 Ileana Corvo1*†
Ileana Corvo1*† Florencia Ferraro2†
Florencia Ferraro2† Alicia Merlino2Kathrin Zuberbühler3
Alicia Merlino2Kathrin Zuberbühler3 Anthony J. O'Donoghue4
Anthony J. O'Donoghue4 Lucía Pastro1Natalia Pi-Denis1
Lucía Pastro1Natalia Pi-Denis1 Tatiana Basika5†Leda Roche1
Tatiana Basika5†Leda Roche1 James H. McKerrow6†Charles S. Craik3Conor R. Caffrey6†
James H. McKerrow6†Charles S. Craik3Conor R. Caffrey6† José F. Tort1
José F. Tort1- 1Departamento de Genética, Facultad de Medicina, Universidad de la República, Montevideo, Uruguay
- 2Laboratorio de Química Teórica y Computacional, Facultad de Ciencias, Universidad de la República, Montevideo, Uruguay
- 3Department of Pharmaceutical Chemistry, Pharmacology, Biochemistry and Biophysics, University of California, San Francisco, San Francisco, CA, United States
- 4Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, San Diego, CA, United States
- 5Departamento de Biología Celular y Molecular, Unidad de Biología Parasitaria, Facultad de Ciencias, Instituto de Higiene, Universidad de la República, Montevideo, Uruguay
- 6Department of Pathology, Center for Discovery and Innovation in Parasitic Diseases, University of California, San Francisco, San Francisco, CA, United States
Cysteine proteases are widespread in all life kingdoms, being central to diverse physiological processes based on a broad range of substrate specificity. Paralogous Fasciola hepatica cathepsin L proteases are essential to parasite invasion, tissue migration and reproduction. In spite of similarities in their overall sequence and structure, these enzymes often exhibit different substrate specificity. These preferences are principally determined by the amino acid composition of the active site's S2 subsite (pocket) of the enzyme that interacts with the substrate P2 residue (Schetcher and Berger nomenclature). Although secreted FhCL1 accommodates aliphatic residues in the S2 pocket, FhCL2 is also efficient in cleaving proline in that position. To understand these differences, we engineered the FhCL1 S2 subsite at three amino acid positions to render it identical to that present in FhCL2. The substitutions did not produce the expected increment in proline accommodation in P2. Rather, they decreased the enzyme's catalytic efficiency toward synthetic peptides. Nonetheless, a change in the P3 specificity was associated with the mutation of Leu67 to Tyr, a hinge residue between the S2 and S3 subsites that contributes to the accommodation of Gly in S3. Molecular dynamic simulations highlighted changes in the spatial distribution and secondary structure of the S2 and S3 pockets of the mutant FhCL1 enzymes. The reduced affinity and catalytic efficiency of the mutant enzymes may be due to a narrowing of the active site cleft that hinders the accommodation of substrates. Because the variations in the enzymatic activity measured could not be exclusively allocated to those residues lining the active site, other more external positions might modulate enzyme conformation, and, therefore, catalytic activity.
Introduction
Fasciola hepatica causes fascioliasis, a zoonotic disease that affects mainly cattle and sheep, and generates major economic loses worldwide. The World Health Organization classified it as an emerging neglected disease of humans (Keiser and Utzinger, 2009). There is an urgent need to develop new control strategies particularly with the increasing number of reports of resistance to the drug of choice, triclabendazole, in both livestock and humans (Cabada et al., 2016; Kelley et al., 2016). As in other parasites, liver fluke secreted proteases help colonize the mammalian host, and contribute to parasite development, survival and reproduction (Dalton et al., 2006; Robinson et al., 2009; Cancela et al., 2010). Papain-like cysteine proteases predominate and comprise an expanded multigenic family recently reported to have at least 14 distinct isoforms (McNulty et al., 2017). Among these, cathepsin L1 (FhCL1) and to a lesser extent- cathepsin L2 (FhCL2) are the most abundant enzymes secreted by adult flukes (Robinson et al., 2008, 2009). Despite their high sequence identity and overall structure conservation, FhCL1 and FhCL2 show distinct substrate specificities, highlighting that they might play different roles in parasite biology. The most notable difference is the ability of FhCL2 to hydrolyze peptides with Pro in the P2 position (Dowd et al., 1997; Stack et al., 2008), an activity similar to that of human cathepsin K (Choe et al., 2006). This property confers on FhCL2 the ability to cleave native collagen (Lecaille et al., 2008). A third collagen-digesting cathepsin, FhCL3, is expressed exclusively in the invasive stage and might mediate the invasion process (Cancela et al., 2008; Corvo et al., 2009, 2013). Irving et al. identified residues with positive selection in the Fasciola cathepsin family, several of which are in the active site cleft and which might account for the different specificities of the enzymes (Irving et al., 2003).
The general mechanism of catalysis of cysteine proteases relies on the nucleophilic attack of the peptide bond of the substrate by a catalytic Cys residue in the active site. The substrate specificity of the enzyme on the other hand is primarily determined by the interactions between the side chains of the residue of the substrate that is accommodated in the S2 subsite of the enzyme active site pocket (Schechter and Berger, 1967; Turk et al., 1998). Previous studies pointed to a role for the particular residues lining the S2 pocket (Table 1 and Figure 1) in conferring specific enzymes with their distinct substrate specificities (Pauly et al., 2003; Choe et al., 2006; Corvo et al., 2013). For example, positions 67 and 205 (papain numbering), respectively situated at the entrance and bottom of the S2 pocket, have an important contribution to the substrate specificities of human cathepsin K and L. The replacement of these residues in human cathepsin K with those present in human cathepsin L render the S2 preferences for short peptides similar to cathepsin L. Likewise, the converse experiment bestows cathepsin K-like specificities on cathepsin L (Lecaille et al., 2007). In Fasciola, both FhCL1 and CL2 have a Leu205 residue but they differ at position 67 where FhCL1 has Leu and FhCL2 has Tyr. However, when FhCL1 Leu67 was replaced with Tyr (as in FhCL2), the ability of the mutated enzyme to cleave peptides with Pro in P2 was not significantly increased (Stack et al., 2008). Interestingly, the same substitution in Fasciola cathepsin L5 (a minor enzyme with substrate preferences similar to FhCL1) was reported to increase its acceptance of peptides containing Pro at P2 (Smooker et al., 2000). Recently, we showed that the mutation of FhCL1 Leu67 to Trp greatly increases its ability to accommodate Gly in S3, making this pocket similar to that of FhCL2 and FhCL3 (Corvo et al., 2013), and suggesting that the ability of FhCL5 (Tyr67) to accommodate the Gly-Pro-Arg peptide was due to a better acceptance of Gly in P3.
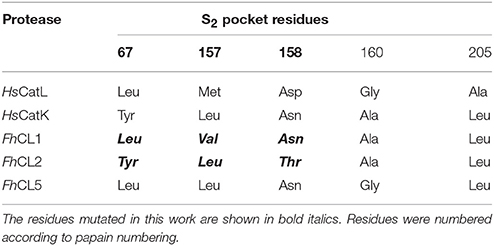
Table 1. Residues found in the S2 pocket of human cathepsin L and K (HsCatL, HsCatK) and adult F. hepatica CLs.
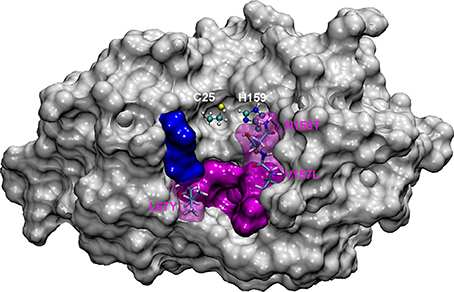
Figure 1. The most representative structure for FhCL1 is shown in gray surface, the catalytic diad Cys25 and His159 is labeled and shown in balls and sticks; pink surface represents the S2 subsite and blue surface represents the S3 subsite. Residues mutated in this work are represented in sticks and labeled.
The contribution of the other S2 residues, however, has not been assessed. Leu157 in FhCL2, which is opposite to the gatekeeper residue Tyr67, was suggested to contribute to P2-Pro interaction by stabilizing the planar ring of Pro (Stack et al., 2008). Also, Thr158 in FhCL2, which is located beside the catalytic His159, was proposed to influence the accessibility to the S2 pocket (Stack et al., 2008). Predictions of per-residue free energy decomposition studies on a FhCL3 enzyme model provided further theoretical support for the contribution of these positions (67 and 157) to substrate binding (Hernández Alvarez et al., 2015). Val157 in FhCL1 is a residue one carbon shorter than the Leu found in FhCL2 and human cathepsin K. This is too short to contribute to aliphatic interactions and led Stack et al. to suggest that the lack of both the Tyr67 and Leu157 gatekeeper residues might account for the reduced preference for P2 Pro by FhCL1 (Stack et al., 2008).
To study how the S2 pocket residues that differ between FhCL1 and FhCL2 contribute to substrate specificity, we engineered the FhCL1 S2 pocket by replacing one or more of the Leu67, Val157 and Asn158 residues in the S2 pocket (Table 1 and Figure 1), with the respective residues present in FhCL2. We analyzed the mutated enzymes by following the hydrolysis of synthetic fluorogenic peptides and by Positional Scanning- Synthetic Combinatorial Libraries of fluorogenic tetrapeptides (PS-SCL) (Choe et al., 2006). To gain further insights into the mechanisms of specificity, we performed molecular dynamic simulations on both native and mutant enzymes. Our results point to subtle differences in the secondary structure and geometry of the FhCL1 mutants that hinder substrate accommodation and might underlie their reduced affinity and catalytic efficiency. The data introduce novel aspects to consider for the rational design of cysteine protease inhibitors which have shown therapeutic utility in the control of trematode infections (Abdulla et al., 2007; Ferraro et al., 2016).
Materials and Methods
Generation of FhCL1 Mutants
Seven FhCL1 variants bearing substitutions at the S2 active site pocket were generated by site-specific mutagenesis using the QuikChange Site-Directed Mutagenesis Kit (Invitrogen). Briefly, different pairs of complementary oligonucleotides containing the base pair substitutions to be introduced in the cathepsin gene sequences were used in a PCR reaction, using as template FhCL1 cloned in the X4-Mfα-ScPas3 expression plasmid (kindly provided by Dr. R.J.S. Baerends and Dr. J.A.K.W. Kiel, Molecular Cell Biology Lab, Groningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, The Netherlands). Double and triple variants were obtained by using plasmids bearing the single (or double) mutations as templates, with the only exception of the FhCL1 V157L/N158T double variant, where a pair of oligonucleotides containing both changes was used (Supplementary Table 1). The plasmids were propagated in bacteria and sequenced to verify the presence of the desired mutations. Then, they were electroporated in the Hansenula polymorpha yeast strain as previously described (Faber et al., 1994).
Production of FhCL1 and Mutants in Yeast
FhCL1 recombinant proenzyme was produced in the yeast Hansenula polymorpha as previously described (Corvo et al., 2009). Briefly, yeast transformants were cultured in 500 ml YEPD broth at 37°C to an OD600 of 4–6, harvested by centrifugation at 2,000 g for 10 min and induced by resuspending in 50 ml of buffered minimal media (0.67% yeast nitrogen base, 0.1 M phosphate buffer, pH 6.0, 1% methanol) for 36 h at 30°C. Recombinant propeptidase was secreted to the culture media, and recovered by 20–30-fold concentration of culture supernatants by ultrafiltration with a 10 kDa cut-off membrane. The proenzyme was autocatalytically activated to the mature form by incubation for 2 h at 37°C in 0.1 M sodium citrate buffer (pH 5.0) containing 2 mM DTT and 2.5 mM EDTA, then dialyzed against PBS, pH 7.3, and stored at −20°C. The protein concentration was assessed by the BCA method (Smith et al., 1985) and the amount of active enzyme was determined by titration against the specific cysteine protease inhibitor E-64c. The mutant enzymes were obtained using the same protocol for the production of FhCL1.
Multiplex Substrate Profiling by Mass Spectrometry (MSP-MS)
The enzymatic activity of FhCL1 and FhCL2 was tested by MSP-MS, a procedure designed for unbiased profiling of protease activity consisting of the cleavage of a library of unmodified tetradecapeptides followed by mass spectrometry identification of all cleavage products (O'Donoghue et al., 2012). Briefly, a highly diversified peptide library consisting of 228 synthetic tetradecapeptides containing all possible amino acid pairs, and near neighbor pairs was used to test enzymatic activity. All peptides had unmodified termini and consist of natural amino acids except Met that was substituted by norleucine and Cys omitted because of potential disulfide bond formation. The library was distributed into two pools consisting of 114 peptides and diluted to 1 μM in 0.1 M sodium phosphate, pH6.0, 1 mM DTT, 1 mM EDTA. An equal volume of FhCL1 or FhCL2 in the same buffer was added to the peptide pools such that the final concentration of the enzyme was 1 nM and 50 nM, respectively. The enzyme concentrations used for this experiment was determined based on the activity of FhCL1 and FhCL2 against fluorescent substrates. An enzyme-free assay was set up as a control. Assays were incubated at room temperature for a total of 4 h and a 25% of the reaction volume was removed after 60 min and 4 h and the enzyme quenched by addition of formic acid to a final concentration of 4%. Samples were desalted using C18 LTS tips (Rainin) and rehydrated using 0.2% formic acid prior to mass spectrometry acquisition. Peptide sequencing an LTQ Orbitrap-XL mass spectrometer (Thermo) under identical running conditions as outlined previously (Winter et al., 2017). Mass spectrometry peak lists were generated using in-house software called PAVA. To identify peptide cleavage products, data searches were performed against the library of 228 peptides using Protein Prospector software (http://prospector.ucsf.edu/prospector/mshome.htm, UCSF). Because, in general, substrate binding pockets beyond four residues on each side of the scissile bond do not contribute to substrate specificity, we focus on the amino acids present in the P4 to P4′ positions. These octapeptide (P4–P4′) cleavage products are provided as Supplemental Data Sheet. For database searching, tolerances of 20 ppm and 0.8 Da were used for parent and fragment ions, respectively. The following variable modifications were selected with a maximum of 2 modifications per peptide: amino acid oxidation (proline, tryptophan, and tyrosine) and N-terminal pyroglutamate conversion from glutamine. Protein Prospector score thresholds were set to 22 and 15 with maximum expectation values of 0.01 and 0.05 for protein and peptide matches, respectively. Peptide cleavage products were imported into iceLogo software v.1.2 to generate protease substrate specificity profiles (Colaert et al., 2009). Octapeptides (P4–P4′) corresponding to the peptide cleavage products were used as the positive dataset, and octapeptides corresponding to all possible cleavage sites in the 228-member library (n = 2.964) minus 154 sites that were discovered in the non-enzyme treated samples, to yield a total of 2,810, were used as the negative dataset (see Supplemental Data Sheet).
Protease Assays Using Synthetic Fluorogenic Peptides
Short fluorogenic peptides are a fast and simple method to measure protease enzymatic activity and are commonly used to study substrate specificity using peptides with different sequences. Here, the protease activity was monitored by the hydrolysis of the fluorophore 7-amino-4-methyl coumarin (AMC) from the synthetic peptide substrates Z-Val-Leu-Lys-AMC and Tos-Gly-Pro-Arg-AMC (Z and Tos correspond to Carboxybenzyl and Tosyl, respectively; the blocking groups that enable cathepsin endopeptidases to position for peptide hydrolisis). The kinetic parameters were determined in a reaction buffer containing 0.1 M sodium phosphate buffer, pH 6.0, 1 mM DTT and 1 mM EDTA at 25°C with final enzyme concentrations in the 10−9 M range. Different substrate concentrations (5–100 μM) were added after a 10 min pre-incubation of the enzyme in reaction buffer and reaction rates were measured in duplicate. The slope of the progress curves were obtained by continuous recording in a FluoStar spectrofluorimeter at 345 nm excitation and 440 nm emission wavelengths, using an AMC standard curve for product concentration calculation. The enzyme concentration was determined by active-site titration with E-64c. The kinetic parameters Vmax and KM were estimated by non-linear regression analysis of the Michaelis–Menten plot using the OriginPro 6.1 software. kcat was calculated as Vmax/[E] where [E] is the active enzyme concentration (fit to the Michelis-Menten equation for FhCL1 hydrolysis of Z-Val-Leu-Lys-AMC peptide is included as an example in Supplementary Table 2). The FhCL2 recombinant enzyme for the kinetic analysis was kindly provided by Prof. John Dalton (School of Biological Sciences, Queen's University Belfast).
P1-P4 Specificity Testing Using a Positional Scanning—Synthetic Combinatorial Library (PS-SCL)
The substrate specificities of the FhCL1 and FhCL1 S2 pocket mutants were studied using a Positional Scanning- Synthetic Combinatorial Library (PS-SCL). This is composed of synthetic peptides with the general structure of acetyl-P4-P3-P2-P1-ACC, a bifunctional fluorophore leaving group similar to AMC. It consists of four libraries (P1, P2, P3, and P4) in which one position P1, P2, P3 or P4 is fixed with one of the 20 amino acids (omitting cysteine and including norleucine), whereas the remaining three positions contain an equimolar mixture of all amino acids. Each library consists of 20 sub-libraries, one for each fixed amino acid containing 8.000 different compounds (Harris et al., 2000; Choe et al., 2006). Assays were performed in triplicate in 0.1 M sodium phosphate buffer pH 6.0, 1 mM DTT, 1 mM EDTA, 0,01% PEG-6000 and 0.5% Me2SO (from the substrates) at 25°C. Aliquots of 12.5 nmol in 0.5 μl from each of the 20 sub-libraries were added to the wells of a 96-well Microfluor-1 flat-bottom plates. The final concentration of each compound of the 8.000 compounds per well was 15.62 nM in a 100 μl final reaction volume. The reaction was started by addition of the enzyme diluted in the above buffer and monitored with a SpectraMax Gemini fluorescence spectrometer (Molecular Devices) with excitation at 380 nm, emission at 460 nm and cutoff at 435 nm.
Homology Modeling and Molecular Dynamic Simulations
Homology models of FhCL1 and FhCL2 were generated with SwissModel (Arnold et al., 2006) using as the principal template the crystal structure of the pro-cathepsin FhCL1 (PDB ID: 2O6X). Template and models were superimposed for visualization with Swiss PDBViewer version 4.1. (Guex and Peitsch, 1997; http://www.expasy.org/spdbv/). Active site residues were identified based on the literature and confirmed by structural alignment with human cathepsin L (PDB ID: 1MHW), human cathepsin K (PDB ID: 1ATK) and papain (PDB ID: 5PAD). The FhCL1 mutants were generated with DS Visualizer. In order to improve structure accuracy, MD simulations were performed using the pmemd module implemented in the AMBER14 package (Case et al., 2014), with the ff14SB force field (Maier et al., 2015). Hydrogen atoms and sodium ions (to neutralize charge) were added to each protein with the leap utility. Each system was placed in a truncated octahedral box of TIP3P explicit water (Jorgensen et al., 1983) that was extended 12 Å outside the protein on all sides. The structures of FhCL1, FhCL2 and the FhCL1 mutants were treated as follows: water and counter ions were relaxed over 2,500 steps (500 steepest descent steps, SD, and 2,000 conjugate-gradient steps, CG) with the protein restrained with a force constant of 500 kcal/molÅ2. Then, the systems were minimized without restraints during 20,000 steps (5,000 SD and 15,000 CG). The cutoff distance for direct calculation of non-bonded interactions was set to 10 Å. Beyond this distance, electrostatic interactions were calculated using the Particle-Mesh-Ewald (PME) method (Essmann et al., 1995). After minimization, each system was gradually heated in a NVT ensemble from 0 to 300 K over 100 ps using the Berendsen coupling algorithm (Berendsen et al., 1984). This procedure was followed by 50 ns of NPT simulations at 300 K and 1 atm pressure using the Monte Carlo barostate algorithm (Faller and De Pablo, 2002). All bonds involving hydrogen atoms were constrained using the SHAKE algorithm (Ryekaert et al., 1997). The equations of motion were integrated with a time step of 2.0 fs and coordinates of the systems were saved every 2 ps. Representative structures of FhCL1, FhCL1 mutants and FhCL2 from the trajectories were obtained through cluster analysis using the average-linkage algorithm (Shao et al., 2007) and used for further inspection of the structures. Clustering, RMSD, RMSF, hydrogen bond and DSSP analysis were performed using the cpptraj module in AmberTools14. For trajectories, visualization and figures of this paper the VMD program was used (Humphrey et al., 1996).
Results and Discussion
FhCL1 and FhCL2 Multiple Substrate Profiling by Mass Spectrometry (MSP-MS)
We employed the MSP-MS method to compare the specificity profile of the two main cathepsins of adult liver flukes. This method has been used to uncover the substrate specificity profile of proteases resulting in the development of optimized fluorescent substrates (O'Donoghue et al., 2015; Winter et al., 2017), peptide inhibitors (Li et al., 2016; Roncase et al., 2017), activity based probes (Lentz et al., 2016) and activity based biomarkers (Ivry et al., 2018). After 1 h incubation of each enzyme with the peptide library, FhCL1 and FhCL2 cleaved at 46 and 54 sites, respectively. Incubation for a further 3 h yielded 79 and 107 sites, respectively (Supplemental Data Sheet). We generated a substrate specificity profile using the P4 to amino acid frequency of the confirmed cleavage sites by comparing it to the amino acid frequency of all possible cleavage sites within the peptide library (n = 2.810). This profile confirmed that the S2-P2 interactions dominated the substrate specificity for both enzymes, as expected for clan CA cysteine proteases (Turk et al., 1998; Pauly et al., 2003; Figures 2A,B). Both enzymes preferentially cleaved at sites where Leu, Ile and norleucine were in the P2 position, however, FhCL1 also cleaved when Val was in the P2 position, while FhCL2 preferred Pro, consistent with previous reports (Dowd et al., 1997; Stack et al., 2008). Both enzymes share a preference for Lys and Arg at the P1 position and Trp and Phe at . When the cleavages sites were directly compared, 59 of the sites were cut by both enzymes, however 48 sites were uniquely cleaved by FhCL2 (Figure 2C). We next investigated the positon of cleaved within the 14-mer peptides and found that neither enzymes have aminopeptidase or carboxypeptidase activity since no peptide bonds in between residues 1 and 2 or between residues 13 and 14 were cleaved. Interestingly, many of the sites that are cleaved by FhCL2 and not by FhCL1 are found between residues 3 and 4, indicating that FhCL2 may have some tripeptidyl-peptidase activity that is absent in FhCL1 (Figure 2D).
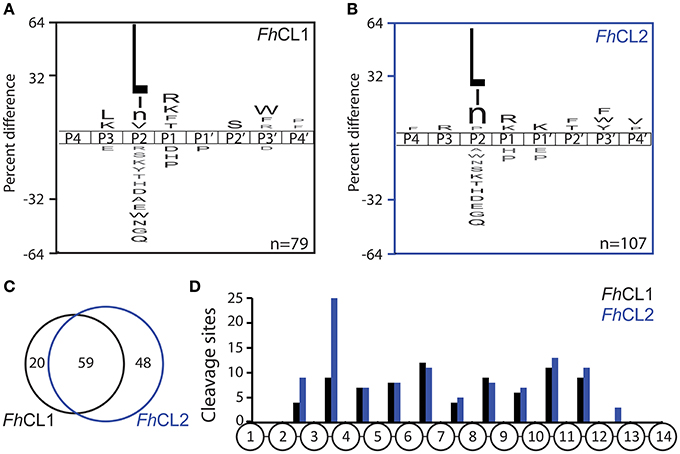
Figure 2. Differential cleavage specificities of FhCL1 and FhCL2 using MSP-MS. A. iceLogo plot illustrating the frequency of amino acids found in the P4-P4′ position of (A) FhCL1 and (B) FhCL2, following incubation with the 228-member peptide library for 4 h. Amino acids that are significantly enriched (top) or de-enriched (bottom) (p ≤ 0.05) are colored black. Lowercase “n” in the amino acid list corresponds to norleucine. The number of cleavage sites used to build the iceLogo plot is indicated in the right lower corner of the plot. (C) Venn diagram showing the number of shared and unique cleavage sites for FhCL1 (black) and FhCL2 (blue). (D) Distribution of the cleavage sites observed within the 14-mer peptides.
Kinetic Analysis of the Cathepsin Mutants With Short Peptide Substrates
To test the contribution of different active site residues involved in the enzymatic activity and amino acid preference of FhCLs we engineered the FhCL1 S2 pocket to resemble that of FhCL2. The changes correspond to key residues at the entrance and wall of the S2 pocket that had been proposed by us and others to be important for substrate recognition (Table 1; Stack et al., 2008; Corvo et al., 2009, 2013). We then employed two different tripeptide substrates that contain Leu or Pro at P2 to evaluate the mutant specificities.
The fluorescent substrate Z-Val-Leu-Lys-AMC is hydrolyzed by FhCL1 and FhCL2 with similar efficiency, however, Tos-Gly-Pro-Arg-AMC is cleaved by FhCL2 with >50-fold higher efficiency mostly due to a higher kcat (Table 2). All the amino acid changes introduced result in a decrease in the hydrolysis of Z-Val-Leu-Lys-AMC, either due to a diminution of the kcat (substitutions Leu67Tyr and Val157Leu), or a reduction in affinity associated with the Asn158Thr mutation (Table 2). These results suggest that the mutated FhCL1 S2 subsite cannot interact properly with the substrate, or does it in an orientation that is unfavorable for catalysis.
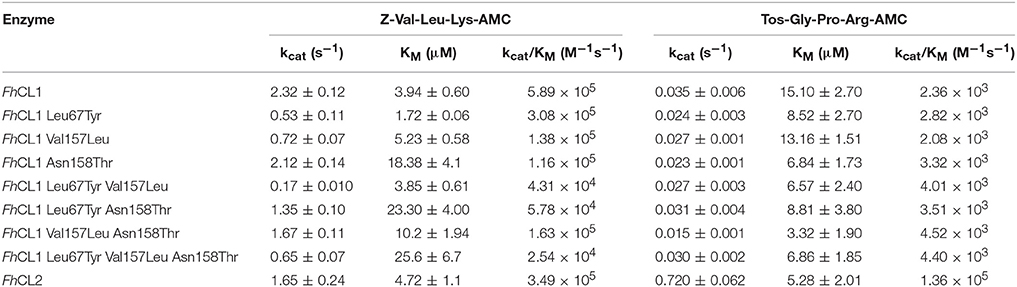
Table 2. Kinetic parameters of FhCL1, FhCL2 and the mutant enzymes over two different substrates.
An increased affinity for Tos-Gly-Pro-Arg-AMC was generally observed in the mutants, but as the kcat is slightly diminished, the specificity remains similar to that of FhCL1. This behavior has also been observed by Stack et al. for the substitution Leu67Tyr in FhCL1 (Stack et al., 2008). In the adult enzyme FhCL5, however, the Leu67Tyr mutation provoked an increase in kcat and activity toward Tos-GPR-AMC (3- to 10-fold as reported by Smooker et al., 2000; Norbury et al., 2012), an effect proposed to be due to the presence of the bulkier Leu at 157 that is also positioned at the entrance of the S2 pocket opposite residue 67. Yet, in our experiments, the substitution Val157Leu in FhCL1, when combined with Leu67Tyr did not show a significant increase in specificity for Tos-Gly-Pro-Arg-AMC.
P2 and P3 Preferences of FhCL1 Mutants Based on PS-SCL
A second step in the characterization of the mutant enzymes was assessing their individual amino acid preferences by PS-SCL (Choe et al., 2006). In Figure 3 we show the specificity profile of FhCL1 and the mutant enzymes at the P2 and P3 position of the substrates. We found that neither of the S2 mutations alone or in combination could significantly modify the FhCL1 preferences at P2. Even the triple mutant, in which the S2 pocket of FhCL1 has exactly the same residues as FhCL2, did not increase significantly its acceptance of Pro in that position (Figure 3H). However, the substitution Leu67Tyr increased the acceptance of Gly in P3 (black colored bar), both in the single change variant (Figure 3B) and in the double or triple substitutions (Figures 3F–H). We corroborated that the P1 preferences were not modified by any of the mutations (data not shown). These results suggest that even when the FhCL1 residues lining the S2 pocket are identical to those in FhCL2, the overall conformation of the subsite remains different as the FhCL1 triple mutant cannot favorably interact with a P2 Pro. However, the S3 specificity of FhCL2 can be mimicked in FhCL1 when replacing Leu for Tyr at position 67, conferring it a preference for Gly that is similar to that observed in FhCL2 and FhCL3 (compare with Figure 3 of Stack et al., 2008; Corvo et al., 2013). In summary, the better accommodation of Gly in P3 as shown by the PS-SCL slightly improves the affinity of the FhCL1 mutants bearing the Leu67Tyr substitution for substrates such as Tos-Gly-Pro-Arg-AMC but the interaction does not promote a more efficient hydrolysis of this peptide.
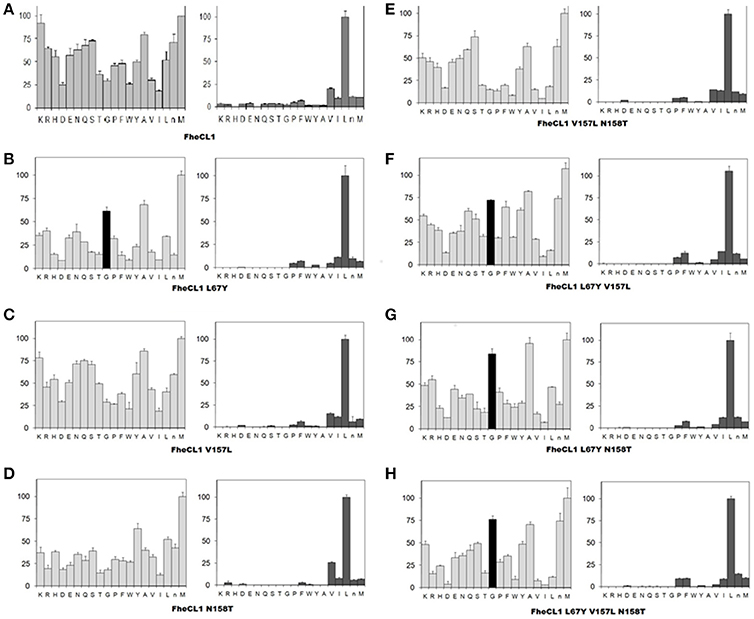
Figure 3. Profiling of the P2 and P3 substrate specificity of FhCL1 enzyme variants using PS-SCL (A–H). The y axis shows the enzymatic activity, against each of the 20 sub-libraries with P2 and P3 position of the substrates fixed, as a percentage relative to the highest activity in each library (the hydrolysis rates for the Leu- and Met- fixed peptide pools at P2 and P3, respectively, are taken as 100%). The x axis shows the different fixed amino acids using the one-letter code (n = norleucine). The error bars display the standard deviation from triplicate experiments.
Molecular Dynamics of the Active Site Configuration
To further investigate the active site determinants, we studied the differences in the overall conformation and the secondary structure between the representative structures of FhCL1 and FhCL2 obtained by hierarchical clustering. Then, we compared the structural features of the native enzymes with those of the FhCL1 mutants. In order to improve structure accuracy we performed molecular dynamics simulations over the structures previously obtained by homology modeling (Corvo et al., 2013). We measured subtle changes in the protein backbones and minor structural variation between the enzymes particularly in the loops 1 and 3 which comprises residues 52–67 and 149–159 of the mature enzyme, respectively (Figure 4 and Supplementary Figure 1). A recent structural study of human cathepsin K and related papain-like proteases showed that loops edging the S1, S2, and S3 pockets of the active site are flexible (Novinec, 2017). In the FhCL1 mutants, loop 1 adopts a 310 helix conformation with a higher frequency than the native enzymes (Figure 4). Similarly, on the other side of the active site, loop 3 also tends to form a 310 helical arrangement while reducing the turn configuration, changes that would result in a more rigid structure (Figure 4). Furthermore, a hydrogen bond, formed between residues Asn60–Gly65 in the S3 pocket of the FhCL1 mutant enzymes, might contribute to forming the helical configuration seen in the proximities of this site. Taken together, our data suggest that minor conformational changes at the edges of the active site may be sufficient to modify these enzymes' activity.
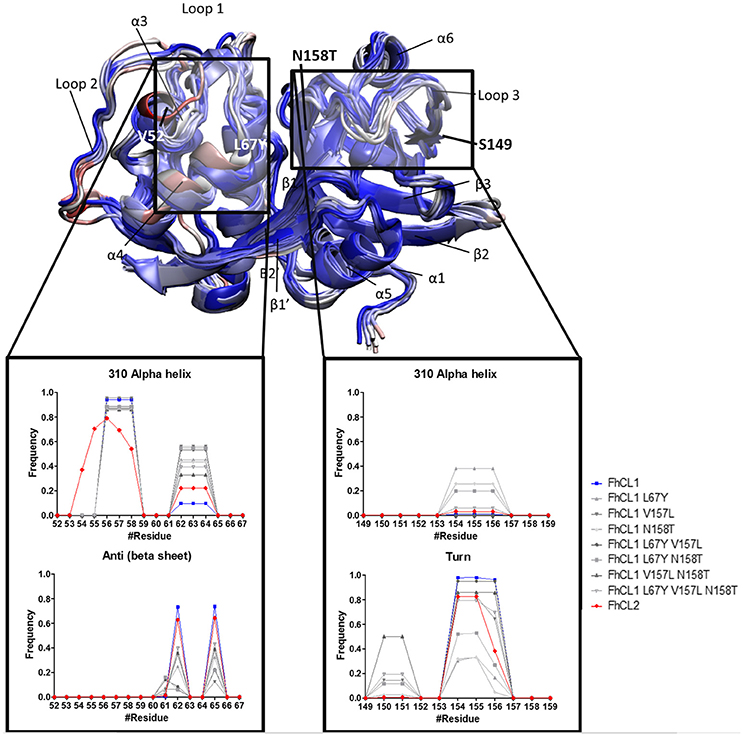
Figure 4. Standardized Definition of Secondary Structure Protein (DSSP) from the S2 and S3 subsites, and surrounding residues. Representative structures of FhCL1, FhCL2 and mutants are shown in new cartoons colored by RMSD of the alpha carbon by residue. Labels indicate main components of the enzyme structure and the rectangles mark the regions analyzed in the graphics. The graphics show the frequency values obtained from DSSP for residues 52–67 (S2 and its surroundings) and 149–159 (S3 and its surroundings).
The S2 site represents the narrowest part of the active site cleft and is considered the major specificity determinant in papain-like proteases (Schechter and Berger, 1967). Besides the residues mutated in this work, a conserved glycine from the S2 wall, Gly65, was predicted to be a main contributor to substrate binding (Hernández Alvarez et al., 2015). Hence, we compared the interatomic distances that define the geometry of this pocket, measuring the distances between Gly65:CA—Thr158:O, Gly65:CA—Thr158:CB and Gly66:O—Val157:O (Figure 5 and Table 3). We also studied the conformation of the S3 subsite, as residue 67 also modulates its architecture, and computed the distances among Asn61:CG—Tyr67:CG (Figure 4 and Table 3). Whereas the S2 bottom and S3 sites of FhCL2 are more constrained than those of FhCL1, the cleft tends to narrow even more in the FhCL1 mutants (around 1.2 Å and 2 Å shorter than FhCL2; Table 3). This may account for their reduced activity toward the Z-Val-Leu-Lys-AMC substrate. Also, a shortening is seen in the width of the less defined S3 pocket when Leu 67 of FhCL1 is replaced by Tyr as in FhCL2 (Table 3). This result supports the experimental data previously seen (Figure 3), because mutants bearing this substitution accept Gly better in S3, a characteristic of the FhCL2 enzyme.
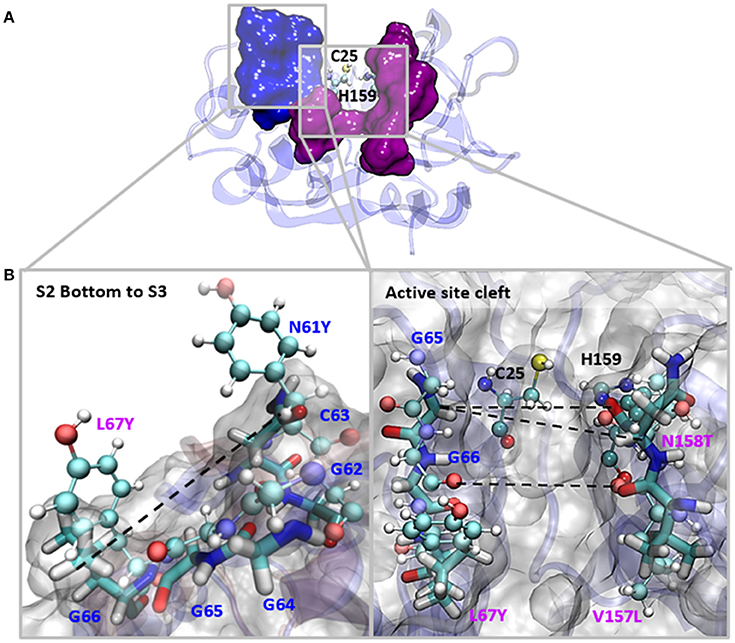
Figure 5. Interatomic distances between residues in the S2 and S3 sites in MD simulations. (A) The most representative structure for FhCL1 is depicted in blue new cartoons, in ball and sticks are depicted Cys25 and His159; blue surface represents the S3 residues and pink surface represents the S2 residues. (B) We zoomed the regions in the squares and show the measured distances with dashed lines. Blue labels show the S3 residues and pink labels show the S2 residues. FhCL1 residues are depicted in sticks and FhCL2 residues are represented in ball and sticks.
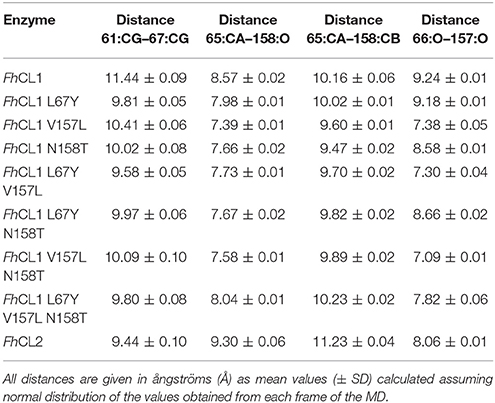
Table 3. Interatomic distances measured as shown in Figure 5 between selected residues in the S2 and S3 sites in MD simulations.
Recent studies support the fact that the width of the active site pockets is important for a productive enzyme-substrate interaction. Specifically, the binding of a substrate in the active site of human cathepsin K stabilizes the enzyme in a conformation that has a wider S2-S1 site, while the free enzyme displays a narrower site that hampers substrate accommodation (Novinec, 2017). Similarly, the reduced width of the S2 and S3 pockets of FhCL1 mutants hinder the binding of substrates into the active site.
In summary, molecular dynamics simulations explain the weak ability of the FhCL1 mutants to cleave after Pro in P2 as their S2 pocket configuration does not resemble that of FhCL2.
Functional Implications
Although active site conformation is very similar in the papain-family of enzymes, our results provide additional evidence to previous data (Novinec, 2017) suggesting that minor structural changes affect enzyme specificity. Here, we have focused on the differences in the S2 active site pocket, finding that despite its central role, it alone cannot fully explain substrate preferences, as seen for human cathepsins (Lecaille et al., 2007). There are clearly residues outside the active site cleft that modulate its conformation, probably belonging to the so-called “protein sectors.” These are dispersed networks of residues spatially contiguous in the structure of a protein that co-evolve and are implicated in structural and functional properties, for example ligand binding specificity and allosteric communication (McLaughlin et al., 2012). For the papain-like cysteine protease family, a single protein sector was identified which shows a continuous spatial distribution of residues around the active site and extends far throughout both domains of the protein (Novinec et al., 2014). Many, but not all, of the sector residues are conserved between the different liver fluke CLs. Our results suggest that the remaining variable residues outside the S2 pocket delineate the architecture of the active site and contribute,via long range dynamic correlation patterns to the correct positioning of the substrate. Further structural predictions corroborated by experimental data could provide evidence for this hypothesis in the future.
Author Contributions
IC, LR, and JT: conceived and designed the mutagenesis experiments; IC, LP, and NP-D: performed the mutagenesis experiments and kinetic measurements; FF and AM: performed the molecular dynamics simulations; IC, AO, KZ, and TB: performed the MSP-MS and the PSSCL experiments; IC, AM, JM, CRC, CSC, and JT: contributed reagents, materials and analysis tools; IC, FF, AM, AO, CRC, LR, and JT: analyzed and interpreted the data; IC and JT: wrote the paper.
Funding
This work was supported by Universidad de la República, Uruguay and the PEDECIBA Program. These studies were funded in part by NIH R01 GM104659 to CSC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. R.J.S. Baerends and Dr. J.A.K.W. Kiel, Molecular Cell Biology Lab, Groningen Biomolecular Sciences and Biotechnology Institute for kindly providing the yeast expression system. We are grateful to Dr. John Dalton for providing the recombinant FhCL2 for the kinetic analysis and Dr. Alberto Rascon and Brian Suzuki for their valuable help with experiments at UCSF.
We also want to thank Comisión Sectorial de Investigación Científica (CSIC) from Uruguay for providing a travel grant to IC to perform experiments in the Center for Discovery and Innovation in Parasitic Diseases, UCSF (now at UCSD).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2018.00040/full#supplementary-material
References
Abdulla, M. H., Lim, K. C., Sajid, M., McKerrow, J. H., and Caffrey, C. R. (2007). Schistosomiasismansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 4:e14. doi: 10.1371/journal.pmed.0040014
Arnold, K., Bordoli, L., Kopp, J., and Schwede, T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. doi: 10.1093/bioinformatics/bti770
Berendsen, H. J. C., Postma, J. P. M., Van Gunsteren, W. F., Haak, A. R. H. J., and Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3584–3690. doi: 10.1063/1.448118
Cabada, M. M., Lopez, M., Cruz, M., Delgado, J. R., Hill, V., White, A. C., et al. (2016). Treatment failure after multiple courses of triclabendazole among patients with fascioliasis in cusco, peru: a case series. PLoS Negl. Trop. Dis. 10:e0004361. doi: 10.1371/journal.pntd.0004361
Cancela, M., Acosta, D., Rinaldi, G., Silva, E., Durán, R., Roche, L., et al. (2008). A distinctive repertoire of cathepsins is expressed by juvenile invasive Fasciola hepatica. Biochimie 90, 1461–1475. doi: 10.1016/j.biochi.2008.04.020
Cancela, M., Ruetalo, N., Dell'Oca, N., da Silva, E., Smircich, P., Rinaldi, G., et al. (2010). Survey of transcripts expressed by the invasive juvenile stage of the liver fluke Fasciola hepatica. BMC Genomics 11:227. doi: 10.1186/1471-2164-11-227
Case, D. A., Babin, V., Berryman, J. T., Kollman, P. A., et al. (2014). AMBER 14. San Francisco, CA: University of California.
Choe, Y., Leonetti, F., Greenbaum, D. C., Lecaille, F., Bogyo, M., Brömme, D., et al. (2006). Substrate profiling of cysteine proteases using a combinatorial peptide library identifies functionally unique specificities. J. Biol. Chem. 281, 12824–12832. doi: 10.1074/jbc.M513331200
Colaert, N., Helsens, K., Martens, L., Vandekerckhove, J., and Gevaert, K. (2009). Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–797. doi: 10.1038/nmeth1109-786
Corvo, I., Cancela, M., Cappetta, M., Pi-Denis, N., Tort, J. F., and Roche, L. (2009). The major cathepsin L secreted by the invasive juvenile Fasciola hepatica prefers proline in the S2 subsite and can cleave collagen. Mol. Biochem. Parasitol. 167, 41–47. doi: 10.1016/j.molbiopara.2009.04.005
Corvo, I., O'Donoghue, A. J., Pastro, L., Pi-Denis, N., Eroy-Reveles, A., Roche, L., et al. (2013). Dissecting the active site of the collagenolytic cathepsin L3 protease of the invasive stage of Fasciola hepatica. PLoS Negl. Trop. Dis. 7:e2269. doi: 10.1371/journal.pntd.0002269
Dalton, J., Caffrey, C. R., Sajid, C. R., Stack, M., Donnelly, C., Loukas, S., et al. (2006). “Proteases in trematode biology,” in Parasitic Flatworms: Molecular Biology, Biochemistry, Immunology and Physiology, eds A. G. Maule and N. J. Marks (Oxford: CAB International), 348–368.
Dowd, A. J., Tort, J. F., Roche, L., Ryan, T., and Dalton, J. P. (1997). Isolation of a cDNA encoding Fasciola hepatica cathepsin L2 and functional expression in Saccharomyces cerevisiae. Mol. Biochem. Parasitol. 88, 163–174.
Essmann, U., Perera, L., Berkowit, M. L., Darden, T., Lee, H., and Pedersen, L. (1995). A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593. doi: 10.1063/1.470117
Faber, K. N., Haima, P., Harder, W., Veenhuis, M., and Ab, G. (1994). Highly-efficient electrotransformation of the yeast Hansenula polymorpha. Curr. Genet. 25, 305–310. doi: 10.1007/BF00351482
Faller, R., and De Pablo, J. J. (2002). Constant pressure hybrid molecular dynamics–Monte Carlo simulations. J. Chem. Phys. 116, 55–59. doi: 10.1063/1.1420460
Ferraro, F., Merlino, A., Dell'Oca, N., Gil, J., Tort, J. F., Gonzalez, M., et al. (2016). Identification of chalcones as Fasciola hepatica cathepsin L inhibitors using a comprehensive experimental and computational approach. PLoS Negl. Trop. Dis. 10:e0004834. doi: 10.1371/journal.pntd.0004834
Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723. doi: 10.1002/elps.1150181505
Harris, J. L., Backes, B. J., Leonetti, F., Mahrus, S., Ellman, J. A., and Craik, C. S. (2000). Rapid and general profiling of protease specificity by using combinatorial fluorogenic substrate libraries. Proc. Natl. Acad. Sci. U.S.A. 97, 7754–7759. doi: 10.1073/pnas.140132697
Hernández Alvarez, L., Naranjo Feliciano, D., Hernández González, J. E., de Oliveira Soares, R., Barreto Gomes, D. E., and Pascutti, P. G. (2015). Insights into the interactions of Fasciola hepatica Cathepsin L3 with a substrate and potential novel inhibitors through in silico approaches. PLoS Negl. Trop. Dis. 9:e0003759. doi: 10.1371/journal.pntd.0003759
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD - Visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Irving, J. A., Spithill, T. W., Pike, R. N., Whisstock, J. C., and Smooker, P. M. (2003). The evolution of enzyme specificity in Fasciola spp. J. Mol. Evol. 57, 1–15. doi: 10.1007/s00239-002-2434-x.
Ivry, S. L., Meyer, N. O., Winter, M. B., Bohn, M. F., Knudsen, G. M., O'Donoghue, A. J., et al. (2018). Global substrate specificity profiling of post-translational modifying enzymes. Protein Sci. 27, 584–594. doi: 10.1002/pro.3352
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W., and Klein, M. L. (1983). Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935. doi: 10.1063/1.445869
Keiser, J., and Utzinger, J. (2009). Food-borne trematodiases. Clin. Microbiol. Rev. 22, 466–483. doi: 10.1128/CMR.00012-09
Kelley, J. M., Elliott, T. P., Beddoe, T., Anderson, G., Skuce, P., and Spithill, T. W. (2016). Current threat of triclabendazole resistance in Fasciola hepatica. Trends Parasitol. 32, 458–469. doi: 10.1016/j.pt.2016.03.002
Lecaille, F., Brömme, D., and Lalmanach, G. (2008). Biochemical properties and regulation of cathepsin K activity. Biochimie 90, 208–226. doi: 10.1016/j.biochi.2007.08.011
Lecaille, F., Chowdhury, S., Purisima, E., Brömme, D., and Lalmanach, A. (2007). The S2 subsites of cathepsins K and L and their contribution to collagen degradation. Protein Sci. 16, 662–670. doi: 10.1110/ps.062666607
Lentz, C. S., Ordonez, A. A., Kasperkiewicz, P., La Greca, F., O'Donoghue, A. J., Schulze, C. J., et al. (2016). Design of selective substrates and activity-based probes for Hydrolase Important for Pathogenesis 1 (HIP1) from Mycobacterium tuberculosis. ACS Infect Dis. 2, 807–815. doi: 10.1021/acsinfecdis.6b00092
Li, H., O'Donoghue, A. J., van der Linden, W. A., Xie, S. X., Yoo, E., Foe, I. T., et al. (2016). Structure and function based design of Plasmodium-selective proteasome inhibitors. Nature. 530, 233–236. doi: 10.1038/nature16936
Maier, J. A., Martinez, C., Kasavajhala, K., Wickstrom, L., Hauser, K. E., and Simmerling, C. (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713. doi: 10.1021/acs.jctc.5b00255
McLaughlin, R. N. Jr., Poelwijk, F. J., Raman, A., Gosal, W. S., and Ranganathan, R. (2012). The spatial architecture of protein function and adaptation. Nature 491, 138–142. doi: 10.1038/nature11500
McNulty, S. N., Tort, J. F., Rinaldi, G., Fischer, K., Rosa, B. A., Smircich, P., et al. (2017). Genomes of Fasciola hepatica from the americas reveal colonization with neorickettsia endobacteria related to the agents of potomac horse and human sennetsu fevers. PLoS Genet. 13:e1006537. doi: 10.1371/journal.pgen.1006537
Norbury, L. J., Hung, A., Beckham, S., Pike, R. N., Spithill, T. W., Craik, C. S., et al. (2012). Analysis of Fasciola cathepsin L5 by S2 subsite substitutions and determination of the P1-P4 specificity reveals an unusual preference. Biochimie 94, 1119–1127. doi: 10.1016/j.biochi.2012.01.011
Novinec, M. (2017). Computational investigation of conformational variability and allostery in cathepsin K and other related peptidases. PLoS ONE 12:e0182387. doi: 10.1371/journal.pone.0182387
Novinec, M., Korenc, M., Caflisch, A., Ranganathan, R., Lenarc, B., and Baici, A. (2014). A novel allosteric mechanism in the cysteine peptidase cathepsin K discovered by computational methods. Nat. Commun. 5, 1–10. doi: 10.1038/ncomms4287
O'Donoghue, A. J., Eroy-Reveles, A. A., Knudsen, G. M., Ingram, J., Zhou, M., Statnekov, J. B., et al. (2012). Global identification of peptidase specificity by multiplex substrate profiling. Nat. Methods. 9, 1095–1100. doi: 10.1038/nmeth.2182
O'Donoghue, A. J., Knudsen, G. M., Beekman, C., Perry, J. A., Johnson, A. D., DeRisi, J. L., et al. (2015). Destructin-1 is a collagen-degrading endopeptidase secreted by Pseudogymnoascus destructans, the causative agent of white-nose syndrome. Proc. Natl. Acad. Sci. U.S.A. 112, 7478–7483. doi: 10.1073/pnas.1507082112
Pauly, T. A., Sulea, T., Ammirati, M., Sivaraman, J., Danley, D. E., Griffor, M. C., et al. (2003). Specificity determinants of human cathepsin S revealed by crystal structures of complexes. Biochemistry 42, 3203–3213. doi: 10.1021/bi027308i
Robinson, M. W., Menon, R., Donnelly, S. M., Dalton, J. P., and Ranganathan, S. (2009). An integrated transcriptomics and proteomics analysis of the secretome of the helminth pathogen Fasciola hepatica: proteins associated with invasion and infection of the mammalian host. Mol. Cell. Proteomics 8, 1891–1907. doi: 10.1074/mcp.M900045-MCP200
Robinson, M. W., Tort, J. F., Lowther, J., Donnelly, S. M., Wong, E., Xu, W., et al. (2008). Proteomics and phylogenetic analysis of the cathepsin L protease family of the helminth pathogen Fasciola hepatica: expansion of a repertoire of virulence-associated factors. Mol. Cell. Proteomics 7, 1111–1123. doi: 10.1074/mcp.M700560-MCP200
Roncase, E. J., Moon, C., Chatterjee, S., González-Páez, G. E., Craik, C. S., O'Donoghue, A. J., et al. (2017). Substrate profiling and high resolution co-complex crystal structure of a secreted C11 protease conserved across commensal bacteria. ACS Chem. Biol. 12, 1556–1565. doi: 10.1021/acschembio.7b00143
Ryekaert, J. P., Ciecotti, G., and Berendsen, H. J. C. (1997). Numerical-integration of cartesian equations of motion of a system with constraints -molecular-dynamics of N-alkanes. J. Comput. Chem. 23, 327–341.
Schechter, I., and Berger, A. (1967). On the size of the active site in proteases, I papain. Biochem. Biophys. Res. Commun. 27, 157–162. doi: 10.1016/S0006-291X(67)80055-X
Shao, J., Tanner, S. W., Thomson, N., and Cheatham, T. E. (2007). Clustering molecular dynamics trajectories: 1. characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 3, 2312–2334. doi: 10.1021/ct700119m
Smith, P. K., Krohn, R. I., Hermanson, G. T., Mallia, A. K., Gartner, F. H., Provenzano, M. D., et al. (1985). Measurement of protein using bicinchoninic acid. Anal Biochem. 150, 76–85.
Smooker, P. M., Whisstock, J. C., Irving, J. A., Siyaguna, S., Spithill, T. W., and Pike, R. N. (2000). A single amino acid substitution affects substrate specificity in cysteine proteinases from Fasciola hepatica. Protein Sci. 9, 2567–2572. doi: 10.1110/ps.9.12.2567
Stack, C. M., Caffrey, C. R., Donnelly, S. M., Seshaadri, A., Lowther, J., Tort, J. F., et al. (2008). Structural and functional relationships in the virulence-associated cathepsin L proteases of the parasitic liver fluke, Fasciola hepatica. J. Biol. Chem. 283, 9896–9908. doi: 10.1074/jbc.M708521200
Turk, D., Guncar, G., Podobnik, M., and Turk, B. (1998). Revised definition of substrate binding sites of papain-like cysteine proteases. Biol. Chem. 379, 137–147.
Keywords: Fasciola hepatica, cathepsin L, active site conformation, S2 pocket, mutagenesis, molecular dynamics simulation
Citation: Corvo I, Ferraro F, Merlino A, Zuberbühler K, O'Donoghue AJ, Pastro L, Pi-Denis N, Basika T, Roche L, McKerrow JH, Craik CS, Caffrey CR and Tort JF (2018) Substrate Specificity of Cysteine Proteases Beyond the S2 Pocket: Mutagenesis and Molecular Dynamics Investigation of Fasciola hepatica Cathepsins L. Front. Mol. Biosci. 5:40. doi: 10.3389/fmolb.2018.00040
Received: 29 January 2018; Accepted: 03 April 2018;
Published: 19 April 2018.
Edited by:
Thomas Simonson, École Polytechnique, FranceReviewed by:
Ernesto Jorge Fuentes, University of Iowa, United StatesEfstratios Stratikos, National Centre of Scientific Research Demokritos, Greece
Copyright © 2018 Corvo, Ferraro, Merlino, Zuberbühler, O'Donoghue, Pastro, Pi-Denis, Basika, Roche, McKerrow, Craik, Caffrey and Tort. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ileana Corvo, aWxlY29ydm9AZ21haWwuY29t
†Present Address: Ileana Corvo, Florencia Ferraro, Laboratorio de I+D de Moléculas Bioactivas, CENUR Litoral Norte – Sede Paysandú, Universidad de la República, Paysandú, Uruguay
Tatiana Basika, Unidad de Biología Molecular, Institut Pasteur de Montevideo, Montevideo, Uruguay
James H. McKerrow and Conor R. Caffrey, Center for Discovery and Innovation in Parasitic Diseases, Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California, San Diego, San Diego, CA, United States