 Yanting Ling1,2†
Yanting Ling1,2† Xuan Chen1,2†
Xuan Chen1,2† Daidai Wu1,2*
Daidai Wu1,2* Qinyi Zhang2,3Shengyi Mao1,2
Qinyi Zhang2,3Shengyi Mao1,2 Guangrong Jin2Jie Liu2
Guangrong Jin2Jie Liu2 Haizhen Zhai2
Haizhen Zhai2 Chao Yang1,2
Chao Yang1,2 Zheng Su1,2
Zheng Su1,2 Nengyou Wu4
Nengyou Wu4- 1University of Science and Technology of China, Hefei, China
- 2Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Guangzhou, China
- 3Fachbereich Erdsystemwissenschaften, Zentrum für Erdsystemforschung und Nachhaltigkeit, Universität Hamburg, Hamburg, Germany
- 4Key Laboratory of Gas Hydrate, Ministry of Natural Resources, Qingdao Institute of Marine Geology, China Geological Survey, Qingdao, China
Pyrite is an important indicator of methane seepage. The effects of organic sulfate reduction (OSR) and sulfate-driven anaerobic oxidation of methane (SD-AOM) on the trace metals in pyrite have aroused interest; however, further studies about how these two pathways control the trace element patterns are still required. Therefore, we studied the geochemical and mineralogical characteristics of sediment samples from the northern South China Sea, aiming to investigate the enrichment mechanisms of trace elements into pyrites in methane seep systems. We applied laser ablation–inductively coupled plasma–mass spectrometry (LA-ICP-MS) to analyze the trace element patterns of pyrites in core Z22–3. We used Sr/Ca-Mg/Ca, (Mo/U) EF values, chromium reducible sulfur (CRS) content, total organic carbon (TOC), δ34S, and δ13C values of bulk sediments to determine the position of sulfate–methane transition zones (SMTZs). Progressively more positive δ34S values (up to −36.54‰) of the CRS and an increase in Mg/Ca indicate the enhancement of SD-AOM, suggesting that the position of SMTZs is located at depths of 181 to 481 and 1,101 to 1,401 cmbsf. Within SMTZs, pyrites exhibit obvious enrichments in Co, Ni, Mo, and Pb and depletion in Mn, Zn, Cu, and V. We discuss the process of methane seepage affecting trace elements in pyrites: the reductive dissolution of iron/manganese oxyhydroxide aggregates fueled by the enhancement of AOM and substitution for ferrous in pyrites contribute to the enrichments of Mo, Co and Ni in pyrites. Elements with faster water exchange kinetics rates than Fe, such as Cu, Pb and Zn, rather precipitate as independent metal sulfides than incorporating into pyrite, thus resulting in the depletion of these trace elements in pyrites. This study provides novel insight into the deep-sea trace element cycle between sediments and authigenic pyrites, establishing geochemical indicators of AOM based on trace element patterns in pyrites.
1 Introduction
Methane-rich fluid seepage caused by the dissociation of gas hydrates under the influence of decreases in pressure or increases in temperature is widespread on continental margins worldwide (Ruppel and Kessler, 2017; Chuvilin et al., 2018). In methane seepage areas, upward-diffusing methane is primarily consumed by sulfates through a consortium of sulfate-reducing bacteria and anaerobic methanotrophic archaea in shallow surface sediments (Boetius et al., 2000). The biogeochemical process is known as sulfate-driven anaerobic oxidation of methane (SD-AOM), through which methane is oxidized into bicarbonate and HS− is released (Equation 1; Hinrichs et al., 1999; Boetius et al., 2000; Orphan et al., 2001; McGlynn et al., 2015; Wegener et al., 2015). HS− subsequently reacts with active iron to form stable authigenic pyrites. Therefore, pyrites serve as an important indicator of the presence of sulfate–methane transition zones (SMTZs) where intense SD-AOM activities occur. However, the formation of pyrite may also be the result of organic sulfate reduction (OSR) that occurs during early sedimentation diagenesis, which uses organic matter and seawater sulfate as substrates and also produces hydrogen sulfide (H2S) (Equation 2; Berner, 1981; Gieskes, 1981; Jørgensen, 1982; Canfield, 1991; Mazumdar et al., 2012).
Many indicators have been used to investigate the location of SMTZs, and the characteristic sulfur isotope of pyrite in marine sediments may be a critical proxy (Peckmann and Thiel, 2004; Borowski et al., 2013). Borowski et al. (2013) gathered geochemical data from modern and Cenozoic sediments to support that the localized enrichment of 34S within sulfide minerals is closely associated with the function of SD-AOM. Previous studies have focused on the distinctively positive δ34S value of pyrites to distinguish the location of SMTZs and assess the influence of SD-AOM on the formation of authigenic pyrites (e.g., Lin et al., 2016; Liu et al., 2022; Wang et al., 2022; Spruzen et al., 2024). During the formation of pyrite, 32S is preferentially consumed instead of 34S, causing the subsequently formed pyrites to be 34S-depleted and leaving the residual sulfide pool rich in 34S (Rickard, 1975). Thus, pyrites derived from OSR, which dominates early pyritization, are 34S-depleted, whereas SD-AOM-derived pyrites are relatively 34S-enriched (Jørgensen, 1982; Jørgensen et al., 2004). However, OSR can cause sulfur isotopic fractionation up to 70‰ (Canfield et al., 2010; Sim et al., 2011); meanwhile, in stable SMTZs or near the surface of the seafloor, the rapid recharge of downward-diffusing seawater sulfate prevents the exhaustion of porewater sulfate in the sediment (Gong et al., 2018). Both conditions can lead to extremely negative δ34S values, making it challenging to differentiate between OSR and SD-AOM based solely on the characteristic sulfur isotope in pyrites.
In recent years, researchers have begun investigating pyrite in situ trace metal enrichment in modern marine sediments (Lin et al., 2022; Chen et al., 2023). Significant advancements have been made in understanding pyrite-associated trace metal distributions, particularly regarding the influence of methane seepage on their geochemical behavior. A prevailing hypothesis suggests that trace metals from seawater, such as Mo, may be scavenged by iron and manganese oxyhydroxides and then transferred to the surface sediments (Tribovillard et al., 2013). Subsequently, during early diagenesis, dissolved trace metals can be trapped by formed pyrite (Huerta-Diaz and Morse, 1992; Burdige, 2007). Lin et al. (2022) suggested that SD-AOM at SMTZs can enhance the sulfidic dissolution of iron and manganese oxyhydroxides and the release of Mn and associated trace metals into the pore water; thus, the incorporation of these trace metals into pyrite is also enhanced. Also, under highly sulfidic conditions, Ni could more easily replace ferrous in pyrite than Co (Smrzka et al., 2019b, 2020), which may indicate the influence of SD-AOM on the process of pyrite adsorbing trace metals. Pyrite formation in marine sediments is controlled by different geochemical processes (predominantly OSR and SD-AOM), so pyrite formed under these two effects may also lead to distinct trace element compositions.
We conducted a comprehensive analysis of sediment samples collected from the South China Sea (SCS) to investigate pyrite trace element enrichment patterns together with sulfur isotopic compositions. Our findings reveal how sulfate reduction processes affect the trace element distribution and sulfur isotopic signatures in pyrite and how these geochemical characteristics indicate methane seepage, which provides potential implications for the future use of pyrite sulfur isotopes and pyrite trace element characteristics as co-indicators of methane seepage.
2 Geological setting
The SCS is one of the largest marginal seas of the western Pacific Ocean, characterized by a special tectono-sedimentary framework under the influence of the Eurasian, western Pacific, and Indo-Australian Plates (Morley, 2012). Tectonic processes like the collision of crustal blocks and rapid uplift of plateaus or highlands have controlled the sediment provenance of the SCS; meanwhile, the East Asian monsoon has controlled erosion rates (Yan et al., 2024). The thick sediment sequences ranging from 1,000 to 7,000 m with rich organic matter and numerous fault-controlled terraces and basins make the SCS a favorable site for gas hydrate formation and accumulation (Wang et al., 2003; Wu et al., 2013). Several gas hydrate-bearing basins have been identified and investigated through many drilling expeditions in the northern SCS, including the southwest Taiwan Basin, the Pearl River Mouth Basin, and the Qiongdongnan Basin.
The research area is located in the Shenhu area of the Pearl River Mouth Basin. Eocene lacustrine mudstones were considered to be the primary petroleum source rocks in this area, while widespread diapirs and small-scale faults serve as migration pathways for gas/fluids (Su et al., 2016). Moreover, the observed well-distributed bottom-simulating reflectors (BSRs) and the discovered abundant seepage carbonates in this region were also compelling evidence of the existence of gas hydrates (Han et al., 2008; Zhang et al., 2015). The first offshore natural gas hydrate production test was successfully conducted in the Shenhu area in 2017 (Li et al., 2018), and the second production test was conducted from October 2019 to April 2020 (Ye et al., 2020). The success of two production tests and the massive gas hydrate recovered during the drilling expedition confirmed that the Shenhu area has potentially rich gas resources.
3 Materials and methods
The samples were retrieved from the seafloor core Z22-3 in the Shenhu area of northern South China Sea by Qingdao Institute of Marine Geology in 2022 (Figure 1). The water depth was 1,650 m, and the core length was 14.3 m. After collecting the samples, the pore water was squeezed from the core, and then the concentration of pore water ions was measured. Subsequently, the samples were sliced into sections at intervals of 2 cm, freeze-dried, and refrigerated at −4 °C for further processes. The samples were ground into powder before bulk sediment analysis and mineral picking.
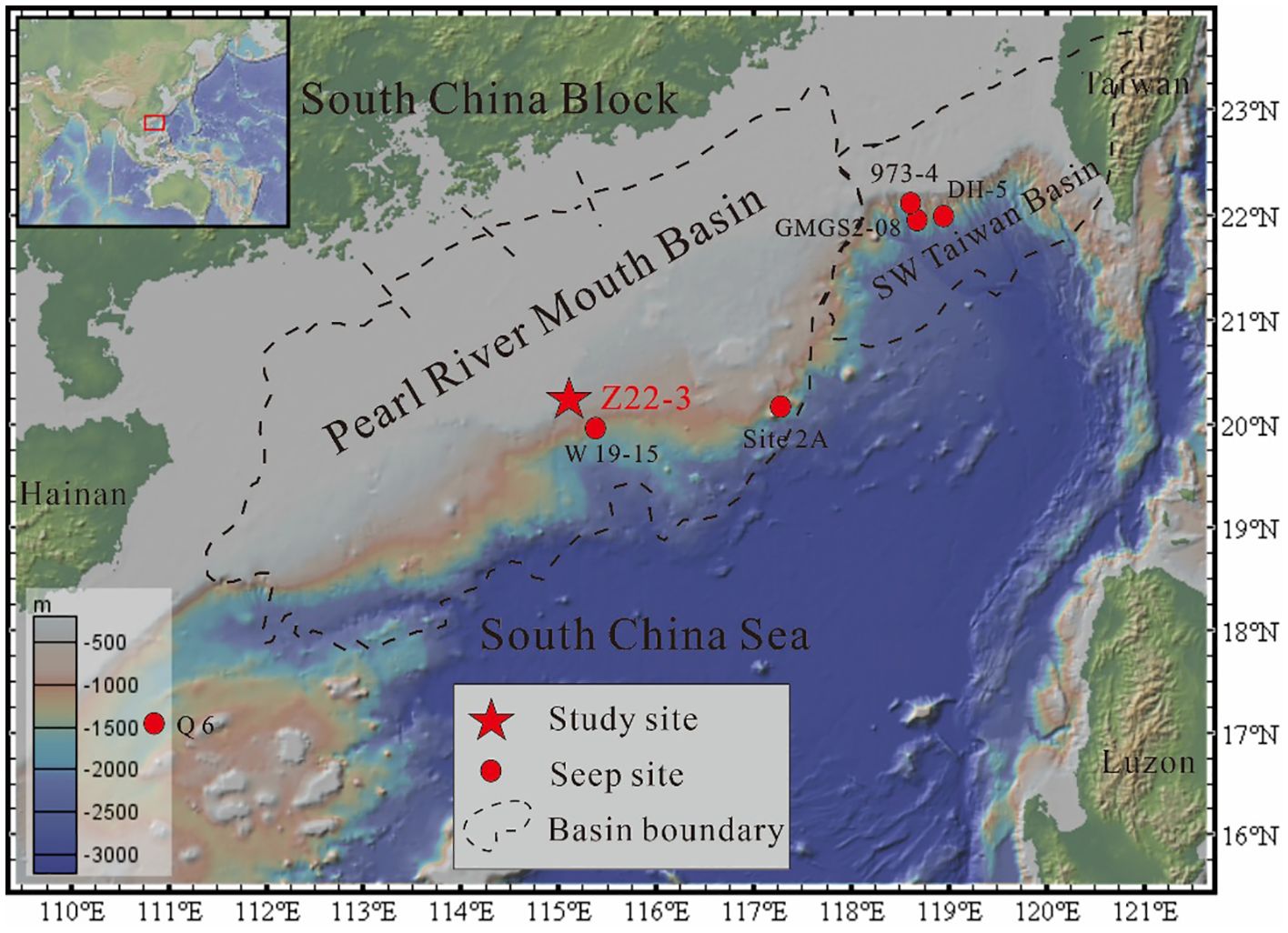
Figure 1. Bathymetric map of core Z22–3 made using GeoMapApp. The basin boundaries were modified according to Zhang B. et al. (2018). Seepage sites were Q6 (Miao et al., 2021a), W19-15 (Gong et al., 2022), Site 2A (Li et al., 2016), GMGS2-08 (Chen et al., 2016), 973-4 (Liu et al., 2020), and DH-5 (Li et al., 2016).
The major elements of sediments were analyzed using X-ray fluorescence spectroscopy (XRF; Thermo ARL ADVANT’X IntelliPower™ 2000, Waltham, MA, USA) at the Analytical and Testing Center, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences. The spectral data were converted into elemental and oxide contents using the UniQuant semi-quantitative analysis software. For trace element measurement of whole-rock sediments, an inductively coupled plasma mass spectrometer (ICP-MS; Agilent 7700e, Santa Clara, CA, USA) was used to analyze the content at Wuhan SampleSolution Analytical Technology Co., Ltd., Wuhan, China. The total organic carbon (TOC) and total inorganic carbon (TIC) contents of sediments, together with their corresponding carbon isotopes, were analyzed at Wuhan SampleSolution Analytical Technology Co., Ltd., Wuhan, China.
Minerals like pyrite, together with their oxidation products, were handpicked from the sediment samples, similar to the chromium reducible sulfur (CRS) procedure. Then, cold field emission scanning electron microscopy (SEM) was used to observe mineral morphology and clarify the species. Minerals were first fixed at the sample stage by conductive glue and then gilded. Further, the sample stage was put into the sample holder and then carefully observed under SEM. The experiment mentioned above was conducted at the Analytical and Testing Center, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences.
The in situ trace element analysis of pyrites was conducted by laser ablation–inductively coupled plasma–mass spectrometry (LA-ICP-MS) at the Wuhan SampleSolution Analytical Technology Co., Ltd., Wuhan, China. The COMPexPro 102 ArF excimer laser (wavelength of 193 nm and maximum energy of 200 mJ) with an Agilent 7900 ICP-MS was used. In this study, the laser energy was 80 mJ, the spot size was 32 µm, and the frequency of the laser was 5 Hz. Laser sampling was performed with helium as carrier gas and argon as make-up gas to adjust the sensitivity. Before entering the ICP, these two gases were mixed together through a T-connector. Data acquisition time for each spot was 50 s and 20–30 s for gas blank analysis. Trace element calibration was conducted under the reference of standard samples including NIST 610, BHVO-2G, BIR-1G, BCR-2G, and GSE-1G. The standard samples are all international standard substances, and the recommended values are replicated from GeoRem (http://georem.mpch-mainz.gwdg.de/). An Excel-based software, ICPMSDataCal, was used to perform off-line selection and integration of background and analyzed signals, time-drift correction, and quantitative calibration for trace element analysis (Liu et al., 2008). Epoxy discs of pyrites were made for the analysis of LA-ICP-MS. Backscattered electron image of pyrites fixed in the epoxy discs is shown in Figure 2. In the backscattered electron (BSE) image, the light-color spherical aggregates are framboids, and the grey substance surrounding them is cementing material. Light-color points without cracks and with a radius larger than the laser beam spot were selected for LA-ICP-MS analysis.
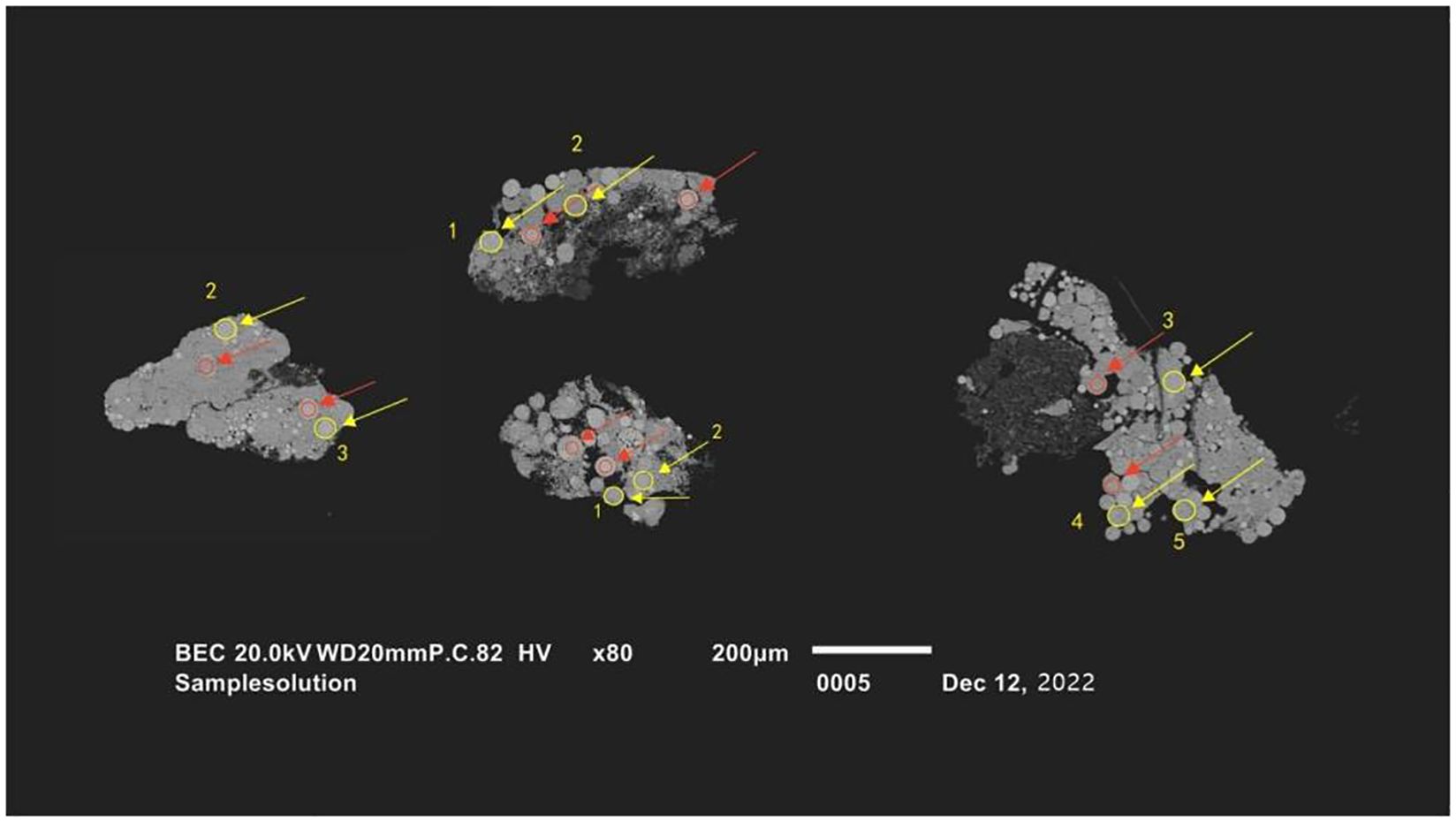
Figure 2. Backscattered electron (BSE) image of pyrite. The yellow ellipses indicate samples analyzed by laser ablation–inductively coupled plasma–mass spectrometry (LA-ICP-MS).
4 Results
4.1 Bulk CS contents and MoEF–UEF
The TOC content of bulk sediments ranged from 0.63% to 1.35%, with the highest value from 181 to 481 cmbsf and an average of 1.2%. TIC exhibited a totally contrary trend, which increased from 181 to 1,101 cmbsf and then gradually decreased below 1,101. The δ13C of TOC exhibited detectably high values of 181 to 481 when the δ13C of TIC ranged from −0.1 to 1.0, and most of the values were positive. δ13CTIC decreased from the top sediment before reaching a peak at 201 cmbsf, and within 181 to 481 and 1,101 to 1,401 cmbsf, the inorganic carbon isotopes exhibited an obviously low value.
CRS showed an abnormally high value within 181 to 481 and 1,101 to 1,401 cmbsf, which indicates large amounts of pyrite formation. The δ34S value of CRS was negative throughout the core, indicating a strongly depleted sulfur isotopic composition. Corresponding to the CRS content, the δ34SCRS values were discernibly negative but exhibited a slightly increasing trend from 181 to 481 cmbsf. The S/C ratio was calculated based on the ratio of CRS/TOC. The S/C ratio showed a familiar trend as CRS, and it remained at a high value in the depths of potential SMTZs. MoEF and UEF were relatively high in 181 to 481 and decreased within 1,101 to 1,401 cmbsf.
4.2 In situ trace element patterns of pyrite
The distribution of trace elements in various types of pyrite was analyzed using LA-ICP-MS. To minimize the potential mixture of different paragenetic phases during LA-ICP-MS analysis, only large pyrite aggregates (>20 μm) were selected for in situ analysis. However, it should be noted that the influence of later-stage phases adhering to framboidal pyrite could not be excluded to some extent. The contents of different trace elements in sediments varied greatly, even by an order of magnitude. The trace element contents of pyrite from core Z22–3 exhibited no consistent trends within individual pyrite aggregates. In comparison with trace element concentrations in sediments, the variations in trace element content within pyrite were significantly more pronounced, displaying distinct enrichment or depletion. Based on this observation, trace elements can be divided into two groups (Group 1: Mn, Zn, Cu, and V; Group 2: Pb, Co, Ni, and Mo).
The contents of the group 1 elements Mn, Zn, Cu, and V in pyrite exhibited obvious depletion compared with sediments (Figure 3). At the Z22–3 study sites, the measured concentrations of Mn (10.56 to 421.36 ppm for pyrite; 441.13 to 1405.56 ppm for sediments), Zn (8.25 to 69.76 ppm for pyrite; 92.1 to 112 ppm for sediments), Cu (0.62 to 58.75 ppm for pyrite; 34.37 to 43.39 ppm for sediments), and V (4.87 to 136.49 ppm for pyrite; 94.72 to 129.08 ppm for sediments) in pyrite were markedly lower than those in corresponding sediments. Notably, Mn displayed the most pronounced depletion, with an average concentration of 53.9 ppm in pyrite, representing only 8.8% of its average sediment concentration (615.67 ppm).
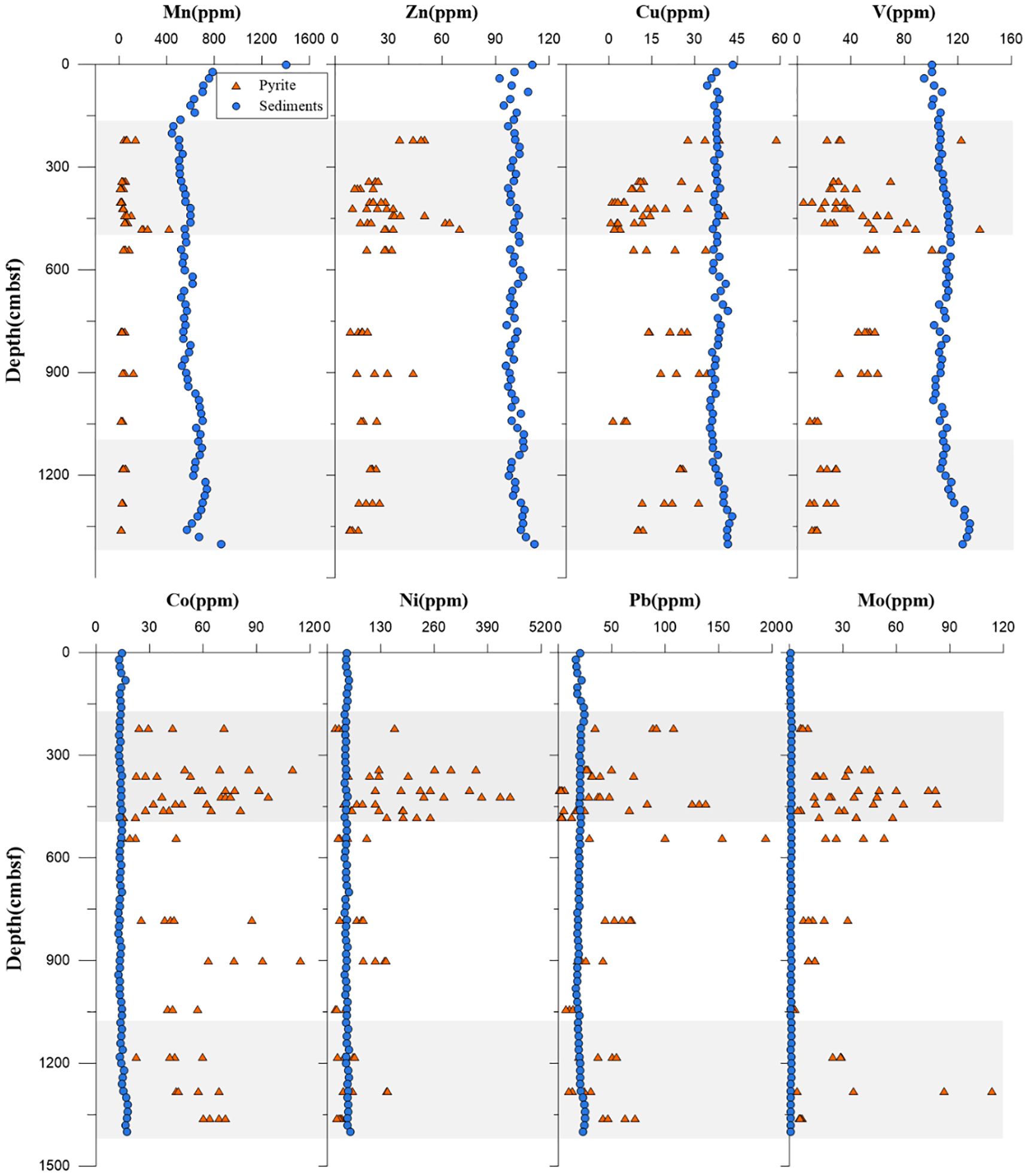
Figure 3. Contents of trace elements (Mn, Zn, Cu, V, Co, Ni, Pb, and Mo) in selected pyrite and bulk sediments. The shaded zone indicates the position of potential SMTZs. SMTZs, sulfate–methane transition zones.
On the contrary, the contents of the group 2 elements Co, Ni, Pb, and Mo in pyrite demonstrated significant enrichments relative to sediment concentrations (Figure 3). The contents of Co (13.87 to 114.51 ppm for pyrite; 12.5 to 17.9 ppm for sediments), Ni (19.05 to 445.5 ppm for pyrite; 41.9 to 55.7 ppm for sediments), Pb (1.38 to 193.68 ppm for pyrite; 16.7 to 25.4 ppm for sediments), and Mo (1.94 to 113.72 ppm for pyrite; 0.26 to 1.3 ppm for sediments) in sediment were significantly higher than those in sediments. Evidently, Ni, Pb, and Mo were significantly enriched in pyrites at the depth of the SD-AOM-affected zone, from which Mo was enriched by approximately 33 times.
4.3 Mg/Ca-Sr/Ca
The geochemical profiles of CaO, Sr, MgO, Mg/Ca, and Sr/Ca ratios are presented in Figure 4. The content ranges of CaO, Sr, and MgO were 5.0% to 14.0% (9.0% on average), 242 to 602 ppm (397 ppm on average), and 1.4% to 1.9% (1.5% on average), respectively. CaO exhibited decreasing tendencies above 181 cmbsf (11.4% on average) and below 1,101 cmbsf (8.1% on average), and it exhibited an increasing trend but remained at a low value at 181–1,101 cm (8.8% on average). The Mg/Ca and Sr/Ca ratios exhibited broadly similar trends. Above 181 cmbsf and from 1,101 cmbsf to the core bottom, these ratios displayed distinct increases, with two pronounced peaks observed within the lower SMTZs.
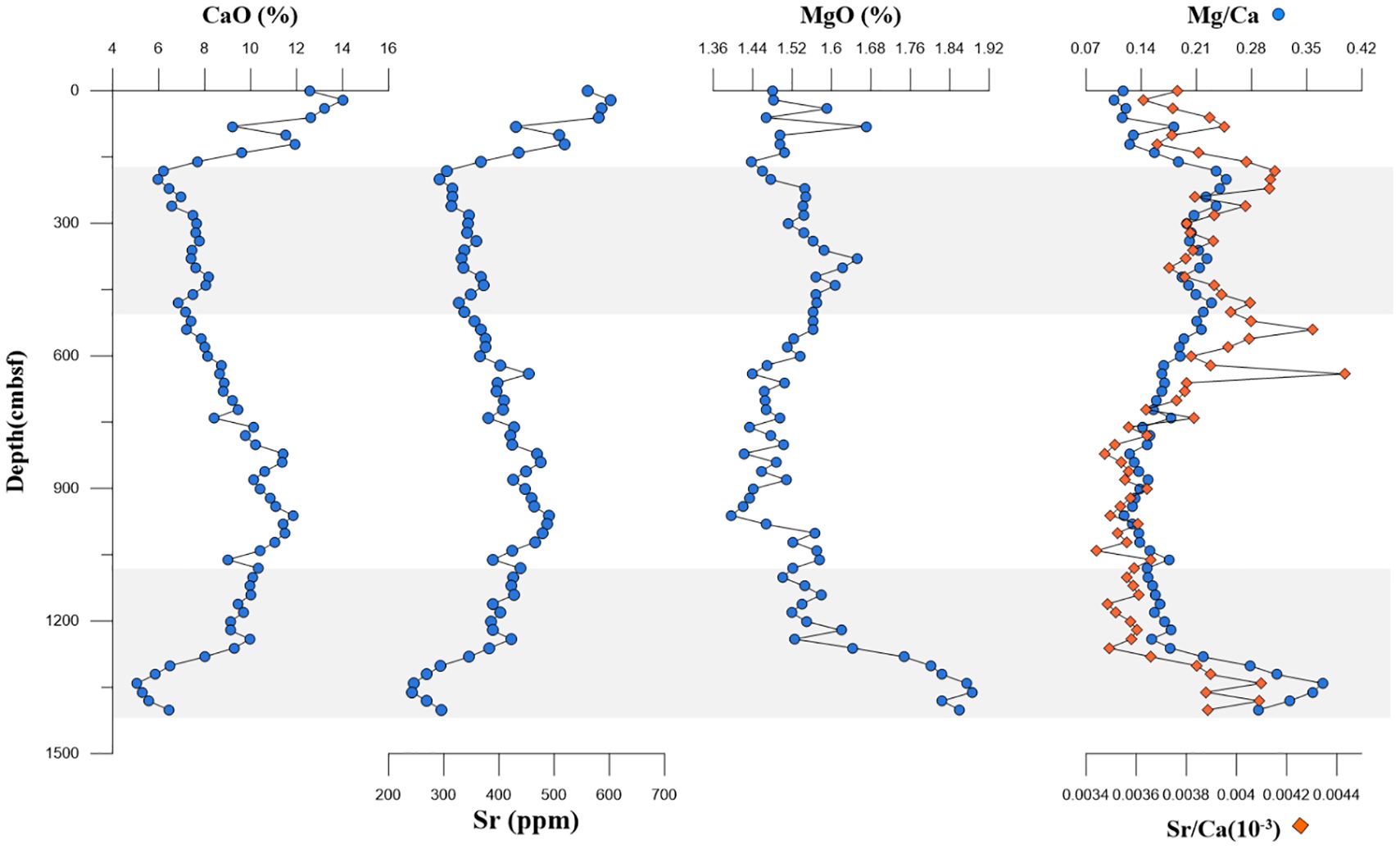
Figure 4. Profile of indicators of carbonate minerals.
4.4 Enrichment factor of selected trace elements
The enrichment factor of each trace element was calculated to determine whether the element was enriched in sediments compared to the detrital background (authigenic enrichment). Enrichment factors (EFs) were calculated according to the formula XEF = [(X/Al)sample/(X/Al)PAAS], where X and Al represent the weight% concentrations of element X and aluminum in the sample and the standard, respectively. EFs are highly sensitive to the selection of the reference element, so Al was used here due to its conservative behavior and little variability (Smrzka et al., 2020). Samples were normalized using the Post-Archean Australian Shale (PAAS) composition summarized by McLennan (2001).
It was defined that if the value of EF is greater than 1, then the element is considered to be enriched in the sample in comparison with the standard. Algeo and Tribovillard (2009) expounded the definition that EF greater than 3 is detectable enrichment and greater than 10 is substantial enrichment. Most of the trace metals discussed here showed enrichment (2.4 to 3.3 for ZnEF; 1.6 to 2.2 for CuEF; 1.5 to 1.8 for VEF; 1.2 to 1.6 for CoEF; 1.7 to 2.3 for NiEF; 2.0 to 2.6 for PbEF; 0.6 to 3.0 for MoEF; 1.6 to 3.3 for UEF); among them, Pb, Zn, and U showed nearly detectable enrichment (2.3 on average for PbEF; 2.7 on average for ZnEF; 2.9 on average for UEF). On the contrary, the EFs of Mn ranged from 0.7 to 1.4, with most of the values less than 1, which showed no enrichment. MnEF, ZnEF, CuEF, VEF, CoEF, and NiEF all showed decreasing trends at the upper 181 cm, while MnEF, ZnEF, and CuEF remained almost stable below 181 cm; the values of VEF, CoEF, and NiEF gradually increased downcore. Furthermore, the values of PbEF and MoEF first decreased and then increased at the upper 181 cm, while UEF showed an increasing trend.
4.5 The morphologies of pyrites
The various morphologies of pyrite that were handpicked throughout this core are displayed in Figure 5. At Z22-3, both the shapes and particle sizes of the pyrite varied greatly (Figures 5A–C). Similar to other methane seepage study sites in the northern South China Sea (Xie et al., 2012; Chang et al., 2022), throughout the core, the octahedral form is the dominant type of pyrite (Figure 5E). Framboids formed by the intermediate between an optimized truncated cubic shape and an octahedron are also found in several depths (Figures 5D, G). Figures 5F, G show the transitions of pyrites from an octahedron to a cubic shape. Furthermore, in some specific depths, subhedral elemental sulfur with a smooth surface under the SEM was observed (Figures 5H, I).

Figure 5. Image of pyrites under the stereoscope and cold field emission scanning electron microscope (SEM). (A–C) Handpicked pyrites under the stereoscope. (D) Framboidal pyrites. (E) Octahedral pyrites. (F, G) Octahedral-cubic pyrites. (H, I) Subhedral elemental sulfur.
5 Discussion
5.1 Geochemical evidence for the position of SMTZs
5.1.1 CRS and sulfur isotope of authigenic pyrites
CRS has been widely used as an indicator of authigenic pyrite content since the method was introduced and continuously advanced (Canfield et al., 1986). In this study, from 181 to 481 cmbsf and below 1,101 cmbsf, the contents of CRS were significantly higher throughout the core, and the δ34S values of the CRS showed a slightly increasing trend (Figure 6). The content of CRS at Z22–3 varied greatly but ranged from 0.2 to 0.5 wt% within SMTZs, which displayed an abnormally high value. The CRS contents in SMTZs of other nearby seepage sites were 1.01–2.60 wt% (Site 2A; Lin et al., 2016), 1.04–1.65 wt% (W19-15; Gong et al., 2022), and 0.38–1.57 wt% (973-4; Liu et al., 2020). The CRS content of our study site was slightly lower than the contents of the aforementioned sites, but still within a reasonable range. Consequently, we suggest that SD-AOM was the main factor causing the increase in CRS and the sulfur isotopic composition of pyrite (Figure 6). However, we also found from our study that the value of δ34SCRS in SMTZs only showed inconspicuous positive bias with an existing extremely negative value ranging from −47.69‰ to −41.0‰. Such a negative value of δ34SCRS can also be found in other gas hydrate-bearing areas like the Gulf of Mexico, Southwestern Taiwan Basin, Qiongdongnan Basin, East Coast of India, and Pearl River Basin (Peketi et al., 2012; Formolo and Lyons, 2013; Li et al., 2016; Lin et al., 2016; Zhang M. et al., 2018; Miao et al., 2021b), which indicates that when using the sulfur isotope of authigenic pyrites as an indicator of SMTZs, more specific constraints are needed.

Figure 6. Depth profiles of bulk sedimentary TOC, TIC with corresponding carbon isotopes (δ13C), chromium reducible sulfur (CRS) contents with corresponding sulfur isotopes (δ34S), MoEF, and UEF. The shaded zone represents potential SMTZs. The data of TOC, δ13CTOC, CRS, δ34SCRS, CRS, and S/C were derived from Zhang et al. (2025). TOC, total organic carbon; TIC, total inorganic carbon; SMTZs, sulfate–methane transition zones.
According to previous studies, due to strong SD-AOM, the values of δ34SCRS can gradually converge to the seawater values or even higher values. However, the extremely negative value of δ34S can be attributed to environmental and depositional situations. 1) “Open” condition: Under the “open” sedimentary condition, the exchange between pore fluids and seawater sulfate is more frequent, so the 32S consumed can be sufficiently supplemented, and the residual 34S-enriched sulfate pool is not effectively utilized (Fike et al., 2015; Peketi et al., 2015). Low sedimentary rate can contribute to the “open” condition by promoting the communication between pore water and underlying seawater, thus enlarging the sulfur isotopic fractionation (Liu et al., 2019, 2021). 2) Limited iron supply: Reactive iron supply is also important to control the sulfur isotope of pyrites. If iron is available at depths in sediments, it can react with the 34S-enriched sulfide and form high δ34S in pyrite (Spruzen et al., 2024). However, if the reactive iron is limited, it can only react with sulfide near the surface, which is not yet rich in 34S, thus forming negative δ34Spy (Formolo and Lyons, 2013). 3) Disproportionation of sulfur intermediates: According to mass balance studies (Markovic et al., 2016; Yao et al., 2020), much of the sulfide produced by microbial sulfate reduction is re-oxidized, resulting in the disproportionation of sulfur intermediates. Canfield and Thamdrup (1994) suggested that the repeated sulfur reduction and disproportionation can increase the sulfur isotopic fractionation between dissolved sulfate and sulfide. The value of sulfur isotope above 181 cmbsf in Z22–3 ranged from −47.7‰ to −18.9‰, much lower than the value of seawater sulfate (≈21‰; Rees et al., 1978). Here, we consider that the open environment and the disproportionation of sulfur intermediates contribute most to the low value of sulfur isotopes in pyrites.
5.1.2 Carbonate minerals indicate methane seepage
The existence of authigenic carbonate minerals is an important piece of evidence for methane seepage (Bayon et al., 2011; Feng et al., 2014; Bayon et al., 2015). The enhanced AOM can increase the alkalinity in pore water to facilitate the precipitation of authigenic carbonate minerals such as calcite, dolomite, and aragonite (Baker and Burns, 1985; Raiswell, 1988). They exist in the form of carbonate rocks on the sea floor or dispersed carbonate minerals in the sediment column (Han et al., 2014; Yang et al., 2014); however, it is hard to detect massive carbonate rocks because they only exist when the seepage intensity is moderate. Bayon et al. (2007) showed that Sr/Ca and Mg/Ca ratios can be used for identifying Sr-rich (aragonite) and Mg-rich (high-Mg calcite and dolomite) carbonate layers in cold seepage sediments. From 181 to 481 cmbsf and from 1,101 cmbsf to the bottom of the core, the marked increase in Sr/Ca and Mg/Ca may indicate the formation of authigenic carbonate minerals like aragonite and calcite. The simple mixing model of four end-members (aragonite, high-Mg calcite, biogenic calcite, and detrital fraction) can be used to identify the proportion of carbonate mineral components. The four-component end-member model was used to calculate the carbonate mineral composition of Z22–3 in this paper, and the samples were plotted in the model using the content of Sr/Ca-Mg/Ca. It was found to fall within the range defined by the model and was distributed on both sides of the high-Mg calcite–aragonite mixing curve. This indicated that the authigenic carbonate minerals in the sediments mainly consisted of aragonite and high-Mg calcite, which was very similar to gas hydrate-bearing sites such as the northern South China Sea (Chen et al., 2016) and Qiongdongnan Basin (Miao et al., 2021a) (Figure 7). Therefore, it was believed that the abnormal Mg/Ca-Sr/Ca in the 181–481-cm and 1,101–1,401-cm layers was related to the carbonate minerals produced by methane seepage. However, different from the value of Mg/Ca-Sr/Ca, the CaO value within the two identified layers showed a lower value in the sediment core. This may be attributed to the low level of authigenic carbonate minerals because of the alteration caused by extra H2S. The hypothesis still needs sufficient mineralogical evidence to be validated.
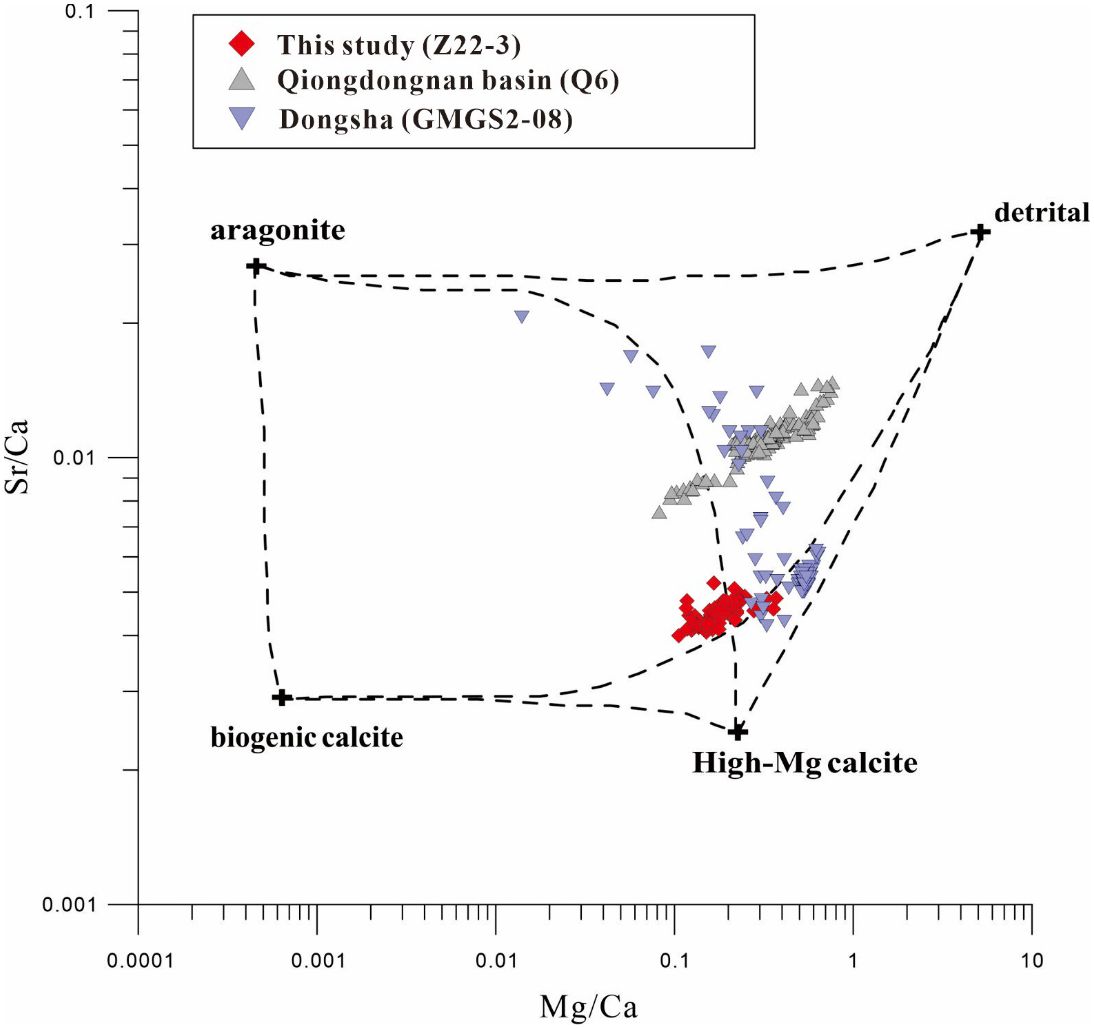
Figure 7. Sr/Ca ratios vs. Ma/Ca ratios for the sediment samples. Published data from Dongsha (Chen et al., 2016) and Qiongdongnan Basin (Miao et al., 2021a) were included for comparison. The end-member ratios of aragonite, high-Mg calcite, biogenic calcite, and the detrital fraction from Bayon et al. (2007) and Yang et al. (2014) were used.
5.1.3 Mo–U covariation reveals redox conditions
AOM caused by gas hydrate dissociation can alter the redox conditions of sediments and underlying seawater, leading to the enrichment of elements like Mo, Cu, and U (Tribovillard et al., 2006; Scott and Lyons, 2012; Smrzka et al., 2019a). These redox-sensitive elements, especially Mo and U, are commonly used to record the paleo-sedimentary environment and seawater chemistry, owing to their stability under oxidizing conditions and their susceptibility to being reduced and transferred to sediments in anoxic water. In oxic seawater, Mo and U primarily exist as MoO42− and U6+ adsorbed onto Mn oxyhydroxides. In anoxic water, MoO42− and U6+ are converted into thiomolybdates (MoS(4−x)Ox2−) and U4+, respectively, when the concentration of H2S reaches a critical level of approximately 10–100 μM (Morford and Emerson, 1999; Tribovillard et al., 2006). At this point, Mo and U are quickly sequestered into organic fraction or precipitated with Fe–Mo sulfide minerals and transferred to sediments (Helz et al., 1996; Tribovillard et al., 2006). As a result, Mo enrichment in marine sediments can occur in two distinct ways: 1) a transient enrichment associated with the recycling of Mn oxyhydroxides at or near the sediment–water interface and 2) a permanent enrichment in the sulfidic zone, where MoS(4−x)Ox2− forms and is sequestered (Scott and Lyons, 2012). Mo was apparently enriched in the 181–481-cmbsf and 1,101–1,401-cmbsf layers to a certain extent (Figure 6). (Mo/U)EF mostly fell on the diagonal line at 0.3 times the present-day seawater, which indicated a suboxic–sulfidic sedimentary environment (Figure 8). Chen et al. (2016) proposed that OSR provided the release of dissolved sulfide and that AOM activated the Mo enrichment in sediments during seepage events. High methane flux induced sulfidic conditions, which allowed the capture of Mo into pyrites through the partition shuttle process (Sato et al., 2012; Bazzaro et al., 2020). These could properly illustrate the enrichment of Mo as well as the depletion of elements like Cu within the 181–481-cmbsf and 1,101–1,401-cmbsf layers (Figures 3, 9).
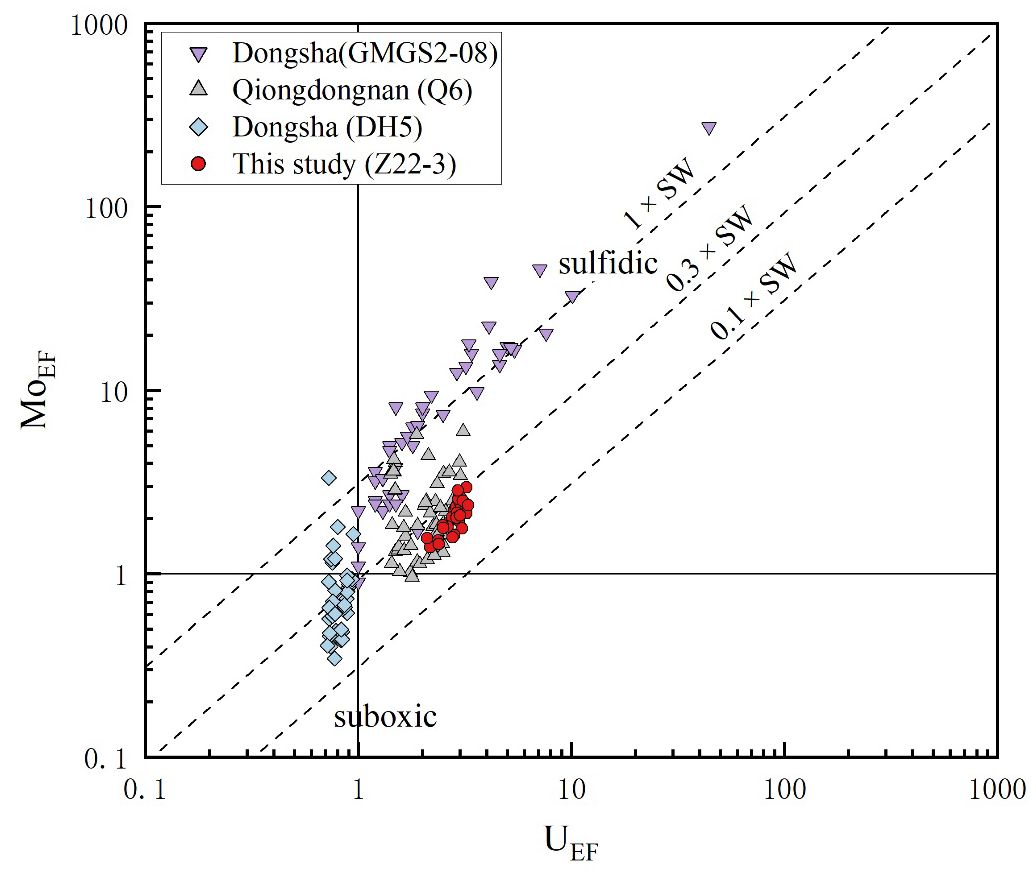
Figure 8. MoEF vs. UEF of the sediment samples. Published data from Dongsha (GMGS2–08) (Chen et al., 2016), Qiongdongnan (Q6) (Miao et al., 2021a), and Dongsha (DH-5) (Li et al., 2016) were included for comparison. The diagonal lines that represent multiples of the Mo/U weight ratio of present-day seawater were taken from Algeo and Tribovillard (2009).
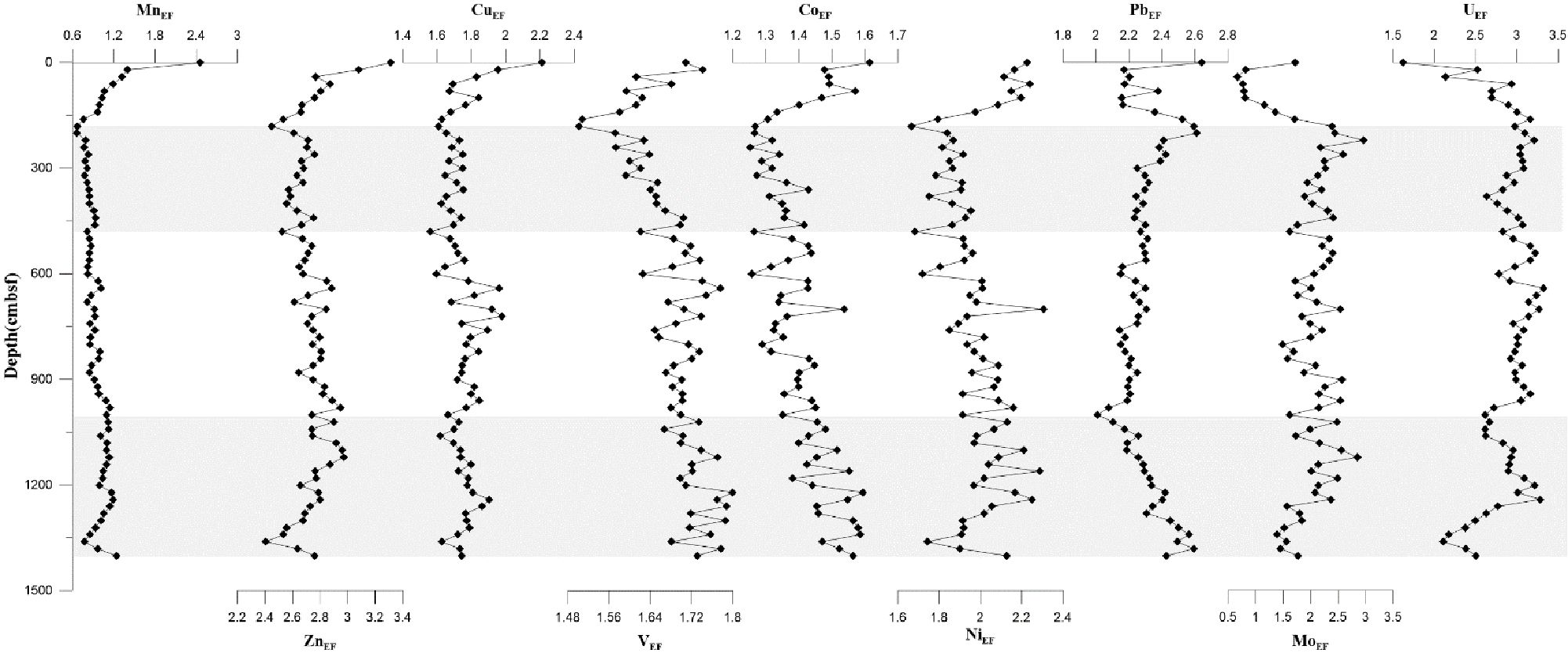
Figure 9. Enrichment factors (EFs) of trace elements (Mn, Zn, Cu, V, Co, Ni, Pb, Mo, and U) in bulk sediments from core Z22-3. The shaded zone indicates the position of potential SMTZs. SMTZs, sulfate–methane transition zones.
5.2 Controls on trace element patterns of authigenic pyrites
5.2.1 Enhancement of Co, Ni, and Mo in pyrite compared to sediments
Trace element distribution of authigenic pyrites reflects the geochemical conditions during the growth of mineral crystals (cf. Rickard, 2012). Compared to their concentrations in seawater, most trace elements in pyrite analyzed herein showed various degrees of enrichment (Figure 3; Table 1). Co, Ni, and Mo exhibited similar patterns for core Z22-3, with higher contents and greater variation of these trace elements in pyrite and lower contents in sediments (Figure 3). Although Co, Ni, and Mo showed great enrichments in pyrites in SMTZs, the enrichment factors of bulk sediments of these elements are not elevated in the same depths. Thus, the element patterns of pyrites may be attributed to early diagenesis rather than the primary trace element contents of the host sediments (Lin et al., 2022).
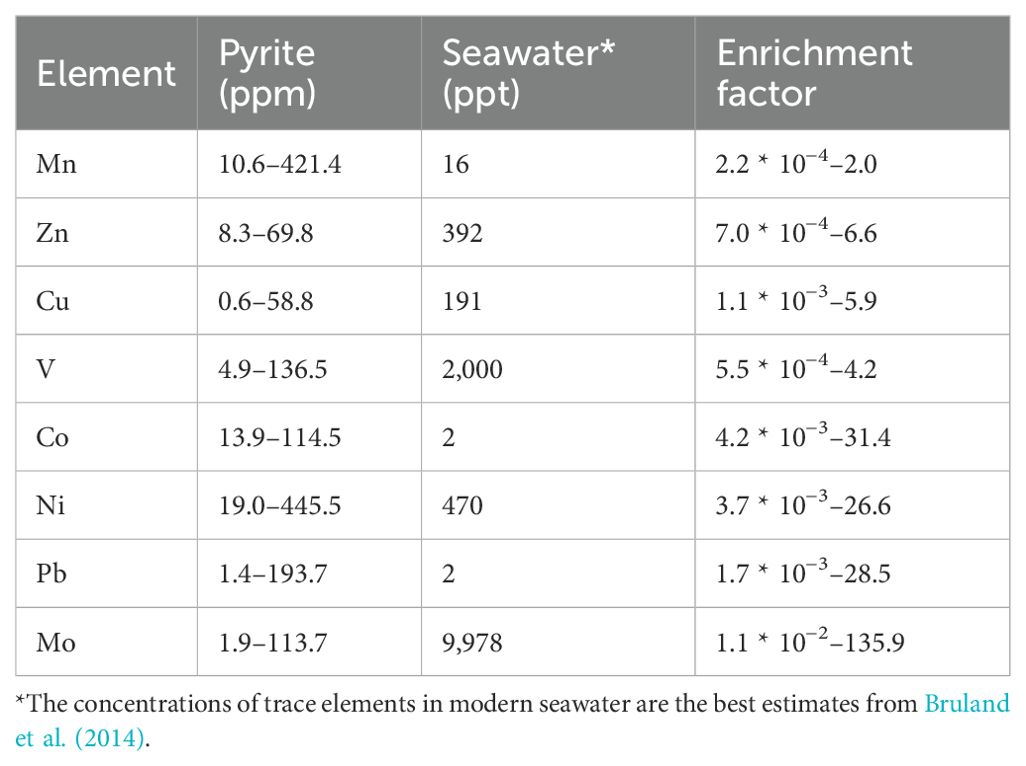
Table 1. Enrichment factors for trace elements in pyrite from core Z22–3 compared to seawater values.
Concerning the mechanism of Mo and the enrichment of other trace metals, two hypotheses have been brought forward: 1) the trace metals were carried to the surface sediments by ascending seepage fluids, resulting in trace metal enrichment in shallow pore fluid and sediments (Nath et al., 2008; Cangemi et al., 2010); 2) the trace metals were predominantly derived from seawater, scavenged by iron and manganese oxyhydroxides in the water column, and then transferred to the surface sediments (Tribovillard et al., 2013). In this study, the latter hypothesis was more consistent with the characteristics of trace metals in sediments at Z22-3, as other trace metals carried by chloride-rich ascending brine (such as Cu, Ni, and Zn) showed greater enrichment than Mo (Figure 3). Tribovillard et al. (2013) proposed that trace elements like Mo, As, and Sb were thought to be actively transferred to sediments through the particle shuttle process by Fe and Mn phases. Then, Mo could be trapped by pyrite formed during early diagenesis, and Mo was reported to have the highest degree of trace metal pyritization (DTMP; Huerta-Diaz and Morse, 1992; Burdige, 2007) among other trace metals, which means that Mo was highly prone to incorporation into pyrite during early diagenesis. In the OSR-affected zone (0 to 181 cmbsf), the contents of Mn in the sediments significantly decreased with increasing depth, while the contents of Mo increased; in the 481–1,101-cmbsf layers, the Mn contents of the sediments at the study site were low and constant, and Mo revealed much higher and more variable contents. In the SD-AOM-affected zone, the enrichment factors of Mo in sediments slightly decreased as the Mo contents in pyrite apparently increased. With the higher H2S concentrations occurring in seawater (H2S ≥ 0.011 mM), the chemical reaction of Mo thiolation is given as follows (Equation 3) (Erickson and Helz, 2000):
Therefore, by combining these results with those of previous studies, we suggest that Mo was most probably adsorbed and carried by iron and manganese oxyhydroxides. Then, in the OSR-affected zone, Mo was dissolved and turned into MoS42−, entering pyrite during early diagenesis under the effect of SD-AOM.
Trace elements such as Ni, Co, As, and Se are commonly lattice-bound within pyrite (Vaughan and Rosso, 2006). Co and Ni commonly substitute for ferrous in pyrite due to their similar ionic radii and charge (Huerta-Diaz et al., 1998; Tribovillard et al., 2006). The ratio between Co and Ni (Co/Ni) in pyrite has been effectively used to investigate the evolution of geochemical conditions during pyrite formation (e.g., Dill and Kemper, 1990; Maslennikov et al., 2009). The Co and Ni contents of the two discussed zones in our study area are plotted in Figure 10, and the slope of the linear trends represents the ratio between Co and Ni (Co/Ni). The OSR-derived pyrite (with a slope of up to 0.44) revealed a higher Co/Ni ratio than the SD-AOM-derived pyrite (small slope of 0.17), but both were higher than the ratio of seawater (ca. 0.004; Bruland et al., 2014). The elevated Co/Ni ratio of pyrite from the OSR-affected zone of the study site indicated stronger enrichment of Co compared to Ni in pyrite at this depth. Ni could be more easily incorporated into pyrite than Co under highly sulfidic conditions (Smrzka et al., 2019a, 2020), in accord with enhanced SD-AOM as a cause for the decreased Co/Ni ratio for the AOM-affected zone. With respect to the Ni and Co contents, both revealed detectable enrichment in SD-AOM-derived pyrites compared with OSR-derived pyrites. A convincing hypothesis indicates that Ni and Co are dispensable metals of enzymes required in the reverse methanogenesis pathway (Hallam et al., 2003; Scheller et al., 2010; Glass and Orphan, 2012); thus, Ni and Co may be taken up through reverse methanogenesis and then accumulate in pyrites.
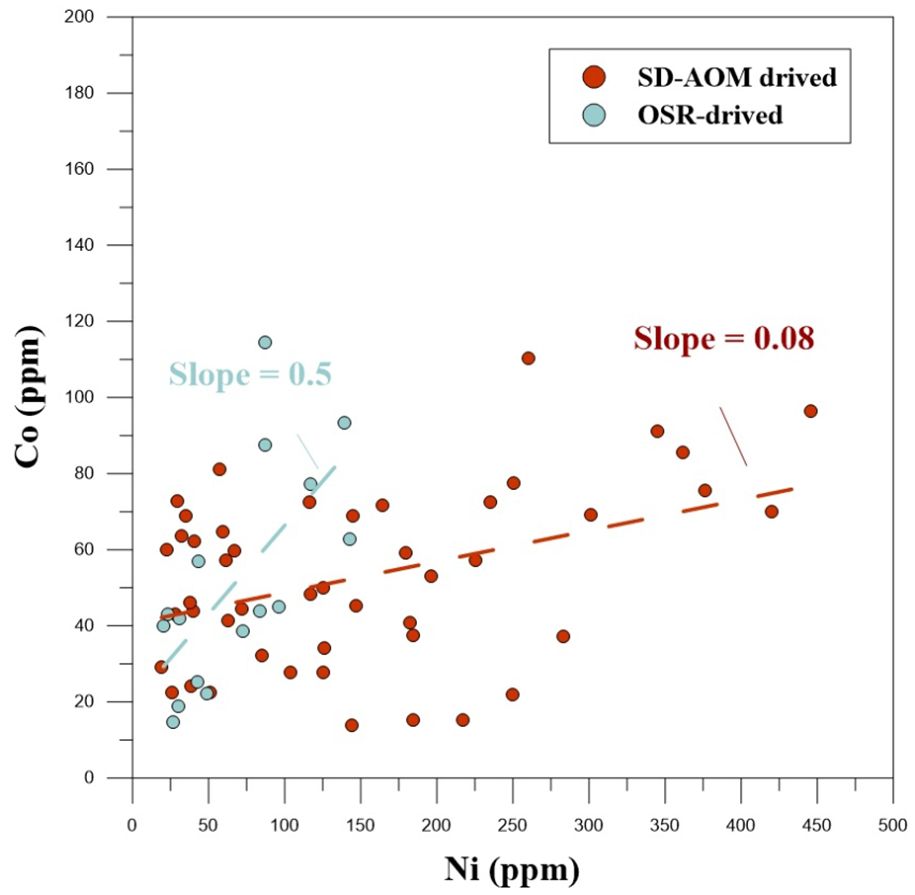
Figure 10. Ni and Co contents in authigenic pyrites. The diagonal lines represent Ni vs. Co.
5.2.2 Decrease of Mn, Zn, Cu, and V and enhancement of Pb in pyrite
In oxic marine environments, Mn typically coexists with metastable Fe–Mn oxyhydroxides (Bayon et al., 2004; Smrzka et al., 2019a, 2020). Due to the strong sorption capacity, Fe–Mn oxyhydroxides can adsorb most ions in the water column or the pore water, such as Mn, Pb, Zn, Cu, and V (Bayon et al., 2004; Lin et al., 2022; Miao et al., 2022). However, the adsorbed trace elements are released into pore water when iron and manganese oxyhydroxides are reductively dissolved by hydrogen sulfide in the sediment (Equations 4, 5) (Berner, 1981; Burdige and Nealson, 1986; Aller and Rude, 1988; Canfield and Thamdrup, 2009):
In this study, we used CRS and sulfur isotopic composition to constrain the depths of SMTZs, and we used (Mo/U)EF to indicate the sulfide conditions, which favor the reductive dissolution of iron and manganese oxyhydroxides and the release of trace elements. No enrichment of elements Mn, Pb, Zn, Cu, and V was observed for bulk sediments (Figure 9), and reduced contents of Mn, Zn, Cu, and V in pyrite from both the OSR- and SD-AOM-affected zones were identified (Figure 3). Some studies have suggested that the water exchange reaction kinetics of metals influence mineral precipitation (Huerta-Diaz and Morse, 1992; Morse and Luther, 1999). Trace elements, such as Cu, Pb, and Zn, which have faster water exchange kinetics rates than Fe, tend to precipitate as independent sulfide rather than incorporate into pyrite. It is likely that at Z22-3, Mn, Cu, and Zn were released and accumulated when SD-AOM rates were high and then precipitated as independent metal sulfides. It is suggested that the weakening of the OSR can limit the release of Zn and Cu in the methane seepage environment, which also contributes to the depletion of pyrites (Yu et al., 2025). However, the enhancement of Pb content in pyrite may indicate that the metal sulfide of Pb has further incorporated into pyrite. V is usually not present in sulfide minerals such as pyrite, and dissolved V in pore water is reduced and precipitates in the insoluble V phase in the presence of hydrogen sulfide (Emerson and Huested, 1991; Wanty and Goldhaber, 1992). In the OSR-affected zone (481 to 1,101 cmbsf), the contents of Mn, Zn, Cu, and V in the pyrites were also very depleted (Figure 3). This is because OSR also leads to a substantial solution of iron and manganese oxyhydroxides, making it difficult to use these depleted elements as proxies to identify SD-AOM and OSR.
6 Conclusion
We analyzed bulk sediment major and trace elements, CRS contents and the associated sulfur isotopes, TOC with the associated carbon isotopes, and in situ trace elements of pyrites to investigate the sediment samples at core Z22–3 from the northern SCS. The positions of SMTZs were defined from 181 to 481 and from 1,101 to 1,401 cmbsf using comprehensive geochemical and mineralogical characteristics. The trace elements exhibited varying degrees of enrichment and depletion in pyrite, as their distribution patterns differed significantly between SD-AOM and OSR. Compared with the trace metal contents in sediment, the trace elements in pyrite at Z22–3 are divided into two groups: 1) Co, Ni, Mo, and Pb contents were enhanced in pyrite; and 2) Mn, Zn, Cu, and V contents were depleted in pyrite. Our results show how methane seepages modulate the process of adsorption of trace elements by pyrite in a suboxic–sulfide environment (Figure 11): first, water column from deep layers carried the trace metals (Co, Mo, Ni, etc.) to the sediment–water interface (SWI), and then these trace metals were adsorbed by aggregates of iron and manganese oxyhydroxides. Subsequently, iron and manganese oxyhydroxides were reduced in suboxic sediments and released Co, Mo, Ni, Mn, etc., which they carried. With the high methane fluxes, the combination of Ni, Mo, Co, and pyrite was enhanced. Mn, Zn, Cu, and V can precipitate as independent metal sulfides due to high water exchange reaction kinetics rates in both the SD-AOM- and OSR-affected zones.
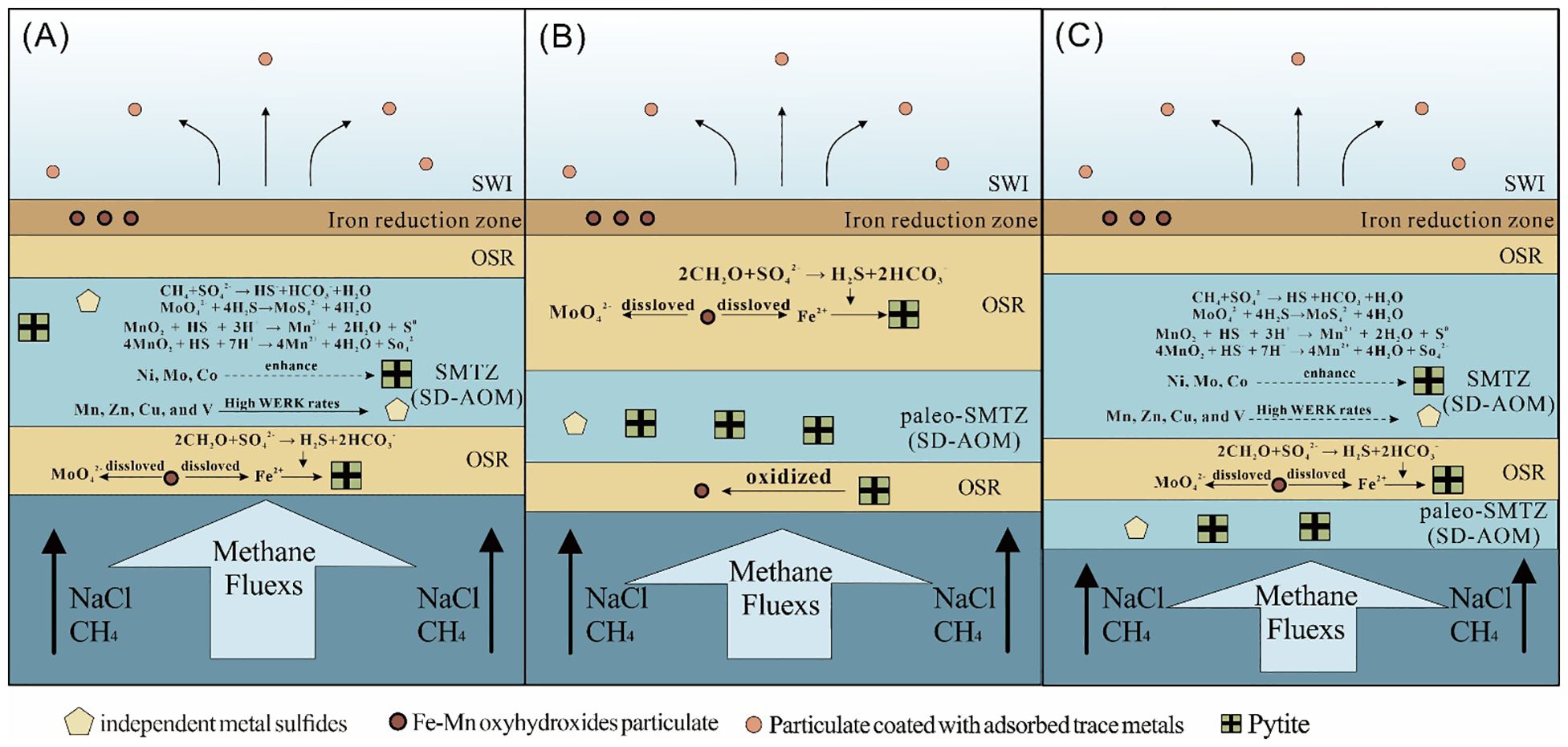
Figure 11. Schematic diagram illustrating trace metals that entered pyrite under methane seepage (based on Hu et al., 2014). (A) Trace elements were carried by water column to sediment–water interface (SWI) and adsorbed by aggregates of iron and manganese oxyhydroxides. In SMTZs, anaerobic oxidation of methane occurred, and manganese oxyhydroxides were reductively dissolved; meanwhile, trace elements were released. Ni, Mo, and Co entered pyrites; Mn, Zn, Cu, and V entered independent metal sulfides. In OSR-affected zone, reductive dissolution of iron and manganese oxyhydroxides also occurred, and pyrites were generated. (B) SD-AOM was no longer active, but pyrites and independent metal sulfides were preserved in SMTZs. In paleo-OSR-affected zone, pyrites were oxidized into oxyhydroxides. At the shallower layers, the current phase of OSR was active. (C) New phase of SD-AOM was active. WERK, water exchange reaction kinetics. SMTZs, sulfate–methane transition zones; SD-AOM, sulfate-driven anaerobic oxidation of methane; OSR, organic sulfate reduction.
Data availability statement
The data that support the findings can be found online in Mendeley Data at https://data.mendeley.com/datasets/bt5h3g26j4/1.
Author contributions
YL: Conceptualization, Investigation, Methodology, Visualization, Writing – original draft, Writing – review & editing. XC: Conceptualization, Investigation, Methodology, Visualization, Writing – original draft. DW: Conceptualization, Funding acquisition, Methodology, Supervision, Writing – review & editing. QZ: Investigation, Methodology, Visualization, Writing – review & editing. SM: Investigation, Supervision, Writing – review & editing. GJ: Funding acquisition, Investigation, Supervision, Writing – review & editing. JL: Investigation, Supervision, Writing – review & editing. HZ: Investigation, Supervision, Writing – review & editing. CY: Funding acquisition, Investigation, Supervision, Writing – review & editing. ZS: Funding acquisition, Investigation, Supervision, Writing – review & editing. NW: Investigation, Resources, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by Guangdong Province Key Areas Research and Development Plan (Grant No. 2023B1111050014), Youth Promotion Project of Natural Science Foundation of Guangdong Province (Grant No. 2023A1515030280), Guangdong Province Local Innovative and Research Teams Project (Grant No. 2019BT02L278-01), the National Natural Science Foundation of China (Grant No. 42302120) and the Open Fund Project of the Key Laboratory of Carbonate Reservoirs, China National Petroleum Corporation (Project No. RIPED-2024-JS-1805).
Acknowledgments
The samples were collected by the Qingdao Institute of Marine Geology, China Geological Survey, in 2022. The authors thank the voyage scientists for their hard work in collecting the research samples. Thanks to the Analytical and Testing Center, Guangzhou Institute of Energy Conversion, Chinese Academy of Sciences, Wuhan SampleSolution Analytical Technology Co., Ltd., and the State Key Laboratory of Biogeology and Environmental Geology, China University of Geosciences (Wuhan, China), for providing the research used in this paper. Thanks to the reviewers for their constructive comments on this paper.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1653453/full#supplementary-material
References
Algeo T. J. and Tribovillard N. (2009). Environmental analysis of paleoceanographic systems based on molybdenum–uranium covariation. Chem. Geol. 268, 211–225. doi: 10.1016/j.chemgeo.2009.09.001
Aller R. C. and Rude P. D. (1988). Complete oxidation of solid phase sulfides by manganese and bacteria in anoxic marine sediments. Geochim. Cosmochim. Acta 52, 751–765. doi: 10.1016/0016-7037(88)90335-3
Baker P. A. and Burns S. J. (1985). Occurrence and formation of dolomite in organic-rich continental margin sediments1. AAPG Bull. 69, 1917–1930. doi: 10.1306/94885570-1704-11D7-8645000102C1865D
Bayon G., Birot D., Ruffine L., Caprais J.-C., Ponzevera E., Bollinger C., et al. (2011). Evidence for intense REE scavenging at cold seeps from the Niger Delta margin. Earth Planet. Sci. Lett. 312, 443–452. doi: 10.1016/j.epsl.2011.10.008
Bayon G., German C. R., Burton K. W., Nesbitt R. W., and Rogers N. (2004). Sedimentary Fe–Mn oxyhydroxides as paleoceanographic archives and the role of aeolian flux in regulating oceanic dissolved REE. Earth Planet. Sci. Lett. 224, 477–492. doi: 10.1016/j.epsl.2004.05.033
Bayon G., Henderson G. M., Etoubleau J., Caprais J.-C., Ruffine L., Marsset T., et al. (2015). U-Th isotope constraints on gas hydrate and pockmark dynamics at the Niger delta margin. Mar. Geol. 370, 87–98. doi: 10.1016/j.margeo.2015.10.012
Bayon G., Pierre C., Etoubleau J., Voisset M., Cauquil E., Marsset T., et al. (2007). Sr/Ca and Mg/Ca ratios in Niger Delta sediments: Implications for authigenic carbonate genesis in cold seep environments. Mar. Geol. 241, 93–109. doi: 10.1016/j.margeo.2007.03.007
Bazzaro M., Ogrinc N., Relitti F., Lucchi R. G., Giani M., Adami G., et al. (2020). Geochemical signatures of intense episodic anaerobic oxidation of methane in near-surface sediments of a recently discovered cold seep (Kveithola trough, NW Barents Sea). Mar. Geol. 425, 106189. doi: 10.1016/j.margeo.2020.106189
Berner R. A. (1981). A new geochemical classification of sedimentary environments. J. Sediment. Res. 51, 359–365. doi: 10.1306/212F7C7F-2B24-11D7-8648000102C1865D
Boetius A., Ravenschlag K., Schubert C. J., Rickert D., Widdel F., Gieseke A., et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407, 623–626. doi: 10.1038/35036572
Borowski W. S., Rodriguez N. M., Paull C. K., and Ussler W. (2013). Are 34S-enriched authigenic sulfide minerals a proxy for elevated methane flux and gas hydrates in the geologic record? Mar. Petroleum Geol. 43, 381–395. doi: 10.1016/j.marpetgeo.2012.12.009
Bruland K. W., Middag R., and Lohan M. C. (2014). 8.2 - Controls of Trace Metals in Seawater, in: Holland H.D. and Turekian K.K. (Eds.), Treatise on Geochemistry (Second Edition). Elsevier, Oxford, 19–51. doi: 10.1016/B978-0-08-095975-7.00602-1
Burdige D. J. (2007). Geochemistry of Marine Sediments (Princeton: Princeton University Press). doi: 10.1515/9780691216096
Burdige D. J. and Nealson K. H. (1986). Chemical and microbiological studies of sulfide-mediated manganese reduction 1. Geomicrobiol. J. 4, 361–387. doi: 10.1080/01490458609385944
Canfield D. E. (1991). Sulfate reduction in deep-sea sediments. Am. J. Sci. 291, 177–188. doi: 10.2475/ajs.291.2.177
Canfield D. E., Farquhar J., and Zerkle A. L. (2010). High isotope fractionations during sulfate reduction in a low-sulfate euxinic ocean analog. Geology 38, 415–418. doi: 10.1130/G30723.1
Canfield D. E., Raiswell R., Westrich J. T., Reaves C. M., and Berner R. A. (1986). The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales. Chem. Geol. 54, 149–155. doi: 10.1016/0009-2541(86)90078-1
Canfield D. E. and Thamdrup B. (1994). The production of 34S-depleted sulfide during bacterial disproportionation of elemental sulfur. Science 266, 1973–1975. doi: 10.1126/science.11540246
Canfield D. E. and Thamdrup B. (2009). Towards a consistent classification scheme for geochemical environments, or, why we wish the term ‘suboxic’ would go away. Geobiology 7, 385–392. doi: 10.1111/j.1472-4669.2009.00214.x
Cangemi M., Di Leonardo R., Bellanca A., Cundy A., Neri R., and Angelone M. (2010). Geochemistry and mineralogy of sediments and authigenic carbonates from the Malta Plateau, Strait of Sicily (Central Mediterranean): Relationships with mud/fluid release from a mud volcano system. Chem. Geol. 276, 294–308. doi: 10.1016/j.chemgeo.2010.06.014
Chang J., Li Y., and Lu H. (2022). The morphological characteristics of authigenic pyrite formed in marine sediments. JMSE 10, 1533. doi: 10.3390/jmse10101533
Chen F., Hu Y., Feng D., Zhang X., Cheng S., Cao J., et al. (2016). Evidence of intense methane seepages from molybdenum enrichments in gas hydrate-bearing sediments of the northern South China Sea. Chem. Geol. 443, 173–181. doi: 10.1016/j.chemgeo.2016.09.029
Chen C., Wang J., Algeo T. J., Zhu J.-M., Wang Z., Ma X., et al. (2023). Sulfate-driven anaerobic oxidation of methane inferred from trace-element chemistry and nickel isotopes of pyrite. Geochim. Cosmochim. Acta 349, 81–95. doi: 10.1016/j.gca.2023.04.002
Chuvilin E., Bukhanov B., Davletshina D., Grebenkin S., and Istomin V. (2018). Dissociation and self-preservation of gas hydrates in permafrost. Geosciences 8, 431. doi: 10.3390/geosciences8120431
Dill H. and Kemper E. (1990). Crystallographic and chemical variations during pyritization in the upper Barremian and lower Aptian dark claystones from the Lower Saxonian Basin (NW Germany). Sedimentology 37, 427–443. doi: 10.1111/j.1365-3091.1990.tb00145.x
Emerson S. R. and Huested S. S. (1991). Ocean anoxia and the concentrations of molybdenum and vanadium in seawater. Mar. Chem. 34, 177–196. doi: 10.1016/0304-4203(91)90002-E
Erickson B. E. and Helz G. R. (2000). Molybdenum(VI) speciation in sulfidic waters:: Stability and lability of thiomolybdates. Geochim. Cosmochim. Acta 64, 1149–1158. doi: 10.1016/S0016-7037(99)00423-8
Feng D., Birgel D., Peckmann J., Roberts H. H., Joye S. B., Sassen R., et al. (2014). Time integrated variation of sources of fluids and seepage dynamics archived in authigenic carbonates from Gulf of Mexico Gas Hydrate Seafloor Observatory. Chem. Geol. 385, 129–139. doi: 10.1016/j.chemgeo.2014.07.020
Fike D. A., Bradley A. S., and Rose C. V. (2015). Rethinking the ancient sulfur cycle. Annu. Rev. Earth Planet. Sci. 43, 593–622. doi: 10.1146/annurev-earth-060313-054802
Formolo M. J. and Lyons T. W. (2013). Sulfur biogeochemistry of cold seeps in the Green Canyon region of the Gulf of Mexico. Geochim. Cosmochim. Acta 119, 264–285. doi: 10.1016/j.gca.2013.05.017
Gieskes J. M. (1981). “Deep-sea drilling interstitial water studies: implications for chemical alteration of the oceanic crust, layers I and II,” in The Deep Sea Drilling Project: A Decade of Progress. Eds. Warme J. E., Douglas R. G., and Winterer E. L. (Tulsa, OK: SEPM Society for Sedimentary Geology). doi: 10.2110/pec.81.32.0149
Glass J. B. and Orphan V. J. (2012). Trace metal requirements for microbial enzymes involved in the production and consumption of methane and nitrous oxide. Front. Microbiol. 3. doi: 10.3389/fmicb.2012.00061
Gong S., Hu Y., Li N., Feng D., Liang Q., Tong H., et al. (2018). Environmental controls on sulfur isotopic compositions of sulfide minerals in seep carbonates from the South China Sea. J. Asian Earth Sci. South China Sea Seep 168, 96–105. doi: 10.1016/j.jseaes.2018.04.037
Gong S., Izon G., Peng Y., Cao Y., Liang Q., Peckmann J., et al. (2022). Multiple sulfur isotope systematics of pyrite for tracing sulfate-driven anaerobic oxidation of methane. Earth Planet. Sci. Lett. 597, 117827. doi: 10.1016/j.epsl.2022.117827
Hallam S. J., Girguis P. R., Preston C. M., Richardson P. M., and DeLong E. F. (2003). Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane-oxidizing archaea. Appl. Environ. Microbiol. 69, 5483–5491. doi: 10.1128/AEM.69.9.5483-5491.2003
Han X., Suess E., Huang Y., Wu N., Bohrmann G., Su X., et al. (2008). Jiulong methane reef: Microbial mediation of seep carbonates in the South China Sea. Mar. Geol. 249, 243–256. doi: 10.1016/j.margeo.2007.11.012
Han X., Suess E., Liebetrau V., Eisenhauer A., and Huang Y. (2014). Past methane release events and environmental conditions at the upper continental slope of the South China Sea: constraints by seep carbonates. Int. J. Earth Sci. (Geol Rundsch) 103, 1873–1887. doi: 10.1007/s00531-014-1018-5
Helz G. R., Miller C. V., Charnock J. M., Mosselmans J. F. W., Pattrick R. A. D., Garner C. D., et al. (1996). Mechanism of molybdenum removal from the sea and its concentration in black shales: EXAFS evidence. Geochim. Cosmochim. Acta 60, 3631–3642. doi: 10.1016/0016-7037(96)00195-0
Hinrichs K. U., Hayes J. M., Sylva S. P., Brewer P. G., and DeLong E. F. (1999). Methane-consuming archaebacteria in marine sediments. Nature 398, 802–805. doi: 10.1038/19751
Hu Y., Feng D., Peckmann J., Roberts H. H., and Chen D. (2014). New insights into cerium anomalies and mechanisms of trace metal enrichment in authigenic carbonate from hydrocarbon seeps. Chem. Geol. 381, 55–66. doi: 10.1016/j.chemgeo.2014.05.014
Huerta-Diaz M. A. and Morse J. W. (1992). Pyritization of trace metals in anoxic marine sediments. Geochim. Cosmochim. Acta 56, 2681–2702. doi: 10.1016/0016-7037(92)90353-K
Huerta-Diaz M. A., Tessier A., and Carignan R. (1998). Geochemistry of trace metals associated with reduced sulfur in freshwater sediments. Appl. Geochem. 13, 213–233. doi: 10.1016/S0883-2927(97)00060-7
Jørgensen B. B. (1982). Mineralization of organic matter in the sea bed—the role of sulphate reduction. Nature 296, 643–645. doi: 10.1038/296643a0
Jørgensen B. B., Böttcher M. E., Lüschen H., Neretin L. N., and Volkov I. I. (2004). Anaerobic methane oxidation and a deep H2S sink generate isotopically heavy sulfides in Black Sea sediments 1. Geochim. Cosmochim. Acta 68, 2095–2118. doi: 10.1016/j.gca.2003.07.017
Li N., Feng D., Chen L., Wang H., and Chen D. (2016). Using sediment geochemistry to infer temporal variation of methane flux at a cold seep in the South China Sea. Mar. Petroleum Geol. 77, 835–845. doi: 10.1016/j.marpetgeo.2016.07.026
Li J., Ye J., Qin X., Qiu H., Wu N., Lu H.-L., et al. (2018). The first offshore natural gas hydrate production test in South China Sea. China Geol. 1, 5–16. doi: 10.31035/cg2018003
Lin Z., Sun X., Chen K., Strauss H., Klemd R., Smrzka D., et al. (2022). Effects of sulfate reduction processes on the trace element geochemistry of sedimentary pyrite in modern seep environments. Geochim. Cosmochim. Acta 333, 75–94. doi: 10.1016/j.gca.2022.06.026
Lin Q., Wang J., Taladay K., Lu H., Hu G., Sun F., et al. (2016). Coupled pyrite concentration and sulfur isotopic insight into the paleo sulfate–methane transition zone (SMTZ) in the northern South China Sea. J. Asian Earth Sci. 115, 547–556. doi: 10.1016/j.jseaes.2015.11.001
Liu J., Antler G., Pellerin A., Izon G., Dohrmann I., Findlay A. J., et al. (2021). Isotopically “heavy” pyrite in marine sediments due to high sedimentation rates and non-steady-state deposition. Geology 49, 816–821. doi: 10.1130/G48415.1
Liu X., Fike D., Li A., Dong J., Xu F., Zhuang G., et al. (2019). Pyrite sulfur isotopes constrained by sedimentation rates: Evidence from sediments on the East China Sea inner shelf since the late Pleistocene. Chem. Geol. 505, 66–75. doi: 10.1016/j.chemgeo.2018.12.014
Liu Y., Hu Z., Gao S., Günther D., Xu J., Gao C., et al. (2008). In situ analysis of major and trace elements of anhydrous minerals by LA-ICP-MS without applying an internal standard. Chem. Geol. 257, 34–43. doi: 10.1016/j.chemgeo.2008.08.004
Liu J., Pellerin A., Izon G., Wang J., Antler G., Liang J., et al. (2020). The multiple sulphur isotope fingerprint of a sub-seafloor oxidative sulphur cycle driven by iron. Earth Planet. Sci. Lett. 536, 116165. doi: 10.1016/j.epsl.2020.116165
Liu J., Pellerin A., Wang J., Rickard D., Antler G., Zhao J., et al. (2022). Multiple sulfur isotopes discriminate organoclastic and methane-based sulfate reduction by sub-seafloor pyrite formation. Geochim. Cosmochim. Acta 316, 309–330. doi: 10.1016/j.gca.2021.09.026
Markovic S., Paytan A., Li H., and Wortmann U. G. (2016). A revised seawater sulfate oxygen isotope record for the last 4 Myr. Geochim. Cosmochim. Acta 175, 239–251. doi: 10.1016/j.gca.2015.12.005
Maslennikov V. V., Maslennikova S. P., Large R. R., and Danyushevsky L. V. (2009). Study of trace element zonation in vent chimneys from the silurian yaman-kasy volcanic-hosted massive sulfide deposit (Southern urals, Russia) using laser ablation-inductively coupled plasma mass spectrometry (LA-ICPMS). Econ. Geol. 104, 1111–1141. doi: 10.2113/gsecongeo.104.8.1111
Mazumdar A., Peketi A., Joao H., Dewangan P., Borole D. V., and Kocherla M. (2012). Sulfidization in a shallow coastal depositional setting: Diagenetic and palaeoclimatic implications. Chem. Geol. 322–323, 68–78. doi: 10.1016/j.chemgeo.2012.06.005
McGlynn S. E., Chadwick G. L., Kempes C. P., and Orphan V. J. (2015). Single cell activity reveals direct electron transfer in methanotrophic consortia. Nature 526, 531–535. doi: 10.1038/nature15512
McLennan S. M. (2001). Relationships between the trace element composition of sedimentary rocks and upper continental crust. Geochem. Geophys. Geosyst. 2, 1021. doi: 10.1029/2000GC000109
Miao X., Feng X., Li J., and Lin L. (2021a). Tracing the paleo-methane seepage activity over the past 20,000 years in the sediments of Qiongdongnan Basin, northwestern South China Sea. Chem. Geol. 559, 119956. doi: 10.1016/j.chemgeo.2020.119956
Miao X., Feng X., Li J., Liu X., Liang J., Feng J., et al. (2022). Enrichment mechanism of trace elements in pyrite under methane seepage. Geochem. Persp. Let. 21, 18–22. doi: 10.7185/geochemlet.2211
Miao X., Feng X., Liu X., Li J., and Wei J. (2021b). Effects of methane seepage activity on the morphology and geochemistry of authigenic pyrite. Mar. Petroleum Geol. 133, 105231. doi: 10.1016/j.marpetgeo.2021.105231
Morford J. L. and Emerson S. (1999). The geochemistry of redox sensitive trace metals in sediments. Geochim. Cosmochim. Acta 63, 1735–1750. doi: 10.1016/S0016-7037(99)00126-X
Morley C. K. (2012). Late Cretaceous–Early Palaeogene tectonic development of SE Asia. Earth Sci. Rev. 115, 37–75. doi: 10.1016/j.earscirev.2012.08.002
Morse J. W. and Luther G. W. (1999). Chemical influences on trace metal-sulfide interactions in anoxic sediments. Geochim. Cosmochim. Acta 63, 3373–3378. doi: 10.1016/S0016-7037(99)00258-6
Nath B., Jean J.-S., Lee M.-K., Yang H.-J., and Liu C.-C. (2008). Geochemistry of high arsenic groundwater in Chia-Nan plain, Southwestern Taiwan: Possible sources and reactive transport of arsenic. J. Contam. Hydrol. Distrib. Geogenic Arsenic Hydrol. Syst.: Controls Challenges 99, 85–96. doi: 10.1016/j.jconhyd.2008.04.005
Orphan V. J., House C. H., Hinrichs K. U., McKeegan K. D., and DeLong E. F. (2001). Methane-consuming archaea revealed by directly coupled isotopic and phylogenetic analysis. Science 293, 484–487. doi: 10.1126/science.1061338
Peckmann J. and Thiel V. (2004). Carbon cycling at ancient methane–seeps. Chem. Geol. Geomicrobiol. Biogeochem. Gas Hydrates Hydrocarbon Seeps 205, 443–467. doi: 10.1016/j.chemgeo.2003.12.025
Peketi A., Mazumdar A., Joao M., Patil D., Usapkar A., and Dewangan P. (2015). Coupled C-S-Fe geochemistry in a rapidly accumulating marine sedimentary system: Diagenetic and depositional implications. Geochem. Geophys. Geosyst. 16, 2865–2883. doi: 10.1002/2015GC005754
Peketi A., Mazumdar A., Joshi R. K., Patil D. J., Srinivas P. L., and Dayal A. M. (2012). Tracing the Paleo sulfate-methane transition zones and H2S seepage events in marine sediments: An application of C-S-Mo systematics. Geochem. Geophys. Geosyst. 13, Q10007. doi: 10.1029/2012GC004288
Raiswell R. (1988). Chemical model for the origin of minor limestone-shale cycles by anaerobic methane oxidation. Geology 16, 641–644. doi: 10.1130/0091-7613(1988)016<0641:CMFTOO>2.3.CO;2
Rees C. E., Jenkins W. J., and Monster J. (1978). The sulphur isotopic composition of ocean water sulphate. Geochim. Cosmochim. Acta 42, 377–381. doi: 10.1016/0016-7037(78)90268-5
Rickard D. T. (1975). Kinetics and mechanism of pyrite formation at low temperatures. Am. J. Sci. 275, 636–652. doi: 10.2475/ajs.275.6.636
Rickard D. T. (2012). Sulfidic sediments and sedimentary rocks, Developments in sedimentology (Oxford: Elsevier Science Ltd).
Ruppel C. D. and Kessler J. D. (2017). The interaction of climate change and methane hydrates. Rev. Geophys. 55, 126–168. doi: 10.1002/2016RG000534
Sato H., Hayashi K., Ogawa Y., and Kawamura K. (2012). Geochemistry of deep sea sediments at cold seep sites in the Nankai Trough: Insights into the effect of anaerobic oxidation of methane. Mar. Geol. 323–325, 47–55. doi: 10.1016/j.margeo.2012.07.013
Scheller S., Goenrich M., Boecher R., Thauer R. K., and Jaun B. (2010). The key nickel enzyme of methanogenesis catalyses the anaerobic oxidation of methane. Nature 465, 606–608. doi: 10.1038/nature09015
Scott C. and Lyons T. W. (2012). Contrasting molybdenum cycling and isotopic properties in euxinic versus non-euxinic sediments and sedimentary rocks: Refining the paleoproxies. Chem. Geol. Special Issue Recent Adv. Trace Metal Appl. to Paleoceanogr. Stud. 324–325, 19–27. doi: 10.1016/j.chemgeo.2012.05.012
Sim M. S., Bosak T., and Ono S. (2011). Large sulfur isotope fractionation does not require disproportionation. Science 333, 74–77. doi: 10.1126/science.1205103
Smrzka D., Feng D., Himmler T., Zwicker J., Hu Y., Monien P., et al. (2020). Trace elements in methane-seep carbonates: Potentials, limitations, and perspectives. Earth Sci. Rev. 208, 103263. doi: 10.1016/j.earscirev.2020.103263
Smrzka D., Zwicker J., Bach W., Feng D., Himmler T., Chen D., et al. (2019a). The behavior of trace elements in seawater, sedimentary pore water, and their incorporation into carbonate minerals: a review. Facies 65, 41. doi: 10.1007/s10347-019-0581-4
Smrzka D., Zwicker J., Misch D., Walkner C., Gier S., Monien P., et al. (2019b). Oil seepage and carbonate formation: A case study from the southern Gulf of Mexico. Sedimentology 66, 2318–2353. doi: 10.1111/sed.12593
Spruzen C., Bradbury H. J., Kast E. R., and Turchyn A. V. (2024). The sulfur isotopic composition of Cenozoic pyrite is affected by methane content and depositional environment. Earth Planet. Sci. Lett. 648, 119097. doi: 10.1016/j.epsl.2024.119097
Su M., Yang R., Wang H., Sha Z., Liang J., Wu N., et al. (2016). Gas hydrates distribution in the Shenhu Area, northern South China Sea: comparisons between the eight drilling sites with gas-hydrate petroleum system (Geologica Acta).
Tribovillard N., Algeo T. J., Lyons T., and Riboulleau A. (2006). Trace metals as paleoredox and paleoproductivity proxies: An update. Chem. Geol. 232, 12–32. doi: 10.1016/j.chemgeo.2006.02.012
Tribovillard N., du Châtelet E. A., Gay A., Barbecot F., Sansjofre P., and Potdevin J.-L. (2013). Geochemistry of cold seepage-impacted sediments: Per-ascensum or per-descensum trace metal enrichment? Chem. Geol. 340, 1–12. doi: 10.1016/j.chemgeo.2012.12.012
Vaughan D. J. and Rosso K. M. (2006). Chemical bonding in sulfide minerals. Rev. Mineral. Geochem. 61, 231–264. doi: 10.2138/rmg.2006.61.5
Wang B., Lei H., and Huang F. (2022). Impacts of sulfate-driven anaerobic oxidation of methane on the morphology, sulfur isotope, and trace element content of authigenic pyrite in marine sediments of the northern South China Sea. Mar. Petroleum Geol. 139, 105578. doi: 10.1016/j.marpetgeo.2022.105578
Wang H., Zhang G., Yang M., Liang J., Liang J., and Zhong G. (2003). Structural circumstance of gas hydrate deposition in the continent margin, the south China Sea. Mar. Geol. Quat. Geol 23, 82–83. doi: CNKI: SUN: HYDZ.0.2003-01-013
Wanty R. B. and Goldhaber M. B. (1992). Thermodynamics and kinetics of reactions involving vanadium in natural systems: Accumulation of vanadium in sedimentary rocks. Geochim. Cosmochim. Acta 56, 1471–1483. doi: 10.1016/0016-7037(92)90217-7
Wegener G., Krukenberg V., Riedel D., Tegetmeyer H. E., and Boetius A. (2015). Intercellular wiring enables electron transfer between methanotrophic archaea and bacteria. Nature 526, 587–590. doi: 10.1038/nature15733
Wu L., Yang S., Liang J., Su X., Fu S., Sha Z., et al. (2013). Variations of pore water sulfate gradients in sediments as indicator for underlying gas hydrate in Shenhu Area, the South China Sea. Sci. China Earth Sci. 56, 530–540. doi: 10.1007/s11430-012-4545-6
Xie L., Wang J. S., and Lin Q. (2012). The characteristics and formation mechanism of authigenic minerals in shallow sediments of Shenhu area, northern South China Sea. Acta Petrol. Et Mineralogica 31, 382–392. doi: 10.3969/j.issn.1000-6524.2012.03.008
Yan Y., Liu L., Clift P. D., Dilek Y., and He A. (2024). Provenance shift of the abyssal plains in the Southwest sub-basin of the South China Sea at ∼ 8 Ma: Tectonics & climate changes implication. Mar. Geol. 475, 107368. doi: 10.1016/j.margeo.2024.107368
Yang K., Chu F., Ye L., Zhang W., Xu D., Zhu J., et al. (2014). Implication of methane seeps from sedimentary geochemical proxies (Sr/Ca & Mg/Ca) in the Northern South China Sea. J. Jilin Univ. (Earth Sci. Edition) 44, 469–479. doi: 10.13278/j.cnki.jjuese.201402106
Yao W., Paytan A., and Wortmann U. G. (2020). Effects of a transient marine sulfur reservoir on seawater δ18OSO4 during the Paleocene-Eocene Thermal Maximum. Geochim. Cosmochim. Acta 269, 257–269. doi: 10.1016/j.gca.2019.10.019
Ye J., Qin X., Xie W., Lu H.-L., Ma B., Qiu H., et al. (2020). The second natural gas hydrate production test in the South China Sea. China Geol. 3, 197–209. doi: 10.31035/cg2020043
Yu X., Miao X., Oppo D., Guan H., Wei J., Li J., et al. (2025). Trace element signatures in sedimentary pyrite as indicators of methane seepage activities. Mar. Petroleum Geol. 173, 107290. doi: 10.1016/j.marpetgeo.2025.107290
Zhang Q., Chen X., Wu D., Jin G., Su Z., and Wu N. (2025). Geochemistry and mineralogy coupling reveals the Fe-S cycle in a receding methane seep. China Geol. 8, 1–17. doi: 10.31035/cg20240110
Zhang G., Liang J., Lu J., Yang S., Zhang M., Holland M., et al. (2015). Geological features, controlling factors and potential prospects of the gas hydrate occurrence in the east part of the Pearl River Mouth Basin, South China Sea. Mar. Petroleum Geol. 67, 356–367. doi: 10.1016/j.marpetgeo.2015.05.021
Zhang M., Lu H., Guan H., Liu L., Wu D., and Wu N. (2018). Methane seepage intensities traced by sulfur isotopes of pyrite and gypsum in sediment from the Shenhu area, South China Sea. Acta Oceanol. Sin. 37, 20–27. doi: 10.1007/s13131-018-1241-1
Keywords: methane seep, South China Sea, pyrite, trace metals, biogeochemical cycle
Citation: Ling Y, Chen X, Wu D, Zhang Q, Mao S, Jin G, Liu J, Zhai H, Yang C, Su Z and Wu N (2025) The process of trace metals enrichment in pyrite under methane seepage in the northern South China Sea. Front. Mar. Sci. 12:1653453. doi: 10.3389/fmars.2025.1653453
Received: 25 June 2025; Accepted: 15 September 2025;
Published: 07 October 2025.
Edited by:
Jun Sun, Tianjin University of Science and Technology, ChinaReviewed by:
Niu Li, Chinese Academy of Sciences (CAS), ChinaXiaoming Miao, Ocean University of China, China
Xiuli Feng, Ocean University of China, China
Copyright © 2025 Ling, Chen, Wu, Zhang, Mao, Jin, Liu, Zhai, Yang, Su and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daidai Wu, d3VkZEBtcy5naWVjLmFjLmNu
†These authors have contributed equally to this work