatment conditions (Table 6), reflecting the proliferative state of A. pacificum.
Proline-rich and glycine-rich proteins are required for the synthesis of cell walls, which are required before division and are involved in many important regulatory processes in plants (Josè and Puigdomènech, 1993; Bocca et al., 2005). Among the DEGs, unigenes encoding proline-rich and glycine-rich proteins were found to be upregulated under G and M treatment conditions. However, under the P and N treatment conditions, these unigenes were not differentially expressed due to their involvement in the environmental stress response (Li et al., 2012; Kim et al., 2005).
Calmodulin is the most important Ca2+ receptor in plants and participates in transcriptional regulation by acting on transcription factors, especially during responses to environmental changes. In this study, CaM and CaM-related proteins such as serine-threonine kinase were upregulated under M and G conditions (Table 7). CaM and the CaM-dependent signaling system is essential for the proliferative capacity of cells (Chafouleas et al., 1982). In mammalian cells, overexpression of CaM revealed a linear relationship between CaM concentration and rate of G1 progression (Joseph and Means, 2000). In contrast, reduction of CaM levels led to the majority of cells being arrested in G2, suggesting that CaM has a critical role in mediating entry into and progression through mitosis (Keith et al., 1988). In addition, three microRNAs in A. pacificum including miR169a, miR169c, and miR169f, which target CaM-dependent kinase II (CaMKII), were found to be upregulated in the log phase (Geng et al., 2015). In A. pacificum, CaM was shown to activate CaMKII. In animals, CaMKII can promote cell proliferation and growth by facilitating replication of the centriole. At the same time, the CaMKII inhibitor can arrest cells in the G2 phase of the cell cycle. Hence, CaMKII may play a major role in regulating the cell cycle and rapid growth during the log phase in A. pacificum.
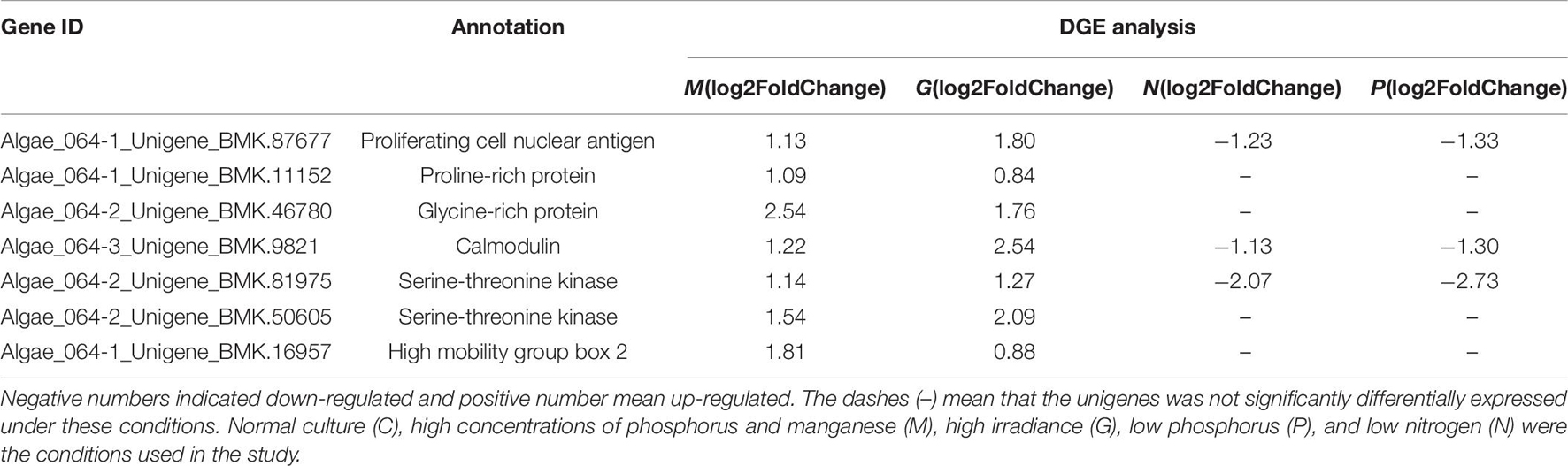
Table 7. Differentially expressed unigenes related to cell division in G, M, P, and N relative to C by DGE analysis.
The high mobility group box (HMGB) proteins are an important component of chromatin and share some functions with histone H1 (Johns, 1982). In A. pacificum, histone-like proteins such as H2A.X are rarely expressed (Hackett et al., 2004). Based on the transcriptome dataset (SRX368254), histones including all four core nucleosomal histones, histone H1, linker histone H1 and H5 family proteins, were found to be expressed at low levels (Zhang et al., 2014). However, HMGB binding affinity is much lower than H1, suggesting that HMGB may interact with the linker region only when histone H1 is absent or weakly expressed (Pallier et al., 2003). In this study, the DEGs encoding HMGB2 were upregulated under M and G conditions, suggesting that HMGB2 may play a critical role in DNA packaging. HMGB2 has been shown to be a component of mitotic chromosomes and is associated with condensed chromosomes in HeLa cells during mitosis (Scaffidi et al., 2002). Thus, HMGB2 may be associated with the condensed chromosomes of A. pacificum during the cell cycle (except DNA replication).
Nitrogen Metabolism Under the Stage of Explosive Growth
Nitrogen metabolism can be regulated by changes in the abundance of mRNA of several components involved in nitrogen uptake and nitrogen assimilation. Nitrate reductase (NR), which can catalyze NO3– reduction to NO2– is critical in nitrogen metabolism. NR activity could be induced by NO3– (Lillo, 2008). As for some research, when the algae experiences N stress, the cell upregulates the enzymes to promote translocation of external nitrogen for supplementing (Li et al., 2020). In this study, the DEGs encoding NR were down regulated under nitrogen-limiting conditions (Table 8), mainly because the external nitrogen has been used up when the cell was collected at 12th day, further resulting in the inhibition of nitrogen assimilation.
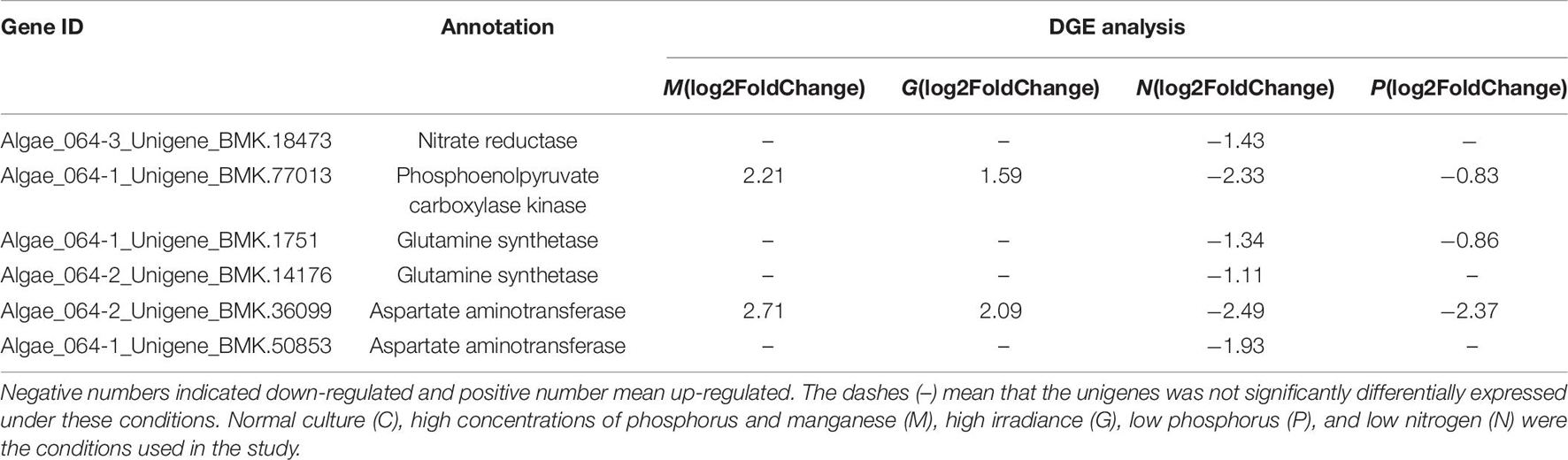
Table 8. Differentially expressed unigenes enriched in nitrogen metabolism in G, M, P, and N relative to C by DGE analysis.
Photosynthesis plays a critical role in nitrogen assimilation by providing ATP and reducing equivalents. Furthermore, large amounts of inorganic nitrogen are used in photosynthesis to produce sugars and amino acids. In C3 plants, PEPC is essential for the provision of carbon skeletons used for amino acid biosynthesis (Oaks and Hirel, 2003). The downregulation of PEPC under nitrogen-limiting conditions could lead to the inhibition of carbon assimilation and affect the efficiency of photosynthesis, which in turn would result in insufficient energy and provision of carbon skeletons for nitrogen assimilation (Champigny and Foyer, 1992).
Transcription Factors
In this study, transcription factors including bZIP, MYB, BTF-3, and EF-Tu were found. The upregulation of BTF-3 and EF-Tu was detected under M and G conditions. BTF-3 is a 27 kDa protein that forms a stable complex with RNA polymerase IIB and is required for transcriptional initiation (Kanno et al., 1992). In higher plants, gene silencing of BTF-3 caused leaf yellowing and abnormal leaf morphology without altering the overall growth of the plant (Yang et al., 2007), suggesting that BTF3 plays a vital role in tissue development. EF-Tu plays a role in protein synthesis by promoting the GTP-dependent binding of aminoacyl-tRNA to the A site of the ribosome (Riis et al., 1990). The mitochondrial EF-Tu gene was found to be differentially expressed during flower development, with the highest EF-Tu expression occurring during relatively early stages of flower development (Lee et al., 2002).
The expression of bZIP and MYB were upregulated under N and P treatment conditions. bZIP and MYB are critical for regulating defense gene expression in plant stress tolerance. In Arabidopsis, most of the transcription factor genes induced under P starvation conditions belong to the MYB superfamily (Bender and Fink, 1998). In addition, in Lotus japonicas and soybean, MYB genes were found to be upregulated under nitrogen-limiting conditions, indicating that MYB has a role in regulating flavonoid biosynthesis in response to nitrate starvation (Rubio et al., 2001; Miyake et al., 2003).
Conclusion
The variety and number of unigenes as well as the differences in gene expression patterns under distinct treatment conditions suggest that complicated and diverse regulatory mechanisms are activated under the stage of explosive growth of A. pacificum. Several common metabolic pathways are induced in A. pacificum, such as the pentose phosphate pathway and photosynthesis under eutrophication conditions and the nitrogen metabolism pathway under nutrition deficient conditions. Based on the DGE analysis, PPP and photosynthesis were activated which enhance the biosynthetic capacity and provide intermediates for cell proliferation. In addition, unigenes related to cell division such as PCNA, CAM, Proline-rich and glycine-rich proteins were detected which can facilitate the cell cycle and promote the growth of A. pacificum (Figure 8). This sequencing dataset and analysis may serve as a valuable resource to study the mechanisms of explosive growth of A. pacificum and to further elucidate the process of HAB.
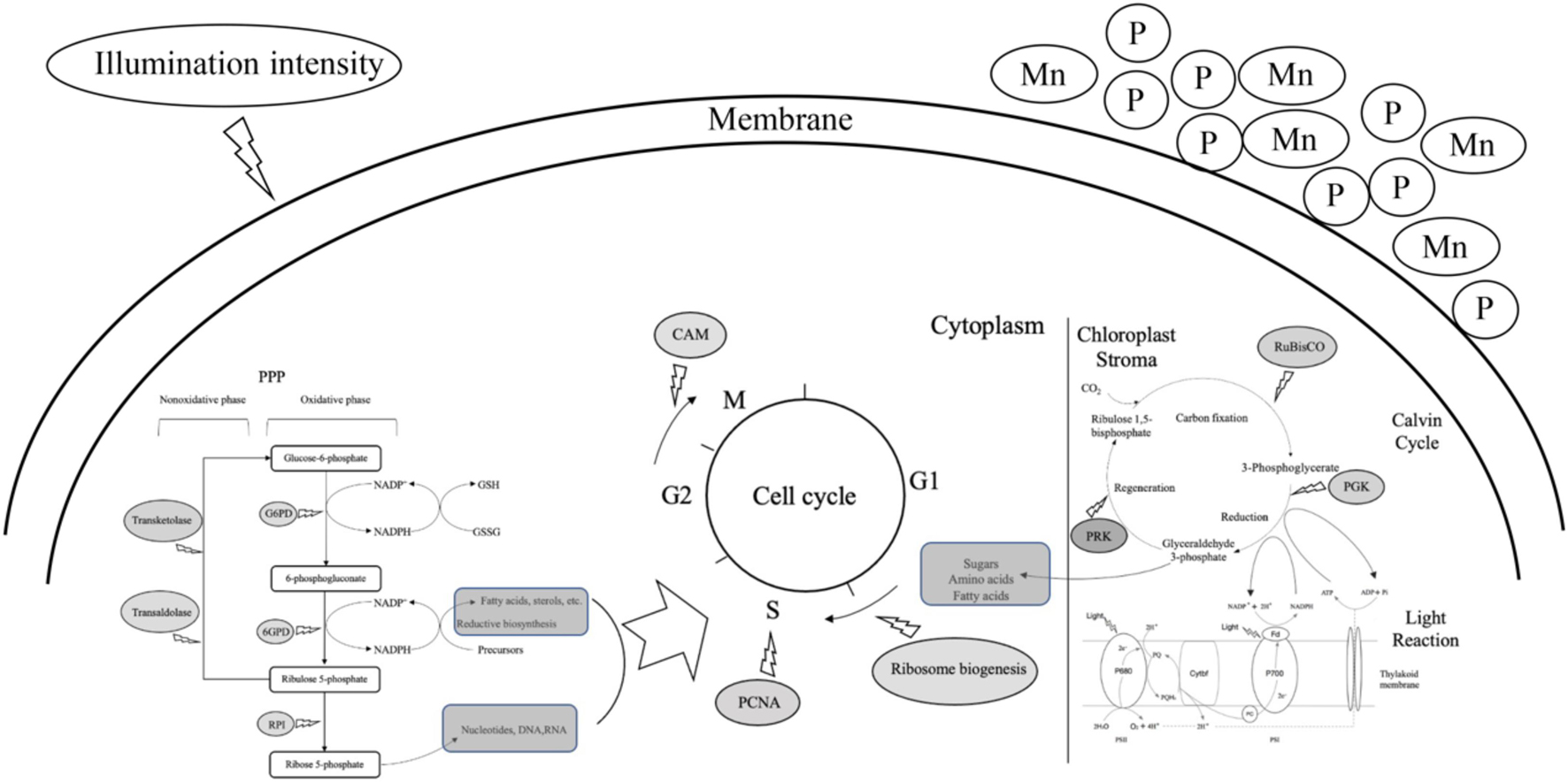
Figure 8. A predictive model of cell proliferation of Alexandrium pacificum under M, G treatment related to algal bloom. The dark gray oval represented the upregulated DEGs identified in this study. The dark gray rounded rectangle represented the intermediates provided for cell proliferation. High concentrations of phosphorus and manganese (M), high irradiance (G), were the conditions used in the study.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI (accession: PRJNA715858).
Author Contributions
YL, ZS, and SZ contributed to conception and design of the study. ES organized the database. ZN performed the statistical analysis. YL wrote the manuscript. ZZ and JQ contributed to the validation experiment. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by National Natural Science Foundation of China (Grant Nos. 41176098 and 41676091).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer FK declared a shared affiliation with several of the authors YL, ZS, ES, and SZ to the handling editor at the time of the review
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2021.751851/full#supplementary-material
Footnotes
References
Armbrust, E. V., Berges, J. A., Bowler, C., Green, B. R., Martinez, D., Putnam, N. H., et al. (2004). The genome of the diatom Thalassiosira pseudonana: ecology, evolution, and metabolism. Science 306, 79–86. doi: 10.1126/science.1101156
Baud, S., Wuillème, S., Dubreucq, B., Almeida, A. D., Vuagnat, C., Lepiniec, L., et al. (2007). Function of plastidial pyruvate kinases in seeds of Arabidopsis thaliana †. Plant J, Cell Mol. Biol. 52, 405–419. doi: 10.1111/j.1365-313X.2007.03232.x
Bender, J., and Fink, G. R. (1998). A Myb homologue, ATR1, activates tryptophan gene expression in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 95, 5655–5660. doi: 10.1073/pnas.95.10.5655
Bocca, S. N., Magioli, C., Mangeon, A., Junqueira, R. M., Cardeal, V., Margis, R., et al. (2005). Survey of glycine-rich proteins (GRPs) in the eucalyptus expressed sequence tag database (FOREST). Genet. Mol. Biol. 28, 608–624. doi: 10.1590/s1415-47572005000400016
Bräutigam, A., and Gowik, U. (2016). Photorespiration connects C3 and C4 photosynthesis. J. Exp. Bot. 67, 2953–2962.
Chafouleas, J. G., Bolton, W. E., Hidaka, H., Iii, A. E. B., and Means, A. R. (1982). Calmodulin and the cell cycle: involvement in regulation of cell-cycle progression. Cell 28, 41–50.
Champigny, M. L., and Foyer, C. (1992). Nitrate activation of cytosolic protein kinases diverts photosynthetic carbon from sucrose to amino Acid biosynthesis: basis for a new concept. Plant Physiol. 100, 7–12. doi: 10.1104/pp.100.1.7
Deberardinis, R. J., Sayed, N., Ditsworth, D., and Thompson, C. B. (2008). Brick by brick: metabolism and tumor cell growth. Curr. Opin. Genet. Dev. 18, 54–61. doi: 10.1016/j.gde.2008.02.003
Donati, G., Montanaro, L., and Derenzini, M. (2012). Ribosome biogenesis and control of cell proliferation: p53 Is not alone. Cancer Res. 72, 1602–1607. doi: 10.1158/0008-5472.can-11-3992
Geng, H., Sui, Z., Zhang, S., Du, Q., Ren, Y., Liu, Y., et al. (2015). Identification of microRNAs in the toxigenic dinoflagellate alexandrium catenella by high-throughput illumina sequencing and bioinformatic analysis. PLoS One 10:e0138709. doi: 10.1371/journal.pone.0138709
Genovesi, B., Laabir, M., Masseret, E., Collos, Y., Vaquer, A., and Grzebyk, D. (2009). Dormancy and germination features in resting cysts of Alexandrium tamarense species complex (Dinophyceae) can facilitate bloom formation in a shallow lagoon (Thau, southern France). J. Plankton Res. 31, 1209–1224. doi: 10.1093/plankt/fbp066
Glibert, P. M., and Terlizzi, D. E. (1999). Cooccurrence of elevated urea levels and dinoflagellate blooms in temperate estuarine aquaculture ponds. Appl. Environ. Microbiol. 65, 5594–5596. doi: 10.1128/AEM.65.12.5594-5596.1999
Hackett, J. D., Scheetz, T. E., Yoon, H. S., Soares, M. B., Bonaldo, M. F., Casavant, T. L., et al. (2004). Insights into a dinoflagellate genome through expressed sequence tag analysis. BMC Genomics 6:80. doi: 10.1186/1471-2164-6-80
Hallegraeff, G. M., Anderson, D. M., Cembella, A. D., and Enevoldsen, H. (2004). Manual on Harmful Marine Microalgae. Paris: UNESCO.
Hibberd, J. M., and Covshoff, S. (2010). The regulation of gene expression required for C4 photosynthesis. Annu. Rev. Plant Biol. 61, 181–207.
Holmgren, A. (1995). Thioredoxin structure and mechanism: conformational changes on oxidation of the active-site sulfhydryls to a disulfide. Structure 3, 239–243. doi: 10.1016/s0969-2126(01)00153-8
Josè, M., and Puigdomènech, P. (1993). Structure and expression of genes coding for structural proteins of the plant cell wall. New Phytol. 125, 259–282. doi: 10.1111/j.1469-8137.1993.tb03881.x
Joseph, J. D., and Means, A. R. (2000). Identification and characterization of two Ca2+/CaM-dependent protein kinases required for normal nuclear division in Aspergillus nidulans. J. Biol. Chem. 275, 38230–38238. doi: 10.1074/jbc.M006422200
Kanno, M., Chalut, C., and Egly, J. M. (1992). Genomic structure of the putative BTF3 transcription factor. Gene 117, 219–228. doi: 10.1016/0378-1119(92)90732-5
Keith, C. H., Lin, D. E., and Parsons, B. H. (1988). Inhibition of mitosis in PtK2 cells by CAPP1-calmodulin. Eur. J. Cell Biol. 47, 94–100.
Kim, Y. O., Jin, S. K., and Kang, H. (2005). Cold-inducible zinc finger-containing glycine-rich RNA-binding protein contributes to the enhancement of freezing tolerance in Arabidopsis thaliana. Plant J. 42, 890–900. doi: 10.1111/j.1365-313X.2005.02420.x
Kochetov, G. A., and Sevostyanova, I. A. (2005). Binding of the coenzyme and formation of the transketolase active center. Int. Union Biochem. Mol. Biol. Life 57, 491–497. doi: 10.1080/15216540500167203
Kroth, P. G., Chiovitti, A., Gruber, A., Martinjezequel, V., Mock, T., Parker, M. S., et al. (2008). A model for carbohydrate metabolism in the diatom Phaeodactylum tricornutum deduced from comparative whole genome analysis. PLoS One 3:e1426. doi: 10.1371/journal.pone.0001426
Kurisu, G., Zhang, H., Smith, J. L., and Cramer, W. A. (2003). Structure of the cytochrome b6f complex of oxygenic photosynthesis: tuning the cavity. Science 302, 1009–1014. doi: 10.1126/science.1090165
Laabir, M. (2011). Influence of temperature, salinity and irradiance on the growth and cell yield of the harmful red tide dinoflagellate Alexandrium catenella colonizing Mediterranean waters. J. Plankton Res. 33, 1550–1563. doi: 10.1093/plankt/fbr050
Lee, J. H., Yoon, H. S., Yun, C. H., Sim, W. S., and Kim, J. K. (2002). Molecular characterization of the mitochondrial elongation factor EF-Tu gene in rice (Oryza sativa L.). Plant Cell Rep. 21, 157–165. doi: 10.1007/s00299-002-0469-5
Lempiäinen, H., and Shore, D. (2009). Growth control and ribosome biogenesis. Curr. Opin. Cell Biol. 21, 855–863. doi: 10.1016/j.ceb.2009.09.002
Levine, A. J., and Puzio-Kuter, A. M. (2010). The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 330, 1340–1344. doi: 10.1126/science.1193494
Li, H., Li, L., Yu, L., Yang, X., and Lin, S. (2020). Transcriptome profiling reveals versatile dissolved organic nitrogen utilization, mixotrophy, and N conservation in the dinoflagellate Prorocentrum shikokuense under N deficiency. Sci. Total Environ. 763:143013. doi: 10.1016/j.scitotenv.2020.143013
Li, H.-M., Tang, H.-J., Shi, X.-Y., Zhang, C.-S., and Wang, X.-L. (2014). Increased nutrient loads from the Changjiang (Yangtze) River have led to increased harmful algal blooms. Harmful Algae 39, 92–101. doi: 10.1016/j.hal.2014.07.002
Li, L., Zhang, C., Xu, D., Schläppi, M., and Xu, Z. Q. (2012). Expression of recombinant EARLI1, a hybrid proline-rich protein of Arabidopsis, in Escherichia coli and its inhibition effect to the growth of fungal pathogens and Saccharomyces cerevisiae. Gene 506, 50–61. doi: 10.1016/j.gene.2012.06.070
Lillo, C. (2008). Signalling cascades integrating light-enhanced nitrate metabolism. Biochem. J. 415, 11–19. doi: 10.1042/BJ20081115
Lin, S. (2006). The smallest dinoflagellate genome is yet to be found: a comment on lajeunesse et al.“symbiodinium (Pyrrhophyta) genome sizes (DNA content) are smallest among dinoflagellates” 1. J. Phycol. 42, 746–748. doi: 10.1111/j.1529-8817.2006.00213.x
Liu, Y., Sui, Z., Zhang, S., Ma, J., and Zhong, J. (2019). Isolation and expression analysis of growth-related genes at different growth stages of dinoflagellate Alexandrium pacificum. J. Ocean Univ. China, 18, 710–718. doi: 10.1007/s11802-019-3849-y
Miclet, E., Stoven, V., Michels, P., Opperdoes, F., Lallemand, J., and Duffieux, F. (2001). NMR spectroscopic analysis of the first two steps of the pentose-phosphate pathway elucidates the role of 6-phosphogluconolactonase. J. Biol. Chem. 276, 34840–34846. doi: 10.1074/jbc.M105174200
Miyake, K., Ito, T., Senda, M., Ishikawa, R., Harada, T., Niizeki, M., et al. (2003). Isolation of a subfamily of genes for R2R3-MYB transcription factors showing up-regulated expression under nitrogen nutrient-limited conditions. Plant Mol. Biol. 53, 237–245. doi: 10.1023/B:PLAN.0000009296.91149.34
Murakami, R., Ifuku, K., Takabayashi, A., Shikanai, T., Endo, T., and Sato, F. (2002). Characterization of an Arabidopsis thaliana mutant with impaired psb O, one of two genes encoding extrinsic 33-kDa proteins in photosystem II. FEBS Lett. 523, 138–142. doi: 10.1016/s0014-5793(02)02963-0
Oaks, A., and Hirel, B. (2003). Nitrogen metabolism in roots. Annu. Rev. Plant Physiol. 36, 345–365. doi: 10.1146/annurev.pp.36.060185.002021
Pallier, C., Scaffidi, P., Chopineau-Proust, S., Agresti, A., Nordmann, P., Bianchi, M. E., et al. (2003). Association of chromatin proteins high mobility group box (HMGB) 1 and HMGB2 with mitotic chromosomes. Mol. Biol. Cell 14, 3414–3426. doi: 10.1091/mbc.e02-09-0581
Parsley, K., and Hibberd, J. M. (2006). The Arabidopsis PPDK gene is transcribed from two promoters to produce differentially expressed transcripts responsible for cytosolic and plastidic proteins. Plant Mol. Biol. 62, 339–349. doi: 10.1007/s11103-006-9023-0
Paul, A., and Hauck, M. (2006). Effects of manganese on chlorophyll fluorescence in epiphytic cyano- and chlorolichens. Flora 201, 451–460. doi: 10.1016/j.flora.2005.09.001
Riis, B., Rattan, S. I. S., Clark, B. F. C., and Merrick, W. C. (1990). Eukaryotic protein elongation factors. Trends Biochem. Sci. 15, 420–424. doi: 10.1016/0968-0004(90)90279-k
Robinson, M., and Oshlack, A. (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, 1–9.
Rubio, V., Linhares, F., Solano, R., Martín, A. C., Iglesias, J., Leyva, A., et al. (2001). A conserved MYB transcription factor involved in phosphate starvation signaling both in vascular plants and in unicellular algae. Genes Dev. 15, 2122–2133. doi: 10.1101/gad.204401
Ruelland, E., and Miginiac-Maslow, M. (1999). Regulation of chloroplast enzyme activities by thioredoxins: activation or relief from inhibition? Trends Plant Sci. 4, 136–141. doi: 10.1016/s1360-1385(99)01391-6
Samland, A. K., and Sprenger, G. A. (2009). Transaldolase: from biochemistry to human disease. Int. J. Biochem. Cell Biol. 41, 1482–1494.
Scaffidi, P., Misteli, T., and Bianchi, M. E. (2002). Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195. doi: 10.1038/nature00858
Schürmann, P. (2003). Redox signaling in the chloroplast: the ferredoxin/thioredoxin system. Antioxid. Redox Signal. 5, 69–78. doi: 10.1089/152308603321223559
Shen, J. R., Qian, M., Inoue, Y., and Burnap, R. L. (1998). Functional characterization of Synechocystis sp. PCC 6803 delta psbU and delta psbV mutants reveals important roles of cytochrome c-550 in cyanobacterial oxygen evolution. Biochemistry 37, 1551–1558. doi: 10.1021/bi971676i
Slekar, K. H., Kosman, D. J., and Culotta, V. C. (1996). The yeast copper/zinc superoxide dismutase and the pentose phosphate pathway play overlapping roles in oxidative stress protection. J. Biol. Chem. 271, 28831–28836. doi: 10.1074/jbc.271.46.28831
Toulza, E., Shin, M. S., Blanc, G., Audic, S., Laabir, M., Collos, Y., et al. (2010). Gene expression in proliferating cells of the dinoflagellate Alexandrium catenella (Dinophyceae). Appl. Environ. Microbiol. 76, 4521–4529. doi: 10.1128/AEM.02345-09
Tsuji, Y., Suzuki, I., and Shiraiwa, Y. (2009). Photosynthetic carbon assimilation in the coccolithophorid Emiliania huxleyi (Haptophyta) : evidence for the predominant operation of the C_3 cycle and the contribution of β-carboxylases to the active anaplerotic reaction. Plant Cell Physiol. 50, 318–329. doi: 10.1093/pcp/pcn200
Uribe, P., Fuentes, D., Valdés, J., Shmaryahu, A., Zúñiga, A., Holmes, D., et al. (2008). Preparation and analysis of an expressed sequence tag library from the toxic dinoflagellate Alexandrium catenella. Mar. Biotechnol. 10, 692–700. doi: 10.1007/s10126-008-9107-8
Wang, B., Xin, M., Wei, Q., and Xie, L. (2018). A historical overview of coastal eutrophication in the China Seas. Mar. Pollut. Bull. 136, 394–400. doi: 10.1016/j.marpolbul.2018.09.044
Wang, D.-Z., and Hsieh, D. P. (2005). Growth and toxin production in batch cultures of a marine dinoflagellate Alexandrium tamarense HK9301 isolated from the South China Sea. Harmful Algae 4, 401–410. doi: 10.1016/j.hal.2004.07.002
Wang, L., Peterson, R. B., and Brutnell, T. P. (2011). Regulatory mechanisms underlying C 4 photosynthesis. New Phytol. 190, 9–20. doi: 10.1111/j.1469-8137.2011.03649.x
Wang, Z. W., Ren, J. L., Zhang, R. F., Xu, D. F., and Wu, Y. (2019). Physical and biological controls of dissolved manganese on the northern slope of the South China Sea. Deep Sea Res. 167, 25–33. doi: 10.1016/j.dsr2.2018.07.006
Xu, J., Xiao, F., Zhang, X., Xu, D., Mou, S., Cao, S., et al. (2012). Evidence of coexistence of C3 and C4 photosynthetic pathways in a green-tide-forming alga, Ulva prolifera. PLoS One 7:410. doi: 10.1371/journal.pone.0037438
Yang, K. S., Kim, H. S., Jin, U. H., Lee, S. S., Park, J. A., Lim, Y. P., et al. (2007). Silencing of NbBTF3 results in developmental defects and disturbed gene expression in chloroplasts and mitochondria of higher plants. Planta 225, 1459–1469. doi: 10.1007/s00425-006-0453-3
Yanjun, S., Hanhua, H. U., Runyu, M. A., Wei, C., and Zhaoling, C. (2003). Nitrogen and phosphorus absorption and growth characteristics of Alexandrium tamarense. Chinese J. Appl. Ecol. 14, 1143–1146.
Zhang, Q. C., Song, J. J., Yu, R. C., Tian, Y., Wang, Y. F., Kong, F. Z., et al. (2013). Roles of mixotrophy in blooms of different dinoflagellates: implications from the growth experiment. Harmful Algae 30, 10–26. doi: 10.1111/evo.12029
Zhang, S., Sui, Z., Chang, L., Kang, K., Ma, J., Kong, F., et al. (2014). Transcriptome de novo assembly sequencing and analysis of the toxic dinoflagellate Alexandrium catenella using the Illumina platform. Gene 537, 285–293. doi: 10.1016/j.gene.2013.12.041
Zhang, Y. J., Yu, C., Wang, Z. H., Han, B. P., and Yang, Y. F. (2006). Effects of limitation of nitrogen and phosphorus on the growth of Alexandrium tamarense. J. Trop. Subtrop. Bot. 13, 21–24.
Keywords: A. pacificum, DGE, explosive growth, pentose phosphate pathway, photosynthesis, cell cycle
Citation: Liu Y, Zhu Z, Qi J, Sui Z, Shang E, Zhang S and Niaz Z (2021) Comparative Transcriptome Profiling Reveals Insights Into the Mechanisms Related to Explosive Growth of Alexandrium pacificum. Front. Mar. Sci. 8:751851. doi: 10.3389/fmars.2021.751851
Received: 02 August 2021; Accepted: 15 October 2021;
Published: 03 November 2021.
Edited by:
Taewoo Ryu, Okinawa Institute of Science and Technology Graduate University, JapanReviewed by:
Fanna Kong, Ocean University of China, ChinaJianhua Fan, East China University of Science and Technology, China
Ting Xue, Fujian Normal University, China
Yiqin Deng, Chinese Academy of Fishery Sciences (CAFS), China
Xupeng Cao, Dalian Institute of Chemical Physics, Chinese Academy of Sciences (CAS), China
Copyright © 2021 Liu, Zhu, Qi, Sui, Shang, Zhang and Niaz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenghong Sui, c3VpemhlbmdoQG91Yy5lZHUuY24=