 Pilar Garcia-Jimenez
Pilar Garcia-Jimenez Rafael R. Robaina
Rafael R. Robaina- Department of Biology, Faculty of Marine Sciences, Institute of Environmental Studies and Natural Resources (IUNAT), University of Las Palmas de Gran Canaria, Las Palmas de Gran Canaria, Spain
Introduction: The discovery of transposable elements (TEs), or transposons, by Barbara McClintock in 1950 revolutionized our understanding of genome dynamics. TEs are recognized for their critical role in genetic variability and evolution. They are categorized into two main classes: class I (retrotransposons), which transpose via an RNA intermediate, and class II, which transpose directly via DNA. TEs significantly influence gene expression through insertions that can disrupt gene function. Consequently, organisms have evolved mechanisms to regulate TE activity, particularly under stress conditions, where TE activation can lead to mutations. In marine macroalgae, TEs are known to shape genome architecture, yet little is known about their dynamics.
Methods: In this study, 17 publicly available but non-annotated algal genomes were analyzed to identify and characterize transposable elements. The Earlgrey pipeline, a powerful tool for TE annotation, was used to quantify their diversity and historical activity. A local script was further employed to investigate the genomic co-localization of TEs with annotated protein-coding sequences.
Results: The analysis revealed significant diversity in TE composition among red, brown, and green algae. Retrotransposons with long terminal repeats (LTRs) were found to be particularly abundant in red algae. Many of these LTRs were located near or within regions encoding proteins, as identified through three protein databases. Notably, these included LTR-specific enzymes such as ribonuclease H, as well as nucleic acid–binding proteins and cation-binding proteins like CCHC-type zinc-finger proteins and haem peroxidase superfamily members, which are involved in stress response pathways.
Discussion: The co-localization of LTRs with stress-responsive protein-coding genes raises intriguing questions about the potential regulatory interplay between TEs and stress adaptation. It remains to be determined whether LTR activity is modulated by the activation of these proteins under stress, or if LTRs have been assimilated into the cellular network to promote protein expression as part of an adaptive response. These findings suggest a promising avenue for exploring the functional integration of TEs into stress resilience mechanisms and highlight their potential role in the evolutionary dynamics of marine algae.
1 Introduction
The first discovery of transposable elements (TEs), also known as selfish genes or transposons, was made by Barbara McClintock in 1950 in the context of research on maize (McClintock, 1950). Since then, our knowledge of their diversity, abundance in genomes, and impact on the evolution and gene expression of organisms has expanded considerably (Bourque et al., 2018).
It is now understood that TEs exhibit a high degree of diversity, with several types and forms. Their classification is complex and dynamic, with the emergence of new forms occurring continuously (Elliott et al., 2021). The classification proposed by Dfam (Storer et al., 2021) distinguishes between class I and class II transposons, class I is distinguished by their requirement for the formation of an intermediate RNA in the transposition process. Several orders of class I elements have been described: long interpersed nuclear elements, short interspersed nuclear elements, Peneleope-like elements, long terminal repeat (LTR) retrotransposons, and Dictyostelium Intermediate Repeat Sequence (DIRS)-like elements (Wicker et al., 2007). Each of these types can carry its own machinery for the transposition mechanism. For example, LTRs carry highly distinctive sequences, including the long tandem repeats at the ends that give them their name. They also carry genes that encode for crucial activities, such as protease, integrase, reverse transcriptase, and ribonuclease H. Each of these genes plays a specific role in the transposition process (Kazazian, 2004; Riehl et al., 2022).
Class II TEs, also termed DNA transposons or “cut-and-paste” transposons are distinguished by their direct transposition. Class II TEs are separated into DDD/DDE-containing DNA TEs, which transpose with the use of an encoded transposase domain and are flanked by terminal inverted repeats (TIRs); miniature inverted-repeat TEs, which are non-autonomous elements flanked by TIRs; rolling circles (also termed Helitrons), which use a “peel-and-paste” mechanism involving an encoded helicase domain and a replication initiator (REP); crypton elements, which transpose using a tyrosine recombinase (YR); and Maverick/Polinton elements, which encode a protein-primed type B DNA polymerase (PolB) among other transposition machinery and are also flanked by TIRs (Wicker et al., 2007).
A substantial corpus of evidence has been amassed concerning their role in genetic variability and evolution in species. The profound alterations that their movement and insertion into disparate regions of the genome can engender are, arguably, self-evident, particularly in light of the considerable TE load observed in genomes and the immediate impact on the expression of a gene or set of genes when a TE is inserted into or in the vicinity of a gene (Akakpo et al., 2020; Domínguez et al., 2020; Kalendar et al., 2021). The examples of transposon modification that have resulted in the creation of the chardonnay grape, the oval tomato, and the grapefruit are illustrative of this phenomenon (Lisch, 2013).
One of the most intriguing phenomena in relation to transposons is the manner in which organisms regulate the dynamics of such a substantial load of elements, as the majority of TEs insertions are expected to be on a spectrum from nearly neutral to highly deleterious, highly detrimental insertions being removed via purifying selection or downregulated to avoid their deleterious effect, whereas beneficial TEs are expected to experience strong positive selection and rapid fixation in populations (LeRouzic et al., 2007). Furthermore, it would be of interest to ascertain whether these mechanisms operate in the stress situations that activate them (Wessler, 1996), as it is assumed that such activation inevitably results in mutations (Lisch, 2013).
TEs have also been identified in marine macroalgae. The considerable advancement in sequencing and bioinformatics techniques has resulted in a notable expansion of knowledge regarding the genomes of macroalgae. This, in turn, has led to the accumulation of an increasing amount of data on their structural components, derived from their study. Consequently, the percentage of TEs varies between the best-characterized species and genomes. For instance, 22% of the Ectocarpus siliculosus genome is composed of TEs, whereas 73% of the Chondrus crispus genome and 48% of the Pyropia yezoensis genome are made up of these elements. A further 35% was identified in Ulva mutabilis (Cock et al., 2010; Collén et al., 2013; DeClerck et al., 2018; Wang et al., 2020).
The dynamics of these processes remain unstudied. It is widely accepted that algae undergo stable phenotypic modifications, based on mutations, under stress. This “phenotypic plasticity” has also been shown to have evolutionary effects on species in general and algae in particular (Fowler-Walker et al., 2006; Pfennig et al., 2010). Our own experience has demonstrated that in vitro culture conditions can disrupt the growth pattern (Robaina et al., 1992) and that deformations can appear in culture tanks (Robledo and García-Reina, 1993). The possibility of a “switch-off” in the nuclear genes of the phycobilisome by the action of transposons in green strains of red algae, such as Kappaphycus alvarezii, has been a topic of interest and has motivated this research.
In this study, we used in silico approaches to examine the predominant gene types in the less-characterized, non-annotated, yet published genomes of red, brown, and green macroalgae, with a particular focus on those of industrial interest and potential utility. Our investigation proceeded with the contiguous sequences of internal transposons and the annotation of affected sequences, to determine which sequences are disrupted or close to the TEs and their gene products in the protein database and to which biological processes they are related. Our goal is to expand new fields of research into the role played by something so prevalent in the genome but so long overlooked in its functional biological relevance.
2 Materials and methods
2.1 Species and genomic data
Genome assembly was taken from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). TE annotation process is computationally and time-intensive and depends heavily on the quality of the genome assembly to be analyzed; in fact, some assemblies could not be analyzed. In the end, 17 non-annotated genomes (six brown, average N50 = 769,437; three green, average N50 = 691,844; and eight red, average N50 = 456,727) could be analyzed, representing species of the different groups with a preference for species of well-known commercial interest, diversity of genome sizes, cultivated vs. collected, etc., whose registration data can be found in Table 1.
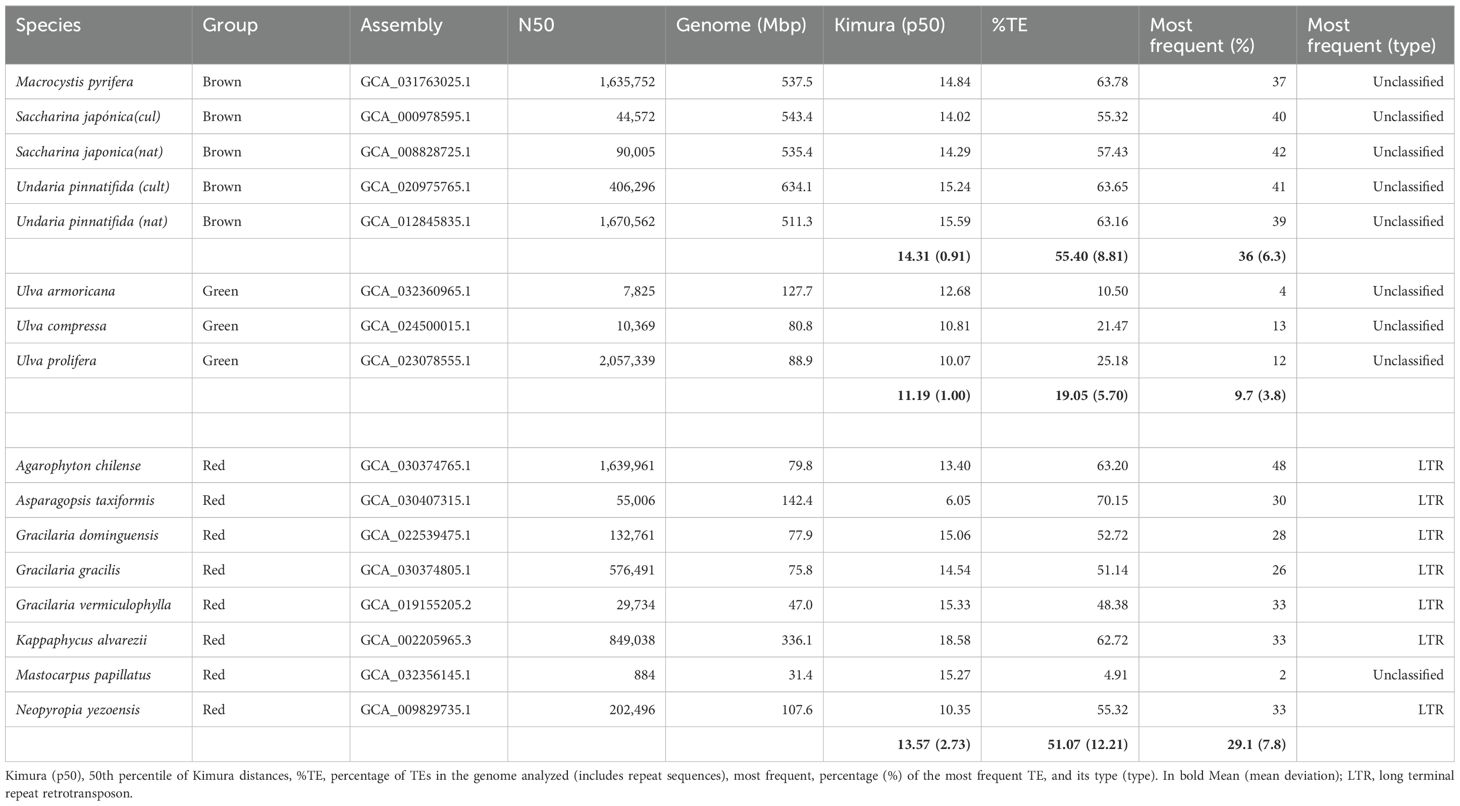
Table 1. Overview of transposon occurrence in different species of brown, green, and red algae constructed from analysis with Eargrey pipeline (Baril et al., 2024), using genome assembly records available in GenBank as a source (https://www.ncbi.nlm.nih.gov/genbank/).
2.2 Key features of TE’s in different brown, green, and red seaweeds
For the annotation of the TEs, the Earlgrey pipeline (default settings) was used to annotate the TEs (Baril et al., 2024). Earlgrey is a multistep pipeline that combines, in turn, different pipelines [i.e., RepeatModeler 2 (Flynn et al., 2020)] and programming languages to obtain a library specific to the target organism, through an automated curation process of the consensus sequences detected in the analyzed genome.
From the results obtained from the Earlgrey analysis for each species, we proceeded to quantify parameters related to the diversity found: the relative activity of the TEs over time or the most abundant TE type for further analysis. Kimura’s distance (Kimura, 1981) is expressed as a percentage that is calculated using the two-parameter Kimura model, which considers both transitions (shifts between purines A↔G or between pyrimidines C↔T) and transversions (shifts between a purine and a pyrimidine, such as A↔C, A↔T, G↔C, and G↔T). A higher Kimura% value indicates greater divergence between sequences, implying that there have been more changes or mutations. Earlgrey returns as a result a distribution of the distances to the consensus sequence for the several types of TEs detected and their clusters. To estimate the characteristics of each distribution obtained, the 50th percentile of each was calculated. Note that the Kimura distance is also an estimate of the activity over time, as it is assumed that larger distances from the consensus sequence represent older TE activity (Baril et al., 2024).
A kernel density estimation (KDE) analysis was performed (Silverman, 1986) to determine the distribution of transposon start positions, following the data in the bed file of the Earlgrey pipeline analysis.
2.3 Analysis of sequences neighboring long terminal repeat retrotransposons in red algae
Because gene annotations were not available for any of the genomes studied, no GFF3 or similar file with their coordinates were available; therefore, we proceed as follows to determine sequences affected by TE: using the data contained in the.bed file derived from the Earlgrey analysis of the red algae that contains the coordinates of each LTR type TE, we were able to extract from the assembly file the sequences neighboring the LTR type retrotransposon, the most common in this group. We built a local script (Supplementary Material S1) in Python (version 3.11.3, Python Software Foundation, https://www.python.org/) that allows us to extract and translate into proteins in the six possible frames the sequences of at least 200 nucleotides, containing between 100 and 200 nucleotides upstream and 100–200 downstream of the sequence where the LTR is inserted.
The sequences obtained were analyzed in the InterProScan database of the Galaxy bioinformatics analysis platform (https://usegalaxy.eu/). InterProScan is a batch tool to query the InterPro database. It provides annotations based on multiple searches of profile and other functional databases; in our case, we used the databases SUPERFAMILY (database for structural and functional annotation for all proteins and genomes), PANTHER (Protein analysis Through Evolutionary Relationships), and Gene3D (structural assignments using the CATH domain structure database), all under default setting.
The generated files were merged and analyzed to obtain the most frequent proteins in the vicinity of the LTRs, according to the InterProScan (IPR) and Gene Ontology (GO) codes generated by InterProScan. Frequency distributions of the more frequent protein (i.e., frequency > 10%) were plotted with the R package ggplot2 (R version 4.4.1, The R Foundation for Statistical Computing).
3 Results
3.1 Key features of transposons in red, green, and brown algae
Figure 1 shows the differences in genome size—the largest in the brown algae, the smallest in green, and a wide range of variation in the red algae species analyzed, whereas the percentage of TEs in the genome is similar between brown and red algae and is lower in green algae.
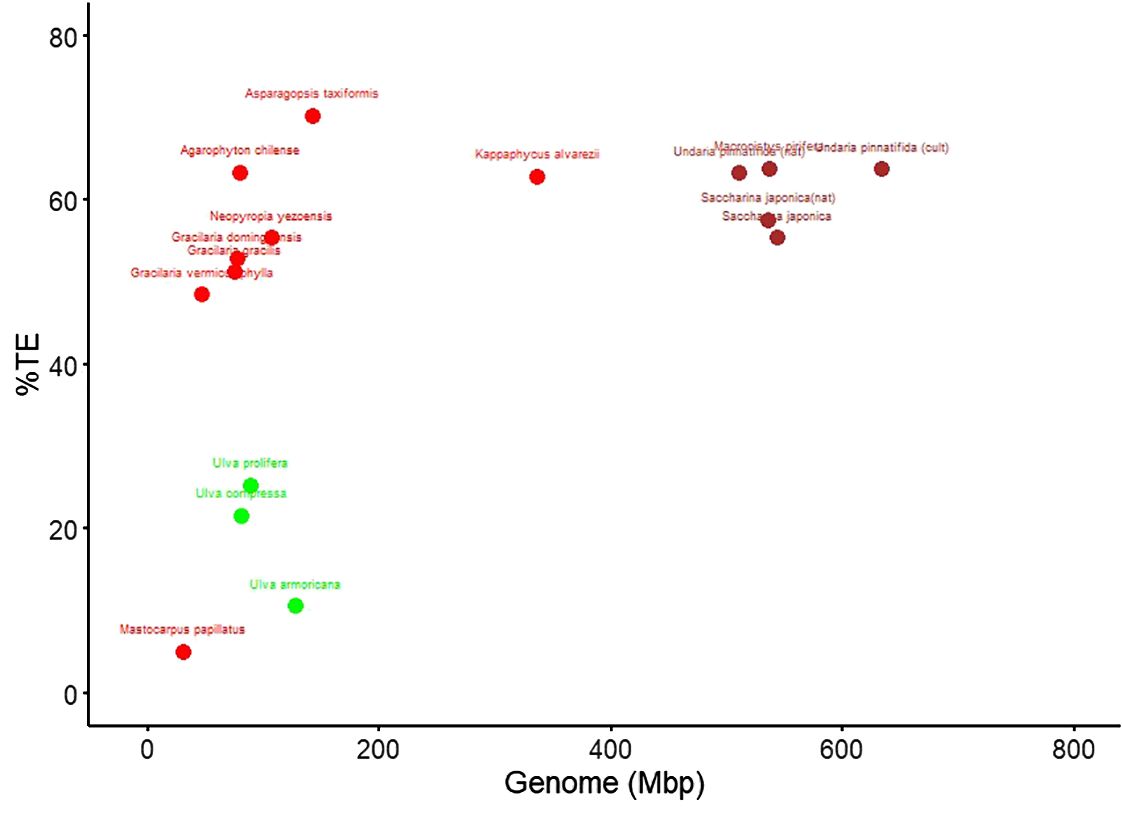
Figure 1. Genome size versus transposable element (TE) load in red, green, and brown algae. Each dot represents one genome; genome size is plotted in megabase pairs (Mbp) and TE content as percentage of the assembly (% TE).
TE diversity seems similar between the groups, although a majority of TE type in brown and green algae is unknown in the databases, whereas the LTR type is dominant in red algae. TE’s activity, measured as relative Kimura distance, is also the lowest in green seaweeds and, therefore, more recent (Table 1). Figure 2 highlights the extreme cases in Table 1; most activity is very recent (i.e., largest peak in both cases is at divergence = 0) but very little retention of aging TE copies in A. taxiformis, whereas K. alvarezii seems to retain degrading elements (which we see by the bars toward the left-hand side of the plot; Figures 2A, B). A. taxiformis presents the highest percentage of TEs (Figures 2C, D).
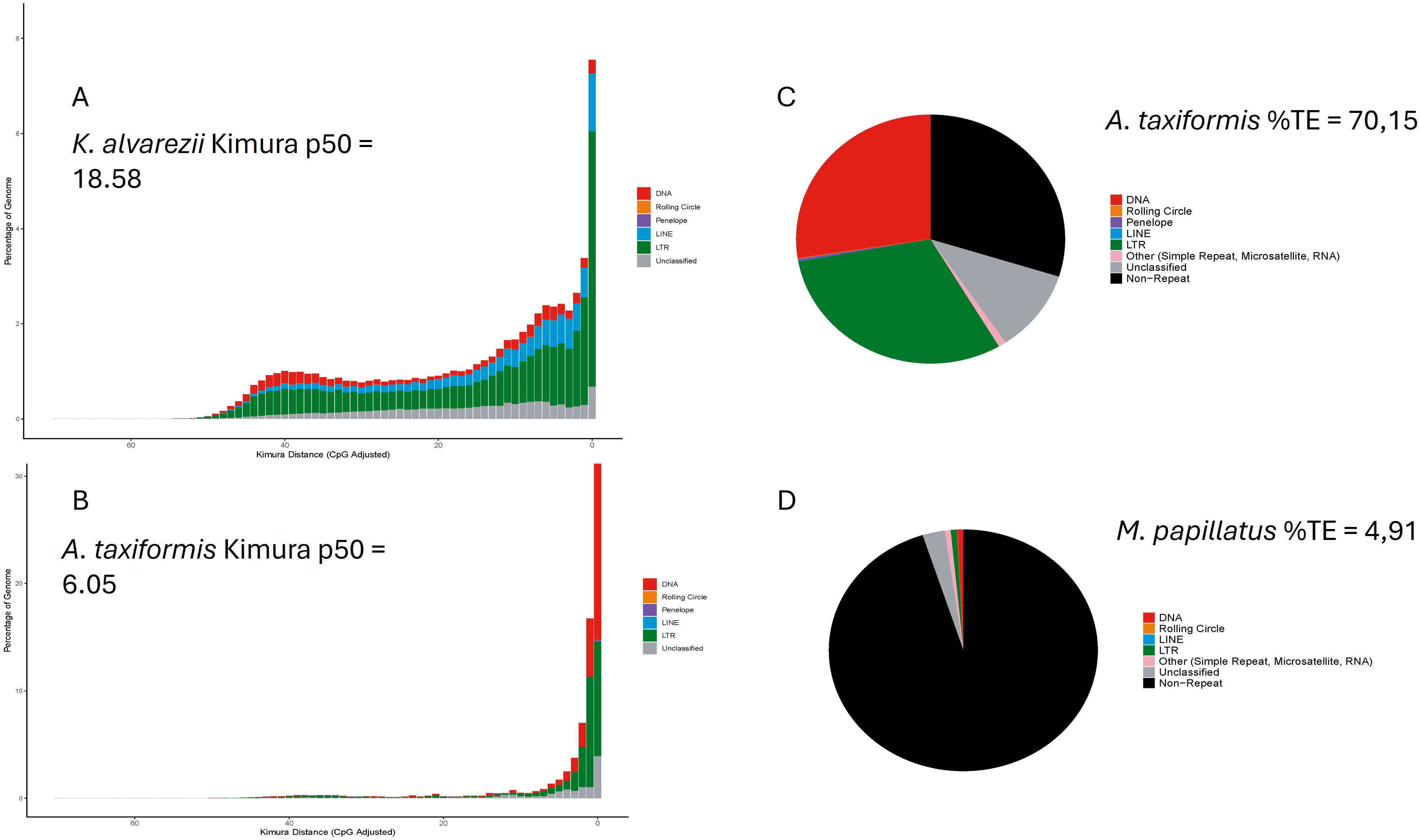
Figure 2. TE dynamics inferred with the Earlgrey pipeline. The largest red-algal genome analyzed, Kappaphycus alvarezii (336.1 Mbp), is dominated by ancient TE activity [p50 = 18.58, (A)], whereas Asparagopsis taxiformis (genome 142.4 Mbp) shows a more recent burst [p50 = 6.05, (B)], having the highest overall TE content (C). Mastocarpus stellatus shows the smallest genome (31.4 Mbp) and TE content (D).
Figure 3 was constructed by KDE analysis of the frequencies of the starting positions of the TEs and shows the accumulation of TEs in the genome and the initial zones of the contigs or scaffolds, depending on the degree of elaboration of the different genomes, with the extreme case of M. stellatus in the insert, where, practically, all the TEs are located at the beginning of the contigs.
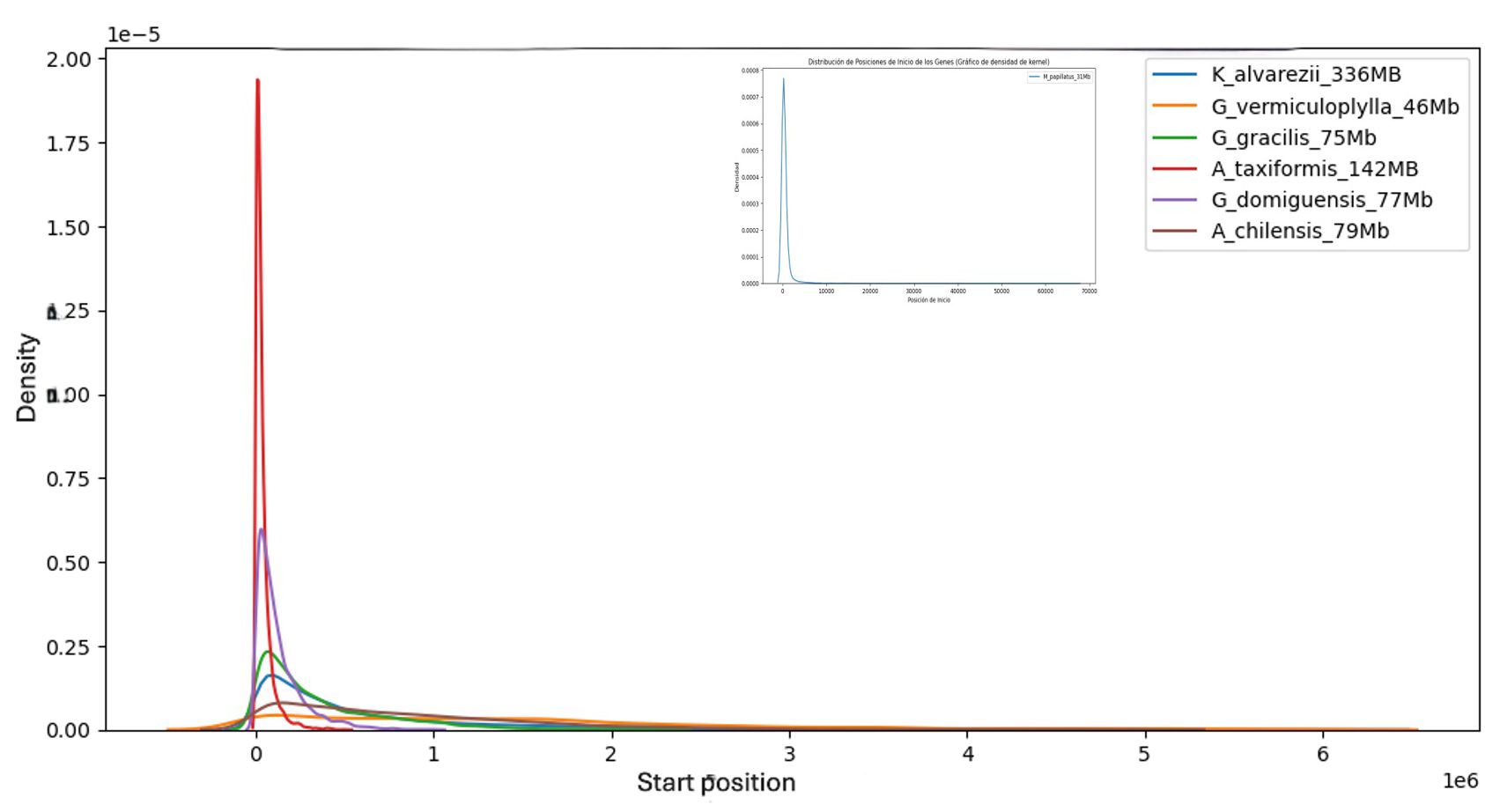
Figure 3. Kernel density estimate of TE start positions along red-algal genome assemblies. Transposable elements cluster toward the extremities of each scaffold or chromosome, a pattern that probably reflects assembly and coverage biases rather than a true positional preference. Mastocarpus is shown in an inset with an expanded x-axis; despite the different scale, it displays the same end-biased distribution.
3.2 Sequences adjacent to LTRs in red algae
Table 2 shows the number of sequences that could be assembled with the nucleotides adjacent to the LTRs, in which K. alvarezii stands out with the highest number and M. stellatus with none, compared to the other species.
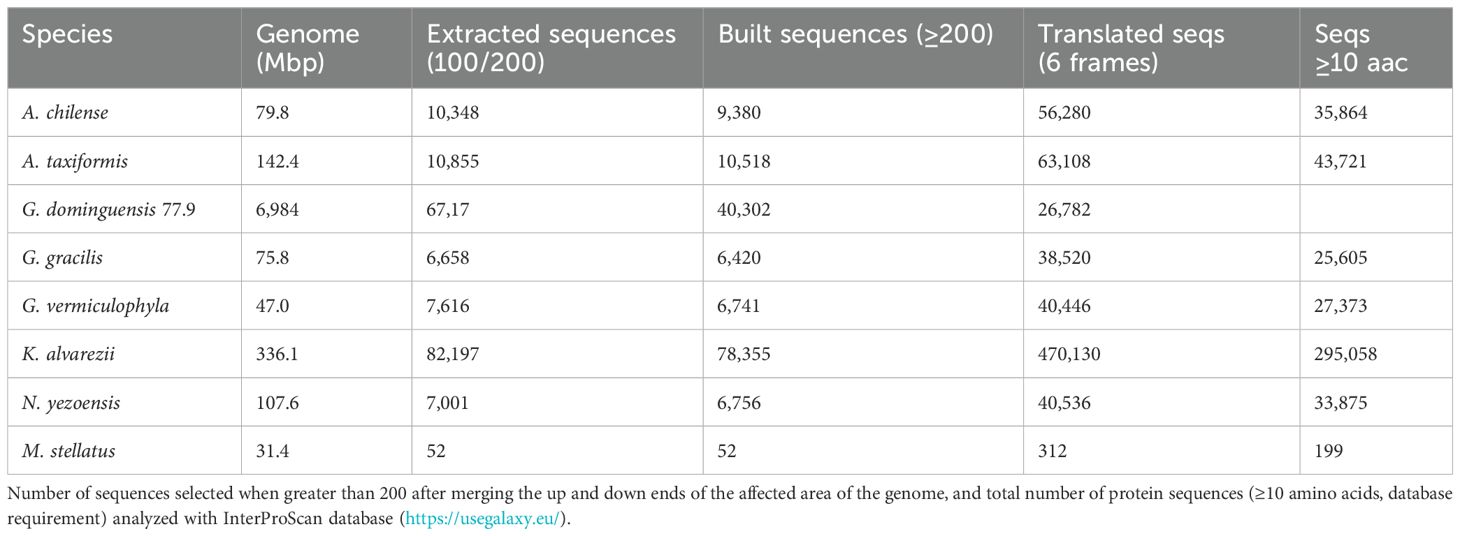
. Table 2 Extracted sequences affected by LTR elements.
Submitting the full gene-product database to InterProScan produced a comprehensive set of annotations (Supplementary Material S1). Applying an E-value cutoff of 1 × 10−5 (1e−5) to whose modal length is 133–amino acid residues (399 bp). Restricting the analysis to annotations found in >10% of these proteins further minimized spurious assignments; the predominant InterPro domains and Gene Ontology terms are summarized in Figures 4A, B, respectively.
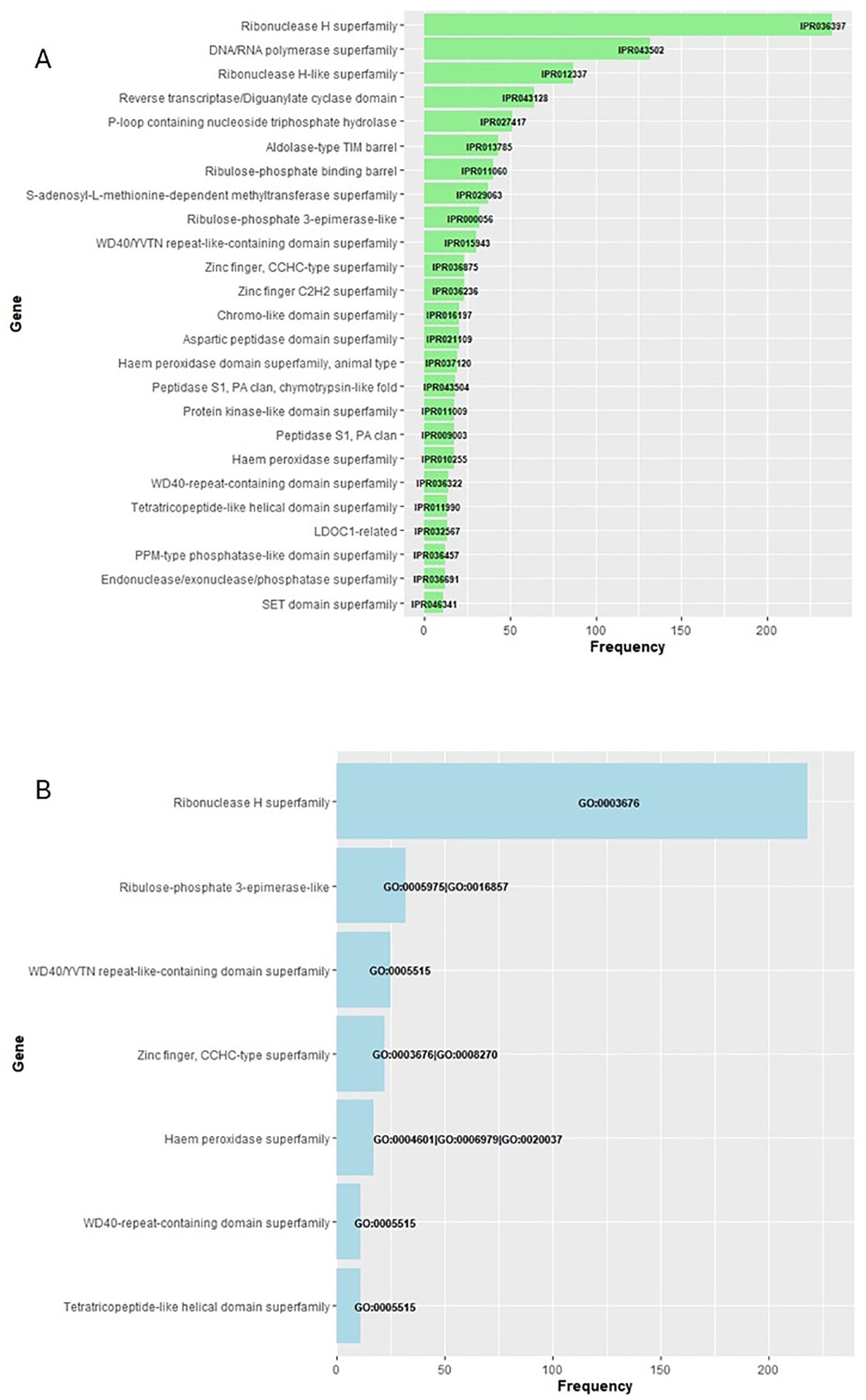
Figure 4. Functional profile of the high-confidence protein set. The full gene-product database was annotated with InterProScan (Supplementary Material S1); hits with E-values < 1 × 10−5 were retained, yielding ~1,900 proteins (modal length = 133 aa, 399 bp). Only annotations present in > 10% of these proteins are shown: (A) InterPro domains and (B) Gene Ontology terms.
4 Discussion
4.1 Genome size and TE content: a variable relationship in our dataset
In general terms, the hypothesis that increased genome size results from the accumulation of repetitive sequences and TEs remains plausible. This is illustrated by the case of Asparagopsis taxiformis, where a relatively recent expansion of TEs (as inferred from Kimura distance) may have contributed to its large genome size, the second largest among the red algae studied, following Kappaphycus alvarezii. The association between genome size and TE content also holds when considering Mastocarpus stellatus, the species with both the smallest genome and the lowest proportion of TEs in our dataset.
However, this pattern does not hold consistently across broader taxonomic comparisons. For example, green algae exhibit relatively low TE content despite their genome sizes, and this discrepancy becomes even more pronounced in brown algae. These species possess genomes that are two to three times larger than those of the green and red algae analyzed yet do not exhibit a proportional increase in TE content. In fact, their TE levels, although high relative to some other organisms, appear to be plateauing. This observation raises the possibility that other factors, such as differences in ploidy, may contribute to genome size variation in these groups.
The number of chromosomes cannot be inferred from the number of contigs or scaffolds, as the latter is typically higher and is also dependent on sequencing methods and devices (Kalendar et al., 2021). Furthermore, references to ploidy in algal genomic analyses render them unsuitable for this purpose (Collén et al., 2013) or are not mentioned at all (Wang et al., 2020). However, there are cytogenetic studies that reported an haploid chromosome numbers of up to 30–32 in brown algae (Laminariales, Dyctiotales, and Fucales) as opposed to 8–10 in Ulva compressa (as Enteromorpha compressa (Godward, 1959; Naylor, 1958; Roberts, 1962), which certainly points to the existence of ploidy in brown algae to explain the discrepancy between %TEs and genome size.
Finally, from the perspective of TEs, cultured samples studied such as S. japonica or U. pinnatifida exhibit comparable key features to those observed in wild samples, in spite of the genetic diversity that is typically higher in the latter, as previously described by (Ye et al., 2015). Given that some TEs are of viral origin, it is plausible to hypothesize that intensive farming practices and the associated spread of pests could facilitate TE transmission and proliferation. However, our observations do not support a clear link between these factors and increased TE content.
4.2 Sequences neighboring LTR transposons
Most transposon sequences identified in the analyzed red algal species are clustered and occupy starting positions typically located at the beginning of the generated sequences. The literature demonstrates that TEs are frequently clustered in regions of heterochromatin near telomeres and centromeres, thereby contributing to the maintenance of these potentially dangerous components in a silenced state (Guo et al., 2021; Lisch, 2009). Furthermore, sequencing techniques can introduce artefacts when locating highly repetitive areas where TEs are found, they may arrange these sequences at initial contigs/scaffolds without necessarily reflecting the structural quality of the genome. It is possible that one or the other, or both, may have been the cause of the observed results (Figure 3).
Notwithstanding their prevalence in these initial positions, LTR-like TEs were also identified throughout the genome, disrupting sequences. This was observed in 1,580 Panther, Gene3D, or Superfamily database hit instances (score 10−5 to 10−54), actually close to 500 (i.e., 0.4% of the built sequences; Table 2), as the three protein databases may contribute with a different annotation but still representing a diverse range of functional categories, including metabolism, nucleic acids, and structural components (Supplementary Material S1).
If we consider all these sequences to be genuinely disrupted, as we will discuss below, the mutation rate may be consistent with the proportion of TEs observed in genomes; however, this seems still very high given the mostly deleterious effects that TEs will have in genomes and considerably higher than that of point mutations, which typically range from 10−8 to 10−9 mutations per site per generations (Lynch et al., 2016).
Published reference genomes for algae have characterized the proportion of TEs (Cock et al., 2010; Collén et al., 2013; DeClerck et al., 2018; Wang et al., 2020) but have not delved into their location or the extent of their relationship with other genes, as we have attempted to do in this work with unannotated genomes. The expectation that the above is the picture would justify studying this phenomenon in depth, using the precise location of annotated genes and their comparison with TE coordinates, to continue with the mechanisms of regulation of their activity.
4.3 Potential regulatory functions of proteins associated with LTR-proximal sequences in TE activity
Close examination of the sequences reveals that the positive hits in protein-coding sequences may came from the upstream or downstream nucleotides (we have not filtered out sequence ambiguities at the extremes) rather than from the constructed sequence involving both ends. Consequently, we must consider that some genes actually are disrupted, whereas, in other cases, the effect may be limited to adjacent genes (i.e., the TE located in the proximity).
In our analysis, we filtered out the most frequently affected proteins, defined as those detected with a frequency greater than 10% of those assigned IPR (1,580, Figure 2A) and GO (939, Figure 2B). Among them, two stand out as particularly significant: firstly, those enzymes that are part of the LTR structure, such as reverse transcriptase or ribonuclease H (Kazazian, 2004; Riehl et al., 2022) and a multitude of disparate proteins whose functions, contingent on their IPR or GO code, encompass cation binding, as exemplified by the zinc-finger proteins CCHC-type, haem peroxidase superfamily, and even the metabolic enzyme ribulose-phosphate-3-epimerase, which alternates this action with that of cation binding. In addition, the WD40 repeat-containing superfamily, WD40/YVTN repeat-like, and tetratricopeptide-like helical domain proteins have been observed to bind to nucleic acids.
Regarding the TE components occurrence, it is noted that TEs often insert into regions where other TEs or incomplete TE insertions are present (Bourque et al., 2018), and this would explain the occurrence of these LTR components. Furthermore, the prevalence of the TE components lends robustness to the analysis performed in this study. As an example, the analysis of the size distribution of the LTRs found in the species that accumulates the most, A. taxiformis, reveals a range between 33 and 33,000 bp, so not all are complete, assuming LTR-TE minimum sequence size of 1,500 bp (Baucom et al., 2009), and more than half (10,021 sequences, 58%) of the LTR-TEs found in A. taxiformis would be LTR-TE incomplete sequences.
One of the most intriguing aspects of the enormous abundance of TEs in eukaryotic genomes is the regulation of their activity. This is because the mobilisation of TEs according to their presence would generate very unstable genotypes with high mutation rates. It is evident that organisms employ a range of mechanisms to regulate the activity of TEs. These include DNA modification, such as methylation, and post-transcriptional action through small interfering RNAs and the degradation of double-stranded RNAs (Guo et al., 2021; Lisch, 2009; Slotkin and Martienssen, 2007).
Among the most frequent proteins encountered in our analysis, WD proteins lack methylase activity in themselves, but they do participate in the assembly of complexes that do possess this activity (Trievel and Shilatifard, 2009). Consequently, their function may be linked to the prevention of LTR expression through the methylation of the region in which they are located. The proximity of these elements to the insertion site may have been a beneficial selective factor within the species insertion polymorphism (Akakpo et al., 2020; Domínguez et al., 2020).
With regard to cation-binding proteins, it seems plausible to suggest that they may also play a role in the regulation of LTR expression. The daily fluctuations in abiotic parameters, including salinity, that seaweeds in coastal intertidal zones experience trigger a cascade of ion exchange events. This is followed by the regulation of metabolism and carbon pools, which allows the accumulation of osmotically active substances, such as (iso)floridoside in red algae (Dickson and Kirst, 1987). It is reasonable to assume that proteins capable of buffering sudden increases in ions would be favored in proximity to each other to prevent changes in DNA and LTR activity. This suggests that LTR activation during stress situations is controlled by surrounding genes, thus preventing its deleterious effects.
In contrast, according to Grandbastien (2015), in organisms exposed to constant stress, TEs may have been integrated in such a way that the more stress-sensitive LTRs trigger the expression of specific proteins, thereby contributing to the plasticity of these organisms in frequently stressful conditions. The key question, therefore, is whether TE expression is being suppressed or whether the TE has been co-opted into the cellular gene regulatory machinery, with its early activation serving as a mechanism for the rapid induction of genes requiring immediate response. This perspective unveils a promising avenue for investigating the functional integration of TEs in cellular resilience, with potential implications for understanding the evolutionary dynamics of stress adaptation.
Regarding the suitability of InterProScan for annotations, it is necessary to clarify that InterProScan provides reliable annotations when a domain is detected, and its effectiveness is limited for underrepresented taxa such as multicellular marine algae (seaweeds). The tool does not typically produce incorrect annotations; rather, its main limitation is that many proteins remain unannotated or are assigned only generic functions. As a result, while the annotations that it provides are trustworthy, they tend to underestimate the functional diversity present in seaweed proteomes (Blum et al., 2020).
In conclusion, our results reveal that seaweed genomes contain substantial amounts of TEs, in proportions comparable to those observed in other eukaryotes. Whereas many TEs in green and brown algae remain uncharacterized, red algae display a predominance of LTR-type elements. Beyond their abundance, the presence of nucleic acid– and cation-binding proteins encoded near LTRs suggests a possible role in modulating TE activity, either through direct regulation or co-option into stress-responsive pathways. These findings point to an underexplored layer of genome regulation in algae and open promising avenues for future research into TE functionality, stress adaptation, and the evolutionary expansion of seaweed genomic complexity.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
PG-J: Investigation, Methodology, Writing – review & editing. RR: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by the University of Las Palmas Gran Canaria and partially by the EU Project I3-4-Seaweeds (I3-2023-101161142).
Acknowledgments
We would like to express our gratitude to Dr. Tobias Baril for his invaluable assistance in using the Earlgrey pipeline and for his insightful recommendations following the review of the manuscript. Furthermore, we would like to express our gratitude to all colleagues who have made their genomic results publicly available, as this has enabled our group to undertake this work. We would like to express our gratitude to the National Center for Biotechnology Information (NCBI) for making their database accessible to the scientific community.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1592442/full#supplementary-material
References
Akakpo R., Carpentier M. C., Hsing Y. I., and Panaud O. (2020). The impact of transposable elements on the structure, evolution and function of the rice genome. New Phytol 226, 44–495. doi: 10.1111/nph.16356
Baril T., Galbraith J., and Hayward A. (2024). Earl grey: A fully automated user-friendly transposable element annotation and analysis pipeline. Mol. Biol. Evol. 41, msae068. doi: 10.1093/molbev/msae068
Baucom R. S., Estill J. C., Chaparro C., Upshaw N., Jogi A., Deragon J. M., et al. (2009). Exceptional diversity, non-random distribution, and rapid evolution of retroelements in the B73 maize genome. PloS Genet. 5, e10007325. doi: 10.1371/journal.pgen.1000732
Blum M., Chang H., Chuguransky S., Grego T., Kandasamy S., Mitchell A., et al. (2020). The InterPro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354. doi: 10.1093/nar/gkaa977
Bourque G., Burns K. H., Gehring M., Gorbunova V., Seluanov A., Hammell M., et al. (2018). Ten things you should know about transposable elements. Genome Biol. 19, 9451–9457. doi: 10.1186/s13059-018-1577-z
Cock J.M., Sterck L., Rouzé P., Scornet D., Allen A. E., Amoutzias G., et al. (2010). The ectocarpus genome and the independent evolution of multicellularity in brown algae. Nature 465, 617–621. doi: 10.1038/nature09016
Collén J., Porcel B., Carré W., Ball S. G., Chaparro C., Tonon T., et al. (2013). Genome structure and metabolic features in the red seaweed chondrus crispus shed light on evolution of the archaeplastida. Proc. Natl. Acad. Sci. United States America 110, 5247–5252. doi: 10.1073/pnas.1221259110
DeClerck O., Kao S. M., Bogaert K. A., Blomme J., Foflonker F., Kwantes M., et al. (2018). Insights into the evolution of multicellularity from the sea lettuce genome. Curr. Biol. 28, 2921–2933. doi: 10.1016/j.cub.2018.08.015
Dickson D. M. J. and Kirst G. O. (1987). Osmotic adjustment in marine eukaryotic algae: the role of inorganic ions, quaternary ammonium, tertiary sulphonium and carbohydrate solutes: I. Diatoms and a rhodophyte. New Phytol. 106, 645–655. doi: 10.1111/j.1469-8137.1987.tb00165.x
Domínguez M., Dugas E., Benchouaia M., Leduque B., Jiménez-Gómez J. M., Colot V., et al. (2020). The impact of transposable elements on tomato diversity. Nat. Commun. 11, 40585. doi: 10.1038/s41467-020-17874-2
Elliott T. A., Heitkam T., Hubley R., Quesneville H., Suh A., Wheeler T. J., et al. (2021). TE hub: A community-oriented space for sharing and connecting tools, data, resources, and methods for transposable element annotation. Mob DNA 12, 16. doi: 10.1186/s13100-021-00244-0
Flynn J. M., Hubley R., Goubert C., Rosen J., Clark A., Feschotte C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. PNAS 117, 9451–9457. doi: 10.1186/s13059-018-1577-z
Fowler-Walker M. J., Wernberg T., and Connell S. D. (2006). Differences in kelp morphology between wave sheltered and exposed localities: morphologically plastic or fixed traits? Mar. Biol. 148, 755–675. doi: 10.1007/s00227-005-0125-z
Godward M. B. E. (1959). Chromosome numbers in the algae II chlorophyta (1). Br. Phycol Bull. 1, 43–46. doi: 10.1080/00071615900650081
Grandbastien M. (2015). LTR retrotransposons, handy hitchhikers of plant regulation and stress response. Biochimica et biophysica acta. 4, 403–416. doi: 10.1016/j.bbagrm.2014.07.017
Guo W., Wang D., and Lisch D. (2021). RNA-Directed DNA Methylation Prevents Rapid and Heritable Reversal of Transposon Silencing under Heat Stress in Zea mays. PloS Genet. 17, e1009326. doi: 10.1371/journal.pgen.1009326
Kalendar R., Sabot F., Rodriguez F., Karlov G., Natali L., and Alix K. (2021). Editorial: mobile elements and plant genome evolution, comparative analyzes and computational tools. Front. Plant Sci. 12, e735134. doi: 10.3389/fpls.2021.735134
Kazazian H. (2004). Mobile elements: drivers of genome evolution. Science 303, 1626–1632. doi: 10.1126/SCIENCE.1089670
Kimura M. (1981). Estimation of evolutionary distances between homologous nucleotide sequences. PNAS. 78 (1), 454–458. doi: 10.1073/PNAS.78.1.454
LeRouzic A., Boutin T. S., and Capy P. (2007). Long-term evolution of transposable elements. PNAS December 4, 19357–19805. doi: 10.1073/pnas.0705238104
Lisch D. (2009). Epigenetic regulation of transposable elements in plants. Annu. Rev. Plant Biol. 60, 43–66. doi: 10.1146/annurev.arplant.59.032607.092744
Lisch D. (2013). How important are transposons for plant evolution? Nat. Rev. Genet. 14, 49–61. doi: 10.1038/nrg3374
Lynch M., Ackerman M. S., Gout J. F., Long H., Sung W., Thomas W. K., et al. (2016). Genetic drift, selection and the evolution of the mutation rate. Nat. Rev. Genet. 17, 704–714. doi: 10.1038/nrg.2016.104
McClintock B. (1950). The origin and behavior of mutable loci in maize. PNAS. 36 (6), 344–355. doi: 10.1073/pnas.36.6.344
Naylor M. (1958). Chromosome numbers in the algae: I. Phaeophtya. Br. Phycol Bull. 1, 34–40. doi: 10.1080/00071615800650051
Pfennig D. W., Wund M. A., Snell-Rood E. C., Cruickshank T., Schlichting C. D., and Moczek A. P. (2010). Phenotypic plasticity’s impacts on diversification and speciation. Trends Ecol. Evol. 25, 459–675. doi: 10.1016/j.tree.2010.05.006
Riehl K., Riccio C., Miska E. A., and Hemberg M. (2022). Transposon ultimate: software for transposon classification, annotation and detection. Nucleic Acids Res. 50, e645. doi: 10.1093/nar/gkac136
Robaina R. R., Garcia-Jimenez P., and Luque A. (1992). The growth pattern and structure of callus from the red alga laurencia sp. (Rhodophyta, ceramiales) compared to shoot regeneration. Botanica Mar 35, 267–725. doi: 10.1515/botm.1992.35.4.267
Roberts M. (1962). Chromosome numbers in the algae: phaeophyta II. Br. Phycol Bull. 2, 165–166. doi: 10.1080/00071616200650111
Robledo D. R. and García-Reina G. (1993). Apical callus formation in solieria filiformis (Gigartinales, rhodophyta) cultured in tanks. Hydrobiologia 260–261, 401–406. doi: 10.1007/BF00049048
Silverman B. W. (1986). Density estimation for statistics and data analysis (London: Chapman & Hall).
Slotkin R.K. and Martienssen R. (2007). Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 8, 272–855. doi: 10.1038/nrg2072
Storer J., Hubley R., Rosen J., Wheeler T. J., and Smit A. F. (2021). The dfam community resource of transposable element families, sequence models, and genome annotations. Mob DNA 12, 25. doi: 10.1186/s13100-020-00230-y
Trievel R. C. and Shilatifard A. (2009). Nature_WDR_inmetilationcomplex_2009. Nat. Struct. Mol. Biol. 16, 678–680. doi: 10.1038/nsmb0709-678
Wang D., Yu X., Xu K., Bi G., Cao M., Zelzion E., et al. (2020). Pyropia yezoensis genome reveals diverse mechanisms of carbon acquisition in the intertidal environment. Nat. Commun. 11, 4028. doi: 10.1038/s41467-020-17689-1
Wessler S. R. (1996). Plant retrotransposons: turned on by stress. Curr. Biol. 6, 959–961. doi: 10.1016/S0960-9822(02)00638-3
Wicker T., Sabot F., Hua-Van A., Bennetzen J. L., Capy P., Chalhoub B., et al. (2007). A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982. doi: 10.1038/nrg2165
Keywords: transposons, seaweed, LTR type, gene products, evolutionary dynamics, stress response
Citation: Garcia-Jimenez P and Robaina RR (2025) Exploring transposons in macroalgae: LTR elements and neighboring genes in red seaweeds. Front. Mar. Sci. 12:1592442. doi: 10.3389/fmars.2025.1592442
Received: 12 March 2025; Accepted: 28 May 2025;
Published: 20 June 2025.
Edited by:
Wenlei Wang, Jimei University, ChinaReviewed by:
Jinlin Liu, Tongji University, ChinaXiaodong Li, Chinese Academy of Sciences (CAS), China
Copyright © 2025 Garcia-Jimenez and Robaina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael R. Robaina, cmFmYWVsLnJvYmFpbmFAdWxwZ2MuZXM=