 Ziyan He1,2,3,4
Ziyan He1,2,3,4 Yang Chen1,2,3,4
Yang Chen1,2,3,4 Yichao Wang1,2,3,4
Yichao Wang1,2,3,4 Kuiyan Liu1,2,3,4
Kuiyan Liu1,2,3,4 Qing Xu5Yang Li6
Qing Xu5Yang Li6 Nansheng Chen1,2,4,7*
Nansheng Chen1,2,4,7*- 1Chinese Academy of Sciences (CAS) Key Laboratory of Marine Ecology and Environmental Sciences, Institute of Oceanology, Chinese Academy of Sciences (CAS), Qingdao, China
- 2Laboratory of Marine Ecology and Environmental Science, Qingdao National Laboratory for Marine Science and Technology, Qingdao, China
- 3 College of Marine Science, University of Chinese Academy of Sciences (CAS), Beijing, China
- 4Center for Ocean Mega-Science, Chinese Academy of Sciences (CAS), Qingdao, China
- 5College of Life Science and Technology, Huazhong Agricultural University, Wuhan, China
- 6Guangdong Provincial Key Laboratory of Healthy and Safe Aquaculture, College of Life Science, South China Normal University, Guangzhou, China
- 7Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, BC, Canada
Pseudo-nitzschia is a species-rich genus where many species can induce harmful algae blooms (HABs) associated with the toxin domoic acid (DA) production. Despite the importance of Pseudo-nitzschia species to coastal environments, their genomic information is rather limited, hindering research on biodiversity and evolutionary analysis. In this study, we report full-length chloroplast genomes (cpDNAs) of nine Pseudo‐nitzschia, among which cpDNAs of eight Pseudo-nitzschia species were reported for the first time. The sizes of these Pseudo-nitzschia cpDNAs, which showed typical quadripartite structures, varied substantially, ranging from 116,546 bp to 158,840 bp in size. Comparative analysis revealed the loss of photosynthesis-related gene psaE in cpDNAs of all Pseudo-nitzschia species except that of P. americana, and the selective loss of rpl36 in P. hainanensis. Phylogenetic analysis showed that all Pseudo-nitzschia strains were grouped into two clades, with clade 1 containing cpDNAs of P. multiseries, P. pungens, P. multistriata, and P. americana, and clade 2 containing cpDNAs of P. hainanensis, P. cuspidata, Pseudo-nitzschia sp. CNS00097, P. delicatissima, and P. micropora. The small size of the P. americana cpDNA was primarily due to its shortened inverted repeat (IR) regions. While psaA and psaB were found in the IR regions of cpDNAs of other eight species, these two genes were found outside of the IR regions of P. americana cpDNA. In contrast, P. hainanensis had the largest size because of expansion of IR regions with each IR region containing 15 protein-coding genes (PCGs). Eleven genetic regions of these Pseudo-nitzschia cpDNAs exhibited high nucleotide diversity (Pi) values, suggesting that these regions may be used as molecular markers for distinguishing different Pseudo-nitzschia species with high resolution and high specificity. Phylogenetic analysis of the divergence of nine Pseudo-nitzschia species indicated that these species appeared at approximately 41 Mya. This study provides critical cpDNA resources for future research on the biodiversity and speciation of Pseudo-nitzschia species.
Introduction
The Bacillariophyta (commonly known as diatoms) represents a diverse group of unicellular eukaryotes found in almost all freshwater and marine habitats (Seckbach and Kociolek, 2011), forming an important part of the basal aquatic food webs (Falkowski and Knoll, 2007). They have significant ecological importance in the carbon and silicate cycles, accounting for approximately 20% of the global photosynthetic carbon fixation (Field et al., 1998). Diatoms are also vital in evolutionary and archeological researches because they are frequently found in subfossil and fossil records because they are silicified microorganisms and their silica shells are resistant to decay (Mann et al., 2017).
Pseudo-nitzschia is a species-rich genus widely distributed in polar, temperate, subtropical and tropical seas, many of which can induce harmful algae blooms (HABs) in coastal and oceanic waters and produce domoic acid (DA), a neurotoxin causing amnesic shellfish poisoning (ASP) (Lelong et al., 2012; Bates et al., 2018). During toxic Pseudo‐nitzschia blooms, DA can be channeled through the food web, causing serious environmental toxicologic threats and significant exposure risks on marine lives and human health (Saeed et al., 2017). Accumulating evidences suggests that Pseudo-nitzschia blooms can occur in many coastal environments (McCabe et al., 2016; Clark et al., 2019; Ajani et al., 2020; Stonik, 2021). As such, a large number of studies have been conducted on Pseudo‐nitzschia, exploring morphology, life history, taxonomy, ecology, toxicity, and physiology (Lelong et al., 2012; Trainer et al., 2012; Bates et al., 2018). To date, 57 Pseudo‐nitzschia species have been described (Guiry and Guiry, 2021), among which 26 species have been found to produce DA (Bates et al., 2018). In the Bohai Sea, the Yellow Sea, the East China Sea, and the South China Sea, 37 Pseudo‐nitzschia taxa have been reported, among which DA has been detected in nine species. (Li et al., 2010; Lu et al., 2012; Li et al., 2017a; Li et al., 2018; Huang et al., 2019; Dong et al., 2020a; Chen et al., 2021).
Due to the high similarity of morphological characters of closely related Pseudo-nitzschia species, morphological characters are often inadequate for distinguishing different Pseudo-nitzschia species (Lelong et al., 2012; Trainer et al., 2012; Bates et al., 2018). The application of molecular markers greatly improved the resolution of Pseudo-nitzschia species (Trainer et al., 2012; Amato et al., 2019). For example, cryptic Pseudo-nitzschia species P. arenysensis and P. dolorosa were successfully separated from the P. delicatissima complex based on comparative analysis of molecular markers including ITS1, 5.8S rDNA, and ITS2 regions (Lundholm et al., 2006; Quijano-Scheggia et al., 2009). However, many common molecular markers (LSU, rbcL, and 18S rDNA) cannot effectively distinguish different Pseudo-nitzschia species due to their limited resolution (Lundholm et al., 2012; Lim et al., 2013; Lim et al., 2016). Other molecular markers including ITS1, 5.8S rDNA, ITS2 regions, and cox1 also have their limitations (Lim et al., 2013; Yuan et al., 2016; Lim et al., 2018).
The chloroplast genomes (cpDNAs) are composed largely of single copy genes, with limited horizontal gene transfer events (Ruck et al., 2014), and cpDNA protein-coding genes (PCGs) are also readily aligned across a wide range of diatoms (Theriot et al., 2015), which facilitate phylogenomic research. Furthermore, cpDNAs can be applied in species identification, and be exploited in developing high-resolution molecular markers, tracking patterns of gene loss, exploring adaptive changes that optimize photosynthesis, addressing questions concerning plastid inheritance and recombination, and synthetic biology (Tonti-Filippini et al., 2017; Shi et al., 2019; Song et al., 2020). Chloroplast genomes have been demonstrated to be valuable for evolutionary analyses even at the family or the genus level (Dong et al., 2020b; Li et al., 2020; Sun et al., 2020). However, to date, only a single cpDNA has been constructed for the entire genus Pseudo-nitzschia (Cao et al., 2016).
Here, we report complete cpDNAs of nine Pseudo‐nitzschia species, among which cpDNAs of eight Pseudo-nitzschia species were reported for the first time. The aim of this study was to ascertain the conservation and diversity of Pseudo‐nitzschia cpDNAs through comparative genomic approaches, and to gain insight into the evolution of Pseudo‐nitzschia species.
Materials and Methods
Sampling, Isolation, Culture Conditions, and Species Identification
Putative Pseudo-nitzschia cells were isolated using micropipette and incubated in L1 seawater culture medium at temperature of 18–20°C, with an irradiance of 30 µmol photons m−2 s−1 and a photoperiod of 12/12 h light/dark. Nine Pseudo-nitzschia strains analyzed in this study were isolated from water samples collected in the Bohai Sea (strains CNS00141, CNS00142, and CNS00159) and the Yellow Sea (strain CNS00130) onboard the research vessel “Beidou” supported by the National Natural Science Foundation of China, Bohai and Yellow Sea Oceanography Expedition (NORC2019-01), the Jiaozhou Bay (strains CNS00133 and CNS00138) onboard the research vessel “Chuangxin” operated by the Jiaozhou Bay Marine Ecosystem Research Station, the East China Sea (strain CNS00150) onboard on the research vessel “Zheyu 2” supported by the Natural Science Foundation of China (NSFC), and the Western Pacific (strains CNS00090 and CNS00097) onboard the research vessel “Kexue” (Figure 1A; Table 1).

Figure 1 Collection localities of nine Pseudo-nitzschia strains (A). Micrographs of Pseudo-nitzschia sp. CNS00097 (B), P. delicatissima CNS00130 (C), P. americana CNS00138 (D), P. pungens CNS00141 (E), P. multistriata CNS00142 (F), and P. multiseries CNS00159 (G). Phylogenetic analysis based on 18S ribosomal DNA (18S rDNA) gene (H). Numbers at the branches represent bootstrap values. Branch lengths are proportional to the genetic distances, which are indicated by the scale bar.
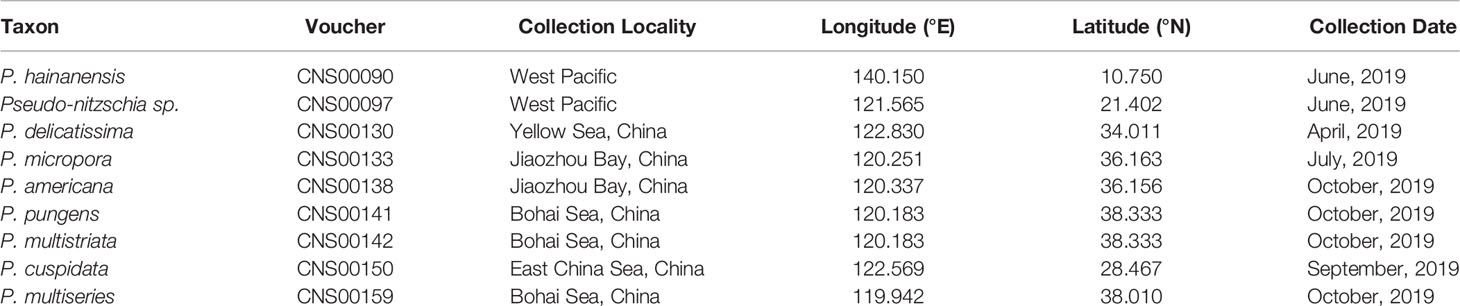
Table 1 Collection locality and date of nine Pseudo-nitzschia strains.
All strains isolated and studied in this project were deposited at the KLMEES of IOCAS (Nansheng Chen,Y2hlbm5AcWRpby5hYy5jbg==). Morphological features of cells were observed by a ZEISS IMAGER A2 microscope (Carl Zeiss AG, Oberkochen, Germany) equipped with differential interference contrast optics. Species were identified based on their morphological features and the similarity of molecular markers to reference molecular markers of known Pseudo-nitzschia species (Table 2).
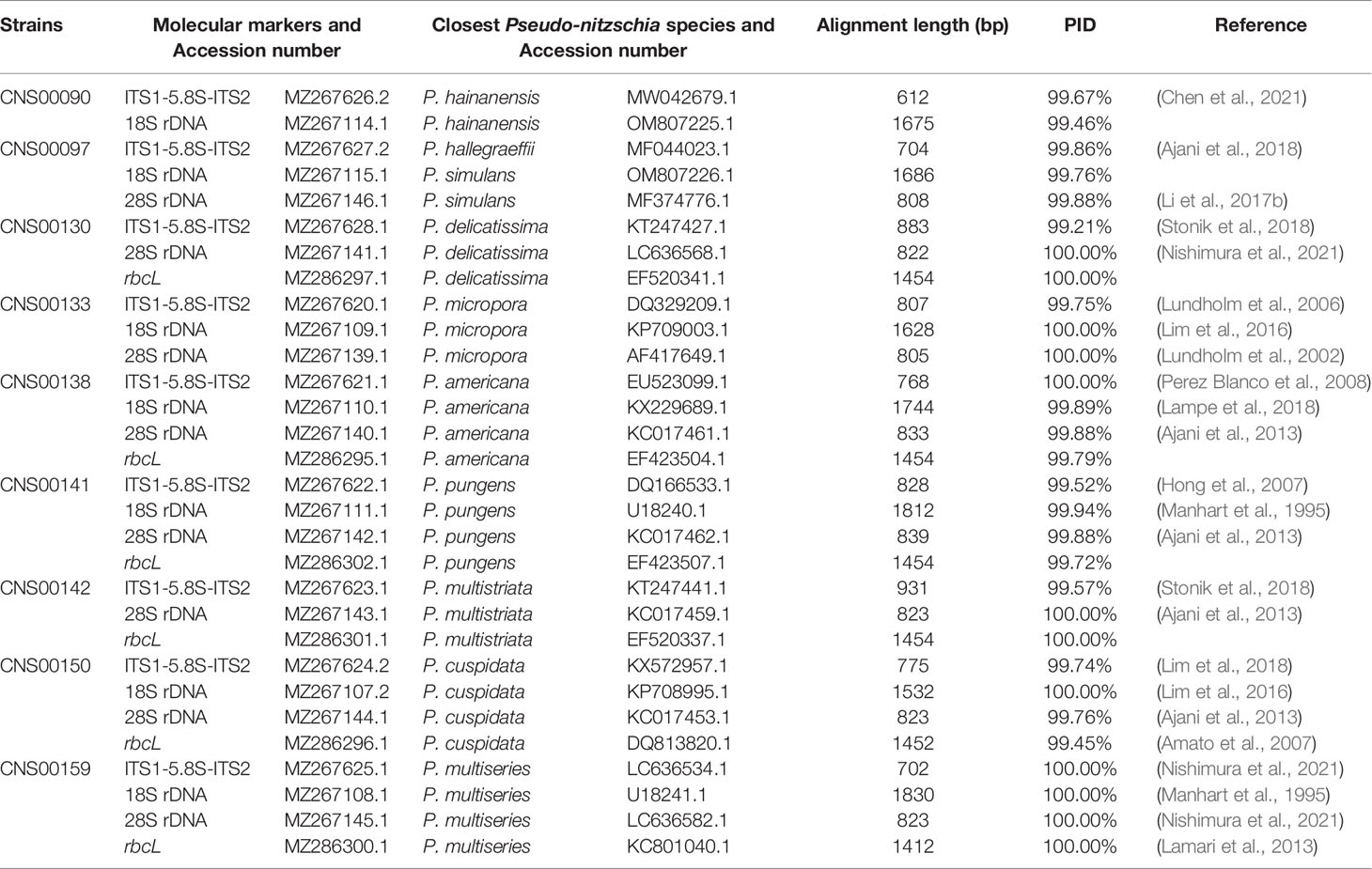
Table 2 Species identification of nine strains based on multiple molecular markers.
DNA Extraction, Sequencing, Molecular Identification, Genome Assembly, and Annotation
DNA samples of nine candidate Pseudo-nitzschia strains were prepared using the modified CTAB method (Doyle and Doyle, 1987), which were used to generate paired-end sequencing libraries of 350 bp in size. Genomic DNAs were sequenced using the Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) at Novogene (Beijing, China). Raw data of 3.78 – 7.33 Gb were generated for each strain with 150 bp paired-end read lengths. Low-quality reads and adapters were removed from the raw data using Trimmomatic (Bolger et al., 2014). Genome size estimation was conducted using Jellyfish (Marcais and Kingsford, 2011) and GenomeScope (Vurture et al., 2017) with k-mer 17. Genome sizes of nine Pseudo-nitzschia strains were estimated to be ranging from 36.6 M (strain CNS00130 and CNS00133) to 252.8 M (strain CNS00159) (Table S1). 1,000,000 clean reads were randomly selected for each strain for Basic Local Alignment Search Tool (BLAST) (Camacho et al., 2009) search against the National Center for Biotechnology Information (NCBI) NT database for estimating bacterial contamination. Bacterial contamination was negligible (< 0.5%) in the DNA samples of all strains (including CNS00090, CNS00130, CNS00133, CNS00141, CNS00142, CNS00150, and CNS00159), except the strains CNS00097 and CNS00138, which contained 58.78% and 7.24% bacterial contamination, respectively (Table S1). Nuclear genome assemblies of nine strains were assembled using SPAdes (Bankevich et al., 2012) using clean data. Genome sequencing depth was estimated based on the base of clean data and nuclear genome size, considering bacterial contamination (Table S1).
Molecular markers including full-length ITS1-5.8S-ITS2, 18S rDNA, 28S rDNA D1-D3, and rbcL were assembled with SPAdes (Bankevich et al., 2012). Quality assessment was done by aligning paired-end reads against each assembled molecular marker using BWA v0.7.17 (Li and Durbin, 2010), and inspected using IGV v2.8.12 (Robinson et al., 2011). The ITS2 regions were identified according to the method described in a previous study (Ajani et al., 2018), using ITS2 sequences of Pseudo-nitzschia dolorosa strains BP3 and 300 (GenBank accession numbers DQ336151 and DQ336153 respectively) as references. The annotation of Pseudo-nitzschia strains were primarily based on ITS1-5.8S-ITS2 sequences and ITS2 sequences and structures (if necessary). The assembled ITS1-5.8S-ITS2 sequence for each strain was used as a query to search the NCBI NT database using BLAST for the target sequence with the highest bitscore (and percentage ≥ 99%). A Pseudo-nitzschia species was annotated as the species from which the reference ITS1-5.8S-ITS2 sequence was supported by publications (Table 2). This annotation was further validated by examining the ITS2 sequences and structures (if necessary), with the focus on the compensatory base changes (CBCs), which was an important indicator for species identification of Pseudo-nitzschia (Li et al., 2017b; Ajani et al., 2018; Chen et al., 2021). Furthermore, the annotation of a Pseudo-nitzschia strain was also checked by examining other molecular markers (including 18S rDNA, 28S rDNA D1-D3, and rbcL) of this strain for consistency (Table 2).
The maximum likelihood (ML) phylogenetic trees of molecular markers were constructed using MEGA7 with 1000 bootstrap replicates (Kumar et al., 2016). Bootstrap values were shown next to the branches (Felsenstein, 1985). The best-fit models were Hasegawa-Kishino-Yano model (HKY + G + I), Kimura 2-parameter model (K2 + G), and General Time Reversible model (GTR + G) for 18S rDNA, 28S rDNA D1-D3, and rbcL, respectively.
Each cpDNA was de novo assembled using GetOrganelle (Jin et al., 2020), which in turn used SPAdes (Bankevich et al., 2012) for assembly, Bowtie2 (Langmead and Salzberg, 2012) for alignment, and BLAST+ (Camacho et al., 2009) for searches. The paths of the cpDNA were viewed using Bandage version 0.8.1 (Wick et al., 2015). Subsequently, complete cpDNAs were examined by aligning sequencing reads against the cpDNAs using the MEM algorithm of BWA v0.7.17 (Li and Durbin, 2010). Alignments were visualized using IGV v2.8.12 (Robinson et al., 2011). Meanwhile, the sequencing depth of cpDNAs were also calculated. ORF finder (https://www.ncbi.nlm.nih.gov/orffinder) and MFannot (https://megasun.bch.umontreal.ca/RNAweasel/) were used to annotate the cpDNAs. The annotated cpDNA sequences were submitted to GenBank under accession numbers MW853965 (P. hainanensis CNS00090), MW853966 (Pseudo-nitzschia sp. CNS00097), MW715816 (P. delicatissima CNS00130), MW722940 (P. micropora CNS00133), MW722941 (P. americana CNS00138), MW722942 (P. pungens CNS00141), MW722943 (P. multistriata CNS00142), MW722944 (P. cuspidata CNS00150), MW722945 (P. multiseries CNS00159). Gene maps of the annotated Pseudo-nitzschia cpDNAs were drawn using the online program OGDRAW (Greiner et al., 2019).
Because psaE was not found in the cpDNAs of eight Pseudo-nitzschia species constructed in this study, alignment of bas1-ftsH regions of nine Pseudo-nitzschia strains was constructed using MEGA7 (Kumar et al., 2016) to examine the gene losses from the cpDNAs. Because the gene psaE could have been transferred from cpDNAs to their corresponding nuclear genomes via endosymbiotic gene transfer (EGT) (Lommer et al., 2010), to ascertain this possibility, we searched for psaE in the assembled genomes of all eight Pseudo-nitzschia strains whose psaE genes were missing using psaE protein sequence of P. americana (CNS00138) as the query using BLAST+ (Camacho et al., 2009). We also searched for potential psaE in Pseudo-nitzschia genomes and transcriptomes downloaded from NCBI (Table S2). This method was successfully applied previously to identify endosymbiotic gene transfer cases in other diatom species (Liu et al., 2021b). To further verify that the loss of the psaE gene was not due to miss assemblies of the genomes and transcriptomes, we PCR amplified an internal segment of psaE (150 bp) by designing the following PCR primers (F: ACTAATTCATCTAAAGCAA; R: TCGTATTCTTAGAAAAG) based on the alignment of psaE genes of P. americana and other diatom species including Nitzschia ovalis (OK505007), Skeletonema tropicum (MW679507), Thalassiosira nordenskioeldii (MW592698). PCR assays were carried out using genomic DNAs of Nitzschia ovalis, Skeletonema tropicum, Thalassiosira nordenskioeldii, and seven Pseudo-nitzschia strains including CNS00130, CNS00133, CNS00138, CNS00141, CNS00142, CNS00150, and CNS00159 as templates. PCR amplification conditions included an initial denaturation at 94°C for 4 min, followed by 34 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 15 s, elongation at 72°C for 15 s, and a final extension at 72°C for 5 min. To verify the quality of all extracted DNA samples, primers DPrbcL1 (AAGGAGAAATHAATGTCT) and DPrbcL7 (AARCAACCTTGTGTAAGTCTC) (Daugbjerg and Andersen, 1997) were used for the amplification of rbcL gene in all DNA samples. PCR amplification conditions for rbcL gene included an initial denaturation at 94°C for 4 min, followed by 34 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, elongation at 72°C for 1.5 min, and a final extension at 72°C for 5 min.
Phylogenetic Analysis and Intergenic Region Analysis
A total of 95 PCGs including atpA; atpB; atpD; atpE; atpF; atpG; atpH; atpI; cbbx; ccs1; ccsA; chlI; clpC; dnaB; ftsH; groEL; lysR; petA; petB; petD; petG; petL; petM; petN; psaA; psaB; psaD; psaF; psaJ; psaL; psbB; psbC; psbD; psbE; psbF; psbH; psbI; psbJ; psbK; psbL; psbN; psbT; psbV; psbX; psbY; psbZ; rbcL; rbcS; rpl1, 2, 3, 4, 5, 6, 11, 12, 13, 14, 16, 18, 19, 20, 23, 24, 29, 31, 32, 34, 35; rpoA; rpoB; rpoC1; rpoC2; rps2, 3, 4, 5, 7, 9, 10, 11, 12, 13, 14, 16, 17, 18, 20; secA; secG; secY; sufB; sufC; tatC; ycf3, which were shared among 65 cpDNAs, including 55 previously published Bacillariophyta cpDNAs (Accession number were included in Table S3), nine Pseudo-nitzschia cpDNAs constructed in this study, and Triparma laevis (AP014625) (an Ochrophyta cpDNA used as an outgroup taxa), were used for phylogenetic analysis. The amino acid sequences of each of the 95 PCGs were individually aligned using MAFFT with default parameters (Katoh and Standley, 2013). The regions that were ambiguously aligned in each alignment were deleted using trimAl 1.2rev59 (Capella-Gutierrez et al., 2009) with the parameters gt = 1, and all amino acid sequences were concatenated using Phyutility (Smith and Dunn, 2008). Phylogenetic trees were constructed with IQ-TREE using default parameters (Trifinopoulos et al., 2016). Ultrafast bootstrap analysis with 1000 replicates of the dataset and approximate Bayes test was performed to estimate statistical reliability (Anisimova et al., 2011; Minh et al., 2013). Annotation information noted in the phylogenetic tree was based on Algaebase (Guiry and Guiry, 2021). In addition, a consensus phylogenetic tree was constructed refer to previous studies (Huang et al., 2015; Garrison et al., 2016), ASTRAL (Mirarab et al., 2014) was used for phylogenetic analysis under default settings based on ML trees of 95 shared PCGs constructed by RAxML (Stamatakis, 2014).
The program TOPD-FMTS version 4.6 (Puigbo et al., 2007) was used to compare the similarity of two trees constructed by IQ-TREE and ASTRAL using two different approaches: splits and disagree from the program with 100 repetitions. Phylogenetic trees of cpDNAs (constructed by IQ-TREE), 18S rDNA, 28S rDNA D1-D3, and rbcL were also analyzed by TOPD-FMTS version 4.6 (Puigbo et al., 2007).
Synteny Analysis and IR Regions Analysis
Synteny analysis of 10 Pseudo-nitzschia cpDNAs was executed using Mauve v2.3.1 using progressive Mauve with default parameters (Darling et al., 2010). The comparative view of representative cpDNAs was performed using circos-0.69 (Krzywinski et al., 2009). The arrangements of genes in nine Pseudo-nitzschia cpDNAs inverted repeat (IR) region were displayed using OGDRAW (Greiner et al., 2019). IRscope (Amiryousefi et al., 2018) was used for the analyses of IR region contraction and expansion at the junctions of cpDNAs.
Comparative cpDNA Analysis and Divergence Hotspots
Ka/Ks rates were calculated using KaKs_Calculator2.0 (Wang et al., 2010) based on 120 protein-coding gene sequences from 10 Pseudo-nitzschia strains. The nucleotide diversity (Pi) values of Pseudo-nitzschia were evaluated by Perl script. Primer 5 was used to design molecular markers of ycf89 (F: ATGRGTTTARATGAWAA R: KRTCATTTGGAATWGGA) and the ML phylogenetic tree of target sequences of ycf89 was constructed by the method mentioned above.
Divergence Time Analysis
MCMCTree in PAML (Yang, 1997) was used to perform Bayesian estimation of species divergence times, based on the 109 PCGs shared by Ectocarpus siliculosus (NC_013498), Proboscia sp. (MG755791), Coscinodiscus radiatus (KC509521), Rhizosolenia setigera (MG755793), Thalassiosira pseudonana (EF067921), Chaetoceros muellerii (MW004650), Attheya longicornis (MG755798), Phaeodactylum tricornutum (EF067920), Fragilariopsis kerguelensis (LR812620), and 10 Pseudo-nitzschia cpDNAs. Divergence times were calculated according to methods described previously (Matari and Blair, 2014) and fossil evidence was used to calibrate the molecular clock analyses (Medlin, 2015). Fossil evidence from Late Cretaceous (Turonian) provided a minimum age of 89.8 Mya on the divergence between Rhizosolenia setigera and Coscinodiscus radiatus (5-95% quantiles = 92–118 Mya), fossil evidence from Late Cretaceous (Campanian) pennate diatoms provided a minimum age of 72.1 Mya on the divergence between Thalassiosira and Bacillariophyceae (5-95% quantiles = 74–100 Mya), and Early Jurassic (Toarcian) diatom fossils provided a minimum age of 174 Mya on the divergence between diatoms and Ectocarpus (5-95% quantiles = 176–202 Mya).
Tree topology was constrained to reflect the ML tree, and a GTR substitution model was used. The Markov chain Monte Carlo (MCMC) process of PAML mcmctree was run to sample 1, 000, 000 times, with sample frequency set to 50, after a burn-in of 500, 000 iterations.
Results
Morphological and Molecular Identification of Pseudo-nitzschia Strains
Nine putative Pseudo-nitzschia strains (CNS00141, CNS00142, CNS00159, CNS00130, CNS00133, CNS00138, CNS00150, CNS00090, and CNS00097) were first annotated based on their morphological characteristics (Hasle, 1994). Their cells were fusiform or lanceolate in shape and tapered at both ends (Figures 1B–G). In general, each cell contained two plastids symmetrically distributed on either side of the transapical axis. Because morphological features of these strains could not be used to adequately determine their taxonomical status, molecular markers constructed in this study were used to facilitate species identification. The Pseudo-nitzschia strains were first annotated using ITS (ITS1-5.8S-ITS2) sequences (Table 2), and the ITS2 regions of these strains differed by at most one base compared to their reference sequences (Table S4), suggesting that there were no compensatory base changes (CBCs), confirming the ITS-based annotation of the Pseudo-nitzschia strains. ITS-based annotation of the Pseudo-nitzschia strains was supported by all other molecular markers including 18S rDNA, 28S rDNA D1-D3, and rbcL, except the strain CNS00097, which was annotated as P. hallegraeffii based on ITS and ITS2 (Tables S4, S5). Based on 18S rDNA sequence (MZ267115) and 28S rDNA D1-D3 (MZ267146), this strain was annotated as P. simulans based on the high similarities to the reference 18S rDNA sequence (OM807226) and 28S rDNA D1-D3 (MF374776), respectively (Tables 2; Table S5), suggesting that the strain CNS00097 might actually represent an unidentified Pseudo-nitzschia species. Thus, we named it Pseudo-nitzschia sp. CNS00097 (Figure 1H; Figure S1). Phylogenetic analysis of these molecular markers including 18S rDNA (Figure 1H), 28S rDNA and rbcL (Figure S1), which were constructed primarily for strain annotation, supported the above annotation.
Construction and Comparative Analysis of Pseudo-nitzschia cpDNAs
Complete cpDNAs were constructed for nine Pseudo-nitzschia strains characterized above. Together with one cpDNA constructed for P. multiseries (KR709240) (Cao et al., 2016), ten cpDNAs corresponding to nine Pseudo-nitzschia species have been constructed altogether (Table 3). Nine newly constructed Pseudo-nitzschia cpDNAs varied substantially, ranging from 116,546 bp (P. americana) to 158,840 bp (P. hainanensis) in length (Figure 2). Interestingly, the lengths of these newly constructed cpDNAs were all substantially longer than that of the recently published cpDNA of P. multiseries, which is 111,539 bp (Cao et al., 2016). Indeed, the length of cpDNA of the P. multiseries strain CNS00159 constructed in this study (123,195 bp) was much longer than the recently published cpDNA of P. multiseries (111,539 bp) (Table 3). Of the nine Pseudo-nitzschia cpDNAs constructed in this study, each had typical four conjoined structures with one long single copy (LSC) (59,316–64,301bp), one short single copy (SSC) (38,030–48,237 bp), and two inverted repeats (IRs) (7,188–23,151 bp). In contrast, a single IR region was present in the recently published P. multiseries cpDNA (Cao et al., 2016) (Table 3), which was the main reason for the shorter length of its cpDNA. LSC, SSC, and two IRs of nine cpDNAs accounted for 40.48–55.03%, 30.37-32.63%, and 12.34-29.15% of the total cpDNA lengths. GC contents of these cpDNAs were rather similar, ranging from 30.69% (P. hainanensis) to 35.67% (Pseudo-nitzschia sp. CNS00097). Coding sequences of these nine cpDNAs showed moderate variations, ranging from 99,196 to 116,087 in length. In contrast, non-coding sequences of these nine cpDNAs varied substantially, ranged from 17,350 to 42,753 in length (Table 3).
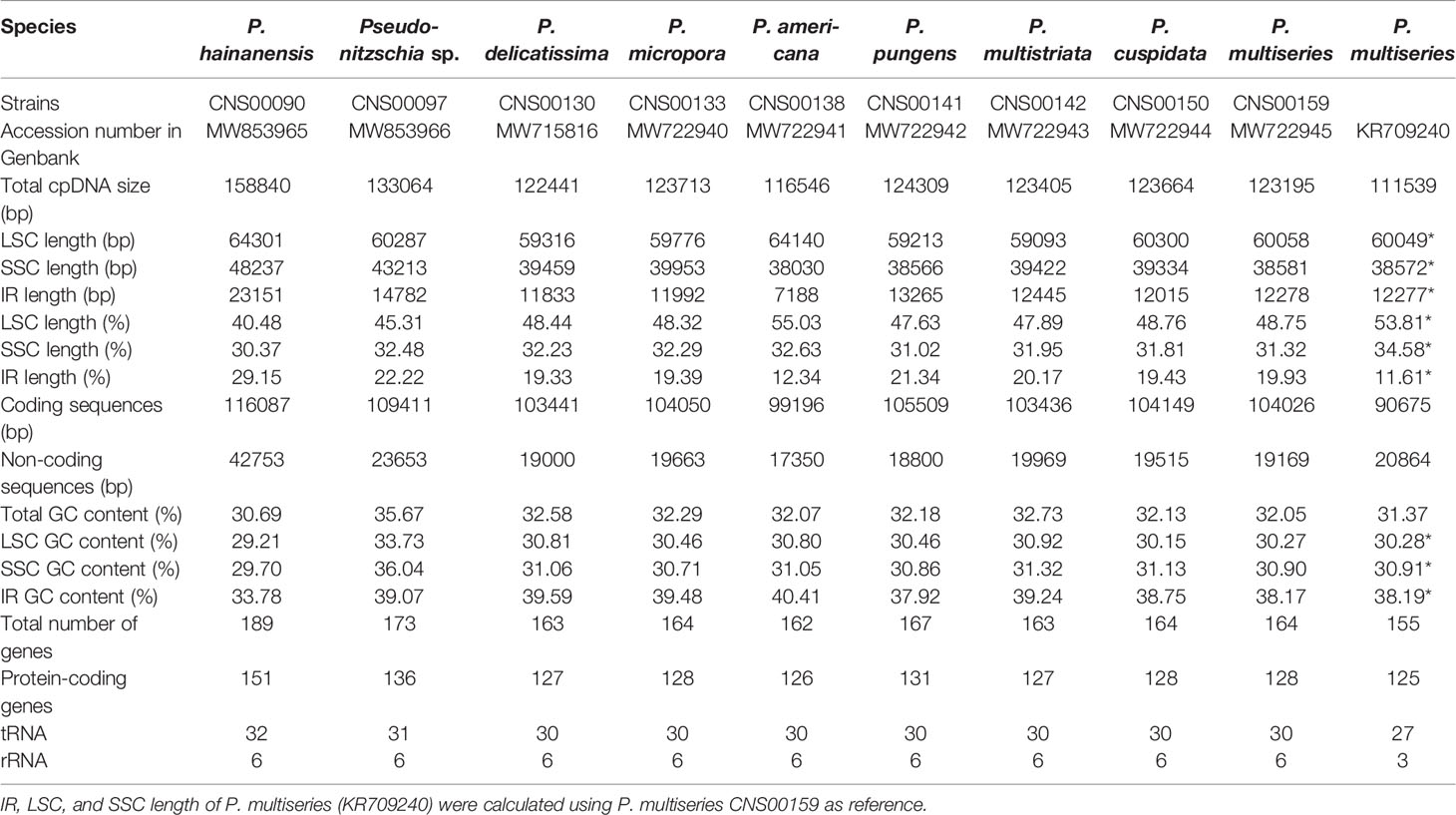
Table 3 Molecular features of cpDNAs of nine Pseudo-nitzschia species.
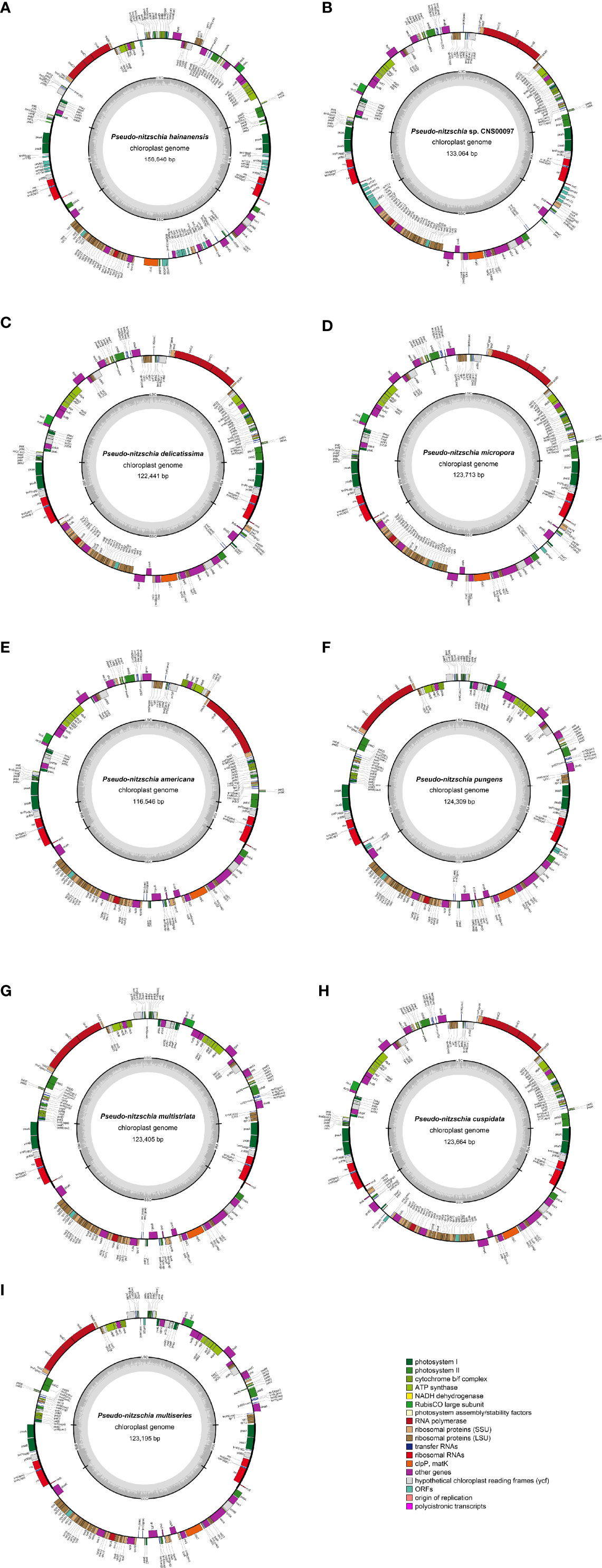
Figure 2 Gene maps of cpDNAs of P. hainanensis CNS00090 (A), Pseudo-nitzschia sp. CNS00097 (B), P. delicatissima CNS00130 (C), P. micropora CNS00133 (D), P. americana CNS00138 (E), P. pungens CNS00141 (F), P. multistriata CN00142 (G), P. cuspidata CNS00150 (H), and P. multiseries CNS00159 (I). The genes drawn outside and inside of the circle are transcribed in clockwise and counterclockwise directions, respectively. Genes were colored based on their functional groups. The inner circle shows the quadripartite structure of the chloroplast: small single copy (SSC), large single copy (LSC) and a pair of inverted repeats (IRa and IRb). The gray ring marks the GC content with the inner circle marking a 50% threshold.
The lengths of intergenic regions of all cpDNAs analyzed in this study were short (Figure 3), which was consistent to previous studies (Yu et al., 2018), confirming that cpDNAs of Bacillariophyta are generally compact with short intergenic regions. In cpDNAs of Pseudo-nitzschia species, the average length of intergenic regions in P. hainanensis cpDNA was obviously larger than those in other species (Figure 3; Table S3). In general, besides P. hainanensis cpDNA, cpDNAs of all other Pseudo-nitzschia species had no significant difference in intergenic region length. Nevertheless, cpDNA of P. multistriata and Pseudo-nitzschia sp. CNS00097 had some large values in the intergenic region (Figure S2; Table S3). These large intergenic regions in the cpDNA of P. hainanensis were responsible for its large cpDNA size (158,840 bp).
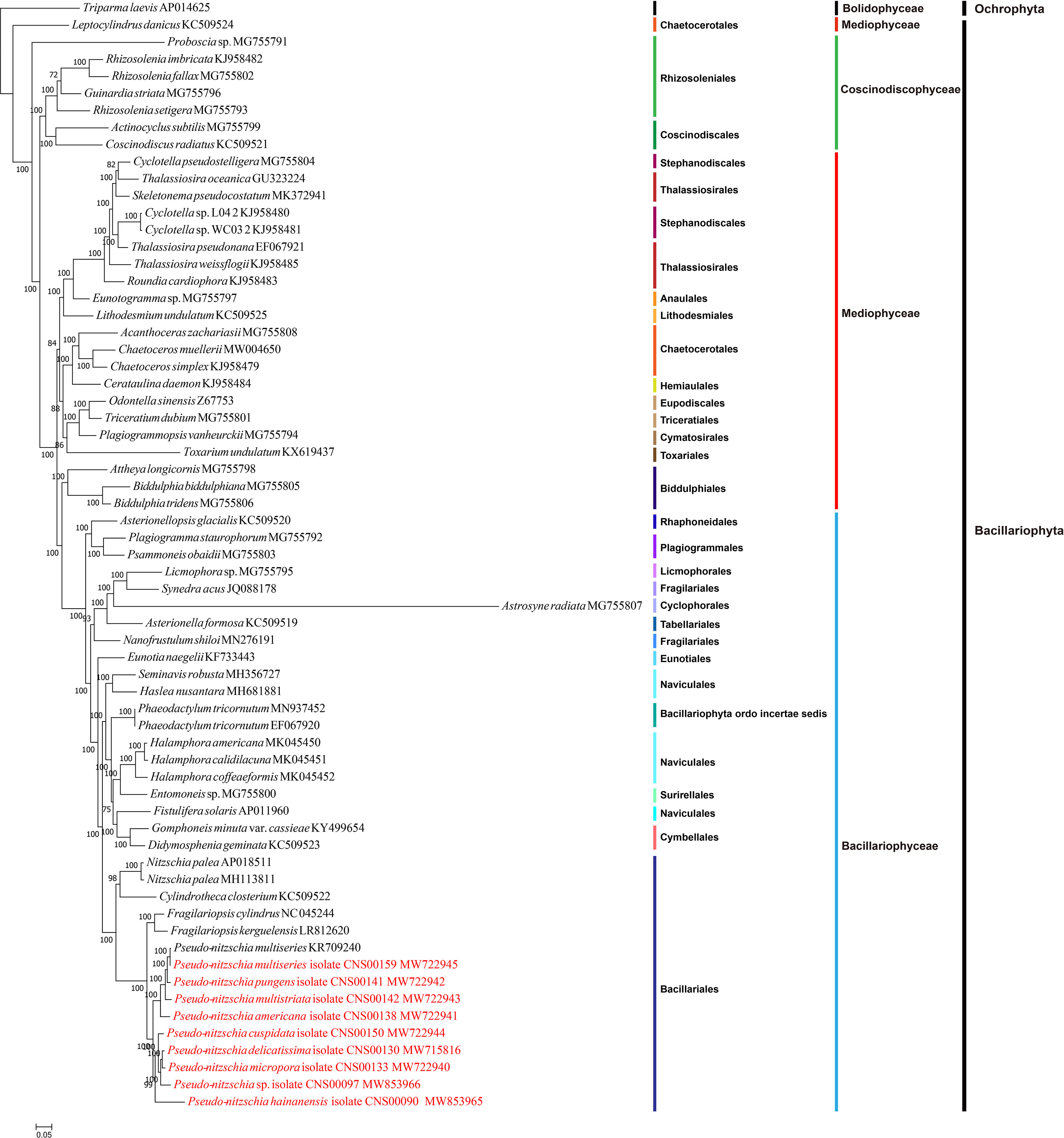
Figure 3 Maximum likelihood (ML) phylogenetic tree based on tandem amino acid sequences of 95 common PCGs from 65 cpDNAs, including 55 previously published Bacillariophyta cpDNAs, nine Pseudo-nitzschia cpDNAs constructed in this study, and Triparma laevis (AP014625) (an Ochrophyta chloroplast genome used as an outgroup taxa). Numbers at the branches represent bootstrap values.
Among the nine newly constructed cpDNAs, P. hainanensis cpDNA was the largest primarily due to its large IR regions. In contrast, the small P. americana cpDNA was primarily due to its shortened IR regions. The differences of gene numbers between Pseudo-nitzschia species were also caused by the numbers of orf genes. No introns were found in all Pseudo-nitzschia cpDNAs, which was not surprising because introns are generally rare in diatom cpDNAs (Ruck et al., 2014). Four pairs of genes overlapping with each other were found in nine cpDNAs, including rpl4-rpl23 (8 bp), psbC-psbD (53 bp), atpD-atpF (4 bp) and sufC-sufB (1 bp). Moreover, a unique pair of overlapping genes orf238-orf126 (7 bp) was found in P. hainanensis (Table 4).
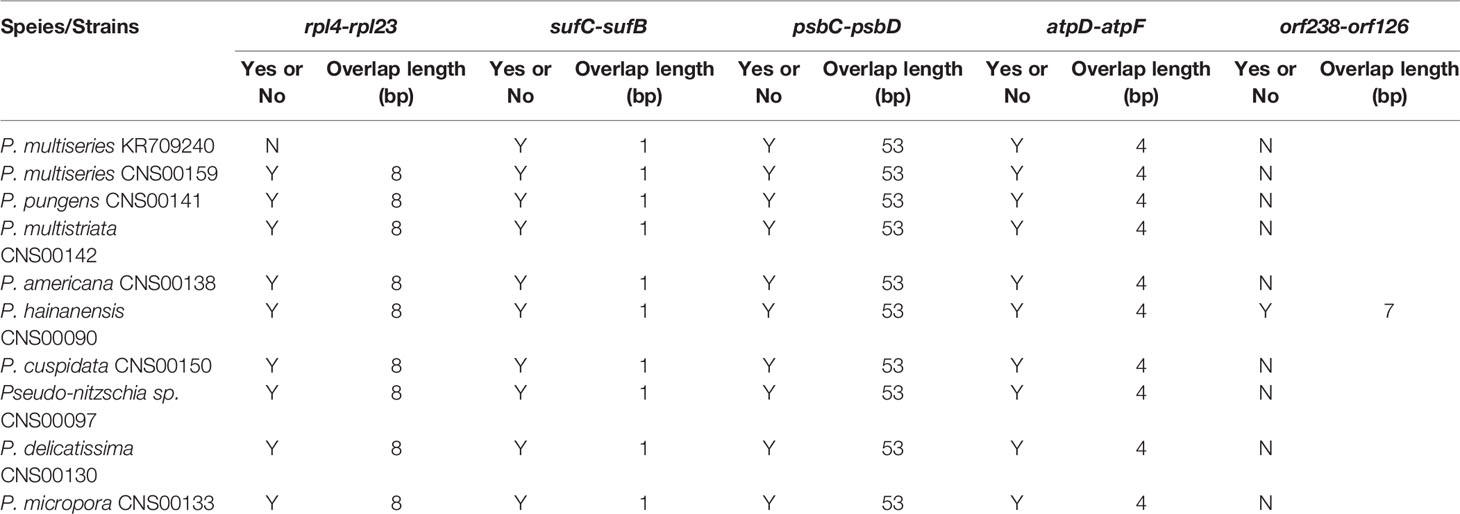
Table 4 Overlapping genes in the cpDNAs of Pseudo-nitzschia species. “Y” or “N” represents whether the two genes were overlap.
Two gene loss events were identified, including psaE loss from the cpDNAs of all Pseudo-nitzschia species except for that of P. americana, and rpl36 loss from the cpDNA of P. hainanensis. To confirm the loss of psaE was not due to the misannotation of this gene, we aligned the genomic region of P. americana cpDNA containing psaE and its upstream gene bas1 and downstream gene ftsH against syntenic regions of other eight Pseudo-nitzschia strains. The genomic spaces between bas1 and ftsH in all eight Pseudo-nitzschia strains were much shorter than that of P. americana (Figure S3A) and no similarities were identified between pasE and the genomic sequences between bas1 and ftsH in the eight strains (Figure S3B), supporting the loss of psaE from this region. To further explore the possibility that the gene psaE transferred to the nuclear genomes of these eight Pseudo-nitzschia strains via EGT, P. americana psaE protein sequence was used as a query to search for potential targets in the assembled genomes based on Illumina reads of each strain. The searches did not find any candidate psaE genes. Further searches using other published Pseudo-nitzschia sequencing data, including nuclear genomes of P. multistriata and P. multiseries, assembled transcriptomes of P. delicatissima and P. pungens, also did not find candidate psaE genes (Table S2). To test the possibility that the genome and transcriptome assemblies might miss the regions containing psaE gene, we carried out PCR reactions using primers (as described in Materials and Methods) designed against an internal region of psaE. PCR experiments of rbcL gene demonstrated the quality of all DNA samples (Figure S3D), and experiments of psaE gene showed that PCR product around 150 bp was only present in P. americana of seven Pseudo-nitzschia species (Figure S3C). PCR amplification of this region was also successful for other diatom species including Nitzschia ovalis, Skeletonema tropicum, and Thalassiosira nordenskioeldii (Figure S3C), providing independent evidence that gene loss had occurred in eight Pseudo-nitzschia species. rpl36 was also not found in the cpDNAs or in the nuclear genome assemblies of P. hainanensis (CNS00090).
Phylogenetic Analysis
To explore the evolution relationship of 10 Pseudo-nitzschia strains and other diatom species, the amino acid sequences of 95 shared PCGs of Bacillariophyta and Ochrophyta were used for constructing a concatenated tree using the maximum likelihood method (Figure 3). In addition, we also constructed a coalescent tree (Figure S4). These two phylogenetic trees showed highly consistent topologies (Split Distance: 0.1290) with three disagreement taxa including Astrosyne radiata, Toxarium undulatum, and Cylindrotheca closterium (Figure S4). As expected, most diatoms species were well grouped into three main clades corresponding to three classes of Coscinodiscophyceae, Mediophyceae, and Bacillariophyceae, respectively. However, interestingly, Leptocylindrus was sister to all other diatoms, and Attheya plus Biddulphia were sister to Bacillariophyceae.
For nine Pseudo-nitzschia strains in this study, phylogenetic trees based on cpDNAs and different molecular markers showed some differences (Figure 1H; Figure S1; Figure 3; Table S6). However, these molecular markers were primarily used for our species identification, and the phylogenetic tree of cpDNAs was the focus of this study. Based on the phylogenetic tree of cpDNAs, ten Pseudo-nitzschia strains could be grouped into two clades based on their phylogenetic relationships (Figure 3; Figure S4), with clade 1 containing two cpDNAs of two P. multiseries strains, and cpDNAs of P. pungens, P. multistriata, and P. americana, and clade 2 containing cpDNAs of P. hainanensis, P. cuspidata, Pseudo-nizschia sp. CNS00097, P. delicatissima, and P. micropora. A previous study suggested a categorization that can separate Pseudo-nitzschia species into two groups by cell width: (1) seriata group (cell width > 3 μm) and (2) delicatissima group (cell width < 3 μm) (Hasle and Syvertsen, 1997). Based on statistics on the cell size of different Pseudo-nitzschia species (Lelong et al., 2012), species in clade 2 (including P. hainanensis, P. cuspidata, P. delicatissima, and P. micropora) were also known to belong to the delicatissima group (cell width < 3 μm). In contrast, P. multiseries and P. pungens belonged to the seriata group (cell width > 3 μm). Furthermore, P. multistriata and P. americana, whose cell widths span both groups, belonged to neither group.
Synteny Analysis of Pseudo-nitzschia cpDNAs
Comparative analysis of cpDNAs of 10 Pseudo-nitzschia strains showed that these cpDNAs can be divided into four groups (Figure 4), compared with the two clades revealed by phylogenetic analysis (Figure 3), suggesting that full-length cpDNA synteny provide higher resolution in distinguishing cpDNAs of Pseudo-nitzschia species. The first group containing the cpDNAs of P. pungens (CNS00141), P. multistriata (CNS00142), and P. multiseries (CNS00159, KR709240), the second group containing the cpDNAs of P. delicatissima (CNS00130), Pseudo-nitzschia sp. (CNS00097), P. micropora (CNS00133), and P. cuspidata (CNS00150), and the third and fourth groups each containing a single strain.
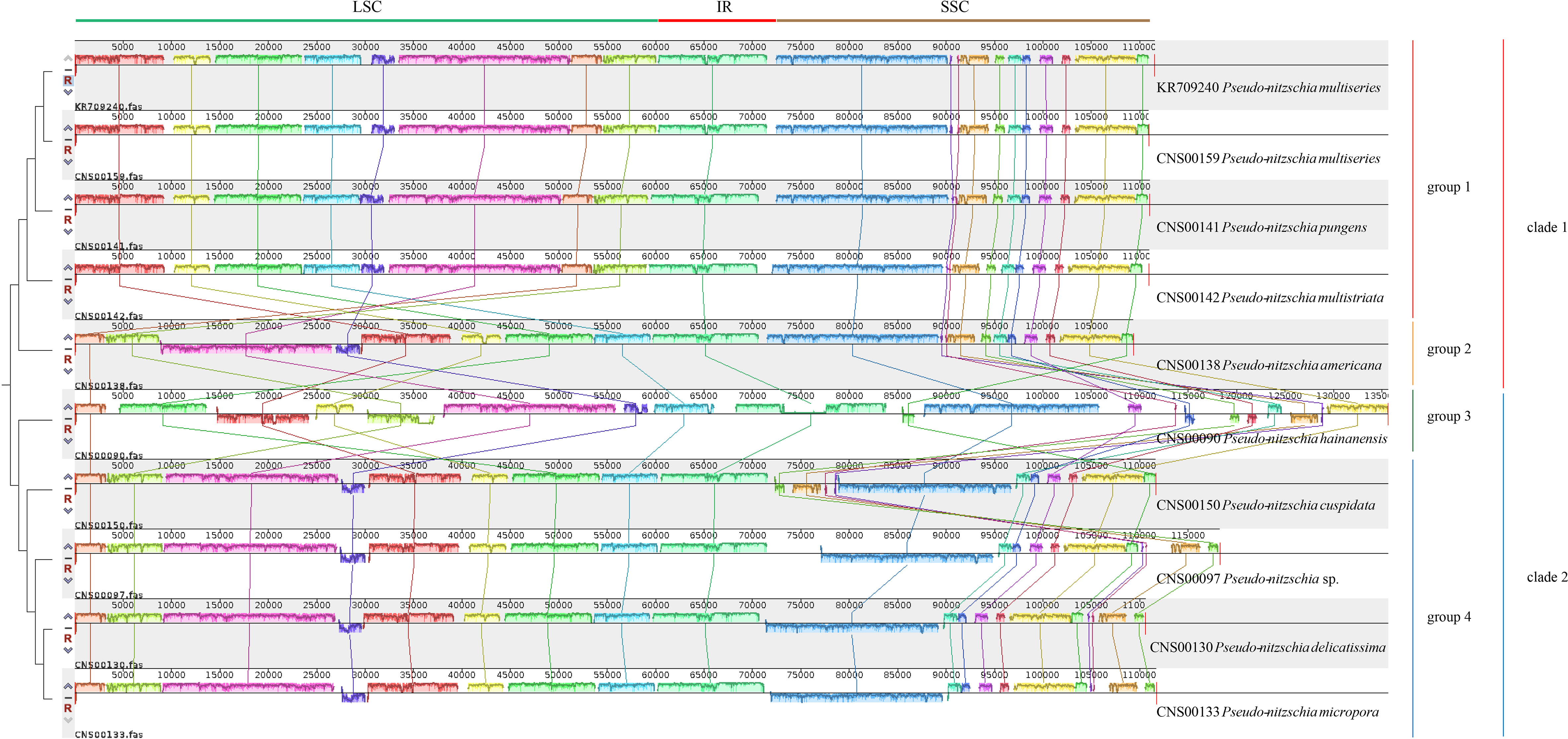
Figure 4 Synteny comparison of 10 Pseudo-nitzschia cpDNAs using Mauve. Rectangular blocks of the same color indicate collinear regions of sequences. Vertical bars inside collinear blocks show degree of sequence identity. The color blocks at the top indicate different collinear regions located roughly in LSC, IR or SSC.
Although within groups, cpDNAs showed high collinearity (Figure 4), such as P. pungens and P. multiseries (Figure 5A), cpDNAs of different groups showed substantial genome rearrangements (Figures 4, 5). For example, between the cpDNAs of P. americana and P. multiseries (CNS00159), multiple inversion and translocation events were identified (Figures 4, 5B). Similarly, multiple inversion and translocation events were also identified between the cpDNAs of P. hainanensis and P. multiseries (CNS00159) (Figures 4, 5C), and between the cpDNAs of P. delicatissima and P. multiseries (Figures 4, 5D).
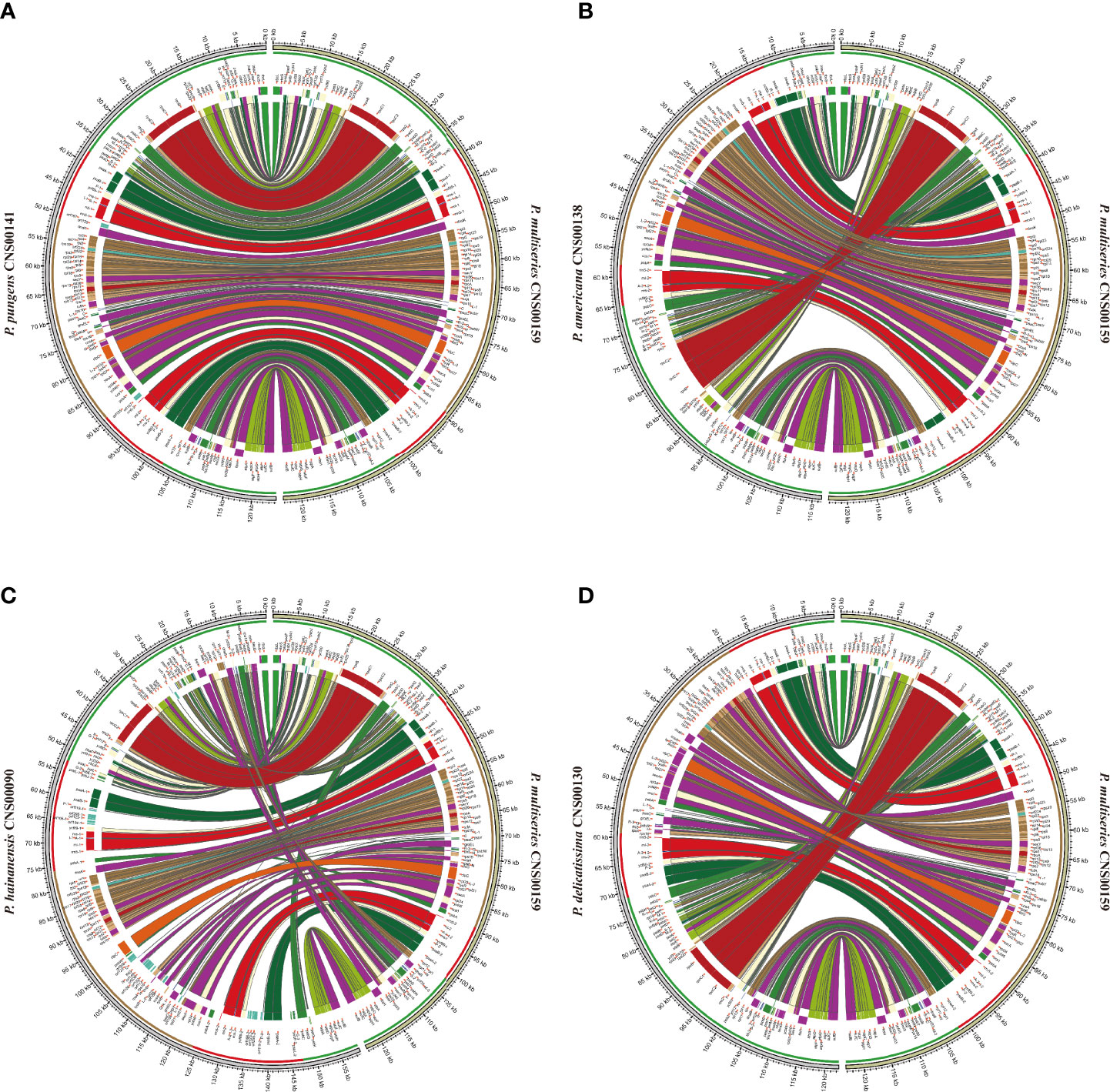
Figure 5 The comparative analysis of cpDNAs of P. multiseries CNS00159 and 4 respective Pseudo-nitzschia species, including P. pungens CNS00141 (A), P. americana CNS00138 (B), P. hainanensis CNS00090 (C), P. delicatissima CNS00130 (D).
Expansion and Contraction of IR Regions
The lengths of the IR regions of cpDNAs of the nine Pseudo-nitzschia species were quite different, ranging from 7,188 bp (P. americana) to 23,151 bp (P. hainanensis). Such large differences in the IR regions may cause differences in the gene content. To test this hypothesis, the arrangements of genes in IR region of nine Pseudo-nitzschia cpDNAs were analyzed (Figure 6A). The topology tree of Figure 6A on the left was constructed based on the phylogenetic tree of cpDNAs. In addition, the junctions JLB (LSC/IRb), JSB (IRb/SSC), JSA (SSC/IRa), and JLA (IRa/LSC) were examined to analyze the contraction and expansion of IR regions of the nine Pseudo-nitzschia species (Figure 6B). Although most IR regions contain nine genes including psaA, psaB, trnP(ugg), ycf89, rns, trnI(gau), trnA(ugc), rnl, and rrn5, many IR regions of these Pseudo-nitzschia species hosts rather different sets of genes (Figure 6; Table S7). The IRa and IRb of the P. americana cpDNA each contained seven genes. Interestingly, these two genes (psaA and psaB) missing from the IRa and IRb regions were located in the LSC region. Thus, compared with the cpDNAs of other eight species constructed in this project that each had two copies of psaA and psaB, the P. americana cpDNA contained a single copy of psaA and psaB. The loss of these two genes in the IRa and IRb regions of the P. americana cpDNA was the main reason for its small size. In contrast, the IRa and IRb regions of the P. hainanensis cpDNA each contained 15 genes (trnG(ucc), psbE, psbF, psbL, psbJ, psaA, psaB, trnP(ugg), ycf89, rns, trnI(gau), trnA(ugc), rnl, rrn5, and psbA) and five orfs (orf119, orf295, orf123, orf166, and orf104) (Figure 6A; Table S7). These 15 genes included all nine genes in other cpDNAs. The addition of six genes and five orfs made the IRa and IRb sizes substantially longer than that of other Pseudo-nitzschia species, which was the main reason for the large size of the cpDNA of P. hainanensis.
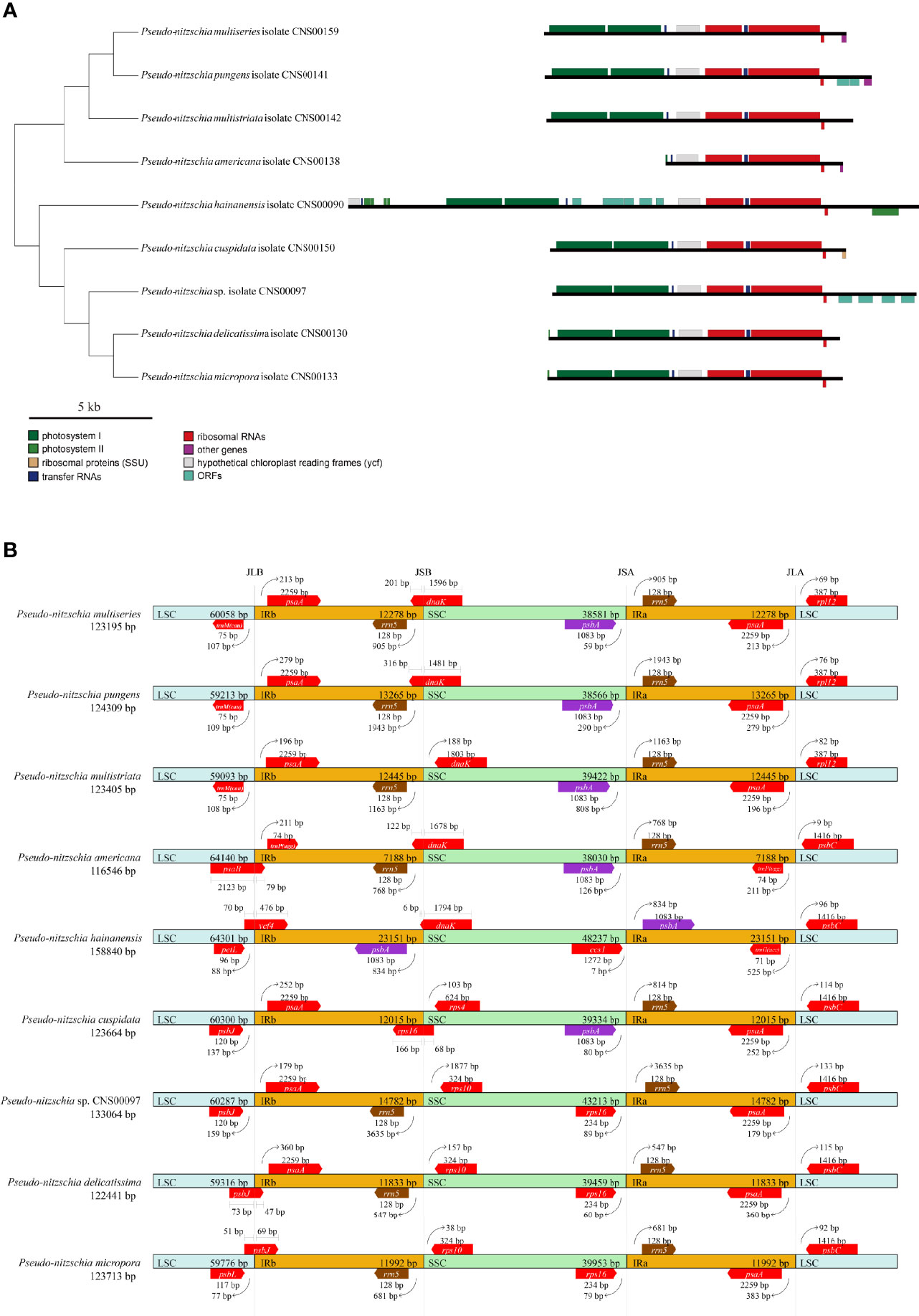
Figure 6 Comparison of the Inverted Repeat region among the nine Pseudo-nitzschia cpDNAs (A). The topology tree on the left were constructed based on phylogenetic tree of cpDNAs. Genes were colored based on their functional groups. Comparison of the junction sites between the Long Single Copy (LSC), Short Single Copy (SSC) and Inverted Repeat (IRa and IRb) regions among the nine Pseudo-nitzschia cpDNAs (B). JLB (IRb/LSC), JSB (IRb/SSC) JSA (SSC/IRa) and JLA (IRa/LSC) denote the junction sites between each corresponding region on the genome.
In addition to the changes that involves gene content contraction (in the P. americana cpDNA) or expansion (in P. hainanensis cpDNA), many other changes have also been observed in the cpDNAs of other Pseudo-nitzschia species (Figure 6A; Table S7). Two orfs (orf167 and orf125) were found to be added to the IRs of P. pungens cpDNA, while four orfs (orf181, orf191, orf173, and orf174) were found to be added to the IRs of Pseudo-nitzschia sp. CNS00097 cpDNA. Moreover, we have found many cases in which genes were found to overlap with junctions. The dnaK gene was found to overlap with the JSB junctions of the cpDNAs of P. multiseries, P. pungens, P. americana, and P. hainanensis, and the rps16 gene was found to overlap with the JSB junction of the P. cuspidata cpDNA (Figure 6B). Similarly, psbB was found to overlap with the JLB junction of the P. americana cpDNA, ycf4 was found to overlap with the JLB junction of the P. hainanaensis cpDNA, and psbJ was found to overlap with the JLB junctions of the cpDNAs of P. delicatissima and P. micropora (Figure 6B; Table S7).
Evolutionary Selection Pressure and Divergence Hotspots
The set of 120 shared protein-coding genes of 10 Pseudo-nitzschia cpDNAs were used to analyze Ka/Ks (Table S8). For these 120 genes, petM showed the highest average Ka/Ks of 0.2231, petN, psbH, and psbL had the lowest average Ka/Ks of 0.0010. All Ka/Ks values were found to be < 1, indicating that all common protein-coding genes in the cpDNAs had purifying selection.
We further examined sequence variability of 150 genes by computing nucleotide diversity (Pi) shared by 10 Pseudo-nitzschia cpDNAs (Figure S5). Among the 10 Pseudo-nitzschia cpDNAs, the Pi values were from 0.0027 (trnP(ugg) and trnR(acg)) to 0.2221 (petF), and the average value of Pi of 150 genes was 0.0847. There were 11 genes ccs1, clpC, dnaB, petF, rpoC2, rps16, secA, secG, secY, thiS, ycf33, ycf41, ycf89, and ycf90 exhibited high Pi values (>0.15). These mutational hotspots can be appropriate loci for developing molecular markers for population genetic studies. Among these 11 genes with high Pi value, the flanking regions of the gene ycf89 were appropriate for designing PCR primers. Phylogenetic trees based on the target sequences suggested that this region could be used as a potential molecular marker. (Figure S6). Primers targeting ycf89, which were described in methods, could be potentially applied to track Pseudo-nitzschia species.
Divergence Time of Pseudo-nitzschia Species
To explore the speciation of Pseudo-nitzschia species, we constructed the time-scale of Pseudo-nitzschia phylogeny (Figure 7). Estimated divergence time result suggested that crown age of Bacillariophyta was dated at approximately 189 Mya. Within the genus Pseudo-nitzschia, all species were divided into two main clades at approximately 41 Mya. P. hainanensis diverged from other Pseudo-nitzschia species at approximately 35 Mya on one of the clades, after which P. cuspidata and Pseudo-nitzschia sp. CNS00097 diverged at about 27 and 19 Mya, and P. delicatissima and P. micropora diverged at about 12 Mya. Within the other clades, the estimated time of divergence between P. americana, P. multistriata, and P. pungens were 30, 21, and 12 Mya, respectively. Thus, comparative analysis of Pseudo-nitzschia cpDNAs suggested that most Pseudo-nitzschia species were generated within the last 40 Mya.
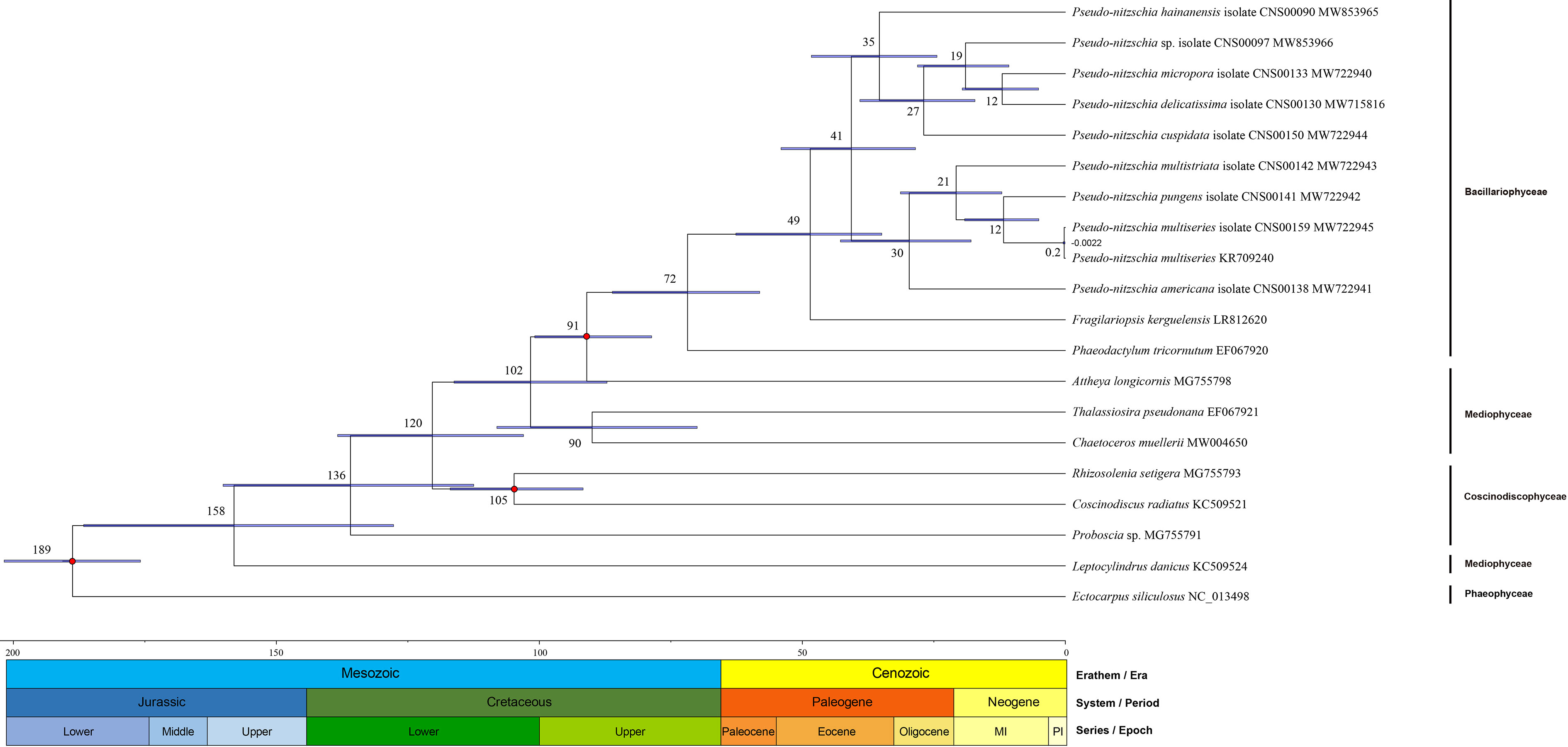
Figure 7 Divergence time of 18 Bacillariophyta species (including 10 Pseudo-nitzschia strains) and one Ochrophyta species. Node values of the tree represent the average and gray bars of every node represent 95% credible interval of divergence time. The values under nodes are the divergence time and range of variation. Color blocks under the tree represent geological time.
Discussion
Through applying high-throughput DNA sequencing technology and bioinformatics analysis software, we have successfully constructed nine cpDNAs for nine Pseudo-nitzschia species, substantially expanding the number of cpDNAs for Pseudo-nitzschia species from one to nine. The availability of these cpDNAs not only facilitated our ability to identify Pseudo-nitzschia species with high resolution, but also provided insight into the evolutionary changes of genes in the cpDNAs, as well as enabling us to ascertain the divergence of Pseudo-nitzschia species.
The identification result of strain CNS00097 was unusual, which could be annotated as P. hallegraeffii based on ITS and ITS2 but could be annotated as P. simulans based on 18S rDNA and 28S rDNA D1-D3 (Table 2; Tables S4, Table S5). A recent study showed that the ITS2 was a molecular marker with higher resolution than 28S rDNA D1-D3 for Pseudo-nitzschia species (Turk Dermastia et al., 2020). Species annotation of the strain Pseudo-nitzschia sp. CNS00097 showed conflicting results when different molecular markers were used, suggesting a unique evolutionary history of Pseudo-nitzschia sp. CNS00097. We are unaware of similar cases in closely related diatoms.
The nine Pseudo-nitzschia cpDNAs revealed in the present study were ranging from 116,546 bp to 158,840 bp in length and had typical quadripartite structure, consisting of LSC, SSC, and two IRs, which were consistent with most published diatom cpDNAs (Sabir et al., 2014; Yu et al., 2018; Hamsher et al., 2019). However, the published P. multiseries cpDNA (KR709240) do not have two IR regions (Cao et al., 2016). One possibility is that there may be errors in cpDNA assembly of P. multiseries (KR709240). Alternatively, the genome assembly was correct, but the cpDNA of this P. multiseries strain lacks an entire copy of the IR region, representing a major genomic difference between this strain and the strain CNS00159 we analyzed. Indeed, the genomic structure of the cpDNA of P. multiseries (KR709240) would be different from all other diatom cpDNAs constructed thus far. The lack of a second copy of the IR region was not without precedent. Chloroplast genomes of many Chlorophyta species (Lemieux et al., 2014; Turmel et al., 2015) and Angiospermae species (Lavin et al., 2005; Ruhlman et al., 2017) have been identified to harbor only a single IRs (Turmel et al., 2017). Therefore, although IR losses were relatively rare, the loss of a second copy of IR from P. multiseries cpDNA was not impossible. More evidence is needed to confirm this possibility in further studies.
Notably, photosynthesis-related gene psaE was lost in all Pseudo-nitzschia species except in P. americana. At present, psaE was identified in most cpDNAs of Bacillariophyta. (Ruck et al., 2014; Ruck et al., 2017; Crowell et al., 2019; Zheng et al., 2019), except in cpDNAs of Fragilariopsis kerguelensis, Rhizosolenia fallax, and Rhizosolenia imbricate (Yu et al., 2018). Therefore, the loss of psaE occurred independently in two classes Coscinodiscophyceae and Bacillariophyceae. It is possible that the loss of photosynthetic genes from cpDNAs may have been the transfer of these cpDNA genes to the nuclear genomes (Sabir et al., 2014). However, this gene psaE was also not found in the nuclear genome assemblies of all nine strains based on the Illumina DNA sequencing data, suggesting that psaE genes have indeed been lost from eight Pseudo-nitzschia species. PsaE is a stromal extrinsic photosystem I (PSI) subunit that forms the docking site of ferredoxin at the acceptor side of PSI (Caspy and Nelson, 2018). Although PsaE was found to be vital in limiting chronic formation of reactive oxygen species, deletion of psaE (hence the loss of PsaE) had little visible effect on photosynthesis of Synechocystis cells, suggesting that PsaE-deficient Synechocystis cells can counteract the chronic photoreduction of oxygen (Jeanjean et al., 2008). We predict that psaE deletion in cpDNAs of eight Pseudo-nitzschia species had little functional consequence on photosynthesis. In addition, rpl36 was lost from P. hainanensis cpDNA. rpl36 was also lost from the cpDNAs of Proboscia sp. and Rhizosolenia fallax (Yu et al., 2018). To date, rpl36 loss has not been found in cpDNAs of other Bacillariophyceae species, the loss of rpl36 in P. hainanensis appears to be a separate event from the rpl36 loss in other two Coscinodiscophyceae species, Proboscia sp. and Rhizosolenia fallax. Perhaps rpl36 loss in P. hainanensis related to the rearrangement of cpDNA and the expansion of the IR regions. Despite the fact that experimental evidence suggests that rpl36 is not essential in Escherichia coli (Ikegami et al., 2005; Baba et al., 2006), studies in Nicotiana tabacum have shown that rpl36 loss results in a severe mutant phenotype (Fleischmann et al., 2011). The impact of rpl36 loss in P. hainanensis needed to be investigated further.
Results from phylogenetic analysis of 65 cpDNAs of diatom species, including nine Pseudo-nitzschia cpDNAs constructed in this study (Figure 3) were in good agreement with that of previous studies (Yu et al., 2018). The ten Pseudo-nitzschia cpDNAs were well separated in the phylogenetic tree, illustrating the power of cpDNAs in resolving different Pseudo-nitzschia species. These species were also nicely resolved in a 28S rDNA D1-D3-based phylogenetic tree (Lim et al., 2018) and in a ITS2-based phylogenetic tree (Chen et al., 2021). Furthermore, it is worth noticing that the position of Pseudo-nitzschia and Fragillariopsis in the different phylogenetic trees, Pseudo-nitzschia and Fragilariopsis formed a cluster in the LSU and ITS2 phylogenetic tree (Lim et al., 2018), while cpDNA-based phylogenetic analysis in this study showed that Fragilariopsis was phylogenetically separated from Pseudo-nitzschia species (Figure 3; Figure S4). More cpDNAs of Pseudo-nitzschia species are needed to consolidate the phylogenetic relationship of Pseudo-nitzschia and Fragilariopsis species. Ten Pseudo-nitzschia cpDNAs were divided into two main clades, species in clade 2 (including P. hainanensis, P. cuspidata, P. delicatissima, and P. micropora) were also known to belong to the delicatissima group with smaller cell width, while P. multiseries and P. pungens in clade 1 belonged to the seriata group with larger cell width (Hasle and Syvertsen, 1997; Lelong et al., 2012). This grouping suggests that the cell size of Pseudo-nitzschia may be related to their evolutionary positions.
Previous studies have shown that P. multiseries, P. pungens, P. multistriata, P. cuspidata, and P. delicatissima were toxigenic, while P. hainanensis, P. americana, and P. micropora have not been detected to be toxigenic (Bates et al., 2019; Chen et al., 2021). Results revealed by the cpDNA sequences-baesd in the phylogenetic tree indicated that toxic species were not clustered. Moreover, a recent study showed that all subclades of the Pseudo-nitzschia genus contain toxic species, and both toxic and non-toxic strains were found within a species (Turk Dermastia et al., 2022), suggesting that molecular mechanisms for toxicity-producing capacity may acquire via HGT (horizontal gene transfer). Another recent study identified a compact gene cluster associated with DA biosynthesis (Brunson et al., 2018), and compact gene cluster were more typically observed in bacteria or fungi (Medema et al., 2015), which could be evidence supporting this HGT hypothesis. Alternatively, genes for producing toxins were selectively lost in evolution.
Comparative analysis of cpDNAs of 10 Pseudo-nitzschia strains showed that these cpDNAs can be divided into two clades (clade 1 and clade 2), each of which contained two groups, a main group and a single cpDNA-containing group (Figure 4). Within the two main groups, cpDNAs showed high collinearity (Figure 4), while cpDNAs of different groups showed substantial genome rearrangements (Figures 4, 5). Their collinearity relationships were generally consistent with their phylogenetic relationships. In the clade 1, cpDNAs of Pseudo-nitzschia species of the main group (group 1) maintain good collinearity after separation from CNS00138 (group 2). Similarly, cpDNAs of Pseudo-nitzschia species in the main group (group 4) of the clade 2 showed high collinearity, while the cpDNA of P. hainanensis (group 3) separated from the cpDNAs of species in group 4 of the clade 2, and its cpDNA underwent a significant structural change. Moreover, the high collinearity between the cpDNA of P. americana in the clade 1 and cpDNAs of the main group species of the clade 2 (including P. cuspidata, Pseudo-nitzschia sp. CNS00097, P. micropora, and P. delicatissima) in the LSC and IR regions (1-70 kb in size) suggested a clear inheritance from their common ancestor. Previous studies had showed genome rearrangements within same genus in diatom, including Thalassiosira and Halamphora (Sabir et al., 2014; Hamsher et al., 2019). However, studies in Angiospermae and Rhodophyta showed that the cpDNAs of species within the same genus were highly conserved (Du et al., 2016; Ng et al., 2017). Interestingly, the study of Halamphora indicated the cpDNAs within this genus may be evolving at 4–7 times faster than those of terrestrial plants (Hamsher et al., 2019), thus faster evolutionary rates may have led to a higher intra-genus diversity in cpDNAs of diatom.
Whole cpDNAs have been used as a super barcode for species identification for Amomum (Cui et al., 2019) and Panax (Ji et al., 2019), because they contain abundant mutation sites. In addition, highly variable regions also can be selected as potential barcode sequences for species identification (Shi et al., 2019; Song et al., 2020). Due to differences in genome structure of Pseudo-nitzschia cpDNAs, a sliding window analysis could not be performed, thus common PCGs were used for nucleotide diversity analysis. As a result, 11 genes ccs1, clpC, dnaB, petF, rpoC2, rps16, secA, secG, secY, thiS, ycf33, ycf41, ycf89, and ycf90 were identified as mutational hotspots. Currently rbcL was a common molecular marker in many studies (D'Alelio and Ruggiero, 2015; Turk Dermastia et al., 2020), but genes with higher Pi value in Pseudo-nitzschia species could be used as a potential molecular marker for the identification and phylogenetic study in the future.
The non-synonymous (Ka) and synonymous (Ks) pattern of nucleotide substitution are valuable in gene evolution studies (Yang and Nielsen, 2000; Yan et al., 2019). Some plants, such as Cardamineae (Yan et al., 2019) and Thuja (Yu et al., 2020), have Ka/Ks ratios > 1 in some genes of cpDNAs, which indicated that these genes suggest a positive selection. However, our results demonstrate the average Ka/Ks of each gene was less than 1. That’s not unusual either, since studies of Isochrysidales (Fang et al., 2020) and Chlorophyceae (Liu et al., 2021a) consistent with our results, their Ka/Ks ratios of shared genes of cpDNAs were also all less than 1. Thus, our results indicating that all common protein-coding genes of 10 Pseudo-nitzschia cpDNAs had purifying selection.
Expansion and contraction in the IR region were common phenomenon in cpDNAs, and expansion of the IR region has resulted in a large number of gene duplications in diatoms (Yu et al., 2018). Among the nine cpDNAs in this study, expansion and contraction in the IR region were also consistent with their phylogenetic relationships, with P. hainanensis separating first from the other species in the clade 2 and showing significant expansion in the IR regions. On the contrary, P. americana first separated from the species in the clade 1 and its IR regions showed significant contraction. While the IR regions of P. hainanensis were longer than that of other species, containing 15 genes and five orfs, the IR regions of P. americana were shorter than that of all other species, containing only seven genes with psaA and psaB that were present in the IR regions of cpDNAs of all other Pseudo-nitzschia species no longer part of its IR regions. In addition to the length of intergenic regions of cpDNAs, the expansion and contraction of IR regions also contribute to the variations of the lengths of cpDNAs. Moreover, examination of the junctions JLB (LSC/IRb), JSB (IRb/SSC), JSA (SSC/IRa), and JLA (IRa/LSC) revealed different types of junctions in nine species with many genes overlapping with the junctions. The different junction types were caused by expansion and contraction in the IR regions and the rearrangements of cpDNAs. Our results were consistent to previous reports which also noted overlaps with junctions in cpDNAs, such as ycf1 and rps19 in the cpDNA of Acanthochlamys bracteate (Wanga et al., 2021), ycf1, rpl12 and ndhF in the cpDNA of Paeonia rockii (Wu et al., 2020).
Diatoms have a rich subfossil and fossil record, and many studies have estimated the divergence time. Previous studies indicated that the origin of the diatoms ranges from 135 to 266 Mya based on multiple calibration points (Medlin et al., 2000; Medlin, 2015). Also, based on a single gene with one calibration point at a time, the average age of the diatom was concluded from 183 to 250 Mya (Sorhannus, 2007). In our result, the crown age of Bacillariophyta was dated at approximately 189 Mya, which was within the range of results obtained in previous studies. Moreover, our result showed that most species within the genus Pseudo-nitzschia were divided into two main clades at approximately 41 Mya, and this time matched the first pulses of diversification of marine diatoms since the early Cenozoic (Cermeño, 2016). Thus, the species diversity of Pseudo-nitzschia may gradually formed since the first pulses in marine diatoms (late Eocene to early Oligocene). To understand their evolutionary history would provide us more useful information to study their diversity and characteristic.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MW853965; https://www.ncbi.nlm.nih.gov/genbank/, MW853966; https://www.ncbi.nlm.nih.gov/genbank/, MW715816; https://www.ncbi.nlm.nih.gov/genbank/, MW722940; https://www.ncbi.nlm.nih.gov/genbank/, MW722941; https://www.ncbi.nlm.nih.gov/genbank/, MW722942; https://www.ncbi.nlm.nih.gov/genbank/, MW722943; https://www.ncbi.nlm.nih.gov/genbank/, MW722944; https://www.ncbi.nlm.nih.gov/genbank/, MW722945.
Author Contributions
ZH and NC designed the research. ZH drafted the manuscript. NC revised the manuscript. YL assisted with the identification. YC assisted with the experiments. ZH, YW, KL, and QX conducted the data analysis. All authors have read and agreed to the submitted version of the manuscript.
Funding
This work was supported by the Strategic Priority Research Program of Chinese Academy of Sciences, Grant No. XDB42000000, Chinese Academy of Sciences; Pioneer Hundred Talents Program (to NC); Taishan Scholar Project Special Fund (to NC); Qingdao Innovation and Creation Plan (Talent Development Program-5th Annual Pioneer and Innovator Leadership Award to NC, 19-3-2-16-zhc).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to colleagues from the Jiaozhou Bay Marine Ecosystem Research Station for their help in field sampling. The samples from the Bohai Sea and Yellow Sea were supported by the National Natural Science Foundation of China, Bohai and Yellow Sea Oceanography Expedition (NORC2019-01). Data acquisition and sample collections from the East China Sea were supported by National Natural Science Foundation of China (NSFC) Open Research Cruise (Cruise No. NORC2019-2), funded by Shiptime Sharing Project of NSFC. This cruise was conducted onboard R/V “Xiang Yang Hong 18” by The First Institute of Oceanography, Ministry of Natural Resources, China. The samples from Western Pacific were supported by the Science & Technology Basic Resources Investigation Program of China (2017FY100804). This cruise was conducted onboard R/V “Science” by The Institute of Oceanology, the Chinese Academy of Sciences, China.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.784579/full#supplementary-material
Supplementary Figure 1 | Maximum likelihood (ML) phylogenetic tree based on 28S rDNA D1-D3 (A) and rbcL (B). Numbers at the branches represent bootstrap values.
Supplementary Figure 2 | Box-plot based on intergenic region of 55 previously published Bacillariophyta cpDNAs and nine Pseudo-nitzschia cpDNAs.
Supplementary Figure 3 | Comparison of the bas1-ftsH region of the nine Pseudo-nitzschia cpDNAs (A). The topology tree on the left were constructed based on phylogenetic tree of cpDNAs. Sequence alignment results near the psaE gene of nine Pseudo-nitzschia cpDNAs based on the bas1-ftsH region (B). The topology tree on the left were constructed based on phylogenetic tree of cpDNAs. The regions of the bas1, psaE, and ftsH were delineated according to P. americana strain CNS00138. PCR results of psaE gene of seven Pseudo-nitzschia species, Skeletonema tropicum, Thalassiosira nordenskioeldii, and Nitzschia ovalis (C). PCR results of rbcL gene of seven Pseudo-nitzschia species, Skeletonema tropicum, Thalassiosira nordenskioeldii, and Nitzschia ovalis (D).
Supplementary Figure 4 | ASTRAL analysis based on 95 common PCGs from 65 cpDNAs, including 55 previously published Bacillariophyta cpDNAs, nine Pseudo-nitzschia cpDNAs constructed in this study, and Triparma laevis (AP014625). Numbers at the branches represent bootstrap values.
Supplementary Figure 5 | Nucleotide diversity of 10 Pseudo-nitzschia cpDNAs. The color blocks at the top indicate different genes located roughly in LSC, IR or SSC.
Supplementary Figure 6 | Maximum likelihood (ML) phylogenetic tree based on target sequences of ycf89 gene of 10 Pseudo-nitzschia strains. Numbers at the branches represent bootstrap values.
Supplementary Table 1 | Statistics of sequencing data and assembly-related information of nine Pseudo-nitzschia strains.
Supplementary Table 2 | Published sequencing data of Pseudo-nitzschia for searching the pasE gene.
Supplementary Table 3 | Mean, maximum and minimum length of intergenic regions of 55 previously published Bacillariophyta cpDNAs and nine Pseudo-nitzschia cpDNAs.
Supplementary Table 4 | ITS2 comparison results of nine Pseudo-nitzschia strains.
Supplementary Table 5 | Molecular markers comparison results of strain CNS00097 with P. simulans and P. hallegraeffi.
Supplementary Table 6 | Topd result based on phylogenetic trees of cpDNAs, 18S rDNA, 28S rDNA D1-D3, and rbcL.
Supplementary Table 7 | Gene located in IRs, JLB, JSB, JSA, and JLA of nine Pseudo-nitzschia cpDNAs. Each gene in the IRs contains two copies.
Supplementary Table 8 | Ka, Ks of 120 shared protein-coding genes of 10 Pseudo-nitzschia strains.
References
Ajani P. A., Larsson M. E., Woodcock S., Rubio A., Farrell H., Brett S., et al. (2020). Fifteen Years of Pseudo-nitzschia in an Australian Estuary, Including the First Potentially Toxic P. delicatissima Bloom in the Southern Hemisphere. Estuarine Coastal Shelf Sci. 236, 106651. doi: 10.1016/j.ecss.2020.106651
Ajani P., Murray S., Hallegraeff G., Lundholm N., Gillings M., Brett S., et al. (2013). The Diatom Genus Pseudo-nitzschia (Bacillariophyceae) in New South Wales, Australia: Morphotaxonomy, Molecular Phylogeny, Toxicity, and Distribution. J. Phycol 49 (4), 765–785. doi: 10.1111/jpy.12087
Ajani P. A., Verma A., Lassudrie M., Doblin M. A., Murray S. A. (2018). A New Diatom Species P. hallegraeffii Sp. Nov. Belonging to the Toxic Genus Pseudo-nitzschia (Bacillariophyceae) From the East Australian Current. PloS One 13 (4), e0195622. doi: 10.1371/journal.pone.0195622
Amato A., Kooistra W. H. C. F., Ghiron J. H. L., Mann D. G., Proschold T., Montresor M. (2007). Reproductive Isolation Among Sympatric Cryptic Species in Marine Diatoms. Protist 158 (2), 193–207. doi: 10.1016/j.protis.2006.10.001
Amato A., Kooistra W. H. C. F., Montresor M. (2019). Cryptic Diversity: A Long-lasting Issue for Diatomologists. Protist 170 (1), 1–7. doi: 10.1016/j.protis.2018.09.005
Amiryousefi A., Hyvonen J., Poczai P. (2018). IRscope: An Online Program to Visualize the Junction Sites of Chloroplast Genomes. Bioinformatics 34 (17), 3030–3031. doi: 10.1093/bioinformatics/bty220
Anisimova M., Gil M., Dufayard J. F., Dessimoz C., Gascuel O. (2011). Survey of Branch Support Methods Demonstrates Accuracy, Power, and Robustness of Fast Likelihood-Based Approximation Schemes. Syst. Biol. 60 (5), 685–699. doi: 10.1093/sysbio/syr041
Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M., et al. (2006). Construction of Escherichia Coli K-12 in-Frame, Single-Gene Knockout Mutants: The Keio Collection. Mol. Syst. Biol. 2 (1), 2006.0008. doi: 10.1038/msb4100050
Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: A New Genome Assembly Algorithm and its Applications to Single-Cell Sequencing. J. Comput. Biol. 19 (5), 455–477. doi: 10.1089/cmb.2012.0021
Bates S. S., Hubbard K. A., Lundholm N., Montresor M., Leaw C. P. (2018). Pseudo-nitzschia, Nitzschia, and Domoic Acid: New Research Since 2011. Harmful Algae 79, 3–43. doi: 10.1016/j.hal.2018.06.001
Bates S. S., Lundholm N., Hubbard K. A., Montresor M., Leaw C. P. (2019). “Toxic and Harmful Marine Diatoms,” in Diatoms: Fundamentals and Applications. Eds. Seckbach J., Gordon R. (New York: Wiley), 389–434. doi: 10.1002/9781119370741.ch17
Bolger A. M., Lohse M., Usadel B. (2014). Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 30 (15), 2114–2120. doi: 10.1093/bioinformatics/btu170
Brunson J. K., McKinnie S. M. K., Chekan J. R., McCrow J. P., Miles Z. D., Bertrand E. M., et al. (2018). Biosynthesis of the Neurotoxin Domoic Acid in a Bloom-Forming Diatom. Science 361 (6409), 1356. doi: 10.1126/science.aau0382
Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., et al. (2009). BLAST+: Architecture and Applications. BMC Bioinf. 10, 421. doi: 10.1186/1471-2105-10-421
Cao M., Yuan X.-L., Bi G. (2016). Complete Sequence and Analysis of Plastid Genomes of Pseudo-nitzschia multiseries (Bacillariophyta). Mitochondrial DNA Part A 27 (4), 2897–2898. doi: 10.3109/19401736.2015.1060428
Capella-Gutierrez S., Silla-Martinez J. M., Gabaldon T. (2009). trimAl: A Tool for Automated Alignment Trimming in Large-Scale Phylogenetic Analyses. Bioinformatics 25 (15), 1972–1973. doi: 10.1093/bioinformatics/btp348
Caspy I., Nelson N. (2018). Structure of the Plant Photosystem I. Biochem. Soc. Trans. 46, 285–294. doi: 10.1042/bst20170299
Cermeño P. (2016). The Geological Story of Marine Diatoms and the Last Generation of Fossil Fuels. Perspect. Phycol 3, 53–60. doi: 10.1127/pip/2016/0050
Chen X. M., Pang J. X., Huang C. X., Lundholm N., Teng S. T., Li A., et al. (2021). Two New and Nontoxigenic Pseudo-nitzschia Species (Bacillariophyceae) From Chinese Southeast Coastal Waters. J. Phycol 57 (1), 335–344. doi: 10.1111/jpy.13101
Clark S., Hubbard K. A., Anderson D. M., McGillicuddy D. J. Jr., Ralston D. K., Townsend D. W. (2019). Pseudo-nitzschia Bloom Dynamics in the Gulf of Maine: 2012–2016. Harmful Algae 88, 101656. doi: 10.1016/j.hal.2019.101656
Crowell R. M., Nienow J. A., Cahoon A. B. (2019). The Complete Chloroplast and Mitochondrial Genomes of the Diatom Nitzschia palea (Bacillariophyceae) Demonstrate High Sequence Similarity to the Endosymbiont Organelles of the Dinotom Durinskia Baltica. J. Phycol 55 (2), 352–364. doi: 10.1111/jpy.12824
Cui Y., Chen X., Nie L., Sun W., Hu H., Lin Y., et al. (2019). Comparison and Phylogenetic Analysis of Chloroplast Genomes of Three Medicinal and Edible Amomum Species. Int. J. Mol. Sci. 20 (16), 4040. doi: 10.3390/ijms20164040
D'Alelio D., Ruggiero M. V. (2015). Interspecific Plastidial Recombination in the Diatom Genus Pseudo-nitzschia. J. Phycol 51 (6), 1024–1028. doi: 10.1111/jpy.12350
Darling A. E., Mau B., Perna N. T. (2010). Progressivemauve: Multiple Genome Alignment With Gene Gain, Loss and Rearrangement. PloS One 5 (6), e11147. doi: 10.1371/journal.pone.0011147
Daugbjerg N., Andersen R. A. (1997). A Molecular Phylogeny of the Heterokont Algae Based on Analyses of Chloroplast-Encoded rbcL Sequence Data. J. Phycol 33 (6), 1031–1041. doi: 10.1111/j.0022-3646.1997.01031.x
Dong H. C., Lundholm N., Teng S. T., Li A., Wang C., Hu Y., et al. (2020a). Occurrence of Pseudo-nitzschia Species and Associated Domoic Acid Production Along the Guangdong Coast, South China Sea. Harmful Algae 98, 101899. doi: 10.1016/j.hal.2020.101899
Dong W., Xu C., Wen J., Zhou S. (2020b). Evolutionary Directions of Single Nucleotide Substitutions and Structural Mutations in the Chloroplast Genomes of the Family Calycanthaceae. BMC Evol. Biol. 20 (1), 96. doi: 10.1186/s12862-020-01661-0
Doyle J. J., Doyle J. L. (1987). A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue. Phytochem. Bull. 19, 11–15. doi: 10.2307/4119796
Du Q. W., Bi G. Q., Mao Y. X., Sui Z. H. (2016). The Complete Chloroplast Genome of Gracilariopsis lemaneiformis (Rhodophyta) Gives New Insight into the Evolution of Family Gracilariaceae. J. Phycol 52 (3), 441–450. doi: 10.1111/jpy.12406
Falkowski P. G., Knoll A. H. (2007). “An Introduction to Primary Producers in the Sea: Who They are, What They do, and When They Evolved,”, in Evolution of Primary Producers in the Sea. Eds. Falkowski P. G., Knoll A. H. (London: Elsevier Academic Press), 1–6. doi: 10.1016/b978-012370518-1/50002-3
Fang J. P., Lin A. T., Yuan X., Chen Y. Q., He W. J., Huang J. L., et al. (2020). The Complete Chloroplast Genome of Isochrysis Galbana and Comparison With Related Haptophyte Species. Algal Res-Biomass Biofuels Bioprod 50, 101989. doi: 10.1016/j.algal.2020.101989
Felsenstein J. (1985). Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 39 (4), 783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x
Field C. B., Behrenfeld M. J., Randerson J. T., Falkowski P. (1998). Primary Production of the Biosphere: Integrating Terrestrial and Oceanic Components. science 281 (5374), 237–240. doi: 10.1126/science.281.5374.237
Fleischmann T. T., Scharff L. B., Alkatib S., Hasdorf S., Schottler M. A., Bock R. (2011). Nonessential Plastid-Encoded Ribosomal Proteins in Tobacco: A Developmental Role for Plastid Translation and Implications for Reductive Genome Evolution. Plant Cell 23 (9), 3137–3155. doi: 10.1105/tpc.111.088906
Garrison N. L., Rodriguez J., Agnarsson I., Coddington J. A., Griswold C. E., Hamilton C. A., et al. (2016). Spider Phylogenomics: Untangling the Spider Tree of Life. Peerj 4, e1719. doi: 10.7717/peerj.1719
Greiner S., Lehwark P., Bock R. (2019). OrganellarGenomeDRAW (OGDRAW) Version 1.3.1: Expanded Toolkit for the Graphical Visualization of Organellar Genomes. Nucleic Acids Res. 47 (W1), W59–W64. doi: 10.1093/nar/gkz238
Guiry M. D., Guiry G. M. (2021). AlgaeBase (Galway: World-wide electronic publication, National University of Ireland). Available at: https://www.algaebase.org.
Hamsher S. E., Keepers K. G., Pogoda C. S., Stepanek J. G., Kane N. C., Kociolek J. P. (2019). Extensive Chloroplast Genome Rearrangement Amongst Three Closely Related Halamphora Spp. (Bacillariophyceae), and Evidence for Rapid Evolution as Compared to Land Plants. PloS One 14 (7), e0217824. doi: 10.1371/journal.pone.0217824
Hasle G. R. (1994). PSEUDO-NITZSCHIA AS A GENUS DISTINCT FROM NITZSCHIA (BACILLARIOPHYCEAE). J. Phycol 30 (6), 1036–1039. doi: 10.1111/j.0022-3646.1994.01036.x
Hasle G., Syvertsen E. (1997). “Marine Diatoms” in Identifying Marine Phytoplankton. Ed. Tomas C. R. (San Diego: Academic Press), 5–385.
Hong D. D., Thu N. T. H., Nam H. S., Hien H. M., Hai L. Q., Ha D. V., et al. (2007). The Phylogenetic Tree of Alexandrium Prorocentrum And Pseudo-Nitzschia of Harmful and Toxic Algae in Vietnam Coastal Waters Based on Sequences of 18srdna, Its1-5.8 s-Its2 Gene Formats and Single Cell-Per Method. Marine Res. Indonesia 32 (2), 203–218. doi: 10.14203/mri.v32i2.456
Huang C. X., Dong H. C., Lundholm N., Teng S. T., Zheng G. C., Tan Z. J., et al. (2019). Species Composition and Toxicity of the Genus Pseudo-nitzschia in Taiwan Strait, Including P. chiniana Sp. Nov. And P. qiana Sp. Nov. Harmful Algae 84, 195–209. doi: 10.1016/j.hal.2019.04.003
Huang C.-H., Sun R., Hu Y., Zeng L., Zhang N., Cai L., et al. (2015). Resolution of Brassicaceae Phylogeny Using Nuclear Genes Uncovers Nested Radiations and Supports Convergent Morphological Evolution. Mol. Biol. Evol. 33 (2), 394–412. doi: 10.1093/molbev/msv226
Ikegami A., Nishiyama K., Matsuyama S., Tokuda H. (2005). Disruption of rpmJ Encoding Ribosomal Protein L36 Decreases the Expression of secY Upstream of the Spc Operon and Inhibits Protein Translocation in Escherichia Coli. Biosci Biotechnol. Biochem. 69 (8), 1595–1602. doi: 10.1271/bbb.69.1595
Jeanjean R., Latifi A., Matthijs H. C. P., Havaux M. (2008). The PsaE Subunit of Photosystem I Prevents Light-Induced Formation of Reduced Oxygen Species in the Cyanobacterium Synechocystis Sp PCC 6803. Biochim. Et Biophys. Acta-Bioenergetics 1777 (3), 308–316. doi: 10.1016/j.bbabio.2007.11.009
Ji Y., Liu C., Yang Z., Yang L., He Z., Wang H., et al. (2019). Testing and Using Complete Plastomes and Ribosomal DNA Sequences as the Next Generation DNA Barcodes in Panax (Araliaceae). Mol. Ecol. Resour 19 (5), 1333–1345. doi: 10.1111/1755-0998.13050
Jin J. J., Yu W. B., Yang J. B., Song Y., dePamphilis C. W., Yi T. S., et al. (2020). GetOrganelle: A Fast and Versatile Toolkit for Accurate De Novo Assembly of Organelle Genomes. Genome Biol. 21 (1), 241. doi: 10.1186/s13059-020-02154-5
Katoh K., Standley D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30 (4), 772–780. doi: 10.1093/molbev/mst010
Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., et al. (2009). Circos: An Information Aesthetic for Comparative Genomics. Genome Res. 19 (9), 1639–1645. doi: 10.1101/gr.092759.109
Kumar S., Stecher G., Tamura K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Lamari N., Ruggiero M. V., d'Ippolito G., Kooistra W. H. C. F., Fontana A., Montresor M. (2013). Specificity of Lipoxygenase Pathways Supports Species Delineation in the Marine Diatom Genus Pseudo-nitzschia. PloS One 8 (8), e73281. doi: 10.1371/journal.pone.0073281
Lampe R. H., Cohen N. R., Ellis K. A., Bruland K. W., Maldonado M. T., Peterson T. D., et al. (2018). Divergent Gene Expression Among Phytoplankton Taxa in Response to Upwelling. Environ. Microbiol. 20 (8), 3069–3082. doi: 10.1111/1462-2920.14361
Langmead B., Salzberg S. L. (2012). Fast Gapped-Read Alignment With Bowtie 2. Nat. Methods 9 (4), 357–359. doi: 10.1038/nmeth.1923
Lavin M., Herendeen P. S., Wojciechowski M. F. (2005). Evolutionary Rates Analysis of Leguminosae Implicates a Rapid Diversification of Lineages During the Tertiary. Systematic Biol. 54 (4), 575–594. doi: 10.1080/10635150590947131
Lelong A., Hégaret H., Soudant P., Bates S. S. (2012). Pseudo-nitzschia (Bacillariophyceae) Species, Domoic Acid and Amnesic Shellfish Poisoning: Revisiting Previous Paradigms. Phycologia 51 (2), 168–216. doi: 10.2216/11-37
Lemieux C., Otis C., Turmel M. (2014). Six Newly Sequenced Chloroplast Genomes From Prasinophyte Green Algae Provide Insights Into the Relationships Among Prasinophyte Lineages and the Diversity of Streamlined Genome Architecture in Picoplanktonic Species. BMC Genomics 15, 857. doi: 10.1186/1471-2164-15-857
Li Y., Dong Y., Liu Y., Yu X., Yang M., Huang Y. (2020). Comparative Analyses of Euonymus Chloroplast Genomes: Genetic Structure, Screening for Loci With Suitable Polymorphism, Positive Selection Genes, and Phylogenetic Relationships Within Celastrineae. Front. Plant Sci. 11. doi: 10.3389/fpls.2020.593984
Li Y., Dong H. C., Teng S. T., Bates S. S., Lim P. T. (2018). Pseudo-nitzschia nanaoensis Sp. Nov. (Bacillariophyceae) From the Chinese Coast of the South China Sea. J. Phycol 54 (6), 918–922. doi: 10.1111/jpy.12791
Li H., Durbin R. (2010). Fast and Accurate Long-Read Alignment With Burrows–Wheeler Transform. Bioinformatics 26 (5), 589–595. doi: 10.1093/bioinformatics/btp698
Li X. Q., Feng Y. Y., Leng X. Y., Liu H. J., Sun J. (2017a). Phytoplankton Species Composition of Four Ecological Provinces in Yellow Sea, China. J. Ocean Univ. China 16 (6), 1115–1125. doi: 10.1007/s11802-017-3270-3
Li Y., Huang C. X., Xu G. S., Lundholm N., Teng S. T., Wu H., et al. (2017b). Pseudo-nitzschia simulans Sp. Nov. (Bacillariophyceae), the First Domoic Acid Producer From Chinese Waters. Harmful Algae 67, 119–130. doi: 10.1016/j.hal.2017.06.008
Li Y., Ma Y. Y., Lu S. H. (2010). Morphological Characteristics of Pseudo-nitzschia americana Complex in Daya Bay, China. Acta Hydrobiol Sin. 34, 851–855. doi: 10.3724/SP.J.1035.2010.00851
Lim H. C., Tan S. N., Teng S. T., Lundholm N., Orive E., David H., et al. (2018). Phylogeny and Species Delineation in the Marine Diatom Pseudo-nitzschia (Bacillariophyta) Using Cox1, LSU, and ITS2 rRNA Genes: A Perspective in Character Evolution. J. Phycol 54 (2), 234–248. doi: 10.1111/jpy.12620
Lim H. C., Teng S. T., Leaw C. P., Lim P. T. (2013). Three Novel Species in the Pseudo-nitzschia pseudodelicatissima Complex: P. batesiana Sp. Nov., P. lundholmiae Sp. Nov., and P. fukuyoi Sp. Nov. (Bacillariophyceae) From the Strait of Malacca, Malaysia. J. Phycol 49 (5), 902–916. doi: 10.1111/jpy.12101
Lim H. C., Teng S. T., Lim P. T., Wolf M., Leaw C. P. (2016). 18s rDNA Phylogeny of Pseudo-nitzschia (Bacillariophyceae) Inferred From Sequence-Structure Information. Phycologia 55 (2), 134–146. doi: 10.2216/15-78.1
Liu K., Chen Y., Cui Z., Liu S., Xu Q., Chen N. (2021b). Comparative Analysis of Chloroplast Genomes of Thalassiosira Species. Front. Marine Sci. 8. doi: 10.3389/fmars.2021.788307
Liu B. W., Zhu H., Dong X. Q., Yan Q. F., Liu G. X., Hu Z. Y. (2021a). Reassessment of Suitable Markers for Taxonomy of Chaetophorales (Chlorophyceae, Chlorophyta) Based on Chloroplast Genomes. J. Eukaryotic Microbiol. 68 (5), e12858. doi: 10.1111/jeu.12858
Lommer M., Roy A.-S., Schilhabel M., Schreiber S., Rosenstiel P., LaRoche J. (2010). Recent Transfer of an Iron-Regulated Gene From the Plastid to the Nuclear Genome in an Oceanic Diatom Adapted to Chronic Iron Limitation. BMC Genomics 11 (1), 718. doi: 10.1186/1471-2164-11-718
Lu S. H., Li Y., Lundholm N., Ma Y. Y., Ho K. C. (2012). Diversity, Taxonomy and Biogeographical Distribution of the Genus Pseudo-nitzschia (Bacillariophyceae) in Guangdong Coastal Waters, South China Sea. Nova Hedwigia 95 (1-2), 123–152. doi: 10.1127/0029-5035/2012/0046
Lundholm N., Bates S. S., Baugh K. A., Bill B. D., Connell L. B., Léger C., et al. (2012). Cryptic and Pseudo-Cryptic Diversity in Diatoms—With Descriptions of Pseudo-nitzschia hasleana Sp. Nov. And P. fryxelliana Sp. Nov. J. Phycol 48 (2), 436–454. doi: 10.1111/j.1529-8817.2012.01132.x
Lundholm N., Daugbjerg N., Moestrup O. (2002). Phylogeny of the Bacillariaceae With Emphasis on the Genus Pseudo-nitzschia (Bacillariophyceae) Based on Partial LSU rDNA. Eur. J. Phycol 37 (1), 115–134. doi: 10.1017/s096702620100347x
Lundholm N., Moestrup Ø., Kotaki Y., Hoef-Emden K., Scholin C., Miller P. (2006). Inter-And Intraspecific Variation of the Pseudo-nitzschia delicatissima Complex (Bacillariophyceae) Illustrated by rRNA Probes, Morphological Data and Phylogenetic Analyses. J. Phycol 42 (2), 464–481. doi: 10.1111/j.1529-8817.2006.00211.x
Manhart J. R., Fryxell G. A., Villac M. C., Segura L. Y. (1995). PSEUDO-NITZSCHIA PUNGENS AND P-MULTISERIES (BACILLARIOPHYCEAE) - NUCLEAR RIBOSOMAL DNAS AND SPECIES-DIFFERENCES. J. Phycol 31 (3), 421–427. doi: 10.1111/j.0022-3646.1995.00421.x
Mann D., Crawford R., Round F. (2017). “Bacillariophyta,” in Handbook of Protists. Eds. Archibald J., Simpson A., Slamovits C. (Cham: Springer).
Marcais G., Kingsford C. (2011). A Fast, Lock-Free Approach for Efficient Parallel Counting of Occurrences of K-Mers. Bioinformatics 27 (6), 764–770. doi: 10.1093/bioinformatics/btr011
Matari N. H., Blair J. E. (2014). A Multilocus Timescale for Oomycete Evolution Estimated Under Three Distinct Molecular Clock Models. BMC Evolutionary Biol. 14 (1), 1–11. doi: 10.1186/1471-2148-14-101
McCabe R. M., Hickey B. M., Kudela R. M., Lefebvre K. A., Adams N. G., Bill B. D., et al. (2016). An Unprecedented Coastwide Toxic Algal Bloom Linked to Anomalous Ocean Conditions. Geophysical Res. Lett. 43 (19), 10366–10376. doi: 10.1002/2016gl070023
Medema M. H., Kottmann R., Yilmaz P., Cummings M., Biggins J. B., Blin K., et al. (2015). Minimum Information About a Biosynthetic Gene Cluster. Nat. Chem. Biol. 11 (9), 625–631. doi: 10.1038/nchembio.1890
Medlin L. K. (2015). A Timescale for Diatom Evolution Based on Four Molecular Markers: Reassessment of Ghost Lineages and Major Steps Defining Diatom Evolution. Vie Milieu-life Environ. 65 (4), 219–238.
Medlin L., Kooistra W., Schmid A.-M. (2000). “A Review of the Evolution of the Diatoms-a Total Approach Using Molecules, Morphology and Geology,” in The Origin and Early Evolution of the Diatoms: Fossil, Molecular and Biogeographical Approaches. Eds. Witkowski A., Sieminska J. (Krakow, Poland: Polish Academy of Sciences), pp., 13–35.
Minh B. Q., Nguyen M. A., von Haeseler A. (2013). Ultrafast Approximation for Phylogenetic Bootstrap. Mol. Biol. Evol. 30 (5), 1188–1195. doi: 10.1093/molbev/mst024
Mirarab S., Reaz R., Bayzid M. S., Zimmermann T., Swenson M. S., Warnow T. (2014). ASTRAL: Genome-Scale Coalescent-Based Species Tree Estimation. Bioinformatics 30 (17), I541–I548. doi: 10.1093/bioinformatics/btu462
Ng P. K., Lin S. M., Lim P. E., Liu L. C., Chen C. M., Pai T. W. (2017). Complete Chloroplast Genome of Gracilaria Firma (Gracilariaceae, Rhodophyta), With Discussion on the Use of Chloroplast Phylogenomics in the Subclass Rhodymeniophycidae. BMC Genomics 18 (1), 40. doi: 10.1186/s12864-016-3453-0
Nishimura T., Murray J. S., Boundy M. J., Balci M., Bowers H. A., Smith K. F., et al. (2021). Update of the Planktonic Diatom Genus Pseudo-nitzschia in Aotearoa New Zealand Coastal Waters: Genetic Diversity and Toxin Production. Toxins 13 (9), 637. doi: 10.3390/toxins13090637
Perez Blanco E., Antoine E., Crassous M.-P., Compere C. (2008). "Detection and Molecular Identification of Pseudo-nitzschia Species in Natural Samples From the French Coasts" in 3rd Congress of the International Society for Applied Phycology Incorporating the 11th International Conference on Applied Phycology 21st-27th of June 2008 (Galway: National University of Ireland).
Puigbo P., Garcia-Vallve S., McInerney J. O. (2007). TOPD/FMTS: A New Software to Compare Phylogenetic Trees. Bioinformatics 23 (12), 1556–1558. doi: 10.1093/bioinformatics/btm135
Quijano-Scheggia S. I., Garcés E., Lundholm N., Moestrup Ø., Andree K., Camp J. (2009). Morphology, Physiology, Molecular Phylogeny and Sexual Compatibility of the Cryptic Pseudo-nitzschia delicatissima Complex (Bacillariophyta), Including the Description of P. arenysensis Sp. Nov. Phycologia 48 (6), 492–509. doi: 10.2216/08-21.1
Robinson J. T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E. S., Getz G., et al. (2011). Integrative Genomics Viewer. Nat. Biotechnol. 29 (1), 24–26. doi: 10.1038/nbt.1754
Ruck E. C., Linard S. R., Nakov T., Theriot E. C., Alverson A. J. (2017). Hoarding and Horizontal Transfer Led to an Expanded Gene and Intron Repertoire in the Plastid Genome of the Diatom, Toxarium undulatum (Bacillariophyta). Curr. Genet. 63 (3), 499–507. doi: 10.1007/s00294-016-0652-9
Ruck E. C., Nakov T., Jansen R. K., Theriot E. C., Alverson A. J. (2014). Serial Gene Losses and Foreign DNA Underlie Size and Sequence Variation in the Plastid Genomes of Diatoms. Genome Biol. Evol. 6 (3), 644–654. doi: 10.1093/gbe/evu039
Ruhlman T. A., Zhang J., Blazier J. C., Sabir J. S. M., Jansen R. K. (2017). Recombination-Dependent Replication and Gene Conversion Homogenize Repeat Sequences and Diversify Plastid Genome Structure. Am. J. Bot. 104 (4), 559–572. doi: 10.3732/ajb.1600453
Sabir J. S. M., Yu M. J., Ashworth M. P., Baeshen N. A., Baeshen M. N., Bahieldin A., et al. (2014). Conserved Gene Order and Expanded Inverted Repeats Characterize Plastid Genomes of Thalassiosirales. PloS One 9 (9), e107854. doi: 10.1371/journal.pone.0107854
Saeed A. F., Awan S. A., Ling S. M., Wang R. Z., Wang S. (2017). Domoic Acid: Attributes, Exposure Risks, Innovative Detection Techniques and Therapeutics. Algal Res-Biomass Biofuels Bioprod 24, 97–110. doi: 10.1016/j.algal.2017.02.007
Shi H., Yang M., Mo C., Xie W., Liu C., Wu B., et al. (2019). Complete Chloroplast Genomes of Two Siraitia Merrill Species: Comparative Analysis, Positive Selection and Novel Molecular Marker Development. PloS One 14 (12), e0226865. doi: 10.1371/journal.pone.0226865
Smith S. A., Dunn C. W. (2008). Phyutility: A Phyloinformatics Tool for Trees, Alignments and Molecular Data. Bioinformatics 24 (5), 715–716. doi: 10.1093/bioinformatics/btm619
Song H., Liu F., Li Z., Xu Q., Chen Y., Yu Z., et al. (2020). Development of a High-Resolution Molecular Marker for Tracking Phaeocystis globosa Genetic Diversity Through Comparative Analysis of Chloroplast Genomes. Harmful Algae 99, 101911. doi: 10.1016/j.hal.2020.101911
Sorhannus U. (2007). A Nuclear-Encoded Small-Subunit Ribosomal RNA Timescale for Diatom Evolution. Marine Micropaleontol 65 (1-2), 1–12. doi: 10.1016/j.marmicro.2007.05.002
Stamatakis A. (2014). RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 30 (9), 1312–1313. doi: 10.1093/bioinformatics/btu033
Stonik I. V. (2021). Long-Term Variations in Species Composition of Bloom-Forming Toxic Pseudo-nitzschia Diatoms in the North-Western Sea of Japan During 1992-2015. J. Marine Sci. Eng. 9 (6), 568. doi: 10.3390/jmse9060568
Stonik I. V., Isaeva M. P., Aizdaicher N. A., Balakirev E. S., Ayala F. J. (2018). Morphological and Genetic Identification of Pseudo-nitzschia H. Peragallo 1900 (Bacillariophyta) From the Sea of Japan. Russian J. Marine Biol. 44 (3), 192–201. doi: 10.1134/s1063074018030100
Sun J., Wang Y., Liu Y., Xu C., Yuan Q., Guo L., et al. (2020). Evolutionary and Phylogenetic Aspects of the Chloroplast Genome of Chaenomeles Species. Sci. Rep. 10 (1), 11466. doi: 10.1038/s41598-020-67943-1
Theriot E. C., Ashworth M. P., Nakov T., Ruck E., Jansen R. K. (2015). Dissecting Signal and Noise in Diatom Chloroplast Protein Encoding Genes With Phylogenetic Information Profiling. Mol. Phylogenet. Evol. 89, 28–36. doi: 10.1016/j.ympev.2015.03.012
Tonti-Filippini J., Nevill P. G., Dixon K., Small I. (2017). What can We do With 1000 Plastid Genomes? Plant J. 90 (4), 808–818. doi: 10.1111/tpj.13491
Trainer V. L., Bates S. S., Lundholm N., Thessen A. E., Cochlan W. P., Adams N. G., et al. (2012). Pseudo-nitzschia Physiological Ecology, Phylogeny, Toxicity, Monitoring and Impacts on Ecosystem Health. Harmful Algae 14, 271–300. doi: 10.1016/j.hal.2011.10.025
Trifinopoulos J., Nguyen L. T., von Haeseler A., Minh B. Q. (2016). W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis. Nucleic Acids Res. 44 (W1), W232–W235. doi: 10.1093/nar/gkw256
Turk Dermastia T., Cerino F., Stankovic D., France J., Ramsak A., Znidaric Tusek M., et al. (2020). Ecological Time Series and Integrative Taxonomy Unveil Seasonality and Diversity of the Toxic Diatom Pseudo-nitzschia H. Peragallo in the Northern Adriatic Sea. Harmful Algae 93, 101773. doi: 10.1016/j.hal.2020.101773
Turk Dermastia T., Dall'Ara S., Dolenc J., Mozetic P. (2022). Toxicity of the Diatom Genus Pseudo-nitzschia (Bacillariophyceae): Insights From Toxicity Tests and Genetic Screening in the Northern Adriatic Sea. Toxins 14 (1). doi: 10.3390/toxins14010060
Turmel M., Otis C., Lemieux C. (2015). Dynamic Evolution of the Chloroplast Genome in the Green Algal Classes Pedinophyceae and Trebouxiophyceae. Genome Biol. Evol. 7 (7), 2062–2082. doi: 10.1093/gbe/evv130
Turmel M., Otis C., Lemieux C. (2017). Divergent Copies of the Large Inverted Repeat in the Chloroplast Genomes of Ulvophycean Green Algae. Sci. Rep. 7 (1), 994. doi: 10.1038/s41598-017-01144-1
Vurture G. W., Sedlazeck F. J., Nattestad M., Underwood C. J., Fang H., Gurtowski J., et al. (2017). GenomeScope: Fast Reference-Free Genome Profiling From Short Reads. Bioinformatics 33 (14), 2202–2204. doi: 10.1093/bioinformatics/btx153
Wanga V. O., Dong X., Oulo M. A., Mkala E. M., Yang J. X., Onjalalaina G. E., et al. (2021). Complete Chloroplast Genomes of Acanthochlamys bracteata (China) and Xerophyta (Africa) (Velloziaceae): Comparative Genomics and Phylogenomic Placement. Front. Plant Sci. 12. doi: 10.3389/fpls.2021.691833
Wang D., Zhang Y., Zhang Z., Zhu J., Yu J. (2010). KaKs_Calculator 2.0: A Toolkit Incorporating Gamma-Series Methods and Sliding Window Strategies. Genom Proteomics Bioinf. 8 (1), 77–80. doi: 10.1016/S1672-0229(10)60008-3
Wick R. R., Schultz M. B., Zobel J., Holt K. E. (2015). Bandage: Interactive Visualization of De Novo Genome Assemblies. Bioinformatics 31 (20), 3350–3352. doi: 10.1093/bioinformatics/btv383
Wu L., Nie L., Xu Z., Li P., Wang Y., He C., et al. (2020). Comparative and Phylogenetic Analysis of the Complete Chloroplast Genomes of Three Paeonia Section Moutan Species (Paeoniaceae). Front. Genet. 11. doi: 10.3389/fgene.2020.00980
Yan C., Du J., Gao L., Li Y., Hou X. (2019). The Complete Chloroplast Genome Sequence of Watercress (Nasturtium Officinale R. Br.): Genome Organization, Adaptive Evolution and Phylogenetic Relationships in Cardamineae. Gene 699, 24–36. doi: 10.1016/j.gene.2019.02.075
Yang Z. (1997). PAML: A Program Package for Phylogenetic Analysis by Maximum Likelihood. Bioinformatics 13 (5), 555–556. doi: 10.1093/bioinformatics/13.5.555
Yang Z., Nielsen R. (2000). Estimating Synonymous and Nonsynonymous Substitution Rates Under Realistic Evolutionary Models. Mol. Biol. Evol. 17 (1), 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Yuan X.-L., Cao M., Bi G.-Q. (2016). The Complete Mitochondrial Genome of Pseudo-nitzschia multiseries (Baciuariophyta). Mitochondrial DNA Part A 27 (4), 2777–2778. doi: 10.3109/19401736.2015.1053061
Yu M. J., Ashworth M. P., Hajrah N. H., Khiyami M. A., Sabir M. J., Alhebshi A. M., et al. (2018). "Evolution of the Plastid Genomes in Diatoms," in Plastid Genome Evolution. Eds. Chaw S. M., Jansen R. K. (London: Academic Press Ltd-Elsevier Science Ltd), 129–155.
Yu T., Huang B. H., Zhang Y., Liao P. C., Li J. Q. (2020). Chloroplast Genome of an Extremely Endangered Conifer Thuja Sutchuenensis Franch.: Gene Organization, Comparative and Phylogenetic Analysis. Physiol. Mol. Biol. Plants 26 (3), 409–418. doi: 10.1007/s12298-019-00736-7
Keywords: diatom, Pseudo-nitzschia, chloroplast genome, inverted region, comparative analysis, phylogenetic analysis, divergence analysis
Citation: He Z, Chen Y, Wang Y, Liu K, Xu Q, Li Y and Chen N (2022) Comparative Analysis of Pseudo-nitzschia Chloroplast Genomes Revealed Extensive Inverted Region Variation and Pseudo-nitzschia Speciation. Front. Mar. Sci. 9:784579. doi: 10.3389/fmars.2022.784579
Received: 28 September 2021; Accepted: 19 April 2022;
Published: 18 May 2022.
Edited by:
Andrew Stanley Mount, Clemson University, United StatesReviewed by:
Maria Valeria Ruggiero, Anton Dohrn Zoological Station, ItalyPeter Von Dassow, Pontificia Universidad Católica de Chile, Chile
Copyright © 2022 He, Chen, Wang, Liu, Xu, Li and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nansheng Chen, Y2hlbm5AcWRpby5hYy5jbg==; Y2hlbm5Ac2Z1LmNh