 Júnia Schultz1*†
Júnia Schultz1*† Sharifah Altalhi1,2†Froukje M. van der Zwan3Nico Augustin4,5
Sharifah Altalhi1,2†Froukje M. van der Zwan3Nico Augustin4,5 Alexandre Soares Rosado1*
Alexandre Soares Rosado1*- 1Biological and Environmental Sciences and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
- 2Biotechnology Department, Taif University, Taif, Saudi Arabia
- 3Physical Science and Engineering Division, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
- 4RD4, MuHS-V, GEOMAR Helmholtz Centre for Ocean Research Kiel, Kiel, Germany
- 5OceanQuest, King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia
Introduction: Hydrothermal vents are among Earth’s most extreme ecosystems, characterized by high temperatures, elevated metal concentrations, and steep chemical gradients that sustain specialized microbial life. Although bacterial and archaeal communities in these environments have been extensively studied, fungal diversity remains poorly understood. The recently discovered Hatiba Mons hydrothermal vent field in the Red Sea Rift provides a unique setting to investigate fungal communities in a hypersaline, metal-rich environment.
Methods: We analyzed fungal diversity in crusts, sediments, and microbial mats collected from five active vent sites at Hatiba Mons. A total of 38 subsamples were obtained using a remotely operated vehicle (ROV) during the KRSE Aegaeo RV cruise in May 2022. DNA was extracted, and the fungal ITS rRNA gene region was sequenced on an Illumina MiSeq platform. Sequence processing and taxonomic assignment were performed with QIIME2 and the UNITE database, while downstream statistical analyses were conducted in R with phyloseq.
Results: Fungal community composition varied significantly across sample types, as shown by Principal Coordinates Analysis (PCoA) and confirmed by PERMANOVA. Ascomycota, Basidiomycota, and Chytridiomycota dominated the assemblages. Functional predictions using FUNGuild revealed diverse ecological roles, including saprotrophic, symbiotic, and pathogenic lifestyles.
Discussion: This study provides the first characterization of fungal communities in the Hatiba Mons hydrothermal system. The distinct taxonomic and functional profiles observed suggest that fungi contribute to biogeochemical cycling and ecosystem dynamics in extreme marine habitats. These findings expand current knowledge of fungal ecology in hydrothermal vents and underscore the importance of including fungi in future deep-sea microbiological research.
Introduction
Hydrothermal vents are extreme environments characterized by high temperatures, elevated concentrations of heavy metals, and steep physicochemical gradients that support diverse and unique microbial communities (Dick, 2019). While bacterial and archaeal diversity in these environments has been extensively explored, fungal diversity remains largely understudied (Salcedo et al., 2023). This knowledge gap is particularly evident in deep-sea hydrothermal vents, where fungi could play significant roles in nutrient cycling, organic matter degradation, metal chelation, and interactions with other organisms (Richards et al., 2012; Burgaud et al., 2010).
The recently discovered Hatiba Mons hydrothermal vent field, located in the Red Sea Rift, offers a novel setting to investigate fungal communities in one of the most chemically distinct marine ecosystems. The Red Sea Rift is a geologically active region formed by the divergence of the Arabian and African plates, leading to the development of hydrothermal vent systems along its mid-ocean ridge axis (Augustin et al., 2021). Unlike other mid-ocean ridge hydrothermal systems, Red Sea vents are characterized by hypersalinity (~40‰ vs. 35‰ global average) (Pearse and Gunter, 1957) and warm bottom water temperatures (21.7°C vs. 2–4°C) Yao and Hoteit, 2018; Berumen et al., 2019) which results from the partial enclosure of the basin, as well as the vent fluids present high metal concentrations, particularly iron and manganese (van der Zwan et al., 2023). These atypical conditions create environmental filters that may select for highly specialized, extremotolerant fungal taxa.
Recent studies have suggested that fungi (mainly representatives of Ascomycota, Basidiomycota and Chytridiomycota) are the most abundant groups observed in deep-sea hydrothermal vents (Velez et al., 2022; Le Calvez et al., 2009), where they exhibit diverse metabolic capabilities, including adaptations to high temperatures and metal-rich environments (Vaksmaa et al., 2023; Shourie and Vijayalakshmi, 2022). In hydrothermal environments, such fungi may contribute to key ecosystem processes including the transformation of hydrothermal precipitates and organic carbon, and facilitation of biogeochemical cycling in synergy with prokaryotic communities. Nevertheless, data on fungal ecology in vent systems remain scarce, especially in deep and underexplored regions like the Red Sea Rift.
The Hatiba Mons vents, a newly described low-temperature (~50°C) hydrothermal system (Augustin et al., 2024; van der Zwan et al., 2023), represent a promising site to expand our understanding of fungal community structure, adaptation, and ecological functions in extreme marine environments. In this study, we present the first high-resolution survey of fungal diversity associated with sediments, crusts, and microbial mats from five active sites within Hatiba Mons. Through a culture-independent metabarcoding approach, we aim to characterize fungal community composition, assess habitat-driven patterns, and explore potential functional guilds that may illuminate the ecological significance and biotechnological potential of deep-sea fungi in vent ecosystems.
Materials and methods
Study site and sample collection
Samples were collected at the active hydrothermal vents located on the Hatiba Mons volcano, in the Red Sea Rift. The active Hatiba hydrothermal fields are situated at ≈1,000 m water depth and are characterized by widespread diffuse venting of low-temperature fluids (up to 40°C). Forty-five individual sites are mainly built of iron-oxyhydroxide mounds and chimneys, surrounded by warm Red Sea bottom water with a temperature of 21.7°C and high salinity (41 psu), flourishing microbial mats, and the absence of vent-specific macrofauna. The Hatiba vent fields are considered the largest active low-T vent field area observed so far (van der Zwan et al., 2023).
A total of 38 subsamples from crusts, sediments, and microbial mats from five hydrothermal sites within Hatiba Mons (Figure 1) were collected aboard the KRSE Aegaeo RV cruise in May 2022 (Table 1). Detailed information of the sampling sites in the Hatiba Mons vent fields is available in van der Zwan et al. (2023). At the Field 8 hydrothermal vent site, three hydrothermal crust samples (KRSE4-2 ROV HYD) were collected using a remotely operated vehicle (ROV). Each crust was subsampled into three non-homogenized parts, resulting in nine subsamples. Push cores (4 cm in diameter and 60 cm in length) were collected from the Kabir Field. Each core was divided into two depth intervals for subsampling: a microbial mat layer (0–5 cm, flocculant to loosely consolidated) and an underlying precipitate layer (5–10 cm, more consolidated). Each interval was subsampled into three non-homogenized parts, resulting in six subsamples per core and 18 across the three cores. A hydrothermal chimney crust sample (KRSE4-4 ROV HYD X) was collected and subsampled into three parts. A microbial mat sample (KRSE4-6 ROV NET) from the Farwah Safaa Ridges was retrieved using an ROV sampler and divided into three non-homogenized zones; replicate aliquots from these zones yielded eight subsamples in total.
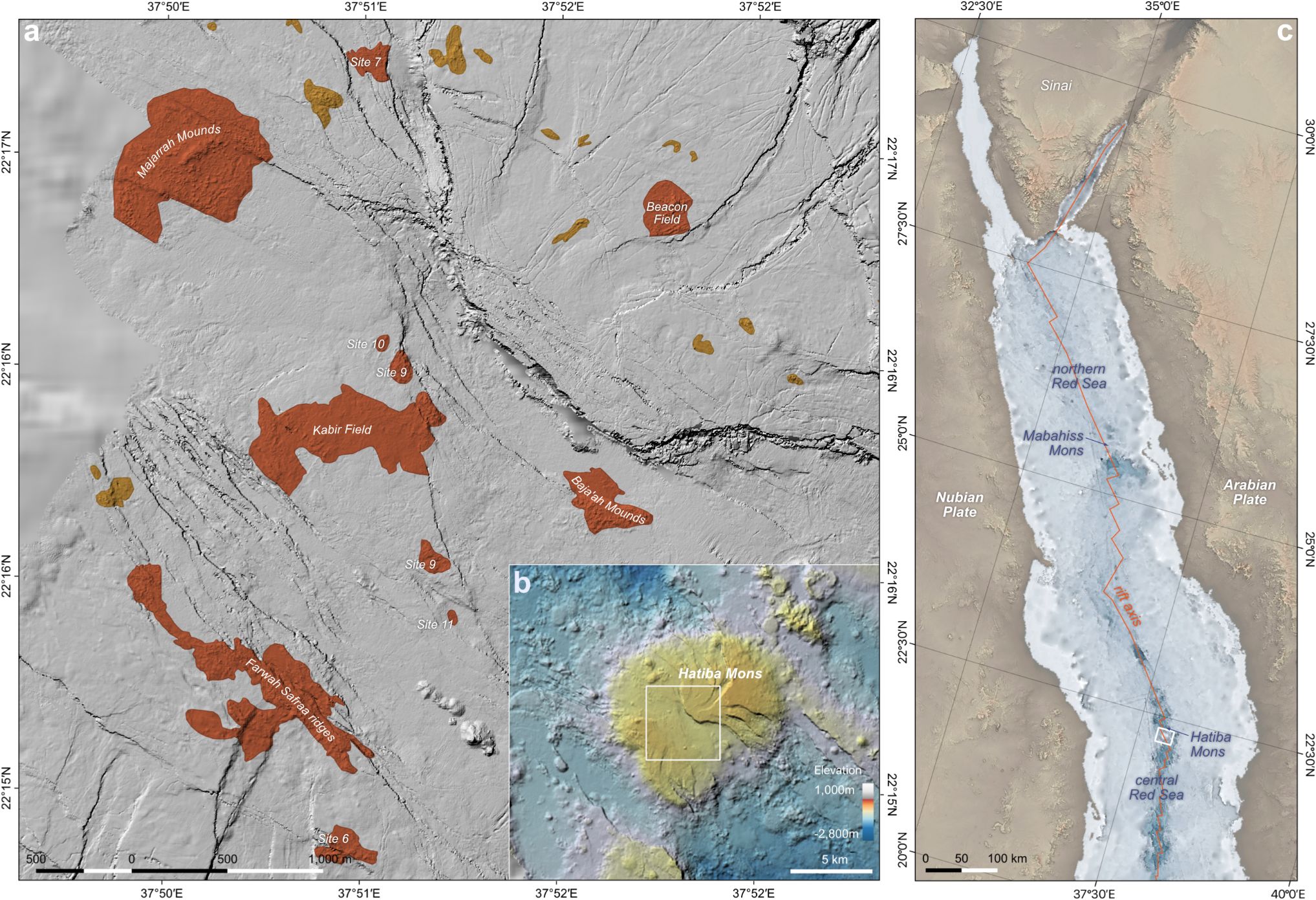
Figure 1. Overview of the hydrothermal vent fields on the summit of Hatiba Mons volcano. (a) AUV map showing confirmed (orange) and inferred (yellow) vent sites of the central summit area. (b) Ship bathymetry of the Hatiba Mons volcano and the close vicinity. (c) The location of Hatiba Mons in the tectonic framework of the northern and central Red Sea.
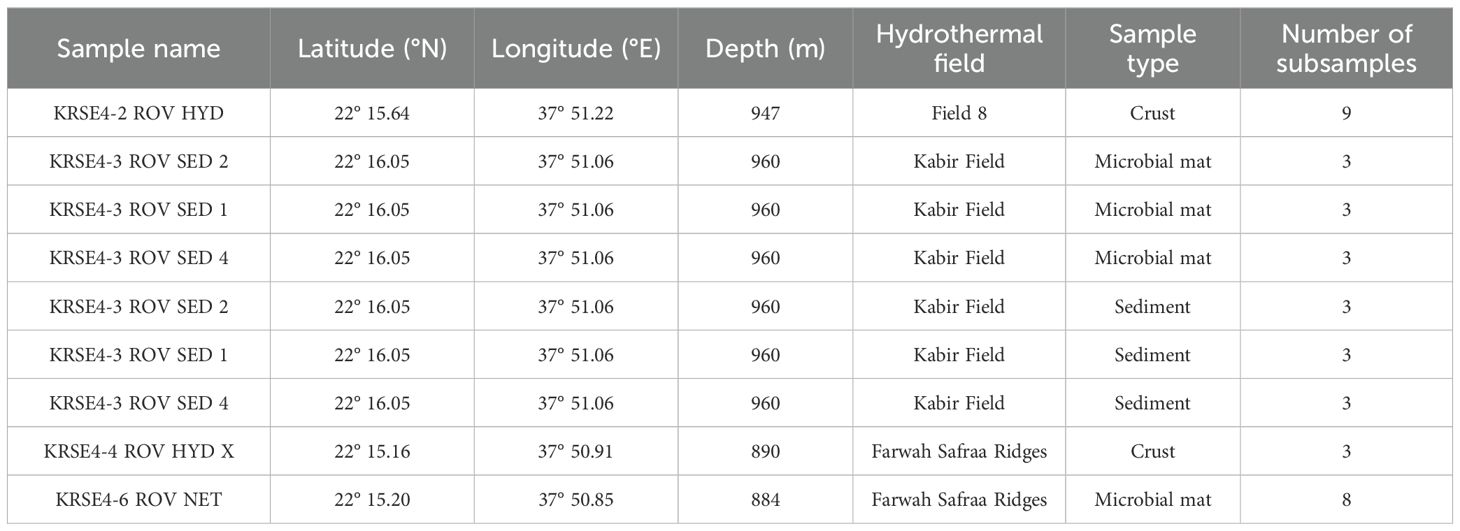
Table 1. Overview of samples collected from Hatiba Mons hydrothermal fields (KRSE Aegaeo RV Cruise, May 2022).
Subsampling and transfers were conducted on board the research vessel as rapidly as possible in a designated clean workspace, adhering to aseptic technique throughout the process. The workspace consisted of a dedicated bench area that was cleaned with 70% ethanol before and during handling, with sterile tools and consumables used throughout. During all sampling procedures, personnel wore gloves and face masks, and work surfaces and gloves were routinely disinfected with ethanol prior to and during processing. All samples were aseptically transferred into sterile Whirl-Pak bags and sterile Falcon tubes by using sterile spatula. Whenever possible, subsampling focused on the interior portions of mats, cores, and crusts, with the exteriors avoided to minimize potential contamination. Herein, field blanks during subsampled were not taken. Following collection, all subsamples were promptly sealed and immediately frozen at −20°C onboard, then subsequently transferred to −80°C in the laboratory for long-term storage.
DNA extraction, amplification and sequencing
The DNA was extracted from each of the 38 samples (~10 g per sample) using a DNeasy PowerMax Soil Kit (Qiagen, Germany), following the manufacturer’s protocol. DNA concentration and integrity were checked using a Qubit dsDNA HS Assay and Qubit fluorometer 4.0 (ThermoFisher Scientific, Waltham, USA) and agarose gel (1%) electrophoresis containing Sybr Safe (ThermoFisher Scientific, Waltham, USA), respectively.
The extracted DNA was subjected to internal transcribed spacer (ITS) rRNA gene sequencing at the MR DNA (Molecular Research LP, Shallowater, USA). The DNA was amplified using a commonly used ITS primer set for fungal studies, ITS1 (5’-CTTGGTCATTTAGAGGAAGTAA-3’) and ITS2 (5’-GCTGCGTTCTTCATCGATGC-3’) (White et al., 1990). Amplification was performed using the HotStarTaq Plus Master Mix Kit (Qiagen, USA), as follows: initial denaturation at 95°C for 5 min, followed by 30 cycles at 95°C for 30 s, 53°C for 40 s, and 72°C for 1 min, and a final extension step at 72°C for 10 min. PCR products were then run in 2% agarose gel, and subsequently used to prepare the barcoded library. Barcoded libraries were quantified using a bioanalyzer and pooled at an equimolar ratio based on their molecular weight and ITS concentrations and later purified using AMPure XP beads. For the ITS sequencing (2 × 300 bp), MiSeq reagent kit v3 (Illumina, USA) was employed, in accordance with the manufacturer’s instructions.
Extraction controls were included throughout the DNA extraction process and gene amplification. These controls were assessed using the Qubit dsDNA HS Assay and Qubit fluorometer 4.0 (ThermoFisher Scientific, Waltham, USA), as well as agarose gel (1%) electrophoresis containing Sybr Safe (ThermoFisher Scientific, Waltham, USA). All extraction and PCR controls consistently showed negative results, with no detectable DNA or PCR product bands on the gel and concentrations below the detection limit. We emphasize here that the blank controls from DNA extraction, which were negative by gel electrophoresis and high-sensitivity assays for quantification, were nevertheless sequenced and sequences were deposited alongside the dataset (Supplementary Table 1). However, reads derived from blank were not included in further data processing steps and were not extracted from the sequences of the biological samples.
Data processing
The ITS raw reads were processed using QIIME2 (v2021.11) (QIIME 2 Development Team, 2021). Raw ITS sequencing reads were initially inspected for quality using FastQC (Andrews, 2010) and the QIIME 2 demux summarize visualization. Reads were then processed and denoised using the DADA2 plugin in QIIME 2 (version 2021.11; Bolyen et al., 2019). As part of the DADA2 workflow, forward and reverse reads were truncated to 220 bases, and reads containing more than two expected errors were discarded. Reads were further truncated at the first base with a Phred quality score below 2. Only paired reads with a minimum overlap of 12 bp were merged, and chimeric sequences were identified and removed using the consensus method (Supplementary Figure 1). This process generated a feature table of Amplicon Sequence Variants (ASVs) and their representative sequences. Taxonomic classification of ASVs was performed using a pre-trained Naive Bayes classifier implemented in QIIME 2, with the UNITE database for fungi (version 7, clustered at 97% similarity, released December 2017; Kõljalg et al., 2013) as the reference database for taxonomic assignment. QIIME2 output files, including the feature table, taxonomy assignments, phylogenetic tree, and sample metadata, were imported in R (v4.4.1) using ‘qiime2R’ (v0.99.6) and ‘phyloseq’ (v1.50.0) packages.
Before all downstream analyses, non-fungal taxa were excluded by subsetting for sequences assigned to the Kingdom Fungi. To evaluate sequencing depth and sampling completeness, rarefaction curves were generated using the rarecurve()function in the vegan R package, based on the ASV abundance table. To visualize the relative abundances of fungal communities at different taxonomic levels, the data were aggregated at the phylum, class, order, genus, and species levels. The relative abundances were calculated by normalizing the read counts within each sample. The most abundant taxa were plotted for each taxonomic level, while low-abundance taxa were grouped under ‘Other’ to simplify interpretation. Stacked bar plots were generated to display the relative abundances across sample groups, using ‘ggplot2’ v3.5. 1. To generate species heatmaps across sample groups, the ASV table was processed using the ‘phyloseq’ v1.50.0 package. The table was collapsed to the species level with the tax_glom() function, and sample counts were normalized to relative abundances within each sample. For visualization, the dataset was aggregated by species and sample groups, summing relative abundances across samples in each group. The top 15 most abundant fungal species, based on cumulative relative abundance, were retained to improve readability. The resulting matrix, with species as rows and sample groups as columns, was visualized using the ‘pheatmap’ v1.0.12 package. Row-wise Z-score scaling highlighted relative differences in species composition. A continuous white-to-dark red color gradient represented low to high abundance, and hierarchical clustering was applied to both rows and columns to reveal distribution patterns.
Alpha-diversity metrics, including Observed richness, Shannon and Simpson diversity index, were calculated using the estimate_richness() function from the ‘phyloseq’ package. The calculated diversity indices were merged with the sample metadata for downstream analysis. Beta-diversity was assessed using the Bray-Curtis dissimilarity index to quantify compositional differences among fungal communities. Principal Coordinates Analysis (PCoA) was conducted to visualize variation in community structure. To statistically evaluate differences in beta-diversity, Permutational Multivariate Analysis of Variance (PERMANOVA) was conducted using the adonis2 function in vegan, with 999 permutations, and the coefficient of determination (R²) was reported to indicate the proportion of variance explained by groupings. To confirm homogeneity of variance, Permutational Analysis of Multivariate Dispersions (PERMDISP) was performed using the betadisper function. If PERMDISP was non-significant (p > 0.05), the assumption of equal variance was met, validating PERMANOVA results. A PCoA plot was generated using ‘ggplot2’ v3.5.1, with sample points colored by group, and 95% confidence ellipses included only if PERMANOVA results were significant.
To characterize the ecological functions of the identified fungal taxa, we utilized the FUNGuild tool v1.1 (Nguyen et al., 2016; available at https://github.com/UMNFuN/FUNGuild). Taxonomic classifications of fungal ASVs were first obtained by exporting taxonomy assignments from QIIME2 (Bolyen et al., 2019). Afterward, the FUNGuild was used to assign ecological functions to the ASV based on its taxonomic information, providing details such as trophic modes, specific functional guilds, and associated confidence rankings. In parallel, the fungal abundance table containing ASV counts across all samples was also exported from QIIME2. Finally, the abundance data were merged with the FUNGuild output, to better understand the fungal communities’ taxonomic composition and functional roles. For visualization of the patterns in fungal functional diversity, two metrics were employed to describe guild composition, in which ASV richness, representing the proportion of unique ASVs assigned to each guild, and sequence richness, representing the proportion of total sequence abundance, were attributed to each guild. These metrics were calculated separately for each sample type and hydrothermal vent site. The results were summarized as percentage values and visualized using ‘ggplot2’ v3.5. 1. It is important to acknowledge that precise community-wide conclusions for ecological function remain challenging due to several factors, such as multiple trophic strategies of the fungal taxa, guild data are still limited for numerous fungal groups (Nilsson et al., 2019), and the ecological roles of marine fungal taxa are underexplored (Velez et al., 2022). Nevertheless, FUNGuild can provide a valuable foundation toward advancing our understanding of deep-sea and hydrothermal vent fungal communities.
Results
Sequence analysis
Raw sequencing yielded a total of 8,271,048 reads (4,135,524 paired-end read pairs) across all samples. After quality control, 228,043 reads were retained from hydrothermal sediment, microbial mats and chimney crust samples collected in low-temperature hydrothermal vent fields in the Hatiba Mons, with 104,274 for microbial mat samples, 58,547 for crust samples and 65,222 for hydrothermal sediments (Supplementary Table 2). A total of 34,940 reads corresponded to KRSE4-2 ROV HYD, 33,137 for KRSE4-3 ROV SED 1, 48,032 for KRSE4-3 ROV SED 2, 46,991 for KRSE4-3 ROV SED 4, 23,607 for KRSE4-4 ROV HYD X, and 41,336 for KRSE4-6 ROV NET.
The retained sequences were classified into 550 ASVs out of which 534 represented fungal taxa (214,707 reads). All of the rarefaction and extrapolation curves of the fungal assemblages in the samples from the different sites reached the asymptote (Supplementary Figure 2), indicating that the data generated from the 38 samples provided a good description of the fungal diversity. However, it is important to note that technical factors, such as DNA extraction method, primer specificity, or PCR bias, may have influenced the recovery and detection of certain fungal groups (Tedersoo et al., 2015; Portela et al., 2025).
Taxonomic profiling of the fungal community
Out of 228,043 reads, a total of 21,4707 were taxonomically assigned to Fungi. In all the studied sites, the fungal community was dominated by the Basidiomycota, followed by Ascomycota. The crust sample from Field 8 exhibited more diversity at the phylum, also harboring Mucuromycota and Zoopagomycota, while the crust from Farwah Safraa Ridges presented only the most abundant fungal phyla and higher abundance of unidentified ASVs. In the crust of Field 8 and sediment samples of Kabir Field, Mortierellomycota was also observed (Figure 2A). At the order level, dominance patterns varied across the hydrothermal fields. For instance, sequences of Poliporales were vastly observed in the microbial crust and mat sample of Farwah Safraa Ridges with ASVs showing 96.0–98.3% identity to Resinoporia piceata, a known terrestrial member of Polyporales, while Filobasidiales and Sacharomycetales were highly represented in the hydrothermal sediments of Kabir Field. High relative sequence abundance of Agaricales were found in the crust samples (Field 8 and Farwah Safraa Ridges). Higher sequence abundance of the other observed orders was found in the Field 8 and Kabir Field (Figure 2B). Farwah Safraa Ridges samples (crust and microbial mat) showed higher sequence abundance of the class Agaricomycetes, followed by Saccharomycetes, while for the samples from Field 8 and Kabir Field, the abundance was more equally distributed between the different classes (Figure 2C). Prevalence of Lipomyces was higher in the samples of Field 8, Farwah Safraa Ridges, and one sample group of Kabir Field, and for the other samples of Kabir, Filobasidium were in higher sequence abundance (Figure 2D). The genus Coprinus was only observed in the crust samples, especially in the Field 8, with ASVs showing 96% identity to known Coprinus sequences, while Kluyveromyces was found only in one sample of the hydrothermal sediment of Kabir Field, and Rigidoporus was uniquely found in Farwah Safraa Ridges crust (Figure 2E). Both Coprinus and Rigidoporus are examples of taxa known from the terrestrial environment and while these, as well as other close relatives of terrestrial taxa may be able to survive marine conditions, some may also persist in the deep sea as spores. Overall, at the species level, nine fungal species were assigned to the ASVs (Figure 2F), whereas each species was found in higher abundance in different samples. For instance, Coprinus cordisporus was abundant in the crusts of Field 8, Filobasidium wieringae, Trichoderma longibrachiatum, Debaryomyces udenni, Malassezia globosa, and Neoascochyta paspali were dominant species in Kabir Field, and Rigidoporus ulmarius were prevalent in the microbial mat of Farwah Safraa Ridges.
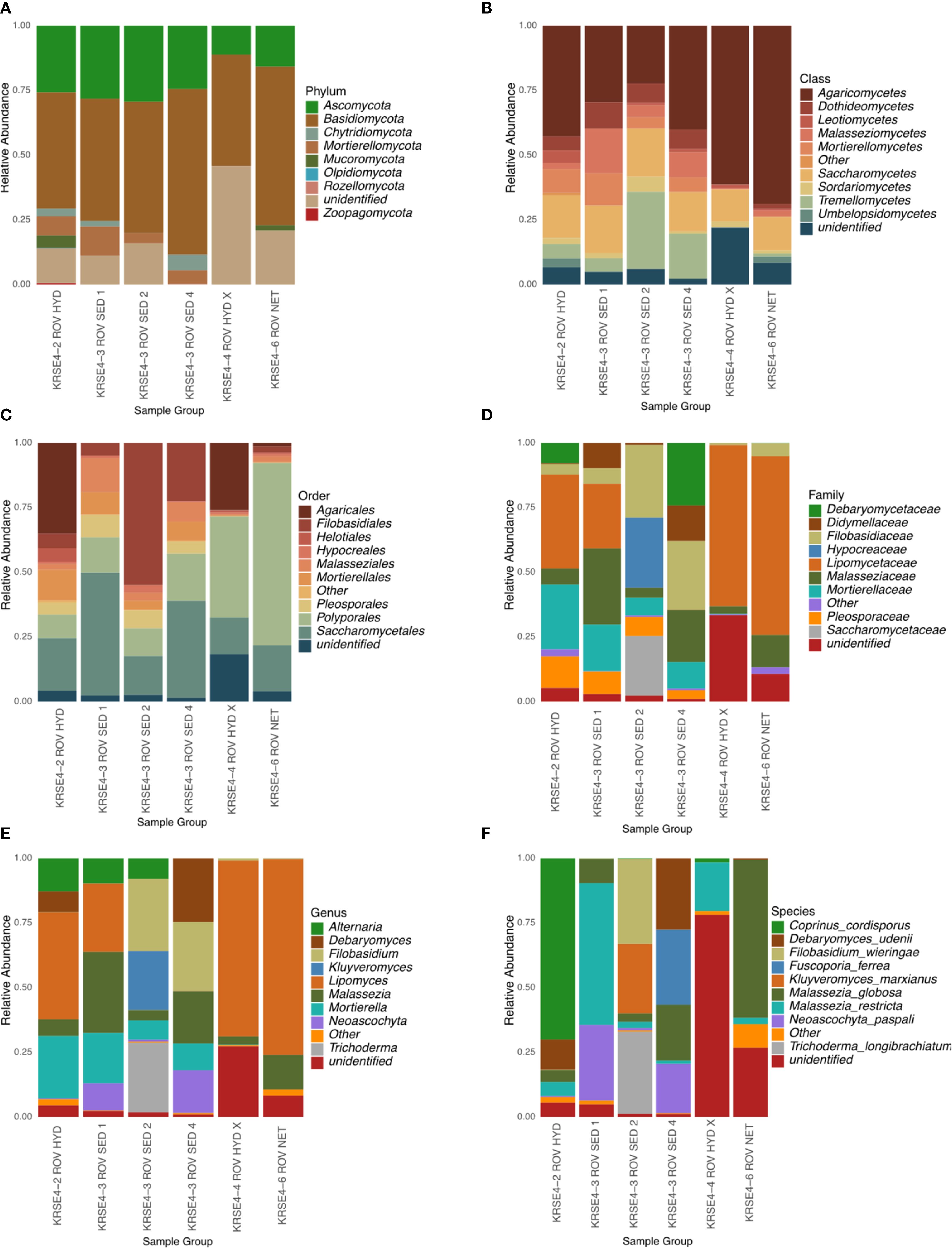
Figure 2. The relative abundance of fungal communities across taxonomic levels in Red Sea hydrothermal vent samples. Composition is displayed at the (A) Phylum, (B) Class, (C) Order, (D) Family, (E) Genus, and (F) Species levels for sediment and mat samples from different ROV deployments. Only the top 10 taxa are shown per level; others are grouped as “Other”, and unclassified reads as “unidentified”. Color schemes are consistent within each level.
Alpha and beta-diversity estimates
Measures of fungal alpha-diversity (number of observed ASV, Shannon and Simpson indexes) were significantly higher in the crust of the Field 8 across the other Red Sea hydrothermal vent type samples and locations. Microbial mat from Farwah Safraa Ridges and samples from Kabir Field showed similar richness and evenness, without significant difference between their fungal communities (Figure 3A). The same pattern was observed when analyzing the fungal beta-diversity explored by Bray–Curtis and weighted Unifrac distances. The PcoA analysis revealed a clear distinction between the crust and the rest of the samples and locations that formed a single cluster (Figure 3B).
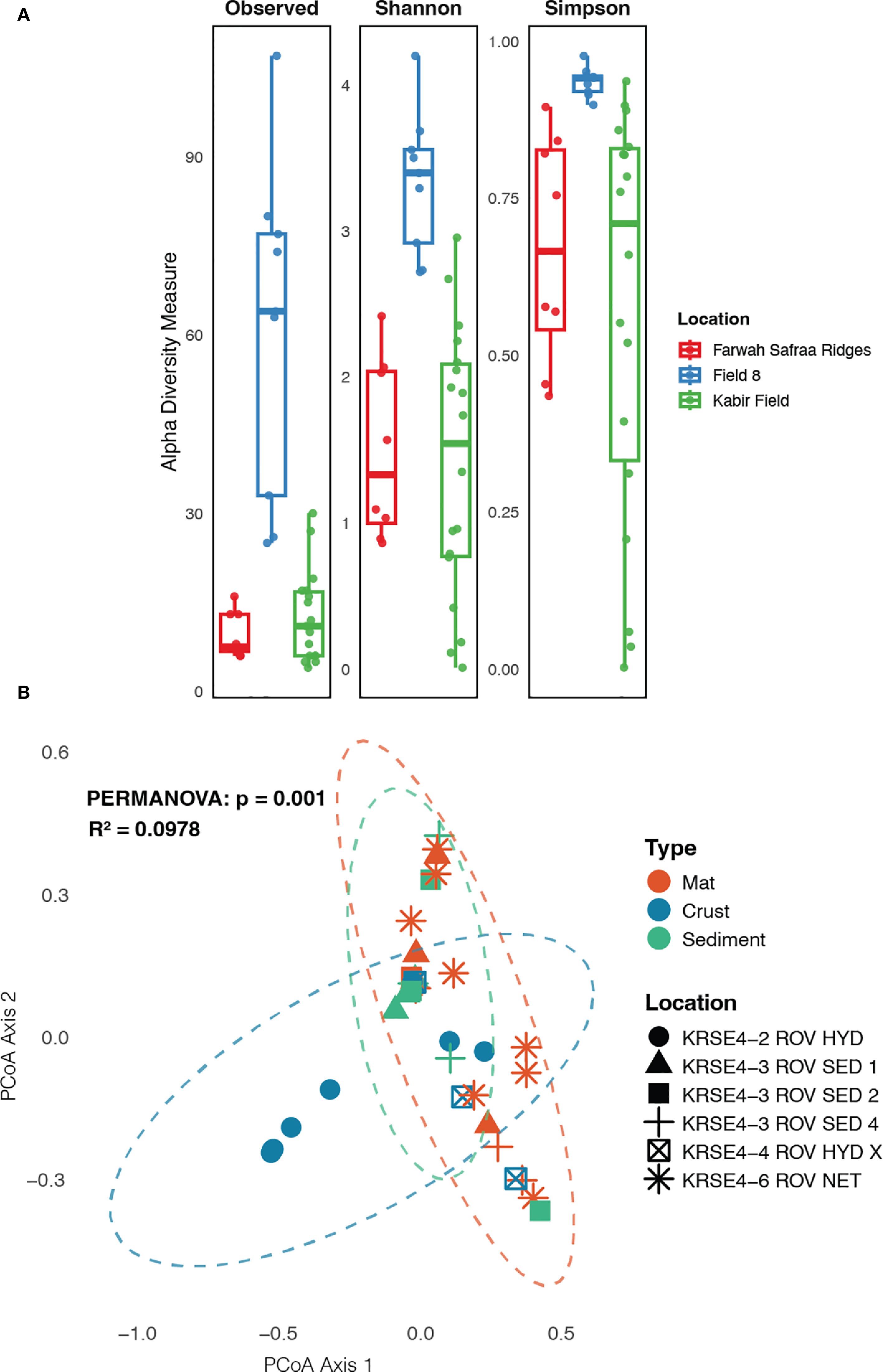
Figure 3. Fungal community structure across samples type and location in the Red Sea hydrothermal field. (A) alpha-diversity indexes (Observed ASV, Shannon and Simpson indexes) across locations, and (B) beta-diversity analysis using PCoA, showing the differences between samples’ type and location within the Red Sea hydrothermal field sites.
Functional guilds of the Red Sea hydrothermal vents fungi
Prediction of the fungal communities’ functions were performed using FUNGuild, and characterized in trophic mode in the sample’s location, and also across the sample type. In all of the samples’ locations and types, the dominance was for unassigned mode, whereas around 50% of the sequences were not assigned to any function. For the assigned reads, it was observed that putative saprotrophs were dominant in all the sites and sample types, except for microbial mat of Kabir Field (sample KRSE4-3 ROV SED 2), whereas it was observed putative pathotroph-saprotroph-symbiotroph was highest enriched with relative to the other trophic modes (Figure 4A). Crust samples, in comparison with hydrothermal sediment and microbial mat, presented the highest abundance for putative symbiotrophs (Figure 4B).
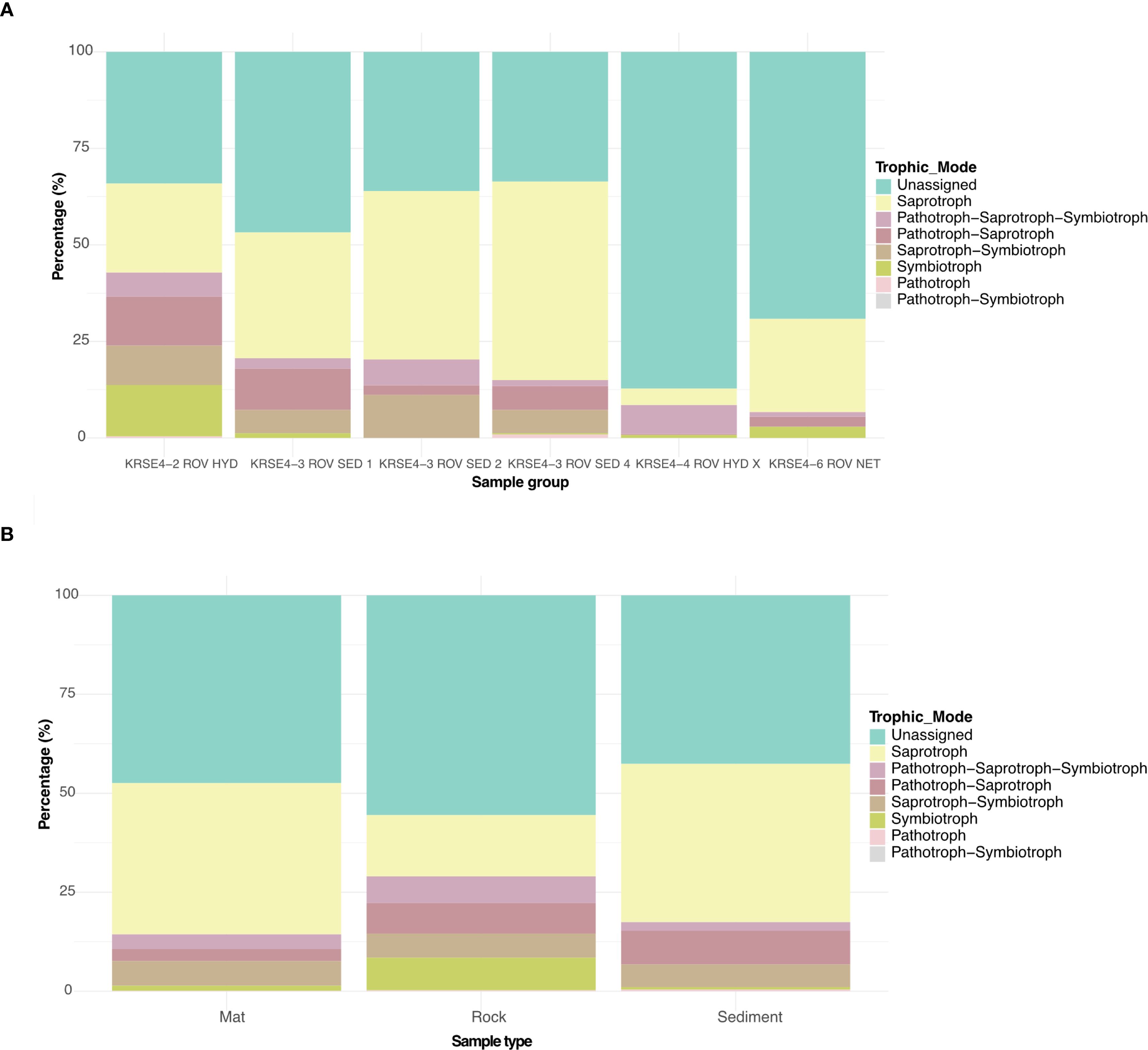
Figure 4. Stacked bar plot showing the relative proportion (y-axis) of the predicted fungal functional guilds across hydrothermal vent sites location (A) and samples type (B) (x-axis). The bars represent the percentage of sequence reads assigned to each trophic category.
Discussion
Studies on the diversity and ecological role of fungi in aquatic environments, specifically from hydrothermal vent systems, have been comparatively scarce compared to terrestrial ecosystems, but have progressed in recent years (Jones et al., 2019; Grossart et al., 2019; da Silva et al., 2022; Asseri et al., 2025). Aiming to contribute to this growing field, our study provides insights into the fungal diversity and community structure in low-temperature hydrothermal vent fields of the Hatiba Mons, Red Sea. Through high-throughput sequencing, we characterized fungal assemblages across distinct substrates, including sediment, microbial mats, and chimney crusts, highlighting variations in taxonomic composition and diversity across hydrothermal vent sites.
Our findings reveal that the fungal communities in the studied hydrothermal vent sites are dominated by Basidiomycota, followed by Ascomycota, which aligns with previous studies on marine fungal diversity (Hassett et al., 2020; Cunliffe, 2023; Varrella et al., 2024). The crust sample from Field 8 exhibited the highest taxonomic diversity, additionally harboring Mucoromycota and Zoopagomycota—groups rarely reported in marine hydrothermal systems. Recent studies have reported representatives of these phyla in marine settings, including deep-sea sediment (Luo et al., 2020), seawater samples and infecting marine animals (Pang et al., 2021; Zhang et al., 2023), suggesting a broader ecological distribution, possibly facilitated by dispersal, ecological plasticity, spores or cryptic lifestyles (Amend et al., 2019). The presence of Mortierellomycota in the crust of Field 8 and sediments of Kabir Field suggests adaptation to specific microhabitats, potentially influenced and shaped by geochemical factors (Tisthammer et al., 2016), such as mineral composition and thermal gradients from the vent chimney. Although members of the Mortierellomycota phylum are widely distributed in soils covered by snow in the temperate zone, and are observed in the winter-active soil microbial community in alpine and subalpine regions (Telagathoti et al., 2022), they have been detected in marine ecosystems, including deep-sea sediments (Xu et al., 2018, 2019; Luo et al., 2020).
At the genus level, we observed a distinct distribution of fungal taxa across substrates. Notably, Rigidoporus was uniquely found in the microbial mat of Farwah Safraa Ridges, suggesting that this taxon may have a role in organic matter turnover within biofilms (Xu et al., 2018). Coprinus was exclusively found in crust samples, particularly in Field 8, while Kluyveromyces was detected only in one hydrothermal sediment sample from Kabir Field. The restricted occurrence of certain genera to specific substrates may reflect ecological specialization or substrate-dependent metabolic capabilities (Jones et al., 2015). Some ASVs recovered in this study were affiliated with terrestrial genera such as Polyporales (87.8–88.9% identity to Resinoporia piceata) and Coprinus (96.0–98.3% identity to known species of the genus Coprinus). Prior work has shown that several terrestrial-affiliated fungi can be isolated from deep marine sediments and tolerate in situ conditions such as salinity and temperature stress (Rédou et al., 2015). Notably, members of Polyporales, including Bjerkandera and Trametes, have previously been isolated from deep-subsurface marine sediments, supporting the possibility that this order includes fungi capable of surviving and possibly adapting to deep-sea conditions (Rédou et al., 2015). These taxa were cultured under hydrostatic pressure and varying salinity, suggesting the possibility of physiological plasticity within these taxa. However, possible sources of contamination cannot be entirely ruled out, and they may also originate from terrestrial sources and persist in the benthos as spores. These observations raise the possibility that certain terrestrial-derived fungi may persist in deep-marine sediments, although their ecological role remains unclear. To confirm the ecological relevance of the metabolically active fungi in the deep sea, integrated approaches must be employed, including DNA-based and mRNA-based approaches and cultivation strategies. Additionally, regarding the detection of taxa related to terrestrial fungi, note that sequences from extraction controls (blanks) were not substracted from the samples’ reads and so sample datasets may contain reagent contaminants as well as biologically plausible sequences (Eisenhofer et al., 2019).
Herein, it is worth mentioning that we employed the ITS1F/ITS2 primer set, a commonly used marker pair targeting the ITS1 region of the fungal ribosomal operon. While widely applied in fungal metabarcoding studies, this primer set is known to have limitations in amplifying early diverging lineages such as Chytridiomycota, a commonly found fungi taxa in marine ecosystems (Hassett and Gradinger, 2016; Asseri et al., 2025), potentially leading to their underrepresentation in our hydrothermal vent dataset. No single primer set can comprehensively capture the full breadth of fungal diversity, and ITS1F/ITS2 represents one of several available options (Grossart et al., 2019; Reynolds et al., 2022). As ITS may fail to resolve cryptic or divergent lineages adapted to extreme conditions, we suggest that future research should incorporate multi-marker strategies or shotgun metagenomic approaches to improve taxonomic resolution (Reynolds et al., 2022).
Diversity patterns were also observed across hydrothermal vent fields, whereas alpha diversity estimates indicate that the crust samples from Field 8 harbor the highest fungal richness and evenness, while microbial mat and sediment samples from Farwah Safraa Ridges and Kabir Field exhibited similar diversity patterns. This elevated richness in crusts may reflect microhabitats enriched in trace metals and mineral gradients, providing niches for metabolically versatile fungi. Previous studies have shown that crusts in hydrothermal vents can serve as hotspots for microbial diversity due to their structural complexity and mineral composition (Sheik et al., 2015). Beta-diversity analyses further support the uniqueness of fungal communities in crust samples, as evidenced by the distinct clustering pattern in the PCoA analysis. The differentiation between crust-associated fungi and those in microbial mats and sediments suggests habitat-driven selection, possibly influenced by temperature fluctuations, mineral availability, and interactions with prokaryotic communities (Biddle et al., 2012). The formation of a single cluster encompassing microbial mat and sediment samples indicates a greater degree of similarity among these communities, potentially due to shared ecological constraints and resource availability.
The FUNGuild analysis evidenced characteristic functional fungal signatures across the low-temperature Red Sea hydrothermal vent sites and across the sample type. For instance, the dominance of putative saprotrophs in all the samples suggests that the fungal community likely plays a central role in organic matter decomposition and recycling of detrital biomass (Breyer and Baltar, 2023). The presence of predicted symbiotrophs in the crust samples from Field 8 may reflect associations with chemoautotrophic prokaryotes or sessile invertebrates, contributing to nutrient exchange and microbial network stability (Orsi et al., 2020). These results align with the findings from Velez et al. (2022) that by analyzing samples from high-temperature hydrothermal vents and oxygen minimum zone, found that saprotrophs were dominant in both systems. Similarly, studies from the Mid-Atlantic Ridge and East Pacific Rise have reported the presence of thermotolerant fungal lineages with potential roles in sulfur and metal cycling (Le Calvez et al., 2009; Tisthammer et al., 2016), further reinforcing the ecological relevance of fungi in vent environments globally. It is important to mention that predictions of ecological function conducted with FUNGuild must be interpreted with caution because the reliability of assignments depends on the accuracy of taxonomic classification, and the presence of ecologically-characterized close relatives in the database used by the tool is a particular challenge when investigating poorly explored deep sea habitats, including the Red Sea.
Beyond ecological insights, the unique environmental pressures of the Red Sea hydrothermal vents may select for fungi with novel enzymatic systems and secondary metabolites of biotechnological interest. Several of the taxa identified here—including filamentous saprotrophs and symbiotrophs—are known from other systems to produce thermostable enzymes, metal-tolerant biocatalysts, and bioactive compounds with potential pharmaceutical or industrial relevance (Wang et al., 2023). Given the unique environmental conditions of hydrothermal vents, these fungi may possess novel metabolic pathways relevant for biotechnological applications, such as enzyme production and bioremediation (Arfi et al., 2022; Vaksmaa et al., 2023).
However, a substantial portion (~50%) of the ASVs in our dataset remained functionally unassigned in the FUNGuild analysis. A possible explanation is that less than 0.001% of the deep ocean (this includes hydrothermal vent systems) has been investigated, being considered one of the least explored biomes on Earth (Bell et al., 2025). Future studies that combine metagenomics, metabolomics, and culturing approaches with marker gene approaches are required to unravel the taxonomic and functional diversity of fungi at Hatiba Mons and their ecological roles.
Overall, this study presents the first assessment of fungal diversity within the Hatiba Mons hydrothermal vent fields, uncovering a complex and previously undocumented fungal community. The distinct taxonomic profiles observed across different substrates underscore the role of habitat heterogeneity in shaping fungal community structures. Notably, the identification of taxa potentially involved in metal cycling and organic matter degradation suggests that fungi actively contribute to essential biogeochemical processes in these vent ecosystems. These insights enhance our understanding of marine fungal ecology and highlight the significance of deep-sea mycological research in elucidating ecosystem dynamics. Furthermore, the discovery of novel and unclassified fungal lineages points to the potential for uncovering unique bioactive compounds, positioning deep-sea fungi as promising candidates for biotechnological applications.
Data availability statement
The raw ITS reads datasets supporting the conclusions of this article are available in the NCBI GenBank database under the accession numbers from SRR29740229 to SRR29740269, in the BioProject PRJNA1133038.
Author contributions
JS: Methodology, Writing – original draft, Formal analysis, Writing – review & editing, Investigation, Visualization, Data curation, Conceptualization. SA: Writing – review & editing, Methodology, Formal analysis, Writing – original draft, Visualization, Data curation. FMvdZ: Data curation, Formal analysis, Methodology, Project administration, Resources, Writing – review & editing. NA: Data curation, Formal analysis, Methodology, Project administration, Resources, Writing – review & editing. ASR: Funding acquisition, Resources, Supervision, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research received financial support from the KAUST Baseline Grant BAS/1/1096-01-01 (awarded to AR). Ship time was sponsored by the KAUST VPR office.
Acknowledgments
The authors would like to express gratitude to the captain, crew and scientific participants of RV Aegaeo expeditions KRSE4-1 and KRSE5 for their support on board during the research expeditions. Particularly we thank Lera Shepard and Nicholas Kontis for their help in the sampling process. The authors appreciate the support provided by the KAUST Coastal and Marine Core Labs for organizing the research campaigns and providing technical support at sea. All research and sampling were permitted by the Kingdom of Saudi Arabia through KAUST Government Affairs.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1649339/full#supplementary-material
References
Amend A., Burgaud G., Cunliffe M., Edgcomb V. P., Ettinger C. L., Gutierrez M. H., et al. (2019). Fungi in the marine environment: Open questions and unsolved problems. mBio 10, e01189–e01118. doi: 10.1128/mBio.01189-18
Andrews S. (2010). FastQC: a quality control tool for high throughput sequence data [Internet]. Cambridge: Babraham Bioinformatics. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Arfi Y., Matei E., Bibi F., Ghigo E., and Armougom F. (2022). Deep-sea fungi: Sources of novel enzymes and bioactive metabolites. Mar. Drugs 20, 55. doi: 10.3390/md20010055
Asseri A. A., Coello-Camba A., and Agustí S. (2025). Fungal planktonic community related to salinity and temperature in an oligotrophic sea. Front. Microbiol. 16. doi: 10.3389/fmicb.2025.1435925
Augustin N., van der Zwan F. M., Devey C. W., and Petersen S. (2021). 13 million years of seafloor spreading throughout the Red Sea Basin. Nat. Commun. 12, 2427. doi: 10.1038/s41467-021-22586-2
Augustin N., van der Zwan F. M., Petersen S., and Devey C. W. (2024). The effect of spreading rate on the volcanic activity and frequency distribution of hydrothermal vent sites in the Red Sea (Bonn: Begutachtungspanel Forschungsschiffe). Available online at: https://www.tib.eu/de/suchen/id/awi%3A340165d5e4c8e59c67c016bac86046153777576a (Accessed August 18, 2025).
Bell K. L. C., Johannes K. N., Kennedy B. R. C., and Poulton S. E. (2025). How little we’ve seen: A visual coverage estimate of the deep seafloor. Sci. Adv. 11, eadp8602. doi: 10.1126/sciadv.adp8602
Berumen M. L., Voolstra C. R., Daffonchio D., Agusti S., Aranda M., Irigoien X., et al. (2019). “The red sea: environmental gradients shape a natural laboratory in a Nascent Ocean,” in Coral reefs of the red sea. Eds. Voolstra C. R. and Berumen M. L. (Springer International Publishing, Cham), 1–10. doi: 10.1007/978-3-030-05802-9_1
Biddle J. F., Sylvan J. B., Brazelton W. J., Tivey M. K., Smith G., Popa R., et al. (2012). Prospects for the study of evolution in the deep biosphere. Front. Microbiol. 2. doi: 10.3389/fmicb.2011.00285
Bolyen E., Rideout J. R., Dillon M. R., Bokulich N. A., Abnet C. C., Al-Ghalith G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Breyer E. and Baltar F. (2023). The largely neglected ecological role of oceanic pelagic fungi. Trends Ecol. Evol. 38 (9), 870–888. doi: 10.1016/j.tree.2023.04.007
Burgaud G., Arzur D., Durand L., Cambon-Bonavita M. A., and Barbier G. (2010). Marine culturable yeasts in deep-sea hydrothermal vents: Species richness and association with fauna. FEMS Microbiol. Ecol. 73, 121–133. doi: 10.1111/j.1574-6941.2010.00881.x
Cunliffe M. (2023). Who are the marine fungi? Environ. Microbiol. 25 (1), 131–134. doi: 10.1111/1462-2920.16240
da Silva M. K., de Souza L. M. D., Vieira R., Ayres Neto A., Lopes F. A.C., Oliveira F. S., et al. (2022). Fungal and fungal-like diversity in marine sediments from the maritime Antarctic assessed using DNA metabarcoding. Sci. Rep. 12, 21044. doi: 10.1038/s41598-022-25310-2
Dick G. J. (2019). The microbiomes of deep-sea hydrothermal vents: Distributed globally, shaped locally. Nat. Rev. Microbiol. 17, 271–283. doi: 10.1038/s41579-019-0160-2
Eisenhofer R., Minich J. J., Marotz C., Cooper A., Knight R., and Weyrich L. S. (2019). Contamination in low microbial biomass microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. doi: 10.1016/j.tim.2018.11.003
Grossart H. P., Van den Wyngaert S., Kagami M., Wurzbacher C., Cunliffe M., and Rojas-Jimenez K. (2019). Fungi in aquatic ecosystems. Nat. Rev. Microbiol. 17, 339–354. doi: 10.1038/s41579-019-0175-8
Hassett B. T. and Gradinger R. (2016). Chytrids dominate arctic marine fungal communities. Environ. Microbiol. 18, 2001–2009. doi: 10.1111/1462-2920.13216
Hassett B. T., Vonnahme T. R., Peng X., Jones E. G., and Heuzé C. (2020). Global diversity and geography of planktonic marine fungi. Bot. Mar. 63, 121–139. doi: 10.1515/bot-2018-0113
Jones E. B. G., Pang K. L., Abdel-Wahab M. A., Scholz B., Hyde K. D., Boekhout T., et al. (2019). An online resource for marine fungi. Fungal Diversity 96, 347–433. doi: 10.1007/s13225-019-00426-5
Jones E. B. G., Suetrong S., Sakayaroj J., Bahkali A. H., Abdel-Wahab M. A., Boekhout T., et al. (2015). Classification of marine ascomycota, basidiomycota, blastocladiomycota and chytridiomycota. Fungal Diversity 73, 1–72. doi: 10.1007/s13225-015-0339-4
Kõljalg U., Nilsson R. H., Abarenkov K., Tedersoo L., Taylor A. F. S., Bahram M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5287. doi: 10.1111/mec.12481
Le Calvez T., Burgaud G., Mahé S., Barbier G., and Vandenkoornhuyse P. (2009). Fungal diversity in deep-sea hydrothermal ecosystems. Appl. Environ. Microbiol. 75, 6415–6421. doi: 10.1128/AEM.00653-09
Luo Y., Wei X., Yang S., Gao Y. H., and Luo Z. H. (2020). Fungal diversity in deep-sea sediments from the Magellan seamounts as revealed by a metabarcoding approach targeting the ITS2 regions. Mycology 11, 214–229. doi: 10.1080/21501203.2020.1799878
Nguyen N. H., Song Z., Bates S. T., Branco S., Tedersoo L., Menke J., et al. (2016). FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 20, 241–248. doi: 10.1016/j.funeco.2015.06.006
Nilsson R. H., Anslan S., Bahram M., Wurzbacher C., Baldrian P., and Tedersoo L. (2019). Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat. Rev. Microbiol. 17, 95–109. doi: 10.1038/s41579-018-0116-y
Orsi W. D., Morard R., Vuillemin A., et al. (2020). Anaerobic metabolism of Foraminifera thriving below the seafloor. ISME J 14, 2580–2594. doi: 10.1038/s41396-020-0708-1
Pang K. L., Hassett B. T., Shaumi A., Guo S. Y., Sakayaroj J., Chiang M. W. L., et al. (2021). Pathogenic fungi of marine animals: A taxonomic perspective. Fungal Biol. Rev. 35, 39–58. doi: 10.1016/j.fbr.2021.03.008
Portela NéstorD., Mena C., Martín M. G., Burstein VerónicaL., Chiapello L. S., and Pesoa S. A. (2025). Effect of DNA extraction method in gut fungal community assessment. Rev. Argent. Microbiología. 57 (4), 1–9. doi: 10.1016/j.ram.2024.12.005
QIIME 2 Development Team (2021). QIIME 2 2021.11 is now available. Available online at: https://forum.qiime2.org/t/qiime-2-2021-11-is-now-available/21457 (Accessed June 2, 2025).
Rédou V., Navarri M., Meslet-Cladière L., Barbier G., and Burgaud G. (2015). Species richness and adaptation of marine fungi from deep-subseafloor sediments. Appl. Environ. Microbiol. 81, 3571–3583. doi: 10.1128/AEM.04064-14
Reynolds N. K., Jusino M. A., Stajich J. E., and Smith M. E. (2022). Understudied, underrepresented, and unknown: Methodological biases that limit detection of early diverging fungi from environmental samples. Mol. Ecol. Resour. 22, 1065–1085. doi: 10.1111/1755-0998.13540
Richards T. A., Jones M. D. M., Leonard G., and Bass D. (2012). Marine fungi: Their ecology and molecular diversity. Annu. Rev. Mar. Sci. 4, 495–522. doi: 10.1146/annurev-marine-120710-100802
Salcedo D. L., Velez P., Hernandez-Monroy A., and Soto L. A. (2023). Insights into the functional role of fungi in deep-sea hydrothermal vents through the analysis of stable isotopes of carbon, nitrogen, and sulfur. Fungal Ecol. 64, 101250. doi: 10.1016/j.funeco.2023.101250
Sheik C. S., Anantharaman K., Breier J. A., Sylvan J. B., Edwards K. J., and Dick G. J. (2015). Spatially resolved sampling reveals dynamic microbial communities in rising hydrothermal plumes across a back-arc basin. ISME J 9 (6), 1434–1445. doi: 10.1038/ismej.2014.228
Shourie A. and Vijayalakshmi U. (2022). Fungal diversity and its role in mycoremediation. Geomicrobiol. J. 39, 426–444. doi: 10.1080/01490451.2022.2032883
Tedersoo L., Anslan S., Bahram M., Põlme S., Riit T., Liiv I., et al. (2015). Shotgun metagenomes and multiple primer pair-barcode combinations of amplicons reveal biases in metabarcoding analyses of fungi. MycoKeys 10, 1–43. doi: 10.3897/mycokeys.10.4852
Telagathoti A., Probst M., Mandolini E., and Peintner U. (2022). Mortierellaceae from subalpine and alpine habitats: new species of Entomortierella, Linnemannia, Mortierella, Podila and Tyroliella gen. nov.. Stud Mycol 103, 25–58. doi: 10.3114/sim.2022.103.02
Tisthammer K. H., Cobian G. M., Amend A. S., and Riquelme M. (2016). High-throughput sequencing reveals fungal diversity in deep-sea hydrothermal vent sediments. Fungal Ecol. 20, 69–77. doi: 10.1016/j.funeco.2015.10.002
Vaksmaa A., Guerrero-Cruz S., Ghosh P., Zeghal E., Hernando-Morales V., and Niemann H. (2023). Role of fungi in bioremediation of emerging pollutants. Front. Mar. Sci. 10. doi: 10.3389/fmars.2023.1070905
Van Der Zwan F. M., Augustin N., Petersen S., Altalhi S. M., Schultz J., Peixoto R. S., et al. (2023). Widespread diffuse venting and large microbial iron-mounds in the Red Sea. Commun. Earth Environ. 4, 496. doi: 10.1038/s43247-023-01169-7
Varrella S., Barone G., Corinaldesi C., Giorgetti A., Nomaki H., Nunoura T., et al. (2024). Fungal abundance and diversity in the mariana trench, the deepest ecosystem on earth. J. Fungi (Basel) 10, 73. doi: 10.3390/jof10010073
Velez P., Salcedo D. L., Espinosa−Asuar L., Gasca−Pineda J., Hernandez Monroy A., and Soto L. A. (2022). Fungal diversity in sediments from deep−sea extreme ecosystem: Insights into low- and high-temperature hydrothermal vents, and an oxygen minimum zone in the southern gulf of California, Mexico. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.802634
Wang Z., Qader M., Wang Y., Kong F., Wang Q., and Wang C. (2023). Progress in the discovery of new bioactive substances from deep-sea associated fungi during 2020–2022. Frontiers in Marine Science 10, 1232891. doi: 10.3389/fmars.2023.1232891
White T. J., Bruns T. D., Lee S. B., and Taylor J. W. (1990). Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis M. A., Gelfand D. H., Sninsky J. J., and White T. J., editors. PCR Protocols: A Guide to Methods and Applications. San Diego: Academic Press; p. 315–322.
Xu W., Gao Y. H., Gong L. F., Li M., Pang K. L., and Luo Z. H. (2019). Fungal Diversity in the Deep Sea Hadal Sediments of the Yap Trench by Cultivation and High Throughput Sequencing Methods based on the ITS rRNA gene. Deep Sea Res. Part I Oceanogr. Res. Pap. 145, 125–136. doi: 10.1016/j.dsr.2019.02.001
Xu W., Gong L. F., Pang K. L., and Luo Z. H. (2018). Fungal diversity in deep-sea sediments of a hydrothermal vent system in the Southwest Indian Ridge. Deep Sea Res. Part I Oceanogr. Res. Pap. 131, 16–26. doi: 10.1016/j.dsr.2017.11.001
Yao F. and Hoteit I. (2018). Rapid Red Sea deep water renewals caused by volcanic eruptions and the north Atlantic oscillation. Sci. Adv. 4, eaar5637. doi: 10.1126/sciadv.aar5637
Keywords: fungi, microbiome, hydrothermal venting, amplicon sequencing, ITS
Citation: Schultz J, Altalhi S, Zwan FMvd, Augustin N and Rosado AS (2025) Diving into the deep: fungal diversity in the newly discovered hydrothermal vents of Hatiba Mons, Red Sea. Front. Mar. Sci. 12:1649339. doi: 10.3389/fmars.2025.1649339
Received: 18 June 2025; Accepted: 29 August 2025;
Published: 25 September 2025.
Edited by:
Paraskevi Polymenakou, Hellenic Centre for Marine Research (HCMR), GreeceReviewed by:
Virginia P. Edgcomb, Woods Hole Oceanographic Institution, United StatesLuis Eduardo Servín Garcidueñas, National Autonomous University of Mexico, Mexico
Leyla Benammar, University of Batna 2, Algeria
Copyright © 2025 Schultz, Altalhi, Zwan, Augustin and Rosado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Júnia Schultz, anVuaWFzY2h1dHpAZ21haWwuY29t; Alexandre Soares Rosado, YWxleGFuZHJlLnJvc2Fkb0BrYXVzdC5lZHUuc2E=
†The authors share first authorship