 Takanori Hidaka
Takanori Hidaka Taku Fujimura
Taku Fujimura Setsuya Aiba
Setsuya Aiba- Department of Dermatology, Tohoku University Graduate School of Medicine, Sendai, Japan
The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that responds to a wide range of chemicals, including chemical carcinogens such as dioxins and carcinogenic polyaromatic hydrocarbons, and induces a battery of genes associated with detoxification, proliferation, and immune regulation. Recent reports suggest that AHR plays an important role in carcinogenesis and maintenance of various types of skin cancers. Indeed, AHR is a susceptibility gene for squamous cell carcinoma and a prognostic factor for melanoma and Merkel cell carcinoma. In addition, the carcinogenic effects of ultraviolet (UV) and chemical carcinogens, both of which are major environmental carcinogenetic factors of skin, are at least partly mediated by AHR, which regulates UV-induced inflammation and apoptosis, the DNA repair system, and metabolic activation of chemical carcinogens. Furthermore, AHR modulates the efficacy of key therapeutic agents in melanoma. AHR activation induces the expression of resistance genes against the inhibitors of V600E mutated B-Raf proto-oncogene, serine/threonine kinase (BRAF) in melanoma and upregulation of programmed cell death protein 1 (PD-1) in tumor-infiltrating T cells surrounding melanoma. Taken together, these findings underscore the importance of AHR in the biology of skin cancers. Development of therapeutic agents that modulate AHR activity is a promising strategy to advance chemoprevention and chemotherapy for skin cancers.
Introduction
Recently, the incident rate of skin cancer has been greatly increasing. The number of patients treated for skin cancers has increased by 44% during the past 5 years (1), and skin cancer has become the most common cancer type in Caucasians (2). Although both genetic and environmental factors contribute to the carcinogenesis of skin cancer, this rapid increase suggests the relative importance of environmental factors. The skin is the outermost interface between the body and the environment and is ineluctably exposed to environmental insults such as ultraviolet radiation (UVR) or air pollutants (3). As UVR and air pollutants can induce carcinogenesis in the skin (4), the skin contains a system that recognizes and detoxifies these carcinogenic insults, the dysregulation of which leads to the initiation of skin cancer. In addition to the increase in carcinogenesis of skin cancer, recent therapeutic aspects of skin cancer have greatly changed. In particular, the emergence of molecular targeted therapies including inhibitors for V600E mutated B-Raf proto-oncogene, serine/threonine kinase (BRAF) and checkpoint inhibitors, which attenuate suppression of the anti-tumor immune response, have drastically improved the outcome of advanced melanoma. These drugs retrogradely elucidated the critical contribution of specific proliferative signals and tumor immunity in the maintenance of melanoma. These recent changes in skin cancers imply the importance of identifying a key molecule that modulates carcinogenesis and maintains skin cancer to improve prevention of and therapy for skin cancers.
The aryl hydrocarbon receptor (AHR) is an evolutionarily conserved, ligand-activated transcription factor, which is a member of the basic helix-loop-helix/PER-ARNT-SIM family (5). Due to its broad capacity to recognize a wide range of chemicals in the environment, AHR is often described as an environmental sensor. Once activated by ligand binding, AHR translocates into the nucleus and dimerizes with ARNT (Ah receptor nuclear translocator). Then the AHR/ARNT heterodimer enhances the expression of its target genes that encode drug-metabolizing cytochrome P450s, including CYP1A1, CYP1A2, and CYP1B1 (6) (Figure 1). These target molecules of AHR facilitate the metabolic degradation of its ligands. In addition to this role in detoxification, recent works have also revealed novel roles for AHR in tumor biology. In various tumors, differential expression of AHR is indeed observed compared to normal tissue. This different expression status of AHR plays a critical role in pro- or anti-tumor activity according to the cell state (7). Regarding skin cancer, a genome-wide association study of cutaneous squamous cell carcinoma (SCC) also identified AHR as a novel susceptibility locus (8). Furthermore, among various solid tumors, the expression level of CYP1A1, CYP1A2, and CYP1B1 is associated with prognosis of melanoma (9). These findings imply that AHR also plays important roles in the biology of skin cancers. In support of this hypothesis, AHR has recently been found to be associated with UVR and air pollutant-induced carcinogenesis of skin cancer (10, 11). Furthermore, AHR may play a role in modulating the efficacy of BRAF inhibitors and checkpoint inhibitors (12, 13) (Figure 2). In the following sections, we introduce the function of AHR in the context of carcinogenesis and maintenance of skin cancer and mainly focus on environmental carcinogens and molecular targeted therapy.
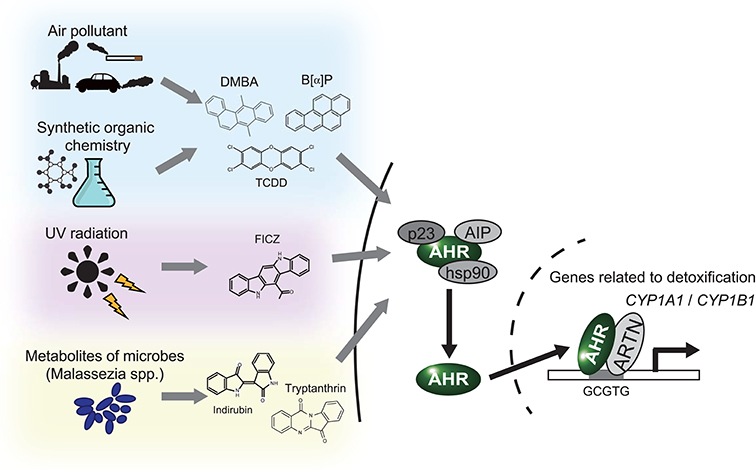
Figure 1. AHR works as an environmental sensor. AHR binds to polycyclic aromatic hydrocarbons and their derivatives derived from environment. Once these ligands binded, AHR isolates from the complex in cytoplasm, translocates into nucleus and activates translation of the target genes, including CYP1A1 and CYP1B1. DMBA, 7,12-Dimethyl benz[a]anthracen; B[a]P, Benzo[a]pyrene; TCDD, 2,3,7,8-Tetrachloro dibenzo-p-dioxin; FICZ, 6-Formylindolo [3,2-b]carbazole.
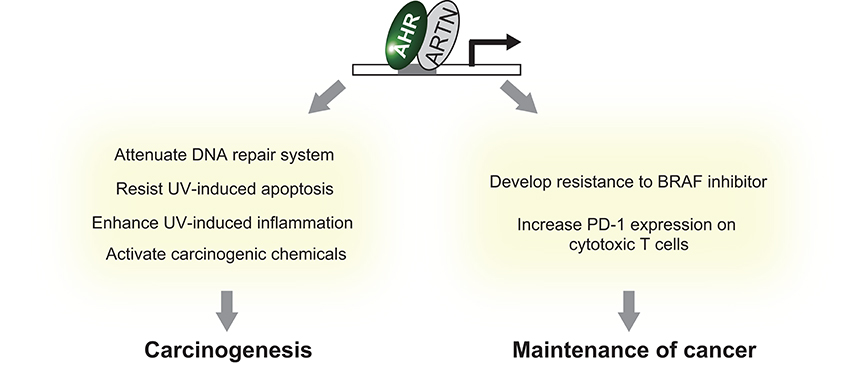
Figure 2. Summary of the effect of AHR activation on skin cancer.
Environmental Factor-Induced Skin Carcinogenesis via AHR Activation
Ultraviolet Radiation
As much as 90% of non-melanoma skin cancers are associated with exposure to UVR (14). UVR causes mutagenesis of DNA and inflammation, which may eventually lead to the formation of skin cancers. UVA radiation (400-320 nm wavelength) excites endogenous chromophores and generates reactive oxygen species, leading to modifications of oxidative bases and generation of 7,8-dihydro-8-oxoguanine at guanine bases (15). In contrast, UVB radiation (320-290 nm wavelength) activates a photochemical reaction and forms photoproducts, including cyclobutane pyrimidine dimers (CPDs) and pyrimidine 6-4 pyrimidones, at adjacent pyrimidine nucleotides (15). To keep genomic integrity, these DNA photoproducts have to be removed by DNA repair system or apoptosis, depending on the extent of DNA damage (16). However, once the incorrect repair of these DNA modifications occurs, it may inhibit polymerases, lead to the arrest of replication or cause misreadings during transcription or replication, which results in the formation of mutations, initiation of carcinogenesis, and skin cancer (15). The importance of DNA repair enzymes is clearly evident as seen by the drastically increased risk of developing UV signature mutations and subsequent skin cancers in Xeroderma Pigmentosum patients who lack one of the DNA repair enzymes (17). In addition to mutagenesis, UVR causes the body a UV stress response, the inflammatory response at the exposed site. Increasing evidence suggests that sustained inflammation induced by UVR plays an important role in cancer initiation and progression (18).
AHR acts as a light sensor in keratinocytes following activation by UVR. UVR (in particular UVB) generates formylindolo (3,2-b)carbazole (FICZ), a tryptophan derivative, in epidermal keratinocytes (19). FICZ functions as a high-affinity ligand for AHR and induces UVR-mediated AHR activation, which is associated with UV-induced skin carcinogenesis. Pollet et al. reported that the chronic irradiation of UVB causes only a half numbers of cutaneous SCCs on AHR−/− mice compared to AHR+/+ littermates, which implies a critical contribution of AHR in carcinogenesis of SCC. As a molecular mechanism, they revealed AHR activation attenuates the clearance of UVB-induced CPDs by repressing global genomic repair in a p27-dependent manner (10). In addition to the role of AHR in the attenuation of DNA repair systems, AHR works as a negative regulator of apoptosis in UVB damaged keratinocytes. Frauenstein et al. reported that chemical inhibition or knockdown of AHR sensitize keratinocytes to UVB-induced apoptosis by decreasing the expression of E2F1 and its target gene checkpoint kinase 1 (CHK1) (20). AHR also promotes the UV stress response (19). Although the concise molecular mechanism of the UV stress response remains largely unknown, the involvement of different tyrosine kinases including the epidermal growth factor receptor (EGFR) and pro-inflammatory molecules has been suggested (21). For instance, FICZ induces the internalization and subsequent activation of EGFR in an AHR-dependent manner, which is also induced by UVB radiation (19). Moreover, AHR activation in keratinocyte induces the expression of various pro-inflammatory molecules. Irradiation of UVR followed by topical application with FICZ on Ahr+/+ mice induces cutaneous expression of a neutrophil directing chemokine (C-X-C motif) ligand 5 (Cxcl5) compared with UV alone, which cannot be observed in Ahr−/− mice (22). In addition, the exposure of FICZ to keratinocyte cell-line induces the activation of AHR and ROS production, which leads to the production of pro-inflammatory cytokine, IL-6 (23). Furthermore, AHR activation induced by UVB irradiation can activate the expression of cyclooxygenase-2, which is pro-inflammatory and associated with the development of skin cancer (19, 24).
These observations imply that AHR activation promotes UVR-induced skin carcinogenesis via attenuation of the DNA repair system and apoptosis and via enhancement of the UV response.
Carcinogenic Chemicals in Air Pollutants
Carcinogenic chemicals are another well-known type of environmental carcinogen that leads to skin cancer. Airborne particulate matter (PM) and ambient air pollution, which contain various carcinogenic chemicals, are considered group 1 human carcinogens by the International Agency for Research on Cancer (25, 26). As the skin is located at the outermost layer of the body, it is continuously exposed to air pollutants, which may increase the risk of skin cancer. Carcinogenic chemicals, including polycyclic aromatic hydrocarbons (PAHs) and dioxins, contained in PM are responsible for PM-induced carcinogenesis (27). Due to their lipophilicity, these chemicals easily penetrate through the skin (28, 29) and are retained in the skin for a long time (30). PAHs and dioxins exert their biological effects via binding to AHR. AHR activation by these chemicals has gained a lot of attention as a mechanism that contributes to skin carcinogenesis. In fact, PAHs and dioxins cause SCC in in vivo animal models. For instance, chronic subcutaneous injection of TCDD to hamster results in formation cutaneous SCC (31). In addition, application of 7,12-Dimethylbenz[a]anthracene (DMBA), a member of the PAH family that is typically found in cigarette smoke, to murine skin causes lesions that are histologically similar to benign papilloma to SCC (32, 33). Whole-exome sequencing analysis has been conducted in this murine model of DMBA-induced SCC to investigate its mutational landscape (34). As a result, the majority of DMBA-induced SCC possesses mutations in oncogenes including Hras, Kras, and Rras2. These mutations in human SCCs are similar to those in head and neck, esophageal, lung, and cervical SCC (34–36). In addition to SCC, the development of melanoma is also accelerated by the application of DMBA in some genetically engineered mouse models of melanoma (37).
In these models of PAH-induced skin carcinogenesis, AHR plays a considerable role. Chronic topical application of organic extracts of airborne particulate matter causes SCCs in a half of AHR +/+ mice but none of AHR−/− mice (11). Benzo[a]pyrene, another PAH contained in PM from cigarettes or air pollutants, can also induce SCC following subcutaneous or topical application to wild-type mice. This carcinogenic property of benzo[a]pyrene is attenuated when applied to Ahr-deficient mice (38). In the case of DMBA-induced carcinogenesis, the mice possessing the 375A allele of Ahr, encoding the high-affinity ligand-binding receptor, develop skin cancers, but the mice possessing the 375V allele, encoding the low-affinity one do not (39); in contrast, there is another report demonstrating no significant differences in carcinogenesis between Ahr+/+ mice and Ahr−/−mice by topical application of DMBA (40). Taken together, these results suggest that AHR activation promotes tumor induction of PAH-induced skin carcinogenesis.
Several studies investigated the mechanism of PAHs-induced carcinogenesis and revealed that AHR-dependent induction of CYP1A1/CYP1B1 expression likely plays a key role (38). In general, CYP1A1 and CYP1B1 enzymes facilitate removal of AHR ligands by degrading them to metabolites with decreased activity and increasing their water solubility (41). In contrast, in the case of carcinogenic PAHs, the same metabolic reaction results in the metabolic activation of PAHs. For instance, CYP1B1-mediated metabolism of DMBA results in the synthesis of DMBA-trans-3,4-diol, which is highly electrophilic and causes damages to DNA (42). Moreover, CYP1A1, CYP1B1 and epoxide hydrolase-mediated metabolism of benzo[a]pyrene results in the synthesis of highly electrophilic benzo[a]pyrene-7,8-diol-9,10-epoxide (43).
Regarding the mechanism of dioxins-induced carcinogenesis and its dependency of AHR, they remain largely unknown, as dioxins are not generally metabolized to be directly genotoxic and there is lack of related articles.
Maintenance of Skin Cancer via AHR Activation
In addition to the carcinogenic role of AHR activation, AHR also greatly contributes to the maintenance of various skin cancers. In non-cutaneous tumors, AHR is an established factor that induces suppression of the anti-tumor immune response, resulting in the escape of tumor cells from immune-mediated cell death (44). Furthermore, AHR affects multiple aspects of cancer biology, including cell survival and proliferation (45). Recent findings show that AHR modulates anti-tumor immunity and proliferative signals in skin cancers. In the following sections, we introduce recent findings regarding how AHR contributes to the maintenance of skin cancers, mainly focusing on melanoma.
Melanoma
Melanoma is believed to be derived from malignant transformation of melanocytes, which are pigment-producing cells that generally reside in skin (46). Studies investigating the melanocytes of Ahr-deficient mice indicated that AHR is essential for proliferation of melanocytes (47). In addition, some reports using melanoma cell lines indicate that AHR activation attenuates tumorigenicity (48, 49); in contrast, others reported that AHR activation promotes tumorigenicity of melanoma (50, 51). These observations suggest the contribution of the AHR system to the biology of melanoma, the details of which have been revealed in recent reports.
In the clinical setting, therapy for melanoma is based on the staging system, which scores clinical and pathological risk factors, including tumor thickness, mitotic rate, and presence of ulceration and metastases (52). In the past, once a melanoma was scored as high grade, patients were considered to have an extremely high mortality rate due to resistance to chemotherapy (53). However, recent development of molecular targeted therapies against the oncogene or checkpoint inhibitors has drastically improved the prognosis of patients with advanced melanoma (54). This improvement indicates the critical importance of BRAF and checkpoint molecules in the maintenance of melanoma. Surprisingly, recent findings have revealed a significant role for AHR in modulating the effect of these critical molecules.
The BRAF V600E mutation is the most prevalent mutation and is present in approximately half of patients with advanced melanoma (55). Specific inhibitors of mutated BRAF have achieved high response rates and improved overall survival (56). Meanwhile, the efficacy of BRAF inhibitors is transient due to acquired resistance, which usually appears within a year after the time of response and results in relapse of melanoma (57, 58). One mechanism of the induction of resistance to BRAF inhibitors is upregulation of genes related to resistance to BRAF inhibitors, including AXL receptor tyrosine kinase (AXL), EGFR, and neuropilin 1 (NRP1) (59, 60). Recently, Corre et al. demonstrated that in a subset of melanoma cells, AHR is constitutively activated, which drives expression of these genes that are related to resistance to BRAF inhibitors (12). In addition, they also reported that co-administration of AHR antagonists with BRAF inhibitors maintains at least partial sensitivity to BRAF inhibitors in melanoma cells.
Melanoma is a solid tumor with a high mutational burden, which induces the generation of neo-antigens and the infiltration of cytotoxic T cells (CTLs) that recognize neo-antigens (61, 62). The level of mutational burden is correlated with that of transcripts related to cytolytic activity of local immune infiltrates (63). To evade the anti-tumor immune response, melanoma cells express molecules associated with checkpoint pathways. Approximately 40% of melanoma biospecimens express programmed death-ligand 1 (PD-L1), one of the molecules associated with the checkpoint pathway (64). When PD-L1 expressed on melanoma cells binds to the PD-1 receptor expressed on CTLs, CTLs become dysfunctional, and melanoma cells escape immune-mediated cell lysis (65, 66). As mentioned, PD-1 blockade by checkpoint inhibitors significantly improves overall survival and progression-free survival compared with classical chemotherapy in patients with advanced melanoma (67). These findings imply the importance of elucidating how melanoma cells upregulate the expression of PD-1 on CTLs. Liu et al. found that tumor-repopulating cells, a subpopulation of cancer cells having stem cell-like property that are tumorigenic and can grow in soft 3D matrices, produce kynurenine, a known AHR ligand of tryptophan metabolism, by type I IFN-induced expression of indolamine 2,3-dioxygenase. Then kynurenine activates AHR in tumor-repopulating cells, which enters them into dormancy, the condition resistant to immune-therapies (68, 69). In addition, released kynurenine is taken up by surrounding CTLs and upregulates PD-1 expression on CTLs in an AHR-dependent manner (13). This finding tells us that the AHR system may be a significant modulator of PD-1-mediated suppression of the anti-melanoma immune response.
Other Cutaneous Carcinomas
Several reports have suggested possible links between the AHR system and tumor biology in Merkel cell carcinoma (MCC) and extramammary Paget's disease (EMPD).
MCC is a rare and aggressive neuroendocrine skin cancer, and ~80% of patients are infected with merkel cell polyomavirus. Univariate analysis of clinical specimens revealed that a longer overall survival is achieved in the group with lower expression of tryptophan 2,3-dioxygenase 2 (TDO2) and AHR in cells surrounding the tumor (70). As TDO2 is an enzyme in the tryptophan-kynurenine metabolic pathway, the TDO2-AHR axis may play a significant role in the pathophysiology of MCC.
Another study of EMPD, an adenocarcinoma of apocrine origin, reported that the epidermis adjacent to EMPD lesions expresses CYP1A1 and CCL20, an interleukin-17-related chemokine. Malassezia yeast, which are often pathogenic in apocrine lesions, produce a metabolite that activates AHR and induces the Th17 immune response. Thus, a possible link may be present between Malassezia metabolite-induced AHR activation and the Th17-skewed tumor immune response in EMPD (71).
Concluding Remarks
As summarized above, AHR was recently found to be a key modulator of UVR- and carcinogenic chemical-induced skin carcinogenesis. In addition, this molecule is associated with the efficacy of BRAF inhibitors and checkpoint inhibitors, which are core therapeutic drugs in melanoma. Taken together, these data underscore the importance of the AHR system in carcinogenesis and maintenance of skin cancers, especially SCC and melanoma. This means that the AHR system is a putative target, particularly for chemoprevention and cancer chemotherapy of skin cancer. The emergence of research investigating the effect of AHR antagonists for various skin cancers is promising and eagerly awaited.
Author Contributions
TH wrote the manuscript. TF and SA supervised and reviewed this work.
Funding
This study was supported in part by the Japan Agency for Medical Research and Development (18cm01643h0001).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Guy GP, Machlin SR, Ekwueme DU, Yabroff KR. Prevalence and costs of skin cancer treatment in the U.S., 2002–2006 and 2007–2011. Am J Prev Med. (2015) 48:183–87. doi: 10.1016/j.amepre.2014.08.036
2. Leiter U, Eigentler T, Garbe C. Epidemiology of skin cancer. Adv Exp Med Biol. (2014) 810:120–40. doi: 10.1007/978-1-4939-0437-2_7
3. Elias PM, Choi EH. Interactions among stratum corneum defensive functions. Exp Dermatol. (2005) 14:719–26. doi: 10.1111/j.1600-0625.2005.00363.x
4. Zegarska B, Pietkun K, Zegarski W, Bolibok P, Wiśniewski M, Roszek K, et al. Air pollution, UV irradiation and skin carcinogenesis: what we know, where we stand and what is likely to happen in the future? Postep dermatologii i Alergol. (2017) 34:6–14. doi: 10.5114/ada.2017.65616
5. Hahn ME, Karchner SI, Shapiro MA, Perera SA. Molecular evolution of two vertebrate aryl hydrocarbon (dioxin) receptors (AHR1 and AHR2) and the PAS family. Proc Natl Acad Sci USA. (1997) 94:13743–8. doi: 10.1073/pnas.94.25.13743
6. Fujii-Kuriyama Y, Mimura J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem Biophys Res Commun. (2005) 338:311–7. doi: 10.1016/j.bbrc.2005.08.162
7. Safe S, Cheng Y, Jin UH. The aryl hydrocarbon receptor (AhR) as a drug target for cancer chemotherapy. Curr Opin Toxicol. (2017) 2:24–9. doi: 10.1016/j.cotox.2017.01.012
8. Chahal HS, Lin Y, Ransohoff KJ, Hinds DA, Wu W, Dai HJ, et al. Genome-wide association study identifies novel susceptibility loci for cutaneous squamous cell carcinoma. Nat Commun. (2016) 7:12048. doi: 10.1038/ncomms12048
9. Lv JW, Zheng ZQ, Wang ZX, Zhou GQ, Chen L, Mao YP, et al. Pan-cancer genomic analyses reveal prognostic and immunogenic features of the tumor melatonergic microenvironment across 14 solid cancer types. J Pineal Res. (2019) 66:1–13. doi: 10.1111/jpi.12557
10. Pollet M, Shaik S, Mescher M, Frauenstein K, Tigges J, Braun SA, et al. The AHR represses nucleotide excision repair and apoptosis and contributes to UV-induced skin carcinogenesis. Cell Death Differ. (2018) 25:1823–36. doi: 10.1038/s41418-018-0160-1
11. Matsumoto Y, Ide F, Kishi R, Akutagawa T, Sakai S, Nakamura M, et al. Aryl hydrocarbon receptor plays a significant role in mediating airborne particulate-induced carcinogenesis in mice. Environ Sci Technol. (2007) 41:3775–80. doi: 10.1021/es062793g
12. Corre S, Tardif N, Mouchet N, Leclair HM, Boussemart L, Gautron A, et al. Sustained activation of the Aryl hydrocarbon Receptor transcription factor promotes resistance to BRAF-inhibitors in melanoma. Nat Commun. (2018) 9: doi: 10.1038/s41467-018-06951-2
13. Liu Y, Liang X, Dong W, Fang Y, Lv J, Zhang T, et al. Tumor-repopulating cells induce PD-1 expression in CD8+ T cells by transferring kynurenine and AhR activation. Cancer Cell. (2018) 33:480–494.e7. doi: 10.1016/j.ccell.2018.02.005
14. Vitaliano PP, Urbach F. The relative importance of risk factors in nonmelanoma carcinoma. Arch Dermatol. (1980) 116:454–6.
15. Cadet J, Sage E, Douki T. Ultraviolet radiation-mediated damage to cellular DNA. Mutat Res. (2005) 571:3–17. doi: 10.1016/j.mrfmmm.2004.09.012
16. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. (2016) 16:20–33. doi: 10.1038/nrc.2015.2
17. DiGiovanna JJ, Kraemer KH. Shining a light on xeroderma pigmentosum. J Invest Dermatol. (2012) 132:785–96. doi: 10.1038/jid.2011.426
18. Garibyan L, Fisher DE. How sunlight causes melanoma. Curr Oncol Rep. (2010) 12:319–26. doi: 10.1007/s11912-010-0119-y
19. Fritsche E, Schäfer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci USA. (2007) 104:8851–6. doi: 10.1073/pnas.0701764104
20. Frauenstein K, Sydlik U, Tigges J, Majora M, Wiek C, Hanenberg H, et al. Evidence for a novel anti-apoptotic pathway in human keratinocytes involving the aryl hydrocarbon receptor, E2F1, and checkpoint kinase 1. Cell Death Differ. (2013) 20:1425–34. doi: 10.1038/cdd.2013.102
21. Muthusamy V, Piva TJ. The UV response of the skin: a review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch Dermatol Res. (2010) 302:5–17. doi: 10.1007/s00403-009-0994-y
22. Smith KJ, Boyer JA, Muku GE, Murray IA, Gowda K, Desai D, et al. Editor's highlight: Ah receptor activation potentiates neutrophil chemoattractant (C-X-C Motif) ligand 5 expression in keratinocytes and skin. Toxicol Sci. (2017) 160:83–94. doi: 10.1093/toxsci/kfx160
23. Tanaka Y, Uchi H, Hashimoto-Hachiya A, Furue M. Tryptophan photoproduct FICZ upregulates IL1A, IL1B, and IL6 expression via oxidative stress in keratinocytes. Oxid Med Cell Longev. (2018) 2018:9298052. doi: 10.1155/2018/9298052
24. Buckman SY, Gresham A, Hale P, Hruza G, Anast J, Masferrer J, Pentland AP. COX-2 expression is induced by UVB exposure in human skin: implications for the development of skin cancer. Carcinogenesis. (1998) 19:723–9.
25. Loomis D, Grosse Y, Lauby-Secretan B, Ghissassi F El, Bouvard V, Benbrahim-Tallaa L, et al. The carcinogenicity of outdoor air pollution. Lancet Oncol. (2013) 14:1262–3. doi: 10.1016/s1470-2045(13)70487-x
26. Burnett RT1, Pope CA III, Ezzati M, Olives C, Lim SS, Mehta S, et al. An integrated risk function for estimating the global burden of disease attributable to ambient fine particulate matter exposure. Environ Health Perspect. (2014) 122:397–403. doi: 10.1289/ehp.1307049
27. Harrison RM, Smith DJT, Kibble AJ. What is responsible for the carcinogenicity of PM2.5? Occup Environ Med. (2004) 61:799–805. doi: 10.1136/oem.2003.010504
28. Sanders CL, Skinner C, Gelman RA. Percutaneous absorption of 7, 10 14C-benzo[a]pyrene and 7, 12 14C-dimethylbenz[a]anthracene in mice. J Environ Pathol Toxicol Oncol. (1984) 7:25–34.
29. Kao J, Patterson FK, Hall J. Skin penetration and metabolism of topically applied chemicals in six mammalian species, including man: an in vitro study with benzo[a]pyrene and testosterone. Toxicol Appl Pharmacol. (1985) 81:502–16. doi: 10.1016/0041-008X(85)90421-1
30. Chu I, Dick D, Bronaugh R, Tryphonas L. Skin reservoir formation and bioavailability of dermally administered chemicals in hairless guinea pigs. Food Chem Toxicol. (1996) 34:267–76. doi: 10.1016/0278-6915(95)00112-3
31. Rao MS, Subbarao V, Prasad JD, Scarpelli DG. Carcinogenicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in the Syrian golden hamster. Carcinogenesis. (1988) 9:1677–9. doi: 10.1093/carcin/9.9.1677
32. Melendez-Colon VJ, Luch A, Seidel A, Baird WM. Cancer initiation by polycyclic aromatic hydrocarbons results from formation of stable DNA adducts rather than apurinic sites. Carcinogenesis. (1999) 20:1885–91.
33. Huang PY, Balmain A. Modeling cutaneous squamous carcinoma development in the mouse. Cold Spring Harb Perspect Med. (2014) 4:a013623. doi: 10.1101/cshperspect.a013623
34. Nassar D, Latil M, Boeckx B, Lambrechts D, Blanpain C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat Med. (2015) 21:946–54. doi: 10.1038/nm.3878
35. McCreery MQ, Halliwill KD, Chin D, Delrosario R, Hirst G, Vuong P, et al. Evolution of metastasis revealed by mutational landscapes of chemically induced skin cancers. Nat Med. (2015) 21:1514–20. doi: 10.1038/nm.3979
36. Ginos MA, Page GP, Michalowicz BS, Patel KJ, Volker SE, Pambuccian SE, et al. Identification of a gene expression signature associated with recurrent disease in squamous cell carcinoma of the head and neck. Cancer Res. (2004) 64:55–63. doi: 10.1158/0008-5472.CAN-03-2144
37. Tormo D, Ferrer A, Gaffal E, Wenzel J, Basner-Tschakarjan E, Steitz J, et al. Rapid growth of invasive metastatic melanoma in carcinogen-treated hepatocyte growth factor/scatter factor-transgenic mice carrying an oncogenic CDK4 mutation. Am J Pathol. (2006) 169:665–72. doi: 10.2353/ajpath.2006.060017
38. Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci USA. (2000) 97:779–82. doi: 10.1073/pnas.97.2.779
39. De Souza VRC, Cabrera WK, Galvan A, Ribeiro OG, De Franco M, Vorraro F, et al. Aryl hydrocarbon receptor polymorphism modulates DMBA-induced inflammation and carcinogenesis in phenotypically selected mice. Int J Cancer. (2009) 124:1478–82. doi: 10.1002/ijc.24066
40. Ide F, Suka N, Kitada M, Sakashita H, Kusama K, Ishikawa T. Skin and salivary gland carcinogenicity of 7,12-dimethylbenz[a]anthracene is equivalent in the presence or absence of aryl hydrocarbon receptor. Cancer Lett. (2004) 214:35–41. doi: 10.1016/j.canlet.2004.04.014
41. Guengerich FP. Cytochrome p450 and chemical toxicology. Chem Res Toxicol. (2008) 21:70–83. doi: 10.1021/tx700079z
42. Kleiner HE, Vulimiri SV, Reed MJ, Uberecken A, DiGiovanni J. Role of cytochrome P450 1a1 and 1b1 in the metabolic activation of 7,12-dimethylbenz[a]anthracene and the effects of naturally occurring furanocoumarins on skin tumor initiation. Chem Res Toxicol. (2002) 15:226–35. doi: 10.1021/tx010151v
43. Shimada T. Xenobiotic-metabolizing enzymes involved in activation and detoxification of carcinogenic polycyclic aromatic hydrocarbons. Drug Metab Pharmacokinet. (2006) 21:257–76. doi: 10.2133/dmpk.21.257
44. Xue P, Fu J, Zhou Y. The aryl hydrocarbon receptor and tumor immunity. Front Immunol. (2018) 9:286. doi: 10.3389/fimmu.2018.00286
45. Feng S, Cao Z, Wang X. Role of aryl hydrocarbon receptor in cancer. Biochim Biophys Acta. (2013) 1836:197–210. doi: 10.1016/j.bbcan.2013.05.001
46. Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. (2007) 445:843–50. doi: 10.1038/nature05660
47. Jux B, Kadow S, Luecke S, Rannug A, Krutmann J, Esser C. The aryl hydrocarbon receptor mediates UVB radiation-induced skin tanning. J Invest Dermatol. (2011) 131:203–10. doi: 10.1038/jid.2010.269
48. O'Donnell EF, Kopparapu PR, Koch DC, Jang HS, Phillips JL, Tanguay RL, et al. The Aryl hydrocarbon receptor mediates leflunomide-induced growth inhibition of melanoma cells. PLoS ONE. (2012) 7: doi: 10.1371/journal.pone.0040926
49. Contador-Troca M, Alvarez-Barrientos A, Barrasa E, Rico-Leo EM, Catalina-Fernández I, Menacho-Márquez M, et al. The dioxin receptor has tumor suppressor activity in melanoma growth and metastasis. Carcinogenesis. (2013) 34:2683–93. doi: 10.1093/carcin/bgt248
50. Villano CM, Murphy KA, Akintobi A, White LA. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces matrix metalloproteinase (MMP) expression and invasion in A2058 melanoma cells. Toxicol Appl Pharmacol. (2006) 210:212–24. doi: 10.1016/j.taap.2005.05.001
51. Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. (2012) 483:603–7. doi: 10.1038/nature11003
52. Coit DG, Thompson JA, Algazi A, Andtbacka R, Bichakjian CK, Carson WE, et al. Melanoma, Version 2.2016, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2016) 14:450–73. doi: 10.6004/jnccn.2016.0051
54. Luther C, Swami U, Zhang J, Milhem M, Zakharia Y. Advanced stage melanoma therapies: detailing the present and exploring the future. Crit Rev Oncol Hematol. (2019) 133:99–111. doi: 10.1016/j.critrevonc.2018.11.002
55. Colombino M, Capone M, Lissia A, Cossu A, Rubino C, De Giorgi V, et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J Clin Oncol. (2012) 30:2522–9. doi: 10.1200/JCO.2011.41.2452
56. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. (2011) 364:2507–16. doi: 10.1056/NEJMoa1103782
57. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. (2012) 380:358–65. doi: 10.1016/S0140-6736(12)60868-X
58. Long GV, Weber JS, Infante JR, Kim KB, Daud A, Gonzalez R, et al. Overall survival and durable responses in patients with BRAF V600-mutant metastatic melanoma receiving dabrafenib combined with trametinib. J Clin Oncol. (2016) 34:871–8. doi: 10.1200/JCO.2015.62.9345
59. Müller J, Krijgsman O, Tsoi J, Robert L, Hugo W, Song C, et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat Commun. (2014) 5:5712. doi: 10.1038/ncomms6712
60. Kong X, Kuilman T, Shahrabi A, Boshuizen J, Kemper K, Song JY, et al. Cancer drug addiction is relayed by an ERK2-dependent phenotype switch. Nature. (2017) 550:270–4. doi: 10.1038/nature24037
61. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. (2013) 500:415–21. doi: 10.1038/nature12477
62. Efremova M, Finotello F, Rieder D, Trajanoski Z. Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol. (2017) 8:1–8. doi: 10.3389/fimmu.2017.01679
63. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. (2015) 160:48–61. doi: 10.1016/j.cell.2014.12.033
64. Rodić N, Anders RA, Eshleman JR, Lin MT, Xu H, Kim JH, et al. PD-L1 expression in melanocytic lesions does not correlate with the BRAF V600E mutation. Cancer Immunol Res. (2015) 3:110–5. doi: 10.1158/2326-6066.CIR-14-0145
65. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
66. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. (2005) 65:1089–96.
67. Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. (2015) 372:320–30. doi: 10.1056/NEJMoa1412082
68. Liu Y, Liang X, Yin X, Lv J, Tang K, Ma J, et al. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat Commun. (2017) 8:15207. doi: 10.1038/ncomms15207
69. Liu Y, Lv J, Liu J, Liang X, Jin X, Xie J, et al. STAT3/p53 pathway activation disrupts IFN-β-induced dormancy in tumor-repopulating cells. J Clin Invest. (2018) 128:1057–73. doi: 10.1172/JCI96329
70. Wardhani LO, Matsushita M, Iwasaki T, Kuwamoto S, Nonaka D, Nagata K Expression of the IDO1/TDO2-AhR pathway in tumor cells or the tumor microenvironment is associated with Merkel cell polyomavirus status and prognosis in Merkel cell carcinoma. Hum Pathol. (2019) 84:52–61. doi: 10.1016/j.humpath.2018.09.003
Keywords: aryl hydrocarbon (Ah) receptor, squamous cell carcinoma, melanoma, ultraviolet, air pollutant, BRAF inhibitor, PD-1
Citation: Hidaka T, Fujimura T and Aiba S (2019) Aryl Hydrocarbon Receptor Modulates Carcinogenesis and Maintenance of Skin Cancers. Front. Med. 6:194. doi: 10.3389/fmed.2019.00194
Received: 06 June 2019; Accepted: 20 August 2019;
Published: 04 September 2019.
Edited by:
Peter Wolf, Medical University of Graz, AustriaReviewed by:
Thomas Haarmann-Stemmann, Leibniz-Institut Für Umweltmedizinische Forschung (IUF), GermanyMasutaka Furue, Kyushu University, Japan
Copyright © 2019 Hidaka, Fujimura and Aiba. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takanori Hidaka, dGFrYW5vcmktaEBtZWQudG9ob2t1LmFjLmpw