 Han Zhang
Han Zhang Lei Wang
Lei Wang Yinbin Qiu
Yinbin Qiu Fahui Gong5
Fahui Gong5 Baoting Nong
Baoting Nong Xinghua Pan
Xinghua Pan- 1Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Southern Medical University, Guangzhou, China
- 2Guangdong Provincial Key Laboratory of Single Cell Technology and Application, Southern Medical University, Guangzhou, China
- 3Guangdong-Hong Kong-Macao Greater Bay Area Center for Brain Science and Brain-Inspired Intelligence, Southern Medical University, Guangzhou, China
- 4Department of Pharmaceutical Engineering, College of Materials and Energy, South China Agricultural University, Guangzhou, China
- 5School of Marine Sciences, Sun Yat-sen University (Zhuhai Campus), Zhuhai, China
- 6Key Laboratory of Gene Engineering of the Ministry of Education, Institute of Healthy Aging Research, School of Life Sciences, Sun Yat-sen University, Guangzhou, China
Current ConoServer database accumulates 8,134 conopeptides from 122 species of cone snail, which are pharmaceutically attractive marine resource. However, many more conopeptides remain to be discovered, and the enzymes involved in their synthesis and processing are unclear. In this report, firstly we screened and analyzed the differentially expressed genes (DEGs) between venom duct (VD) and venom bulb (VB) of C. caracteristicus, and obtained 3,289 transcripts using a comprehensive assembly strategy. Then using de novo deep transcriptome sequencing and analysis under a strict merit, we discovered 194 previously unreported conopeptide precursors in Conus caracteristicus. Meanwhile, 2 predicted conopeptides from Consort were verified using liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS). Furthermore, we demonstrated that both VD and VB of C. caracteristicus secreted hundreds of different conotoxins, which showed a high diversity among individuals of the species. Finally, we identified a protein disulfide isomerase (PDI) gene, which, functioning for intramolecular disulfide-bond folding, was shared among C. caracteristicus, C. textile, and C. bartschi and was the first PDI identified with five thioredoxin domains. Our results provide novel insights and fuel further studies of the molecular evolution and function of the novel conotoxins.
Introduction
Cone snails, which are carnivorous marine gastropod mollusks, produce a cocktail of venom peptides, known as conotoxins or conopeptides, that are used in predation, defense, or competition (Gao et al., 2017). A drop of Conus venom may contain hundreds of different peptides, each of which is generally comprised of 12–50 residues and multiple pairs of disulfide bonds (Terlau and Olivera, 2004). The signal peptide region of the conopeptide is relatively well conserved (Kaas et al., 2008, 2010, 2012). Based on sequence similarities with signal peptides in the ConoServer database1, conopeptides are currently classified into 27 gene superfamilies (A, B1, B2, B3, C, D, E, F, G, H, I1, I2, I3, J, K, L, M, N, O1, O2, O3, P, Q, S, T, V, and Y) (Kaas et al., 2008, 2010, 2012; Puillandre et al., 2012; Ye et al., 2012; Aguilar et al., 2013; Dutertre et al., 2013; Luo et al., 2013; Lu et al., 2014; Peng et al., 2016; Fu et al., 2018; Yao et al., 2019; Li et al., 2020) and 13 additional putative gene superfamilies (Espiritu et al., 2001; Biggs et al., 2010; Kaas et al., 2010, 2012). More than 30 additional novel superfamilies have recently been reported (Lavergne et al., 2015; Peng et al., 2016; Prashanth et al., 2016). Due to the high selectivity for various ion channels and nerve receptors, conotoxins have become an important tool in neuroscience research and have a great potential for use in the development of novel drugs (Wermeling, 2005; Han et al., 2008; Lebbe et al., 2014; Pan, 2019; Shen, 2019).
A variety of interpretations on the genetic diversity of conotoxins were proposed, such as gene duplication, gene loss, code shift mutations, early termination, and recombination (Espiritu et al., 2001; Stanley, 2008; Puillandre et al., 2010, 2014; Chang and Duda, 2012). Conotoxin folding occurs mainly in the endoplasmic reticulum, where hundreds of different cysteine-rich polypeptides are folded effectively (Tayo et al., 2010; Lu et al., 2014; Zhang et al., 2019). How these domains are correctly oxidized and folded in the poison gland tissues remains unclear, but it is known that molecular chaperones in the endoplasmic reticulum are involved in toxin folding (Gao et al., 2017). In vitro folding of these toxins usually results in low folding yield, misfolding, or accumulation of polymer products (Bulaj and Olivera, 2008). Cone snails can secrete a variety of different toxins in a short period of time (Duda and Palumbi, 2004; Puillandre et al., 2014). To date, more than 15 different post-translational-modification enzymes have been implicated in the post-translational modification of conotoxins (Buczek et al., 2005). Several previous studies have examined the molecular relationship between PDIs (protein disulfide isomerase) and conotoxin diversity (Hatahet and Ruddock, 2009; Safavi-Hemami et al., 2010; Wang et al., 2017).
To the best of our knowledge, an in-depth comparison of conopeptides among individual Conus caracteristicus, a vermivorous species, has yet to be performed. Furthermore, it is interesting that previous studies have identified hundreds of unique toxins or conopeptides from a single Conus specimen using genomic or proteomic methods (Biass et al., 2009; Davis et al., 2009; Kaas et al., 2010; Lavergne et al., 2015; Peng et al., 2021). Besides the diversity on gene or protein sequence level, the high toxin diversity is attributed to the oxidative folding of proteases in the venom (Buczek et al., 2005; Bulaj and Olivera, 2008). It is assumed that many novel natural conopeptides and post-translational modification enzymes involved in oxidative conotoxin folding remain to be discovered (Peng et al., 2016).
Here, we report a set of novel conopeptides isolated from the venom duct (VD) and venom bulb (VB) of vermivorous C. caracteristicus. We also present conopeptide diversity among individuals and verify these superfamilies using liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS). During this process, we have accidentally identified a new protein disulfide isomerase (PDI) gene, which is highly homologous to sequences in C. textile and C. bartschi. Our results increase the available data concerning marine conopeptides and may provide insight into their potential applications.
Results
Differentially Expressed Genes Between Venom Bulb and Venom Duct Revealed by Transcriptomic Sequencing
The cytochrome c oxidase subunit I gene is often used as a DNA barcode for animal species identification (Hebert et al., 2003; Simion et al., 2018). We generated transcriptomic sequences of the VD and VB from three individuals (Ca-1, Ca-2, and Ca-3): Ca-1-VD, Ca-1-VB, Ca-2-VD, Ca-2-VB, Ca-3-VD, and Ca-3-VB. A filter stat summary of the transcriptome data of the three individuals was given in Supplementary Table 1. Illumina sequencing of Ca-1-VD, Ca-1-VB, Ca-2-VD, Ca-2-VB, Ca-3-VD, and Ca-3-VB generated approximately 11.09, 10.64, 9.71, 8.57, 11.74, and 10.03 Gb of clean data, respectively. The percentage of Q30 clean bases in Ca-1-VD, Ca-1-VB, Ca-2-VD, Ca-2-VB, Ca-3-VD, and Ca-3-VB were 95.75, 95.75, 96.21, 96.24, 95.85, and 95.86%, respectively.
We then combined the COI (mitochondrial cytochrome oxidase subunit I) nucleotide sequences from these transcriptomes with 32 COI nucleotide sequences from 11 Conus species obtained from the NCBI EST database: C. tribblei (Barghi et al., 2015), C. lenavati (Li et al., 2017), C. episcopatus (Dutertre et al., 2013), C. marmoreus (Safavi-Hemami et al., 2014), C. geographus (Liu et al., 2012), C. eburneus (Liu et al., 2009), C. quercinus (Chang and Duda, 2012), C. flavidus (Kaas et al., 2012), and C. caracteristicus. The genetic distances among three previously published COI sequences (KJ549883.1, KJ549747.1 and MN389186.1) and six of the COI sequences collected from the six specimens of the species C. caracteristicus were 0.000–0.064. Combined the appearance and color of C. caracteristicus, we determined Ca-1, Ca-2, and Ca-3 as C. caracteristicus (Supplementary Figure 1A).
Using a comprehensive assembly strategy, we analyzed the differentially expressed genes between the VB group and the VD group; triplicates (three individuals) were set in each group. The correlation coefficient of the transcriptomes within each group (VB or VD) was greater than 0.9 (Figure 1A). We identified 3,289 transcripts that were at least 2-fold differentially expressed between VD and VB (P < 0.01); moreover, 1,207 transcripts were upregulated in the VD group, and 2,082 transcripts were upregulated in the VB group (Figure 1B). Venn diagrams showed that 10,058 genes from the assembled transcripts were commonly annotated in Nt (non-redundant nucleotide sequences), Swiss-Prot (blastx), Swiss-Prot (blastp), and Nr (non-redundant proteins) (Supplementary Figure 1B).
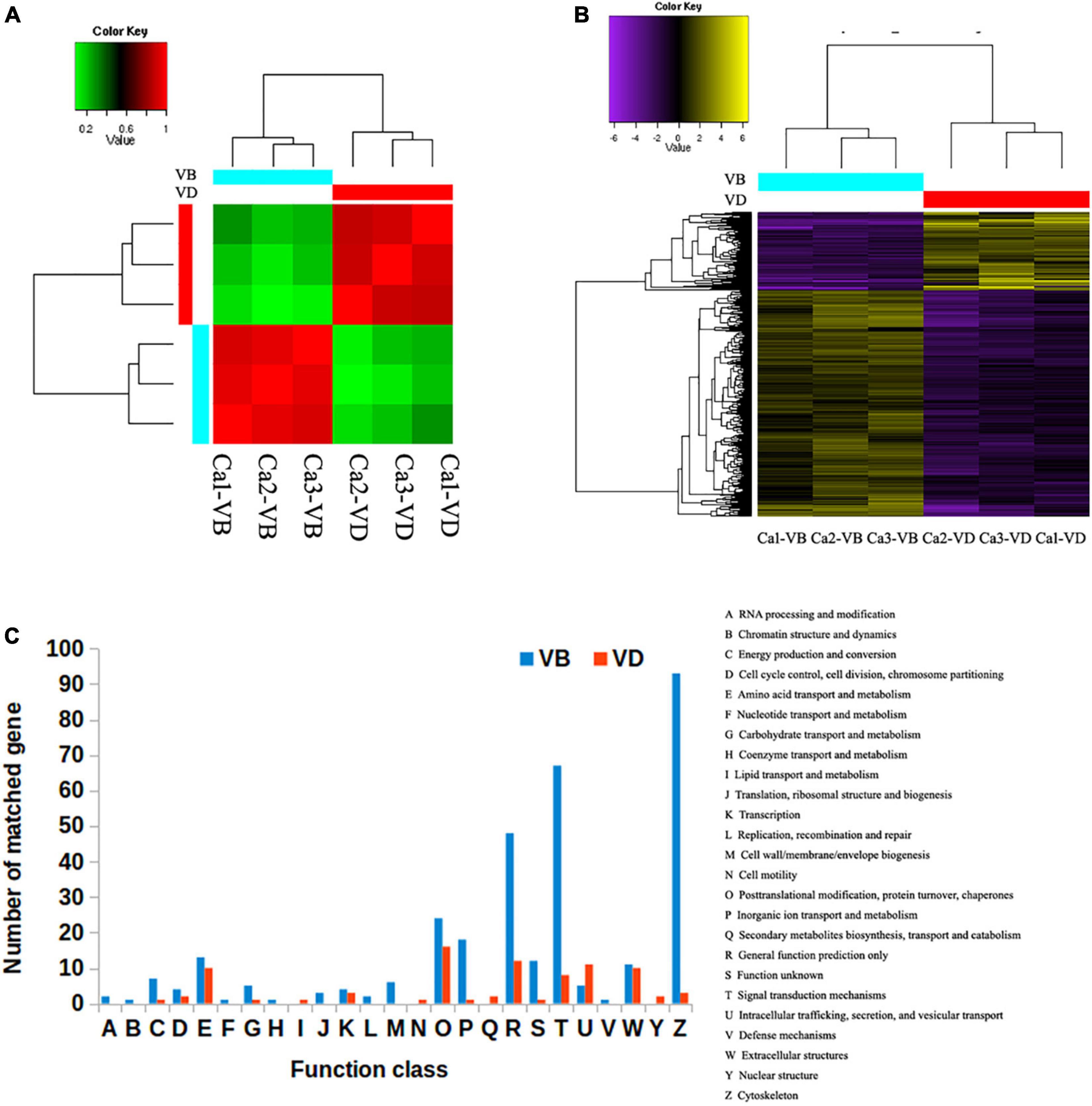
Figure 1. The differentially expressed genes between venom duct (VD) and venom bulb (VB). (A), Sample correlation matrix. (B), Heatmap of differentially expressed genes. (C), KOG annotations of differentially expressed transcripts between the venom ducts and venom bulbs of Conus. caracteristicus.
The Gene Ontology (GO) Consortium terms most enriched in the VB group were cytoskeletal protein binding, contractile fiber, and supramolecular polymer, all of which are closely related to the cytoskeleton (Supplementary Figure 2). In contrast, signaling receptor ligand precursor processing, calcium ion binding, and serine hydrolase activity were the main terms enriched in the VD group (Supplementary Figure 3). These GO terms are closely related to the cleavage of a peptide bond in a precursor.
In order to compare 3,289 differentially expressed transcripts with the genome annotation data of C. betulinus, we downloaded the EST data (ID: PRJNA290540) associated with the article (Peng et al., 2021) and compared it through BLASTN. The threshold value is set to 1e-5. We found that 163 reported EST sequences were detected in our VD group, and 409 reported EST sequences were detected in our VP group. These corresponding sequences deserve more follow-up attention. To further categorize possible functions, we searched the 3,289 differentially expressed transcripts against the EuKaryotic Orthologous Groups (KOG) database. We successfully annotated 328 gene sequences from the VD and 85 gene sequences from the VB with KOG functions in 25 functional categories. The most significantly enriched category in the VB was cytoskeleton (93 sequences), followed by signal transduction mechanisms (67 sequences) and general function prediction only (48 sequences). In contrast, the most significantly enriched category in the VD was post-translational modification, followed by protein turnover, chaperones (16 sequences), general function prediction only (12 sequences), intracellular trafficking, secretion, and vesicular transport (11 sequences) (Figure 1C).
Compared with the VB cells, there were complex regulatory networks related to peptide synthesis and processing in the VD cells. Thus, although both organs produce venom, they have subtly different functions in defense.
Putative Conopeptide Precursors Predicted by ConoSorter
To identify novel conotoxin genes, the six samples (Ca-1-VD, Ca-1-VB, Ca-2-VD, Ca-2-VB, Ca-3-VD, and Ca-3-VB.) were individually assembled. The total numbers of potential transcripts (unigenes) were in Ca-1-VD, Ca-1-VB, Ca-2-VD, Ca-2-VB, Ca-3-VD, and Ca-3-VB 185,317, 111,583, 119,348, 104,157, 158,697, and 120,070, respectively. Then, using ConoSorter (see Materials and Methods), various numbers of conopeptide precursor sequences were annotated: 371 (Ca-1-VD), 268 (Ca-1-VB), 250 (Ca-2-VD), 270 (Ca-2-VB), 379 (Ca-3-VD), and 200 (Ca-3-VB) (Supplementary Table 2). We used venn diagrams to illustrate the relationships among the annotated conopeptide precursor sequences (Figure 2A). Across the three individuals, 39 peptides were shared in the VD and 51 were shared in the VB (Figure 2B). This clearly indicates that conopeptides are not only diverse within a single tissue of one individual but are also heterogeneous among individuals.
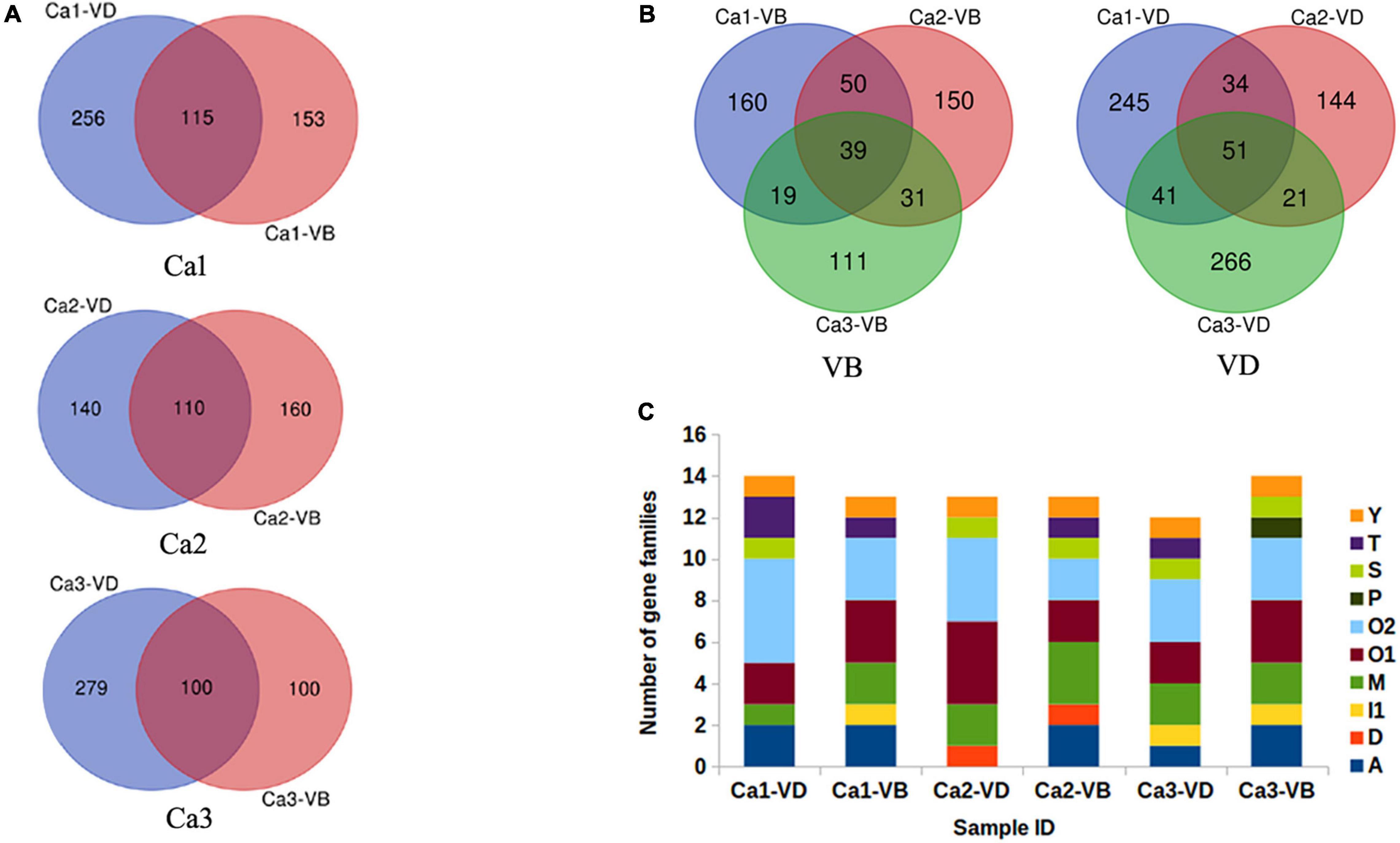
Figure 2. The annotated predicted conopeptides. (A), Relationships of the annotated candidate conopeptides between VD and VB in each individual, as predicted by ConoSorter. (B), Relationships of the annotated candidate conopeptides between VD and VB in the three individuals, as predicted by ConoSorter. (C), The identified candidate conopeptide precursors previously reported in Conus caracteristicus.
The ConoServer database (See Text Footnote 1) indicates that C. caracteristicus produces 138 mature conotoxins in 15 gene superfamilies (A, B1, D, I1, I2, I3, L, M, O1, O2, O3, T, J, S, Y). A small proportion of the conopeptides obtained in this study have previously been reported in known gene superfamilies. In Ca-1-VD, 16 conopeptide precursors in six gene superfamilies (A, M, O1, O2, T, Y) were previously identified. In Ca-1-VB, 15 conopeptide precursors in seven gene superfamilies (A, M, O1, O2, S, T, Y) were previously identified. Similarly, we identified 14 previously reported conopeptide precursors in five gene super families in Ca-2-VD (D, O1, O2, S, Y) and 15 previously reported conopeptide precursors in seven gene super families in Ca-2-VB (A, M, O1, O2, T, S, Y). In Ca-3-VD, 16 conopeptide precursors in seven gene superfamilies (A, M, O1, O2, S, T, Y) were previously identified. In Ca-3-VB, 16 conopeptide precursors in six gene superfamilies (A, M, O1, O2, P, S) were previously identified (Figure 2C).
Most of the conopeptides obtained herein were reported for the first time from C. caracteristicus (Supplementary Table 2). In total, 2,194 candidate toxin genes were identified in C. caracteristicus, more than 15 times the number of toxin genes identified to date. In particular, Lt9a (Pi et al., 2006) in the P gene superfamily was identified for the first time in C. caracteristicus. After removing duplicate sequences and reported sequences, we collectively identified 1,330 candidate conopeptide precursors. With the 1,330 sequences, the software ConoPrec in Conosever (See Text Footnote 1) was used to retain the sequences with signal peptide, pre-peptide and mature peptide. As a result, we finally obtained 249 relatively reliable gene sequences of natural conotoxin, and the remaining 1,081 sequences were considered as of candidate conotoxin. Of the 249 sequences, 55 sequences were recorded as conopeptide precursors and 194 were non-recorded conopeptide precursors (Supplementary Table 3).
Conopeptide Precursors Verified by LC-MS/MS
VD proteins were sequenced using a Thermo LTQ Orbitrap Elite (for details see the Materials and Methods). From the six samples (Ca-1-VD, Ca-1-VB, Ca-2-VD, Ca-2-VB, Ca-3-VD, and Ca-3-VB), LC-MS/MS sequencing generated 215, 222, 219, 244, 216, and 202 Mb of raw ion current traces, respectively.
The total ion current traces from Ca-2-VD and Ca-3-VB are shown in Figures 3A,B. No target conopeptide was detected in other mass spectra, and no conopeptide fragment was identified in the mass spectrometry data of Ca-1-VD, Ca-1-VB, Ca-2-VB, and Ca-3-VD. The identification of the peptide sequences using MS confirmed the expression of the associated conopeptide genes (Figures 3C,D). According to the conservation of signal peptides, the Ca2-VD-c49774_g1_5_3 were not a member of any known gene superfamilies by evolutionary analysis and may thus belong to as-yet undescribed gene superfamilies (Figure 4). The Ca3-VB-c93596_g1_5_1 (Ca6.14) was classified into the O1 gene superfamily. In addition, we also found another 14 O1 superfamily member by sequence alignment with 249 sequences obtained above.
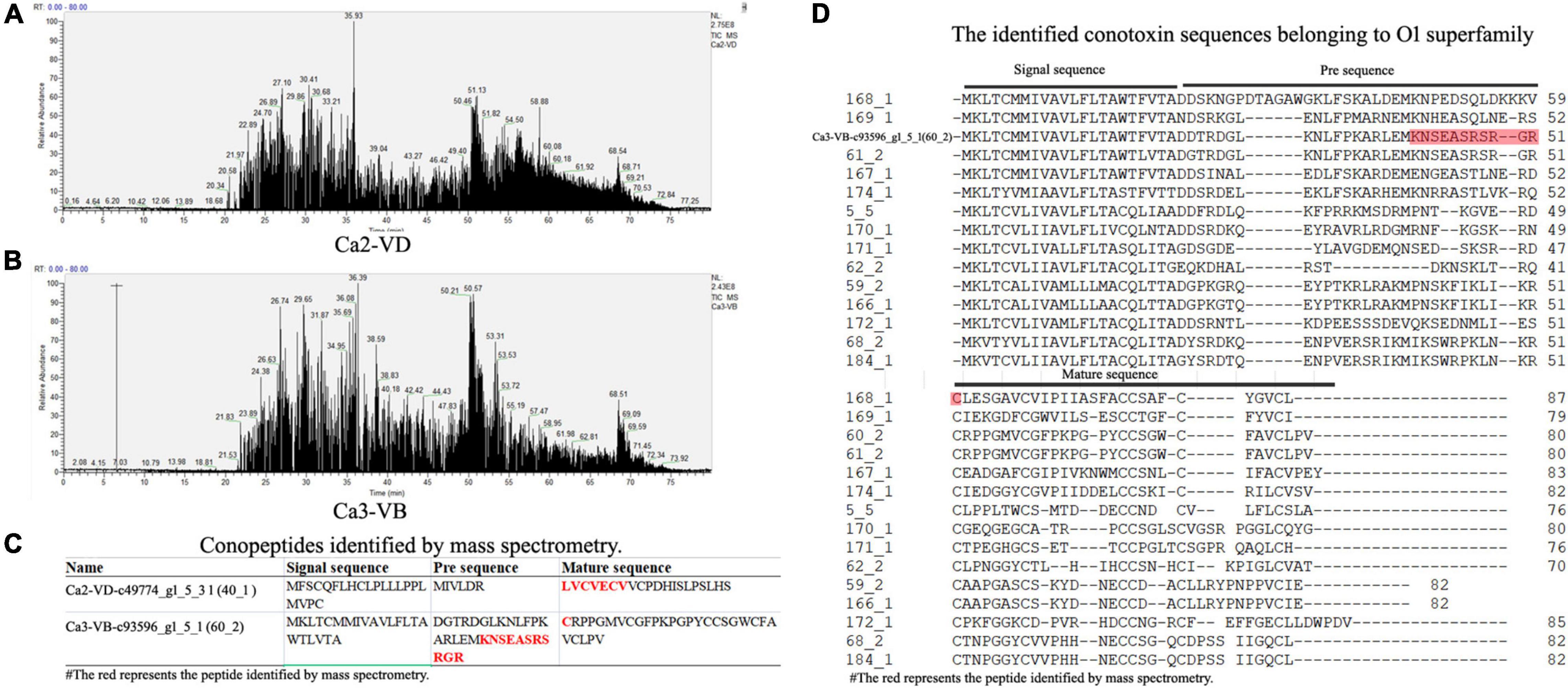
Figure 3. The results of the LC-MS/MS analysis. (A), Total ion current traces of the VD from Conus caracteristicus Ca2. (B), Total ion current traces of VB from C. caracteristicus Ca3. (C), The sequence fragments identified by LC-MS/MS. (D), Sequence alignment of 15 O1 superfamily conotoxins.
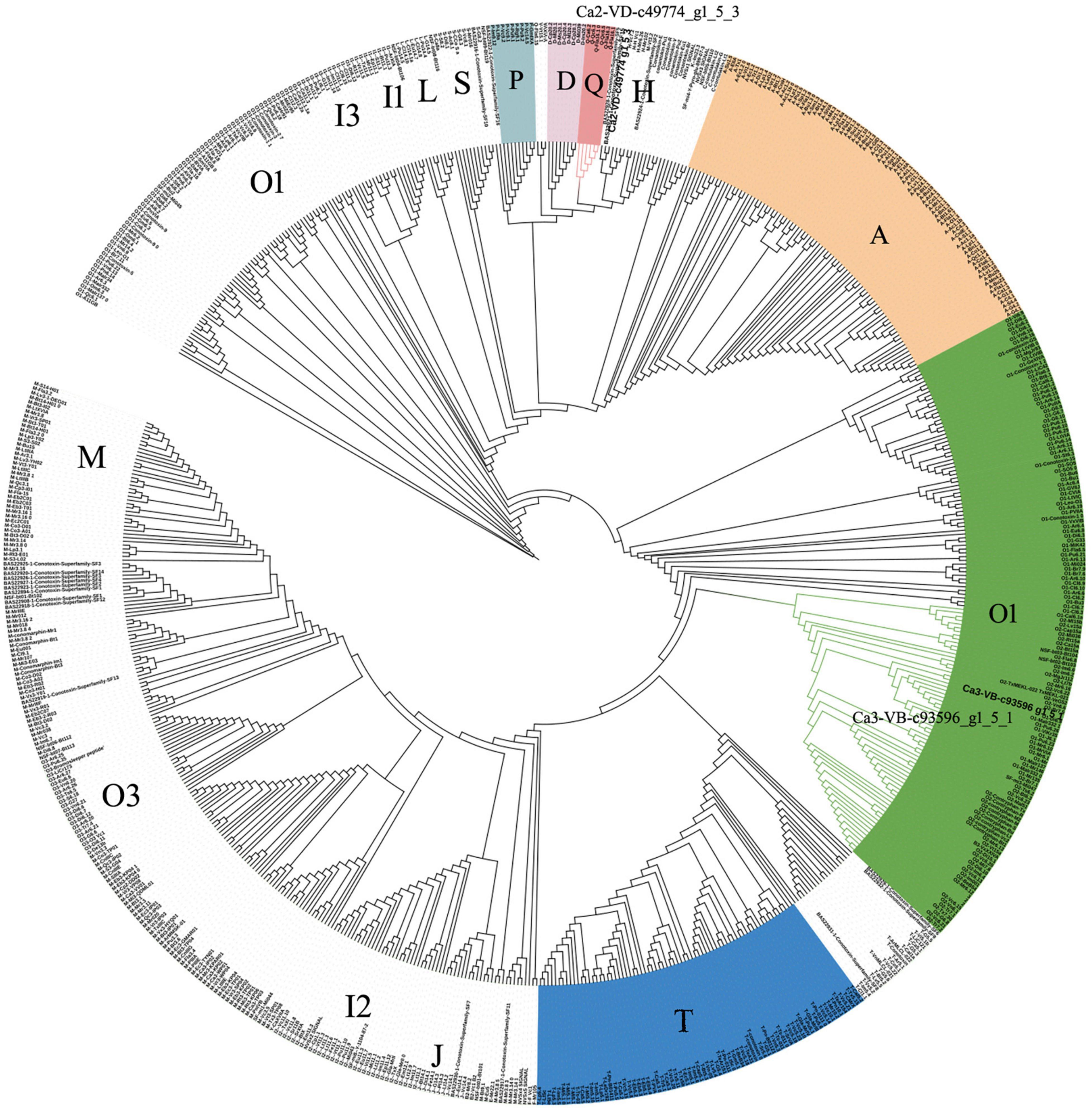
Figure 4. Phylogenetic analysis of signal sequences from novel precursor conopeptides Ca2-VD-c49774_g1_5_3 and previously reported signal sequences. Amino acid sequences were aligned, and a guide tree was constructed from the distance matrix using the CLUSTALW 2.1 multiple sequence alignment algorithm.
Because there was no enrichment of target conopeptides when the samples were processed, eventually only two conopeptides were identified using LC-MS/MS. The abundances of the observed conopeptides were relatively high, suggesting that these were the primary conotoxins used by C. caracteristicus. Due to the low ratio of conopeptides to total proteins, much fewer protein sequences were identified by MS than gene sequences by RNA-seq.
Identification of csPDIA5, a New Protein Disulfide-Isomerase
Protein disulfide isomerase is one of the most critical post-translational modification enzymes affecting conopeptide secondary structure, maturation, and functional activity (Buczek et al., 2005; Bulaj and Olivera, 2008). PDIs are highly diverse both within and among Conus species (Figueroa-Montiel et al., 2016; Safavi-Hemami et al., 2016). We designed PCR primers based on PDI-A5-like, a PDI coding transcript sequence from C. caracteristicus, to isolate cDNA from various Conus species. Putative specific PDI products (length > 2,000 bp) were successfully obtained from C. caracteristicus and C. bartschi (Supplementary Figure 4). Based on homologous sequence alignment, we also extracted the highly homologous sequence of PDI from assembled transcripts of C. textile.
The full-length sequences, obtained using Sanger sequencing (Supplementary Document 1), differed slightly. The distance matrix for the multiple sequence alignments showed that sequence homology among different Conus species was greater than 95% but was less than 80% among Conus species, P. canaliculata, and A. californica (Supplementary Document 2). Based on evolutionary phylogenetic analysis of the three sequences, as well as the characteristics of their thioredoxin-like domains, we identified the novel conotoxin-specific PDI (csPDIA5) (Figures 5, 6). Importantly, these csPDIA5s shared five thioredoxin-like domains: ‘CGYC,’ ‘CGHC,’ ‘CGHC,’ ‘CGHC,’ and ‘CGHC’. For example, csPDIA5 from C. textile had a total length of 2,301 bp and encoded 767 amino acids. In addition, the active site ‘CGH(G)C’ included the conserved sequence ‘GY(F)PTL(F)K(Y)YF,’ and the 3-terminal was rich in proline (Supplementary Figure 5).
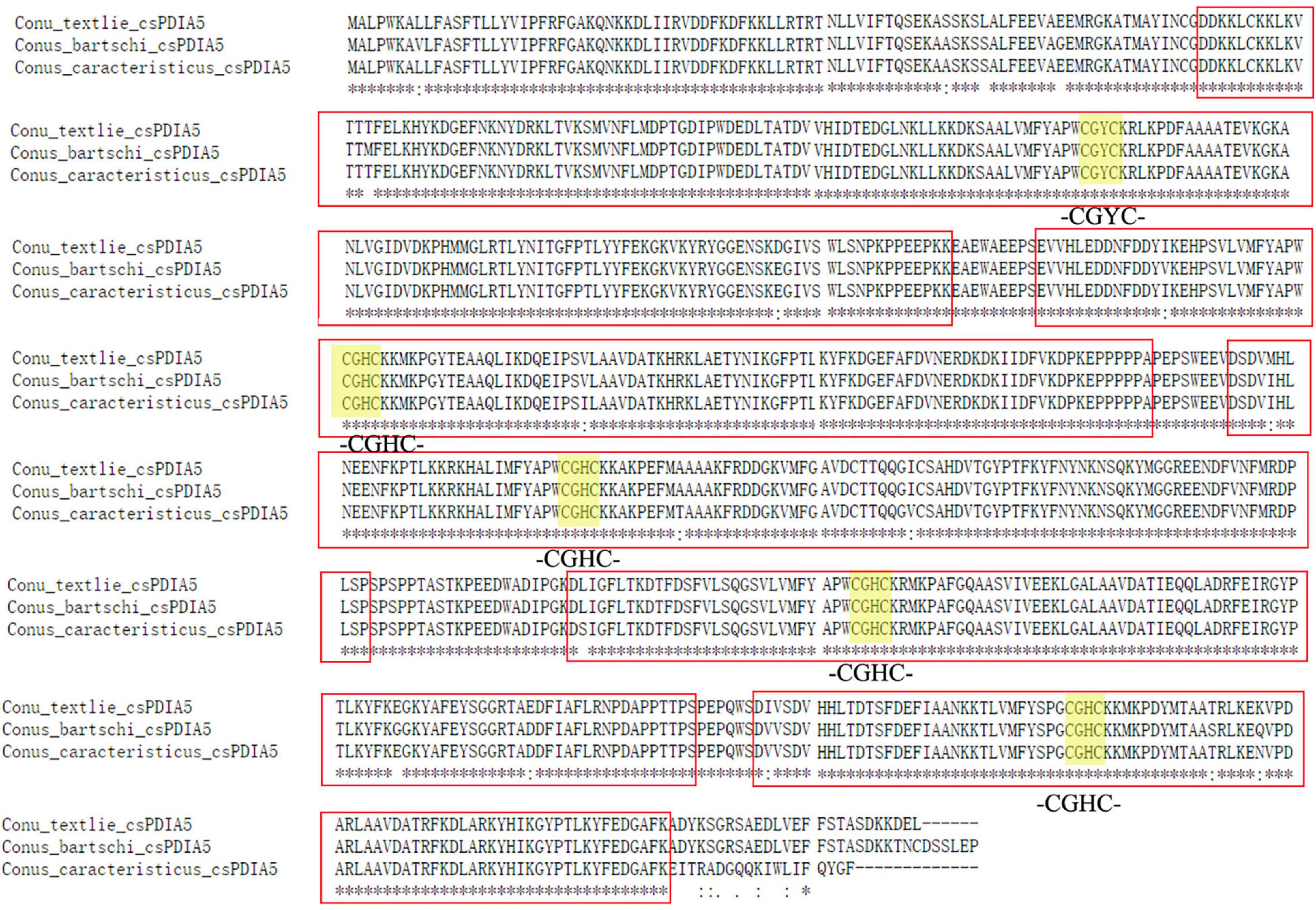
Figure 5. Sequence analysis of csPDIA5 from Conus textile, C. caracteristicus, and C. bartschi.
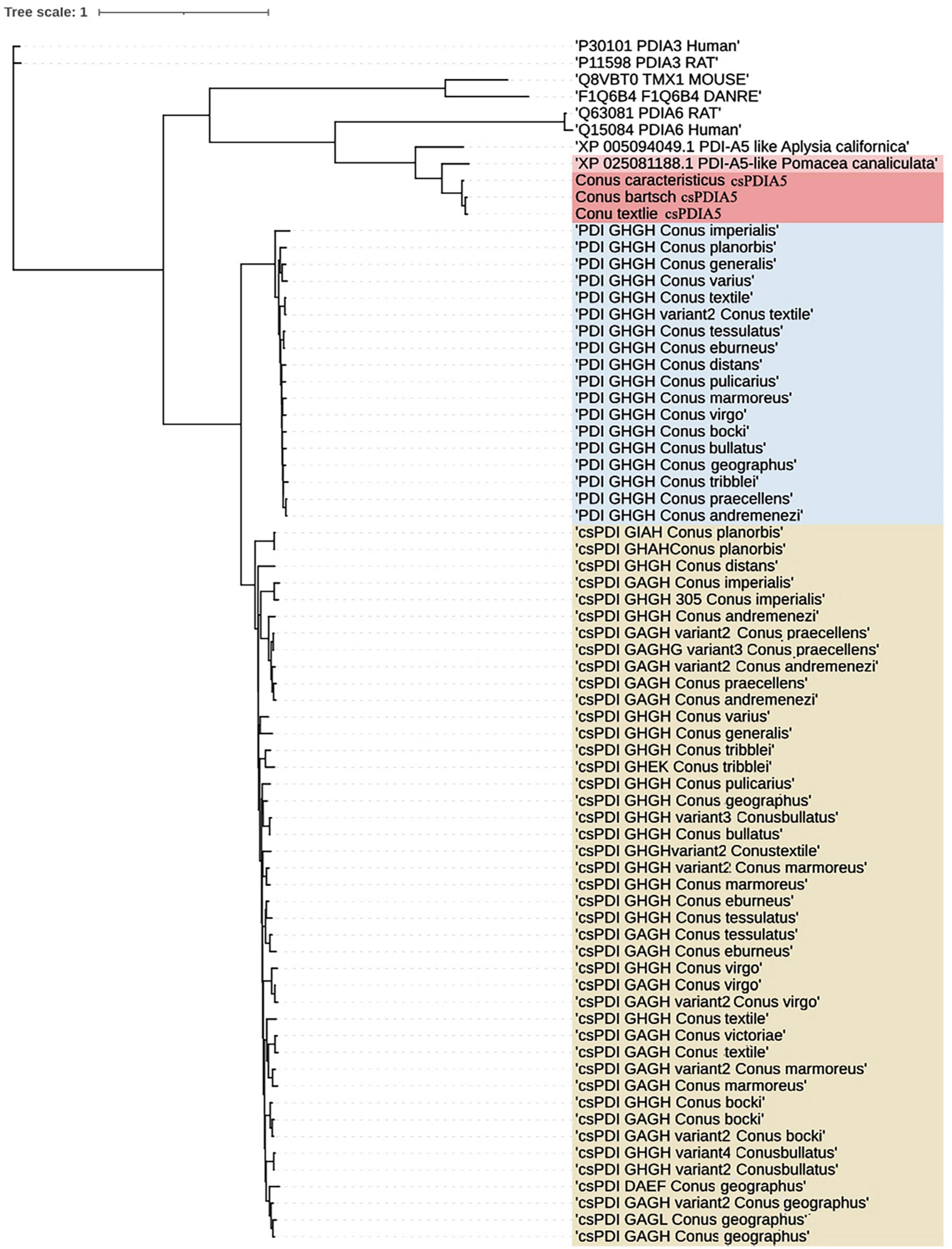
Figure 6. Phylogenetic analysis of members of the PDI gene family. Trees were reconstructed using the neighbor-joining algorithm in UGENE.
Analysis of the Secondary and Tertiary Structure of the csPDIA5 Protein
The secondary protein domains were predicted using SMART software, and the domains of csPDI (GHGH), PDI (GHGH), and csPDIA5 were compared (Figure 7A). We found that csPDIA5 included five thioredoxin domains. However, previous studies have shown that PDI generally contains only three thioredoxin domains (Figueroa-Montiel et al., 2016; Safavi-Hemami et al., 2016). For example, the PDI sequence of another cone snail was highly similar to the human P4HB gene, with only two “CGXC” active sites (Wang et al., 2017).
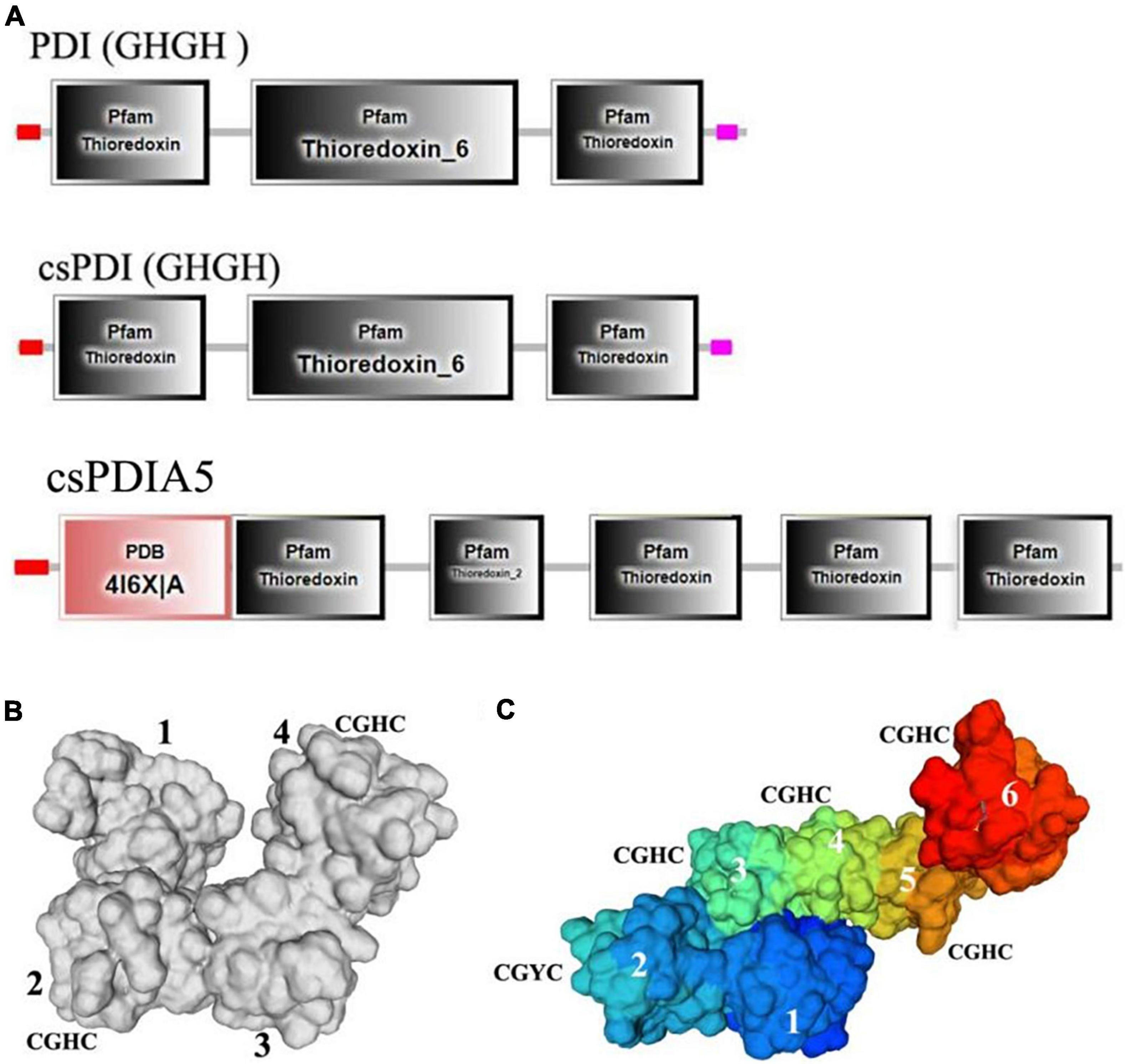
Figure 7. Comparisons of the secondary and tertiary domains among csPDIA5, PDI GHGH, and csPDI GHGH from C. textile. (A), Comparisons of the tertiary structures of csPDIA5, PDI GHGH, and csPDI GHGH. (B), The tertiary structure of csPDIA5; (C), The tertiary structure of csPDI GHGH.
Protein structure determines protein properties and functions. To further explore csPDIA5 domains, we compared the tertiary structures of csPDI to that of the csPDIA5 protein from C. textile (Figures 7B,C). We found that csPDIA5 incorporated six functional structural regions, including five ‘CGH(G)C’ sites, while csPDI included four domains with two “CGHC” sites.
Discussion
Transcriptomics and proteomics are different approaches to gene identification, each with special features. The combination of these two approaches has contributed greatly to the discovery of novel conotoxins (Violette et al., 2012; Jin et al., 2015). Transcriptome sequencing is useful for the identification of rare transcripts, while MS protein sequencing reveals the final secreted peptides. The number of conotoxin genes in VD and VB varies from tens to thousands in previous studies, indicating the potentially high diversity of conotoxin genes and products with each individuals (Lavergne et al., 2015; Peng et al., 2016; Yao et al., 2019; Zhang et al., 2019, 2021). In this study we initially found 1,330 candidate conopeptide precursors on mRNA levels. It seems that the shared number (51) in the VB samples is too big, when compared with previously reported data in other Conus species (such as only few in Peng et al., 2016). It is speculated that there are two reasons. The first is the difference in the methods for data analysis. Peng et al. used BLASTX search and HMMER analysis to predict the assumed conopeptide sequence, and then manually checked it with Conoprec, while we used ConoSort and Conoprec. The second reason is that there are conotoxin differences between different sizes of cono snails and between different species. In addition, not only de novo RNA-seq assembly without additional verification may lead to some errors (Xie et al., 2014), but also machine learning method is regarded as imperfect source with over-classification, thus often gives misleading results (Safavi-Hemami et al., 2015). Using a more strict and conservative restriction for new gene calling, we identified 249 relatively reliable natural conotoxin genes and the remaining 1,081 sequences were considered as candidate genes. Among them, 194 were non-recorded conopeptide precursors.
Sequence alignment and evolutionary tree analysis showed that we identified one putative novel superfamilies. The P gene superfamily, which has been identified in other cone snails (Pi et al., 2006), was herein reported for the first time from C. caracteristicus.
One concern is that we only identified two conopeptides using LC-MS/MS. This may due to the fact that when preparing the total protein, we did not collect the crude venom by centrifugation, but directly processed the protein. Some major unrelated proteins may mask the signal of conotoxin.
Our initial design idea was to study both conotoxins and related post-translational-modification enzymes. In this report, the analysis of two complete organs (VD and VB) from three C. caracteristicus individuals revealed a high level of intraspecies variation among conopeptide precursors in this vermivorous species. Importantly, the analysis of differentially expressed genes showed that the most significantly enriched signaling pathways in the VD were peptide synthesis and processing, which reflected the function of this organ in the synthesis and processing of many conotoxins rich in disulfide bonds.
Post-translational-modification enzymes play a critical role in the maturation and oxidative folding of conopeptides, which are cysteine-rich (Bulaj and Olivera, 2008). PDIs are the primary regulators of this type of post-translational modification (Buczek et al., 2005; Bulaj and Olivera, 2008). Transcriptome sequencing and bioinformatics analysis showed that genes in the disulfide-isomerase family were upregulated compared to any other molecular chaperone genes (Zhang et al., 2019). In human PDIs, the most common active domain is ERDJ5, which includes four catalytic active sites in the thioredoxin domain (Cunnea et al., 2003; Benham, 2012). Herein, we identified the csPDIA5 family, which includes a thioredoxin domain with five catalytic active sites, in a cone snail for the first time. Cone snails are neogastropods in the genus Conus. The mesogastropod P. canaliculate lacks a highly homologous csPDIA5 gene. In addition, we did not identify any highly homologous csPDIA5 genes in any other species in the UniProt protein database or the NCBI nucleic acid database. Finally, it is possible that the csPDIA5 gene family may be unique to the neogastropod Conus genus.
The PDIs and csPDIs are ubiquitously expressed in the VD and play a major role in catalyzing the oxidation of cysteines into their native disulfides (Wang et al., 2007; O’Brien et al., 2018). csPDIs are preferentially expressed in the VD with very low expression levels in other tissues (Safavi-Hemami et al., 2016). In addition, the combinatorial effect of cone snail endoplasmic reticulum oxidoreductin-1 (Conus Ero1) and csPDI provided higher folding yields than Ero1 and PDI in vitro (O’Brien et al., 2018). The active domains of both PDI and csPDI is thioredoxin domains, and usually there are only two domains in each PDI/csPDI identified by now. Notably, we for the first time found csPDIA5 with five thioredoxin domains. We speculate that some cone snails may have evolved the csPDIA5 protein to meet the extreme needs of the oxidative folding process during conopeptide synthesis. In addition, it is worth to further investigate in the future the effects of PDI, csPDI and csPDIA5 on folding rates of conotoxins in vitro, the synergistic effect of other post-translational-modification enzymes in the oxidative folding of conotoxins.
Conclusion
In conclusion, we found 194 previously unreported conopeptide precursors in Conus caracteristicus, identified two conotoxins at the protein level. In addition, we demonstrated that conotoxin diversity among both VD and VB of C. caracteristicus. Finally, we identified the first protein disulfide isomerase (PDI) with five thioredoxin domains. We expect that additional conopeptides will be identified. Nevertheless, this work provides transcriptomic and proteomic data to support future investigations of Conus gene evolution and novel toxin function. Further explorations of the effects of csPDIA5 on the oxidative folding and functional activities of conotoxins may update the current in vitro approaches for the production of conotoxins in pharmaceutics and pharmacologic field.
Materials and Methods
RNA Extraction
Three adult specimens of C. caracteristicus and one specimen of C. textile were collected in the South China Sea. Four VDs and three VBs were extracted and identified as Ct, Ca-1-VD, Ca-2-VD, Ca-3-VD, Ca-1-VB, Ca-2-VB, and Ca-3-VB. VDs and VBs were separated immediately after dissection. Each sample was divided into two parts, and both were stored at −80°C until use. One part was used for RNA extraction, while the other part was used for LC-MS/MS. Total RNA was extracted using TRIzol (Invitrogen), following the manufacturer’s instructions. Agilent 2100 Bioanalyzer and Agilent RNA 6000 nano kit (Agilent Technologies, Santa Clara, CA, United States) were used for the concentration and integrity of the extracted RNA.
Transcriptome Sequencing and Bioinformatics Analysis
The transcriptomes of the C. caracteristicus individuals were paired-end sequenced on an Illumina HiSeq platform. The size of the target fragment was 125 bp. The NGS QC Toolkit (Patel and Jain, 2012) and Trimmomatic-0.60 (Bolger et al., 2014) were used for quality control. The reads were processed to ensure data quality for transcriptome assembly. The filtering criteria were as follows: (1) Remove the adaptor-polluted reads (Reads containing more than 5 adapter-polluted bases were regarded as adaptor-polluted reads and would be filtered out); (2) Remove leading low quality or N bases (below quality 3) (LEADING:3); (3) Remove trailing low quality or N bases (below quality 3) (TRAILING:3); (4) Scan the read with a 4-base wide sliding window, cutting when the average quality per base drops below 15 (SLIDINGWINDOW:4:15); (5) Drop reads below the 100 bases long (MINLEN:100). Both reads of paired-end sequencing data were fifiltered if either read of the paired-end reads was adaptor polluted. The clean reads were checked using FastQC2.
Trinity version 2.33 was used for de novo assembly. For alignment: clean data mapped to assembled transcripts using Bowtie2 version 2.2.34. The alignment rate of more than 75% are considered to meet the downstream analysis criteria. The longest sequences are defined as potential transcripts (unigenes). The open reading frames (ORFs) were predicted by TransDecoder-4.1.05. Then all ORFs and unigenes were used as queries to align against sequences in the databases of Swiss-Prot6, EuKaryotic Orthologous Groups (KOG)7 using the BLAST algorithm with a cut-off e-value of < 10–5.
R packages edgeR was used for differential gene analysis between three VDs and three VBs. The log2fold-change was set to 2, and the P value was set to 0.05. The six unigenes (Ca-1-VD, Ca-2-VD, Ca-3-VD, Ca-1-VB, Ca-2-VB, and Ca-3-VB) were used downstream to conotoxin genes.
Screening New Conotoxins Using ConoSorter
ConoSorter is a machine learning program for the large-scale identification of conopeptide precursors based on their signal, pro- and mature region sequence composition (Lavergne et al., 2013). First, ConoSorter directly analyzes unique transcripts. We chose sequences 40-147 amino acids long. Second, predicted conopeptide precursors with hydrophobicity ≥65% were selected. The predicted genes obtained here are considered to be candidate conotoxin gene databases. Finally, based on the predictions of ConoPrec (Kaas et al., 2012), the sequences with signal peptides, pre-peptides and mature peptides were retained.
Protein Fractionation and Preparation for LC-MS/MS Analysis
The VD samples were stored at −80°C. Sample preparation and analysis were performed following the requirements of the Thermo Scientific EASY-nLC 1,000 system and the Thermo LTQ Orbitrap Elite mass spectrometer, as well as the methods summarized in our previous studies (Zhang et al., 2019). The samples were alkylated at 25 °C for 45 min in the presence of 25 mM iodoacetamide in the dark and then were solubilized in lysis buffer [8 M urea, pH 8.00] containing 5 mM DTT at 60°C for 45 min. At last, the obtained protein solutions were reconstituted in 50 mM ammonium bicarbonate with 0.5 M urea, pH 7.8, and then digested (trypsin: protein = 1:100) for 10 h at 37 °C. The pH of the samples was adjusted to 7.8. The running time for each sample was 50 min.
Proteomic Data Analysis
The total ion current traces of the VD and VB were visualized using Thermo Xcalibur 2.2 sp1.48. pFind Studio 3.1.58 was used for peaklist generation and sequence identification against candidate conopeptide databases (Supplementary Table 2; Chi et al., 2018). For the alkylated samples, the fixed modification was set to carbamidomethyl (+ 57.021 Da). The MS instrument was CID-ITMS (Collision-induced dissociation- Ion Trap Mass Spectrometer). Precursor tolerance was set to 20 ppm and fragment tolerance was set to 20 ppm when searching the databases. The dynamic modification was set to oxidation (+ 15.995 Da, methionine). The maximum modifications per peptide was 4. Precursor tolerance was set to 20 ppm, and fragment tolerance was set to 20 ppm. Up to three missed cleavages were allowed. The enzyme was trypsin, and the FDR (False Discovery Rate) was set to 1%. The peptide mass was 600–10,000 da, and the peptide length was 6–100 amino acids.
Polymerase Chain Reaction and Sanger Sequencing of the New Protein Disulfide Isomerase
From the blast annotation results from Ca-1-VD unigene, we extracted a PDI transcript of more than 2,000 bp. We designed upstream and downstream primers to amplify the full-length sequence from the cDNA template of C. caracteristicus and C.bartschi. The upper-primer was ATGGCGTTGCCCTGGAAAG, and the lower-primer was TCTGATAAAAAGACGAATTGTGA. The 50 μL PCR (Polymerase Chain Reaction) amplification system: 10 × LA Buffer 5 μl, LA Taq DNA Polymerase 0.5 μl, Upper-Primer (10 μM) 1 μl, Lower-Primer (10 μM) 1μl, dNTP 1 μl, cDNA 1∼10 ng (1 μl), Sterile Water 40.5 μl. PCR reaction procedure: 94°C 2 min; 94°C 30 s, 56°C 30 s, 72°C 2 min 20 s, 35 cycle; 72°C 10 min. The effective length for Sanger sequencing was only about 800 bp. We also designed two intermediate sequencing primers (TGATGGGTCTGAGAACCCTGTACA and TGTTTGGGGCTGTGGACTGCAC) based on the full-length gene sequence. An Applied Biosystems 3730 Series Genetic Analyzer was used for Sanger sequencing.
Bioinformatics Analysis of the New Protein Disulfide Isomerase
The open reading frames (ORFs) in the DNA sequences were identified using ORF Finder9. SMART10 was used to predict the secondary domain of the target protein. SWISS-MODEL in SMART is a fully automated protein structure homology-modeling server, accessible via the Expasy web server11. Typically four steps are followed to build a homology model12 : (i) identification of structural template(s), (ii) alignment of target sequence and template structure(s), (iii) model-building, and (iv) evaluation of model quality. The template 3boa.1.A was choose. The 3boa.1.A was crystal structure of yeast protein disulfide isomerase. The sequences of the new PDIs obtained using PCR and Sanger sequencing were compared and analyzed with the CLUSTALW 2.1 (Sievers et al., 2011). All distances were between 0.0 and 1.0.
Phylogenetic Tree
We analyzed signal sequences from the three novel precursor conopeptides identified herein, signal sequences from the gene superfamilies in ConoServer, and 35 previously identified superfamilies (Bernáldez et al., 2013; Lavergne et al., 2015; Prashanth et al., 2016). The nucleic acid sequences of PDI and csPDI were obtained from a previous study (Safavi-Hemami et al., 2016). The remaining sequences (from mouse, Danio rerio, rat, P. canaliculata, A. californica, and human) were obtained from the NCBI and UniProt databases. The rat disulfide-isomerase gene TMX1 was used as the outgroup for the neighbor-joining phylogenetic tree. Amino acid sequences were aligned using CLUSTALW 2.1 (Sievers et al., 2011). All distances were between 0.0 and 1.0.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
HZ, LW, and XP designed the research. HZ and YQ performed the research. HZ, FG, and BN analyzed the data. HZ and XP wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is in part supported by grants from National Key R&D Program of China (2018YFA0507800), National Nature Science Foundation of China (81770173, 32071452), Guangdong Natural Science Foundation (2019B1515120033), Open Fund Programs of Shenzhen Bar Laboratory (SZBL2020090501003), and Pearl River Talents Program Local Innovative and Research Teams (2017BT01S131).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to Sun Yat-sen University and the Shenzhen Science and Technology Industry and Information Committee (ZD20111108120A) for partial material support. We also thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.792908/full#supplementary-material
Footnotes
- ^ http://www.conoserver.org
- ^ http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- ^ https://github.com/trinityrnaseq/trinityrnaseq
- ^ http://bowtie-bio.sourceforge.net/bowtie2
- ^ https://github.com/TransDecoder
- ^ ftp://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz
- ^ ftp://ftp.ncbi.nih.gov/pub/COG/KOG/kyva
- ^ http://pfind.org/software/pFind/index.html
- ^ http://www.bioinformatics.org/sms2/orf_find.html
- ^ http://smart.embl.de/
- ^ http://swissmodel.expasy.org/
- ^ https://swissmodel.expasy.org/interactive
References
Aguilar, M. B., Ortiz, E., Kaas, Q., López-Vera, E., Becerril, B., Possani, L. D., et al. (2013). Precursor De13.1 from Conus delessertii defines the novel G gene superfamily. Peptides 41, 17–20. doi: 10.1016/j.peptides.2013.01.009
Barghi, N., Concepcion, G. P., Olivera, B. M., and Lluisma, A. O. (2015). High conopeptide diversity in Conus tribblei revealed through analysis of venom duct transcriptome using two high-throughput sequencing platforms. Mar. Biotechnol. 17, 81–98. doi: 10.1007/s10126-014-9595-7
Benham, A. M. (2012). The protein disulfide isomerase family: key players in health and disease. Antioxid. Redox Signal. 16, 781–789. doi: 10.1089/ars.2011.4439
Bernáldez, J., Román-González, S. A., Martínez, O., Jiménez, S., Vivas, O., Arenas, I., et al. (2013). A Conus regularis conotoxin with a novel eight-cysteine framework inhibits CaV2.2 channels and displays an anti-nociceptive activity. Mar. Drugs. 11, 1188–1202. doi: 10.3390/md11041188
Biass, D., Dutertre, S., Gerbault, A., Menou, J. L., Offord, R., Favreau, P., et al. (2009). Comparative proteomic study of the venom of the piscivorous cone snail Conus consors. J. Proteom. 72, 210–218. doi: 10.1016/j.jprot.2009.01.019
Biggs, J. S., Watkins, M., Puillandre, N., Ownby, J. P., Lopez-Vera, E., Christensen, S., et al. (2010). Evolution of Conus peptide toxins: analysis of Conus californicus Reeve, 1844. Mol. Phylogenet. Evol. 56, 1–12. doi: 10.1016/j.ympev.2010.03.029
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Buczek, O., Bulaj, G., and Olivera, B. M. (2005). Conotoxins and the posttranslational modification of secreted gene products. Cell Mol. Life Sci. 62, 3067–3079. doi: 10.1007/s00018-005-5283-0
Bulaj, G., and Olivera, B. M. (2008). Folding of conotoxins: formation of the native disulfide bridges during chemical synthesis and biosynthesis of Conus peptides. Antioxid. Redox Signal. 10, 141–155. doi: 10.1089/ars.2007.1856
Chang, D., and Duda, T. F. Jr. (2012). Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol. Biol. Evol. 29, 2019–2029. doi: 10.1093/molbev/mss068
Chi, H., Liu, C., Yang, H., Zeng, W.-F., Wu, L., Zhou, W.-J., et al. (2018). Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat. Biotechnol. 36, 1059–1061. doi: 10.1038/nbt.4236
Cunnea, P. M., Miranda-Vizuete, A., Bertoli, G., Simmen, T., Damdimopoulos, A. E., Hermann, S., et al. (2003). ERdj5, an endoplasmic reticulum (ER)-resident protein containing DnaJ and thioredoxin domains, is expressed in secretory cells or following ER stress. J. Biol. Chem. 278, 1059–1066. doi: 10.1074/jbc.M206995200
Davis, J., Jones, A., and Lewis, R. J. (2009). Remarkable inter- and intra-species complexity of conotoxins revealed by LC/MS. Peptides 30, 1222–1227. doi: 10.1016/j.peptides.2009.03.019
Duda, T. F. Jr., and Palumbi, S. R. (2004). Gene expression and feeding ecology: evolution of piscivory in the venomous gastropod genus Conus. Proc. Biol. Sci. 271, 1165–1174. doi: 10.1098/rspb.2004.2708
Dutertre, S., Jin, A. H., Kaas, Q., Jones, A., Alewood, P. F., and Lewis, R. J. (2013). Deep venomics reveals the mechanism for expanded peptide diversity in cone snail venom. Mol. Cell Proteom. 12, 312–329. doi: 10.1074/mcp.M112.021469
Espiritu, D. J., Watkins, M., Dia-Monje, V., Cartier, G. E., Cruz, L. J., and Olivera, B. M. (2001). Venomous cone snails: molecular phylogeny and the generation of toxin diversity. Toxicon 39, 1899–1916. doi: 10.1016/s0041-0101(01)00175-1
Figueroa-Montiel, A., Ramos, M. A., Mares, R. E., Dueñas, S., Pimienta, G., Ortiz, E., et al. (2016). In Silico Identification of Protein Disulfide Isomerase Gene Families in the De Novo Assembled Transcriptomes of Four Different Species of the Genus Conus. PLoS One 11:e0148390. doi: 10.1371/journal.pone.0148390
Fu, Y., Li, C., Dong, S., and Wu, Y. (2018). Discovery Methodology of Novel Conotoxins from Conus Species. Mar. Drugs. 16:417. doi: 10.3390/md16110417
Gao, B., Peng, C., Yang, J., Yi, Y., Zhang, J., and Shi, Q. (2017). Cone Snails: a Big Store of Conotoxins for Novel Drug Discovery. Toxins 9:397.
Han, T. S., Teichert, R. W., Olivera, B. M., and Bulaj, G. (2008). Conus venoms - a rich source of peptide-based therapeutics. Curr. Pharm. Des. 14, 2462–2479. doi: 10.2174/138161208785777469
Hatahet, F., and Ruddock, L. W. (2009). Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850. doi: 10.1089/ars.2009.2466
Hebert, P. D., Ratnasingham, S., and deWaard, J. R. (2003). Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc. Biol. Sci. 270, S96–S99. doi: 10.1098/rsbl.2003.0025
Jin, A. H., Vetter, I., Himaya, S. W., Alewood, P. F., Lewis, R. J., and Dutertre, S. (2015). Transcriptome and proteome of Conus planorbis identify the nicotinic receptors as primary target for the defensive venom. Proteomics 15, 4030–4040. doi: 10.1002/pmic.201500220
Kaas, Q., Westermann, J. C., and Craik, D. J. (2010). Conopeptide characterization and classifications: an analysis using ConoServer. Toxicon 55, 1491–1509. doi: 10.1016/j.toxicon.2010.03.002
Kaas, Q., Westermann, J. C., Halai, R., Wang, C. K., and Craik, D. J. (2008). ConoServer, a database for conopeptide sequences and structures. Bioinformatics 24, 445–446. doi: 10.1093/bioinformatics/btm596
Kaas, Q., Yu, R., Jin, A. H., Dutertre, S., and Craik, D. J. (2012). ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 40, D325–D330. doi: 10.1093/nar/gkr886
Lavergne, V., Dutertre, S., Jin, A. H., Lewis, R. J., Taft, R. J., and Alewood, P. F. (2013). Systematic interrogation of the Conus marmoreus venom duct transcriptome with ConoSorter reveals 158 novel conotoxins and 13 new gene superfamilies. BMC Genom. 14:708. doi: 10.1186/1471-2164-14-708
Lavergne, V., Harliwong, I., Jones, A., Miller, D., Taft, R. J., and Alewood, P. F. (2015). Optimized deep-targeted proteotranscriptomic profiling reveals unexplored Conus toxin diversity and novel cysteine frameworks. Proc. Natl. Acad. Sci. U.S.A. 112, E3782–E3791. doi: 10.1073/pnas.1501334112
Lebbe, E. K., Peigneur, S., Wijesekara, I., and Tytgat, J. (2014). Conotoxins targeting nicotinic acetylcholine receptors: an overview. Mar. Drugs 12, 2970–3004. doi: 10.3390/md12052970
Li, Q., Barghi, N., Lu, A., Fedosov, A. E., Bandyopadhyay, P. K., Lluisma, A. O., et al. (2017). Divergence of the Venom Exogene Repertoire in Two Sister Species of Turriconus. Chem. Biol. Drug Des. 9, 2211–2225. doi: 10.1093/gbe/evx157
Li, X., Chen, W., Zhangsun, D., and Luo, S. (2020). Diversity of Conopeptides and Their Precursor Genes of Conus Litteratus. Mar. Drugs 18:464.
Liu, Z., Li, H., Liu, N., Wu, C., Jiang, J., Yue, J., et al. (2012). Diversity and evolution of conotoxins in Conus virgo, Conus eburneus, Conus imperialis and Conus marmoreus from the South China Sea. Toxicon 60, 982–989. doi: 10.1016/j.toxicon.2012.06.011
Liu, Z., Xu, N., Hu, J., Zhao, C., Yu, Z., and Dai, Q. (2009). Identification of novel I-superfamily conopeptides from several clades of Conus species found in the South China Sea. Peptides 30, 1782–1787. doi: 10.1016/j.peptides.2009.06.036
Lu, A., Yang, L., Xu, S., and Wang, C. (2014). Various conotoxin diversifications revealed by a venomic study of Conus flavidus. Mol. Cell. Proteomics. 13, 105–118. doi: 10.1074/mcp.M113.028647
Luo, S., Christensen, S., Zhangsun, D., Wu, Y., Hu, Y., Zhu, X., et al. (2013). A novel inhibitor of α9α10 nicotinic acetylcholine receptors from Conus vexillum delineates a new conotoxin superfamily. PLoS One 8:e54648. doi: 10.1371/journal.pone.0054648
O’Brien, H., Kanemura, S., Okumura, M., Baskin, R. P., Bandyopadhyay, P. K., Olivera, B. M., et al. (2018). Ero1-Mediated reoxidation of protein disulfide isomerase accelerates the folding of cone snail toxins. Int. J. Mol. Sci. 19:3418. doi: 10.3390/ijms19113418
Pan, X. (2019). Molecular basis for pore blockade of human Na(+) channel Na(v)1.2 by the μ-conotoxin KIIIA. Mar. Drugs 363, 1309–1313.
Patel, R. K., and Jain, M. (2012). NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. doi: 10.1371/journal.pone.0030619
Peng, C., Huang, Y., Bian, C., Li, J., Liu, J., Zhang, K., et al. (2021). The first Conus genome assembly reveals a primary genetic central dogma of conopeptides in C. betulinus. Cell Discov. 7:11. doi: 10.1038/s41421-021-00244-7
Peng, C., Yao, G., Gao, B. M., Fan, C. X., Bian, C., Wang, J., et al. (2016). High-throughput identification of novel conotoxins from the Chinese tubular cone snail (Conus betulinus) by multi-transcriptome sequencing. Gigascience 5:17. doi: 10.1186/s13742-016-0122-9
Pi, C., Liu, J., Peng, C., Liu, Y., Jiang, X., Zhao, Y., et al. (2006). Diversity and evolution of conotoxins based on gene expression profiling of Conus litteratus. Genomics 88, 809–819. doi: 10.1016/j.ygeno.2006.06.014
Prashanth, J. R., Dutertre, S., Jin, A. H., Lavergne, V., Hamilton, B., Cardoso, F. C., et al. (2016). The role of defensive ecological interactions in the evolution of conotoxins. Mol. Ecol. 25, 598–615. doi: 10.1111/mec.13504
Puillandre, N., Bouchet, P., Duda, T. F. Jr., Kauferstein, S., Kohn, A. J., Olivera, B. M., et al. (2014). Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol. Phylogenet. Evol. 78, 290–303. doi: 10.1016/j.ympev.2014.05.023
Puillandre, N., Koua, D., Favreau, P., Olivera, B. M., and Stöcklin, R. (2012). Molecular phylogeny, classification and evolution of conopeptides. J. Mol. Evol. 74, 297–309. doi: 10.1007/s00239-012-9507-2
Puillandre, N., Watkins, M., and Olivera, B. M. (2010). Evolution of Conus peptide genes: duplication and positive selection in the A-superfamily. J. Mol. Evol. 70, 190–202. doi: 10.1007/s00239-010-9321-7
Safavi-Hemami, H., Bulaj, G., Olivera, B. M., Williamson, N. A., and Purcell, A. W. (2010). Identification of Conus peptidylprolyl cis-trans isomerases (PPIases) and assessment of their role in the oxidative folding of conotoxins. J. Biol. Chem. 285, 12735–12746. doi: 10.1074/jbc.M109.078691
Safavi-Hemami, H., Gajewiak, J., Karanth, S., Robinson, S. D., Ueberheide, B., Douglass, A. D., et al. (2015). Specialized insulin is used for chemical warfare by fish-hunting cone snails. Proc. Natl. Acad. Sci. U.S.A. 112, 1743–1748. doi: 10.1073/pnas.1423857112
Safavi-Hemami, H., Hu, H., Gorasia, D. G., Bandyopadhyay, P. K., Veith, P. D., Young, N. D., et al. (2014). Combined proteomic and transcriptomic interrogation of the venom gland of Conus geographus uncovers novel components and functional compartmentalization. Mol. Cell Proteom. 13, 938–953. doi: 10.1074/mcp.M113.031351
Safavi-Hemami, H., Li, Q., Jackson, R. L., Song, A. S., Boomsma, W., Bandyopadhyay, P. K., et al. (2016). Rapid expansion of the protein disulfide isomerase gene family facilitates the folding of venom peptides. Proc. Natl. Acad. Sci. 113, 3227–3232. doi: 10.1073/pnas.1525790113
Shen, H. (2019). Structures of human Na(v)1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308. doi: 10.1126/science.aaw2999
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Simion, P., Belkhir, K., François, C., Veyssier, J., Rink, J. C., Manuel, M., et al. (2018). A software tool ‘CroCo’detects pervasive cross-species contamination in next generation sequencing data. BMC Biol. 16:28. doi: 10.1186/s12915-018-0486-7
Stanley, S. M. (2008). Predation defeats competition on the seafloor. Paleobiology 34, 1–21. doi: 10.1666/07026.1
Tayo, L. L., Lu, B., Cruz, L. J., and Yates, J. R. III (2010). Proteomic analysis provides insights on venom processing in Conus textile. J. Proteom. Res. 9, 2292–2301. doi: 10.1021/pr901032r
Terlau, H., and Olivera, B. M. (2004). Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol. Rev. 84, 41–68. doi: 10.1152/physrev.00020.2003
Violette, A., Biass, D., Dutertre, S., Koua, D., Piquemal, D., Pierrat, F., et al. (2012). Large-scale discovery of conopeptides and conoproteins in the injectable venom of a fish-hunting cone snail using a combined proteomic and transcriptomic approach. J. Proteom. 75, 5215–5225. doi: 10.1016/j.jprot.2012.06.001
Wang, L., Wang, X., Ren, Z., Tang, W., Zou, Q., Wang, J., et al. (2017). Oxidative Folding of Conopeptides Modified by Conus Protein Disulfide Isomerase. Protein J. 36, 407–416. doi: 10.1007/s10930-017-9738-6
Wang, Z. Q., Han, Y. H., Shao, X. X., Chi, C. W., and Guo, Z. Y. (2007). Molecular cloning, expression and characterization of protein disulfide isomerase from Conus marmoreus. FEBS J. 274, 4778–4787. doi: 10.1111/j.1742-4658.2007.06003.x
Wermeling, D. P. (2005). Ziconotide, an intrathecally administered N-type calcium channel antagonist for the treatment of chronic pain. Pharmacotherapy 25, 1084–1094. doi: 10.1592/phco.2005.25.8.1084
Xie, Y., Wu, G., Tang, J., Luo, R., Patterson, J., Liu, S., et al. (2014). SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 30, 1660–1666. doi: 10.1093/bioinformatics/btu077
Yao, G., Peng, C., Zhu, Y., Fan, C., Jiang, H., Chen, J., et al. (2019). High-throughput identification and analysis of novel conotoxins from three vermivorous cone snails by transcriptome sequencing. Mar. Drugs 17:193. doi: 10.3390/md17030193
Ye, M., Khoo, K. K., Xu, S., Zhou, M., Boonyalai, N., Perugini, M. A., et al. (2012). A helical conotoxin from Conus imperialis has a novel cysteine framework and defines a new superfamily. J. Biol. Chem. 287, 14973–14983. doi: 10.1074/jbc.M111.334615
Zhang, H., Fu, Y., Wang, L., Liang, A., Chen, S., and Xu, A. (2019). Identifying novel conopepetides from the venom ducts of Conus litteratus through integrating transcriptomics and proteomics. J. Proteom. 192, 346–357. doi: 10.1016/j.jprot.2018.09.015
Keywords: conopeptides, Conus, protein disulfide isomerase (PDI), conotoxin, transcriptome
Citation: Zhang H, Wang L, Qiu Y, Gong F, Nong B and Pan X (2022) Discovery of 194 Unreported Conopeptides and Identification of a New Protein Disulfide Isomerase in Conus caracteristicus Using Integrated Transcriptomic and Proteomic Analysis. Front. Mar. Sci. 9:792908. doi: 10.3389/fmars.2022.792908
Received: 11 October 2021; Accepted: 31 January 2022;
Published: 01 March 2022.
Edited by:
Thomas Bartholomäus Brück, Technical University of Munich, GermanyReviewed by:
Wong Yue Him, Shenzhen University, ChinaQiong Shi, Beijing Genomics Institute (BGI), China
Copyright © 2022 Zhang, Wang, Qiu, Gong, Nong and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Han Zhang, emhhbmdoMzU1QG1haWwyLnN5c3UuZWR1LmNu; Xinghua Pan, cGFudmljdG9yQHNtdS5lZHUuY24=