Abstract
The presence of seagrass shapes surface sediments and forms a specific environment for diverse and abundant microbial communities. A severe decline of Cymodocea nodosa, a widespread seagrass species in the Mediterranean Sea, has been documented. To characterise and assess the changes in microbial community composition during the decline of a Cymodocea nodosa meadow, Illumina MiSeq sequencing of the V4 region of the 16S rRNA gene was performed. Samples of surface sediments from two sites, one without any vegetation and one with a declining Cymodocea nodosa meadow, were collected at monthly intervals from July 2017 to October 2018. Microbial communities were stratified by sediment depth and differed between the vegetated and the nonvegetated site. Although the Cymodocea nodosa meadow declined to a point where almost no leaves were present, no clear temporal succession in the community was observed. Taxonomic analysis revealed a dominance of bacterial over archaeal sequences, with most archaeal reads classified as Nanoarchaeota, Thermoplasmatota, Crenarchaeota, and Asgardarchaeota. The bacterial community was mainly composed of Desulfobacterota, Gammaproteobacteria, Bacteroidota, Chloroflexi, Planctomycetota, and Campylobacterota. Our results show that sediment microbial communities are remarkably stable and may resist major disturbances such as seagrass meadow decline.
Introduction
Shallow coastal sediments are often colonized by seagrasses, which cover approximately 0.1 to 0.2% of the global ocean (Duarte, 2002). Seagrasses penetrate the sediment with their roots and rhizomes forming extensive meadows. The presence of seagrass meadows shapes surface sediments and provides a specific environment for diverse and abundant microbial communities (Duarte et al., 2005). Sediments colonized by seagrasses are considered hotspots for microbial activity as seagrass meadows enrich the underlying sediment with organic matter (Duarte et al., 2005). High organic matter content is mainly achieved by releasing dissolved organic carbon from seagrass roots and by trapping organic particles from the water column (Duarte, 2002). Moreover, seagrasses stabilize the underlying sediment, promoting the accumulation of organic matter and sediment particles (Fonseca and Kenworthy, 1987; Terrados and Duarte, 2000; van Katwijk et al., 2010). In addition, seagrass beds can also increase the availability of organic matter through the decomposition of detached leaves, roots and rhizomes (Jensen et al., 2007; Liu et al., 2017).
Studies of marine sediment microbial communities primarily focus on changes in microbial abundance and activity with sediment depth (Jørgensen and Marshall, 2016; Petro et al., 2017; Starnawski et al., 2017; Orsi, 2018). Depth-dependent changes in taxonomic composition have been well described differentiating surface sediment communities dominated by Bacteria, especially Proteobacteria, from deeper communities characterized by Archaea (Orcutt et al., 2011; Chen et al., 2017; Petro et al., 2017). Coastal surface sediments colonized by seagrass are not as well investigated due to studies focusing primarily on rhizosphere communities and only occasionally including sediment communities for comparison (Cúcio et al., 2016; Rabbani et al., 2021). Communities in the rhizosphere are not species-specific and differ from those in the sediment (Cúcio et al., 2016; Ettinger et al., 2017; Zhang et al., 2020). One of the main differences is the higher relative abundance of Desulfobacterota, one of the most abundant sulphate reducing bacteria in seagrass sediments, in contrast to the rhizosphere, which is characterized by Epsilonproteobacteria (Ettinger et al., 2017). When sediment microbial communities were described, the main focus was on the differences between vegetated and nonvegetated sites (Zheng et al., 2019; Sun et al., 2020). In addition, these studies showed that communities differ even with respect to the meadow edge (Ettinger et al., 2017). However, little is known about the response of these communities to seagrass decline. As only limited information is available on the succession of microbial communities in seagrass sediments it is hard to predict how and if seagrass decline influences the underlying sediment communities. It was reported that the sulphate-reducing community in seagrass sediments changes over time (Smith et al., 2004) and that seagrass sediment microbial communities change according to nutrient availability (Guevara et al., 2014). Furthermore, seagrass restoration was also found to alter the sediment microbial community (Bourque et al., 2015). These studies suggest that a temporal community pattern may be observed in sediment communities of seagrass meadows and that these communities could also change as a result of seagrass decline.
In the Mediterranean Sea, Cymodocea nodosa is a widespread seagrass species declining in coastal areas (Ruiz Fernandez et al., 2009; Tuya et al., 2014; Orlando-Bonaca et al., 2015). The rhizosphere and epiphytic communities of C. nodosa have been described (Cúcio et al., 2016; Korlević et al., 2021a), however, little is known about sediment communities underlying C. nodosa meadows. The aim of the present study was to characterize the taxonomic composition of sediment communities of a C. nodosa meadow and to assess the temporal dynamics of these communities. As the studied meadow experienced a major decline (Najdek et al., 2020), we investigated whether this event affected the sediment microbial community structure.
Materials and methods
Sampling
Sediment cores were sampled in a declining C. nodosa meadow (vegetated site) and at an adjacent area without any vegetation (nonvegetated site) both located in the Bay of Saline, east coast of the northern Adriatic Sea (45°7'5'' N, 13°37'20'' E). (Figure 1). One sediment core from each site was collected monthly from July 2017 to October 2018 (Supplementary Table S1) by diving using 15 cm long plastic core samplers. Sediment samples were immediately transported on ice to the laboratory and stored at −80°C until further processing. A detailed description of the study site, the decline of the C. nodosa meadow and the dynamics of environmental conditions during the decline are provided in Najdek et al. (2020). Briefly, at the beginning of the study the seagrass C. nodosa formed a large and dense meadow at the vegetated site. Seagrass roots and rhizomes penetrated into slightly gravelly sandy mud, while shoots and leaves were present from the southwestern coastal area up to the central part of the bay which was without any vegetation. Following the regular vegetation minimum in November 2017, shoots and leaves started to decline, while roots and rhizomes persisted longer. At the end of the study, after a severe meadow decline at the vegetated site only very small patches persisted along the shoreline.
Figure 1
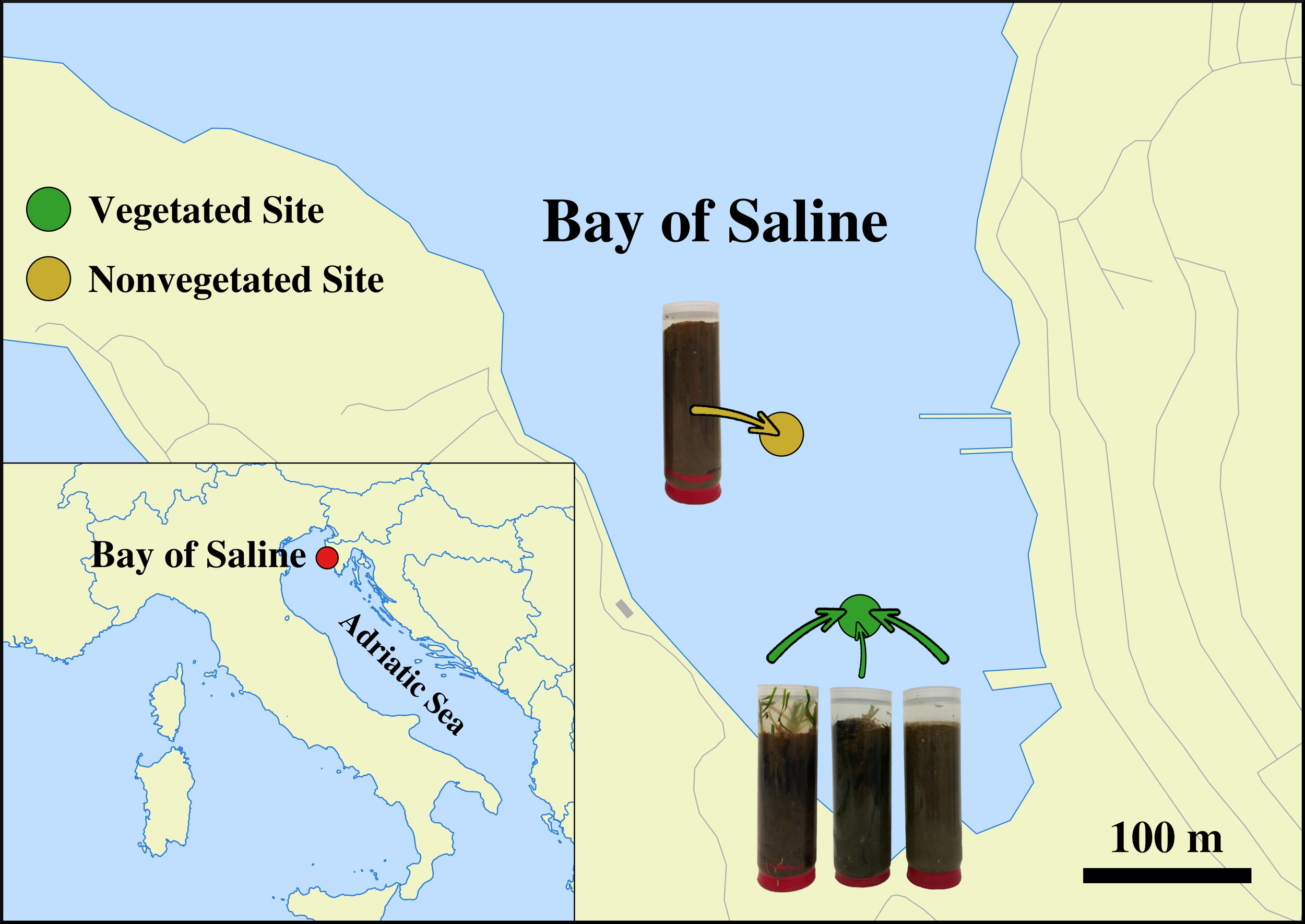
Location of the vegetated (declining Cymodocea nodosa meadow) and nonvegetated site in the Bay of Saline, northern Adriatic Sea, together with visual representations of vegetated and nonvegetated sediment cores (© OpenStreetMap contributors, www.openstreetmap.org/copyright).
DNA isolation
Total DNA from sediment samples was extracted following a modified (Pjevac et al., 2018) isolation protocol of Zhou et al. (1996). Prior to DNA isolation, cores were cut into four different 1 cm sections: top (0 – 1 cm), bottom (7 – 8 cm), and two middle sections: upper middle (1 – 3 cm) and lower middle (3 – 6 cm) section. Sediment samples were weighted (2 g) avoiding roots and rhizomes from vegetated cores, mixed with 5.4 ml of extraction buffer (100 mM Tris [pH 8.0], 100 mM sodium EDTA [pH 8.0], 100 mM Na3PO4 [pH 8.0], 1.5 M NaCl, 1% CTAB) and 10 µl of proteinase K (20 mg ml-1) and incubated by horizontal shaking at 225 rpm at 37°C for 30 min. Thereafter 1.2 ml of 10% SDS was added and the mixture incubated again by horizontal shaking at 225 rpm at 65°C for 60 min. The supernatant was collected after centrifugation at 3220 × g at room temperature for 10 min and mixed with an equal volume of chloroform:isoamyl alcohol (1:1). The aqueous phase was retrieved after centrifugation at 3220 × g at room temperature for 10 min. The extraction procedure with the organic solvent mixture was repeated twice. After the final extraction 0.6 volumes of isopropanol were added to precipitate the DNA. The mixture was incubated at 22°C for 60 min and centrifuged at 3220 × g at room temperature for 45 min. The obtained pellet was washed twice with 10 ml of chilled 70% ethanol, centrifuged at 3220 × g at room temperature for 10 min after each washing, and finally resuspended in 100 μl of deionized water.
Illumina 16S rRNA sequencing
The V4 region of the 16S rRNA gene was sequenced using a two-step PCR approach described previously (Korlević et al., 2021b). Briefly, the V4 region was amplified using the 515F (5'-GTGYCAGCMGCCGCGGTAA-3') and 806R (5'-GGACTACNVGGGTWTCTAAT-3') primers from the Earth microbiome project (https://earthmicrobiome.org/protocols-and-standards/16s/), which contained a sequence tag on the 5' end (Caporaso et al., 2011; Caporaso et al., 2012; Apprill et al., 2015; Parada et al., 2016). Purified samples were sent for Illumina MiSeq sequencing (2 × 250 bp) at IMGM Laboratories (Martinsried, Germany) where the second PCR of the two-step PCR approach was performed using primers targeting the tag region incorporated in the first PCR. These primers also contained adapter and sample-specific index sequences. For each sequencing batch, a positive and a negative control were also sequenced. The positive control consisted of a mock community composed of uniformly mixed DNA from 20 different bacterial strains (ATCC MSA-1002, ATCC, USA), while PCR reactions without DNA template served as the negative control. Sequences obtained in this study have been deposited in the European Nucleotide Archive at EMBL-EBI under the accession numbers SAMEA11293274 – SAMEA11293412 and SAMEA6648825.
Sequence and data analysis
Sequences were analysed on the computer cluster Isabella (University Computing Centre, University of Zagreb) using version 1.45.2 of mothur (Schloss et al., 2009) according to the MiSeq Standard Operating Procedure (MiSeq SOP; https://mothur.org/wiki/miseq_sop) (Kozich et al., 2013) and recommendations given by the Riffomonas project (https://riffomonas.org) to foster data reproducibility. Alignment and classification were performed using the 138.1 release of the SILVA SSU Ref NR 99 database (https://www.arb-silva.de) (Quast et al., 2013; Yilmaz et al., 2014). A cut-off of 97% was used to cluster sequences into operational taxonomic units (OTUs).
Pipeline data processing and visualization were done using R (version 3.6.3) (R Core Team, 2020) combined with packages vegan (version 2.5.7) (Oksanen et al., 2020) tidyverse (version 1.3.1) (Wickham et al., 2019) and multiple other packages (Neuwirth, 2014; Xie, 2014; Xie, 2015; Xie et al., 2018; Xie, 2019; Edwards, 2020; Wilke, 2020; Xie et al., 2020; Xie, 2021a; Xie, 2021b; Allaire et al., 2021; Zhu, 2021). Observed number of OTUs, Chao1, ACE, exponential of the Shannon diversity index and Inverse Simpson diversity index were calculated after normalization to the minimum number of reads per sample to account for different sequencing depths using vegan’s function rrarefy (Oksanen et al., 2020). Chao1 and ACE estimators were calculated using vegan’s function estimateR, while Shannon and Inverse Simpson diversity indices were obtained using vegan’s function diversity (Oksanen et al., 2020). To express both diversity indices in terms of effective number of OTUs the exponential of the Shannon diversity index was retrieved (Jost, 2006). The proportions of shared community members between different sediment layers and the two sites were expressed as the Bray-Curtis similarity coefficient calculated on the OTU data table using vegan’s function vegdist and transformed from dissimilarities to similarities (Legendre and Legendre, 2012; Borcard et al., 2018; Oksanen et al., 2020). The Principal Coordinate Analysis (PCoA) was performed on Bray-Curtis dissimilarities based on OTU abundances using the function wcmdscale (Legendre and Legendre, 2012; Oksanen et al., 2020). Differences between communities of different layers, sites, years, and decay periods were tested by performing the Analysis of Similarities (ANOSIM) using vegan’s function anosim and 1000 permutations (Oksanen et al., 2020). When differences between years or decay periods were tested samples were grouped based on sampling year (2017 and 2018) and decay of roots and rhizomes (before and after decay). The period prior to the decay included samples retrieved from the begining of the study until and including February 2018, while the period after the decay included samples taken after February 2018. To calculate the proportion of OTU community variation explained by environmental variables (redox potential [Eh], oxygen [O2], hydrogen sulfide [H2S], sulfur [S0], organic matter content, and prokaryotic abundance) reported in Najdek et al. (2020) vegan’s function RsquareAdj was applied on the results of the distance-based Redundancy Analysis (db-RDA) (Borcard et al., 2018; Oksanen et al., 2020). To calculate the db-RDA vegan’s function capscale on OTU data and explanatory environmental variables was performed. The analysis was computed using the Bray-Curtis dissimilarity index and the Lingoes correction for negative eigenvalues (Legendre and Legendre, 2012; Borcard et al., 2018; Oksanen et al., 2020). In addition, differences between richness estimators, diversity indices, and relative sequence abundances were tested by performing the Mann-Whitney U test (function wilcox.test), when two groups were compared, or the Kruskal-Wallis H test (function kruskal.test) followed by a pairwise comparison using the Mann-Whitney U test (function pairwise.wilcox.test), when more than two groups were compared. Bonferroni correction was applied to address the problem of multiple comparisons.
In total 3.3 million sequences were obtained after quality curation and exclusion of sequences without known relatives (no relative sequences), and eukaryotic, chloroplast, and mitochondrial sequences. Altogether, 68 samples from the vegetated site and 68 from the nonvegetated site were analysed. The number of reads per sample ranged from 9,722 to 55,381 (Supplementary Table S1). Even with the highest sequencing effort the rarefaction curves did not level off as commonly observed in high-throughput 16S rRNA amplicon sequencing approaches (Supplementary Figures S1, S2). After quality curation and exclusion of sequences as mentioned above, reads were clustered into 89,488 different OTUs. Normalization to the minimum number of sequences (9,722) described earlier resulted in 64,335 distinct OTUs ranging from 1,774 to 3,576 OTUs per sample (Supplementary Figure S3). Based on the positive control, a sequencing error rate of 0.01% was calculated which is in line with previously reported values for high-throughput sequencing data (Kozich et al., 2013; Schloss et al., 2016). Following quality curation, the negative controls yielded on average 34.2 ± 62.6 sequences. The detailed analysis procedure is available in a Github repository (https://github.com/MicrobesRovinj/Markovski_SalineSediment16S_FrontMarSci_2022).
Results
To assess the richness and diversity of microbial communities in sediments of the Bay of Saline the observed number of OTUs, Chao1, ACE, exponential of the Shannon diversity index, and Inverse Simpson diversity index were calculated (Figure 2). The observed number of OTUs was similar between the vegetated (2,746.7 ± 398.4 OTUs) and the nonvegetated site (2,883.0 ± 353.1 OTUs) and showed no statistical difference (p = 0.06). Interestingly, both the highest and lowest number of OTUs were observed at the vegetated site, more specifically the highest number was found in the top layer (2,976.1 ± 262.0 OTUs) and lowest in the bottom layer (2,500.4 ± 462.7 OTUs). These layers were also the only ones showing statistical difference at the vegetated site (Figure 2 and Supplementary Table S2). In contrast, the observed number of OTUs at the nonvegetated site was similar across sediment layers and did not show significant differences (Figure 2 and Supplementary Table S3), although the lowest value was also observed in the bottom layer (2,700.8 ± 378.8 OTUs). During the study period, the observed number of OTUs was variable, with no clear temporal trend observed (Supplementary Figure S3). Chao1, ACE, exponential of the Shannon diversity index and the Inverse Simpson diversity index of sediment communities at the site with and without vegetation were very similar, with no estimate or index showing a statistically significant difference (all p > 0.1). In addition, the Chao1 and ACE richness estimators also showed no significant differences between sediment layers (Figure 2 and Supplementary Tables S2, S3). In contrast, diversity indices at the vegetated site showed a difference between the top and bottom layer and between the upper middle and bottom layer, with exponential of the Shannon diversity index also showing a significant difference between the top and lower middle layer (Figure 2 and Supplementary Table S2). At the nonvegetated site, the different sediment layers showed no statistical difference in either richness or diversity (Figure 2 and Supplementary Table S3). Temporal variability in richness estimates and diversity indices was high at both sites, with no clear trend (Supplementary Figures S3, S4).
Figure 2
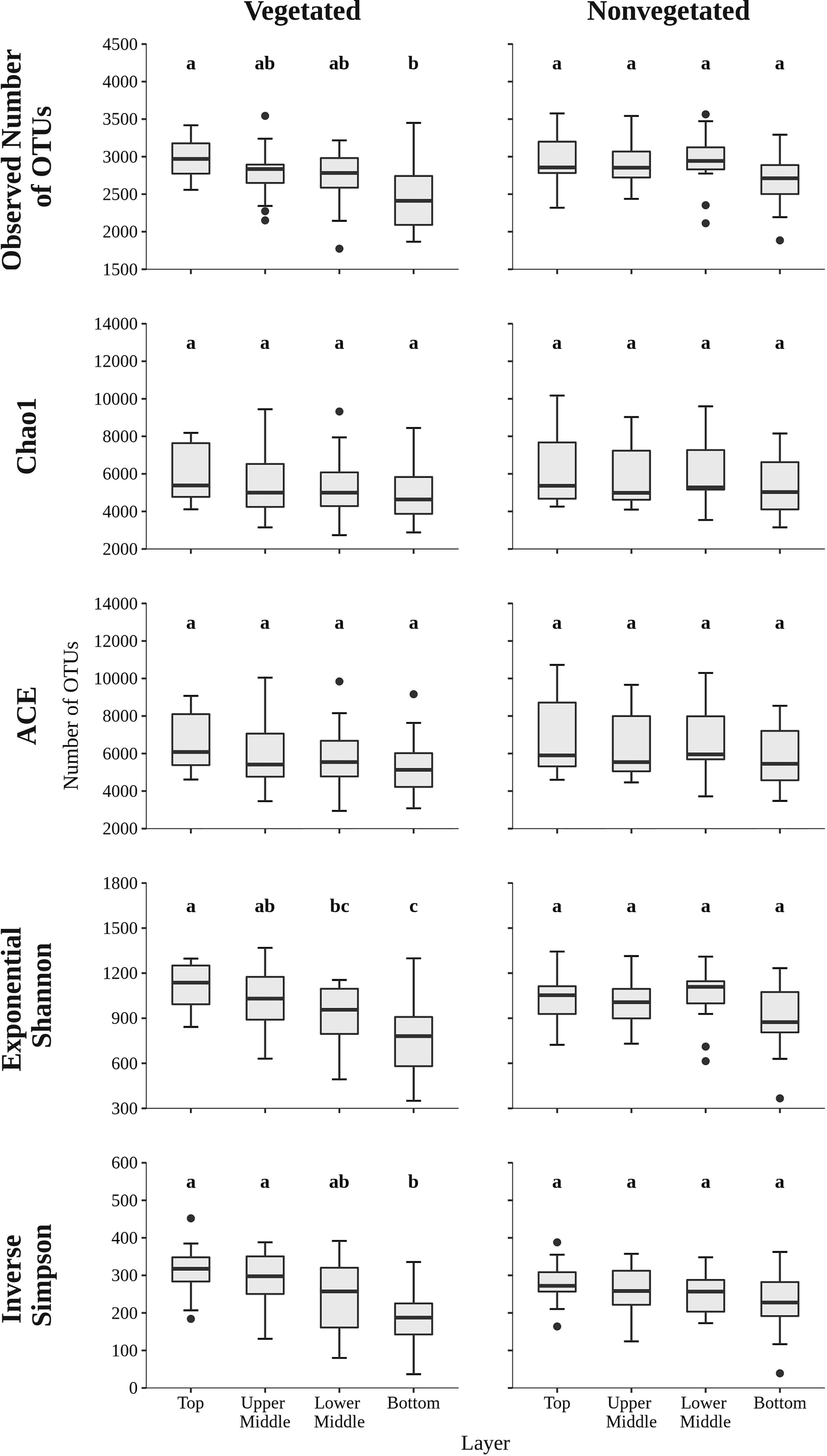
The observed number of OTUs, Chao1, ACE, exponential of the Shannon diversity index, and Inverse Simpson diversity index of sediment microbial communities sampled in different sediment layers of the vegetated and nonvegetated site in the Bay of Saline. Different letters correspond to statistically significant differences.
To evaluate the dynamics of sediment microbial communities Principal Coordinate Analyses (PCoA) of Bray-Curtis distances based on OTU community data were performed. PCoA of all samples differentiated communities based on sediment depth along the first axis, whereas samples from the vegetated and nonvegetated site were separated along the second axis (Figure 3). ANOSIM confirmed that sediment communities in the Bay of Saline differed between sediment layers with some overlap (R = 0.48, p < 0.001), while the communities of the vegetated and nonvegetated site showed a higher degree of overlap (R = 0.27, p < 0.001). When communities of different sediment layers were analysed separately, a clearer differentiation between communities of the vegetated and nonvegetated site was observed (R = 0.45 – 0.49, all p < 0.001). Interestingly, when samples from the same layer of the vegetated and nonvegetated site were compared, the top layers of the sediment showed the highest degree of similarity (Bray-Curtis, 0.64), while the lowest degree of similarity was observed in samples from the upper middle and bottom layers (Bray-Curtis, 0.59) (Figure 3 and Supplementary Figure S5). When samples from each site were analysed separately, the previously observed differentiation of samples based on sediment depth was noted (Figure 4) (ANOSIM; vegetated, R = 0.50, p< 0.001 and nonvegetated R = 0.49, p< 0.001) with the highest degree of similarity observed between samples from middle layers (Bray-Curtis; vegetated, 0.71 and nonvegetated, 0.71) and between lower middle and bottom layers (Bray-Curtis; vegetated, 0.69 and nonvegetated, 0.71) (Supplementary Figure S5). Also, to determine the main environmental parameters governing community changes OTU data were linked to a set of environmental variables reported by Najdek et al. (2020) using db-RDA. Only a small proportion () of the observed community variation could be explained by the environmental variables. To determine whether there is a temporal succession in the community pattern, samples from each layer and site were analysed separately to exclude the effects of sediment depth and vegetation, which have been shown to primarily influence sediment community structure (Figure 4). No grouping of samples by month was observed in any of the layers and sites analysed. Although Najdek et al. (2020) described a sharp decline in above ground biomass in the same meadow since the beginning of 2018, we did not detect a clearly defined grouping of samples based on sampling year in all the analysed layers (ANOSIM; vegetated, R = 0.06 – 0.26, p = 0.05 – 0.18 and nonvegetated, R = 0.03 – 0.18 p = 0.05 – 0.29). In addition, we also analysed the samples according to the reported decline of roots and rhizomes, as belowground biomass showed a later onset of decline than the aboveground biomass (Najdek et al., 2020). However, this analysis also did not reveal a grouping in any of the tested layers (ANOSIM; vegetated, R = 0.07 – 0.19, p = 0.05 – 0.18 and nonvegetated, R = 0.16 – 0.20, p = 0.05 – 0.06). Furthermore, as with the community analysis, taxonomic classification of all samples also did not indicate a temporal succession but a fairly stable community composition was detected in all layers both at the vegetated and nonvegetated site (Supplementary Figure S6).
Figure 3
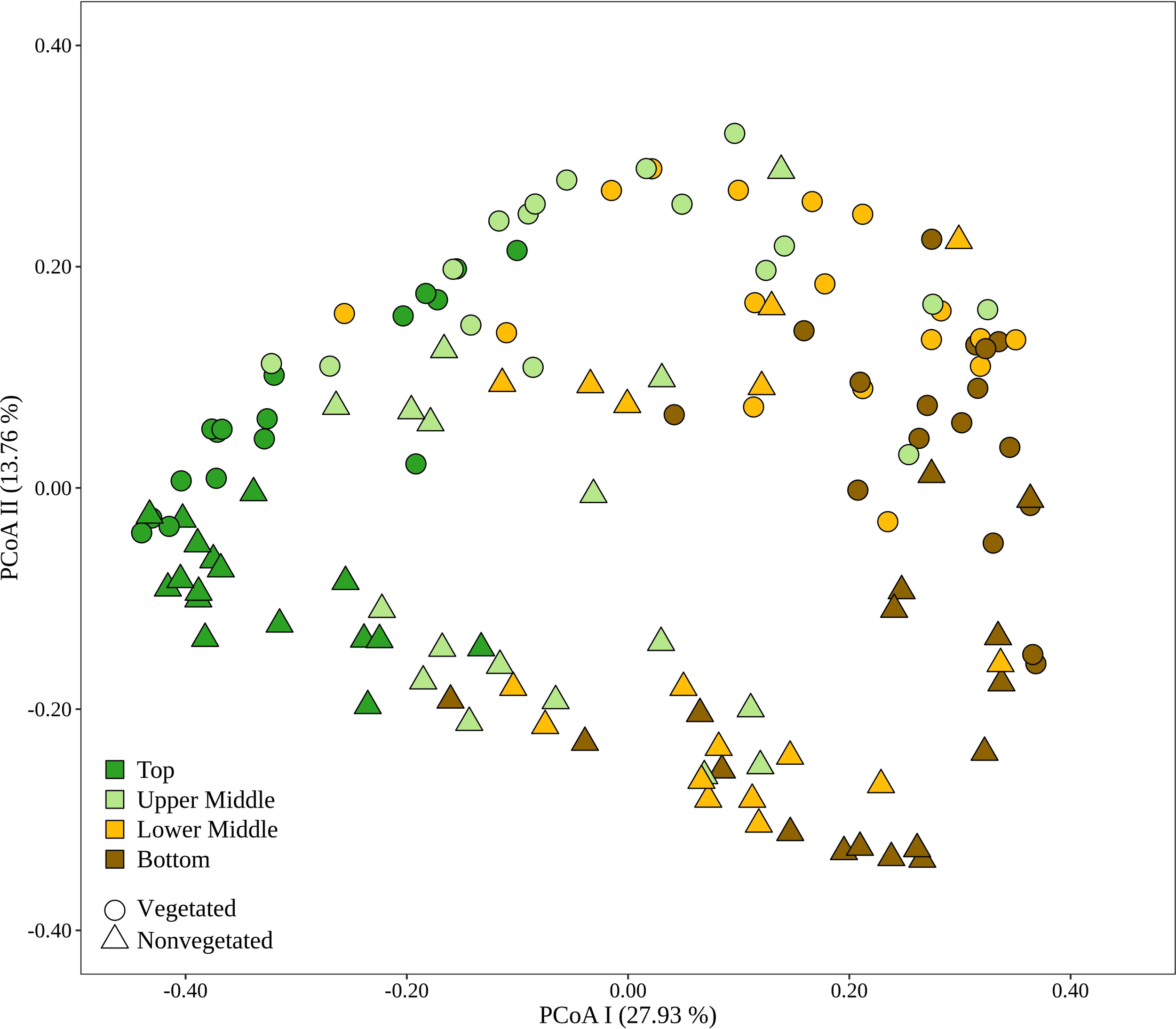
Principal Coordinates Analysis (PCoA) of Bray-Curtis dissimilarities based on OTU abundances of sediment microbial communities sampled in the Bay of Saline. Samples from different sites are labelled with different symbols while samples from different sediment layers are indicated by colour. The proportion of explained variation by each axis is shown on the corresponding axis in parentheses.
Figure 4
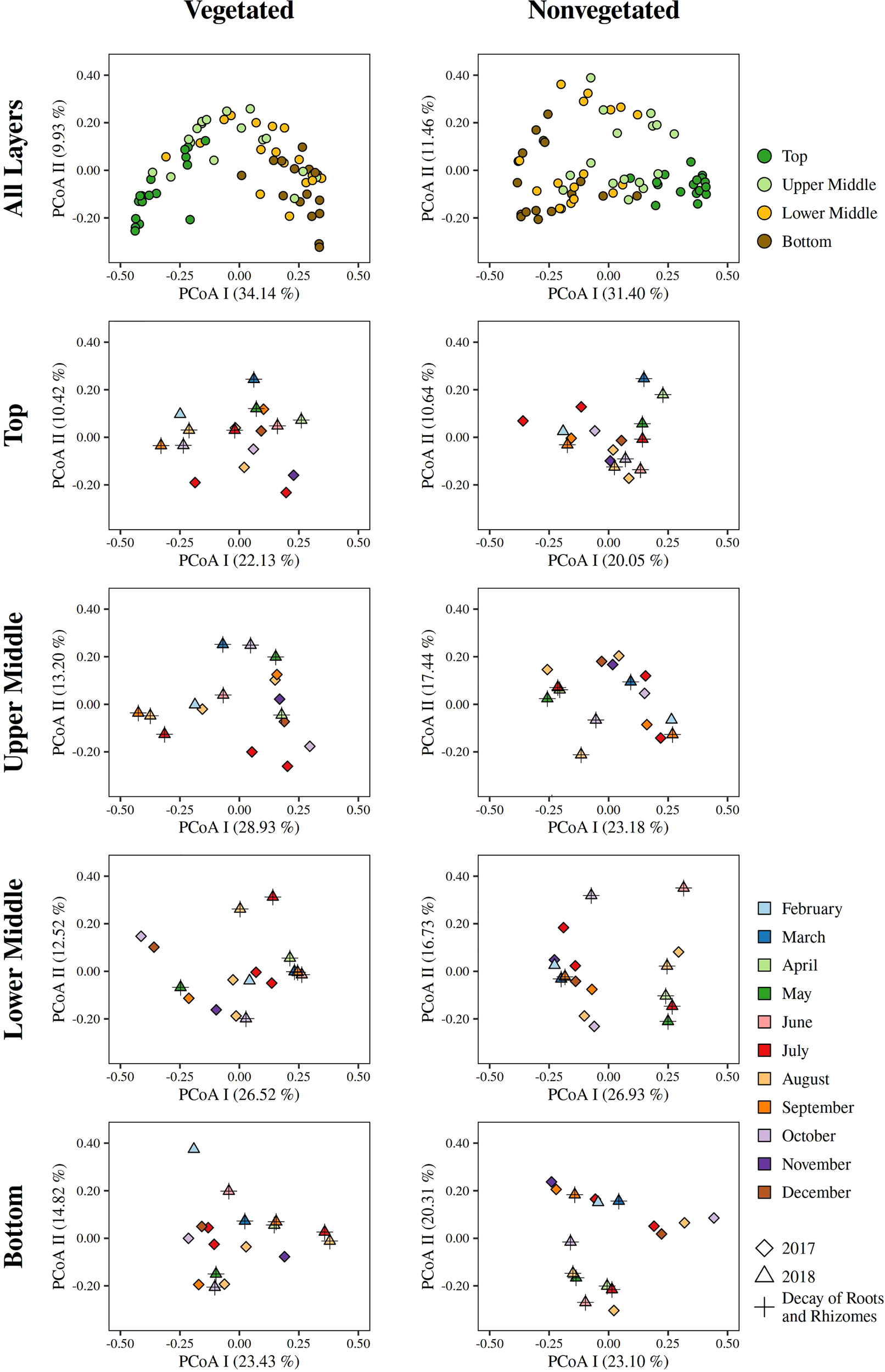
Principal Coordinates Analyses (PCoA) of Bray-Curtis dissimilarities based on OTU abundances of sediment microbial communities, of all and individual sediment layers, sampled at the vegetated and nonvegetated site in the Bay of Saline. The proportion of explained variation by each axis is shown on the corresponding axis in parentheses.
Archaeal sequences comprised 9.5 ± 4.7% of all reads. Sequences classified as Archaea increased in relative abundance from the top (4.5 ± 1.6%) to the bottom sediment layer (14.1 ± 4.0%). The archaeal community was comprised of Nanoarchaeota, Thermoplasmatota, Crenarchaeota, and Asgardarchaeota (Figure 5). Nanoarchaeota comprised 3.6 ± 1.3% of all sequences and were evenly distributed across the different sediment layers, whereas all other archaeal phyla showed a depth-related pattern. All Nanoarchaeota related sequences were classified as Woesearchaeales, with 28.2 ± 13.5% of sequences further classified as SCGC AAA011-D5. A particularly pronounced depth-related pattern was found in Thermoplasmatota. Sequences classified as Thermoplasmatota comprised 4.1 ± 1.2% of all sequences in the bottom sediment layer and only 0.7 ± 0.6% in the top layer. The majority of sequences related to this group was further classified as Marine Benthic Group D and DHVEG-1. Crenarchaeota comprised 1.8 ± 2.3% of all reads. This group had a higher relative sequence abundance at the nonvegetated (2.7 ± 2.8%) than at the vegetated site (1.0 ± 0.9%) (p < 0.0001). The vast majority of Crenarchaeota related sequences were classified as Bathyarcheia. Out of all reads, Asgardarchaeota comprised 0.9 ± 0.7% of sequences that could all be further classified as Lokiarchaeia.
Figure 5
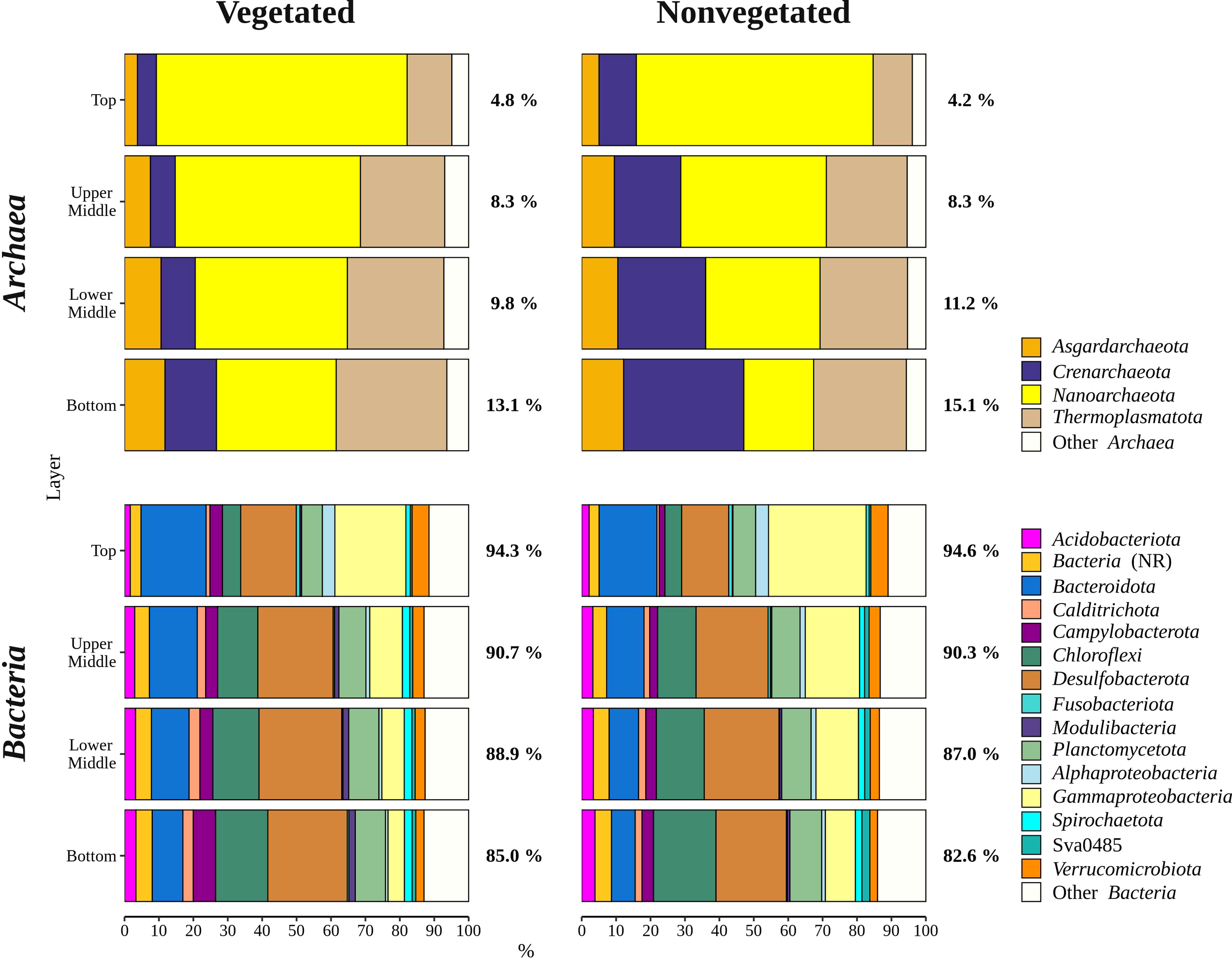
Taxonomic classification and relative contribution of the most abundant bacterial and archaeal (≥ 3%) sequences in sediment communities sampled at the vegetated and nonvegetated site in the Bay of Saline. NR – sequences without known relatives.
Overall, bacterial sequences (90.5 ± 4.7%) dominated over archaeal ones and were mainly comprised of Desulfobacterota, Gammaproteobacteria, Bacteroidota, Chloroflexi, Planctomycetota, and Campylobacterota (Figure 5). Of all reads, Desulfobacterota was the most abundant taxon in the middle (upper middle, 19.4 ± 2.0% and lower middle, 20.2 ± 3.2%) and bottom layers (18.3 ± 3.1%) (Figures 5, 6). Desulfobacterota consisted mainly of Desulfosarcinaceae, Desulfatiglandaceae, Desulfocapsaceae, Desulfobulbaceae, and uncultured members of the order Syntrophobacterales (Figure 6). Sequences classified as Desulfocapsaceae showed affinity for the top sediment layer, where they comprised 26.3 ± 8.2% of Desulfobacterota reads compared to the bottom layer where they constituted only 3.8 ± 3.3% of Desulfobacterota reads. Desulfosarcinaceae and Desulfobulbaceae varied depending on the site. In the whole microbial community, Desulfosarcinaceae reads were more abundant at the vegetated (8.6 ± 2.7%) than nonvegetated site (6.1 ± 2.7%) (p < 0.0001), while sequences classified as Desulfobulbaceae were less represented at the vegetated (0.8 ± 0.7%) than at the nonvegetated site (1.3 ± 0.7%) (p < 0.0001).
Figure 6
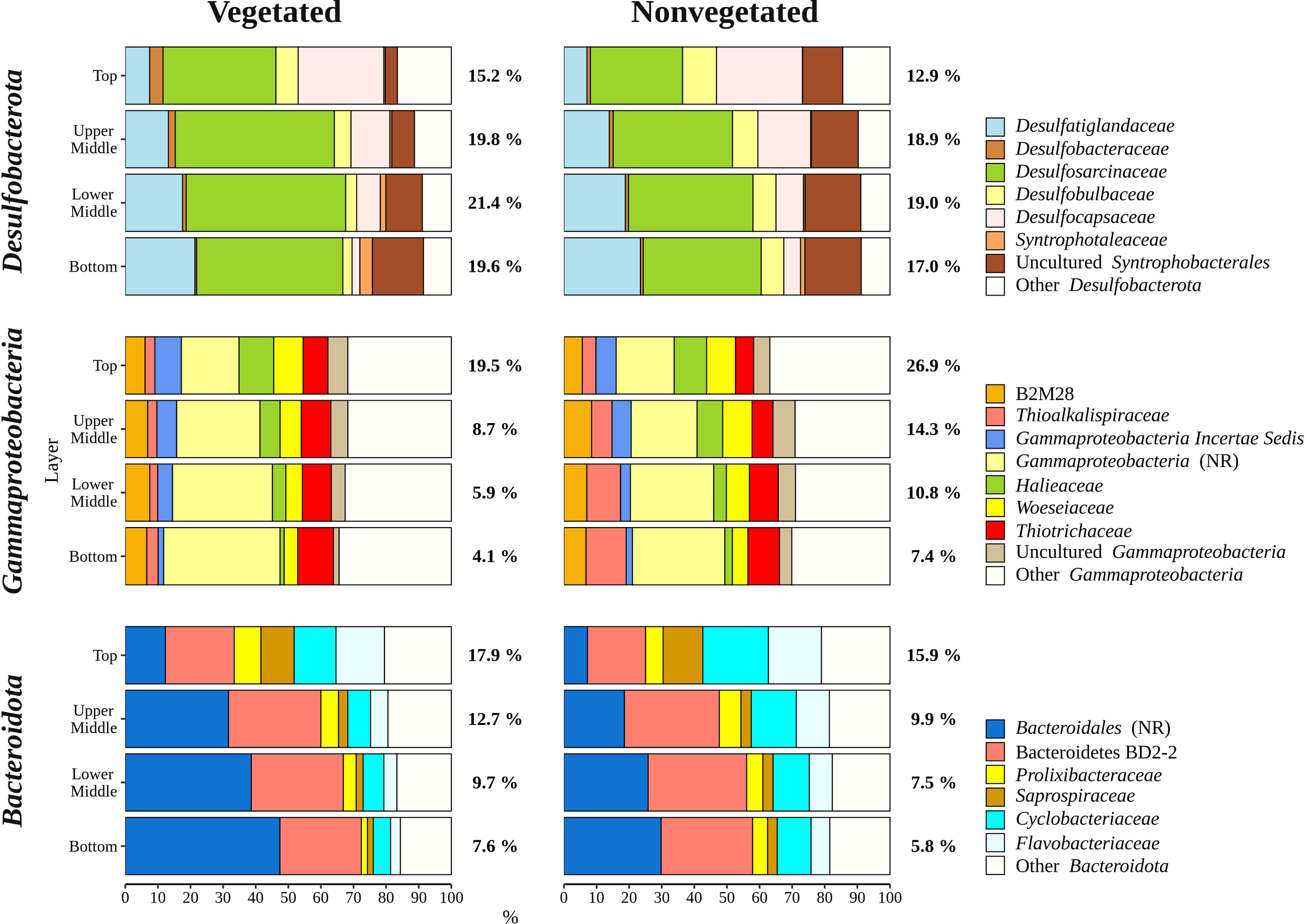
Taxonomic classification and relative contribution of the most abundant (≥ 2%) taxonomic groups within Desulfobacterota, Gammaproteobacteria, and Bacteroidota in sediment communities sampled at the vegetated and nonvegetated site in the Bay of Saline. NR – sequences without known relatives.
Gammaproteobacteria comprised most of the Proteobacteria sequences (87.6 ± 4.1%) and made up the majority of all reads in the top sediment layer (23.2 ± 6.2%) (Figures 5, 6). This group was represented with more sequences at the nonvegetated (14.8 ± 8.9%) than at the vegetated site (9.5 ± 7.4%) (p< 0.001). Out of all gammaproteobacterial sequences, 25.2 ± 8.3% of reads could not be further classified than to the class Gammaproteobacteria (Figure 6). Sequences that could be further classified were mainly assigned to Thiotrichaceae, B2M28, Woeseiaceae, Halieaceae and Thioalkalispiraceae (Figure 6). The observed difference between the relative abundance in Gammaproteobacteria at the two sites was particularly pronounced for Thioalkalispiraceae. Sequences of this group were more abundant at the nonvegetated (1.1 ± 0.8%) than at the vegetated site (0.3 ± 0.3%) (p < 0.0001).
Sequences classified as Bacteroidota were more abundant in the top sediment layer (16.9 ± 2.7%) with their relative abundance decreasing with sediment depth and reaching a minimum in the bottom layer (6.7 ± 2.2%) (Figures 5, 6). A higher relative abundance of Bacteroidota sequences was observed at the vegetated site (12.0 ± 4.5%) than at the nonvegetated site (9.8 ± 4.6%) (p < 0.01). Bacteroidota were mainly composed of sequences without known relatives within Bacteroidales, Bacteroidetes BD2-2, Cyclobacteriaceae, Flavobacteriaceae, Prolixibacteraceae and Saprospiraceae (Figure 6). In contrast to Bacteroidota, sequences classified as Chloroflexi increased with sediment depth (top layer, 4.8 ± 2.0% and bottom layer, 13.8 ± 2.7%) (Figures 5, 7). Chloroflexi were mainly composed of Anaerolineaceae, while SBR1031, uncultured Anaerolineae, sequences without known relatives within Anaerolineae and Dehalococcoidia, AB-539-J10, and Ktedonobacteraceae made up the remainder of the Chloroflexi community (Figure 7).
Figure 7
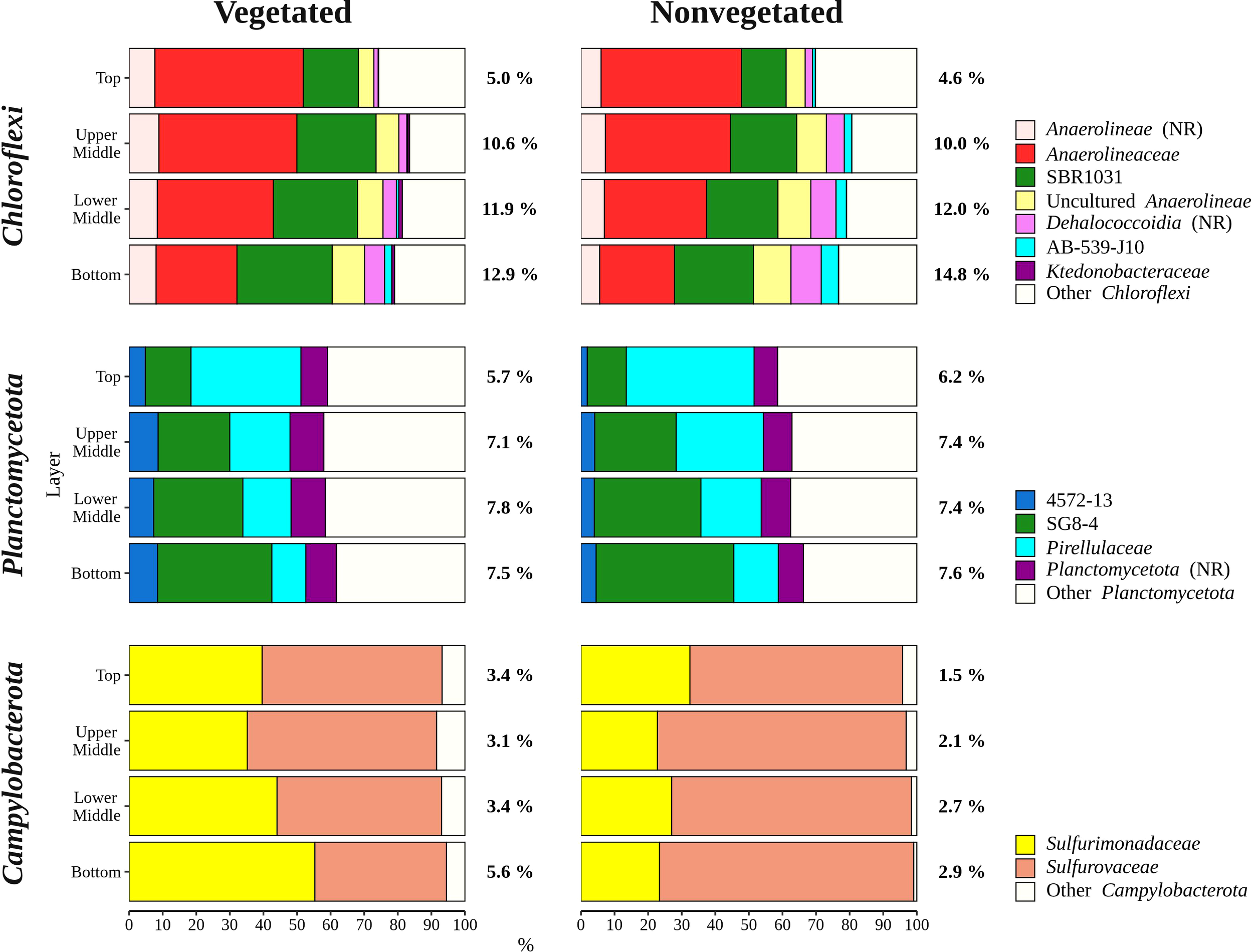
Taxonomic classification and relative contribution of the most abundant (≥ 1%) taxonomic groups within Chloroflexi, Planctomycetota, and Campylobacterota in sediment communities sampled at the vegetated and nonvegetated site in the Bay of Saline. NR – sequences without known relatives.
Planctomycetota were evenly represented in the middle (upper middle, 7.3 ± 0.9% and lower middle, 7.6 ± 0.9%) and bottom layers (7.5 ± 1.0%), and less abundant in the top layer (6.0 ± 0.7%), showing no difference between the sites (vegetated, 7.0 ± 1.2% and nonvegetated, 7.1 ± 1.0%) (Figures 5, 7). The Planctomycetota community consisted mainly of SG8-4, Pirellulaceae, 4572-13, and sequences that could not be further classified (no relative Planctomycetota) (Figure 7). A high proportion of Planctomycetota reads (39.1 ± 5.1%) were assigned to other Planctomycetota, indicating a high diversity within this group. Campylobacterota comprised on average 3.1 ± 3.0% of all sequences (Figures 5, 7). Overall, no pattern related to sediment depth was observed for this group. Slightly higher values were characteristic for the vegetated (3.9 ± 3.5%) than the nonvegetated site (2.3 ± 2.3%) (p < 0.001). When differences between sites were tested for all sediment layers, only the difference in the top layer between the two sites (vegetated, 3.4 ± 1.8% and nonvegetated, 1.5 ± 1.6%) was significant (p < 0.01). Reads related to Campylobacterota could be further classified into two families, Sulfurimonadaceae and Sulfurovaceae (Figure 7). Of these two families, Sulfurimonadaceae showed an area-related difference in relative abundance. Higher values were found at the vegetated (2.3 ± 3.4%) than at the nonvegetated site (0.7 ± 1.8%) (p < 0.0001). Sulfurimonadaceae consisted of the genus Sulfurimonas, while Sulfurovaceae consisted of the genus Sulfurovum.
Discussion
Sediments of seagrass meadows harbour diverse, abundant, and active microbial communities (Smith et al., 2004; Duarte et al., 2005; Sun et al., 2015). Although research on microbial communities of seagrass meadows mainly focused on rhizosphere communities, some studies also included the underlying and surrounding sediment (Jensen et al., 2007; Cúcio et al., 2016; Zhang et al., 2020). As with most sediments, a vertical structuring has been found in the microbial communities of seagrass meadow sediments (Sun et al., 2020). Furthermore, a difference between prokaryotic communities of seagrass meadow sediments and nonvegetated sediments has been observed (Ettinger et al., 2017; Zheng et al., 2019). Temporal studies of these communities are generally rare, and little is known about how microbial communities in seagrass meadow sediments change with meadow decline and loss. In this study, we assessed the microbial communities in the sediment of a declining C. nodosa meadow to gain further insights into the taxonomic composition, vertical structuring, and dynamics of microbial communities in seagrass meadow sediments while at the same time comparing them with bare sediments of a nearby site.
Shannon and Simpson indices account for both richness and evenness and are less sensitive to rare taxa than richness estimators such as ACE and Chao1 (Bent and Forney, 2008). We found no difference in richness (Chao1 and ACE) between sediment layers, suggesting that the observed rare taxa did not play a key role in the vertical structuring of the sediment community in the Bay of Saline (Figure 2). In contrast, diversity indices at the vegetated site showed a depth related pattern (Figure 2). Diversity was highest in the first centimetre of the sediment and differed from the deepest layer (7 – 8 cm). This is consistent with previous studies of marine sediments that describe a decrease in community diversity from the surface to deeper sediment layers, even at small scales within the first few meters (Petro et al., 2017; Hoshino et al., 2020). Seagrasses are known to stabilize the sediment and reduce sediment resuspension (Terrados and Duarte, 2000; van Katwijk et al., 2010). It is possible that the presence of the seagrass, especially roots and rhizomes, increase diversity differences between the top and bottom layer by stabilizing the sediment. In contrast, mechanical mixing may homogenise the sediment together with microbial cells causing more similar microbial diversity in different layers. In addition, seagrass meadows increase the organic matter content of the sediment through the decay of dead tissue (Jensen et al., 2007; Liu et al., 2017), which may have further contributed to the observed differences between sediment layers. Vertical structuring of sediment communities is typically achieved through burial, which is accompanied by selection based on successive changes in environmental conditions (Petro et al., 2017; Kirkpatrick et al., 2019; Marshall et al., 2019). Specific environmental conditions surrounding roots and rhizomes may act as a filter during burial, separating the top from the bottom layer. In contrast, the sediment of the nonvegetated site remained vertically more stable in terms of richness and diversity.
Another component known to differentiate communities in marine sediments besides depth stratification is site-specificity (Polymenakou et al., 2005; Hamdan et al., 2013), which is even more pronounced in seagrass meadows where sediment microbial communities differ not only between the vegetated and nonvegetated area, but also towards the edge of the seagrass patch (Ettinger et al., 2017). In this study, we also observed a grouping of samples according to the two sites (Figure 3), while the microbial communities of both the vegetated and nonvegetated site were stratified according to sediment depth. This is in line with Sun et al. (2020) who noted that the seagrass Zostera marina and Zostera japonica influence the vertical organisation of microbial communities in the sediment. Although the microbial communities at the vegetated site were distinct from the ones at the nonvegetated site, a high degree of overlap was present. Given that the two sampling sites were in close proximity to each other, a high degree of similarity is not surprising. The microbial communities in the Bay of Saline most likely originate from the same source and only through burial undergo a specific selection characteristic for each site. This type of community structuring (Hamdan et al., 2013; Walsh et al., 2016; Petro et al., 2019) is further supported by the highest degree of similarity between the vegetated and nonvegetated site observed in the top sediment layer. Also, such a high similarity of the top sediment layer may be attributed to imports of seagrass detritus to the nonvegetated site. As one of the main carbon sources in C. nodosa meadows (Holmer et al., 2004), seagrass detritus may easily be transported to the adjacent nonvegetated site forming similar communities in the top sediment layer. To assess the temporal dynamics of the microbial community, we analysed each sediment layer and site separately to exclude the influence of sediment depth and site-specificity. Because microbial communities of surface sediments have shorter generation times and higher biomass than communities at deeper sediment strata, and seagrass meadow sediments are hotspots for microbial activity (Duarte et al., 2005; Starnawski et al., 2017), successional changes during the decline of a seagrass meadow could be expected. Surprisingly, the decline of the C. nodosa meadow in the Bay of Saline appeared to have little or no effect on the microbial community, as we did not observe any grouping of communities according to month, year, or meadow condition (Figure 4). In addition, no temporal patterns were observed in the taxonomic composition, richness, or diversity of the microbial community. Such a stable community structure and low proportion of community variation explained by the available environmental variables (Najdek et al., 2020) could be caused by a greater proportion of dormant or dead microbial cells remaining in the sediment, leading to a perceived taxonomic stability (Luna et al., 2002; Jones and Lennon, 2010; Cangelosi and Meschke, 2014; Carini et al., 2016; Torti et al., 2018; Bradley et al., 2019). Taxonomic identification by molecular methods such as sequencing of the 16S rRNA gene cannot distinguish between active and dormant cells, nor whether the cell is alive or dead (Cangelosi and Meschke, 2014). Indeed, it has been reported that in coastal marine sediments dead cells account for 70% of all bacterial cells, while among living bacterial cells only 4% grow actively (Luna et al., 2002). Furthermore, it is possible that the change in community composition may be delayed given that microbial communities in marine sediments often have very long generation times (Jørgensen and Marshall, 2016; Starnawski et al., 2017) and that some recognizable remnants of roots and rhizomes were still observed at the end of the study (Najdek et al., 2020). High metabolic versatility of microbial community members which allows functional continuity to be maintained despite changes in composition (Louca et al., 2018), may also allow for some degree of compositional stability despite changing environmental conditions. Indeed, a decoupling of microbial composition and biogeochemical processes has been observed in sediments. Bowen et al. (2011) have shown that microbial communities in sediments are able to resist compositional changes despite significant variations in external nutrient supply, while Marshall et al. (2021) found that the composition of the nitrogen cycling community might change but these compositional changes are not reflected in functional changes.
The archaeal community of both sites was comprised of Nanoarchaeota, Thermoplasmatota, Crenarchaeota, and Asgardarchaeota which are all typical sediment members (Zheng et al., 2019; Sun et al., 2020). We found a nearly threefold increase in the relative abundance of Archaea in the deepest sediment layer compared to the top layer (Figure 5). This is not surprising as it has been well documented that Bacteria dominate the upper sediment layers while at deeper layers the distribution between Bacteria and Archaea is more uniform (Chen et al., 2017). A particularly pronounced increase in relative abundance with depth was observed for Thermoplasmatota at both sites. It is possible that oxygen penetration in the uppermost sediment layer caused such a pronounced change as representatives of the Marine Benthic Group D and DHVEG-1, accounting for the majority of sequences within the phylum Thermoplasmatota (Rinke et al., 2019), are known to be restricted to anoxic environments (Lloyd et al., 2013).
The main difference between the archaeal community of the vegetated and nonvegetated site was the increased presence of Crenarchaeota in the nonvegetated sediment (Figure 5). This difference resulted from a much greater increase in the relative abundance of Bathyarcheia with increasing depth at the nonvegetated site (Figure 5). In a study comparing archaeal communities in the sediment of a Zostera marina meadow with those of bare sediment, a higher presence of Bathyarchaeota was found in the vegetated sediment, which is not consistent with our results (Zheng et al., 2019). This discrepancy could have been caused by patchiness and different sampling strategies. In contrast to the three samples per vegetated and nonvegetated sediment in the study of Zheng et al. (2019), we analysed sixty-eight samples from each site. Bathyarcheia, formerly known as the Miscellaneous Crenarchaeotal Group (MCG), are typically present in deeper sediment layers as they are well adapted to energy limitation (Kubo et al., 2012). Since seagrasses are known to directly and indirectly enrich the underlying sediment with organic matter (Terrados and Duarte, 2000; Duarte, 2002; Duarte et al., 2005; Jensen et al., 2007; van Katwijk et al., 2010; Liu et al., 2017), it is possible that the presence of C. nodosa caused the observed lower relative abundance of this group in the sediment at the vegetated site.
The sediment bacterial community of both sites consisted of taxonomic groups commonly found in marine sediments such as Desulfobacterota, Gammaproteobacteria, Bacteroidota, Chloroflexi, and Planctomycetota (Walsh et al., 2016; Hoshino et al., 2020), along with Campylobacterota, characteristic of seagrass meadows (Jensen et al., 2007). These major groups showed different patterns in relative abundance depending on sediment depth (Figures 6, 7). The proportion of Gammaproteobacteria and Bacteroidota decreased with sediment depth, while the relative abundance of Chloroflexi increased (Figure 6). Although the proportion of Desulfobacterota remained similar in all sediment layers, Desulfocapsaceae, a major constituent of the Desulfobacterota community, decreased with sediment depth (Figure 6). Gammaproteobacteria and Desulfobacterota (formerly known as Deltaproteobacteria), were reported to decrease with sediment depth, while Chloroflexi increased (Petro et al., 2017). Also, Smith et al. (2004) documented no vertical trend in sulphate-reducing prokaryotes (Desulfobacterota) over a similarly small depth range. Reduction of sulphate is one of many processes that affects pH in sediments, while oxygen penetration controls the depth of pH minima (Silburn et al., 2017). Because Desulfocapsaceae are neutrophilic (Galushko and Kuever, 2021) it is possible that depletion of oxygen below the first centimetre and an increase in hydrogen sulphide with sediment depth (Najdek et al., 2020) contributed to the observed vertical trend of this group. The pronounced decline in Gammaproteobacteria after the top centimetre could also be attributed to the oxygen penetration depth observed in the Bay of Saline (Najdek et al., 2020) coinciding with the abrupt change in the relative abundance of this class. Oxygen availability could also influence the vertical distribution of Chloroflexi and Planctomycetota (Figure 7), as these phyla are known to be prevalent in anoxic sediments (Hoshino et al., 2020). In addition to oxygen availability, the decline of Gammaproteobacteria and Bacteroidota with sediment depth may also be related to the lower availability of fresh organic matter in deeper layers (Middelburg, 1989), as both of these groups are known to break down and assimilate fresh detritus in coastal sediments (Gihring et al., 2009).
The differences in taxonomic composition of microbial communities from the vegetated and nonvegetated site were not as pronounced as those influenced by sediment depth. Gammaproteobacteria made up a large proportion of the microbial community at the nonvegetated site, and as with vertical structuring, their higher presence at this site could be explained by oxygen availability. This class contains representatives with a wide range of metabolisms, including aerobic species (Gutierrez, 2019), which could benefit from the higher oxygen availability at the nonvegetated site (Najdek et al., 2020). Indeed, a study by Ettinger et al. (2017) also found a higher presence of Gammaproteobacteria in the sediment outside a seagrass meadow. The most pronounced difference in the taxonomic composition of this class between the vegetated and nonvegetated site is the higher relative abundance of Thioalkalispiraceae in the nonvegetated sediment (Figure 6). This higher relative abundance could be due to differences in organic matter content. In fact, Thioalkalispiraceae are known to be chemolithoautotrophs (Mori et al., 2011; Mori and Suzuki, 2014) and thus may rely on inorganic compounds rather than organic matter supplied by the seagrass. Slight differences were also observed in the Desulfobacterota community between the vegetated and nonvegetated site. Desulfosarcinaceae were more pronounced at the vegetated site, while Desulfobulbaceae were more pronounced at the nonvegetated site (Figure 6). Although both families have been associated with the rhizosphere of seagrasses (Cúcio et al., 2016), our results are consistent with previous studies that reported a high presence of Desulfosarcinaceae in vegetated sediments and higher relative abundances of Desulfobulbaceae in the nonvegetated sediment (Smith et al., 2004; García-Martínez et al., 2009). The most abundant Desulfobacterota familiy at both the vegetated and nonvegetated site was Desulfosarcinaceae. The high metabolic versatility of this group (Watanabe et al., 2020) may have lead to its even greater proliferation at the vegetated site (Figure 6) where high concentrations of different carbon substrates may become available during decomposition of organic matter. In contrast to Gammaproteobacteria and Desulfobacterota, a higher relative abundance of Bacteroidota at the vegetated site may be influenced by the presence of the plant itself. Seagrass cell walls contain polysaccharides like cellulose (Pfeifer and Classen, 2020) and Bacteroidota have been identified as decomposers of macromolecules such as cellulose (Thomas et al., 2011). The differences between the vegetated and nonvegetated sediment communities were also reflected in the higher proportion of Campylobacterota related sequences at the vegetated site. Campylobacterota, formerly known as Epsilonproteobacteria, are known to be closely associated with roots and rhizomes of seagrasses, particularly Sulfurimonadaceae (Jensen et al., 2007). In this study, the family Sulfurimonadaceae also contributed highly to Campylobacterota at the vegetated site (Figure 7). This high contribution may be caused by close proximity of the sampled sediment to roots and rhizomes. The seagrass selects the rhizosphere microbial community from the surrounding bulk sediment by enrichment of certain taxa and depletion of others (Cúcio et al., 2016; Zhang et al., 2020; Zhang et al., 2022). It may be possible that roots and rhizomes to some degree also alter the composition of the community in the surrounding sediment in their close proximity forming the observed structure of Campylobacterota in our samples.
Taken together, sediment microbial communities in the Bay of Saline were depth stratified, and differed between the vegetated and nonvegetated site, however, remained temporally stable. Although the C. nodosa meadow experienced a sharp decline during the investigation period, no pronounced change in the microbial community was observed. The characterization of the sediment microbial community of the declining C. nodosa meadow in the Bay of Saline forms the basis for further studies based on methods that can differentiate active communities or methods that can provide insight into the prevailing metabolic processes during the period of seagrass decline.
Funding
This study was funded by the Croatian Science Foundation through the MICRO-SEAGRASS project (project number IP-2016-06-7118). GH was supported by the Austrian Science Fund (FWF) through the ARTEMIS project (project number P28781-B21).
Acknowledgments
We would like to thank the University Computing Centre of the University of Zagreb for access to the computer cluster Isabella, Margareta Buterer for technical support and Paolo Paliaga for help during sampling.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repositories andaccession numbers can be found in the article.
Author contributions
MN, GH and MK designed the study. MM, MN and MK performed sampling and laboratory analyses. MM and MK analyzed the data. MM prepared the manuscript with editorial help from MN, GH and MK. All authors contributed to the article and approved the final submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2022.966070/full#supplementary-material
References
1
Allaire J. J. Xie Y. McPherson J. Luraschi J. Ushey K. Atkins A. et al . (2021). rmarkdown: Dynamic documents for R.
2
Apprill A. McNally S. Parsons R. Weber L. (2015). Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat. Microb. Ecol.75, 129–137. doi: 10.3354/ame01753
3
Bent S. J. Forney L. J. (2008). The tragedy of the uncommon: Understanding limitations in the analysis of microbial diversity. ISME J.2, 689–695. doi: 10.1038/ismej.2008.44
4
Borcard D. Gillet F. Legendre P. (2018). Numerical ecology with R. 2nd ed (New York: Springer-Verlag). doi: 10.1007/978-3-319-71404-2
5
Bourque A. S. Vega-Thurber R. Fourqurean J. W. (2015). Microbial community structure and dynamics in restored subtropical seagrass sediments. Aquat. Microb. Ecol.74, 43–57. doi: 10.3354/ame01725
6
Bowen J. L. Ward B. B. Morrison H. G. Hobbie J. E. Valiela I. Deegan L. A. et al . (2011). Microbial community composition in sediments resists perturbation by nutrient enrichment. ISME J.5, 1540–1548. doi: 10.1038/ismej.2011.22
7
Bradley J. A. Amend J. P. LaRowe D. E. (2019). Survival of the fewest: Microbial dormancy and maintenance in marine sediments through deep time. Geobiology17, 43–59. doi: 10.1111/gbi.12313
8
Cangelosi G. A. Meschke J. S. (2014). Dead or alive: Molecular assessment of microbial viability. Appl. Environ. Microbiol.80, 5884–5891. doi: 10.1128/AEM.01763-14
9
Caporaso J. G. Lauber C. L. Walters W. A. Berg-Lyons D. Huntley J. Fierer N. et al . (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J.6, 1621–1624. doi: 10.1038/ismej.2012.8
10
Caporaso J. G. Lauber C. L. Walters W. A. Berg-Lyons D. Lozupone C. A. Turnbaugh P. J. et al . (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A.108, 4516–4522. doi: 10.1073/pnas.1000080107
11
Carini P. Marsden P. J. Leff J. W. Morgan E. E. Strickland M. S. Fierer N. (2016). Relic DNA is abundant in soil and obscures estimates of soil microbial diversity. Nat. Microbiol.2, 16242. doi: 10.1038/nmicrobiol.2016.242
12
Chen X. Andersen T. J. Morono Y. Inagaki F. Jørgensen B. B. Lever M. A. (2017). Bioturbation as a key driver behind the dominance of bacteria over archaea in near-surface sediment. Sci. Rep.7, 2400. doi: 10.1038/s41598-017-02295-x
13
Cúcio C. Engelen A. H. Costa R. Muyzer G. (2016). Rhizosphere microbiomes of european seagrasses are selected by the plant, but are not species specific. Front. Microbiol.7. doi: 10.3389/fmicb.2016.00440
14
Duarte C. (2002). The future of seagrass meadows. Environ. Conserv.29, 192–206. doi: 10.1017/S0376892902000127
15
Duarte C. M. Holmer M. Marbà N. (2005). “Plant microbe interactions in seagrass meadows,” in Interactions between macro- and microorganisms in marine sediments. Eds. KristensenE.HaeseR. R.KostkaJ. E. (Washington: American Geophysical Union), 31–60. doi: 10.1029/CE060p0031
16
Edwards M. S. (2020). Lemon: Freshing up your ‘ggplot2’ plots.
17
Ettinger C. L. Voerman S. E. Lang J. M. Stachowicz J. J. Eisen J. A. (2017). Microbial communities in sediment from Zostera marina patches, but not the Z. marina leaf or root microbiomes, vary in relation to distance from patch edge. PeerJ5, e3246. doi: 10.7717/peerj.3246
18
Fonseca M. S. Kenworthy W. J. (1987). Effects of current on photosynthesis and distribution of seagrasses. Aquat. Bot.27, 59–78. doi: 10.1016/0304-3770(87)90086-6
19
Galushko A. Kuever J. (2021). “ Desulfocapsaceae,” in Bergey’s manual of systematics of Archaea and Bacteria. Ed. WhitmanW. B. (John Wiley & Sons, in association with Bergey’s manual trust), 1–6. doi: 10.1002/9781118960608.fbm00332
20
García-Martínez M. López-López A. Calleja M. L. Marbà N. Duarte C. M. (2009). Bacterial community dynamics in a seagrass (Posidonia oceanica) meadow sediment. Estuaries Coasts32, 276–286. doi: 10.1007/s12237-008-9115-y
21
Gihring T. M. Humphrys M. Mills H. J. Huette M. Kostka J. E. (2009). Identification of phytodetritus-degrading microbial communities in sublittoral Gulf of Mexico sands. Limnol. Oceanogr.54, 1073–1083. doi: 10.4319/lo.2009.54.4.1073
22
Guevara R. Ikenaga M. Dean A. L. Pisani C. Boyer J. N. (2014). Changes in sediment bacterial community in response to long-term nutrient enrichment in a subtropical seagrass-dominated estuary. Microb. Ecol.68, 427–440. doi: 10.1007/s00248-014-0418-1
23
Gutierrez T. (2019). “Marine, aerobic hydrocarbon-degrading Gammaproteobacteria: Overview,” in Taxonomy, genomics and ecophysiology of hydrocarbon-degrading microbes. Ed. McGenityT. J. (Cham: Springer-Verlag), 143–152. doi: 10.1007/978-3-030-14796-9_22
24
Hamdan L. J. Coffin R. B. Sikaroodi M. Greinert J. Treude T. Gillevet P. M. (2013). Ocean currents shape the microbiome of Arctic marine sediments. ISME J.7, 685–696. doi: 10.1038/ismej.2012.143
25
Holmer M. Duarte C. M. Boschker H. T. S. Barrón C. (2004). Carbon cycling and bacterial carbon sources in pristine and impacted Mediterranean seagrass sediments. Aquat. Microb. Ecol.36, 227–237. doi: 10.3354/ame036227
26
Hoshino T. Doi H. Uramoto G.-I. Wörmer L. Adhikari R. R. Xiao N. et al . (2020). Global diversity of microbial communities in marine sediment. Proc. Natl. Acad. Sci. U.S.A.117, 27587–27597. doi: 10.1073/pnas.1919139117
27
Jørgensen B. B. Marshall I. P. (2016). Slow microbial life in the seabed. Annu. Rev. Mar. Sci.8, 311–332. doi: 10.1146/annurev-marine-010814-015535
28
Jensen S. I. Kühl M. Priemé A. (2007). Different bacterial communities associated with the roots and bulk sediment of the seagrass Zostera marina. FEMS Microbiol. Ecol.62, 108–117. doi: 10.1111/j.1574-6941.2007.00373.x
29
Jones S. E. Lennon J. T. (2010). Dormancy contributes to the maintenance of microbial diversity. Proc. Natl. Acad. Sci. U.S.A.107, 5881–5886. doi: 10.1073/pnas.0912765107
30
Jost L. (2006). Entropy and diversity. Oikos113, 363–375. doi: 10.1111/j.2006.0030-1299.14714.x
31
Kirkpatrick J. B. Walsh E. A. D’Hondt S. (2019). Microbial selection and survival in subseafloor sediment. Front. Microbiol.10, 956. doi: 10.3389/fmicb.2019.00956
32
Korlević M. Markovski M. Zhao Z. Herndl G. J. Najdek M. (2021a). Seasonal dynamics of epiphytic microbial communities on marine macrophyte surfaces. Front. Microbiol.12. doi: 10.3389/fmicb.2021.671342
33
Korlević M. Markovski M. Zhao Z. Herndl G. J. Najdek M. (2021b). Selective DNA and protein isolation from marine macrophyte surfaces. Front. Microbiol.12, 665999. doi: 10.3389/fmicb.2021.665999
34
Kozich J. J. Westcott S. L. Baxter N. T. Highlander S. K. Schloss P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol.79, 5112–5120. doi: 10.1128/AEM.01043-13
35
Kubo K. Lloyd K. G. F Biddle J. Amann R. Teske A. Knittel K. (2012). Archaea of the miscellaneous crenarchaeotal group are abundant, diverse and widespread in marine sediments. ISME J.6, 1949–1965. doi: 10.1038/ismej.2012.37
36
Legendre P. Legendre L. (2012). Numerical ecology. 3rd ed (Amsterdam: Elsevier).
37
Liu S. Jiang Z. Deng Y. Wu Y. Zhao C. Zhang J. et al . (2017). Effects of seagrass leaf litter decomposition on sediment organic carbon composition and the key transformation processes. Sci. China Earth Sci.60, 2108–2117. doi: 10.1007/s11430-017-9147-4
38
Lloyd K. G. Schreiber L. Petersen D. G. Kjeldsen K. U. Lever M. A. Steen A. D. et al . (2013). Predominant archaea in marine sediments degrade detrital proteins. Nature496, 215–218. doi: 10.1038/nature12033
39
Louca S. Polz M. F. Mazel F. Albright M. B. N. Huber J. A. O’Connor M. I. et al . (2018). Function and functional redundancy in microbial systems. Nat. Ecol. Evol.2, 936–943. doi: 10.1038/s41559-018-0519-1
40
Luna G. M. Manini E. Danovaro R. (2002). Large fraction of dead and inactive bacteria in coastal marine sediments: Comparison of protocols for determination and ecological significance. Appl. Environ. Microbiol.68, 3509–3513. doi: 10.1128/AEM.68.7.3509-3513.2002
41
Marshall A. Longmore A. Phillips L. Tang C. Hayden H. Heidelberg K. et al . (2021). Nitrogen cycling in coastal sediment microbial communities with seasonally variable benthic nutrient fluxes. Aquat. Microb. Ecol.86, 1–19. doi: 10.3354/ame01954
42
Marshall I. P. G. Ren G. Jaussi M. Lomstein B. A. Jørgensen B. B. Røy H. et al . (2019). Environmental filtering determines family-level structure of sulfate-reducing microbial communities in subsurface marine sediments. ISME J.13, 1920–1932. doi: 10.1038/s41396-019-0387-y
43
Middelburg J. J. (1989). A simple rate model for organic matter decomposition in marine sediments. Geochim. Cosmochim. Acta53, 1577–1581. doi: 10.1016/0016-7037(89)90239-1
44
Mori K. Suzuki K.-I. (2014). “The family Thioalkalispiraceae,” in The prokaryotes: Gammaproteobacteria. Eds. RosenbergE.DeLongE. F.LoryS.StackebrandtE.ThompsonF. (Berlin, Heidelberg: Springer-Verlag), 653–658. doi: 10.1007/978-3-642-38922-1_399
45
Mori K. Suzuki K.-I. Urabe T. Sugihara M. Tanaka K. Hamada M. et al . (2011). Thioprofundum hispidum sp. nov., an obligately chemolithoautotrophic sulfur-oxidizing gammaproteobacterium isolated from the hydrothermal field on suiyo seamount, and proposal of Thioalkalispiraceae fam. nov. in the order Chromatiales. Int. J. Syst. Evol. Microbiol.61, 2412–2418. doi: 10.1099/ijs.0.026963-0
46
Najdek M. Korlević M. Paliaga P. Markovski M. Ivančić I. Iveša L. et al . (2020). Dynamics of environmental conditions during the decline of a Cymodocea nodosa meadow. Biogeosciences17, 3299–3315. doi: 10.5194/bg-17-3299-2020
47
Neuwirth E. (2014). RColorBrewer: ColorBrewer palettes.
48
Oksanen J. Blanchet F. G. Friendly M. Kindt R. Legendre P. McGlinn D. et al . (2020). vegan: Community ecology package.
49
Orcutt B. N. Sylvan J. B. Knab N. J. Edwards K. J. (2011). Microbial ecology of the dark ocean above, at, and below the seafloor. microbiol. Mol. Biol. Rev.75, 361–422. doi: 10.1128/MMBR.00039-10
50
Orlando-Bonaca M. Francé J. Mavrič B. Grego M. Lipej L. Flander-Putrle V. et al . (2015). A new index (MediSkew) for the assessment of the Cymodocea nodosa (Ucria) Ascherson meadow’s status. Mar. Environ. Res.110, 132–141. doi: 10.1016/j.marenvres.2015.08.009
51
Orsi W. D. (2018). Ecology and evolution of seafloor and subseafloor microbial communities. Nat. Rev. Microbiol.16, 671–683. doi: 10.1038/s41579-018-0046-8
52
Parada A. E. Needham D. M. Fuhrman J. A. (2016). Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol.18, 1403–1414. doi: 10.1111/1462-2920.13023
53
Petro C. Starnawski P. Schramm A. Kjeldsen K. (2017). Microbial community assembly in marine sediments. Aquat. Microb. Ecol.79, 177–195. doi: 10.3354/ame01826
54
Petro C. Zäncker B. Starnawski P. Jochum L. M. Ferdelman T. G. Jørgensen B. B. et al . (2019). Marine deep biosphere microbial communities assemble in near-surface sediments in Aarhus Bay. Front. Microbiol.10, 758. doi: 10.3389/fmicb.2019.00758
55
Pfeifer L. Classen B. (2020). The cell wall of seagrasses: Fascinating, peculiar and a blank canvas for future research. Front. Plant Sci.11, 588754. doi: 10.3389/fpls.2020.588754
56
Pjevac P. Meier D. V. Markert S. Hentschker C. Schweder T. Becher D. et al . (2018). Metaproteogenomic profiling of microbial communities colonizing actively venting hydrothermal chimneys. Front. Microbiol.9, 680. doi: 10.3389/fmicb.2018.00680
57
Polymenakou P. Bertilsson S. Tselepides A. Stephanou E. (2005). Bacterial community composition in different sediments from the Eastern Mediterranean Sea: A comparison of four 16S ribosomal DNA clone libraries. Microb. Ecol.50, 447–462. doi: 10.1007/s00248-005-0005-6
58
Quast C. Pruesse E. Yilmaz P. Gerken J. Schweer T. Yarza P. et al . (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res.41, D590–D596. doi: 10.1093/nar/gks1219
59
Rabbani G. Yan B. C. Lee N. L. Y. Ooi J. L. S. Lee J. N. Huang D. et al . (2021). Spatial and structural factors shape seagrass-associated bacterial communities in Singapore and Peninsular Malaysia. Front. Mar. Sci.8. doi: 10.3389/fmars.2021.659180
60
R Core Team (2020). R: A language and environment for statistical computing (Vienna, Austria: R foundation for statistical computing).
61
Rinke C. Rubino F. Messer L. F. Youssef N. Parks D. H. Chuvochina M. et al . (2019). A phylogenomic and ecological analysis of the globally abundant marine group II archaea (Ca. Poseidoniales ord. nov.). ISME J.13, 663–675. doi: 10.1038/s41396-018-0282-y
62
Ruiz Fernandez J. M. Boudouresque C. Enríquez S. (2009). Seagrass ecosystems and Mediterranean seagrasses. Botanica Marina52, 369–381. doi: 10.1515/BOT.2009.058
63
Schloss P. D. Jenior M. L. Koumpouras C. C. Westcott S. L. Highlander S. K. (2016). Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ4, e1869. doi: 10.7717/peerj.1869
64
Schloss P. D. Westcott S. L. Ryabin T. Hall J. R. Hartmann M. Hollister E. B. et al . (2009). Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol.75, 7537–7541. doi: 10.1128/AEM.01541-09
65
Silburn B. Kröger S. Parker E. R. Sivyer D. B. Hicks N. Powell C. F. et al . (2017). Benthic pH gradients across a range of shelf sea sediment types linked to sediment characteristics and seasonal variability. Biogeochemistry135, 69–88. doi: 10.1007/s10533-017-0323-z
66
Smith A. C. Kostka J. E. Devereux R. Yates D. F. (2004). Seasonal composition and activity of sulfate-reducing prokaryotic communities in seagrass bed sediments. Aquat. Microb. Ecol.37, 183–195. doi: 10.3354/ame037183
67
Starnawski P. Bataillon T. Ettema T. J. G. Jochum L. M. Schreiber L. Chen X. et al . (2017). Microbial community assembly and evolution in subseafloor sediment. Proc. Natl. Acad. Sci. U.S.A.114, 2940–2945. doi: 10.1073/pnas.1614190114
68
Sun Y. Song Z. Zhang H. Liu P. Hu X. (2020). Seagrass vegetation affect the vertical organization of microbial communities in sediment. Mar. Environ. Res.162, 105174. doi: 10.1016/j.marenvres.2020.105174
69
Sun F. Zhang X. Zhang Q. Liu F. Zhang J. Gong J. (2015). Seagrass (Zostera marina) colonization promotes the accumulation of diazotrophic bacteria and alters the relative abundances of specific bacterial lineages involved in benthic carbon and sulfur cycling. Appl. Environ. Microbiol.81, 6901–6914. doi: 10.1128/AEM.01382-15
70
Terrados J. Duarte C. M. (2000). Experimental evidence of reduced particle resuspension within a seagrass (Posidonia oceanica) meadow. J. Exp. Mar. Biol. Ecol.243, 45–53. doi: 10.1016/S0022-0981(99)00110-0
71
Thomas F. Hehemann J.-H. Rebuffet E. Czjzek M. Michel G. (2011). Environmental and gut Bacteroidetes: The food connection. Front. Microbiol.2, 93. doi: 10.3389/fmicb.2011.00093
72
Torti A. Jørgensen B. B. Lever M. A. (2018). Preservation of microbial DNA in marine sediments: Insights from extracellular DNA pools. Environ. Microbiol.20, 4526–4542. doi: 10.1111/1462-2920.14401
73
Tuya F. Ribeiro-Leite L. Arto-Cuesta N. Coca J. Haroun R. Espino F. (2014). Decadal changes in the structure of Cymodocea nodosa seagrass meadows: Natural vs. human influences. Estuar. Coast. Shelf Sci.137, 41–49. doi: 10.1016/j.ecss.2013.11.026
74
van Katwijk M. M. Bos A. R. Hermus D. C. R. Suykerbuyk W. (2010). Sediment modification by seagrass beds: Muddification and sandification induced by plant cover and environmental conditions. Estuar. Coast. Shelf Sci.89, 175–181. doi: 10.1016/j.ecss.2010.06.008
75
Walsh E. A. Kirkpatrick J. B. Rutherford S. D. Smith D. C. Sogin M. D’Hondt S. (2016). Bacterial diversity and community composition from seasurface to subseafloor. ISME J.10, 979–989. doi: 10.1038/ismej.2015.175
76
Watanabe M. Fukui M. Galushko A. Kuever J. (2020). “ Desulfosarcina ,” in Bergey’s manual of systematics of Archaea and Bacteria. Ed. WhitmanW. B. (John Wiley & Sons, in association with Bergey’s manual trust), 1–6. doi: 10.1002/9781118960608.gbm01020.pub2
77
Wickham H. Averick M. Bryan J. Chang W. McGowan L. D. François R. et al . (2019). Welcome to the tidyverse. J. Open Source Software4, 1686. doi: 10.21105/joss.01686
78
Wilke C. O. (2020). Cowplot: Streamlined plot theme and plot annotations for ‘ggplot2’.
79
Xie Y. (2014). “knitr: A comprehensive tool for reproducible research in R,”, in Implementing reproducible computational research. Eds. StoddenV.LeischF.PengR. D. (New York: Chapman and Hall/CRC), 3–32.
80
Xie Y. (2015). Dynamic documents with R and knitr. 2nd ed (Boca Raton, Florida: Chapman and Hall/CRC).
81
Xie Y. (2019). TinyteX: A lightweight, cross-platform, and easy-to-maintain LaTeX distribution based on TeX Live. TUGboat40, 30–32.
82
Xie Y. (2021a). knitr: A general-purpose package for dynamic report generation in R.
83
Xie Y. (2021b). Tinytex: Helper functions to install and maintain ‘TeX Live’, and compile ‘LaTeX’ documents.
84
Xie Y. Allaire J. J. Grolemund G. (2018). R Markdown: The definitive guide. 1st ed (Boca Raton, Florida: Chapman and Hall/CRC).
85
Xie Y. Deriveux C. Riederer E. (2020). R Markdown cookbook. 1st ed (Boca Raton, Florida: Chapman and Hall/CRC).
86
Yilmaz P. Parfrey L. W. Yarza P. Gerken J. Pruesse E. Quast C. et al . (2014). The SILVA and “All-species living tree project (LTP)” taxonomic frameworks. Nucleic Acids Res.42, D643–D648. doi: 10.1093/nar/gkt1209
87
Zhang X. Liu S. Jiang Z. Wu Y. Huang X. (2022). Gradient of microbial communities around seagrass roots was mediated by sediment grain size. Ecosphere13, e3942. doi: 10.1002/ecs2.3942
88
Zhang X. Zhao C. Yu S. Jiang Z. Liu S. Wu Y. et al . (2020). Rhizosphere microbial community structure is selected by habitat but not plant species in two tropical seagrass beds. Front. Microbiol.11, 161. doi: 10.3389/fmicb.2020.00161
89
Zheng P. Wang C. Zhang X. Gong J. (2019). Community structure and abundance of archaea in a Zostera marina meadow: A comparison between seagrass-colonized and bare sediment sites. Archaea2019, e5108012. doi: 10.1155/2019/5108012
90
Zhou J. Bruns M. A. Tiedje J. M. (1996). DNA Recovery from soils of diverse composition. Appl. Environ. Microbiol.62, 316–322. doi: 10.1128/aem.62.2.316-322.1996
91
Zhu H. (2021). kableExtra: Construct complex table with ‘kable’ and pipe syntax.
Summary
Keywords
sediment microbial communities, Cymodocea nodosa , seagrass meadow decline, northern Adriatic Sea, Illumina 16S rRNA sequencing
Citation
Markovski M, Najdek M, Herndl GJ and Korlević M (2022) Compositional stability of sediment microbial communities during a seagrass meadow decline. Front. Mar. Sci. 9:966070. doi: 10.3389/fmars.2022.966070
Received
10 June 2022
Accepted
05 August 2022
Published
05 September 2022
Volume
9 - 2022
Edited by
Daniel P. R. Herlemann, Estonian University of Life Sciences, Estonia
Reviewed by
Katharina Kesy, University of Greifswald, Germany; Yinzhao Wang, Shanghai Jiao Tong University, China
Updates
Copyright
© 2022 Markovski, Najdek, Herndl and Korlević.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marino Korlević, marino.korlevic@irb.hr
This article was submitted to Aquatic Microbiology, a section of the journal Frontiers in Marine Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.