 Helena Tossavainen1
Helena Tossavainen1 Vytas Raulinaitis2
Vytas Raulinaitis2 Linda Kauppinen3
Linda Kauppinen3 Ulla Pentikäinen3,4,5
Ulla Pentikäinen3,4,5 Hannu Maaheimo6
Hannu Maaheimo6 Perttu Permi1,3*
Perttu Permi1,3*- 1Department of Chemistry, Nanoscience Center, University of Jyvaskyla, Jyvaskyla, Finland
- 2Program in Structural Biology and Biophysics, Institute of Biotechnology, University of Helsinki, Helsinki, Finland
- 3Department of Biological and Environmental Science, University of Jyvaskyla, Jyvaskyla, Finland
- 4Institute of Biomedicine, University of Turku, Turku, Finland
- 5Turku Centre for Biotechnology, Turku, Finland
- 6VTT Technical Research Centre of Finland Ltd., Espoo, Finland
Lysostaphin from Staphylococcus simulans and its family enzymes rapidly acquire prominence as the next generation agents in treatment of S. aureus infections. The specificity of lysostaphin is promoted by its C-terminal cell wall targeting domain selectivity toward pentaglycine bridges in S. aureus cell wall. Scission of these cross-links is carried out by its N-terminal catalytic domain, a zinc-dependent endopeptidase. Understanding the determinants affecting the efficiency of catalysis and strength and specificity of interactions lies at the heart of all lysostaphin family enzyme applications. To this end, we have used NMR, SAXS and molecular dynamics simulations to characterize lysostaphin structure and dynamics, to address the inter-domain interaction, the enzyme-substrate interaction as well as the catalytic properties of pentaglycine cleavage in solution. Our NMR structure confirms the recent crystal structure, yet, together with the molecular dynamics simulations, emphasizes the dynamic nature of the loops embracing the catalytic site. We found no evidence for inter-domain interaction, but, interestingly, the SAXS data delineate two preferred conformation subpopulations. Catalytic H329 and H360 were observed to bind a second zinc ion, which reduces lysostaphin pentaglycine cleaving activity. Binding of pentaglycine or its lysine derivatives to the targeting domain was found to be of very low affinity. The pentaglycine interaction site was located to the N-terminal groove of the domain. Notably, the targeting domain binds the peptidoglycan stem peptide Ala-D-γ-Glu-Lys-D-Ala-D-Ala with a much higher, micromolar affinity. Binding site mapping reveals two interaction sites of different affinities on the surface of the domain for this peptide.
Introduction
The gram-positive pathogen Staphylococcus aureus causes numerous diseases, ranging from minor skin abscesses to meningitis and even toxic shock syndrome (Lowy, 1998). S. aureus bacteremia is estimated to be fatal in up to 30 % of cases (van Hal et al., 2012) and annually incur billions in overall medical costs just in the US (Lee et al., 2013) and hundreds of millions of additional hospitalization costs in the EU (Köck et al., 2010). The problem is exacerbated by the pathogen being carried by one third of human population (Chambers and Deleo, 2009) and its vigor to develop resistance against drugs mobilized for its treatment. Hence, hospital-acquired S. aureus cases are particularly severe.
For seven decades treatment of S. aureus infections primarily relied on antibiotics interfering with the synthesis of its cell wall, specifically, the penicillin methicillin and the glycopeptide vancomycin. Inhibitors of protein synthesis have been recently added to the arsenal of therapeutic means (Brickner et al., 2008). However, the historic propensity of bacteria to rapidly develop resistance to small molecules impels the conception of new treatment paradigms (Taubes, 2008). Today S. aureus appears as one of the 12 most health-threatening pathogens in the list of resistant bacteria compiled by WHO to promote development of new antibiotics (WHO Mediacentre, 2017).
Pentaglycine cross-bridges of peptidoglycan (PG) are a distinct feature of the S. aureus cell wall and provide innate selectivity as targets (Vollmer et al., 2008). Some members of the Staphylococcus genus have taken advantage of this feature and secrete bactericidal enzymes which cleave this cross-bridge to lyse competing S. aureus (Schindler and Schuhardt, 1964; Browder et al., 1965; Sugai et al., 1997), with their own intrinsic resistance engineered by serine replacements of the glycine residues (Thumm and Götz, 1997; Tschierske et al., 1997). Scission of pentaglycine is executed by endopeptidases of the lysostaphin family, which also includes S. aureus inherent autolytic enzymes that maintain the integrity of the cell wall during cell life cycle (Ramadurai et al., 1999; Ercoli et al., 2015; Raulinaitis et al., 2017a).
A modular organization is common to many peptidoglycan hydrolases (Szweda et al., 2012). Mature lysostaphin consists of an N-terminal catalytic (CAT) domain, and a C-terminal cell wall targeting (CWT) domain (Baba and Schneewind, 1996). Its X-ray crystal structure has been reported recently (Sabala et al., 2014). The CAT domain forms a barrel fold typical for the MEROPS M23 family zinc-dependent endopeptidases, which bear conserved catalytic as well as Zn2+-coordinating amino acid residues (Rawlings et al., 2008). A fourteen-residue linker connects the CAT domain to the CWT domain. The latter assumes the fold corresponding to SH3b domains (Lu et al., 2006). The SH3b domains are the bacterial counterpart of the ubiquitous SH3 domains found in eukaryotes (Ponting et al., 1999; Whisstock and Lesk, 1999). These are small interaction modules present in proteins of signaling pathways, which interact with proline-rich sequences in binding partners (Saksela and Permi, 2012). The canonical PXXP binding site on the surface of SH3 domains is partially blocked in the SH3b domains, and the pentaglycine interaction site of SH3b domains has been located to a groove structure formed by a 20-residue N-terminal extension not present in the smaller SH3 domains (Lu et al., 2006; Hirakawa et al., 2009; Gu et al., 2014). Despite the linker, evidence for CAT–CWT domain interaction in vitro has been presented, although the same study underscored the benefits of flexibility between the domains in vivo (Lu et al., 2013).
Surprisingly little is known about the details of the interaction between lysostaphin and PG. Biochemical characterization has revealed that the CWT domain is indispensable for lysostaphin specificity, and that an intact pentaglycine cross-bridge is crucial for the CWT–PG interaction (Gründling and Schneewind, 2006). The interaction can be inhibited by PG fragments containing multiple cross-linked murein subunits, but not with smaller murein mono- or dimers. Lipoproteins, cell wall-anchored proteins and wall teichoic acids appear not to have a role in the interaction (Gründling and Schneewind, 2006). Similarly, PG binding of Staphylococcus capitis ALE-1 CWT domain, which is highly similar in sequence and structure to the lysostaphin CWT domain, is dependent on the length and amino acid composition of the cross-bridge, showing strong preference for Gly5 bridges (Lu et al., 2006). The complex structure of lysostaphin CWT domain with pentaglycine peptide is a landmark in the characterization of the SH3b–PG interaction (PDB ID 5LEO). In this structure pentaglycine resides in the N-terminal groove, consistent with previous mutational, computational and NMR studies.
Medical applications of recombinant lysostaphin are advancing and some have entered clinical trials (Nelson et al., 2012). Lysostaphin CWT domain fusion proteins have also been considered as a promising source of alternative chemotherapeutics (Schmelcher et al., 2012; Osipovitch and Griswold, 2015; Jagielska et al., 2016). In addition, lysostaphin has been engineered to suppress its immunogenic potential (Blazanovic et al., 2015). It is clear that the key factor in the success of all lysostaphin family enzyme applications is understanding the determinants affecting efficiency of catalysis and strength and specificity of interactions. To this end, we have used NMR, SAXS and molecular dynamics (MD) simulations to characterize lysostaphin structure and dynamics, to address the inter-domain interaction, the enzyme-substrate interaction as well as the catalytic properties of pentaglycine cleavage in solution.
Materials and Methods
Production and Purification of Lysostaphin and Its CWT Domain
Gene for mature lysostaphin (residues 251-493) was synthesized de novo at GenScript (NJ, USA) and cloned into pGEX-2T vector (GE Healthcare Life Sciences) at the BamH1 and EcoR1 cloning sites. Resulting plasmid was transformed into E. coli BL21 (DE3) strain. DNA encoding the CWT domain (residues 402-493) was amplified (primers 5′-CGCGGATCCTGGAAAACCAATAAATATGGCACGC-3′ and 3′-CGCGAATTCTTATTTGA TGGTGCCCCACAGG-5′) using this plasmid as template and was cloned into pGEX-2T vector separately at the BamH1 and EcoR1 cloning sites as well.
Protein expression and purification was carried out as described in detail previously (Raulinaitis et al., 2017a). Overnight cell cultures were inoculated into a fresh medium. LB medium was used for non-labeled protein production and when labeled protein was required for NMR data collection, cells were grown in minimal resource M9 medium (40 mM Na2HPO4, 22 mM KH2PO4, 12 mM NaCl, 1 mM MgSO4, 100 μM CaCl2, 10 μM FeSO4, 0.001 % w/w biotin, and 0.1 mg/ml ampicillin, pH 7.0). Upon cells reaching OD600, protein expression was induced with isopropyl β-D-1-thiogalactopyranoside (IPTG), 0.5 mM final concentration, and further incubated for 3 h. Harvested cells were disrupted by sonication, and soluble fraction was loaded on Glutathione Sepharose 4 Fast Flow resin (GE Healthcare). Lysostaphin was released from resin-bound glutathione tag by thrombin and protein was further purified by gel filtration on HiLoad 16/60 Superdex 75 column (GE Healthcare Life Sciences). Removal of innately bound metal ions was carried out with EDTA, which was thereafter dialyzed away in PBS buffer and protein was concentrated with Amicon®; 5000 MWCO centrifugal filters.
NMR Spectroscopy
Spectra for chemical shift assignment and structure determination were acquired at 308 K on a Varian INOVA 800 MHz spectrometer equipped with a cryogenically cooled triple resonance probehead equipped with an actively shielded z-gradient. Resonance assignment was carried out with the following set of experiments: 1H, 15N HSQC, 1H, 13C HSQC, 1H, 13C CT-HSQC, HNCACB, CBCA(CO)NH, (H)CC(CO)NH, H(CCO)NH, HBHA(CO)NH, (HB)CB(CGCD)HD, (HB)CB(CGCDCE)HE, 1H, 15N NOESY-HSQC, 1H, 13C NOESY-HSQC in 7% D2O/93% H2O, and 1H, 13C HSQC-NOESY in 100% D2O. The latter five spectra were employed for the assignment of the aromatic residues as well as the source of distance restraints for structure determination. Additional structural restraints were derived from the chemical shifts in the form of TALOS-N (Shen and Bax, 2013) dihedral angle restraints and applied for residues in secondary structures as well as H2O to D2O exchange experiment in the form of hydrogen bond restraints for amides experiencing slow exchange. Histidine tautomeric state could be derived from the peak pattern in a 1H, 15N HMBC spectrum or the Cδ2-Cε1 chemical shift difference for four out of nine histidines (Barraud et al., 2012). Spectra were processed with NMRPipe (Delaglio et al., 1995) and analyzed with Sparky (T. D. Goddard and D. G. Kneller, University of California, San Francisco). The automated NOE peak assignment-structure determination routine of CYANA 2.1 (López-Méndez and Güntert, 2006) was used to generate an ensemble of 20 lowest target energy structures which were further refined with AMBER 16 (Case et al., 2005) in explicit solvent. Fifteen lowest AMBER energy structures were chosen to represent the solution state structure of lysostaphin.
The pentaglycine binding site on the CWT domain was determined by comparing the 1H, 15N, and 1H, 13C HSQC spectra of full-length lysostaphin and CWT domain acquired in the presence and absence of a large amount, ~170 molar excess, of KGGGGG and GGGGGK hexapeptides (Sigma-Aldrich). Binding characteristics of a PG peptide mimic were determined by titration of the CWT domain with A-d-EKGGGGGA-d-EK-d-A and comparing 1H, 15N HSQC spectra acquired at approximate peptide to CWT domain molar ratios of 0, 0.5, 1, 2, 5, 10, and 24. Similarly, the pentapeptide A-D-γ-EK-D-A-D-A (Bachem) and muramyl dipeptide N-acetylmuramyl-l-alanyl-D-isoglutamine hydrate (Carbosynth Ltd) were titrated to a CWT domain sample, and 1H, 15N HSQC spectra were collected at approximate peptide to CWT domain molar ratios of 0, 0.5, 1, 2, 4, 8, 16, 32, 45, and 60. CSPs, calculated as Δδ = (ΔδH2+(0.154 × ΔδN)2)1/2, were plotted as a function of the molar ratio of titrated ligand to CWT domain. Dissociation constants for individual residues were obtained by nonlinear least squares fitting, as implemented in the program xcrvfit (www.bionmr.ualberta.ca/bds/software/xcrvfit). Site-specific dissociation constants were calculated as averages of dissociation constants of individual residues within the site. In all binding site studies protein to zinc concentration ratio was 1:1.
CAT domain–SH3 domain interaction was probed by replacing zinc with paramagnetic manganese (1:1 protein to cation ratio) and observing peak bleaching in a 1H, 15N HSQC spectrum.
15N T1, T2 and heteronuclear NOE spectra were acquired for one- and three-zinc bound lysostaphin in the presence and absence of substrate pentaglycine on a Bruker AVANCE III HD 800 MHz spectrometer, equipped with a TCI 1H/13C/15N cryoprobe. For T1 relaxation delays of 20, 60, 100, 200, 400, 600, 900, 1,400, 2,000, and 2,600 ms were used. For T2 the delays were multiples of loop length 16.96 ms, with the loop counter set to 1, 2, 3, 4, 5, 6, 7, 8, 10, and 12. The recycle delay was set to 3.1 and 2.6 s for the T1 and T2 experiments, respectively. All spectra were acquired as pseudo 3D spectra and Fourier transformed with NMRPipe. Peak intensities were fitted to decaying exponential function as implemented in Sparky. Heteronuclear NOE values were calculated as the intensity ratios of peaks from a pair of spectra measured with and without 1H presaturation during the recycle delay. The relaxation delay was set to 5 s. The CWT domain relaxation spectra were recorded on a Bruker AVANCE III 500 MHz spectrometer. For T1, relaxation delays of 20, 60, 100, 200, 400, 600, 800, 1,200, 1,600, and 2,000 ms. The recycle delay was set to 2.5. For T2, the loop counter was set to 1, 2, 4, 8, 10, 12, 14, and 16 and the recycle delay was 1.5 s. Heteronuclear NOE spectra were acquired as for full-length lysostaphin.
Pentaglycine cleavage by lysostaphin was monitored by 1H NMR as described previously (Raulinaitis et al., 2017a). Reactions took place in phosphate buffer saline (PBS), pH 7.2, with >90% D2O by volume as a solvent and final volumes were 600 μL. Initial pentaglycine (Sigma-Aldrich) and lysostaphin concentrations were 1 mM and 2.25 μM, respectively. Metal ions (ZnCl2, CoCl2, CuCl2, and MnCl2) were added as needed. Residual activity of lysostaphin “apo” form was taken into account in activity calculations. Measurements were carried out by using a Bruker Avance III 600 MHz spectrometer, equipped with a QCI 1H/13C/15N/31P cryoprobe and a SampleJet automated sample changer. The preheater of the sample changer was used for sample incubation when product formation was measured as a time series with 3-h intervals for 48 h. Single time-point samples were incubated in an incubator for 21 h and reactions were quenched by heating at 90°C for 15 min before proton spectra acquisitions at 37°C. The data were processed with Bruker TopSpin 3.5 software.
MD Simulations
MD simulations in explicit solvent were performed with AMBER 16 (Case et al., 2005) using the ff14SB force field. Lysostaphin CYANA ensemble structures 1, 5, 7, and 14 were selected as targets of simulations. The topology and coordinate files were generated with the LEaP program in AMBER 16. The zinc ion was modeled using the cationic dummy atom approach (Pang, 1999; Pang et al., 2000). The protein molecule was placed in a cubic box with a minimum solute-box distance of 10 Å, and solvated with TIP3P water molecules. Nine chloride ions were added to maintain electrical neutrality. After minimization, heating and equilibration of the system, the production 30-ns MD simulations were performed with periodic boundary conditions at 300 K. The temperature was maintained by using the Langevin thermostat, whereas the pressure was kept at 1 bar using the Berendsen barostat (Berendsen et al., 1984). The time step was set to 2 fs. Long-range electrostatic interactions were treated using the Particle Mesh Ewald method (Darden et al., 1993) with a cut-off of 10 Å. Bond lengths involving hydrogen atoms were constrained by SHAKE (Ryckaert et al., 1997). Analyses of the trajectories were carried out with CPPTRAJ (Roe and Cheatham, 2013) and VMD (Humphrey et al., 1996).
SAXS
BM29 beamline, ESRF, Grenoble, France was used for collecting the SAXS data on a PILATUS 1M image plate using sample to detector distance of 2.9 m and a wavelength of 1.0 Å (momentum transfer range 0.01 < q < 5 nm−1). The measurements were carried out at 288 K in PBS buffer. Three different protein concentrations (1.0, 3.0, and 5.0 mg/ml) were used in the data acquisition. PRIMUS (Konarev et al., 2003) in ATSAS software package (Franke et al., 2017) was utilized in the data processing. To assess the conformational variability of lysostaphin, EOM (Tria et al., 2015) was used. In EOM calculations, lysostaphin CAT domain (residues 251-384) and CWT domain (residues 410-493) of the NMR structure reported here, were used as rigid bodies, and residues 385-409 as random coil. 10 000 randomized conformations were generated for lysostaphin based on amino acid sequence and 3D-structure of CAT and CWT domains of lysostaphin using both random coil and native options in EOM. The scattering profiles of these randomly generated conformations were compared, and representative 20 structures whose scattering curve fits to the experimental scattering curve were selected by EOM algorithm. EOM run was repeated 100 times to provide statistical data about lysostaphin radius of gyration (Rg) and maximum dimensions (Dmax) distribution.
Results and Discussion
Lysostaphin Catalytic Groove Embracing Loops are Highly Mobile
The solution NMR structure of lysostaphin was determined based on distance restraints from NOESY spectra, φ/ψ dihedral restraints derived from chemical shifts using TALOS-N (Shen and Bax, 2013) and hydrogen bond restraints obtained from a H/D exchange experiment (Figure 1A and Table 1). In line with the X-ray crystal structure (Sabala et al., 2014), lysostaphin is structurally arranged into two domains. The domains are connected by a fourteen-residue linker, which is devoid of persistent structure. The domains have random mutual orientations in the ensemble of structures. The CAT domain shares the structural features of a MEROPS M23 metallopeptidase domain with a β-sheet core and four loops forming the bottom and the rims of the catalytic groove, respectively (Rawlings et al., 2008). In one end of the groove reside the catalytic residues surrounding a zinc cation. The CWT domain has the SH3b domain, barrel-like all β-sheet fold.
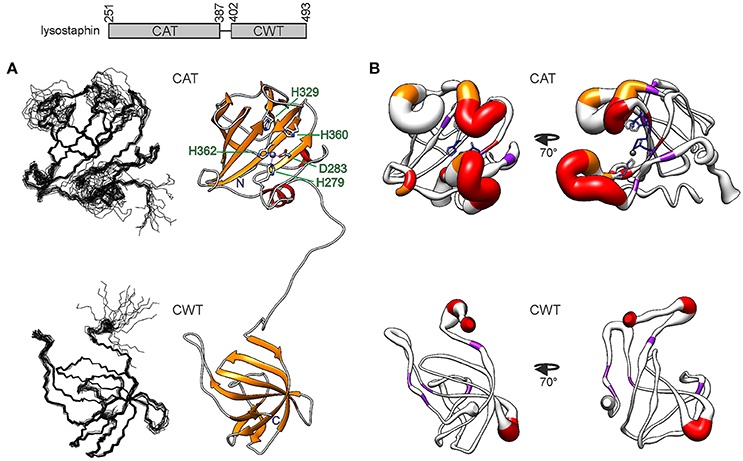
Figure 1. Structure of S. simulans lysostaphin. On top, the domain structure is depicted. The N-terminal CAT domain is composed of 137 residues and the C-terminal CWT domain of 92 residues. The domains are linked by a flexible 14-residue linker. (A) Solution NMR structure of lysostaphin. On the left, the two domains are represented as ensembles of 15 superimposed structures. On the right, a representative member of the ensemble is shown in a ribbon diagram. The catalytic site residues in the CAT domain are labeled. Zinc is shown as a gray sphere. (B) CAT (top) and CWT domain dynamics. The heavy atom RMSF obtained from the MD simulation is depicted as the radius of the ribbon. Residues which lack or display strongly attenuated amide peak in the 1H, 15N HSQC spectrum are colored in red and orange, respectively. Residues which experience μs-ms dynamics are colored in purple. Catalytic site residues in the CAT domain are colored according to their heavy atom RMSF, blue depicting low RMSF values and lighter colors higher RMSF values. The structure on the left has the same orientation as the structures in (A), whereas the structure on the right has been rotated 70 degrees around the vertical axis.
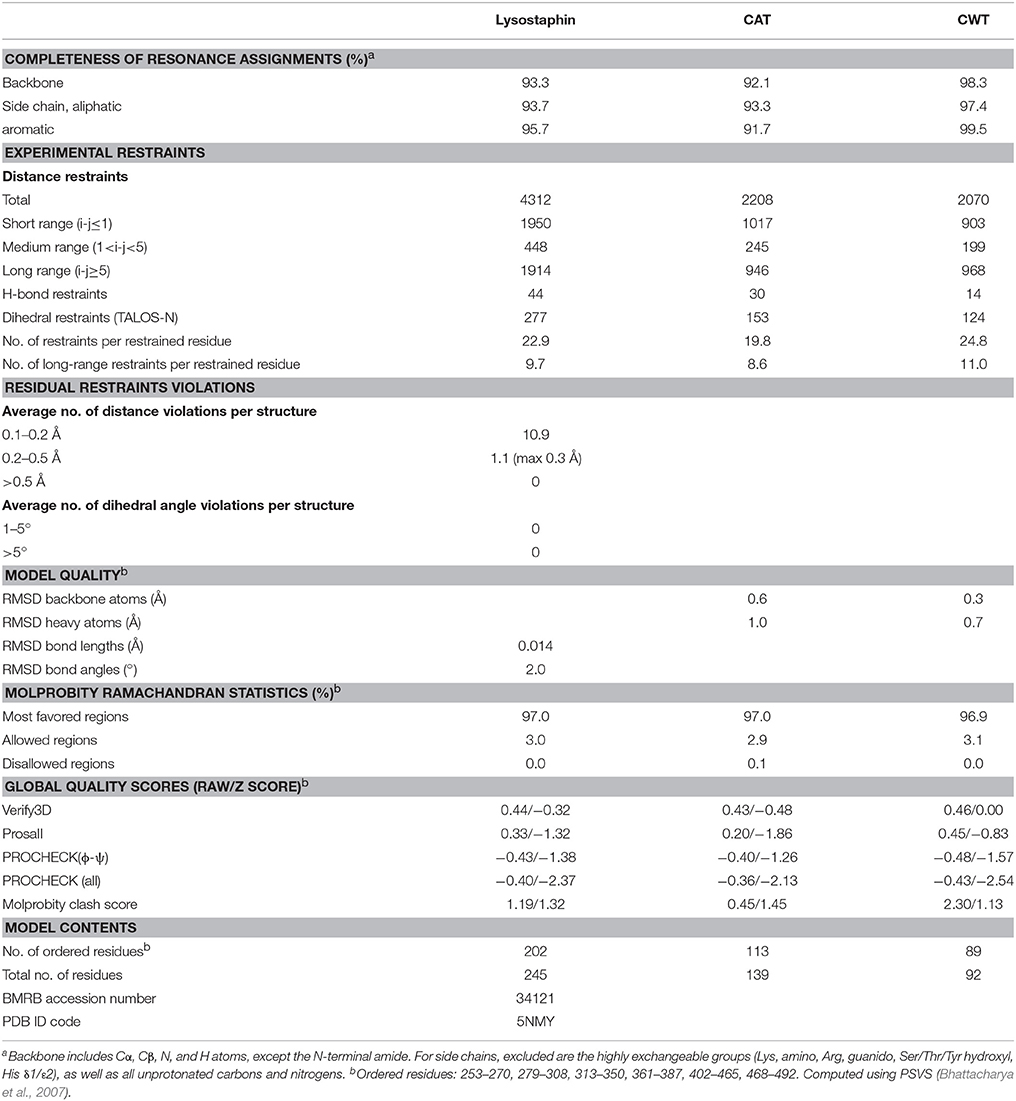
Table 1. NMR restraints and structural statistics for the ensemble of 15 lysostaphin conformers of least restraint violations tabulated for the whole enzyme and domains separately.
While the core of the CAT domain is well-defined, and nicely superimposable with the crystal structure (RMSDs of 0.7 and 1.3 Å for backbone and heavy atoms, respectively), the catalytic groove embracing loops and the catalytic histidines are conformationally dispersed, which results in a poorer overall structural match in these regions. The conformational spread in the NMR ensemble is the result of solvent exposure and flexibility of residues in these regions (Figure 1B and Supplementary Figure 1). Fast amide proton exchange with solvent is likely to be present in the loops, because these do not display or have severely broadened peaks in a 1H, 15N HSQC spectrum. Additionally, the catalytic histidines probably exhibit solvent and/or conformational exchange, because their signals were not observed in a histidine-region 1H, 15N HMBC spectrum, showing cross-peaks between Nδ1, Nε2, Hδ2, and Hε1. Although carbon-bound Hδ2 or Hε1 do not exchange with solvent, they can be influenced by solvent or conformational exchange elsewhere in the aromatic ring. This manifests as additional line-broadening of histidine ring protons and disappearance of catalytic histidine correlations from the HMBC. In addition to exchange, micro- to milli-second time scale motions are present in the catalytic groove (Figure 2 and Table 2), as identified by high R1R2 products derived from 15N relaxation measurements (Kneller et al., 2002).
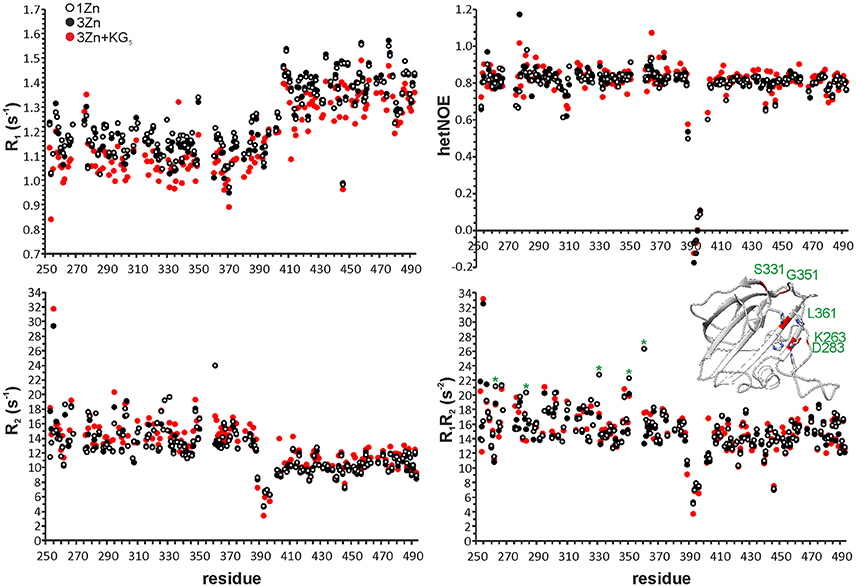
Figure 2. 15N R1, R2, heteronuclear NOE and R1R2 data of one-zinc (white circles), three-zinc (black circles) and three-zinc lysostaphin in the presence of ~140 molar excess of KG5 (red circles). Error bars have been omitted for clarity. Average errors are: 1Zn CAT 0.01 s−1 (R1), 0.19 s−1 (R2), 0.03 (HetNOE) and 0.29 s−2 (R1R2); 1Zn CWT 0.01 s−1 (R1), 0.10 s−1 (R2), 0.01 (HetNOE) and 0.16 s−2 (R1R2); 3Zn CAT 0.01 s−1 (R1), 0.20 s−1 (R2), 0.03 (HetNOE) and 0.27 s−2 (R1R2); 3Zn CWT 0.01 s−1 (R1), 0.11 s−1 (R2), 0.01 (HetNOE), and 0.16 s−2 (R1R2); 3Zn+KG5 CAT 0.02 s−1 (R1), 0.26 s−1 (R2), 0.06 (HetNOE) and 0.40 s−2 (R1R2); 3Zn+KG5 CWT 0.01 s−1 (R1), 0.08 s−1 (R2), 0.04 (HetNOE) and 0.14 s−2 (R1R2). In the R1R2 panel residues experiencing a large change upon addition of excess zinc are marked with asterisks and highlighted in red in the CAT domain structure.
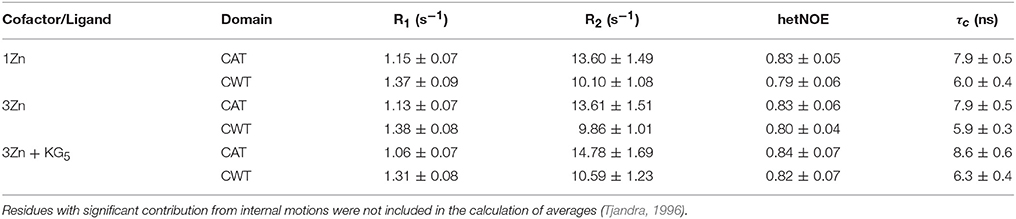
Table 2. Average R1, R2 and hetNOE values and standard deviations derived from 15N relaxation experiments at 800 MHz, 35°C.
Catalytic groove conformational dynamics is evident also from MD simulations. The root mean square fluctuation, RMSF, emphasizes the flexible nature of three of the groove embracing loops (Figure 1B and Supplementary Figure 1). Consistent with the NMR ensemble, the outer half of the N-terminal loop, residues G270-G277, is highly dynamic, whereas the inner half is restrained by transient backbone hydrogen bonds within the loop, Y267-G281, G268-H279 as well as the H279Nε2–Zn2+ interaction. The loop encompassing residues N368-Q376 is the least flexible. It is transiently stabilized by backbone S371-T374 and side chain S369-T374 hydrogen bonds, the former twisting the loop into a three-residue 310 helix.
The MD simulation reveals several possible mechanisms contributing to the disappearance of the catalytic histidine HMBC signals (Supplementary Figure 1). Firstly, while the two histidine rings remain approximately coplanar during the simulation, they slide relative to each other. Secondly, the aromatic ring of F285, next to H360, is able to rotate. Thirdly, the loop closest to the catalytic histidines, residues T353-Y355 thereof in particular, adopt a multitude of conformations at a fluctuating distance from the catalytic histidines. Conformational exchange between all these different structural arrangements is likely to broaden the HMBC signal beyond detection. Moreover, perhaps most importantly, H329 and H360 are transiently accessible to solvent, leading to signal broadening through exchange between tautomeric, ionic and H/D states. Notably, the outward orientation of H360 aromatic ring in some of the NMR ensemble structures appears to be energetically unfavorable as during the MD simulations the ring turns toward the catalytic groove.
Of the M23 family endopeptidases, for which the structure has been determined, lysostaphin CAT domain has the highest amino acid sequence identity with the sequences of LytM and LytU, 49 and 44%, respectively. LytM and LytU are S. aureus autolytic endopeptidases taking part in PG remodeling by cleaving the pentaglycine cross-bridges. The three structures are remarkably similar. Backbone atom RMSDs over the β-sheet core are 0.7 and 0.9 Å for LytM and LytU, respectively, when overlaid with the lysostaphin CAT domain. Besides structural correspondence they share a similar dynamic behavior with a stable core and mobile catalytic site and surrounding loops (Raulinaitis et al., 2017a,b).
The CWT domain is compact, stabilized by extensive hydrogen bonding within loops as well as between loops and the rest of the molecule, and high similarity is found between the solution and crystal structures. Backbone and heavy atom RMSDs are 0.5 and 1.1 Å, respectively, for all residues in the closest matching pair. Some dynamic features are, however, observed. Micro- to millisecond time scale motions are present in five residues, K412, T428, R476, G486, and L488 and four residues, G401, K406, N466, S467 are prone to fast exchange with solvent, all in loops (Figure 1B).
Lysostaphin Domains Do Not Interact in Vitro
No inter-domain NOE peaks were observed in the NOESY spectra, strongly indicating that the two domains do not possess a stable interaction interface. The lack of chemical shift differences between the isolated CWT domain and the CWT domain in full-length lysostaphin (Supplementary Figure 2) provides additional solid evidence for non-interacting domains. Nevertheless, we further inspected the possibility of domain interaction by replacing the CAT-bound zinc cation with manganese and analyzing peak bleaching in a 1H, 15N HSQC spectrum. Unpaired electrons in isotropic paramagnetic probes such as manganese induce transverse paramagnetic relaxation enhancement (PRE) leading to line-broadening, affecting peak intensity, up to 35 Å from the paramagnetic center depending on the paramagnetic probe (Clore and Iwahara, 2009). In Mn2+-bound lysostaphin peaks disappeared or broadened only in the CAT domain (Supplementary Figure 3), at a maximum average distance of 23.6 Å from the paramagnetic center, leaving peaks from the far-most backside of this domain intact. Conceivably, as the linker allows such an arrangement, the domains could interact through this backside CAT domain region without the CWT domain peaks experiencing line-broadening.
The PRE experiment remaining inconclusive, we turned to the 15N relaxation data and overall rotational correlation times (τc), which can also be used to untangle the confines of inter-domain mobility. The τcs derived from 15N R2, R1 rates are notably different for the CAT and CWT domains, 7.8 and 6.0 ns at 35°C, respectively (Table 2). Also, the τc of the CWT domain in full-length lysostaphin is significantly higher than that of the isolated CWT domain, 4.1 ns. These indicate that there is a clear motional hindrance by the neighboring domain in full-length lysostaphin. There still is, however, substantial flexibility, considering that a unified complex encompassing both domains has a predicted τc of 13.4 ns.
To further investigate the structure and flexibility of lysostaphin in solution, SAXS data were collected. An ensemble optimization (EOM) method was employed to analyse the SAXS data. In EOM calculations, lysostaphin CAT (residues 251-384) and CWT (410-493) domains of the NMR structure reported here were used as rigid bodies, and residues 385-409 as random coil. EOM runs yielded reproducible ensembles neatly fitting the experimental data with χ2-values around 1.4 (Figure 3C). The results clearly show that lysostaphin adopts two different conformations in solution; a more favored compact subpopulation with average Rg and Dmax of 27 and 82 Å, respectively, and a less favored extended subpopulation, with average Rg and Dmax of 37 Å and 110 Å (Figures 3A,B,D). The Dmax of the predominant conformation is consistent with an average domain separation of 22–23 Å. This is considerably shorter than the length encompassed by a fully extended conformation of the linker containing two prolines, ~42 Å. This raises the possibility of the presence of secondary structure in the linker or interactions of the linker with one or other domain. Neither is, however, supported by secondary chemical shifts or NOE peaks. Moreover, significantly lower R1R2 products and heteronuclear NOEs found for linker residues K389-W402 with respect to the values in the CAT and CWT domains indicate that the linker is highly flexible. However, two sets of peaks were assigned for linker residues V394-P398, indicating the presence of two preferred conformations within this stretch, likely due to P398 cis/trans isomerism. This potentially offers a functional benefit to lysostaphin as it allows sampling of a larger conformational space and readjustment of domain orientations upon scission of pentaglycine bridges near its anchoring site (Aitio et al., 2012).
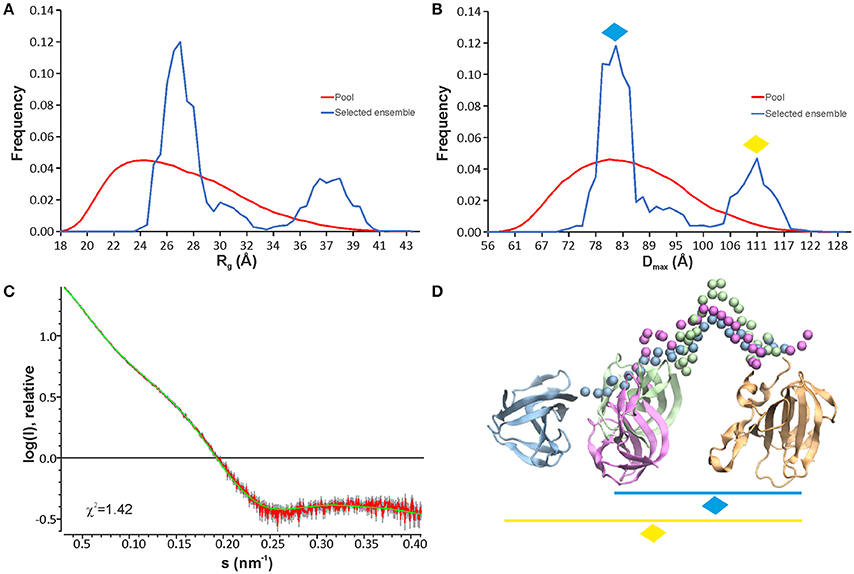
Figure 3. EOM analyses of lysostaphin SAXS data show that two conformation ensembles are present in solution as seen from Rg (A) and Dmax (B) distributions. The pool represents the structures calculated based on the sequence and NMR structures of lysostaphin CAT and CWT domains. The selected models represent those structures within the pool that fit to the experimental scattering curve. (C) A typical fit obtained from the selected ensemble of structures to the experimental scattering of lysostaphin. (D) Three representative models of lysostaphin obtained from EOM calculations. The models are superimposed by the CAT domain (tan). CWT domains of three independent models are shown in green, magenta and blue. Green and magenta CWT domains belong to the compact conformations (marked with a blue diamond in B,D) and blue CWT domain to the extended conformations (marked with a yellow diamond in B,D). The CAT and the CWT domains of the NMR structure were used as rigid bodies in EOM calculations. Spheres represent amino acids in a random coil linker.
A dynamic inter-domain interaction in lysostaphin has been presented previously (Lu et al., 2013). Although the long flexible linker indeed allows the spatial proximity of the domains, there are no indications of such interaction in our data. The linker easily allows cleavage of adjacent and nearby pentaglycine bridges in PG near the bridge that serves as an anchor. Considering the architecture and dimensions of the PG lattice (Kim et al., 2015), formation of a CAT and CWT “envelope” around the very same target pentapeptide, which is also fixated between perpendicular peptidoglycan stem peptides that it connects, is very improbable due to steric restrictions. The current data remain in support of the flexible arrangements between partly independent domains.
Binding of a Second Zinc by the Catalytic Histidines Reduces Lysostaphin Catalytic Activity
Proton NMR allows for direct and real-time reaction monitoring devoid of intricacies of cellular suspension (Raulinaitis et al., 2017a). Scission of pentaglycine by lysostaphin was monitored for 48 h by 1H NMR and under conditions of this study its catalytic rate appeared to be limited only by saturation with the substrate (Supplementary Figure 4). The substrate turnover rate by the enzyme in the first 3 h of the reaction is estimated to be in the order of 0.006 s−1.
We used the method to assess lysostaphin catalytic activity dependence on different metal cofactors (Figure 4). Samples were first incubated for 21 h and then quenched by heating, followed by acquisition of a 1H spectrum. Results indicate that metal cations Zn2+, Co2+, and Cu2+ are suitable cofactors of lysostaphin, whereas Mn2+ is a weak cofactor. The activity increases when Cu2+ is present in excess. This behavior might be explained by Cu2+ being a better cofactor with a lower affinity, the enzyme reaching full metal cation saturation at higher cation concentrations. M23 family endopeptidases are recognized as zinc-dependent enzymes, yet other metal ions have been found to partially restore their activity (Firczuk et al., 2005; Wang et al., 2011) and recently Co2+ ions have been shown to yield a hyperactive LytU (Raulinaitis et al., 2017a). A partial inhibition of lysostaphin activity is observed in the excess of zinc. Even under the inhibition by an extra zinc ion, the enzyme retains its fold, as demonstrated by its 1H, 15N HSQC spectrum displaying features of a structured protein, very similar to those of the one-zinc form (Supplementary Figure 5). Notably, chemical shift perturbations, CSPs, are observed for peaks of residues in the catalytic groove as well as the surrounding loops. Changes in the dynamics are also observed (Figure 2). The majority of the largest changes occur for residues having the largest exchange contribution in the one-zinc form, located in the vicinity of the catalytic site. At these sites, the R1R2 products decrease, which indicates reduction of exchange in the μs-ms timescale, and structure stabilization. Loop amides remain unprotected from fast solvent exchange, though, as no additional peaks appear into the 1H, 15N HSQC spectrum. Conclusively, two additional cross-peak sets appear into a 1H, 15N HMBC spectrum with 15N chemical shifts corresponding to those of a zinc-coordinating nitrogen (Banci et al., 1998). Hence, binding of a second zinc, coordinated by the catalytic histidines, reduces catalytic activity.
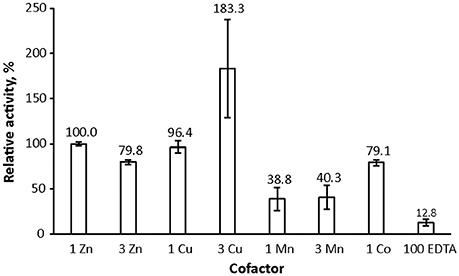
Figure 4. Lysostaphin activity dependence on zinc, cobalt, copper and manganese cofactors. Lysostaphin was incubated with pentaglycine (2.25 μM and 1 mM concentrations, respectively) in PBS buffer, pH 7.2, for 21 h at 37°C. Total cofactor ratios to protein were 1:1 and 3:1, EDTA excess was 70-fold. Purified enzyme, despite extensive treatment with EDTA during purification to remove any cofactors bound to the enzyme, nevertheless displayed residual activity. Activity was adjusted to one zinc-bound lysostaphin as 100% reference point. Bars represent standard error of the mean (S.E.M) of at least three independent measurements.
The inhibitory effect of second zinc binding was also demonstrated and explained by the arrest of catalytic histidines in the autolytic LytU, where the process may have a regulatory role (Raulinaitis et al., 2017a). Whereas it is conceivable for an intracellular autolysin, such regulation by inhibition would be irrational for an extracellular bactericide like lysostaphin and is likely physiologically irrelevant.
Pentaglycine Interacts Transiently With the N-Terminal Groove in the CWT Domain
NMR suits particularly well to study weak or transient molecular interactions. We performed CWT domain binding site mapping with hexapeptides G5K and KG5 (Figure 5A and Supplementary Figure 6). The presence of lysine significantly enhanced the solubility of the peptide, which was essential in order to reach the large excess of ligand needed to achieve noticeable CSPs. The two pentaglycine derivatives produced similar amide peak CSPs when present in ~170 molar excess. Ligand interaction with CWT was independent of the presence of the CAT domain, as deduced by the similar CSPs observed for the CWT domain in full-length lysostaphin in the presence of ~140 molar excess of KG5 (data not shown).
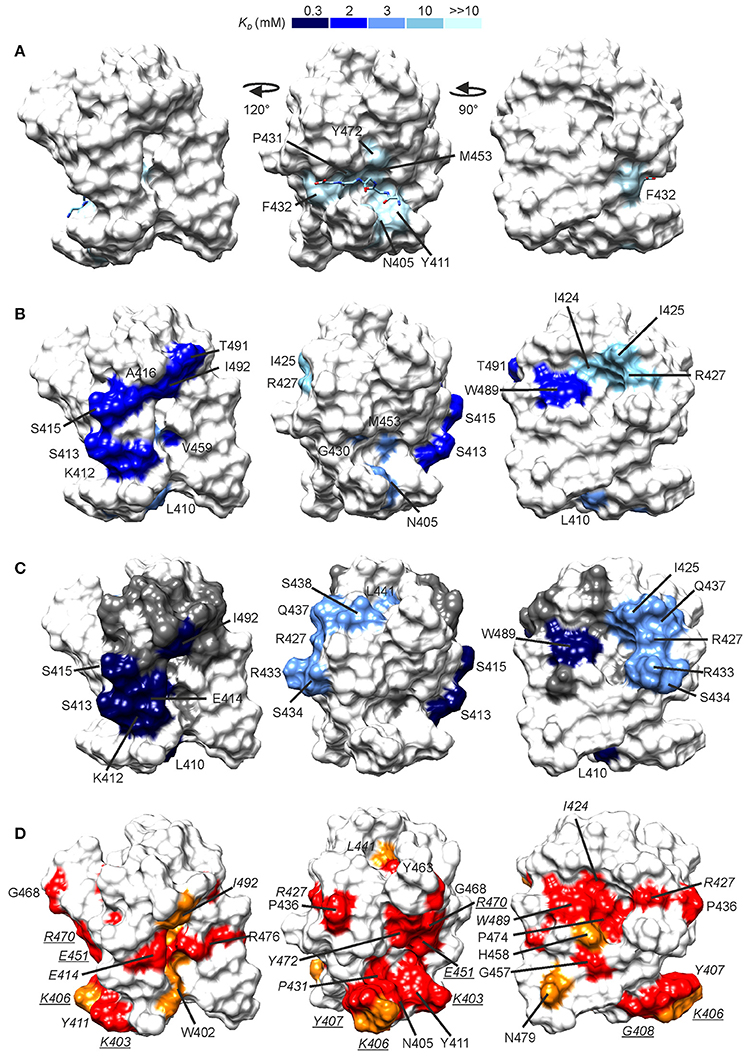
Figure 5. Peptide binding sites as revealed by NMR titrations. CSPs induced by G5K (A), A-d-EK-GGGGG-A-d-EK-d-A (B), and A-d-γ-EK-d-A-d-A (C) mapped onto the CWT domain structure. The surfaces are colored by the associated dissociation constant, the scale of which is given on the top of the figure. In (A), the location of the bound pentaglycine is given from a structural superimposition of the free NMR structure and the X-ray CWT–substrate complex with PDB ID 5LEO. In (C), between the high- and low affinity sites lies a group of residues which demonstrated a curved peak movement (see Supplementary Figure 4).These are shown in gray. (D) Conserved residues in S. aureus-targeting CWT domains mapped onto the lysostaphin CWT domain structure. Conservation was determined from a sequence alignment with ClustalW (Larkin et al., 2007) of lysostaphin CWT-like S. aureus-targeting sequences in the UniProt database (The Uniprot Consortium, 2017). In red are shown fully conserved residues and in orange residues with strongly similar properties. Conserved residues for which large CSPs were observed are labeled in italics, whereas conserved residues for which smaller CSPs were observed (0.06–0.09 ppm, not colored in A-C for clarity) are labeled in underlined italics. Sequence alignment is given in Supplementary Figure 5.
Largest peak shifts, Δδ ≥ 0.10 ppm, in the presence of G5K were observed for N405, Y411, G430-F432, V452, M453, and Y472. Residues in strands β1 and β2 and the loop in between (K403-K412) form the bottom of the pentaglycine binding groove, and residues G430-F432 from the loop between strands β3 and β4 and Y472 in strand β7 the ceiling of the groove. Binding site mapping demonstrates CSPs for a delimited set of residues, without secondary, rearrangement-induced perturbations. Our data are nicely in accordance with the crystal structure of the lysostaphin CWT–pentaglycine complex (PDB ID 5LEO), which reveals that only very small structural rearrangements, namely few sidechain reorientations, occur upon binding of pentaglycine. The dissociation constant of the interaction is estimated to be ultra-weak, > 10 mM, in line with those observed for other pentapeptide–SH3b-type CWT domain interactions (Gu et al., 2014; Benešík et al., 2017).
The 15N relaxation data show interesting differences between free and pentaglycine-bound three-zinc lysostaphin. Both domains in the complex have larger τcs than those in the free three-zinc form, 8.6 vs. 7.9 ns for the CAT and 6.3 vs. 5.9 ns for the CWT domains (Table 2). The difference is significant and cannot be attributed to the change in molecular weight only, considering that an increase of ~430 Da in molecular weight is predicted to increase τc by 0.2 ns only. Exchange between the complex and free forms could be the source of the larger τcs, and indeed many local changes in the R1R2 products are observed. However, there is an equal number of positive and negative changes with values larger than < R1R2> ±1 SD, and the average R1R2 products remain the same for both domains. We gather that the additional rise can be interpreted in terms of transient inter-domain interaction in the presence of the ligand, or as a change in the degree of anisotropy of molecular tumbling induced by peptide or inter-domain interaction. Future studies in alignment media will help to derive information about relative domain orientation and inter-domain motions. The linker region shows similar dynamics in the absence and presence of the peptide, revealing that this stretch remains flexible, loosely connecting the domains.
The CWT Domain Shows Higher Affinity Toward the Stem Peptide
We reasoned that in vivo the CWT–pentaglycine interaction is likely to be bolstered by auxiliary contacts from the glycan backbone or the stem peptides in the PG lattice (for a description of the chemical structure of PG see Vollmer et al., 2008). We found that the CWT domain had no significant affinity for the muramyl dipeptide N-acetylmuramyl-l-A-d-γ-Q hydrate as deduced by the very small CSPs observed at ~80 molar excess of the peptide (data not shown).
Titration of the CWT domain with a synthetic peptide A-d-EKGGGGGA-d-EK-d-A, which would resemble the stem-cross-bridge-stem sequence in native PG, resulted, instead, in significant CSPs at considerably lower concentration ratios, ~24 molar excess. In addition to CSPs of residues in the pentaglycine groove, CSPs were observed for two groups of peaks with distinct binding affinities. If viewing the CWT domain the pentaglycine groove laying horizontally toward the viewer, the N-terminus of the bound pentaglycine on the right (PDB ID 5LEO), then K412-S413, S415-A416, V459, T491, and I492 form an elongated patch on the right side of the domain (Figure 5B). K412 is located between the pentaglycine and the elongated sites. These residues have an average KD of ~2.3 mM. Residues I424-I425 and R427 form a small patch on the left side of the CWT domain with an average KD of ~10.5 mM. W489 is located between the elongated and the small sites, but with a KD of ~2.3 mM belongs to the elongated site. The peptide is not large enough to reach simultaneously the three surfaces. Considering that residues in the pentaglycine site have a much higher affinity toward this peptide than to pentaglycine or KG5/G5K (average KD of ~3.2 mM) we suspect that one peptide molecule binds via its tail to the elongated site, and boosts the affinity of the pentaglycine site for the pentaglycine moiety of the peptide.
Titration with a shorter peptide A-d-γ-EK-d-A-d-A resulted in a complex peak behavior with three distinct patterns of peak position movement (Figure 6). Cross-peaks of residues L410, K412-S415, V452, W489, and I492 moved linearly upon peptide addition and reached saturation at ~32 molar excess of peptide. The dissociation constant was determined to be ~0.3 mM, notably lower than that for pentaglycine or the longer peptide. These residues mostly overlap with the elongated site of the previous titration (Figure 5C). Cross-peaks of the second group of residues also moved linearly, but began prominent movement and reached saturation at later steps of the titration, resulting in a dissociation constant of ~3 mM. These residues form a patch on the surface, which covers the small site of the previous titration (I425, R427, and L473) and has extensions of polar residues (R433, S434, Q437, and S438). The third group of peaks exhibited non-linear peak movement. For some of these residues the CSP are as large as for those in the two other groups, which rules out unspecific multiple binding as the cause of nonlinearity. Instead, as these residues are located between the first two surface patches, and include residues in the hydrophobic core, a conformational rearrangement brought about binding to the high-affinity site might be responsible for the curved titrations. Core residues V461 and V475 are probably involved in this rearrangement because their methyl peaks substantially shift in the 1H spectra along the titration (Supplementary Figure 7).
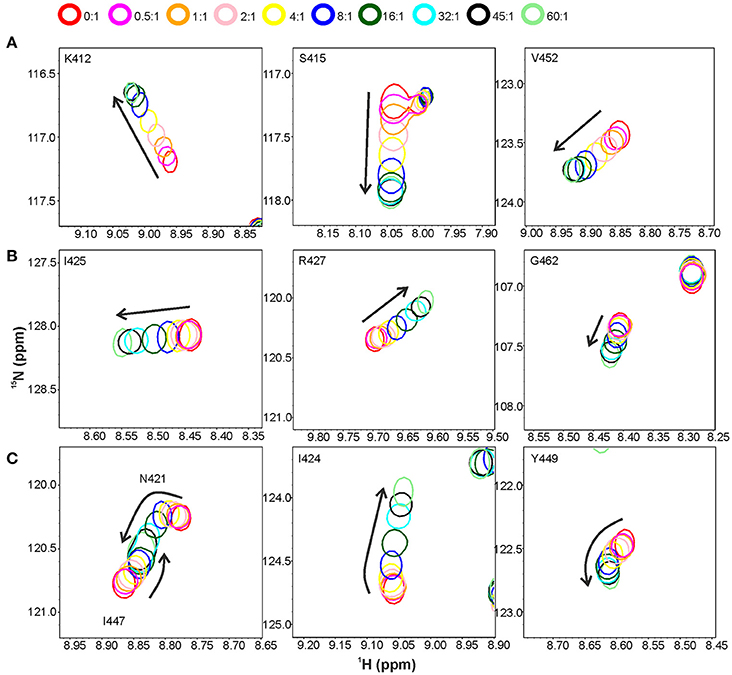
Figure 6. Titration of the CWT domain with the peptide A-D-γ-EK-D-A-D-A. Shown are overlaid sections of 1H, 15N HSQC spectra recorded at peptide to protein concentration ratios of 0:1 (red contours), 0.5:1 (magenta), 1:1 (orange), 2:1 (pink), 4:1 (yellow), 8:1 (blue), 16:1 (dark green), 32:1 (cyan), 45:1 (black) and 60:1 (light green). Three types of peak movement were observed: (A) linear, with saturation at ~32:1 concentration ratio, (B) linear, with saturation at ~60:1 concentration ratio, and (C) non-linear peak movement.
The importance of R296 and W358 in ALE-1 SH3b binding to S. aureus PG has been demonstrated by selective mutations (Lu et al., 2006). Binding of ALE-1 R296A and W358A mutants was reduced 3- and 2-fold, respectively, as compared to wild type binding. These residues correspond to R427 and W489 of lysostaphin CWT, belonging to the lower- and higher affinity sites, respectively, although structurally very close on the surface of CWT. R427 and W489 are strictly conserved in S. aureus-targeting SH3b domains (Figure 5D, Supplementary Figure 8). Based on the titration results and residue conservation, other residues which are likely to have an impact on CWT PG binding other than those in the pentaglycine binding groove are E414 and I492 of the high-affinity site.
We generated model structures of the CWT domain in complex with pentaglycine-stem peptide fragments (Supplementary Figure 9) with the assumption that the pentaglycine moiety occupies the N-terminal binding groove as in the structure 5LEO. The models indicate that the stem peptides on either side of pentaglycine seem to be too short to reach the CSPs observed in the presence of the A-d-γ-EK-d-A-d-A peptide. Also, CSPs in the region of the linkage between pentaglycine and the stem peptide are missing from the CWT domain surface. We gather that the observed CSPs might arise from a structural rearrangement occurring upon binding (see before), or be the result of stem peptide contacts present within the PG architecture, that is from stem peptides not directly linked to the pentaglycine recognized by the N-terminal groove. According to the parallel-stem architecture proposed by Kim et al. (2015) for S. aureus PG, contacts with stem peptides from further up or down in the densely packed glycan chains are probable.
These results clearly support the idea of formation of affinity-heaving accessory contacts with the PG network. Altogether it is advisable to consider using larger PG fragments in SH3b interaction studies in order to overcome the very low affinity of these domains toward pentaglycine (Gu et al., 2014; Benešík et al., 2017).
CWT–PG association studies have demonstrated that CWT binding to S. aureus cells cannot be blocked with excess of pentaglycine (Gründling and Schneewind, 2006), which is in accord with the extremely low affinity found for the pentaglycine–CWT interaction in the present study. The authors also demonstrated that inhibition of binding was achieved only with PG fragments containing multiple cross-linked murein subunits, not with smaller murein mono- or dimers. This suggests that the CWT domain affinity for S. aureus PG has to be higher than that observed here for the five-residue peptide, ~300 μM. It also suggests that the PG lattice structure is needed for maximal affinity, only cross-linked fragments with multiple murein moieties being capable of reproducing this structure. In other words, binding to the pre-arranged PG lattice is likely to be entropically more favorable than binding, through induced folding, to a disordered peptide nonetheless containing the same interaction sites.
Conclusions
This study reveals details on structure and dynamics within and between the lysostaphin domains, on metal ion preference of the CAT domain, as well as characterizes substrate binding sites and affinities of the CWT domain. Remarkably, the affinity of the CWT domain was found to be much higher toward the stem peptide than toward pentaglycine. Also, the CWT domain affinity for pentaglycine increased when the latter was annexed to a stem peptide-like moiety. These findings underscore the importance of auxiliary contacts in CWT PG recognition. The stem peptide analogs interacted with two surfaces on both sides of the N-terminal pentaglycine binding groove of the CWT domain. Residues E414, R427, W489, and I492 are predicted to be crucial to the PG interaction. Taking into account the PG architecture, rather than just the pentaglycine bridge, is imperative to appreciate the factors contributing to the catalytic activity of the CAT domain, to recognize the structural components influencing the interaction between lysostaphin and PG and to engineer novel lysostaphin- or CWT-based antimicrobials.
Data Availability Statement
The assigned chemical shifts and coordinates of lysostaphin have been deposited in the BioMagResBank (http://www.bmrb.wisc.edu/) and the PDB (https://www.rcsb.org/) with accession codes 34121 and 5NMY, respectively.
Author Contributions
PP conceived and designed the research. VR and LK produced and purified the proteins. VR and HM performed enzyme activity studies. UP performed SAXS measurements and SAXS data analysis. HT carried out MD simulations. HT, LK, and PP conducted CWT domain NMR studies. HT and PP performed full-length lysostaphin NMR studies. All authors wrote the paper.
Funding
The study was supported by the grants from Academy of Finland (number 288235 to PP and 28348 to UP) and Sigrid Juselius foundation to PP and the Helsinki Graduate Program in Biotechnology and Molecular Biology and Centre for International Mobility (CIMO) to VR.
Conflict of Interest Statement
HM was employed by company VTT Technical Research Centre of Finland Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge CSC-IT Center for Science Ltd. for the allocation of computational resources. ESRF is thanked for providing the beamline BM29 access.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2018.00060/full#supplementary-material
References
Aitio, O., Hellman, M., Skehan, B., Kesti, T., Leong, J. M., Saksela, K., et al. (2012). Enterohaemorrhagic Escherichia coli exploits a tryptophan switch to hijack host f-actin assembly. Structure 20, 1692–1703. doi: 10.1016/j.str.2012.07.015
Baba, T., and Schneewind, O. (1996). Target cell specificity of a bacteriocin molecule: a C-terminal signal directs lysostaphin to the cell wall of Staphylococcus aureus. EMBO J. 15:4789–4797.
Banci, L., Benedetto, M., Bertini, I., Del Conte, R., Piccioli, M., and Viezzoli, M. S. (1998). Solution structure of reduced monomeric Q133M2 copper, zinc superoxide dismutase (SOD). Why is SOD a dimeric enzyme? Biochemistry 37, 11780–11791. doi: 10.1021/bi9803473
Barraud, P., Schubert, M., and Allain, F. H. (2012). A strong 13C chemical shift signature provides the coordination mode of histidines in zinc-binding proteins. J. Biomol. NMR 53, 93–101. doi: 10.1007/s10858-012-9625-6
Benešík, M., Nováček, J., Janda, L., Dopitová, R., Pernisová, M., Melková, K., et al. (2017). Role of SH3b binding domain in a natural deletion mutant of Kayvirus endolysin LysF1 with a broad range of lytic activity. Virus Genes 54, 130–139. doi: 10.1007/s11262-017-1507-2
Berendsen, H. J. C., Postma, J. P. M., van Gunsteren, W. F., DiNola, A., and Haak, J. R. (1984). Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690. doi: 10.1063/1.448118
Bhattacharya, A., Tejero, R., and Montelione, G. T. (2007). Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795. doi: 10.1002/prot.21165
Blazanovic, K., Zhao, H., Choi, Y., Li, W., Salvat, R. S., Osipovitch, D. C., et al. (2015). Structure-based redesign of lysostaphin yields potent antistaphylococcal enzymes that evade immune cell surveillance. Mol. Ther. Methods. Clin. Dev. 2:15021. doi: 10.1038/mtm.2015.21
Brickner, S. J., Barbachyn, M. R., Hutchinson, D. K., and Manninen, P. R. (2008). Linezolid (ZYVOX), the first member of a completely new class of antibacterial agents for treatment of serious gram-positive infections. J. Med. Chem. 51, 1981–1990. doi: 10.1021/jm800038g
Browder, H. P., Zygmunt, W. A., Young, J. R., and Tavormina, P. A. (1965). Lysostaphin: enzymatic mode of action. Biochem. Biophys. Res. Commun. 19, 383–389. doi: 10.1016/0006-291X(65)90473-0
Case, D. A., Cheatham, T. E., Darden, T., Gohlke, H., Luo, R., Merz, K. M., et al. (2005). The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688. doi: 10.1002/jcc.20290
Chambers, H. F., and Deleo, F. R. (2009). Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 7, 629–641. doi: 10.1038/nrmicro2200
Clore, G. M., and Iwahara, J. (2009). Theory, practice, and applications of paramagnetic relaxation enhancement for the characterization of transient low-population states of biological macromolecules and their complexes. Chem. Rev. 109, 4108–4139. doi: 10.1021/cr900033p
Darden, T., York, D., and Pedersen, L. (1993). Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092. doi: 10.1063/1.464397
Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. doi: 10.1007/BF00197809
Ercoli, G., Tani, C., Pezzicoli, A., Vacca, I., Martinelli, M., Pecetta, S., et al. (2015). LytM proteins play a crucial role in cell separation, outer membrane composition, and pathogenesis in nontypeable Haemophilus influenzae. MBio 6:e02575. doi: 10.1128/mBio.02575-14
Firczuk, M., Mucha, A., and Bochtler, M. (2005). Crystal structures of active LytM. J. Mol. Biol. 354, 578–590. doi: 10.1016/j.jmb.2005.09.082
Franke, D., Petoukhov, M. V., Konarev, P. V., Panjkovich, A., Tuukkanen, A., Mertens, H. D. T., et al. (2017). ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225. doi: 10.1107/S1600576717007786
Gründling, A., and Schneewind, O. (2006). Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus. J. Bacteriol. 188, 2463–2472. doi: 10.1128/JB.188.7.2463-2472.2006
Gu, J., Feng, Y., Feng, X., Sun, C., Lei, L., Ding, W., et al. (2014). Structural and biochemical characterization reveals LysGH15 as an unprecedented “EF-hand-like” calcium-binding phage lysin. PLoS Pathog. 10:e1004109. doi: 10.1371/journal.ppat.1004109
Hirakawa, H., Akita, H., Fujiwara, T., Sugai, M., and Kuhara, S. (2009). Structural insight into the binding mode between the targeting domain of ALE-1 (92AA) and pentaglycine of peptidoglycan. Protein Eng. Des. Sel. 22, 385–391. doi: 10.1093/protein/gzp014
Humphrey, W., Dalke, A., and Schulten, K. (1996). VMD - visual molecular dynamics. J. Mol. Graph. 14, 33–38. doi: 10.1016/0263-7855(96)00018-5
Jagielska, E., Chojnacka, O., and Sabała, I. (2016). LytM fusion with SH3b-like domain expands its activity to physiological conditions. Microb. Drug Resist. 22, 461–469. doi: 10.1089/mdr.2016.0053
Kim, S. J., Chang, J., and Singh, M. (2015). Peptidoglycan architecture of Gram-positive bacteria by solid-state NMR. Biochim. Biophys. Acta 1848, 350–362. doi: 10.1016/j.bbamem.2014.05.031
Kneller, J. M., Lu, M., and Bracken, C. (2002). An effective method for the discrimination of motional anisotropy and chemical exchange. J. Am. Chem. Soc. 124, 1852–1853. doi: 10.1021/ja017461k
Köck, R., Becker, K., Cookson, B., van Gemert-Pijnen, J. E., Harbarth, S., Kluytmans, J., et al. (2010). Methicillin-resistant Staphylococcus aureus (MRSA): burden of disease and control challenges in Europe. Euro Surveill. 15:19688. doi: 10.2807/ese.15.41.19688-en
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003). PRIMUS : a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282. doi: 10.1107/S0021889803012779
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Lee, B. Y., Singh, A., David, M. Z., Bartsch, S. M., Slayton, R. B., Huang, S. S., et al. (2013). The economic burden of community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA). Clin. Microbiol. Infect. 19, 528–536. doi: 10.1111/j.1469-0691.2012.03914.x
López-Méndez, B., and Güntert, P. (2006). Automated protein structure determination from NMR spectra. J. Am. Chem. Soc. 128, 13112–13122. doi: 10.1021/ja061136l
Lowy, F. D. (1998). Staphylococcus aureus infections. N. Engl. J. Med. 339, 520–532. doi: 10.1056/NEJM199808203390806
Lu, H., Gu, M. G., Huang, Q., Huang, J. J., Lu, W. Y., Lu, H., et al. (2013). Hydrogen/deuterium exchange mass spectrometry and site-directed disulfide cross-linking suggest an important dynamic interface between the two lysostaphin domains. Antimicrob. Agents Chemother. 57, 1872–1881. doi: 10.1128/AAC.02348-12
Lu, J. Z., Fujiwara, T., Komatsuzawa, H., Sugai, M., and Sakon, J. (2006). Cell wall-targeting domain of glycylglycine endopeptidase distinguishes among peptidoglycan cross-bridges. J. Biol. Chem. 281, 549–558. doi: 10.1074/jbc.M509691200
Nelson, D. C., Schmelcher, M., Rodriguez-Rubio, L., Klumpp, J., Pritchard, D. G., Dong, S., et al. (2012). Endolysins as antimicrobials. Adv. Virus Res. 83, 299–365. doi: 10.1016/B978-0-12-394438-2.00007-4.
Osipovitch, D. C., and Griswold, K. E. (2015). Fusion with a cell wall binding domain renders autolysin LytM a potent anti-Staphylococcus aureus agent. FEMS Microbiol. Lett. 362, 1–7. doi: 10.1093/femsle/fnu035
Pang, Y. P. (1999). Novel zinc protein molecular dynamics simulations: steps toward antiangiogenesis for cancer treatment. J. Mol. Model. 5, 196–202. doi: 10.1007/s008940050119
Pang, Y. P., Xu, K., Yazal, J. E., and Prendergas, F.G. (2000). Successful molecular dynamics simulation of the zinc-bound farnesyltransferase using the cationic dummy atom approach. Protein Sci. 9, 1857–1865.
Ponting, C. P., Aravind, L., Schultz, J., Bork, P., and Koonin, E. V. (1999). Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J. Mol. Biol. 289, 729–745. doi: 10.1006/jmbi.1999.2827
Ramadurai, L., Lockwood, K. J., Nadakavukaren, M. J., and Jayaswal, R. K. (1999). Characterization of a chromosomally encoded glycylglycine endopeptidase of Staphylococcus aureus. Microbiology (Reading, Engl.) 145 (Pt 4), 801–808. doi: 10.1099/13500872-145-4-801
Raulinaitis, V., Tossavainen, H., Aitio, O., Juuti, J. T., Hiramatsu, K., Kontinen, V., et al. (2017a). Identification and structural characterization of LytU, a unique peptidoglycan endopeptidase from the lysostaphin family. Sci. Rep. 7:6020. doi: 10.1038/s41598-017-06135-w
Raulinaitis, V., Tossavainen, H., Aitio, O., Seppala, R., and Permi, P. (2017b). 1H, 13C and 15N resonance assignments of the new lysostaphin family endopeptidase catalytic domain from Staphylococcus aureus. Biomol. NMR Assign. 11, 69–73. doi: 10.1007/s12104-016-9722-7
Rawlings, N. D., Morton, F. R., Kok, C. Y., Kong, J., and Barrett, A. J. (2008). MEROPS: the peptidase database. Nucleic Acids Res. 36, D320–D325. doi: 10.1093/nar/gkm954
Roe, D. R., and Cheatham, T. E. (2013). PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9, 3084–3095. doi: 10.1021/ct400341p
Ryckaert, J.-P., Ciccotti, G., and Berendsen, H. J. C. (1997). Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. doi: 10.1016/0021-9991(77)90098-5
Sabala, I., Jagielska, E., Bardelang, P. T., Czapinska, H., Dahms, S. O., Sharpe, J. A., et al. (2014). Crystal structure of the antimicrobial peptidase lysostaphin from Staphylococcus simulans. FEBS J. 281, 4112–4122. doi: 10.1111/febs.12929
Saksela, K., and Permi, P. (2012). SH3 domain ligand binding: what's the consensus and where's the specificity? FEBS Lett. 586, 2609–2614. doi: 10.1016/j.febslet.2012.04.042
Schindler, C. A., and Schuhardt, V. T. (1964). Lysostaphin: a new bacteriolytic enzyme for the Staphylococcus. Proc. Natl. Acad. Sci. U.S.A. 51, 414–421. doi: 10.1073/pnas.51.3.414
Schmelcher, M., Powell, A. M., Becker, S. C., Camp, M. J., and Donovan, D. M. (2012). Chimeric phage lysins act synergistically with lysostaphin to kill mastitis-causing Staphylococcus aureus in murine mammary glands. Appl. Environ. Microbiol. 78, 2297–2305. doi: 10.1128/AEM.07050-11
Shen, Y., and Bax, A. (2013). Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J. Biomol. NMR 56, 227–241. doi: 10.1007/s10858-013-9741-y
Sugai, M., Fujiwara, T., Akiyama, T., Ohara, M., Komatsuzawa, H., Inoue, S., et al. (1997). Purification and molecular characterization of glycylglycine endopeptidase produced by Staphylococcus capitis EPK1. J. Bacteriol. 179, 1193–1202. doi: 10.1128/jb.179.4.1193-1202.1997
Szweda, P., Schielmann, M., Kotlowski, R., Gorczyca, G., Zalewska, M., and Milewski, S. (2012). Peptidoglycan hydrolases-potential weapons against Staphylococcus aureus. Appl. Microbiol. Biotechnol. 96, 1157–1174. doi: 10.1007/s00253-012-4484-3
The Uniprot Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169. doi: 10.1093/nar/gkw1099
Thumm, G., and Götz, F. (1997). Studies on prolysostaphin processing and characterization of the lysostaphin immunity factor (Lif) of Staphylococcus simulans biovar staphylolyticus. Mol. Microbiol. 23, 1251–1265. doi: 10.1046/j.1365-2958.1997.2911657.x
Tjandra, N. (1996). Anisotropic rotational diffusion of perdeuterated HIV protease from 15N NMR relaxation measurements at two magnetic fields. J. Biomol. NMR 8, 273–284. doi: 10.1007/BF00410326
Tria, G., Mertens, H. D., Kachala, M., and Svergun, D. I. (2015). Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217. doi: 10.1107/S205225251500202X
Tschierske, M., Ehlert, K., Strandén, A. M., and Berger-Bächi, B. (1997). Lif, the lysostaphin immunity factor, complements FemB in staphylococcal peptidoglycan interpeptide bridge formation. FEMS Microbiol. Lett. 153, 261–264. doi: 10.1111/j.1574-6968.1997.tb12583.x
van Hal, S. J., Jensen, S. O., Vaska, V. L., Espedido, B. A., Paterson, D. L., and Gosbell, I. B. (2012). Predictors of mortality in Staphylococcus aureus bacteremia. Clin. Microbiol. Rev. 25, 362–386. doi: 10.1128/CMR.05022-11
Vollmer, W., Blanot, D., and de Pedro, M. A. (2008). Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167. doi: 10.1111/j.1574-6976.2007.00094.x
Wang, X., Yang, X., Yang, C., Wu, Z., Xu, H., and Shen, Y. (2011). Crystal structure of outer membrane protein NMB0315 from Neisseria meningitidis. PLoS ONE 6:e26845. doi: 10.1371/journal.pone.0026845
Whisstock, J. C., and Lesk, A. M. (1999). SH3 domains in prokaryotes. Trends Biochem. Sci. 24, 132–133. doi: 10.1016/S0968-0004(99)01366-3
WHO Mediacentre (2017). WHO Publishes List of Bacteria For Which New Antibiotics are Urgently Needed. Available online at: http://www.who.int/mediacentre/news/releases/2017/bacteria-antibiotics-needed/en/
Keywords: lysostaphin, NMR structure, pentaglycine, peptidoglycan, protein dynamics, SH3b domain, Staphylococcus aureus, substrate binding
Citation: Tossavainen H, Raulinaitis V, Kauppinen L, Pentikäinen U, Maaheimo H and Permi P (2018) Structural and Functional Insights Into Lysostaphin–Substrate Interaction. Front. Mol. Biosci. 5:60. doi: 10.3389/fmolb.2018.00060
Received: 19 April 2018; Accepted: 12 June 2018;
Published: 03 July 2018.
Edited by:
Maria Rosaria Conte, King's College London, United KingdomReviewed by:
Filippo Prischi, University of Essex, United KingdomVladimir I. Polshakov, Lomonosov Moscow State University, Russia
Copyright © 2018 Tossavainen, Raulinaitis, Kauppinen, Pentikäinen, Maaheimo and Permi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Perttu Permi, cGVydHR1LnBlcm1pQGp5dS5maQ==