Abstract
Metagenomic sequencing facilitates large-scale constitutional analysis and functional characterization of complex microbial communities without cultivation. Recent advances in long-read sequencing techniques utilize long-range information to simplify repeat-aware metagenomic assembly puzzles and complex genome binning tasks. However, it remains methodologically challenging to remove host-derived DNA sequences from the microbial community at the read resolution due to high sequencing error rates and the absence of reference genomes. We here present Symbiont-Screener (https://github.com/BGI-Qingdao/Symbiont-Screener), a reference-free approach to identifying high-confidence host’s long reads from symbionts and contaminants and overcoming the low sequencing accuracy according to a trio-based screening model. The remaining host’s sequences are then automatically grouped by unsupervised clustering. When applied to both simulated and real long-read datasets, it maintains higher precision and recall rates of identifying the host’s raw reads compared to other tools and hence promises the high-quality reconstruction of the host genome and associated metagenomes. Furthermore, we leveraged both PacBio HiFi and nanopore long reads to separate the host’s sequences on a real host-microbe system, an algal-bacterial sample, and retrieved an obvious improvement of host assembly in terms of assembly contiguity, completeness, and purity. More importantly, the residual symbiotic microbiomes illustrate improved genomic profiling and assemblies after the screening, which elucidates a solid basis of data for downstream bioinformatic analyses, thus providing a novel perspective on symbiotic research.
1 Introduction
Powered by advanced biotechnologies and big data analytics, genome sequences become the significant biological basis and genomic resources of modern life science. Simultaneous genome sequencing of the host-symbiont ecosystem reveals the bioinformatic information of both the host species and associated microbial communities, thus prompting the symbiotic studies to move from gene-centric to genome-centric fields (Xie et al., 2016; Xie et al., 2020).
However, valuable insights into the construction and dynamics of the symbiotic ecosystem require successful separation of the host, symbiont, and contaminant data (Cornet and Baurain, 2022). Complicated and laborious experimental approaches have been developed to isolate host sequences from prokaryotic contamination (Arimoto et al., 2019; Cheng et al., 2019; Wang et al., 2020). Current bioinformatic methods rely on the species differentiation in statistical features (Woyke et al., 2006; Alneberg et al., 2014), or nucleotide and protein similarity of pre-assemblies to known genomes or public databases (Coghlan et al., 2019). Unfortunately, most methods designed for next-generation sequencing (NGS) short reads provide incomplete or inaccurate information and are facing challenges such as strong dependence on public data libraries or pre-assembly quality. It has been demonstrated that public genomic data may contain foreign sequences, leading to erroneous genetic characteristics (Neimark, 2015; Steinegger and Salzberg, 2020; Douvlataniotis et al., 2020). Moreover, those sequences that cannot be aligned to the reference genomes usually provide more critical findings, for instance, the identification of novel COVID-19 variants (Ricker et al., 2012; Kim et al., 2020; Cheng et al., 2022).
Short-read assemblies cannot resolve the highly nonuniform coverage of the composing species and the presence of long intra-genomic and inter-genomic repeats (Kolmogorov et al., 2020), resulting in uncompleted draft genomes with gaps (Fraser et al., 2002; Xu et al., 2020). Third-generation sequencing (TGS) long reads, including Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), provide unique long-range information, which straightforwardly simplifies complicated biomedical problems (Nagarajan and Pop, 2013; Rhoads and Au, 2015; Qi et al., 2022). The reconstruction of the complete genome sequence of hosts and microbes enables the analysis of the symbiotic relationship and microbial diversity, including the detection of horizontal gene transfer of mobile elements, large-scale structural rearrangements, and search for biosynthetic gene clusters (Chin et al., 2013; Bertrand et al., 2019). Long-read metagenomic classifiers such as Centrifuge (Kim et al., 2016), Kraken2 (Wood et al., 2019), and MetaMaps (Dilthey et al., 2019) can build indexed databases according to acknowledged reference genomes and NCBI taxonomy, and assign corresponding long reads to the host. Meanwhile, MetaProb (Girotto et al., 2016), BusyBee (Laczny et al., 2017) and MetaBCC-LR (Wickramarachchi et al., 2020) do not require references to classify long reads based on the unsupervised clustering results of k-mer coverage or oligonucleotide composition, but cannot indicate which cluster belongs to the host. But the relatively high sequencing error rates of TGS data might be greater than the genetic difference between organisms, resulting in a low capture ratio of the host’s data and large computational consumption (Bharti and Grimm, 2019). It becomes almost impossible to classify highly similar sequences shared by both the host and symbionts, for instance, symbiotic algae in a floating island of seaweeds (Thiel and Gutow, 2005; Rothäusler et al., 2012). Besides, it is even more complex for the de novo projects without sufficient priori knowledge, that is, lack of reference genomes or libraries.
The combination of TGS’s unprecedented read lengths and the trio’s global inherited information has been demonstrated to improve the genome assembly and further reconstruct haplotypes (Koren et al., 2018; Ebert et al., 2021; Xu et al., 2021). This idea also motivates us to introduce a reference-free and alignment-free way to solve the screening problem prior to assembly. Laboratory cultivation of sexually reproducing diploid host with associated microorganisms enables us to gather the parent-offspring pedigree information without loss of symbiotic microbial information. In this work, we established a novel screening model of TGS raw reads according to the transmissibility of heterozygous variants in the trio of host species, the stability of symbiotic relations, and the randomness of contaminant sources. Based on this model, Symbiont-Screener selects high-confidence host’s reads. Then it captures more host sequences by an unsupervised clustering algorithm. The final data, in which most of the foreign genomes have been screened out, can recover a high-quality host genome. On the other hand, the residual microbial long reads can enhance the variation profiling, metagenomic assemblies, and taxonomic binning.
2 Methods
2.1 Trio-based screening model
The design of Symbiont-Screener focuses on the stepwise purification of the host’s sequences with sufficient precision and recall rates to reconstruct the genome by combining the advantage of long read lengths with trio-binning markers and minimizing the effect of high error rates (Figure 1). The mixed sample possibly comprises steady symbionts and random DNA contaminants induced by laboratory pollution or artificial experimental errors other than the host genome. Among them, symbionts sharing highly similar sequences with the host are the most difficult to be isolated. According to the species sources and the relation of parent-offspring trios, the possible foreign genomes in the offspring’s data can be categorized into four types: the offspring-only (OC), perhaps random contaminants; shared by father and offspring (POC); shared by mother and offspring (MOC); and shared by all three (SC), perhaps steady symbionts. Theoretically, the trio-specific markers inherited from contaminated parents allow the identification and reconstruction of the host chromosomes, and meanwhile, SC can be filtered out since their markers will no longer occur in the parent-specific marker libraries after set operations. The set difference, however, cannot remove parent-specific foreign genomes, POC or MOC. On the other hand, the intersection of marker libraries for all individuals ascertains the host autosomes alongside with SC. Nevertheless, the foreign species shared only by one parent (POC or MOC) will be discarded. At last, none of the markers will be enriched in the sequences of OC. Parental samples of the trio are required to be collected and sequenced to provide paternal- and maternal-specific markers. In principle, the set operations of characteristic marker libraries can eliminate most of the foreign species if the host heterozygosity, read length, and read sing-base accuracy meet certain thresholds.
Figure 1
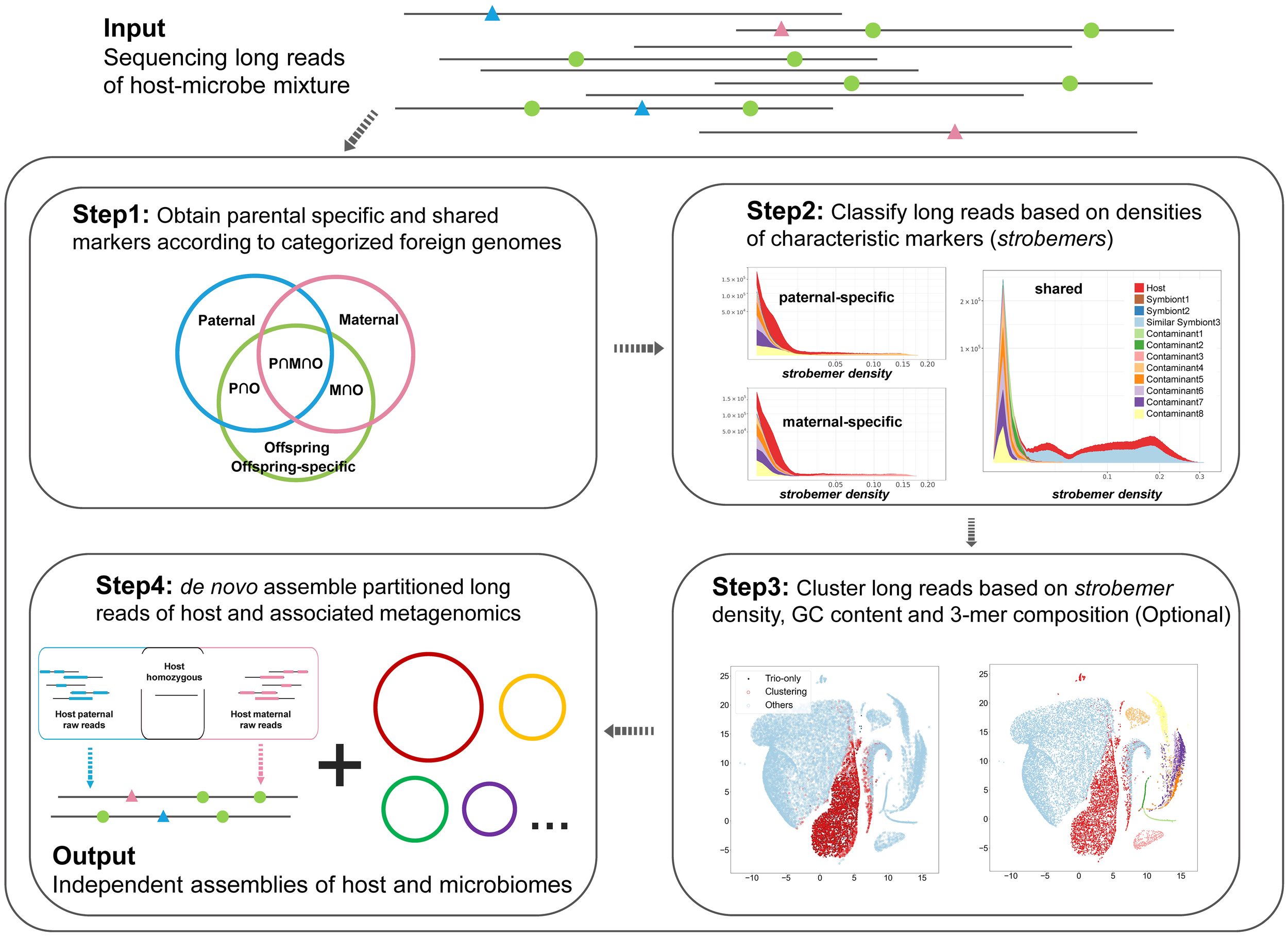
Workflow of Symbiont-Screener. The input sequencing long reads of the host-microbe mixture can be categorized based on whether they contain characteristic markers or not. The tool starts by calculating and matching the parent-specific and shared strobemers. The second step is to classify long reads according to the species differentiation of characteristic marker densities, and detect the high-confidence host’s data, which satisfies the relatively more accurate PacBio reads (single-base accuracy ≥95%). Then, for ONT long reads with higher error rates (single-base accuracy<95%), all the long reads including those without any matched markers are clustered by the features of characteristic strobemer density, GC content, and trinucleotide composition. Next, the clusters which belong to the host are detected by high-confidence long reads preselected in Step2, and other remaining clusters are labeled as metagenomic long reads. The final output includes a high-quality haplotype-collapsed or two haplotype-resolved assemblies for the diploid host as well as complete metagenome-assembled assemblies for the host-associated microbiomes.
The read length of current PacBio or ONT data meets the requirement of this model, although the high sequencing error rate remains the greatest obstacle. Theoretically, the expected number of characteristic k-mers (discriminability) in a single read is proportional to the read length, heterozygosity ratio, and kth power of single-base sequencing accuracy. In addition to the use of error-tolerate strobemers (Sahlin, 2021), we identify raw long-read data based on the genomic feature, which are further clustered with the unsupervised Bayesian Gaussian Mixture model (BGMM) method after principal component analysis (PCA) dimensionality reduction. In practice, the parent-specific and shared strobemers, the species differentiation in GC content and oligonucleotide frequencies consist of the main features in the algorithm. The procedure of genome binning is equivalent to the read clustering based on the Mahalanobis distances to centers in the 36-dimensional feature space. Moreover, the characteristic markers can be used to identify which read cluster belongs to the host.
2.2 Generation of characteristic markers
According to the above model, characteristic markers existing in the paternal group other than the maternal are defined as paternal-only makers, while those only existing in the maternal group are defined as maternal-only. Meanwhile, markers shared by both parents are defined as shared. We removed markers in low- and high-frequency regions and then ran set operations to calculate characteristic markers (Supplementary Figure 1). Supplementary Figure 2 shows the marker category of reference assemblies for the host, symbionts, and contaminants, which reflects the feasibility of this screening model in identifying different types of foreign species.
Plots of normalized densities of parent-specific and shared markers demonstrate that only host long reads synchronously own abundant parent-specific and shared markers (Supplementary Figure 3). Thus, the high-confidence host reads can be determined by the following formula
where x1,iand x2,i refer to the parent-only maker densities in the ith long read, x3,i refers to the shared maker density, while C1 and C2 refer to different thresholds.
Algorithm 1. ReadSourceType
Input Long read LR, marker set POK, MOK, and SK
Output the source type of read LR
1 flag_pok = KmerLookup ( LR , POK )
2 flag_mok = KmerLookup ( LR , MOK )
3 flag_sk = KmerLookup ( LR , SK )
4 if ( ( flag_pok || flag_mok ) && flag_sk )
5 return Host
6 else if ( flag_pok && ! ( flag_mok && flag_sk ) )
7 return POC
8 else if ( flag_mok && ! ( flag_pok && flag_sk ) )
9 return MOC
10 else if ( ! ( flag_pok && flag_mok && flag_sk ) )
11 return OC
12 else if ( ! ( flag_pok || flag_mok ) && flag_sk ) )
13 return SC
14 end
2.3 strobemer vs. k-mer
Characteristic markers can be k-mers as used in conventional trio-binning and genome-binning approaches (Koren et al., 2018; Ebert et al., 2021; Xu et al., 2021), or error-tolerant strobemers (Sahlin, 2021). The utilization of k-mers can statistically capture parent-specific markers in raw data as long as the reads are sufficiently long and the host homologous chromosomes have enough heterozygous sites (Koren et al., 2018). However, the captured ratio is still severely limited by the sequencing errors for such a complex application of screening. Therefore, we chose strobemer implementation for the error-tolerant indexing and matching to provide more evenly distributed matches with higher genome coverage and less sensitive to sequencing errors, especially for insertions and deletions (Sahlin, 2021). In k-mer mode, we applied meryl (Rhie et al., 2020) to generating and counting 21-mers. In strobemer mode, we first used Jellyfish (Marcais and Kingsford, 2011) to build large canonical k-mers (k=40) of two contaminated parental datasets, and then transformed them to strobemers [randstrobes (20,10,10,30)] by custom C++ scripts.
To benchmark the effect of k-mers and strobemers, we generated random sequences with a fixed length of 100 kbp and varied the mutation (error) rates. The mutation spots were randomly selected, in which the error probabilities of substitutions, insertions, and deletions were equal. Benchmarking of matched markers under different sampling protocols with mutation rates of 1%, 5%, and 10% was listed in Supplementary Table 1. Each test was independently run 100 times. Either minstrobes or randstrobes (two types of strobemers) match more mutation spots than k-mers, especially for higher error rates. This benchmark is available at https://github.com/BGI-Qingdao/strobemer_cpptest. Supplementary Figure 4 shows the precision-recall curve with trio-binning strobemers and k-mers for three simulated datasets. The implementation of strobemers obtains relatively better performance on average.
2.4 Unsupervised clustering of remaining reads
The following procedure of screening relies on the raw read clustering analogous to the genomic binning. The high-dimensional feature space for the unsupervised clustering of long reads consists of characteristic marker densities as Feature 1-3, the GC content and canonical 3-mer frequencies as Feature 4-36 (Supplementary Table 2). The preprocessing of PCA decomposes those features into new K independent variables of a 36-dimensional matrix. We assume that the K principal components belong to the same parametric family of Gaussian distribution but with various parameters (mean, variance). The preprocessing of whitening has been employed to reduce information redundancy. The BGMM algorithm is selected according to the applicable geometry and running speed from the comparison of clustering algorithms in scikit-learn (Pedregosa et al., 2011). Briefly, the Bayesian framework infers the posterior distribution of the parameters , and the expectation–maximization algorithm is utilized to update these parameters.
where are the weight, mean and variance of the ith component. Application Programming Interface from scikit-learn (Pedregosa et al., 2011) was used to implement these steps. This unsupervised machine learning algorithm is irrelevant to training. Therefore, no reference genomes or high-quality public databases are needed.
We illustrated the differences in screening results before and after clustering in the k-mer mode in Figure 2A and Supplementary Figure 5. Overall, the clustering increased classification F1-scores by improving the recall rates but sacrificing fractional precision. Note that we only used long reads longer than 5,000 bp for clustering since the inaccurate sequence statistics of shorter reads might mislead the results. The error-tolerant strobemer mode usually did not need further clustering, as the recall rate was already satisfactory. We did not cluster the k-mer-based result of the real ONT dataset, because the performance was adequate and extra clustering could not improve it further.
Figure 2
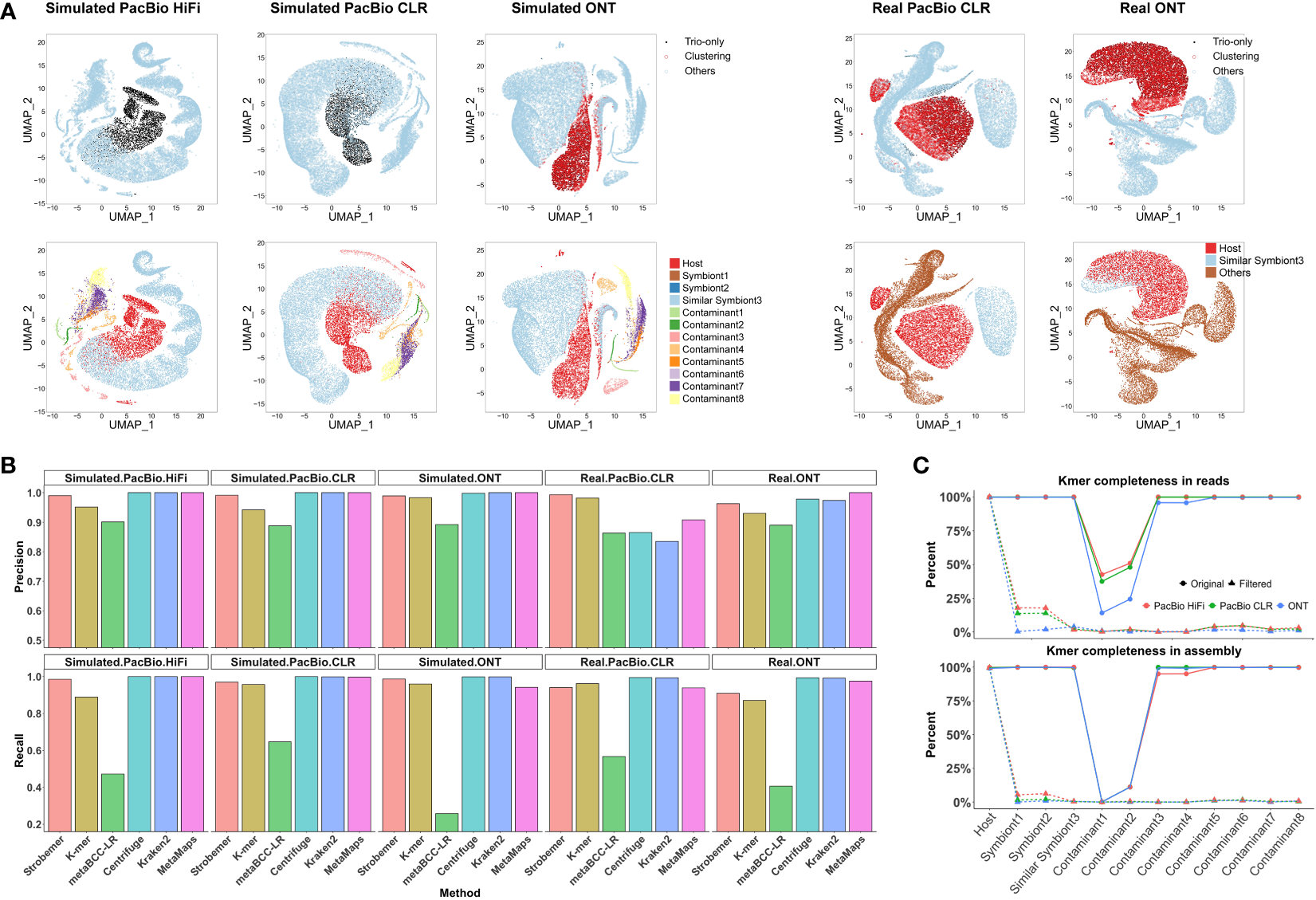
Performance of screening over different benchmarking datasets. (A) Visualization of identified host reads by trio-binning markers and unsupervised clustering after dimension reduction. We arbitrarily selected 20,000 long reads to cancel image blurring. Each point refers to one long read. For each dataset, top figure is colored by screening results via Symbiont-Screener, while bottom is colored by the original species. Note that the simulated PacBio HiFi and CLR reads with higher single-base accuracy are classified by trio-binning markers only. The other three datasets are further identified by clustering. (B) Comparisons of screening precision and recall rates over five simulated and real PacBio and ONT long-read datasets. Note that for the de novo tool, MetaBCC-LR, we benchmarked the result based on the extracted clusters with more than 5,000 reads and precision >0.5 as the host clusters cannot be identified without the reference genome. (C) Distinct k-mer completeness for each species in long-read data (top) and final assemblies (bottom) after screening. The low k-mer completeness of Contaminant 1 and 2 before filtering is because of the 1× input. On average, distinct k-mers for other foreign species including Symbiont3 sharing highly similar sequences with the host are reduced from 99.6% to 0.5% in raw long-read data and from 99.4% to 0.2% in assemblies, while >98.2% of host’s k-mers are retained in both raw data and following assemblies.
Besides, the randomness of BGMM clustering may affect the final classification of host reads. Thus, we independently ran the clustering procedure multiple times (default 10) and achieved consensus results. The best host read cluster was annotated according to the criterion that whether this cluster contained the most preselected high-confidence host reads with the smallest variance. The final host group was automatically produced by merging raw reads, which repeatedly occurred in the best and second-best clusters. Supplementary Figure 6 illustrates the frequencies of occurrence of different species in the best and second-best clusters for 10 runs of BGMM clustering. In the dumbbell-shaped overall profile, the foreign genomic data aggregate in the low-frequency region while the host’s stay in the high-frequency region. Users can determine the frequency threshold based on the position of the second peak in the high-frequency region to extract host data for de novo projects.
2.5 Reconstruction of the host and metagenomic genomes
TGS assemblers, for instance, Canu, Flye, and metaFlye (Koren et al., 2017; Kolmogorov et al., 2019; Kolmogorov et al., 2020) can reconstruct the host chromosomes and symbiotic microbial genomes. We employed default parameters to assemble PacBio HiFi, PacBio CLR, and ONT data types. Note that although the trio-binning markers can partition paternal and maternal reads, we did not show the result of haplotype-resolved assemblies because the host sequencing coverage depth in our datasets is insufficient.
2.6 Benchmarking datasets
We used the following five long-read datasets for evaluation.
Dataset1: Simulated PacBio HiFi dataset: human chromosome 19 of HG002 as the host, two bacteria from the Unified Human Gastrointestinal Genome (UHGG) collection (Almeida et al., 2021) as inter-phyla symbionts, additional eight UHGG bacteria as random contaminants, and mouse chromosome 7 as a symbiont sharing similar sequences with the host genome. PacBio HiFi long reads were simulated by PBSIM2 (Ono et al., 2021) based on the reference genomes with an average read length of 10 kbp and an average error rate of 1% (Supplementary Table 3).
Dataset2: Simulated PacBio CLR dataset: same composition as Dataset1. The average read length is 10 kbp and the average error rate is 5%.
Dataset3: Simulated ONT dataset: same composition as Dataset1. The average read length is 30 kbp and the average error rate is 15%.
Dataset4: Real PacBio RSII CLR dataset: human chromosome 19 of HG002 as the host, two bacteria of mock microbial datasets from the ZymoBIOMICS Microbial Community Standards as inter-phyla symbionts, other eight bacteria and yeasts as random contaminants. In this case, we challenged the chimpanzee chromosome 21 as a symbiont owning highly similar sequences (Supplementary Table 4). The human HG002 raw reads were downloaded from NCBI GIAB, and those mapped to chromosome 19 of the HG002 reference assembly (GCA_011064465.1) were extracted (Shumate et al., 2020). The chimpanzee raw reads were downloaded from (Logsdon et al., 2021), and those mapped to the chromosome 21 reference (GCA_000001515.5) were extracted. The mock raw reads were downloaded from (McIntyre et al., 2019).
Dataset5: Real ONT PromethION dataset: same composition as Dataset4. The human and chimpanzee raw reads were downloaded and extracted in the same way. The mock data were downloaded from (Nicholls et al., 2019).
We used default error profiles (substitution: insertion: deletion=6: 50: 54 for PacBio and 23:31:46 for ONT) provided by PBSIM2 for all simulations. 50× host (human chromosome 19) data were simulated using the HG002 reference (GCA_011064465.1), while 50× mouse chromosome 7 data were simulated based on the GRCm39 reference (GCA_000001635.9). Other bacteria were simulated based on UHGG references with various coverages. For the real PacBio and ONT data, we also gained 50× human and 50× chimpanzee raw reads, and maintained the same proportions of 10 bacteria and yeasts in the mock sequencing data.
All the long reads were annotated by their species names before binning to benchmark the precision and recall rates of screening. The recall rate is defined as the ratio of the number of correctly identified host reads to the total number of host reads in the mixed input. The precision rate is defined as the ratio of the number of correctly identified host reads to the total number of final extracted reads.
2.7 Evaluation methods
To assess the effect of screening on the host genome assembly for three simulated datasets, we generated species-specific non-repeating canonical 21-mers (distinct k-mers) according to the reference genomes. We calculated the completeness and contamination rates in the mixed and purified long-read raw data and assemblies, respectively (Supplementary Table 5). The completeness is defined as the ratio of distinct k-mer numbers belonging to each species in reads or assemblies to the total number in the reference. The contamination rate is defined as the ratio of the distinct k-mer number belonging to other species to the total distinct k-mer number of the host reference assembly.
The host reference assembly was used to validate the assemblies before and after screening. The assembly statistics were reported by QUAST (Gurevich et al., 2013) with default parameters except -m 1000.
The metagenomic analysis of host-associated microbial community is supposed to be improved after screening if we ignore the random contamination. However, the evaluation of this effect does not apply to our benchmarking datasets.
2.8 Parameters of other tools
We used recommended or default parameters for MetaProb, BusyBee, MetaBCC-LR, Centrifuge, Kraken2, and MetaMaps. However, MetaProb and BusyBee cannot support the large data size. We performed parameter sweeps for MetaBCC-LR to obtain its best performance (–sample-count 1%, –bin-size 10). Note that the host clusters generated by MetaBCC-LR cannot be detected as the host without references. In addition, the host data are usually split into several clusters. Thus, we extracted all clusters with more than 5,000 reads and precision >0.5 for evaluation. Centrifuge, Kraken2, and MetaMaps are reference-based classifiers. We first built indexed databases according to host and foreign reference assemblies, as well as NCBI taxonomy. Next, we classified datasets and extracted all reads assigned to the host species for benchmarking.
3 Results
3.1 Screening of simulated and real datasets
We applied Symbiont-Screener to three simulated and two real long-read datasets with varying read lengths and error rates, covering PacBio HiFi, PacBio CLR, and ONT types. Each dataset consists of a host species, inter-phyla symbionts, a symbiont with similar sequences, and several random microbial contaminants.
Figure 2A shows the host’s data identified by characteristic markers and further clustered by other genomic features. Compared with the corresponding ground truth, the parent-specific and shared strobemers precisely detect host long reads for three simulated datasets. Limited by the sequencing errors, the simulated ONT dataset requires species differentiation in GC content and trinucleotide frequencies to obtain more host data as supplementary features. Long reads sufficiently close to the preselected reads in the space after dimension reduction are also marked as host’s and extracted for the following assembly. Real long-read datasets for symbiotic samples are rare. Thus, we chose chimpanzee to imitate an indistinguishable symbiont, sharing approximately 98% of the genome with the host, human, which is a conundrum of alignment-based screening. Although a few foreign reads are misidentified by trio-binning markers in the real PacBio dataset, they are further corrected by genomic signatures. The relatively higher error rate accompanied by the high sequence similarity leads to the lower accuracy of screening for real ONT dataset. Besides, we evaluated the contribution of each feature to the final clustering results (Supplementary Figure 7). For three simulated datasets, the relative importance score for characteristic strobemers is 24.4% on average, among which the parent-specific markers are more important. None of their contributions are negligible in the clustering, which proves those features are highly complementary.
We have also tested several state-of-the-art reference-free or reference-based tools for screening using the same datasets. Figure 2B shows the classification precision and recall of the strobemer mode, k-mer mode, MetaBCC-LR, Centrifuge, Kraken2, and MetaMaps. Hampered by the computing performance, neither MetaProb nor BusyBee can support clustering the entire data. Overall, Symbiont-Screener outperforms MetaBCC-LR for all five datasets. The implementation of trio-binning strobemers and clustering allows Symbiont-Screener to surmount the obstacle of high error rates, thus extracting more host long reads. By contrast, Centrifuge, Kraken2, and MetaMaps benefit from accurate and complete reference genomes as well as taxonomic relationships, and obtain relatively higher precision and recall rates, especially for the simulated datasets. Nevertheless, for more complex relations in the real PacBio dataset where the similar symbiont, chimpanzee shares approximately 98% of the genome with the host, the highest F1-scores are found in both modes of Symbiont-Screener. It indicates that the trio-binning information is a qualified substitute for reference genomes or public databases if they are not available.
3.2 Effect on host assembly
The effect of screening on the final assembly was first assessed by the species-specific genomic non-repeating canonical 21-mers. The distinct k-mer completeness ratio indicated the ability to reconstruct the whole host genome. Meanwhile, the distinct k-mer contamination ratio represented the assembly accuracy.
For three simulated datasets, the filtered long reads obtained up to 99.4% of the host’s 42,988,682 distinct k-mers regardless of repeats or non-ACGT bases on average (Figure 2C), thus ensuring the high quality of genome assembly (~99.2% completeness). On the contrary, k-mer completeness was significantly reduced to 0.3% in raw reads and 0.1% in final assembly for all ten bacteria after screening. For the challenging symbiont with highly similar sequences, the most difficult component to be cleaned, only 2.4% of k-mers were retained in the results. They could not support the foreign genome reconstruction in the following host assembly.
The QUAST-based evaluations also reflected the advantage after a nearly perfect screening. Supplementary Table 7 showed the Canu assembly statistics for three simulated and two real datasets. The assembled total length and unaligned length were significantly reduced as the foreign genomes were removed after the screening, while the genome fraction and misassemblies remained almost the same. The comparison was nearly consistent with that of Flye assemblies as shown in Supplementary Table 8.
3.3 Application to a red seaweed with symbionts
We applied Symbiont-Screener to an economically important red seaweed, Neoporphyra haitanensis, to demonstrate the success of screening results in the natural world. Previous studies have shown a complex relationship between the host algae and the associated metagenomes, involving the microbial components, functional microbial lineages, and the exchange of diverse chemical currencies, which mainly rely on the sequence alignments of short-read reads or genome assemblies (Brawley et al., 2017; Wang et al., 2022). Here, we sequenced 51.6 Gb ONT long reads (read N50 = 25.2 kb) and 23.2 Gb PacBio HiFi reads (read N50 = 18.3 kb) and separate the host seaweed sequences from the symbionts. Symbiont-Screener employed the characteristic strobemers to automatically identify host raw reads without reference genomes. The 108,837,094 characteristic strobemers were generated by the trio pedigree relations of the lab-cultured parents and offspring. The clustering procedure further gathered host long reads to overcome the limit of high sequencing error rate. Finally, the whole identified host raw reads were assembled by Flye, while the remaining associated bacteria were assembled by metaFlye, respectively. We regarded all foreign genomes as symbiotic bacteria. We also applied metaFlye to the assembly of the mixed data for comparison.
The total length of the reconstructed seaweed genome was 45,172,822 bp, consisting of 59 contigs (Supplementary Table 9). The assembly contiguity reached a chromosomal level with a contig N50 of 7,218,067 bp, compared to the previously published closely-related species (Chen et al., 2022). The 81.454% genome fraction against the 53.3 Mbp closely-related genome implied the assembly completeness. Only 2,084,431 bp were unaligned. The significant difference of the chromatin contacts from pair-wise Hi-C reads confirmed the thorough isolation of the host, which further constructed 5 complete chromosomes (Figure 3A). On the other hand, the metagenome-assembled genomes involved 608 contigs with a contig N50 of 4,376,601 bp. The total genome size was 260,496,743 bp, of which only 3.829% could be aligned to the closely-related reference genome. GC-depth plot is an alternative method to benchmark the screening result. Multiple peaks in the preliminary assembly of the whole mixed data indicated different species with various GC contents and covered read depths (Figure 3B). In contrast, the purified host assembly after screening presented a more concentrated peak with a more convergent distribution of GC ratio.
Figure 3
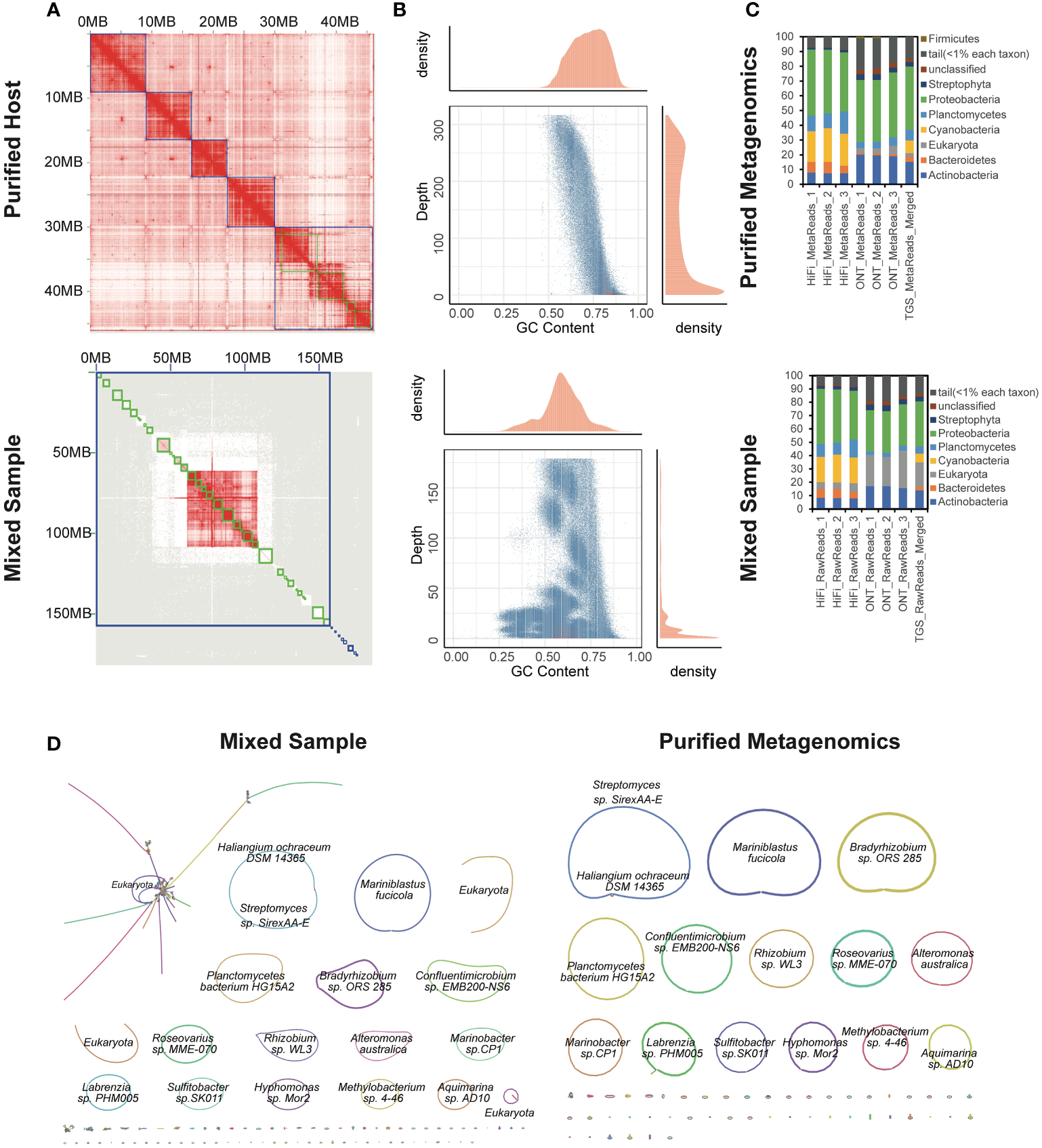
Application to an algal-bacterial ecosystem. (A) Hi-C interaction heatmaps of purified (top) and mixed (bottom) assemblies. (B) GC-Coverage plots for simulated PacBio HiFi assemblies of purified (top) and mixed (bottom) data. (C) Differences in the taxonomic profiling of associated microbial community structures at the phylum level based on the indexed RefSeq database between purified (top) and mixed (bottom) data. (D) Bandage visualization of assembly graphs with connections for the mixed and purified data. Note that the primary assembled sequences are annotated by Kraken2.
Additionally, the metagenomes were binned and taxonomically annotated by Kraken2 using the indexed NCBI RefSeq database k2_pluspfp_20200919, which illustrated that the sequences annotated as eukaryote were dramatically eliminated after screening (Figure 3C). The profiling results also disclosed a bias of different sequencing platforms, possibly due to the more misalignments induced by the relatively higher error rates for ONT. Bandage (Wick et al., 2015) were used to visualize the de novo assembly graphs with sequence connections. There were totally 14 complete, closed and circularized metagenomes were assembled (Figure 3D). Although the 322,277,090 bp-long mixed assembly reconstructed an additional circularized genome, it was annotated as eukaryote. We investigated the mixed assembly and found 84.4 Mbp contigs could be aligned to the closely-related reference of the host seaweed, implying the host assembly errors. The high-quality genomes of Neoporphyra haitanensis and associated bacteria might provide a comprehensive approach for elucidating genome coevolution and the influence of symbiotic metagenomes to the adaptation of Pyropia to intertidal zone habitats.
3.4 Performance
We benchmarked the performance of Symbiont-Screener on a Linux system with Intel Core Processor (Broadwell, IBRS), 15 CPU cores and 30 threads. We individually recorded the CPU and memory usage for each assembly and calculated the percentage of saved consumption after screening. Supplementary Figure 8 recorded the computational consumption for three simulated PacBio HiFi, PacBio CLR and ONT datasets, representing that the screening result saves considerable CPU and memory usage.
4 Discussion
We introduce a novel but accurate model for screening that classifies reliable host raw long reads from the mixed sample according to the trio-binning information, which is computationally efficient without the requirement of reference genomes or sequence alignments. Symbiont-Screener further utilizes other supplementary features to directly cluster error-prone long reads. The multi-dimensional clustering system is open-ended and accepts additional features such as remaining genomic markers after sterilization to avoid overreliance on the presence or accuracy of one specific feature. Moreover, the trio-binning markers support the haplotype-resolved partitioning and genome assembly of extracted host’s long reads. We did not show the haplotype-resolved assemblies due to the insufficient sequencing coverage depth of host’s data.
The application of this algorithm requires parental sequencing data with or without symbionts and contamination for sexually reproducing diploid or allotetraploid species. Therefore, the patient’s parents need to provide their clean or contaminated DNA samples for the microbial pathogen identification in clinical applications. For samples of animals or plants collected from the wild, sexual reproduction in the field or laboratory culture is required to eliminate symbionts and random contaminants. If parental data are unobtainable, then reference assemblies of closely-related species if available, or parental lines for cross-bred crops can be used to mark long reads corresponding to the conserved genomic regions instead.
Statements
Data availability statement
The simulated PacBio HiFi, PacBio CLR and ONT datasets have been deposited in the CNGB Sequence Archive CNSA, (https://db.cngb.org/cnsa) under the accession number CNP0001829. We downloaded real PacBio CLR and ONT ultra-long data of HG002/NA24385 as host from GIAB (https://ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/HG002_NA24385_son/PacBio_MtSinai_NIST and https://ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data/AshkenazimTrio/HG002_NA24385_son/Ultralong_OxfordNanopore). The real PacBio and ONT datasets for chimpanzee are available in the NCBI under the accession number PRJNA659034. PacBio data for the mock microbial community from ZymoBIOMICS Microbial Community Standards are extracted, which are publicly available from (McIntyre et al., 2019). The ONT data for the same mock standard are obtained from (Nicholls et al., 2019). The algal-bacterial data have been deposited in the CNSA under the accession number CNP0003571. But restrictions apply to the availability of these algal-bacterial data, which are not publicly available. Data are however available from the corresponding author upon request. The RefSeq-based database used for the Kraken2 analysis can be downloaded at https://genome-idx.s3.amazonaws.com/kraken/k2_pluspfp_20200919.tar.gz. The source code used in this manuscript is available at https://github.com/BGI-Qingdao/Symbiont-Screener.
Author contributions
MX and LG performed software design and implementation. MX, LG, YQ, CS, XCL, JC, and JH contributed to data modeling, data curation, and benchmarking. MX wrote the draft manuscript, and LG, LD, and GF contributed to manuscript editing. XL and GF supervised the project. MX and GF secured funding. All authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 32100514); and the National Key Research and Development Program of China (Grant No. 2018YFD0900301-05).
Acknowledgments
We thank Kristoffer Sahlin for help with implementation of strobemers. We thank Xiaobin Liu for fruitful discussions during the development and performance test. The data that support the findings of this study have been deposited into CNGB Nucleotide Sequence Archive (CNSA) (Guo et al., 2020) of China National GeneBank DataBase (CNGBdb) (Chen et al., 2020) with accession numbers CNP0001829.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1087447/full#supplementary-material
References
1
Almeida A. Nayfach S. Boland M. Strozzi F. Beracochea M. Shi Z. J. et al . (2021). A unified catalog of 204,938 reference genomes from the human gut microbiome. Nat. Biotechnol.39, 105–114. doi: 10.1038/s41587-020-0603-3
2
Alneberg J. Bjarnason B. S. De Bruijn I. Schirmer M. Quick J. Ijaz U. Z. et al . (2014). Binning metagenomic contigs by coverage and composition. Nat. Methods11, 1144–1146. doi: 10.1038/nmeth.3103
3
Arimoto A. Hikosaka-Katayama T. Hikosaka A. Tagawa K. Inoue T. Ueki T. et al . (2019). A draft nuclear-genome assembly of the acoel flatworm praesagittifera naikaiensis. Gigascience8. doi: 10.1093/gigascience/giz023
4
Bertrand D. Shaw J. Kalathiyappan M. Ng A. H. Q. Kumar M. S. Li C. et al . (2019). Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol.37, 937–944. doi: 10.1038/s41587-019-0191-2
5
Bharti R. Grimm D. G. (2019). Current challenges and best-practice protocols for microbiome analysis. Briefings Bioinf.22, 178–193. doi: 10.1093/bib/bbz155
6
Brawley S. H. Blouin N. A. Ficko-Blean E. Wheeler G. L. Lohr M. Goodson H. V. et al . (2017). Insights into the red algae and eukaryotic evolution from the genome of porphyra umbilicalis (Bangiophyceae, rhodophyta). Proc. Natl. Acad. Sci. U.S.A.114, E6361–e6370. doi: 10.1073/pnas.1703088114
7
Chen H. Chu J. S. Chen J. Luo Q. Wang H. Lu R. et al . (2022). Insights into the ancient adaptation to intertidal environments by red algae based on a genomic and multiomics investigation of neoporphyra haitanensis. Mol. Biol. Evol.39. doi: 10.1093/molbev/msab315
8
Cheng X.-W. Li J. Zhang L. Hu W.-J. Zong L. Xu X. et al . (2022). Identification of SARS-CoV-2 variants and their clinical significance in hefei, China. Front. Med.8. doi: 10.3389/fmed.2021.784632
9
Cheng S. Xian W. Fu Y. Marin B. Keller J. Wu T. et al . (2019). Genomes of subaerial zygnematophyceae provide insights into land plant evolution. Cell179, 1057–1067.e14. doi: 10.1016/j.cell.2019.10.019
10
Chen F. Z. You L. Yang F. Wang L. N. Guo X. Q. Gao F. et al . (2020). CNGBdb: China national GeneBank DataBase. Hereditas42, 799–809. doi: 10.16288/j.yczz.20-080
11
Chin C. S. Alexander D. H. Marks P. Klammer A. A. Drake J. Heiner C. et al . (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods10, 563–569. doi: 10.1038/nmeth.2474
12
Coghlan A. Tyagi R. Cotton J. A. Holroyd N. Rosa B. A. Tsai I. J. et al . (2019). Comparative genomics of the major parasitic worms. Nat. Genet.51, 163–174. doi: 10.1038/s41588-018-0262-1
13
Cornet L. Baurain D. (2022). Contamination detection in genomic data: More is not enough. Genome Biol.23, 60. doi: 10.1186/s13059-022-02619-9
14
Dilthey A. T. Jain C. Koren S. Phillippy A. M. (2019). Strain-level metagenomic assignment and compositional estimation for long reads with MetaMaps. Nat. Commun.10, 3066. doi: 10.1038/s41467-019-10934-2
15
Douvlataniotis K. Bensberg M. Lentini A. Gylemo B. Nestor C. E. (2020). No evidence for DNA N6-methyladenine in mammals. Sci. Adv.6, eaay3335. doi: 10.1126/sciadv.aay3335
16
Ebert P. Audano P. A. Zhu Q. Rodriguez-Martin B. Porubsky D. Bonder M. J. et al . (2021). Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science.372 doi: 10.1126/science.abf7117
17
Fraser C. M. Eisen J. A. Nelson K. E. Paulsen I. T. Salzberg S. L. (2002). The value of complete microbial genome sequencing (you get what you pay for). J. Bacteriol184, 6403–5; discusion 6405. doi: 10.1128/JB.184.23.6403-6405.2002
18
Girotto S. Pizzi C. Comin M. (2016). MetaProb: accurate metagenomic reads binning based on probabilistic sequence signatures. Bioinformatics32, i567–i575. doi: 10.1093/bioinformatics/btw466
19
Guo X. Chen F. Gao F. Li L. Liu K. You L. et al . (2020). CNSA: a data repository for archiving omics data. Database2020. doi: 10.1093/database/baaa055
20
Gurevich A. Saveliev V. Vyahhi N. Tesler G. (2013). QUAST: Quality assessment tool for genome assemblies. Bioinformatics29, 1072–1075. doi: 10.1093/bioinformatics/btt086
21
Kim D. Lee J. Y. Yang J. S. Kim J. W. Kim V. N. Chang H. (2020). The architecture of SARS-CoV-2 transcriptome. Cell181, 914–921.e10. doi: 10.1016/j.cell.2020.04.011
22
Kim D. Song L. Breitwieser F. P. Salzberg S. L. (2016). Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res.26, 1721–1729. doi: 10.1101/gr.210641.116
23
Kolmogorov M. Bickhart D. M. Behsaz B. Gurevich A. Rayko M. Shin S. B. et al . (2020). metaFlye: Scalable long-read metagenome assembly using repeat graphs. Nat. Methods17, 1103–1110. doi: 10.1038/s41592-020-00971-x
24
Kolmogorov M. Yuan J. Lin Y. Pevzner P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol.37, 540–546. doi: 10.1038/s41587-019-0072-8
25
Koren S. Rhie A. Walenz B. P. Dilthey A. T. Bickhart D. M. Kingan S. B. et al . (2018). De novo assembly of haplotype-resolved genomes with trio binning. Nat. Biotechnol.36, 1174–1182. doi: 10.1038/nbt.4277
26
Koren S. Walenz B. P. Berlin K. Miller J. R. Bergman N. H. Phillippy A. M. (2017). Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res.27, 722–736. doi: 10.1101/gr.215087.116
27
Laczny C. C. Kiefer C. Galata V. Fehlmann T. Backes C. Keller A. (2017). BusyBee web: Metagenomic data analysis by bootstrapped supervised binning and annotation. Nucleic Acids Res.45, W171–w179. doi: 10.1093/nar/gkx348
28
Logsdon G. A. Vollger M. R. Hsieh P. Mao Y. Liskovykh M. A. Koren S. et al . (2021). The structure, function and evolution of a complete human chromosome 8. Nature.593, 101–107 doi: 10.1038/s41586-021-03420-7
29
Marcais G. Kingsford C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics27, 764–770. doi: 10.1093/bioinformatics/btr011
30
McIntyre A. B. R. Alexander N. Grigorev K. Bezdan D. Sichtig H. Chiu C. Y. et al . (2019). Single-molecule sequencing detection of N6-methyladenine in microbial reference materials. Nat. Commun.10, 579. doi: 10.1038/s41467-019-08289-9
31
Nagarajan N. Pop M. (2013). Sequence assembly demystified. Nat. Rev. Genet.14, 157–167. doi: 10.1038/nrg3367
32
Neimark J. (2015). Line of attack. Science347, 938–940. doi: 10.1126/science.347.6225.938
33
Nicholls S. M. Quick J. C. Tang S. Loman N. J. (2019). Ultra-deep, long-read nanopore sequencing of mock microbial community standards. GigaScience8. doi: 10.1093/gigascience/giz043
34
Ono Y. Asai K. Hamada M. (2021). PBSIM2: a simulator for long-read sequencers with a novel generative model of quality scores. Bioinformatics37, 589–595. doi: 10.1093/bioinformatics/btaa835
35
Pedregosa F. Varoquaux G. Gramfort A. Michel V. Thirion B. Grisel O. et al . (2011). Scikit-learn: Machine learning in Python. J. Mach. Learn. Res.12, 2825–2830. doi: 10.48550/arXiv.1201.0490
36
Qi Y. Gu S. Zhang Y. Guo L. Xu M. Cheng X. et al . (2022). MetaTrass: A high-quality metagenome assembler of the human gut microbiome by cobarcoding sequencing reads. iMeta, 1, e46. doi: 10.1002/imt2.46
37
Rhie A. Walenz B. P. Koren S. Phillippy A. M. (2020). Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol.21, 245. doi: 10.1186/s13059-020-02134-9
38
Rhoads A. Au K. F. (2015). PacBio sequencing and its applications. Genomics Proteomics Bioinf.13, 278–289. doi: 10.1016/j.gpb.2015.08.002
39
Ricker N. Qian H. Fulthorpe R. R. (2012). The limitations of draft assemblies for understanding prokaryotic adaptation and evolution. Genomics100, 167–175. doi: 10.1016/j.ygeno.2012.06.009
40
Rothäusler E. Gutow L. Thiel M. (2012). Floating Seaweeds and Their Communities. In: Wiencke, C., Bischof, K. (eds) Seaweed Biology. Ecological Studies. (Berlin, Heidelberg: Springer) 219. doi: 10.1007/978-3-642-28451-9_17
41
Sahlin K. (2021). Effective sequence similarity detection with strobemers. Genome Res.31, 2080–2094. doi: 10.1101/gr.275648.121
42
Shumate A. Zimin A. V. Sherman R. M. Puiu D. Wagner J. M. Olson N. D. et al . (2020). Assembly and annotation of an ashkenazi human reference genome. Genome Biol.21, 129. doi: 10.1186/s13059-020-02047-7
43
Steinegger M. Salzberg S. L. (2020). Terminating contamination: large-scale search identifies more than 2,000,000 contaminated entries in GenBank. Genome Biol.21, 115. doi: 10.1186/s13059-020-02023-1
44
Thiel M. Gutow L. (2005). The ecology of rafting in the marine environment. II. the rafting organisms and community. Oceanography Mar. Biol.43, 279–418. doi: 10.1201/9781420037449.ch7
45
Wang J. Tang X. Mo Z. Mao Y. (2022). Metagenome-assembled genomes from pyropia haitanensis microbiome provide insights into the potential metabolic functions to the seaweed. Front. Microbiol.13. doi: 10.3389/fmicb.2022.857901
46
Wang D. Yu X. Xu K. Bi G. Cao M. Zelzion E. et al . (2020). Pyropia yezoensis genome reveals diverse mechanisms of carbon acquisition in the intertidal environment. Nat. Commun.11, 4028. doi: 10.1038/s41467-020-17689-1
47
Wickramarachchi A. Mallawaarachchi V. Rajan V. Lin Y. (2020). MetaBCC-LR: metagenomics binning by coverage and composition for long reads. Bioinformatics36, i3–i11. doi: 10.1093/bioinformatics/btaa441
48
Wick R. R. Schultz M. B. Zobel J. Holt K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics31, 3350–3352. doi: 10.1093/bioinformatics/btv383
49
Wood D. E. Lu J. Langmead B. (2019). Improved metagenomic analysis with kraken 2. Genome Biol.20, 257. doi: 10.1186/s13059-019-1891-0
50
Woyke T. Teeling H. Ivanova N. N. Huntemann M. Richter M. Gloeckner F. O. et al . (2006). Symbiosis insights through metagenomic analysis of a microbial consortium. Nature443, 950–955. doi: 10.1038/nature05192
51
Xie M. Ren M. Yang C. Yi H. Li Z. Li T. et al . (2016). Metagenomic analysis reveals symbiotic relationship among bacteria in microcystis-dominated community. Front. Microbiol.7. doi: 10.3389/fmicb.2016.00056
52
Xie H. Yang C. Sun Y. Igarashi Y. Jin T. Luo F. (2020). PacBio long reads improve metagenomic assemblies, gene catalogs, and genome binning. Front. Genet.11. doi: 10.3389/fgene.2020.516269
53
Xu M. Guo L. Du X. Li L. Peters B. A. Deng L. et al . (2021). Accurate haplotype-resolved assembly reveals the origin of structural variants for human trios. Bioinformatics.37. 2095–2102 doi: 10.1093/bioinformatics/btab068
54
Xu M. Guo L. Gu S. Wang O. Zhang R. Peters B. A. et al . (2020). TGS-GapCloser: A fast and accurate gap closer for large genomes with low coverage of error-prone long reads. GigaScience9. doi: 10.1093/gigascience/giaa094
Summary
Keywords
symbiosis, decontamination, metagenomic sequencing, long reads, bioinformatics, alignment-free, reference-free, de novo assembly
Citation
Xu M, Guo L, Qi Y, Shi C, Liu X, Chen J, Han J, Deng L, Liu X and Fan G (2023) Symbiont-screener: A reference-free tool to separate host sequences from symbionts for error-prone long reads. Front. Mar. Sci. 10:1087447. doi: 10.3389/fmars.2023.1087447
Received
02 November 2022
Accepted
16 January 2023
Published
30 January 2023
Volume
10 - 2023
Edited by
Neil Ross McEwan, Robert Gordon University, United Kingdom
Reviewed by
Matthew Hegarty, Aberystwyth University, United Kingdom; Shuyuan Wang, Harbin Medical University, China
Updates
Copyright
© 2023 Xu, Guo, Qi, Shi, Liu, Chen, Han, Deng, Liu and Fan.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guangyi Fan, fanguangyi@genomics.cn; Xin Liu, liuxin@genomics.cn
†These authors have contributed equally to this work
This article was submitted to Microbial Symbioses, a section of the journal Frontiers in Marine Science
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.