Abstract
The microbiome is a crucial influencer in animal development, immune function and health, and it has complex and dynamic interactions with the environment, but little is known about the microbial signatures of inner body fluids. Recent evidence suggests that inner body fluids could be an indicator of the environmental interactions that fish experience. In the present study, we provide a comparative analysis of the microbial profile found in small-spotted catsharks’ blood plasma and seminal plasma and how microbial signatures vary between aquarium and wild animals. In the blood plasma microbiome, the habitat did not affect the α- and β-diversity, while in seminal plasma, both α- and β-diversity differed between both habitats. Proteobacteria are the main bacteria dominated independently the inner body fluid and habitats. No core microbiome was identified at the genus level, with only Pseudomonas and Cloacibacterium present in both inner body fluids and habitats. Of the 14 genera identified in blood plasma, only four were shared between habitats (making up 45.17% and 51.03% of relative abundance for wild and aquarium, respectively). Similarly, of the 100 genera identified in seminal plasma, only 41 were shared between habitats (84.79% and 64.83%, respectively). Moreover, in the seminal plasma, using ANCOM approaches, Serratia, Salinisphaera and Cutibacterium were found significantly enriched in aquarium animals. None potentially pathogenic bacteria were identified in the blood samples, while Coxiella, Prevotella, Coprococcus, Haemophilus and Phocoenobacter were potentially pathogenic bacteria identified in the seminal plasma samples. In summary, this study provides evidence of a circulating blood and seminal plasma microbiome in healthy small-spotted catsharks. Furthermore, dynamic changes were observed in the microbiome of these inner body fluids, which differed between the aquarium and wild habitats.
1 Introduction
As with other vertebrates, the fish microbiome is fundamental to the health of its host and has complex and dynamic interactions with the environment (Luna et al., 2022). In this context, it is well known that environmental stressors can compromise fish health and fitness (Uren-Webster et al., 2020). Certainly, animals under managed care offer an ideal framework for identifying microbial components that may have significant associations with host metabolism and overall health. It is also important during the identification of potential drivers influencing microbiome assembly, such as environmental changes (Clavere-Graciette et al., 2022). Certainly, large aquaria, designed using recirculation systems to mimic natural marine environments as closely as possible, including host resident microbes, can be particularly useful (Patin et al., 2018; Clavere-Graciette et al., 2022). It is now widely accepted that all multicellular organisms associated with microbes (bacteria, protists, fungi, viruses and protist) that contribute to the biology of their respective host have variable functional impacts, ranging from beneficial to unimpactful to detrimental (Bosch and McFall-Ngai, 2011; Human Microbiome Project Consortium, 2012; Moran, 2015). Indeed, host-associated microbiomes are considered one functional form (biological unit) (Knowlton and Rohwer, 2003; Bosch and McFall-Ngai, 2011; Bang et al., 2018; Jaspers et al., 2019), but there is little literature focused on the microbe communities in large aquaria and their effects on animal physiology (Rowe et al., 2020; Perry et al., 2021; Clavere-Graciette et al., 2022). Likewise, microbial structure in wild animals has been poorly studied, although natural fish populations are increasingly subject to multiple anthropogenic stressors that threaten their conservation status (Perry et al., 2020; Rowe et al., 2020; Uren-Webster et al., 2020).
In recent decades, numerous studies have explored oral, gill, skin, cloacal, and gut microbial microbiomes (Perry et al., 2020; Sehnal et al., 2021; Clavere-Graciette et al., 2022). In contrast, little is known about the existence of a microbiome signature in inner body fluids. For instance, recent data support the existence of a microbial profile in the blood of various domesticated mammals and birds (Sze et al., 2014; Mandal et al., 2016; Vientós-Plotts et al., 2017; Scarsella et al., 2020); however, this statement is not enterily clear in relation to healthy human individuals due to opposite results found in the literature (Whittle et al., 2018; Castillo et al., 2019; D’Aquila et al., 2021; Tan et al., 2023) and, although due to opposite results recently published (Tan et al., 2023). In fish, very little is known about the microbial profile found in blood (Grimes et al., 1985; Buck, 1990; Mylniczenko et al., 2007; Tao et al., 2014; Tarnecki et al., 2018). The bacteria that can access the circulatory compartment could potentially be used as an indicator of the environmentally-derived taxa (Castillo et al., 2019). Likewise, a reproductive microbial profile could significantly affect the reproductive function and fitness of males and females, although these are rarely considered from an ecological or evolutionary perspective (Weinberg, 1974; Karin et al., 2006; Culligan and Sleator, 2016; Taylor et al., 2018; Theodosopoulos et al., 2019; Rowe et al., 2020). Understanding microbial signatures of reproductive fluids is an emerging and critical component of wildlife conservation (Comizzoli et al., 2021). Actually, metagenomic study of the reproductive microbiome is being considered a pivotal tool for species conservation, as it will allow the study of evolutionary and adaptive mechanisms that could be involved in the lack of reproductive efficiency and potentially, its intervention could collaborate in the preservation of endangered species (Contreras et al., 2023).
However, the key elements connecting microbial communities in reproductive fluids and reproductive success are not defined (Koh et al., 2019; Rowe et al., 2020).
In the present study, we compare the microbial profile found in the peripheral blood and seminal plasma of small-spotted male catsharks (Scyliorhinus canicula). In addition, we explored how microbial signatures of these inner body fluids vary between wild and aquarium (Oceanogràfic, Ciudad de las Artes y las Ciencias, Valencia, Spain) animals.
2 Materials and methods
2.1 Sample collection
Small-spotted catsharks were sampled in January 2020. Blood and seminal samples were collected from 17 wild small-spotted catsharks from the Mediterranean Sea and 7 aquarium-housed small-spotted catsharks in collaboration with the Oceanogràfic. Ubication (wild vs aquarium), date of sampling, body weight, length, width and clasper length for all catsharks are provided in Supplementary Table 1. Wild animals were donated by local fisheries in the Valencian Community and were part of accidental captures destined for commercial fisheries from the ports of Valencia (39°26′45″N 0°19′12″W), Jávea (38°47′21″N 0°09′47″E) and Cullera (39°09′58″N 0°15′10″W). Mediterranean Sea water parameters, measured by Valencia buoy (39°52′N0°20′E) during the sampling period were 14.6-19°C temperature and 34-37 g/l salinity (http://www.puertos.es/es-es/oceanografia/Paginas/portus.aspx). Animals were maintained in a closed and recirculation system under controlled conditions, monitored water quality (17–21°C, 5.1 mg/l oxygen, 36 g/l salinity and 7.6–8.2 pH), fixed photoperiod (12:12 h) and disinfection using UV light and ozone. Animals were kept isolated from females for at least 1 year before the experiment started. Aquarium animals’ diet was based on frozen-thawed whole fish: herring (Clupea harengus), mackerel (Scomber scombrus) and squid (Loligo sp). All animals from the study were classified as adults, displaying calcified claspers ensuring reproductive maturity (Kousteni et al., 2010). All wild animals were determined healthy based on physical examination, body conditions and confirmed in vitro sperm quality (Muñoz-Baquero et al., 2021). Aquarium animals were also determined healthy based on physical exams and clinical history. Each sampling day and biometrics were recorded for all the small-spotted catsharks, the weight ranged from 165 to 370 g (mean ± SD was 281.2 ± 48.6 g). The maturity of the sharks was determined by their size according to Ebert and Dando, 2020, together with the gonad development and clasper calcification length ranged from 39 to 50 cm (mean ± SD was 44.9 ± 2.7 cm), and clasper length ranged from 2.9 to 5 cm (mean ± SD was 3.7 ± 0.5 cm), considering also the total width.
Peripheral blood and semen samples from wild and aquarium animals were collected on the same day across 2 sessions. Therefore, during the collection of samples from the aquarium sharks instructed by the veterinarian, small-spotted catsharks were also collected from local fisheries on the same day. Peripheral blood was collected from the ventral coccygeal vein by caudal venipuncture, using 23-gauge needles and transferred to lithium-heparin tubes (Figure 1). Semen samples were collected by applying external pressure on the ampulla of the vas deferens, using a 5 ml syringe for semen collection (Figure 1). In aquarium animals, to rule out possible contamination with skin microbiome during the sampling, an sterile zone was created, both at the venipuncture and cloacal sites, by washing the area with sterile elasmobranch Ringer’s solution (22 g/l urea and 9 g/l NaCl in distilled water) and sterile gauzes (Mylniczenko and Clauss, 2017), following the Elasmobranch Husbandry Guidelines. Moreover, semen samples were checked for possible urine and fecal contamination under the microscope. After the collection of the samples from aquarium animals, the small-spotted catsharks were released back into the quarantine tank, and their recovery was carefully monitored by the aquarium veterinarian. Samples were immediately transferred to the laboratory at 4°C in a dark container after collection. For wild animals, firstly, disinfection of area with an ethanol-soaked (96°) cloth was carried out, followed by washing as was done for the aquarium animals. Samples were transferred to the laboratory under the same conditions, within a maximum of 4 h post-fishing.
Figure 1

Pictures demonstrating blood and semen samples collection from aquarium (i) and wild (ii) small-spotted catshark (Scyliorhinus canicula): (A) (i) an aquarium small-spotted catshark in holding tank and (ii) a wild small-spotted catshark in lab with clasper detail (white arrows), (B) (i, ii) blood sampling, (C) (i, ii) semen sampling, Semen samples were collected by abdominal massage.
Only animals from which both semen (100 µl) and blood (100 µl) were obtained were included in the study. A total of 7 pooled samples (4 for wild and 3 for aquarium animals) were established, mixing 100 µl of each individual sample. The 4 pooled samples from the wild animals included 5, 3, 3, and 6 animals for Valencia, Cullera (2 pooled samples) and Jávea ports, respectively. Regarding the samples from the aquarium animals, due to the limited number of animals (n=7), 3 pooled samples were obtained. The 2 first pooled samples included 3 and 3 animals. The third pooled sample includes the remaining animals, 1 from the first sampling session and 1 from the second pool with an excess in semen production (more than 200 µl). The sample pooling strategy is described in Supplementary Table 1. Pooling samples can serve as an effective method for screening bacterial nucleic acids (Federer, 1994; Furstenau et al., 2020). When both, blood and seminal plasma were available from each individual sample, a random selection of 3 samples (for the aquarium group) or 4 samples (for the wild group) was made to establish a single pooled sample per group. This was achieved by combining 100 µl of each individual sample.
Both blood and semen pooled samples were centrifuged at 3000 g and 7400 g, respectively, for 10 min at 4°C. After centrifugation, the seminal plasma fraction was verified under a microscope ensuring no spermatozoa. All plasma samples were directly flash-frozen in liquid nitrogen and stored at −80°C until further processing.
2.2 DNA extraction, library preparation and sequencing
Total DNA from generated pool samples was extracted using the NucliSens easyMag automated system (bioMérieux, Marcy l’Etoile, France) according to the manufacturer’s instructions. The amplification and sequencing protocol started with the microbial genomic DNA (5 ng/μl in 10 mM Tris pH 8.5) as the starting point. The V3–V4 region of the bacterial 16S rRNA gene sequences was amplified using the primer pair S-D-Bact-0341-b-S-17 -S-D-Bact-0785-a-A-21 (Klindworth et al., 2013) containing the gene-specific and Illumina adapter overhang nucleotide sequences. Primer sequences were forward primer: (5′- TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-CCTACGGGNGGCWGCAG-3’; reverse primer: 5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-GACTACHVGGGTATCTAATCC-3’). After 16S rRNA gene amplification, the multiplexing step was performed using the Nextera XT Index Kit (FC-131-1096). One-μl of the PCR product on a Bioanalyzer DNA 1000 chip (Agilent Technologies, USA, Santa Clara) was run to verify the size, with ∼550 bp as the expected size on the Bioanalyzer trace. Thus, the libraries were sequenced using a 2 × 300 bp paired-end run (MiSeq Reagent kit v3, MS-102-3001) on a MiSeq Sequencer, following the manufacturer’s instructions (Illumina, Inc., San Diego, CA, USA). In addition, a negative control was employed during library preparation to ensure accuracy and identify any potential background or contamination issues.
DNA-free water (UltraClean PCR Water, MO BIO Laboratories, Carlsbad, CA, USA) was employed as the input for the post-extraction steps of the protocol, starting with library generation, to assess contamination in downstream processes.
2.3 Bioinformatics analysis
Raw sequencing data were processed by QIIME2 v2021.4. The DADA2 pipeline included into QIIME2 was used to carry out denoising, filtering, and chimera removal of the sequences and reads were derreplicated in Amplicon Sequence Variants (ASVs). Each ASV was taxonomical assigned using the SILVA v138 database (Quast et al., 2013; Campos et al., 2022). Sequences classified as Chloroplast or Mitochondria were filtered out from the analysis. Identification of contaminant ASVs was performed using the decontam (v1.8.0) R package (Davis et al., 2018). The negative control exhibited the presence of 4 bacterial genera (Supplementary Table 2). R Decontam package did not detected rRNA contaminant gene reads among the datasets (Supplementary Figure 1). These low numbers of contaminant reads did not have any effect on the statistical analyses.
The α, β and taxonomic diversity analyses were mainly based on the QIIME2 pipeline. For the α-diversity, the richness and the richness with their relative abundance (evenness) were measured by Chao1 and Shannon indexes, respectively. The significance of differences among wild and aquarium sharks for seminal plasma and serum plasma was evaluated by Kruskal–Wallis test. Box-and-whisker plots for α-diversity were generated using Graphad Prism 8. A Venn diagram was drawn up to show the shared, and unique features among groups, based on the occurrence of features in a sample group regardless of their relative abundance and an interactive Venn software was used for Venn diagram construction (Heberle et al., 2015). Analysis of the composition of microbiomes (ANCOM) was run to determine if samples changed significantly between habitats by measuring the W value corresponding to the number of times an ASV abundance is significantly different for a group of samples (Mandal et al., 2015). The Bray-curtis distance, unweighted and weighted Unifrac values were calculated to estimate β-diversity between samples. Principal Coordinate Analysis (PCoA) plots were obtained for each sample’s habitats from Bray-Curtis distances using ClustVist software (Metsalu and Vilo, 2015). Microbial taxa that are shared by all groups was considered as core microbiome.
The processed 16S rRNA gene sequencing data were analyzed using PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) (Douglas et al., 2020). This allowed for the prediction of the microbial communities functional capabilities on the taxonomic composition inferred from the 16S rRNA gene data. To identify biomarker functional potentials between groups, the LEfSe (Linear discriminant analysis Effect Size) method was employed through the website http://huttenhower.sph.harvard.edu/galaxy/. Additionally, the Functional Annotation of PROkaryotic TAXa (FAPROTAX) database (Louca et al., 2016; Zhou et al., 2020) was implemented to identify potential pathogens based on the 16S rRNA gene sequences.
3 Results
A total of 37 and 410 unique sequences were used for taxonomic assignment for peripheral blood and seminal plasma samples, respectively (Table 1). For peripheral blood plasma, there were no differences in α-diversity indexes between the wild and aquarium animals (Figure 2A), while for the seminal plasma, lower richness (Chao1 index) for the wild animals was observed (Figure 2B). To observe differences regarding the β-diversity between samples, a PCoA ordination plot was performed using Bray-Curtis distances (Figure 3), where the two first principal components (PCs) explain the 29.8% and 25.5% for blood plasma and the 19.6% and 16.7% for seminal plasma of the observed variance, respectively. Moreover, results of PERMANOVA (Permutational multivariate analysis of variance) on unweighted and weighted UniFrac distances were used to compare microbiome taxonomic composition between body fluids and between the aquarium and wild animals. In pairwise comparisons of inner body fluids, no significant differences were found between the wild and aquarium small-spotted catsharks in blood plasma. However, there was a significant difference in seminal plasma for unweighted UniFrac distance (blood plasma: p = 0.474 and p = 0.774 and, for wild and aquarium animals respectively; and seminal plasma: p = 0.102 and p = 0.036, for wild and aquarium animals, respectively).
Table 1
| Peripheral Blood Plasma | Seminal Fluid | |
|---|---|---|
| Total raw reads | 375,009 | 334,893 |
| Average sequences length (bp) | 376.7 ± 64.35 | 412.8 ± 65.73 |
| Average number of sequences per sample | 46,876.12 | 41,861.65 |
| Minimum number of sequences per sample | 32,709 | 11,195 |
| Maximum number of sequences per sample | 60,524 | 107,421 |
| Unique sequences (a) | 38,456 | 155,169 |
| ASVs generated | 3,485 | 989 |
| Sequences removed | 37,972 | 37,463 |
| Mapped Sequences (b) | 484 | 117,706 |
| ASVs generated for taxonomic assignment | 37 | 410 |
General features of 16S rRNA amplicon sequencing of peripheral blood and seminal plasma microbiota.
(a) Sequences obtained after a series of data processing steps such as data filtering, denoising, merging and chimera removing.
(b) Against the reference dataset SILVA.
Figure 2
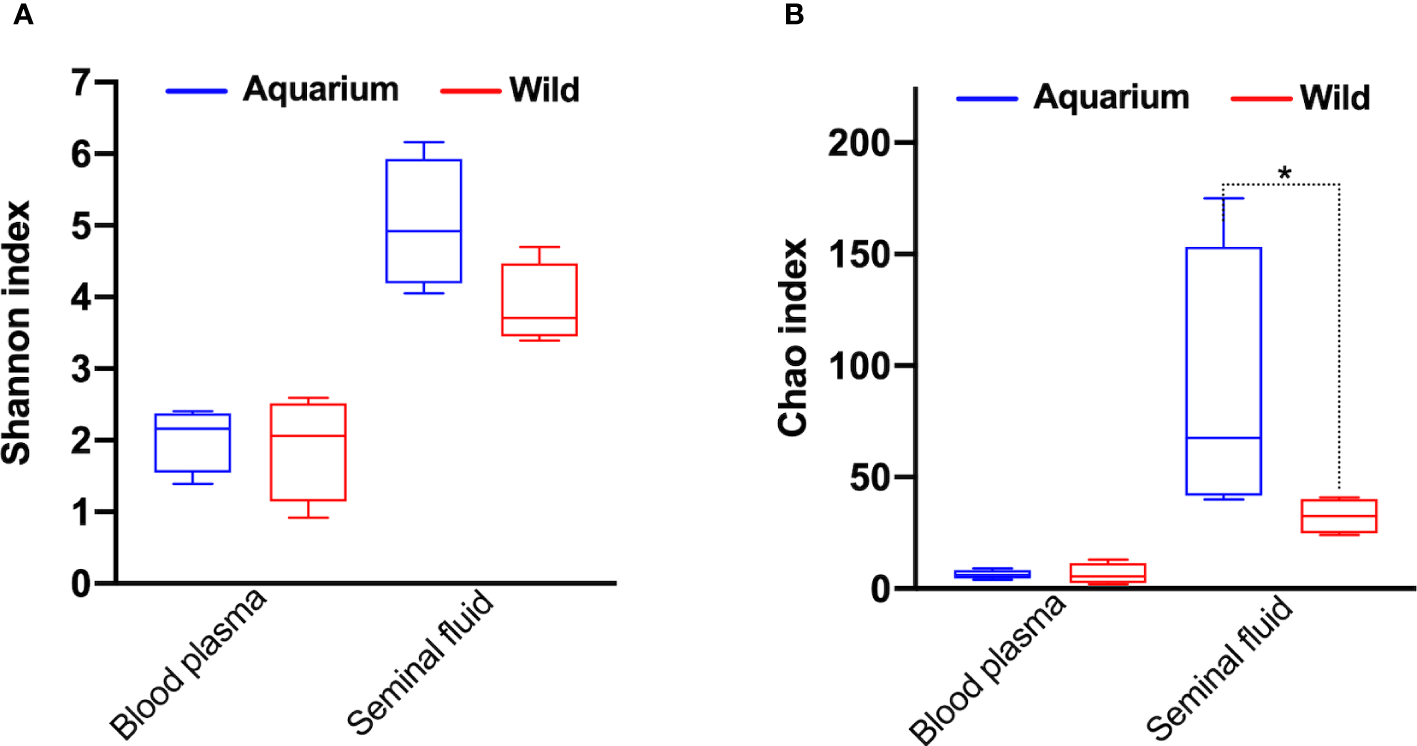
α‐Diversity metrics for (A) Shannon and (B) Chao diversity indices across inner body fluids (peripheral blood plasma and seminal plasma) of aquarium and wild small-spotted catshark (Scyliorhinus canicula). * indicates significant differences (p< 0.05) between aquarium and wild animals in the same inner body fluid analysed.
Figure 3
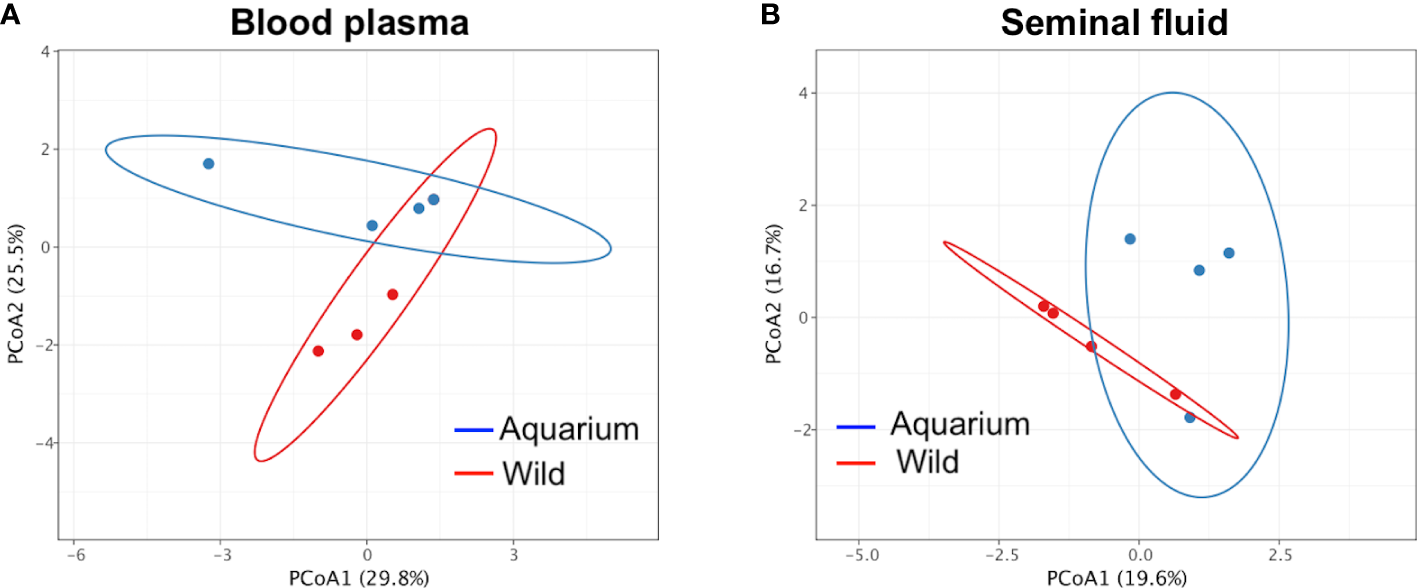
Principal coordinate analysis (PCoA) of β‐diversity comparison using Bray–Curtis distances across inner body fluids of wild and aquarium small-spotted catshark (Scyliorhinus canicula). For (A) peripheral blood plasma and (B) seminal plasma samples.
At the phyla level, peripheral blood plasma microbiome in wild animals was dominated by Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes, while in aquarium small-spotted catsharks, Firmicutes were not present (Figures 4A, B). In the seminal plasma, the microbial composition did not differ between wild and aquarium animals, being dominated in both habitats by Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes (Figures 4C, D). Across both groups of animals (aquarium and wild), Proteobacteria was the predominant phylum independently of the inner body fluid. To examine the existence of an identifiable common core microbiome, defined as the group of members shared among the microbial community, a Venn diagram was represented by overlapping areas in the circles at the genus level of the bacterial community. Thus, no core microbiome could be identified in both inner body fluids (Figure 5A), with only Pseudomonas and Cloacibacterium being present blood plasma and seminal fluid in both wild and aquarium animals (Figure 5B). In peripheral blood plasma, 4 shared genera were identified between both experimental groups (wild and aquarium) (Figure 5C), while in seminal plasma, a total of 41 shared genera were identified between wild and aquarium animals (Figure 5D). At finer taxonomic levels, such as genus, the blood microbiome was distinct between wild and aquarium individuals (Table 2). Differences were observed in several genera present only in wild animals and other genera present only in aquarium catsharks (Table 2). Likewise, the seminal plasma fluid microbiome differs between wild and aquarium organisms (Table 3— seminal plasma microbiome at an average relative abundance of more than 1% in at least one sample group). ANCOM approaches demonstrated that Salinisphaera, Serratia, and Cutibacterium were significantly enriched in the aquarium small-spotted catshark seminal plasma (W=32, 11, and 11, respectively). In addition, these findings were observed for several genera that were exclusively present in wild animals and other genera that were exclusively present in aquarium animals (Table 3).
Figure 4
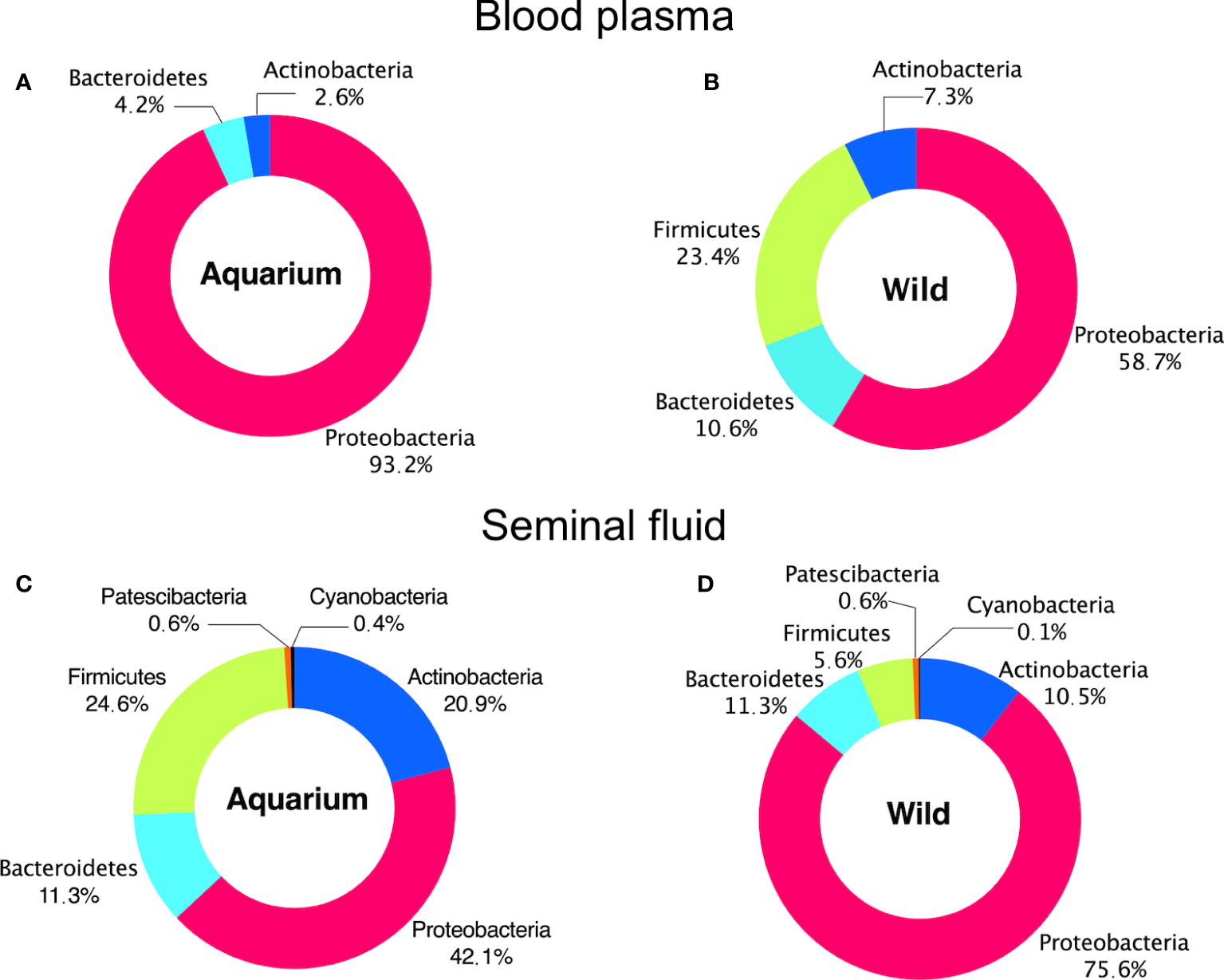
Phylum distribution in different inner body fluids from aquarium and wild small-spotted catshark (Scyliorhinus canicula). For (A, B) peripheral blood plasma and (C, D) seminal plasma samples.
Figure 5
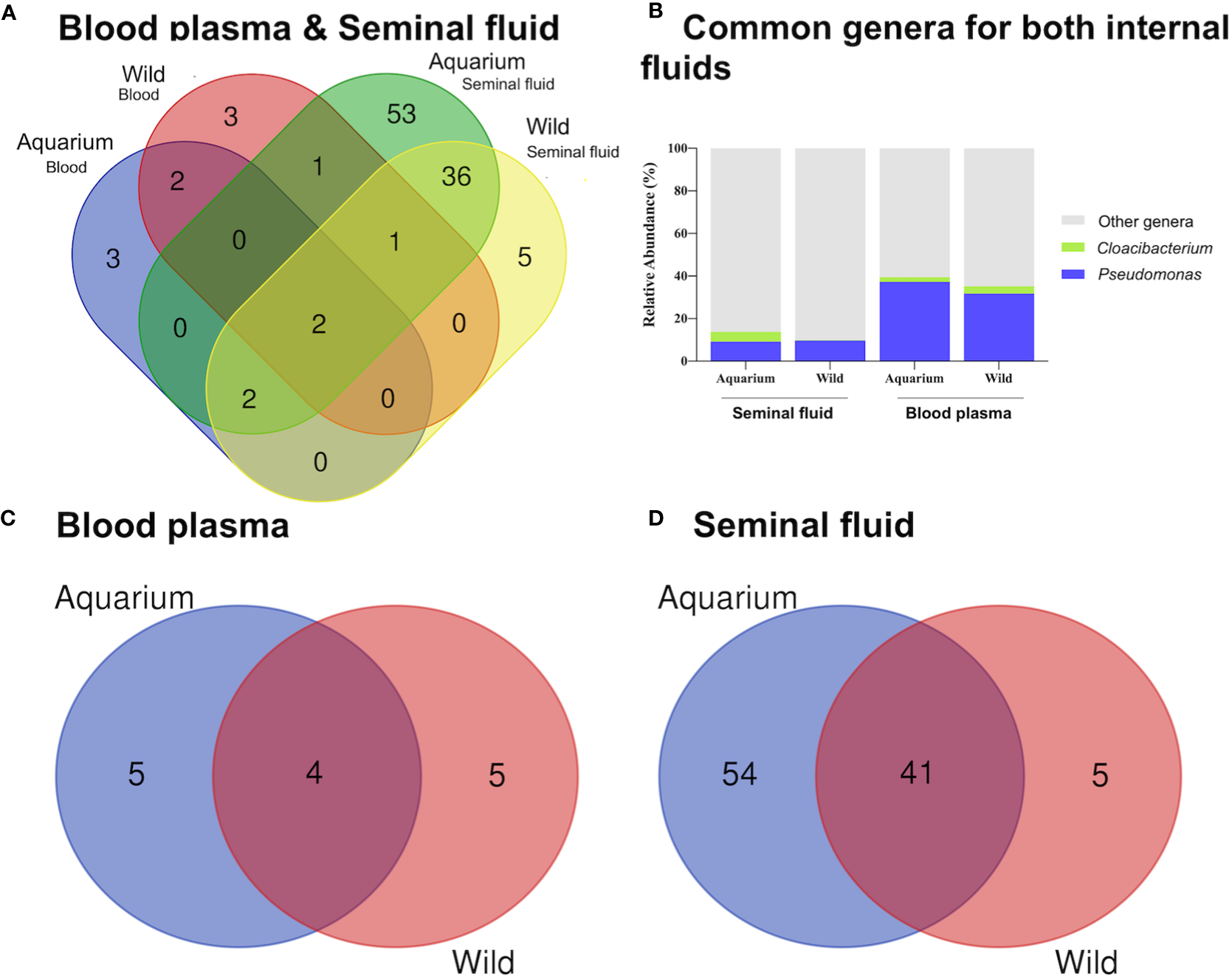
Venn diagram representation of shared and unique genera in different inner body fluids from aquarium and wild small-spotted catshark (Scyliorhinus canicula) using Venny (https://bioinfogp.cnb.csic.es/tools/venny_old/venny.php). For (A) both inner body fluids, (B) Shared genera (C) peripheral blood plasma and (D) seminal plasma samples.
Table 2
| Phylum | Family | Genera | Habitat | |||
|---|---|---|---|---|---|---|
| Aquariumm (%) | Wild (%) | |||||
| Proteobacteria | Pseudomonadaceae | Pseudomonas | 37.28 | 31.72 | ||
| Beijerinckiaceae | Methylobacterium-Methylorubrum | 29.82 | – | |||
| Xanthobacteraceae | Rhodopseudomonas | 7.46 | – | |||
| Bradyrhizobium | 3.07 | 3.45 | ||||
| Rhodobacteraceae | Paracoccus | – | 10.34 | |||
| Burkholderiaceae | Pandoraea | 6.14 | – | |||
| Sphingomonadaceae | Novosphingobium | 7.02 | – | |||
| Yersiniaceae | Serratia | 4.39 | – | |||
| Firmicutes | Bacillaceae | Anoxybacillus | – | 13.10 | ||
| Monoglobaceae | Monoglobus | – | 9.66 | |||
| Thermicanaceae | Thermicanus | – | 8.28 | |||
| Bacteroidota | Dysgonomonadaceae | Dysgonomonas | 2.63 | 12.41 | ||
| Weeksellaceae | Cloacibacterium | 2.19 | 3.45 | |||
| Actinobacteriota | Nocardiaceae | Gordonia | – | 7.59 | ||
Relative abundance of bacteria at the genus level in peripheral blood plasma from aquarium and wild small-spotted catshark (Scyliorhinus canicula), based on the ANCOM test.
(–) indicates undetectable levels.
Table 3
| Phylum | Family | Genera | Habitat | |
|---|---|---|---|---|
| Aquarium (%) | Wild (%) | |||
| Proteobacteria | Pseudomonadaceae | Pseudomonas | 9.15 | 9.69 |
| Saccharospirillaceae | Oleispira | 6.07 | 1.40 | |
| Yersiniaceae | Serratia* | 2.47 | 2.07 | |
| Moraxellaceae | Enhydrobacter | 2.36 | 0.77 | |
| Acinetobacter | 2.21 | 2.47 | ||
| Salinisphaeraceae | Salinisphaera* | 2.12 | – | |
| Marinobacteraceae | Marinobacter | 2.07 | 0.28 | |
| Pseudoalteromonadaceae | Pseudoalteromonas | 2.06 | 29.68 | |
| Comamonadaceae | Comamonas | 1.91 | 0.91 | |
| Sphingomonadaceae | Novosphingobium | 1.32 | 0.09 | |
| U.m. family Sphingomonadaceae | 0.52 | 2.00 | ||
| Halomonadaceae | Halomonas | 1.20 | 0.23 | |
| Rhizobiaceae | Allorhizobium-Neorhizobium-Pararhizobium | 0.37 | 1.29 | |
| U.m. family Rhizobiaceae | 0.23 | 1.31 | ||
| Vibrionaceae | Vibrio | 0.01 | 14.84 | |
| Photobacterium | – | 6.58 | ||
| Firmicutes | Staphylococcaceae | Staphylococcus | 4.75 | 0.05 |
| Streptococcaceae | Streptococcus | 1.84 | 0.39 | |
| Enterococcaceae | Enterococcus | 1.74 | – | |
| Ruminococcaceae | Subdoligranulum | 1.24 | – | |
| Faecalibacterium | 1.56 | – | ||
| Aerococcaceae | Aerococcus | 1.55 | – | |
| Globicatella | 1.71 | – | ||
| Erysipelotrichaceae | Holdemanella | 1.34 | – | |
| Lachnospiraceae | Blautia | 1.48 | – | |
| [Eubacterium]_hallii_group | 1.30 | – | ||
| Lachnoclostridium | 1.25 | – | ||
| Bacillaceae | Geobacillus | 0.69 | 1.24 | |
| Mycoplasmataceae | Mycoplasma | – | 3.18 | |
| Bacteroidota | Weeksellaceae | Cloacibacterium | 4.64 | 0.23 |
| U.m. family Weeksellaceae | 1.52 | – | ||
| Chryseobacterium | 0.30 | 1.97 | ||
| Rikenellaceae | Alistipes | 1.30 | – | |
| Flavobacteriaceae | U.m. family Flavobacteriaceae | – | 2.24 | |
| Actinobacteriota | Propionibacteriaceae | Cutibacterium* | 7.36 | 4.44 |
| Corynebacteriaceae | Corynebacterium | 4.35 | 1.12 | |
| Lawsonella | 4.13 | 0.05 | ||
| Microbacteriaceae | U.m. family Microbacteriaceae | 1.53 | – | |
| Rubrobacteriaceae | Rubrobacter | 0.07 | 1.45 | |
| Promicromonosporaceae | Promicromonospora | – | 1.72 | |
Relative abundance of more than 1% in at least one sample group of bacteria at the genus level in seminal plasma from aquarium and wild small-spotted catshark (Scyliorhinus canicula).
*indicates significant differences in abundance levels between groups based on the ANCOM test. (–) indicates undetectable levels.
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis revealed the detection of a total of 194 pathways across all small-spotted catshark samples. The PICRUSt results elucidated slight differences in the composition of metabolic functions in the blood plasma samples (Supplementary Table 4). The significant metabolic functions identified were glucose and glucose-1-phosphate degradation (GLUCOSE1PMETAB-PWY), L-methionine biosynthesis III (HSERMETANA.PWY), urate biosynthesis/inosine 5-phosphate degradation (PWY.5695), and pyruvate fermentation to isobutano (PWY.7111). It is worth noting that pyruvate fermentation to isobutano is an engineered pathway, not occurring naturally in any known organism, but constructed through metabolic engineering in a living cell. Comparing the seminal plasma samples, no significant differences in the composition of metabolic functions were observed between aquarium and wild animals (Supplementary Table 4).
Furthermore, FAPROTAX analysis revealed that the relative abundance of dominant functions exhibited varied compositions among different samples. A total of 16 and 27 categories of microbial functions linked to the bacterial communities were identified in the blood and seminal plasma samples, respectively (Supplementary Table 5). The predominant microbial functions assigned were chemoheterotrophy and aerobic chemoheterotrophy for both inner fluids. Additionally, nitrate reduction and fermentation were observed in the seminal plasma samples. In order to assess the risks of potential pathogens for aquatic wildlife (e.g., aquatic animals and plants), we focused on groups relevant to pathogens in this study. No pathogenic groups were identified in the blood samples, whether from aquarium or wild animals (Figure 6A). In the seminal plasma, pathogenic groups were detected, including animal parasites or symbionts (in 2 pooled samples from aquarium animals and 1 pooled sample from wild animals, representing 2.36%) and intracellular parasites (in 1 pooled sample from aquarium animals, representing 0.44%) (Figure 6B). The potentially pathogenic bacteria detected were Coxiella, Prevotella, Coprococcus, Haemophilus and Phocoenobacter.
Figure 6
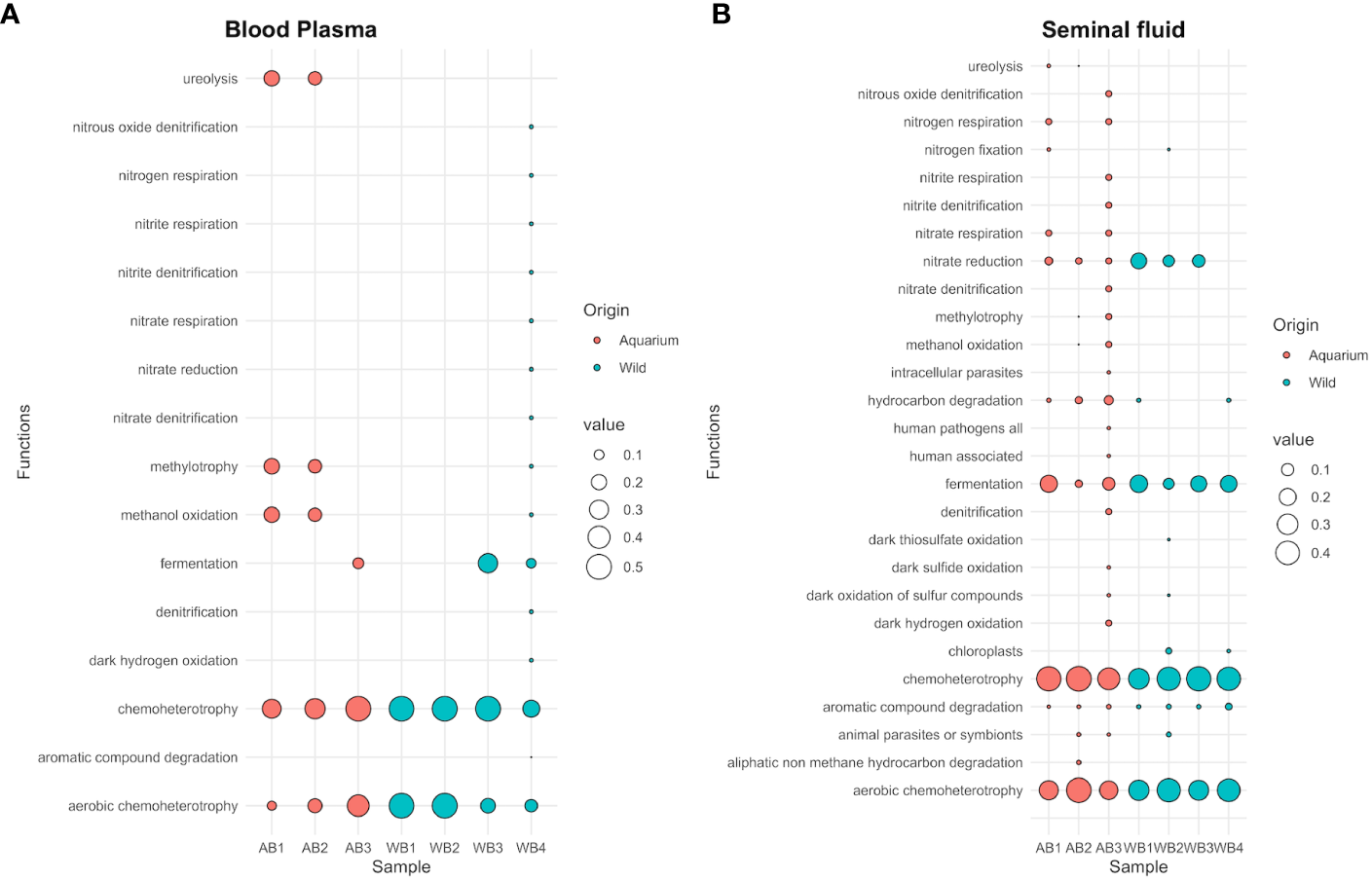
Bubble plot showing the relative abundance of functional annotations within the microbial communities for both aquarium and wild small-spotted catshark (Scyliorhinus canicula) samples. The plot includes data from (A) peripheral blood plasma and (B) seminal plasma samples.
4 Discussion
Our results are consistent with previous studies that have reported bacterial colonization in the blood of healthy marine life, terrestrial animals, and humans (Vientós-Plotts et al., 2017; Tarnecki et al., 2018; Castillo et al., 2019; Anhê et al., 2020). This supports the existence of a blood plasma microbiota in healthy small-spotted catsharks. In this study, no statistical associations were observed between the pathogen community and blood samples, regardless of the sample’s origin. However, it is important to note that functional annotations based on the FAPROTAX database provide inferred functions relying on 16S rRNA fragments, which may not be as precise as a comprehensive shotgun metagenomic study. This consideration should be taken into account when evaluating the accuracy of pathogen assignment in this study. These findings are consistent with the observations made during medical evaluations of sharks from both Oceanogràfic and the wild. In fact, the sharks at Oceanogràfic had been housed in the aquarium facility for at least two years and were determined to be healthy. The sharks from the wild were also examined post-mortem, with accidental capture considered as the cause of death. These results support the hypothesis that the microbial profile found in blood plasma is a credible biological phenomenon with no pathological implications. Previous studies on healthy Chondrichthyes have shown the occurrence of positive bacterial cultures in their hosts without any clinical signs, considering non-sterile blood as their baseline condition (Grimes et al., 1985; Buck, 1990; Mylniczenko et al., 2007; Tao et al., 2014; Tarnecki et al., 2018).
Admittedly, we found a similar α- and β-diversity between wild and aquarium animals in blood plasma. Consequently, we can rule out the possibility of external contamination (skin bacteria) in the wild animals where an aseptic field was created by washing with sterile gauze without previous cleaning using alcohol. At the phylum level, blood plasma microbial composition differs between both experimental groups, being dominated in aquarium animals only by Proteobacteria, while in addition, Proteobacteria, Firmicutes, Actinobacteria and Bacteroidota were also present in wild animals. In the white-spotted eagle ray, Proteobacteria, Firmicutes, Actinobacteria and Bacteroidota also dominate the microbiota of the cloaca, gills and skin (Clavere-Graciette et al., 2022). Likewise, a substantial environmental influence on microbiome structuring has been documented in marine animals, including the common thresher shark (Alopias vulpinus, Doane et al., 2017), the black-tip reef shark (Carcharhinus melanopterus, Pogoreutz et al., 2019), killer whales (Orcinus orca, Hooper et al., 2019), humpback whales (Megaptera novaeangliae, Apprill et al., 2014). Although the environment was not explicitly studied, our findings suggest that the environment has an evident influence on the blood microbiome, confirming the recent results of Clavere-Graciette et al. (2022) in several body compartments in white-spotted eagle rays. Proteobacteria has been repeatedly described as the most abundant phylum in marine organisms, environments and water (Pogoreutz et al., 2019; Ruiz-Rodríguez et al., 2020; Serra et al., 2021; Clavere-Graciette et al., 2022). In fact, the shark skin microbiome is predominantly composed of this genus (Pogoreutz et al., 2019; Clavere-Graciette et al., 2022). Likewise, Proteobacteria were also the dominant phylum in the gut microbiome of fish and sharks (Givens et al., 2015; Huang et al., 2020) and has been identified as the predominant phylum in teleost fish and water in the Mediterranean Sea (Crespo et al., 2013; Ruiz-Rodríguez et al., 2020). However, other phyla identified in wild teleosts from the Mediterranean Sea were Fusobacteria and Tenericutes, phyla not identified in this study (Ruiz-Rodríguez et al., 2020). These results would support previous studies claiming that the autochthonous microbes are not a passive reflection of their habitat communities, i.e., each species has its own microbiota (Sullam et al., 2015; Ruiz-Rodríguez et al., 2020; Clavere-Graciette et al., 2022). Of the 14 genera detected in blood microbiome in wild or aquarium, we only found 4 shared genera (making up 45.17% and 51.03% of relative abundance for wild and aquarium sharks, respectively), with Pseudomonas, Bradyrhizobium, Dysgonomonas and Cloacibacterium. The Pseudomonas genus contains well-known pathogenic species, such as P. aeruginosa, but also contains species with probiotics potential, widely used in the aquaculture industry (Sehnal et al., 2021). Moreover, Pseudomonas and Cloacibacterium have been identified in the blood of healthy captive Red Drum (Tarnecki et al., 2018). In addition, Pseudomonas has been associated with protein biosynthesis and degradation, involved in nitrate and nitrite ammonification and denitrification, leading to ammonia assimilation and urea decomposition (Doane et al., 2022), as a common characteristic from chondrichthyan metabolism in wild and aquarium environments. Additionally, wild and aquarium animals showed significant differences in blood plasma. For example, Paracoccus, Anoxybacillus, Monoglobus, Thermicanus, and Gordonia genera were only present in wild animals. At the same time, Methylobacterium-Methylorubrum, Rhodopseudomonas, Pandoraea, Novosphingobium, and Serratia genera were only present in aquarium catsharks and these differences have been previously associated with the environment and diet influences in microbiome compositional shifts (Clavere-Graciette et al., 2022). Firmicutes was the only phylum present in the wild blood plasma microbiome, not present in aquarium sharks. The presence of Firmicutes is normally associated with the gastrointestinal bacteria present in the blood of wild animals (Clavere-Graciette et al., 2022). Regarding this fact, we cannot ignore possible bacterial contamination related to the capture of wild animals, as the substrate contact originated during the trawling, in contact with a highly perfused tissue such as gills, or to the possible breach of the gastrointestinal barrier for bacterial blood contamination.
Here we provide the first description of the taxonomic composition of the seminal plasma present in fish. We found a significantly lower α-diversity in seminal plasma microbiomes in wild organisms compared to aquarium counterparts, but with a similar β-diversity (microbiome composition). As with blood samples, we can rule out the potential external contamination (ampulla bacteria) during sampling in the aquarium animals. Previous studies also described a higher richness and diversity (fecal and skin samples) in wild counterparts compared to under managed care (Uren-Webster et al., 2018: 2020). Nevertheless, contradictory results have recently been found for the white-spotted eagle ray (Clavere-Graciette et al., 2022). These discrepancies could be explained in part by the differences in microbial community composition across body sites (Minich et al., 2020; Clavere-Graciette et al., 2022). In the seminal fluid, the diverse microbiota was predominantly composed of 4 main phyla, which include Proteobacteria, Firmicutes, Bacteroidetes and Actinobacteria (Koh et al., 2019), also found in a study based on denaturing gradient gel electrophoresis.
The taxonomic composition of each sampling habitat carried a unique signature. At the genus level, collectively, of the 100 genera detected, only 41 were shared between wild and aquarium catsharks. It should be noted that different species reared in the same aquatic environment vary in their microbiome (Li et al., 2015; Sullam et al., 2015; Reverter et al., 2017). Notably, a total of 54 genera (making up 35.2% of the relative abundance) were present only in aquarium animals (Supplementary Table 3), while 5 genera (Photobacterium, Mycoplasma, Lactobacillus, U.m. family Flavobacteriaceae and Promicromonospora, making up 14.6% of the relative abundance) were only present in the wild small-spotted catsharks (Supplementary Table 3). Such compositional differences may be environmentally driven, in agreement with the results of a previous study by Clavere-Graciette et al. (2022) on white-spotted eagle rays (Aetobatus narinari).
Major taxonomic composition shifts were observed (ANCOM approaches) between wild and aquarium small-spotted catsharks in Serratia, Salinisphaera and Cutibacterium genera, being higher in managed care sharks, a factor associated with dysbiosis in other species (Garcia-Segura et al., 2022). Salinisphaera was ubiquitous in the Malaysian Mahseer sperm microbiota (Koh et al., 2019). Correspondingly, Cutibacterium has been identified in rays and skates (Gonçalves et al., 2020; Mika et al., 2021; Garcia-Segura et al., 2022). Even if this observation has not been put forward in fish studies, significant differences between fertile and infertile men were found in the relative presence of the Cutibacterium genus (Garcia-Segura et al., 2022). Likewise, Serratiahas not been put forward in fish studies, although ovine and porcine seminal plasma samples have tested positive for Serratia genera (Althouse and Lu, 2005; Tvrdá et al., 2022). There is some congruence in the identity of bacterial taxa we found in the seminal plasma with other marine fish belonging to different species. Briefly, as part of the seminal plasma microbiome shared between wild and aquarium small-spotted catsharks, an ASV identified as Mycoplasma and Vibrio are found in other marine fish species (Ciric et al., 2019; Ruiz-Rodríguez et al., 2020). Finally, within the shared seminal plasma microbiome of wild and aquarium sharks, an ASV identified as Staphylococcus has been identified in the skin and sting stripes of both wild and aquarium animals (Gonçalves et al., 2020). Notably, Staphylococcus is predominantly composed of the sperm microbiota in the Malayan mahseer; although their function is still unclear, but appears to play an essential role in spermatogenesis in fish testis (Kousteni et al., 2010). The higher diversity (in both α- and β-) found in the seminal plasma from both habitats, may be due to the anatomy of the reproductive tract in males, which is directly linked to the exterior through the genital pore, connecting the seminal ampullae with the cloaca (García-Salinas et al., 2021). It is well established that body sites have unique microbial signatures and are differentially influenced by the environment (Minich et al., 2020; Clavere-Graciette et al., 2022). Nevertheless, there is little information about the microbiome of internal fluids in fish; therefore, a meaningful discussion about the effects of specific genera is not yet possible. For instance, further research is needed to understand the role of Pseudomonas and Cloacibacterium, as both genera were present in the inner fluids of both wild and aquarium spotted sharks.
PICRUSt and FAPROTAX have been recently used to compare the microbiota of dolphins under human care to those of wild animals (Wan et al., 2022). Our results show that, based on the PICRUSt, the blood microbiota in aquarium animals is rich in the glucose-1-phosphate degradation pathway and L-methionine III biosynthesis. This enrichment can be mainly explained the daily food accessibility of the aquarium animals that increased availability of substrates necessary for L-methionine synthesis and glucose metabolism, resulting in heightened activity of genes and enzymes involved in these processes within the bacteria. A recent study by Pinchaud et al. (2022) observed that the urate biosynthesis/5-phosphate-inosine degradation pathways were more prevalent in mice gut microbiota when the animals were fed a lipid-rich diet. Based on the FAPROTAX, our results show that the predominant microbial functions assigned were chemoheterotrophy and aerobic chemoheterotrophy for both inner fluids, while nitrate reduction and fermentation were also predominant in the seminal plasma samples. Further experiments such as metabolomics are needed to verify the functions and roles that these enriched pathways play in aquarium small-spotted catsharks. The potential pathogens mentioned here mainly focus on aquatic wildlife. However, the database for pathogens of marine species is still lacking (Peng et al., 2021). Phylogenetic analysis identified five genera of potentially pathogenic bacteria in the seminal plasma of small-spotted catsharks-associated bacterial communities: Coxiella, Prevotella, Coprococcus, Haemophilus, and Phocoenobacter. All of these are recognized potential pathogens for both animals and humans (Louca et al., 2016). Given the absence of evident signs of illness during sampling and the fact that all males exhibit normal in vitro sperm quality (Muñoz-Baquero et al., 2021), the presence of potential pathogens in the seminal plasma may indicate a commensal relationship with the ejaculates of small-spotted catsharks. The findings demonstrated the potential significance of the male genital tract microbiota and its potential implications for the fertility and pathophysiology of small-spotted catsharks.
There are certain limitations to consider in the study. The environmental microbiome analysis for the wild group was not conducted to the unavailability of accurate water samples. Wild small-spotted catsharks inhabit a wide geographical range, spanning from shallow waters to 550 meters. The lack of this information in the analysis prevented us from confirming whether the bacterial composition is influenced by the environment. Additionally, our study only utilized pooled samples, which limited our ability to explore individual microbiota variations. Regarding the wild individual samples, the four-hour refrigerator storage may have led to some modifications in their composition. These changes could have affected our ability to detect similarities in ASV levels between samples. In the case of aquarium animals, both blood and seminal plasma samples may have been minimally contaminated by skin bacteria, even though the area was thoroughly disinfected with Elasmobranch’s Ringer solution, as the use of alcohol was not recommended.
In summary, this study provides evidence of a circulating blood and seminal plasma microbiome in healthy small-spotted catsharks. Furthermore, dynamic changes were observed in the microbiome of these inner body fluids, which differed between the wild and aquarium habitats. Additional studies are necessary to identify the physiological traits that contribute to bacterial colonization in different ecosystems among small-spotted catsharks. Moreover, further investigation into the potential association between bacteria found in seminal plasma and male fertility is warranted.
Statements
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, PRJNA830653, https://www.ncbi.nlm.nih.gov/, PRJNA974610.
Ethics statement
All the procedures involving aquarium animals in this study were approved by the Animal Care and Welfare Committee of the Oceanogràfic Valencia (Reference number: OCE-18–19) following the aquarium animal care protocol and policies. In the case of wild animals, they were fresh accidental captures with commercial value, donated by local fisheries.
Author contributions
Conceptualization: MM-B and FM-J. Methodology: MM-B, FM-J, LM-P, GM-R, IG-V, LL-R and GD’A. Investigation and data analysis: LL-R, GD’A and FM-J. Writing—original draft: MM-B and FM-J. Writing—review and editing: MM-B, LL-R, FG-V, DG-P, GD’A and FM-J. Supervision: FM-J. All authors contributed to the article and approved the submitted version.
Funding
LL-R was supported by a research grant from the Generalitat Valenciana-Fondo Social Europeo (ACIF/2020/376).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2023.1151119/full#supplementary-material
Supplementary Figure 1Negative control using 16S rRNA gene amplicon sequencing (V4 region).
References
1
Althouse G. C. Lu K. G. (2005). Bacteriospermia in extended porcine semen. Theriogenology63, 573–584. doi: 10.1016/j.theriogenology.2004.09.031
2
Anhê F. F. Jensen B. A. H. Varin T. V. Servant F. Van Blerk S. Richard D. et al . (2020). Type 2 diabetes influences bacterial tissue compartmentalisation in human obesity. Nat. Metab.2, 233–242. doi: 10.1038/s42255-020-0178-9
3
Apprill A. Robbins J. Eren A. M. Pack A. A. Reveillaud J. Mattila D. et al . (2014). Humpback whale populations share a core skin bacterial community: towards a health index for marine mammals? PloS One9, e90785. doi: 10.1371/journal.pone.0090785
4
Bang C. Dagan T. Deines P. Dubilier N. Duschl W. J. Fraune S. et al . (2018). Metaorganisms in extreme environments: do microbes play a role in organismal adaptation? Zool. (Jena)127, 1–19. doi: 10.1016/j.zool.2018.02.004
5
Bosch T. C. G. McFall-Ngai M. J. (2011). Metaorganisms as the new frontier. Zool. (Jena)114, 185–190. doi: 10.1016/j.zool.2011.04.001
6
Buck J. D. (1990). Potentially pathogenic marine vibrio species in seawater and marine animals in the Sarasota, Florida, area. J. Coast. Res.6, 943–948.
7
Campos P. M. Darwish N. Shao J. Proszkowiec-Weglarz M. (2022). Research note: choice of microbiota database affects data analysis and interpretation in chicken cecal microbiota. Poultry Sci.101, 101971. doi: 10.1016/j.psj.2022.101971
8
Castillo D. J. Rifkin R. F. Cowan D. A. Potgieter M. (2019). The healthy human blood microbiome: fact or fiction? Front. Cell. infect. Microbiol.9, 148. doi: 10.3389/fcimb.2019.00148
9
Ciric M. Waite D. Draper J. Jones J. B. (2019). Characterisation of gut microbiota of farmed Chinook salmon using metabarcoding. Arch. Biol. Sci.71 (4), 577–587. doi: 10.2298/ABS190402040C
10
Clavere-Graciette A. G. McWhirt M. E. Hoopes L. A. Bassos-Hull K. Wilkinson K. A. Stewart F. J. et al . (2022). Microbiome differences between wild and aquarium whitespotted eagle rays (Aetobatus narinari). Anim. Microbiome4, 34. doi: 10.1186/s42523-022-00187-8
11
Comizzoli P. Power M. L. Bornbusch S. L. Muletz-Wolz C. R. (2021). Interactions between reproductive biology and microbiomes in wild animal species. Anim. Microbiome3, 87. doi: 10.1186/s42523-021-00156-7
12
Contreras M.J. Núñez-Montero K. Bruna P. Zárate A. Pezo F. García M. Leal K. Barrientos L . (2023). Mammals’ sperm microbiome: current knowledge, challenges, and perspectives on metagenomics of seminal samples. Front. Microbiol. 14, 1167763. doi: 10.3389/fmicb.2023.1167763
13
Crespo B. G. Pommier T. Fernández-Gómez B. Pedrós-Alió C. (2013). Taxonomic composition of the particle-attached and free-living bacterial assemblages in the Northwest Mediterranean Sea analyzed by pyrosequencing of the 16S rRNA. MicrobiologyOpen2, 541–552. doi: 10.1002/mbo3.92
14
Culligan E. P. Sleator R. D. (2016). Advances in the microbiome: applications to clostridium difficile infection. J. Clin. Med.5 (9), 83. doi: 10.3390/jcm5090083
15
D’Aquila P. Giacconi R. Malavolta M. Piacenza F. Bürkle A. Villanueva M. M. et al . (2021). Microbiome in blood samples from the general population recruited in the MARK-AGE project: a pilot study. Front. Microbiol.12, 707515. doi: 10.3389/fmicb.2021.707515
16
Davis N. M. Proctor D. M. Holmes S. P. Relman D. A. Callahan B. J. (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome6, 226. doi: 10.1186/s40168-018-0605-2
17
Doane M. P. Haggerty J. M. Kacev D. Papudeshi B. Dinsdale E. A. (2017). The skin microbiome of the common thresher shark (Alopias vulpinus) has low taxonomic and gene function β-diversity: skin microbiome of the common thresher shark. Environ. Microbiol. Rep.9, 357–373. doi: 10.1111/1758-2229.12537
18
Doane M. P. Johnson C. J. Johri S. Kerr E. N. Morris M. M. Desantiago R. et al . (2022). The epidermal microbiome within an aggregation of leopard sharks (Triakis semifasciata) has taxonomic flexibility with gene functional stability across three time-points. Microbial Ecol.7, 1–18. doi: 10.1007/s00248-022-01969-y
19
Douglas G. M. Maffei V. J. Zaneveld J. R. Yurgel S. N. Brown J. R. Taylor C. M. et al . (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol.38, 685–688. doi: 10.1038/s41587-020-0548-6
20
Ebert D. A. Dando M. (2020). Field guide to sharks, rays & chimaeras of Europe and the Mediterranean (US: Princeton University Press).
21
Federer W. T. (1994). Pooling and other designs for analysing laboratory samples more efficiently. J. R. Stat. Society: Ser. D. (The Statistician)43 (3), 413–422. doi: 10.2307/2348577
22
Furstenau T. N. Cocking J. H. Hepp C. M. Fofanov V. Y. (2020). Sample pooling methods for efficient pathogen screening: practical implications. PloS One15 (11), e0236849. doi: 10.1371/journal.pone.0236849
23
García-Salinas P. Gallego V. Asturiano J. F. (2021). Reproductive anatomy of chondrichthyans: notes on specimen handling and sperm extraction. II. sharks and chimaeras. Animals11, 2191. doi: 10.3390/ani11082191
24
Garcia-Segura S. Del Rey J. Closa L. Garcia-Martínez I. Hobeich C. Castel A. B. et al . (2022). Seminal microbiota of idiopathic infertile patients and its relationship with sperm DNA integrity. Front. Cell Dev. Biol.10, 937157. doi: 10.3389/fcell.2022.937157
25
Givens C. Ransom B. Bano N. Hollibaugh J. (2015). Comparison of the gut microbiomes of 12 bony fish and 3 shark species. Mar. Ecol. Prog. Ser.518, 209–223. doi: 10.3354/meps11034
26
Gonçalves E. S. F. Dos Santos H. F. de Assis L. D. C. Lutfi D. S. Vianna M. Rosado A. S. (2020). Skin and stinger bacterial communities in two critically endangered rays from the south Atlantic in natural and aquarium settings. MicrobiologyOpen9, e1141. doi: 10.1002/mbo3.1141
27
Grimes D. J. Brayton P. Colwell R. R. Gruber S. H. (1985). Vibrios as autochthonous flora of neritic sharks. Syst. Appl. Microbiol.6, 221–226. doi: 10.1016/S0723-2020(85)80056-4
28
Heberle H. Meirelles G. V. da Silva F. R. Telles G. P. Minghim R. (2015). InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinf.16, 169. doi: 10.1186/s12859-015-0611-3
29
Hooper R. Brealey J. C. van der Valk T. Alberdi A. Durban J. W. Fearnbach H. et al . (2019). Host-derived population genomics data provides insights into bacterial and diatom composition of the killer whale skin. Mol. Ecol.28, 484–502. doi: 10.1111/mec.14860
30
Huang Q. Sham R. C. Deng Y. Mao Y. Wang C. Zhang T. et al . (2020). Diversity of gut microbiomes in marine fishes is shaped by host-related factors. Mol. Ecol.29, 5019–5034. doi: 10.1111/mec.15699
31
Human Microbiome Project Consortium (2012). Structure, function and diversity of the healthy human microbiome. Nature486, 207–214. doi: 10.1038/nature11234
32
Jaspers C. Fraune S. Arnold A. E. Miller D. J. Bosch T. C. G. Voolstra C. R. (2019). Resolving structure and function of metaorganisms through a holistic framework combining reductionist and integrative approaches. Zool. Jena Ger.133, 81–87. doi: 10.1016/j.zool.2019.02.007
33
Karin M. Lawrence T. Nizet V. (2006). Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell124, 823–835. doi: 10.1016/j.cell.2006.02.016
34
Klindworth A. Pruesse E. Schweer T. Peplies J. Quast C. Horn M. et al . (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res.41, e1. doi: 10.1093/nar/gks808
35
Knowlton N. Rohwer F. (2003). Multispecies microbial mutualisms on coral reefs: the host as a habitat. Am. Nat.162, S51–S62. doi: 10.1086/378684
36
Koh I. C. C. Badrul Nizam B. H. Muhammad Abduh Y. Abol Munafi A. B. Iehata S. (2019). Molecular characterization of microbiota associated with sperm of Malaysian mahseer tor tambroides. Evolutionary Bioinf. Online15, 117693431985082. doi: 10.1177/1176934319850821
37
Kousteni V. Kontopoulou M. Megalofonou P. (2010). Sexual maturity and fecundity of scyliorhinus canicula (Linnaeus 1758) in the Aegean Sea. Mar. Biol. Res.6, 390–398. doi: 10.1080/17451000903233771
38
Li T. Long M. Gatesoupe F. J. Zhang Q. Li A. Gong X. (2015). Comparative analysis of the intestinal bacterial communities in different species of carp by pyrosequencing. Microbial Ecol.69, 25–36. doi: 10.1007/s00248-014-0480-8
39
Louca S. Parfrey L. W. Doebeli M. (2016). Decoupling function and taxonomy in the global ocean microbiome. Science353, 1272–1277. doi: 10.1126/science.aaf4507
40
Luna G. M. Quero G. M. Kokou F. Kormas K. (2022). Time to integrate biotechnological approaches into fish gut microbiome research. Curr. Opin. Biotechnol.73, 121–127. doi: 10.1016/j.copbio.2021.07.018
41
Mandal R. K. Jiang T. Al-Rubaye A. A. Rhoads D. D. Wideman R. F. Zhao J. et al . (2016). An investigation into blood microbiota and its potential association with bacterial chondronecrosis with osteomyelitis (BCO) in broilers. Sci. Rep.6, 25882. doi: 10.1038/srep25882
42
Mandal S. Van Treuren W. White R. A. Eggesbø M. Knight R. Peddada S. D. (2015). Analysis of composition of microbiomes: a novel method for studying microbial composition. Microbial Ecol. Health Dis.26, 27663. doi: 10.3402/mehd.v26.27663
43
Metsalu T. Vilo J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res.43, W566–W570. doi: 10.1093/nar/gkv468
44
Mika K. Okamoto A. S. Shubin N. H. Mark Welch D. B. (2021). Bacterial community dynamics during embryonic development of the little skate (Leucoraja erinacea). Anim. Microbiome3, 72. doi: 10.1186/s42523-021-00136-x
45
Minich J. J. Petrus S. Michael J. D. Michael T. P. Knight R. Allen E. E. (2020). Temporal, environmental, and biological drivers of the mucosal microbiome in a wild marine fish, scomber japonicus. mSphere5, e00401-20. doi: 10.1128/mSphere.00401-20
46
Moran M. A. (2015). The global ocean microbiome. Science350, 8455. doi: 10.1126/science.aac8455
47
Muñoz-Baquero M. Marco-Jiménez F. García-Domínguez X. Ros-Santaella J. L. Pintus E. Jiménez-Movilla M. et al . (2021). Comparative study of semen parameters and hormone profile in small-spotted catshark (Scyliorhinus canicula): aquarium-housed vs. wild-captured. Animals11, 2884. doi: 10.3390/ani11102884
48
Mylniczenko N. Clauss T. (2017). “Pharmacology of elasmobranchs: updates and techniques,” in The elasmobranch husbandry manual II: recent advances in the care of sharks, rays and their relatives. Eds. SmithM. D.WarmoltsD.ThoneyD.HueterR.MurrayM.EzcurraJ. (Columbus, OH: Ohio Biological Survey), 289–302.
49
Mylniczenko N. D. Harris B. Wilborn R. E. Young. F. A. (2007). Blood culture results from healthy captive and free-ranging elasmobranchs. J. Aquat. Anim. Health19, 159–167. doi: 10.1577/H06-039.1
50
Patin N. V. Pratte Z. A. Regensburger M. Hall E. Gilde K. Dove A. D. M. et al . (2018). Microbiome dynamics in a Large artificial seawater aquarium. Appl. Environ. Microbiol.84, e00179-18. doi: 10.1128/AEM.00179-18
51
Peng S. Hao W. Li Y. Wang L. Sun T. Zhao J. et al . (2021). Bacterial communities associated with four blooming scyphozoan jellyfish: potential species-specific consequences for marine organisms and humans health. Front. Microbiol.12, 647089. doi: 10.3389/fmicb.2021.647089
52
Perry W. B. Lindsay E. Payne C. J. Brodie C. Kazlauskaite R. (2020). The role of the gut microbiome in sustainable teleost aquaculture. Proc. R. Soc. London Ser. B.287, 20200184. doi: 10.1098/rspb.2020.0184
53
Perry C. T. Pratte Z. A. Clavere-Graciette A. Ritchie K. B. Hueter R. E. Newton A. L. et al . (2021). Elasmobranch microbiomes: emerging patterns and implications for host health and ecology. Anim. Microbiome3, 61. doi: 10.1186/s42523-021-00121-4
54
Pinchaud K. Hafeez Z. Auger S. Chatel J. M. Chadi S. Langella P. et al . (2022). Impact of dietary arachidonic acid on gut microbiota composition and gut-brain axis in Male BALB/C mice. Nutrients14, 5338. doi: 10.3390/nu14245338
55
Pogoreutz C. Gore M. A. Perna G. Millar C. Nestler R. Ormond R. F. et al . (2019). Similar bacterial communities on healthy and injured skin of black tip reef sharks. Anim. Microbiome1, 9. doi: 10.1186/s42523-019-0011-5
56
Quast C. Pruesse E. Gerken J. Schweer T. Yarza P. Peplies J. et al . (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res.41, D590–D596. doi: 10.1093/nar/gks1219
57
Reverter M. Sasal P. Tapissier-Bontemps N. Lecchini D. Suzuki M. (2017). Characterisation of the gill mucosal bacterial communities of four butterflyfish species: a reservoir of bacterial diversity in coral reef ecosystems. FEMS Microbiol. Ecol.93, 1–10. doi: 10.1093/femsec/fix051
58
Rowe M. Veerus L. Trosvik P. Buckling A. Pizzari T. (2020). The reproductive microbiome: an emerging driver of sexual selection, sexual conflict, mating systems, and reproductive isolation. Trends Ecol. Evol.35, 220–234. doi: 10.1016/j.tree.2019.11.004
59
Ruiz-Rodríguez M. Scheifler M. Sanchez-Brosseau S. Magnanou E. West N. Suzuki M. et al . (2020). Host species and body site explain the variation in the microbiota associated to wild sympatric Mediterranean teleost fishes. Microbial Ecol.80, 212–222. doi: 10.1007/s00248-020-01484-y
60
Scarsella E. Sandri M. Monego S. D. Licastro D. Stefanon B. (2020). Blood microbiome: a new marker of gut microbial population in dogs? Vet. Sci.7, 198. doi: 10.3390/vetsci7040198
61
Sehnal L. Brammer-Robbins E. Wormington A. M. Blaha L. Bisesi J. Larkin I. et al . (2021). Microbiome composition and function in aquatic vertebrates: small organisms making big impacts on aquatic animal health. Front. Microbiol.12, 567408. doi: 10.3389/fmicb.2021.567408
62
Serra C. R. Oliva-Teles A. Enes P. Tavares F. (2021). Gut microbiota dynamics in carnivorous European seabass (Dicentrarchus labrax) fed plant-based diets. Sci. Rep.11, 447. doi: 10.1038/s41598-020-80138-y
63
Sullam K. E. Rubin B. E. R. Dalton C. M. Kilham S. S. Flecker A. S. Russell J. A. (2015). Divergence across diet, time and populations rules out parallel evolution in the gut microbiomes of trinidadian guppies. ISME J.9, 1508–1522. doi: 10.1038/ismej.2014.231
64
Sze M. A. Tsuruta M. Yang S. W. Oh Y. Man S. F. Hogg J. C. et al . (2014). Changes in the bacterial microbiota in gut, blood, and lungs following acute LPS instillation into mice lungs. PloS One9, e111228. doi: 10.1371/journal.pone.0111228
65
Tan C. C. Minghao C. Ko K. K. Chen H. Liu J. Loh M. et al . (2023). No evidence for a common blood microbiome based on a population study of 9,770 healthy humans. Nat. Microbiol. 8, 973–985. doi: 10.1038/s41564-023-01350-w
66
Tao Z. Bullard S. A. Arias C. R. (2014). Diversity of bacteria cultured from the blood of lesser electric rays caught in the northern gulf of Mexico. J. Aquat. Anim. Health26, 225–232. doi: 10.1080/08997659.2014.922513
67
Tarnecki A. M. Rhody N. R. Walsh C. J. (2018). Health characteristics and blood bacterial assemblages of healthy captive red drum: implications for aquaculture and fish health management. J. Aquat. Anim. Health30, 339–353. doi: 10.1002/aah.10047
68
Taylor M. J. Bordenstein S. R. Slatko B. (2018). Microbe profile: wolbachia: a sex selector, a viral protector and a target to treat filarial nematodes. Microbiol. (Reading England)164, 1345–1347. doi: 10.1099/mic.0.000724
69
Theodosopoulos A. N. Hund A. K. Taylor S. A. (2019). Parasites and host species barriers in animal hybrid zones. Trends Ecol. Evol.34, 19–30. doi: 10.1016/j.tree.2018.09.011
70
Tvrdá E. Lovíšek D. Gálová E. Schwarzová M. Kováčiková E. Kunová S. et al . (2022). Possible implications of bacteriospermia on the sperm quality, oxidative characteristics, and seminal cytokine network in normozoospermic men. Int. J. Mol. Sci.23, 8678. doi: 10.3390/ijms23158678
71
Uren-Webster T. M. Consuegra S. Hitchings M. Garcia de Leaniz C. (2018). Interpopulation variation in the Atlantic salmon microbiome reflects environmental and genetic diversity. Appl. Environ. Microbiol.18, e00691. doi: 10.1128/AEM.00691-18
72
Uren-Webster T. M. Rodriguez-Barreto D. Consuegra S. Garcia de Leaniz C. (2020). Cortisol-related signatures of stress in the fish microbiome. Front. Microbiol.11, 1621. doi: 10.3389/fmicb.2020.01621
73
Vientós-Plotts A. I. Ericsson A. C. Rindt H. Grobman M. E. Graham A. Bishop K. et al . (2017). Dynamic changes of the respiratory microbiota and its relationship to fecal and blood microbiota in healthy young cats. PloS One12, e0173818. doi: 10.1371/journal.pone.0173818
74
Wan X. Li J. Tian R. McLaughlin R.W. Hao Y. Wu J. Wang Z et al . (2022). The effects of human care on the blowhole and gut microbiotas of two cohabiting dolphin species based on a year-round surveillance. Front. Mar. Sci. 9, 1024117. doi: 10.3389/fmars.2022.1024117
75
Weinberg E. D. (1974). Iron and susceptibility to infectious disease. Science184, 952–956. doi: 10.1126/science.184.4140.952
76
Whittle E. Leonard M. O. Harrison R. Gant T. W. Tonge D. P. (2018). Multi-method characterization of the human circulating microbiome. Front. Microbiol.9, 3266. doi: 10.3389/fmicb.2018.03266
77
Zhou L. Liu L. Chen W. Sun J. Hou S. Kuang T. X. et al . (2020). Stochastic determination of the spatial variation of potentially pathogenic bacteria communities in a large subtropical river. Environ. Pollut.264, 114683. doi: 10.1016/j.envpol.2020.114683
Summary
Keywords
microbial community, sharks, aquaria, aquatic animals, chondrichthyan, 16S rRNA
Citation
Muñoz-Baquero M, Lorenzo-Rebenaque L, García-Vázquez FA, García-Párraga D, Martínez-Priego L, De Marco-Romero G, Galán-Vendrell I, D’Auria G and Marco-Jiménez F (2023) Unveiling microbiome signature in inner body fluids: comparison between wild and aquarium small-spotted catshark (Scyliorhinus canicula). Front. Mar. Sci. 10:1151119. doi: 10.3389/fmars.2023.1151119
Received
25 January 2023
Accepted
13 June 2023
Published
29 June 2023
Volume
10 - 2023
Edited by
Senjie Lin, University of Connecticut, United States
Reviewed by
Santosh Thapa, Baylor College of Medicine, United States; Jinsong Zheng, Chinese Academy of Sciences (CAS), China
Updates
Copyright
© 2023 Muñoz-Baquero, Lorenzo-Rebenaque, García-Vázquez, García-Párraga, Martínez-Priego, De Marco-Romero, Galán-Vendrell, D’Auria and Marco-Jiménez.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francisco Marco-Jiménez, fmarco@dca.upv.es
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.