 Pratima Gautam
Pratima Gautam Vanessa Molina2
Vanessa Molina2 Matthew First
Matthew First Ivan Erill
Ivan Erill Kathleen D. Cusick
Kathleen D. Cusick- 1Department of Biological Sciences, University of Maryland Baltimore County, Baltimore, MD, United States
- 2Precise Systems, Inc, Lexington Park, MD, United States
- 3Chemistry Division, U.S. Naval Research Laboratory, Washington, DC, United States
Copper-based antimicrobial paints are frequently used to inhibit the biofouling of marine vessels. However, some bacterial species can overcome this copper challenge and colonize the surfaces. The early adherent bacterial population of marine vessels plays an important role because of its ability to produce extracellular polymeric substances (EPSs), forming a thin layer of organic matter that traps nutrients from the water and protects other colonizers by blocking the toxic antifouling (AF) coatings. It is of interest to study the factors that drive the initial colonization of copper surfaces. We used a metagenomic sequencing approach to analyze the microbial diversity and potential functional abilities of early biofilm communities on copper surfaces and discovered enriched copper-specific functional traits in early colonizers compared to the bacterial community of the surrounding seawater. The functional analysis of early biofilm colonizers on copper surfaces in marine coastal environments provides insights into molecular mechanisms that support biofilm formation on copper surfaces. The taxa associated with copper resistance traits were found to dominate initial microbial communities on copper surfaces, allowing these organisms to survive on copper surfaces despite copper toxicity. Our analysis reveals the dominance of the genera Allomuricauda and Ruegeria, carrying several copper resistance genes, as the early colonizers of copper surfaces.
Introduction
In the marine environment, biofilms provide shelter and sustenance to the microorganisms, which receive protection from environmental changes, substrates for growth at relatively higher concentrations than found in the planktonic environment, and conditions favorable to genetic exchange and survivability (Davey and O Toole, 2000). Biofilm formation creates significant challenges in the marine industry (Weber and Esmaeili, 2023), causing an increase in drag forces that require additional shaft power (Schultz et al., 2011). Microbial attachment to vessel surfaces leads to higher maintenance costs and efforts, and risks introducing previously unknown biomes into new ecosystems (Yebra et al., 2004). The colonization of submerged surfaces is affected by various factors, including environmental characteristics, substrate types, and the biological properties of colonizers (Caruso, 2020; Kimkes and Heinemann, 2020).
Biofouling starts with the adhesion of bacterial cells and the formation of biofilms; these bacterial biofilms alter the surface and enable subsequent attachment of organisms such as diatoms, algae, and barnacles (Dang and Lovell, 2000). To limit biofouling, copper is widely used as a biocide due to its antimicrobial properties (Amara et al., 2018). However, various microbial species are able to colonize copper-treated surfaces. The early colonizing population plays a crucial role in this process, as it can create a barrier between the interaction of toxic biocides and subsequent colonizers (Dang and Lovell, 2000; Chen et al., 2013).
Previous studies of microbial colonization on specific substrates have demonstrated that substrate type significantly influences the composition of biofilm communities (Muthukrishnan et al., 2014; Ding et al., 2019). On copper surfaces, microbial communities may develop resilience to copper toxicity through increased extracellular matrix production, copper resistance and detoxification processes, and the activation of stress-response pathways (Zhang et al., 2019b). However, there are significant knowledge gaps in terms of the mechanisms employed by early colonizers to establish themselves on copper surfaces. Identifying these mechanisms is critical to understanding how biofilms initiate and evolve under the selective pressures of copper toxicity.
Prior research on early marine copper surface colonization has mainly focused on profiling microbial communities within the biofilm environment (Chen et al., 2013; Muthukrishnan et al., 2014; Zhang et al., 2019a; Briand et al., 2022). The functional research into copper-associated biofilm is limited to predictive functional approaches that use 16S rRNA sequencing (Catao et al., 2019). Functional studies of copper-associated biofilm have primarily focused on long-term seawater exposure. Studies on marine biofilms on copper surfaces demonstrate that long-term seawater exposure drives the selection of specific functional genes, especially those involved in membrane transport (Zhang et al., 2019b).
Many bacteria have evolved distinct regulatory mechanisms to cope with high copper concentrations. These mechanisms include resistance-nodulation-cell-division (RND) efflux pumps, P-type ATPases, metallothioneins, cation diffusion facilitator (CDF) family copper transporters, and oxidation of copper ions (Cooksey, 1994; Franke et al., 2003; Rensing and Grass, 2003; Pal et al., 2017).
In this study, we used a shotgun metagenomics approach to identify early bacterial colonizers of copper surfaces and functional gene profiles from biofilm and seawater communities, with a focus on copper resistance pathways. Our analysis revealed that taxa contributing copper resistance traits dominate the early stage of biofilm. This suggests the significance of early colonizers in establishing the necessary conditions to mitigate copper toxicity and facilitate the adherence of other fouling organisms. Our work establishes the fundamental groundwork for future research comparing functional signatures from various copper-related surfaces, thereby developing strategies against marine biofouling.
Methods
Sample collection and processing
The copper panels (90–10 Cu-Ni alloy, 4 X 4 inches, 0.25 inch) were deployed for 7 days at Key West, Florida (October 2023). The copper panels were suspended in an ambient seawater, continuous flow-through trough (Supplementary Figure 1). At the time of deployment, the seawater temperature was 25°C, pH was 8.02, salinity was 36 and DO was 7.5 mg/L. These parameters remained relatively stable until sampling. A thin layer of biofilm was observed and scraped off the surface after 7 days of immersion. The seawater samples from the same area were collected by filtering 1 liter of seawater through a sterile 0.22 µm membrane filter. After collection, the biofilm and seawater were suspended in a nucleic acid stabilizer, DNA/RNA shield (Zymo Research). Samples were immediately frozen at -20°C until shipped.
DNA extraction and quality control
DNA from biofilm was extracted using a DNeasy Power biofilm kit (Qiagen), and seawater samples were extracted using a DNeasy Blood and Tissue kit (Qiagen). After extraction, DNA was quantified using a nanodrop spectrophotometer, and the amplification of bacterial 16S rRNA gene fragments was validated by polymerase chain reaction (PCR) using universal PCR primers (Gautam and Cusick, 2023).
Quality control, library preparation, and sequencing
DNA concentration and fragment size were quantified using the Agilent Femto Pulse capillary system. The sequencing libraries were prepared following the HiFi SMRTbell library preparation protocol. The fragment lengths of the prepared libraries were validated using the Femto Pulse system, revealing that the fragment lengths of water samples were smaller than those of biofilm samples. To account for this size difference, water and biofilm libraries were processed at a 1:5 ratio, respectively, and sequencing was performed on a single SMRT cell. Libraries were sequenced on an 8M SMRTcell on a Sequel II platform using circular consensus sequencing protocol. After sequencing, the reads were demultiplexed to obtain high-quality HiFi FASTQ reads for all samples.
Taxonomic annotation of HiFi reads
The taxonomic classification and functional annotation of the reads were performed using established tools compiled by Pacific Biosciences in their GitHub repository for long-read metagenomic workflows (Portik et al., 2022). The reads were analyzed utilizing the Diamond and MEGAN analysis pipeline (Portik et al., 2022).
A local database of NCBI NR protein sequences was created by using the Diamond alignment tool (Buchfink et al., 2021). The HiFi sequence reads were aligned with the compiled protein database using the Diamond BLASTX option. For this analysis, we used the default parameters provided by the Diamond tool, selecting the -top5 hits parameter. An alignment was considered significant if it was within the top 5 percent of the high-scoring hit range. Given the long-read data, we included the frameshift-aware and range-culling parameters for Diamond alignment.
The aligned sequences were processed for taxonomic binning using the MEGAN 7 software (Huson et al., 2016). MEGAN utilizes NCBI reference taxonomy to generate and assign taxonomy to reads by applying an interval union Lowest Common Ancestor (LCA) parameter. It determines the taxon assignment for each read by analyzing sequence conservation throughout the taxonomic tree. We labeled a node using the total number of aligned bases, considering the length of long reads, as the alignment coverage can vary significantly among different reads (Huson et al., 2018).
MEGAN default parameters were used for the analysis, except for the minimum support parameter, which was set to 0.01 (which provides optimal tradeoff between precision and recall for HiFi metagenomics data) (Portik et al., 2022), and abundance, set to be reported as the number of aligned bases. Thus, a taxon was reported if at least 0.01% of the aligned bases were assigned to that node. If the minimum support value was not met, MEGAN assigned the reads to its lowest common ancestor where the support parameter value was satisfied. The total number of aligned bases was reported as count data. The relative abundance at each taxonomic category was calculated by normalizing counts to the read counts from the water samples (minimum number of counts among all samples).
The taxonomic assignments were stored as read-match archive (RMA) files, which were used for interactive analysis and visualization in the MEGAN user interface. Comparative analyses of aligned base counts at different taxonomic levels were performed to estimate the relative abundance of taxa across the two environments.
SEED subsystems of copper with MEGAN
The assignment of reads to functional categories was determined by their best-scoring match to a reference protein with a known function. SEED-based functional annotations were used to extract reads associated with copper. Copper-associated genes were identified as a part of the membrane transport subsystem (including sub-subsystems: copper transport & blue copper proteins, the copper transport system, and copper uptake system), and the stress response, virulence subsystem (sub-systems: copper homeostasis) of SEED. These copper-associated reads were used for copper gene-specific homology search.
Copper gene-specific functional profiling with protein homology search
Copper-associated genes and their regulators were identified by literature review and were used as the proteins of interest for a homology search (Cusick et al., 2021; Gautam et al., 2023). Profile Hidden Markov models (HMMs) of these proteins of interest were obtained from the PFAM and TIGRFAM (Haft et al., 2003; Mistry et al., 2021). These HMMs were compiled into a local database using the HMMER suite.
The HMMER esl-translate tool was utilized to obtain the six-frame translation of the individual copper-associated reads. Translated reads were scanned against the compiled HMM database to identify protein homologs using the hmmscan tool. The copper-associated proteins of interest and their respective protein families are shown in Table 1. The search process was automated using a Python script. The hit count for copper-associated proteins was evaluated as a function of limiting e-value in 8 representative bacterial species to define an e-value threshold in a previous study (Gautam et al., 2023). The hits with e-values below 10E-30 were considered significant and thus classified as sequence homologs. The copper-associated protein hits from biofilm and water samples were analyzed to identify copper gene abundance levels. The abundance of specific copper genes across two samples was normalized with the same scaling factor as taxonomic abundance to account for the differences in the total sequence between biofilm and water.
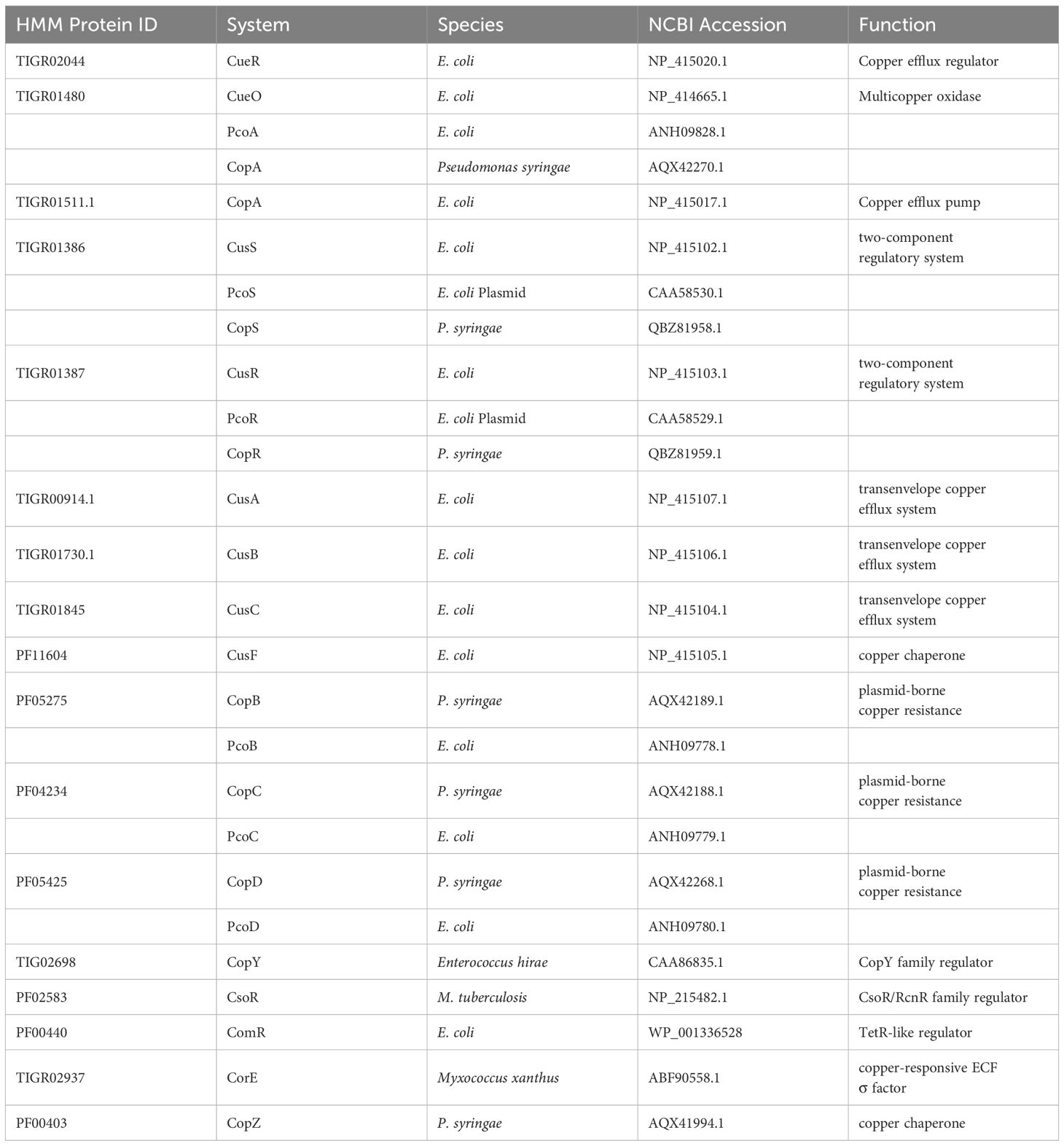
Table 1. Copper-associated protein models used for homology search.
The Wilcoxon rank-sum test method enabled comparison between the overall distributions of copper gene abundance within biofilm samples and water samples (Non-parametric distribution verified by Shapiro-Wilk test p<0.05). A binomial test was used to detect copper genes, which showed statistically significant count differences between the two samples. The Benjamini-Hochberg correction method with a 5% false discovery rate threshold was applied to adjust p-values accounting for multiple hypothesis testing. The statistical analyses and data visualization processes were performed using R with the ggplot2 package for graphical representation and data manipulation.
Taxonomic assignment of copper genes
The reads that mapped to copper systems in SEED classification were assigned taxonomy with the DIAMOND and MEGAN pipeline to identify the bacterial genera carrying copper resistance genes. The taxa associated with the copper gene profile in the biofilm were then compared to the overall bacterial profile to assess the role of these bacterial genera carrying copper-resistant genes in the biofilm environment.
Results and discussion
Bacterial diversity analysis at phylum and genus taxonomic levels
Biofilm and water DNA were sequenced using PacBio circular consensus sequencing (CCS) protocol to obtain HiFi reads (Supplementary Table 1). Bacterial reads accounted for over 90% of the total sequences and were thus isolated from overall reads. To identify bacterial reads, sequences were first aligned to the NCBI database, which were then assigned taxonomy using MEGAN. Reads specific to bacteria were extracted for downstream analysis. Taxonomic composition within the two samples was assessed by calculating the relative abundance, which is defined as the proportion of aligned bases assigned to a specific taxon relative to the total bases in the dataset. The relative abundance of bacterial taxa was examined at the phylum and genus taxonomic levels.
Taxonomic analysis revealed distinct variations in microbial composition between the biofilm and seawater samples. At a phylum level, the biofilm and water samples were quite similar: Pseudomonadota dominated in both communities, followed by Bacteroidota. The overall distribution at a phylum level confirms previously described composition in marine biofilm environments, showing a greater abundance of phyla such as Planctomycetota, and Cyanobacteriota, known to be associated with organisms such as marine microalgae, macroalgae, sponges, protozoa, and other invertebrates (Izumi et al., 2013; Kaboré et al., 2020; Mutalipassi et al., 2021) (Supplementary Figure 2).
The biofilm microbial community mainly consisted of the genus Allomuricauda (11.09%) (phylum Bacteroidota), followed by members of Ruegeria (5.54%) (phylum Pseudomonadota). Both genera were also found in the seawater environment, but their abundance levels remained lower than those in the biofilm (Allomuricauda 2.52% and Ruegeria 1.78%). Additional dominant genera identified in the biofilm included Ulvibacterium (phylum Bacteroidota), Kordiimonas (phylum Pseudomonadota), Winogradskyella (phylum Bacteroidota), Congregibacter (phylum Pseudomonadota), Ekhidna (phylum Bacteroidota), Alkalimarinus (phylum Pseudomonadota), and Tateyamaria (phylum Pseudomonadota) (Figure 1). Some of these genera could not be detected in seawater at the used sequencing depth, suggesting they may be present at a relative abundance below the detection limit in the seawater.
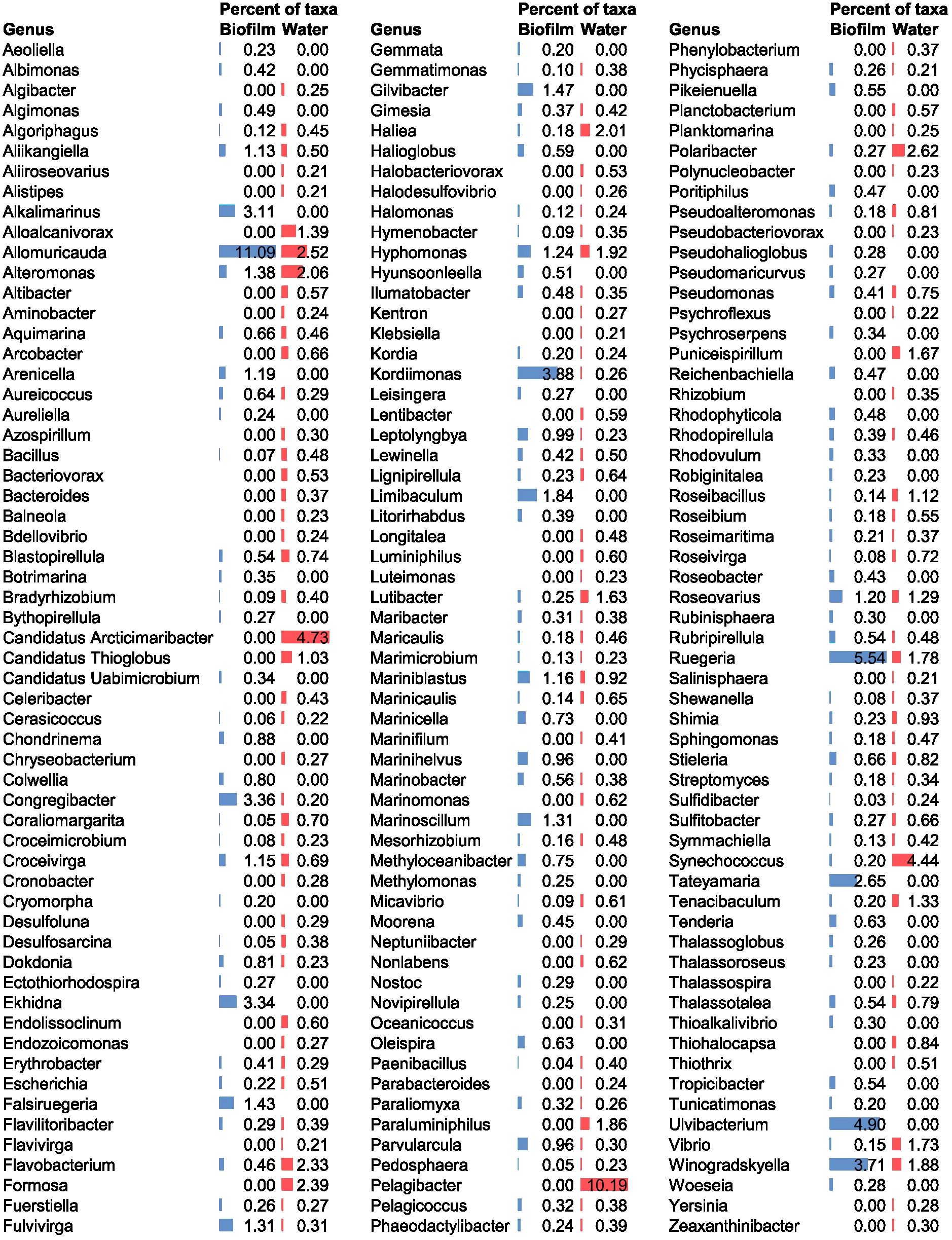
Figure 1. Microbial community composition at a genus level in biofilm and seawater communities. The percent of taxa refers to the relative abundance within each community (blue bars represent biofilm, red bars represent seawater). Genera with a relative abundance >0.20% for one or both samples are shown in the figure.
In contrast, the dominant genus in seawater was Pelagibacter (phylum Pseudomonadota), representing approximately 10% of the community profile (Figure 1).
The genus Allomuricauda (recently reclassified as Flagellimonas) was previously identified as a key microbial genus associated with copper surface colonization (Zhang et al., 2019b; Novoa et al., 2024), and harbors copper-associated genes from both the Cus and Cop systems (Zhang et al., 2019b). Allomuricauda is found in diverse marine habitats, demonstrating its ecological versatility and ability to survive under different environmental stress conditions (Wu et al., 2013), including copper rich biofilm environment.
Ruegeria also exhibits copper-related adaptations, including a specific strain isolated from red algae, that harbors multiple chromosome-borne and plasmid-borne copper resistance operons, establishing its association with copper-enriched habitats (Yin et al., 2023). Some species of Ruegeria, including R. pomeroyi DSS-3 (previously classified as Silicibacter pomeroyi) are moderate copiotrophs, as indicated by their genome analysis, thus suggesting higher abundance as seen in the biofilm environment (Lauro et al., 2009; Christie-Oleza and Armengaud, 2010). Other dominant genera from the biofilm such as Kordimonas, Winogradskyella, and Congregibacter are known to secrete an EPS matrix, which supports their survival and adaptability in the biofilm (Decho and Gutierrez, 2017; Nagar et al., 2021; Ye et al., 2022). In addition to copper genes, these traits may play a significant role in the dominance of these genera in the biofilm.
Previous studies have found that facultative anaerobic microbes dominate mature copper surface biofilm (Zhang et al., 2019b). Our study on early-stage biofilm was dominated by representatives from primarily aerobic genera, including Allomuricauda, Ruegeria, Winogradskyella, Alkalimarinus Tateyamaria, and the obligate aerobe, Kordimonas. Other genera, such as Congregibacter and Ekhidna, are facultative anaerobes, allowing them to thrive in environments with oxygen gradients, such as biofilms. Our results highlight the dominance of taxa involved in aerobic metabolism in the initial biofilm environment.
Enrichment of copper gene panel
SEED-based functional annotations from MEGAN were used to extract reads associated with copper. The reads annotated to copper-associated genes in the SEED subsystems category were used to identify specific copper resistance proteins by homology search.
We performed a HMMER homology search using the profile HMM of proteins involved in bacterial copper homeostasis to identify its sequence homologs from the sequencing reads.
Bacterial copper regulatory mechanism involves genes responsible for efflux transport, detoxification, and chaperone functions. Copper homeostasis is primarily maintained with the chromosome-borne Copper efflux (Cue) system, which includes genes involved in cytoplasmic efflux and copper oxidases, and Copper sensing (Cus) system that includes the periplasmic efflux complexes, and copper chaperone (Rensing and Grass, 2003). Several bacteria that can survive in copper-rich environments may harbor additional one or many copies of plasmid-encoded genes that confer copper resistance, such as Pco and Cop systems, which include genes encoding copper oxidases and membrane proteins (Rensing and Grass, 2003; Cusick et al., 2021).
We found a statistically significant difference in copper-related gene abundance between biofilm and water samples (Wilcoxon Rank test, p < 0.05). The statistically significant difference in individual copper gene homologs between biofilm and water samples was confirmed using a binomial test (Figure 2B). Copper-related genes, including copA, cusB, and cusA exhibited statistically significant enrichment in the biofilm metagenome (Binomial test, adjusted p-value < 0.05) (Figures 2A, B).
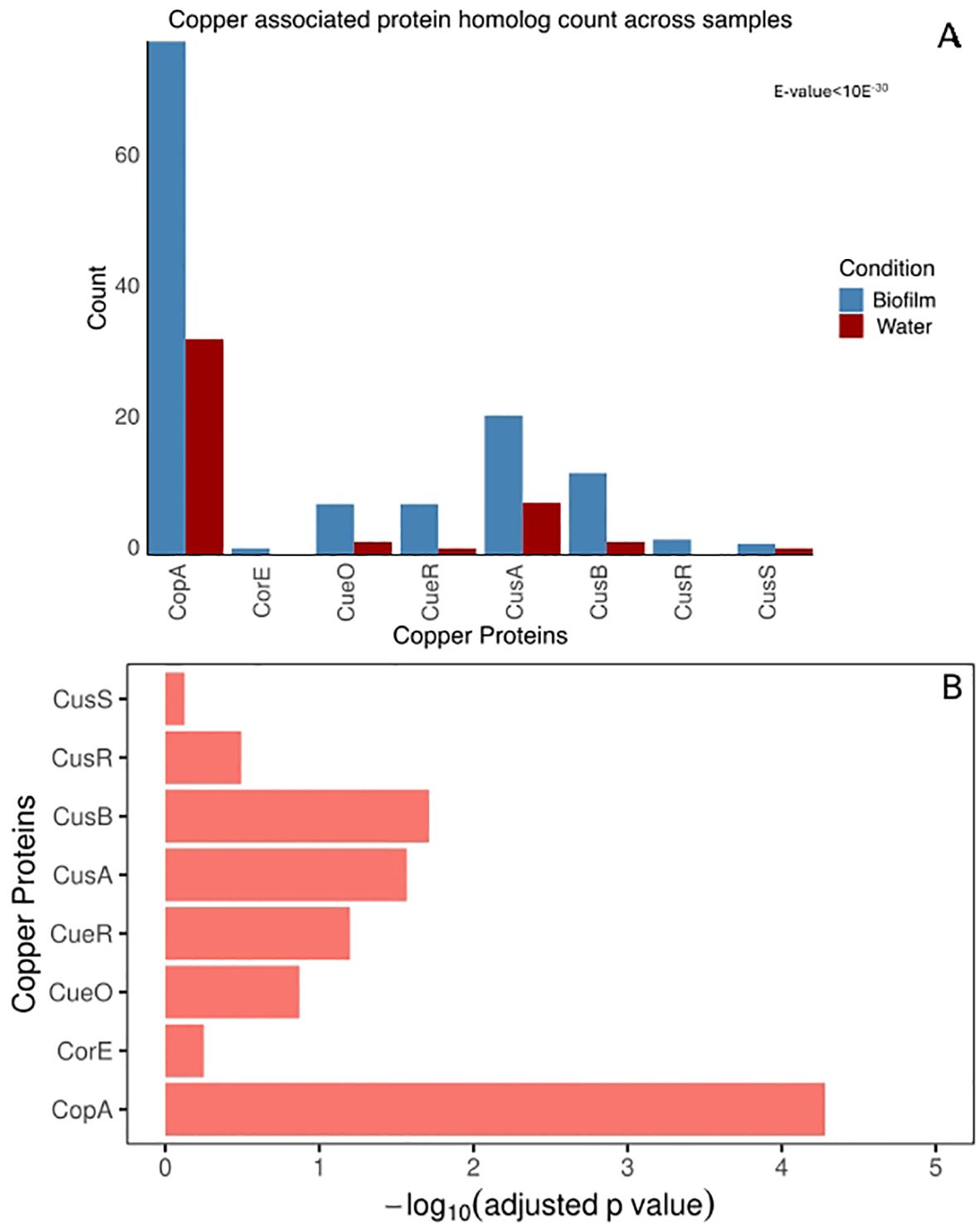
Figure 2. Copper associated protein homolog hit count. (A) Number of homologs to copper-associated proteins from biofilm and water metagenome. The copper-associated system homolog count for biofilm and seawater communities was plotted for each copper protein. The total homolog count to each protein family is normalized with a scaling factor of the smallest number of aligned base counts among the two samples obtained from sequencing. (B) Adjusted P-value testing for significant enrichment in the biofilm, compared to water for each copper protein. The p-value is calculated by binomial test, corrected with BH correction with FDR of 5%.
Among all copper-related genes, the P-type ATPase copA, which exports Cu(I) from the cytoplasm to the periplasm, was the most abundant and exhibited the most significant difference between the two samples. Additionally, the biofilm showed significant enrichment of the periplasmic copper efflux system cusA and cusB genes, enabling proton-driven periplasmic copper ion translocation across the outer membrane (Franke et al., 2003). This observed enrichment of the copper protein from biofilm metagenome highlights the critical role of bacterial copper efflux mechanisms within this environment.
Taxonomic assignment to copper-associated genes
The reads annotated as copper-associated genes from the SEED annotations of the metagenome were taxonomically classified using the Diamond and Megan pipeline to get insights into the bacteria responsible for copper genes in the biofilm environment. This was done by using Diamond and Megan taxonomic analysis on copper-associated reads.
Taxonomic analysis of the biofilm metagenome revealed key contributors to copper surface colonization. We found that copper-related genes found in the biofilm were predominantly linked to the bacterial genera Allomuricauda and Ruegeria (Figure 3). These taxa accounted for over 45% of the biofilm copper-related genes identified through metagenomic analysis. These two taxa also emerged as the most dominant taxa from the overall taxonomic classification. This highlights that the taxa contributing copper genes dominate the early biofilm environment on copper surfaces, suggesting that this allows these organisms to survive on copper surfaces despite copper toxicity and thereby supports the attachment of other fouling organisms.
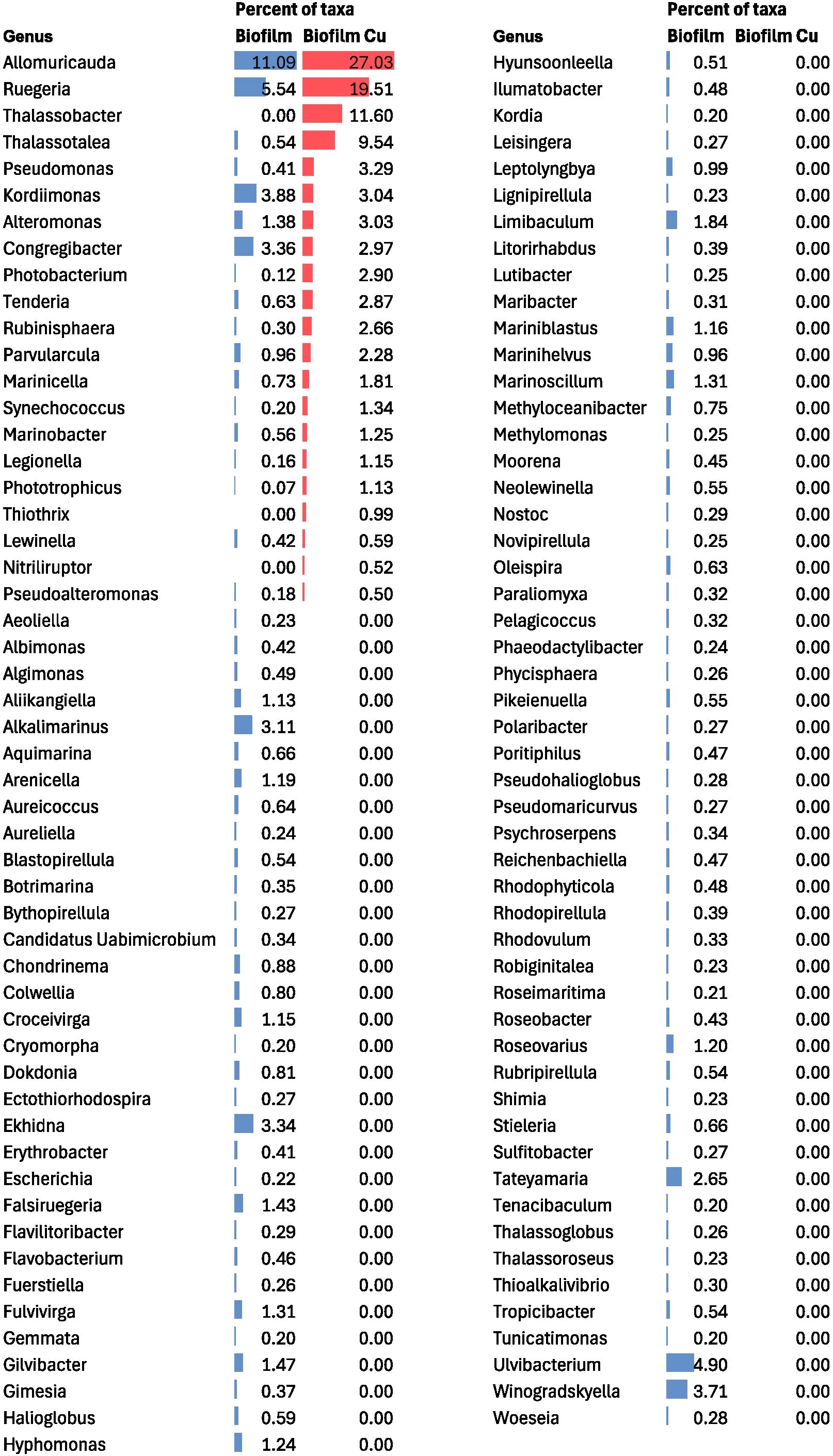
Figure 3. Microbial community composition in biofilm community from total bacterial reads and copper specific reads shown at a genus level. The percent of taxa refers to the relative abundance within each community (shown as data bars: blue bars represent taxa related to total biofilm sequences, red bars represent copper specific sequences). The genera with a relative abundance >0.20% for one or both samples are included.
Overall, our observation suggests the enrichment of copper proteins involved in efflux function in the biofilm (Figure 2). The copper toxicity mitigated by efflux via actively pumping the copper ions out of the cell may lead to an increased localized copper concentration near the biofilm and expose the nearby taxa to higher copper stress. Previous studies have shown spatial gradients of copper within the biofilm when biofilm is grown in the presence of copper. These differences in copper concentration across biofilm have been mainly attributed to the presence of biofilm matrix, which could affect the diffusion of copper within biofilm (Hu et al., 2007; Chen et al., 2011). Considering the observed enrichment of efflux proteins in biofilm metagenomes, we hypothesize that copper efflux proteins may additionally lead to localized copper accumulation. Studies on bacterial multidrug efflux pumps have shown evidence of increased local concentration of antibiotics, thereby affecting growth of bacterial population in the surrounding microenvironment (Wen et al., 2018). The biofilm environment thus may also experience higher localized copper concentrations because of active efflux mechanisms, thereby explaining the dominance of taxa contributing copper genes.
Conclusion
The genetic pathways encoded within microbes of the marine biofilm environment, and the biological processes actively expressed in marine biofilm habitat, referred to as the functional potential and the functional activity respectively, remain poorly understood. To address this gap, we explored the functional potential by characterizing the microbial diversity and copper specific gene profile of early-stage marine copper-associated biofilms. By comparing the functional profiles associated with copper genes of copper-substrate grown biofilms with those of ambient seawater, functional signatures unique to early colonizers were identified.
The biofilm community was enriched with genes associated with copper resistance, particularly those encoding efflux complexes. These included two efflux proteins: the ATPase-driven P-type copA and the RND transporter proteins of the cusABC system (Franke et al., 2003; Rensing and Grass, 2003). The CopA P-type ATPase exports Cu(I) ions from the cytoplasm to the periplasm (Christopher et al., 2000), while the RND efflux proteins mediate the proton-driven translocation of copper ions across the outer membrane via a periplasmic efflux system (Franke et al., 2003).
The genera Allomuricauda and Ruegeria are dominant bacterial genera that contributed significant copper resistance genes in the biofilm metagenome, even though their presence was less abundant in the seawater metagenome. These two genera accounted for more than 45% of the copper-related genes in the early colonization profile, highlighting that the taxa contributing copper traits dominate early copper surface microbial communities.
While previous studies examined taxonomic characterization of the biofilm communities, we performed the first metagenomic functional analysis of early biofilm colonizers on copper surfaces in marine coastal environments, providing insights into molecular mechanisms that support biofilm formation on copper surfaces. Results from this study creates a foundation for subsequent studies to analyze functional signatures beyond copper systems associated with biofilm formation, that can support the development of new genetic strategies against marine biofouling. This study is limited to profiling the taxonomic composition of microbes and their functional potential of copper-associated biofilm and its surrounding seawater. In this approach, gene abundance has been used as a proxy for microbial activity within the community. It does not, however, determine which genes are actively expressed in the biofilm and seawater communities. As a result, this approach does not offer a clear insight into gene function. To fill this gap, our research must be broadened to incorporate RNA-level analyses that can reflect the active biological processes within the biofilm. Future studies should integrate transcriptomic and proteomic approaches, which can provide direct evidence of actively transcribed genes and quantify proteins. These insights can provide a comprehensive understanding of microbial interactions within the biofilm and can expand on microbial function, metabolic, and regulatory interactions. Additionally, these findings of functional enrichment of copper resistance genes in the copper-associated biofilm could be integrated along with studies characterizing genes related to biofilm formation and surface adhesion, like EPS production, to identify conserved functional activity that can aid in designing effective antifouling strategies against these pathways.
Data availability statement
The sequence data generated from this study have been deposited in the NCBI Sequence Read Archive under BioProject accession PRJNA1263017.
Author contributions
PG: Investigation, Formal Analysis, Methodology, Validation, Visualization, Data curation, Software, Writing – review & editing, Writing – original draft. VM: Formal Analysis, Methodology, Writing – review & editing. MF: Resources, Project administration, Writing – review & editing, Methodology, Funding acquisition. IE: Writing – review & editing, Software, Resources, Validation, Supervision, Methodology. KC: Project administration, Supervision, Methodology, Conceptualization, Validation, Funding acquisition, Writing – review & editing, Resources.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by UMBC internal funding. Portions of this work were funded through the U.S. Naval Research Laboratories under its base funds to the Nanoscience Institute, but the paper does not represent the U.S. Navy, the Office of Naval Research, or the U.S. Government.
Acknowledgments
We thank Olga Shevchenko and Bruce Brewster Kingham at the University of Delaware DNA Sequencing & Genotyping Center for their assistance in sample quality control validation and Pacbio HiFi sequencing. The hardware used in the computational studies is part of the UMBC High Performance Computing Facility (HPCF). The facility is supported by the U.S. National Science Foundation through the MRI program (grant nos. CNS-0821258, CNS-1228778, OAC-1726023, and CNS-1920079) and the SCREMS program (grant no. DMS-0821311), with additional substantial support from the University of Maryland, Baltimore County (UMBC).
Conflict of interest
Author VM was employed by Precise Systems, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmars.2025.1661535/full#supplementary-material
References
Amara I., Miled W., Slama R. B., and Ladhari N. (2018). Antifouling processes and toxicity effects of antifouling paints on marine environment. A review. Environ. Toxicol. Pharmacol. 57, 115–130. doi: 10.1016/j.etap.2017.12.001
Briand J.-F., Pollet T., Misson B., Garnier C., Lejars M., Maintenay M., et al. (2022). Surface characteristics together with environmental conditions shape marine biofilm dynamics in coastal NW mediterranean locations. Front. Mar. Sci. 8, 746383. doi: 10.3389/fmars.2021.746383
Buchfink B., Reuter K., and Drost H. (2021). Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–36+. doi: 10.1038/s41592-021-01101-x
Caruso G. (2020). Microbial colonization in marine environments: overview of current knowledge and emerging research topics. J. Mar. Sci. Eng. 8 (2), 78. doi: 10.3390/jmse8020078
Catao E., Pollet T., Misson B., Garnier C., Ghiglione J., Barry-Martinet R., et al. (2019). Shear stress as a major driver of marine biofilm communities in the NW mediterranean sea. Front. Microbiol. 10. doi: 10.3389/fmicb.2019.01768
Chen C. L., Maki J. S., Rittschof D., and Teo S. L. M. (2013). Early marine bacterial biofilm on a copper-based antifouling paint. International Biodeterioration & Biodegradation 71, 71–76. doi: 10.1016/j.ibiod.2013.04.012
Chen G., Chen X., Yang Y., Hay A., Yu X., and Chen Y. (2011). Sorption and distribution of copper in unsaturated pseudomonas putida CZ1 biofilms as determined by X-ray fluorescence microscopy. Appl. Environ. Microbiol. 77, 4719–4727. doi: 10.1128/AEM.00125-11
Christie-Oleza J. and Armengaud J. (2010). In-depth analysis of exoproteomes from marine bacteria by shotgun liquid chromatography-tandem mass spectrometry: the ruegeria pomeroyi DSS-3 case-study. Mar. Drugs 8, 2223–2239. doi: 10.3390/md8082223
Christopher R., Bin F., Rakesh S., Bharati M., and Barry P. R. (2000). CopA: an escherichia coli cu(I)-translocating P-type ATPase. Proc. Natl. Acad. Sci. United. States Am. 97, 652. doi: 10.1073/pnas.97.2.652
Cooksey D. A. (1994). Molecular mechanisms of copper resistance and accumulation in bacteria. FEMS Microbiol. Rev. 14, 381–386. doi: 10.1111/j.1574-6976.1994.tb00112.x
Cusick K., Iturbide A., Gautam P., Price A., Polson S., MacDonald M., et al. (2021). Enhanced copper-resistance gene repertoire in Alteromonas macleodii strains isolated from copper-treated marine coatings. PloS One 16, 1–30. doi: 10.1371/journal.pone.0257800
Dang H. and Lovell C. R. (2000). Bacterial Primary Colonization and Early Succession on Surfaces in Marine Waters as Determined by Amplified rRNA Gene Restriction Analysis and Sequence Analysis of 16S rRNA Genes. Appl. Environ. Microbiol. 66 (2), 467–475. doi: 10.1128/AEM.66.2.467-475.2000
Davey M. E. and O Toole G. A. (2000). Microbial biofilms: from ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 64 (4), 847–867. doi: 10.1128/MMBR.64.4.847-867.2000
Decho A. and Gutierrez T. (2017). Microbial extracellular polymeric substances (EPSs) in ocean systems. Front. Microbiol. 8. doi: 10.3389/fmicb.2017.00922
Ding W., Zhang W., Alikunhi N. M., Batang Z., Pei B., Wang R., et al. (2019). Metagenomic analysis of zinc surface-associated marine biofilms. Microbial. Ecol. 77, 406–416. doi: 10.1007/s00248-018-01313-3
Franke S., Grass G., Rensing C., and Nies D. H. (2003). Molecular analysis of the copper-transporting efflux system CusCFBA of Escherichia coli. J. Bacteriol. 185, 3804–3812. doi: 10.1128/JB.185.13.3804-3812.2003
Gautam P. and Cusick K. D. (2023). Development of a real-time quantitative PCR assay for detection and quantification of the marine bacterium Alteromonas macleodii from coastal environments. J. Microbiol. Methods 204, 10. doi: 10.1016/j.mimet.2022.106629
Gautam P., Erill I., and Cusick K. D. (2023). Linking copper-associated signal transduction systems with their environment in marine bacteria. Microorganisms 11, 20. doi: 10.3390/microorganisms11041012
Haft D., Selengut J., and White O. (2003). The TIGRFAMs database of protein families. Nucleic Acids Res. 31, 371–373. doi: 10.1093/nar/gkg128
Hu Z., Jin J., Abruña H., Houston P., Hay A., Ghiorse W., et al. (2007). Spatial distributions of copper in microbial biofilms by scanning electrochemical microscopy. Environ. Sci. Technol. 41, 936–941. doi: 10.1021/es061293k
Huson D., Albrecht B., Bagci C., Bessarab I., Górska A., Jolic D., et al. (2018). MEGAN-LR: new algorithms allow accurate binning and easy interactive exploration of metagenomic long reads and contigs. Biol. Direct. 13. doi: 10.1186/s13062-018-0208-7
Huson D., Beier S., Flade I., Górska A., El-Hadidi M., Mitra S., et al. (2016). MEGAN community edition - interactive exploration and analysis of large-scale microbiome sequencing data. PloS Comput. Biol. 12. doi: 10.1371/journal.pcbi.1004957
Izumi H., Sagulenko E., Webb R. I., and Fuerst J. A. (2013). Isolation and diversity of planctomycetes from the sponge Niphates sp., seawater, and sediment of Moreton Bay, Australia. Antonie. Van. Leeuwenhoek. 104, 533–546. doi: 10.1007/s10482-013-0003-5
Kaboré O., Godreuil S., and Drancourt M. (2020). Planctomycetes as host-associated bacteria: A perspective that holds promise for their future isolations, by mimicking their native environmental niches in clinical microbiology laboratories. Front. Cell. Infect. Microbiol. 10. doi: 10.3389/fcimb.2020.519301
Kimkes T. E. P. and Heinemann M. (2020). How bacteria recognise and respond to surface contact. FEMS Microbiology Reviews 44, 106–122. doi: 10.1093/femsre/fuz029
Lauro F., McDougald D., Thomas T., Williams T., Egan S., Rice S., et al. (2009). The genomic basis of trophic strategy in marine bacteria. Proc. Of Natl. Acad. Sci. United. States Am. 106, 15527–15533. doi: 10.1073/pnas.0903507106
Mistry J., Chuguransky S., Williams L., Qureshi M., Salazar G., Sonnhammer E., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D4D9. doi: 10.1093/nar/gkaa913
Mutalipassi M., Riccio G., Mazzella V., Galasso C., Somma E., Chiarore A., et al. (2021). Symbioses of cyanobacteria in marine environments: ecological insights and biotechnological perspectives. Mar. Drugs 19. doi: 10.3390/md19040227
Muthukrishnan T., Abed R., Dobretsov S., Kidd B., and Finnie A. (2014). Long-term microfouling on commercial biocidal fouling control coatings. Biofouling 30, 1155–1164. doi: 10.1080/08927014.2014.972951
Nagar S., Antony R., and Thamban M. (2021). Extracellular polymeric substances in Antarctic environments: A review of their ecological roles and impact on glacier biogeochemical cycles. Polar. Sci. 30. doi: 10.1016/j.polar.2021.100686
Novoa E., Deshmukh U., and Oren A. (2024). Reclassification of Allomuricauda and Muricauda species as members of the genus Flagellimonas Bae et al., 2007 and emended description of the genus Flagellimonas. Int. J. Syst. Evol. Microbiol. ;74. doi: 10.1099/ijsem.0.006286
Pal C., Asiani K., Arya S., Rensing C., Stekel D. J., Larsson D. G. J., et al. (2017). Chapter seven - metal resistance and its association with antibiotic resistance. Adv. Microbial. Physiol. 70, 261–313. doi: 10.1016/bs.ampbs.2017.02.001
Portik D. M., Brown C. T., and Pierce-Ward N. T. (2022). Evaluation of taxonomic classification and profiling methods for long-read shotgun metagenomic sequencing datasets. BMC Bioinf. 23, 39. doi: 10.1186/s12859-022-05103-0
Rensing C. and Grass G. (2003). Escherichia coli mechanisms of copper homeostasis in a changing environment. FEMS Microbiol. Rev. 27, 197–213. doi: 10.1016/S0168-6445(03)00049-4
Schultz M. P., Bendick J. A., Holm E. R., and Hertel W. M. (2011). Economic impact of biofouling on a naval surface ship. Biofouling 27 (1), 87–98. doi: 10.1080/08927014.2010.542809
Weber F. and Esmaeili N. (2023). Marine biofouling and the role of biocidal coatings in balancing environmental impacts. Biofouling 39, 661–681. doi: 10.1080/08927014.2023.2246906
Wen X., Langevin A., and Dunlop M. (2018). Antibiotic export by efflux pumps affects growth of neighboring bacteria. Sci. Rep. 8. doi: 10.1038/s41598-018-33275-4
Wu Y. H., Yu P. S., Zhou Y. D., Xu L., Wang C. S., Wu M., et al. (2013). Muricauda Antarctica sp. nov., a marine member of the Flavobacteriaceae isolated from Antarctic seawater. Int. J. Syst. Evol. Microbiol. 63, 3451–3456. doi: 10.1099/ijs.0.048355-0
Ye Y., Hao Z., Yue Y., Ma L., Ye M., and Du Z. (2022). Characterization of Kordiimonas marina sp. nov. and Kordiimonas laminariae sp. nov. and Comparative Genomic Analysis of the Genus Kordiimonas, A Marine-Adapted Taxon. Front. Mar. Sci. 9. doi: 10.3389/fmars.2022.919253
Yebra D. M., Kiil S., and Dam-Johansen K. (2004). Antifouling technology—past, present and future steps towards efficient and environmentally friendly antifouling coatings. Prog. Organic. Coat. 50, 75–104. doi: 10.1016/j.porgcoat.2003.06.001
Yin Q.-J., Zhu F.-C., Tang H.-Z., Chen X.-Y., Liu X., Tang L.-C., et al. (2023). Complete genome sequence of marine Roseobacter lineage member Ruegeria sp. YS9 with five plasmids isolated from red algae. Mar. Genomics 67, 100997. doi: 10.1016/j.margen.2022.100997
Zhang Y., Ma Y., Duan J., Li X., Wang J., and Hou B. (2019a). Analysis of marine microbial communities colonizing various metallic materials and rust layers. Biofouling. 35 (4), 429–442. doi: 10.1080/08927014.2019.1610881
Keywords: copper, surface colonization, taxonomic profiling, functional profiling, metagenomics, biofilm, seawater, long-read sequencing
Citation: Gautam P, Molina V, First M, Erill I and Cusick KD (2025) Determining the taxonomic and functional profile of marine bacterial copper systems involved in marine early copper surface colonization. Front. Mar. Sci. 12:1661535. doi: 10.3389/fmars.2025.1661535
Received: 07 July 2025; Accepted: 08 August 2025;
Published: 29 August 2025.
Edited by:
Yu Zhang, Shanghai Jiao Tong University, ChinaReviewed by:
Khaled Mohammed Geba, Menoufia University, EgyptHafiz Zeshan Wadood, Lahore Garrison University, Pakistan
Copyright © 2025 Gautam, Molina, First, Erill and Cusick. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kathleen D. Cusick, a2N1c2lja0B1bWJjLmVkdQ==