 Ting-Yi Su
Ting-Yi Su Paul M. Harrison
Paul M. Harrison- Department of Biology, McGill University, Montreal, QC, Canada
Prions in eukaryotes have been linked to diseases, evolutionary capacitance, large-scale genetic control, and long-term memory formation. Prion formation and propagation have been studied extensively in the budding yeast Saccharomyces cerevisiae. Here, we have analysed the conservation of sequence and of prion-like composition for prion-forming proteins and for other prion-like proteins from S. cerevisiae, across three evolutionary levels. We discover that prion-like status is well-conserved for about half the set of prion-formers at the Saccharomycetes level, and that prion-forming domains evolve more quickly as sequences than other prion-like domains do. Such increased mutation rates may be linked to the acquisition of functional roles for prion-forming domains during the evolutionary epoch of Saccharomycetes. Domain scores for prion-like composition in S. cerevisiae are strongly correlated with scores for such composition weighted evolutionarily over the dozens of fungal species examined, indicating conservation of such prion-like status. Examples of notable prion-like proteins that are highly conserved both in sequence and prion-like composition are discussed.
Introduction
Prions are proteinaceous infectious particles that were originally identified as the causative agents (made from the prion protein PrP) of devastating neurological diseases in mammals. Prions propagate alternative protein states, through co-option of further copies of the same proteins. In budding yeast (Saccharomyces cerevisiae), propagation of these alternative states can be sustained during budding, mating, and artificial laboratory protocols. Such yeast prions have been linked to diverse phenomena including evolutionary capacitance, disease-like states, and large-scale genetic control. The first well-characterized yeast prions, that underlie the [PSI+] and [URE3] prions, are propagating amyloids of the proteins Sup35p and Ure2p, respectively. The protein Sup35p is part of the translation termination complex. [PSI+] prion formation reduces translation termination efficiency and increases non-sense-codon read-through levels (Cox, 1965; Shorter and Lindquist, 2005). This read-through has been shown to have a potential role in uncovering cryptic genetic variation (True and Lindquist, 2000; True et al., 2004). [URE3] causes upregulation of poor nitrogen source usage, even when rich sources are available (Lacroute, 1971; Wickner, 1994; Wickner et al., 2004). Prion variants sometimes behave as budding-yeast diseases (Nakayashiki et al., 2005; McGlinchey et al., 2011). The [MOT3+] prion has been shown to have a possible role in control of transitions to multicellularity (Holmes et al., 2013). The stress-inducible cytoskeleton-linked budding-yeast protein Lsb2 can form a metastable prion in response to high temperatures (Chernova et al., 2017). There are now >10 known amyloid-based prions of S. cerevisiae (Harbi et al., 2012; Harbi and Harrison, 2014). Prion-forming proteins have also been discovered in the fungus Podospora anserina and the fission yeast Schizosaccharomyces pombe (Saupe, 2011; Sideri et al., 2017). Almost all amyloid-based budding yeast prion-forming regions have a high degree of intrinsic disorder and share a bias for asparagine (N) and/or glutamine (Q) residues (Harbi and Harrison, 2014; Harrison, 2017). Glutamine and asparagine have differing influences on prion formation: Ns promote benign prion formation, whereas excess Q can lead to toxic non-amyloid conformer formation (Halfmann et al., 2011). Several algorithms have been developed that annotate protein regions with high potential prion-forming propensity (Espinosa Angarica et al., 2013; Ross et al., 2013; Lancaster et al., 2014; Zambrano et al., 2015). Prion-like proteins in yeast and other organisms have more recently been linked to other processes, such as the formation of stress granules and other membraneless biomolecular condensates (Jain et al., 2016; Franzmann et al., 2018).
The original mammalian PrP domain is not biased for Ns and Qs, and is deeply conserved since a PrP founder gene emerged in early chordate evolution (Harrison et al., 2010; Ehsani et al., 2011; Westaway et al., 2011). The [PSI+] prion has an N/Q bias that is conserved across Ascomycota and Basidiomycota, which diverged >1 billion years ago (Harrison et al., 2007). A large population of yeast-prion-like proteins emerged early in the evolution of Saccharomycetes, as a result of mutational trends to form more polyasparagine runs, thus providing an evolutionary “test set” from which several prion-forming domains seem to have developed (An and Harrison, 2016). Eukaryotic proteomes often bear large numbers of these prion-like domains. The slime mold Dictyostelium has >20% of its proteins containing prion-like domains (Malinovska et al., 2015; An et al., 2016) and there is evidence it has evolved a mechanism for subvertion of prion formation (Malinovska and Alberti, 2015; Malinovska et al., 2015). Other organisms that have high levels of prion-like proteins include Drosophila melanogaster, Plasmodium falciparum, and the leech Helobdella robusta (An et al., 2016; Pallarès et al., 2018). Several other yeast-prion-like proteins have links to human neurodegenerative pathomechanisms (Sun et al., 2011; Kim et al., 2013; Pokrishevsky et al., 2016) or to long-term memory formation in Aplysia and Drosophila (Si et al., 2010; Khan et al., 2015). Predicted prions have been detected in all the domains of life (Espinosa Angarica et al., 2013), including thousands in viruses and phages (Tetz and Tetz, 2017, 2018), and tens of thousands in bacteria (Harrison, 2019). Possible bacterial prion-forming proteins have also been detected experimentally (Yuan et al., 2014; Shahnawaz et al., 2017; Yuan and Hochschild, 2017; Molina-García et al., 2018). A survey of over 800 bacterial proteomes discovered >2,000 potential bacterial prions linked to diverse functional roles such as cell adaptability and invasion (Iglesias et al., 2015; Pallarès and Ventura, 2017). Bacterial prion-like proteins have a characteristic pattern of multi-phylum distribution coupled to sparse, intermittent conservation across their evolutionary range (Harrison, 2019). About 5% of compositionally-biased dark matter in the TrEMBL protein database (i.e., regions that cannot be assigned as either structured or intrinsically disordered) are predicted to be prion-like domains (Harrison, 2018).
Here, we examine the conservation of sequence and of prion-like composition for sets of prion-forming and prion-like proteins from the budding-yeast S. cerevisiae. We discover that prion-like composition in S. cerevisiae is strongly correlated with prion-like composition when weighted evolutionarily over dozens of fungal species, for both prion-formers and other prion-like proteins. However, sequence-wise prion-forming domains generally evolve more quickly than other prion-like domains.
Methods
Data
The UniProt (Boeckmann et al., 2003) set of reference fungal proteomes was downloaded from www.uniprot.org in June 2017, and collated into sets at three evolutionary levels relative to the budding yeast S. cerevisiae, as illustrated in Figure 1A.
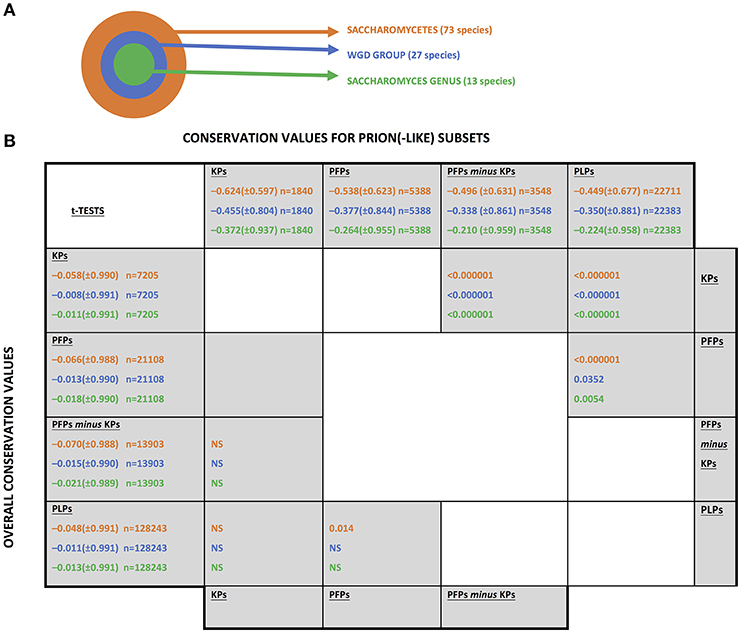
Figure 1. The fungal levels examined for conservation of prion(-like) proteins from Saccharomyces cerevisiae. (A) A schematic of the three taxonomic levels examined. “WGD” stands for “whole genome duplication.”(B) Conservation values of the different sets of prion-like or prion-forming domains. The abbreviations for the sets are as listed in section Methods. The mean conservation values for prion-forming or prion-like domains are listed along the top of the table, with the values for the whole sequences listed along the left-hand column. The standard deviations are in brackets. The number of coulmns in each sample is indicated (n). They are colour-coded as in part (A). t-Tests were performed to compare the mean conservation values for each set, in the upper diagonal half for the prion(-like) domains and in the lower diagonal for the whole sequences of the same proteins. “NS” means “not significant”.
Sets of Prion-Forming and Prion-Like Proteins
Prion-forming proteins for S. cerevisiae were taken from the PrionHome database (Harbi et al., 2012; Harbi and Harrison, 2014). Two groups were analysed: (i) the set of bona fide known prions (the “KP” set), and (ii) a larger set made from these known prions plus other “prionogenic” amyloid-forming proteins (in total called the “PFP” set). The prion protein Mod5p, which underlies the [MOD+] prion state (Suzuki et al., 2012), is not included in the analysis, since it is not N/Q-rich. The PFP set includes the prionogenic proteins from the analysis of Alberti et al. (2009) that have been shown to form prions, through the SUP35C prion assay in conjunction with evidence for in vivo amyloid formation by the full-length proteins from other assays. The PFP protein set is as follows (UniProt IDs and standard gene names, the KP set is asterisked): P05453, Sup35*; P09547, Swi1*; P14922, Cyc8*; P23202, Ure2*; P25367, Rnq1*; P32432, Sfp1*; Q08972, New1*; P54785, Mot3*; Q02629, NUP100*; P32588, Pub1*; P40070, Lsm4; P14907, Nsp1; P18494, Gln3; P32770, Nrp1; P38180, YBL081W; P38216, YBR016W; P38429, Sap30; P38691, Ksp1; P40356, Pgd1; P53894, Cbk1; Q05166, Asm4; Q08925, Mrn1; Q12139, YPR022C; Q12221, Puf2; Q12224, Rlm1; Q12361, Gpr1; P40356, Med3; P12383, Pdr1; Q05672, Rbs1; P40956, Gts1; Q99383, Hrp1; Q06449, Pin3*.
A set of additional prion-like proteins was also generated. To do this, we applied the PLAAC prion prediction program (Toombs et al., 2012; Ross et al., 2013; Lancaster et al., 2014) to S. cerevisiae, and also applied it to all the complete fungal proteomes for further calculations of prion-like conservation as detailed below. PLAAC uses a Hidden Markov Model trained on the composition of known prion-forming domains, which all have a pronounced bias for N and/or Q residues. For PLAAC, we used PRDscore values ≥0.0 as prion propensity scores. Any other sequences analysed that yield negative values or “N/A” in the output from PLAAC are set equal to 0.0 for the purposes of this analysis. The boundaries for the predicted prion domain from PLAAC were used, if they have not been experimentally determined. The LLR score (which is the highest overall score achieved within a scanning window) has been used in previous studies to pick out the stretch of sequence in a protein that is most likely to form prions (Saupe, 2011; An and Harrison, 2016; Sideri et al., 2017; Harrison, 2019). We used PRDscore in preference to LLR score since it better reflects the overall degree of bias toward a prion-like composition (i.e., longer prion-like domains have higher PRDscore values).
Multiple Sequence Alignment and Calculation of Conservation Scores
Orthologs in other fungal proteomes were detected for each S. cerevisiae protein examined using BLASTP and the bi-directional best hits method, with an expectation value threshold of 0.001 and default parameters otherwise (Altschul et al., 1997). Multiple sequence alignments of orthologs were performed using the default KMAD program (Lange et al., 2016), which is designed for optimal sequence alignment of intrinsically disordered proteins (Narasumani and Harrison, 2018), such as the N/Q-rich prion-forming proteins, which are the focus of this study. Sequence conservation was calculated using the program AL2CO with default parameters (Pei and Grishin, 2001). Mean conservation values were calculated as before for our analysis of folding-on-binding proteins (Narasumani and Harrison, 2015). Evolutionarily-weighted prion-propensity scores were derived according to the equation:
where Pi is the prion propensity score for the ith ortholog, n is the number of orthologs and wi is the evolutionary weighting for the ith ortholog sequence. This weighted score indicates the degree of conservation of the prion propensity score, but taking into account the differing divergences of the orthologs. Prion propensity scores = 0.0 are included in the summation. These evolutionary weightings were calculated in either of two ways. Firstly a “PC” (“%identity”) weighting was calculated wi = (1 – %identity/100.0), where %identity is from the alignment of the S. cerevisiae protein and the ith ortholog. Then a “BS” (“bitscore”) weighting was calculated wi = (ith bitscore / self-bitscore) where the ith bitscore is the bitscore from the alignment of the S. cerevisiae protein and the ith ortholog, and the self-bitscore is the bitscore from aligning the S. cerevisiae protein to itself. The difference in the results from using these two weightings is minimal (as can be seen in Table 1). Generally, results for the PC evolutionary weighting are reported and discussed in the section Results and Discussion.
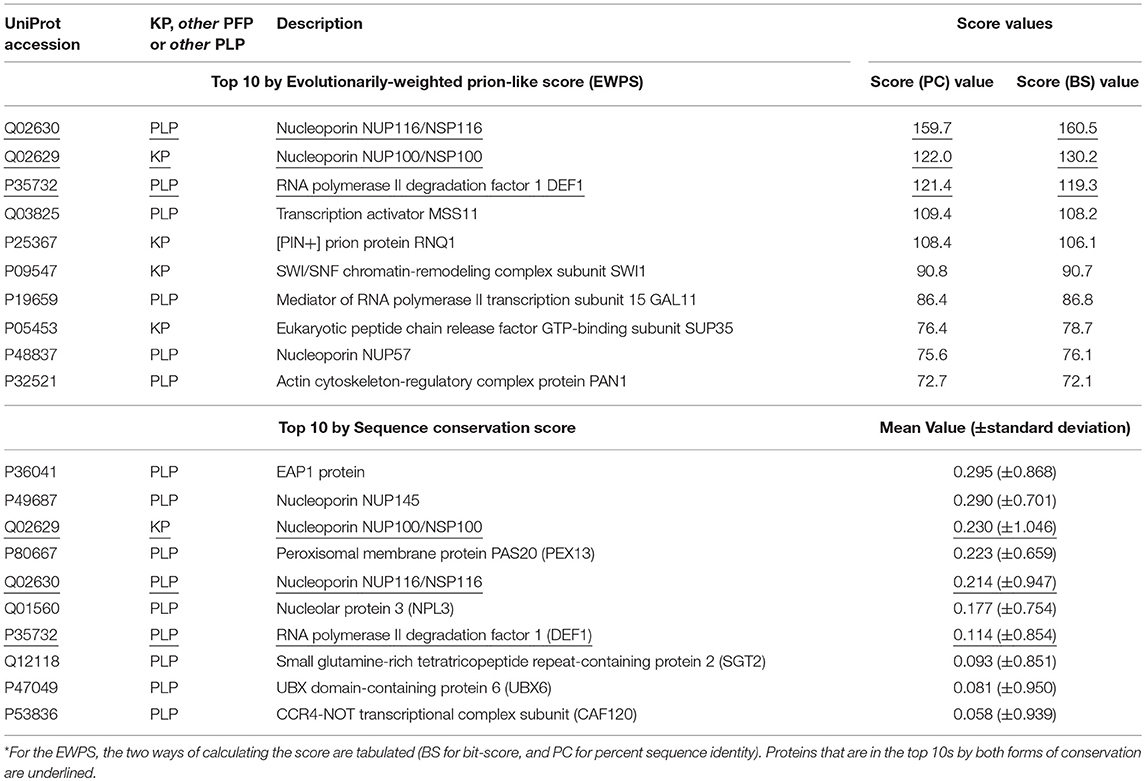
Table 1. Top 10 prion-like or prion-forming proteins by conservation across Saccharomycetes of prion-like composition, or of sequence*.
Conservation of sequence and of prion-like status was examined at three evolutionary levels that are all centred on the S. cerevisiae species: (i) across the class Saccharomycetes; (ii) across the set of yeasts that descend from a common ancestor that underwent a whole-genome duplication (termed the “WGD group”); (iii) across the Saccharomyces genus (Figure 1A). This set of three levels was chosen because of the surge in formation of prion-like domains that has occurred since the last common ancestor of Saccharomycetes (An and Harrison, 2016). Also, the whole genome duplication may influence the conservation of prion status.
Results and Discussion
Overall Conservation Trends
Sequence conservation was analysed for known prion-forming regions and for other prion-like protein domains (PLPs) defined by the program PLAAC (Lancaster et al., 2014). The complete set of sequence conservation values is tabulated in Supplementary File 1. Prion-forming domains are considered either as a “known prion” domains (KPs) set, or a larger set of prion-forming domains (PFPs), of which the KPs are a subset. Mean conservation was compared for each of these three sets of domains (but using [PFPs-minus-KPs] when comparing to KPs) at three different evolutionary levels (Figure 1A). In general, regardless of evolutionary level, the KPs are significantly less conserved than the PFPs, which are significantly less conserved than the PLPs (Figure 1B, upper diagonal half). In comparison, there is virtually no significant difference detected for mean conservation values for the whole sequences of the proteins in which these domains reside (these mean conservation values include the components of the conservation that come from the prion-like/prion-forming domains; Figure 1B, lower diagonal half). Thus, these trends are due to mutation patterns in the prion-like/prion-forming areas themselves. So, for proteins with more evidence of prion-forming ability, the sequences have faster evolutionary rates. Such increased mutation rates may be linked to the acquisition of functional roles for prion-forming domains during the evolutionary epoch of Saccharomycetes. That is, out of the large “test set” of prion-like N/Q-rich domains that has formed during Saccharomycetes evolution (An and Harrison, 2016), we suggest that those that have become functional have mutated more quickly. These results also tally well with previous observations that intrinsically disordered regions often evolve more quickly than structured regions (Brown et al., 2002, 2011) (prion-forming regions tend to be highly intrinsically disordered; Harbi and Harrison, 2014; Harrison, 2017).
Individual Conservation Behaviour of Prion-Forming Domains
Mean conservation values were also calculated for each individual prion-forming domain, at the three evolutionary levels (Figure 2). The domains were grouped into thirds based on the ranking of their mean conservation values, as colour-coded in the figure. More than half of the prion-forming domains (59%) are maintained in the same third of the list across the three evolutionary levels (Saccharomycetes, the WGD group and the Saccharomyces genus). This indicates a substantial consistency in conservation across different evolutionary epochs in prion-forming domain evolution. Only one domain, in the protein NRP1, a domain from the Alberti et al. data set (Alberti et al., 2009), moves between the three thirds of the listings, to become the most conserved prion-like domain across the Saccharomyces genus (underlined in Figure 2). This is a putative RNA-binding protein that localizes to stress granules, which has not been studied extensively (Buchan et al., 2008). The five most conserved PFPs across the Saccharomyces genus sequence-wise also include: the GLFG-motif nucleoporin NUP100, a bona fide prion-former which is part of the nuclear pore complex (Halfmann et al., 2012); GLN3, a transcriptional activator of genes regulated by nitrogen catabolite repression; RBS1, a protein involved in assembly of the RNA polymerase III (Pol III) complex; MED3/PGD1, a subunit of the RNA polymerase II mediator complex. NUP100 is also a significant protein-interaction hub for other prion-like proteins (Harbi and Harrison, 2014). About a third of its interactors were found to be prion-like, with most of these being nucleoporins, including NUP116 and ASM4 (Harbi and Harrison, 2014). NUP116, which is the paralog of NUP100 arising from the whole-genome duplication that occurred during Saccharomycetes evolution, can be induced to form aggregate foci at low levels by over-expression of the prion domain of NUP100 (Halfmann et al., 2012). ASM4 is a member of the set of prion-forming proteins from the experiments by Alberti et al. (2009).
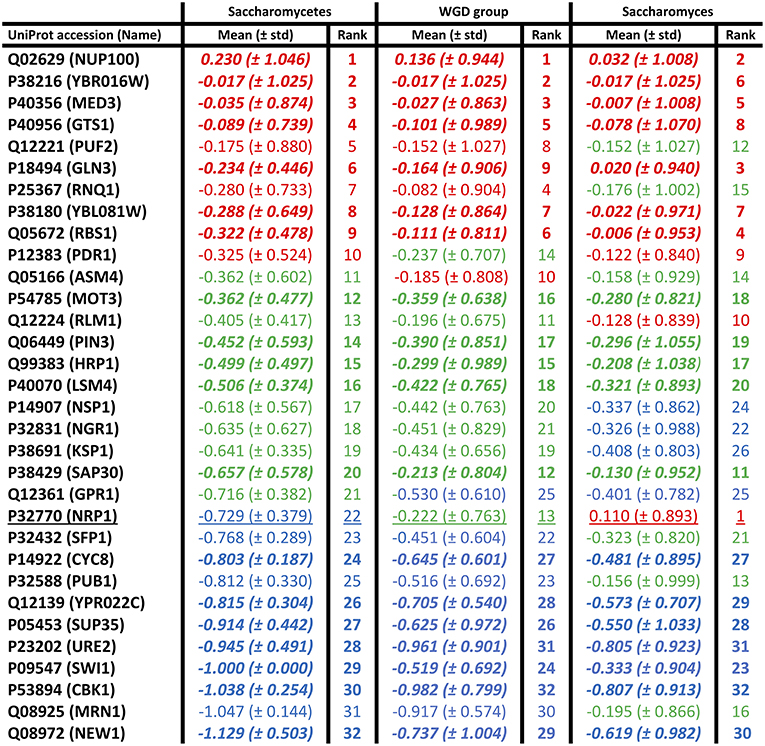
Figure 2. Individual mean conservation values for prion-forming domains. These are tabulated for each domain, with the top, middle, and bottom thirds colour-coded red, green, and blue, but with the ordering of the first column maintained across the figure. The numerical ranking is also listed for each domain at each level. The domains that are conserved in the same thirds at all levels are in bold italic. There is only one domain that moves between all three thirds of the list (underlined). The standard deviations are in brackets.
Conservation of Prion-Like Composition
Prion-like status has been demonstrated experimentally to be largely composition-dependent, although special roles in prion propagation have been determined for specific parts of prion-determinant sequence or specific repeat patterns (Toombs et al., 2010; MacLea et al., 2015; Shattuck et al., 2017). Domains of prion-like composition also have roles in formation of stress granules and other biomolecular condensates (Jain et al., 2016; Franzmann et al., 2018). Methods (such as PLAAC) to annotate prion-like regions largely rely on detection of regions of proteins that are compositionally similar to known cases of prion-forming regions (Lancaster et al., 2014; Cascarina et al., 2017). We used PLAAC to analyse the conservation of prion-like composition in orthologs of prion-forming and prion-like proteins of S. cerevisiae. An evolutionarily-weighted prion score (EWPS) was calculated, which is a prion score made from terms for other orthologs/species weighted according to how far away from S. cerevisiae an ortholog/species is (as described in section Methods). Thus, orthologs from more similar species are given lower weightings, while those from more dissimilar species are given higher ones. We discover that this score is highly correlated with the S. cerevisiae prion score (SCPS) for both PFPs and PLPs (Figure 3A). The complete set of conservation values for prion-like composition are tabulated in Supplementary File 1.
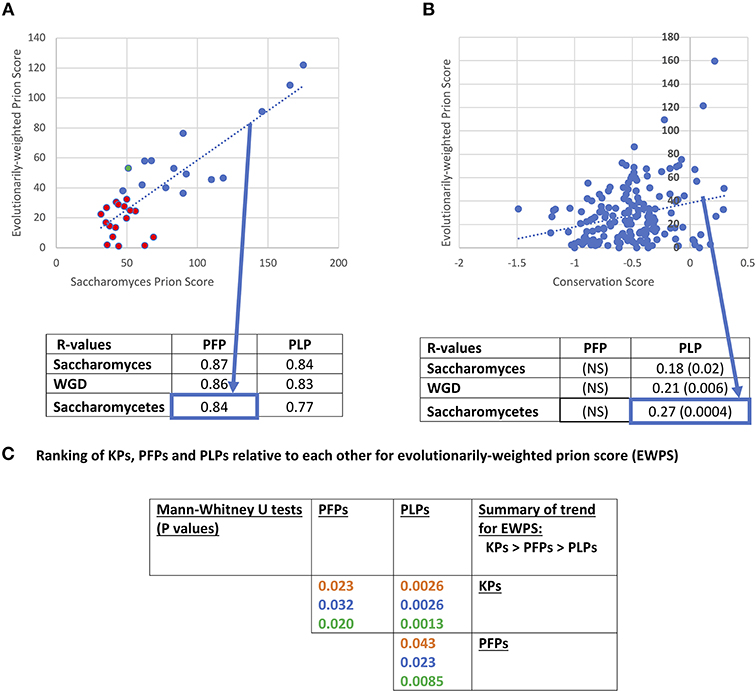
Figure 3. Evolutionarily-weighted prion score (EWPS). (A) Scatterplot of the EWPS vs. the Saccharomyces cerevisiae prion score (SCPS) for the prion-forming protein set at the Saccharomycetes level. Only one has an EWPS > SCPS (green point). Seventeen of the domains have EWPS < the lowest value of SCPS for a known prion domain (=35.6 for PIN3). Below the plot is a tabulation of R-values of these correlations for the PFP and PLP sets at the three evolutionary levels. These are all significant at P < 0.000001. The percent sequence identity method for calculating the EWPS has been used, but the difference in results obtained with the bitscore method in all cases is minimal. “NS” stands for “not significant.” (B) Scatterplot of the EWPS vs. the conservation score at the Saccharomycetes level for the prion-like protein set. Below the plot is a tabulation of R-values for the PFP and PLP sets at the three evolutionary levels. The significant P-values are in brackets. (C) Ranking of KPs, PFPs and PLPs relative to each other for evolutionarily-weighted prion score (EWPS). Tabulation of results of Mann-Whitney U-tests for comparison of the EWPS scores of the PFP, PLP and KP sets. Colour coding is as in Figure 1.
Previously, it was shown that there was a major surge in formation of prion-like regions since the last common ancestor of Saccharomycetes, and that this seems to be linked to increased formation of runs of asparagine residues (An and Harrison, 2016). Thus, this is the most appropriate level at which to analyse patterns of conservation. At this level, R-values for correlation of EWPS and SCPS are = 0.84 for PFPs, and = 0.77 for PLPs, and are maintained at levels of R >0.84 for the WGD group and the Saccharomyces genus. However, for only one prion-former is the EWPS > SCPS (Figure 3A). This is probably because the PLAAC algorithm is trained on S. cerevisiae proteins, and does not necessarily imply that the prion-forming ability of the proteins is generally less in these other species. However, about half (15/32, 47%) have values that are above the lowest level observed for SCPS for any known prion.
Is this conservation of prion-like status correlated with sequence conservation? Consistent with the observations for decreased conservation for PFPs relative to PLPs, we find no correlation between sequence conservation score and EWPS for the prion-formers (Figure 3B). However, there are significant correlations for the prion-like proteins at all levels between EWPS and conservation score, with the most significant value at the Saccharomycetes level. This implies that the prion-like composition of the proteins is partly conserved in specific sequence motifs. In comparison, no significant correlations were observed for prion-forming or prion-like proteins between sequence conservation and the SCPS, implying that the correlations are derived from evolutionary information.
We also checked whether the EWPS has significantly different behaviour for the different sets of KP, PFP and PLP sets (Figure 3C). As would be expected since the PLAAC algorithm was trained on the PFP set, the un-weighted SCPSs are significantly higher for the prion-formers compared to the other prion-like proteins (P-values < 0.000001, Mann-Whitney U-test). However, they are also significantly higher for known prions compared to the rest of the PFP set (P-values < 0.000001, MWU-test). This may be a sort of knock-on effect, since most of the other PFP set members were found in a large-scale analysis to detect prion-forming domains, wherein selection for testing was guided by an earlier algorithm trained on a handful of bona fide known prions (Alberti et al., 2009). This indicates that further experimental procedures that do not cause such a bias or feedback will be necessary to discover more prion domains that might look quite different.
Prion-Like Proteins That Are Highly Conserved in Sequence and Prion-Like Composition
We examined which proteins in the combined KP, PFP and PLP sets have the highest conservation in terms of sequence and of prion-like composition in Saccharomycetes (Table 1). Only three of the top ten sorted on the evolutionarily-weighted prion score are known prions, the rest being PLPs. Nucleoporins (components of the nuclear pore complex) figure prominently (3/10), with the two most conserved being the known prion-former NUP100, and the prion-like protein NUP116 (which was discussed above). Of course, the conservation of the prion-like domains of nucleoporins may be for other reasons, such as for facilitating interactions between pore proteins.
In the top 10 sorted by sequence conservation score, we also see nucleoporins figuring prominently (3/10). The most conserved prion-forming/prion-like sequence in this list is the EAP1 protein, that competes with eIF4G for binding to eIF4E and accelerates mRNA degradation by promotion of decapping. Three proteins are in the top ten by both methods (underlined in Table 1, NUP100, NUP116 and the RNA polymerase degradation factor DEF1), indicating that their prion-like composition is conserved in sequence motifs.
Concluding Remarks
This analysis can be used to guide further experiments aimed at finding prion-forming proteins, both from S. cerevisiae and from other Saccharomycetes species. Of course, promising candidates from other species that are evolutionarily conserved can be studied in S. cerevisiae as a model system, using well-established techniques that have previously been applied to candidate prion-formers from other fungi (Edskes and Wickner, 2013), bacteria (Yuan and Hochschild, 2017), the sea hare Aplysia californica (Si et al., 2003), the plant Arabidopsis thaliana (Chakrabortee et al., 2016), and from humans (Kim et al., 2013). Well-conserved prion-formers may have functional roles linked to prion-like aggregation that have been maintained across many millions of years of evolution. For example, the yeast protein PUB1 has prion-like aggregation that is conserved in the prion-like domain of its human co-ortholog TIA1 (Li et al., 2014; An et al., 2016), which functions as a stress granule component (Gilks et al., 2004). Despite this, PUB1 is one of the prion-forming domains that evolves at a faster rate sequence-wise across Saccharomycetes, possibly indicating the influence of complex selection pressures on its mutation rates (Figure 2).
Data Availability
All datasets generated for this study are included in the manuscript/Supplementary Files.
Author Contributions
T-YS performed data analysis, prepared figures, and edited the paper. PH conceived the study, performed data analysis, prepared figures and tables, and wrote the paper.
Funding
This research was funded by the Natural Sciences Engineering Research Council of Canada. Some of the calculations in this paper were performed on Compute Canada resources.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2019.00054/full#supplementary-material
Supplementary File 1. List of prion-forming and prion-like proteins with their conservation values as studied in this research article.
References
Alberti, S., Halfmann, R., King, O., Kapila, A., and Lindquist, S. (2009). A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell 137, 146–158. doi: 10.1016/j.cell.2009.02.044
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
An, L., Fitzpatrick, D., and Harrison, P. M. (2016). Emergence and evolution of yeast prion and prion-like proteins. BMC Evol. Biol. 16:24. doi: 10.1186/s12862-016-0594-3
An, L., and Harrison, P. M. (2016). The evolutionary scope and neurological disease linkage of yeast-prion-like proteins in humans. Biol. Direct 11:32. doi: 10.1186/s13062-016-0134-5
Boeckmann, B., Bairoch, A., Apweiler, R., Blatter, M. C., Estreicher, A., Gasteiger, E., et al. (2003). The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 31, 365–370. doi: 10.1093/nar/gkg095
Brown, C. J., Johnson, A. K., Dunker, A. K., and Daughdrill, G. W. (2011). Evolution and disorder. Curr. Opin. Struct. Biol. 21, 441–446. doi: 10.1016/j.sbi.2011.02.005
Brown, C. J., Takayama, S., Campen, A. M., Vise, P., Marshall, T. W., Oldfield, C. J., et al. (2002). Evolutionary rate heterogeneity in proteins with long disordered regions. J. Mol. Evol. 55, 104–110. doi: 10.1007/s00239-001-2309-6
Buchan, J. R., Muhlrad, D., and Parker, R. (2008). P bodies promote stress granule assembly in Saccharomyces cerevisiae. J. Cell Biol. 183, 441–455. doi: 10.1083/jcb.200807043
Cascarina, S. M., Paul, K. R., and Ross, E. D. (2017). Manipulating the aggregation activity of human prion-like proteins. Prion 11, 323–331. doi: 10.1080/19336896.2017.1356560
Chakrabortee, S., Kayatekin, C., Newby, G. A., Mendillo, M. L., Lancaster, A., and Lindquist, S. (2016). Luminidependens (LD) is an Arabidopsis protein with prion behavior. Proc. Natl. Acad. Sci. U.S.A. 113, 6065–6070. doi: 10.1073/pnas.1604478113
Chernova, T. A., Kiktev, D. A., Romanyuk, A. V., Shanks, J. R., Laur, O., Ali, M., et al. (2017). Yeast short-lived actin-associated protein forms a metastable prion in response to thermal stress. Cell Rep. 18, 751–761. doi: 10.1016/j.celrep.2016.12.082
Cox, B. (1965). [PSI], a cytoplasmic suppressor of super-suppression in yeast. Heredity 20, 505–521.
Edskes, H. K., and Wickner, R. B. (2013). The [URE3] prion in Candida. Eukaryotic Cell 12, 551–558. doi: 10.1128/EC.00015-13
Ehsani, S., Tao, R., Pocanschi, C. L., Ren, H., Harrison, P. M., and Schmitt-Ulms, G. (2011). Evidence for retrogene origins of the prion gene family. PLoS ONE 6:e26800. doi: 10.1371/journal.pone.0026800
Espinosa Angarica, V., Ventura, S., and Sancho, J. (2013). Discovering putative prion sequences in complete proteomes using probabilistic representations of Q/N-rich domains. BMC Genomics 14:316. doi: 10.1186/1471-2164-14-316
Franzmann, T. M., Jahnel, M., Pozniakovsky, A., Mahamid, J., Holehouse, A. S., Nüske, E., et al. (2018). Phase separation of a yeast prion protein promotes cellular fitness. Science 359:eaao5654. doi: 10.1126/science.aao5654
Gilks, N., Kedersha, N., Ayodele, M., Shen, L., Stoecklin, G., Dember, L. M., et al. (2004). Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 15, 5383–5398. doi: 10.1091/mbc.e04-08-0715.
Halfmann, R., Alberti, S., Krishnan, R., Lyle, N., O'Donnell, C. W., King, O. D., et al. (2011). Opposing effects of glutamine and asparagine govern prion formation by intrinsically disordered proteins. Mol. Cell 43, 72–84. doi: 10.1016/j.molcel.2011.05.013
Halfmann, R., Wright, J. R., Alberti, S., Lindquist, S., and Rexach, M. (2012). Prion formation by a yeast GLFG nucleoporin. Prion 6, 391–399. doi: 10.4161/pri.20199
Harbi, D., and Harrison, P. M. (2014). Interaction networks of prion, prionogenic and prion-like proteins in budding yeast, and their role in gene regulation. PLoS ONE 9:e100615. doi: 10.1371/journal.pone.0100615
Harbi, D., Parthiban, M., Gendoo, D. M., Ehsani, S., Kumar, M., Schmitt-Ulms, G., et al. (2012). PrionHome: a database of prions and other sequences relevant to prion phenomena. PLoS ONE 7:e31785. doi: 10.1371/journal.pone.0031785
Harrison, L. B., Yu, Z., Stajich, J. E., Dietrich, F. S., and Harrison, P. M. (2007). Evolution of budding yeast prion-determinant sequences across diverse fungi. J. Mol. Biol. 368, 273–282. doi: 10.1016/j.jmb.2007.01.070
Harrison, P. M. (2017). fLPS: Fast discovery of compositional biases for the protein universe. BMC Bioinformatics 18:476. doi: 10.1186/s12859-017-1906-3
Harrison, P. M. (2018). Compositionally biased dark matter in the protein universe. Proteomics 18:e1800069. doi: 10.1002/pmic.201800069
Harrison, P. M. (2019). Evolutionary behaviour of bacterial prion-like proteins. PLoS ONE14:e0213030. doi: 10.1371/journal.pone.0213030
Harrison, P. M., Khachane, A., and Kumar, M. (2010). Genomic assessment of the evolution of the prion protein gene family in vertebrates. Genomics 95, 268–277. doi: 10.1016/j.ygeno.2010.02.008
Holmes, D. L., Lancaster, A. K., Lindquist, S., and Halfmann, R. (2013). Heritable remodeling of yeast multicellularity by an environmentally responsive prion. Cell 153, 153–165. doi: 10.1016/j.cell.2013.02.026
Iglesias, V., de Groot, N. S., and Ventura, S. (2015). Computational analysis of candidate prion-like proteins in bacteria and their role. Front. Microbiol. 6:1123. doi: 10.3389/fmicb.2015.01123
Jain, S., Wheeler, J. R., Walters, R. W., Agrawal, A., Barsic, A., and Parker, R. (2016). ATPase-Modulated Stress Granules Contain a Diverse Proteome and Substructure. Cell 164, 487–498. doi: 10.1016/j.cell.2015.12.038
Khan, M. R., Li, L., Pérez-Sánchez, C., Saraf, A., Florens, L., Slaughter, B. D., et al. (2015). Amyloidogenic oligomerization transforms Drosophila Orb2 from a translation repressor to an activator. Cell 163, 1468–1483. doi: 10.1016/j.cell.2015.11.020
Kim, H. J., Kim, N. C., Wang, Y. D., Scarborough, E. A., Moore, J., Diaz, Z., et al. (2013). Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495, 467–473. doi: 10.1038/nature11922
Lacroute, F. (1971). Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J. Bacteriol. 106, 519–522.
Lancaster, A. K., Nutter-Upham, A., Lindquist, S., and King, O. D. (2014). PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics 30, 2501–2502. doi: 10.1093/bioinformatics/btu310
Lange, J., Wyrwicz, L. S., and Vriend, G. (2016). KMAD: knowledge-based multiple sequence alignment for intrinsically disordered proteins. Bioinformatics 32, 932–936. doi: 10.1093/bioinformatics/btv663
Li, X., Rayman, J. B., Kandel, E. R., and Derkatch, I. L. (2014). Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol. Cell 55, 305–318. doi: 10.1016/j.molcel.2014.05.027
MacLea, K. S., Paul, K. R., Ben-Musa, Z., Waechter, A., Shattuck, J. E., Gruca, M., et al. (2015). Distinct amino acid compositional requirements for formation and maintenance of the [PSI(+)] prion in yeast. Mol. Cell. Biol. 35, 899–911. doi: 10.1128/MCB.01020-14
Malinovska, L., and Alberti, S. (2015). Protein misfolding in Dictyostelium: using a freak of nature to gain insight into a universal problem. Prion 9, 339–346. doi: 10.1080/19336896.2015.1099799
Malinovska, L., Palm, S., Gibson, K., Verbavatz, J. M., and Alberti, S. (2015). Dictyostelium discoideum has a highly Q/N-rich proteome and shows an unusual resilience to protein aggregation. Proc. Natl. Acad. Sci. U.S.A. 112, E2620–E2629. doi: 10.1073/pnas.1504459112
McGlinchey, R. P., Kryndushkin, D., and Wickner, R. B. (2011). Suicidal [PSI+] is a lethal yeast prion. Proc. Natl. Acad. Sci. U.S.A. 108, 5337–5341. doi: 10.1073/pnas.1102762108
Molina-García, L., Gasset-Rosa, F., Álamo, M. M., de la Espina, S. M., and Giraldo, R. (2018). Addressing intracellular amyloidosis in bacteria with RepA-WH1, a prion-like protein. Methods Mol. Biol. 1779, 289–312. doi: 10.1007/978-1-4939-7816-8_18
Nakayashiki, T., Kurtzman, C. P., Edskes, H. K., and Wickner, R. B. (2005). Yeast prions [URE3] and [PSI+] are diseases. Proc. Natl. Acad. Sci. U.S.A. 102, 10575–10580. doi: 10.1073/pnas.0504882102
Narasumani, M., and Harrison, P. M. (2015). Bioinformatical parsing of folding-on-binding proteins reveals their compositional and evolutionary sequence design. Sci. Rep. 5:18586. doi: 10.1038/srep18586
Narasumani, M., and Harrison, P. M. (2018). Discerning evolutionary trends in post-translational modification and the effect of intrinsic disorder: analysis of methylation, acetylation and ubiquitination sites in human proteins. PLoS Comput. Biol. 14:e1006349. doi: 10.1371/journal.pcbi.1006349
Pallarès, I., de Groot, N. S., Iglesias, V., Sant'Anna, R., Biosca, A., Fernàndez-Busquets, X., et al. (2018). Discovering putative prion-like proteins in Plasmodium falciparum: a computational and experimental analysis. Front. Microbiol. 9:1737. doi: 10.3389/fmicb.2018.01737
Pallarès, I., and Ventura, S. (2017). The transcription terminator Rho: a first bacterial prion. Trends Microbiol. 25, 434–437. doi: 10.1016/j.tim.2017.03.008
Pei, J., and Grishin, N. V. (2001). AL2CO: calculation of positional conservation in a protein sequence alignment. Bioinformatics 17, 700–712. doi: 10.1093/bioinformatics/17.8.700
Pokrishevsky, E., Grad, L. I., and Cashman, N. R. (2016). TDP-43 or FUS-induced misfolded human wild-type SOD1 can propagate intercellularly in a prion-like fashion. Sci. Rep. 6:22155. doi: 10.1038/srep22155
Ross, E. D., Maclea, K. S., Anderson, C., and Ben-Hur, A. (2013). A bioinformatics method for identifying Q/N-rich prion-like domains in proteins. Methods Mol. Biol. 1017, 219–228. doi: 10.1007/978-1-62703-438-8_16
Saupe, S. J. (2011). The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Semin. Cell Dev. Biol. 22, 460–468. doi: 10.1016/j.semcdb.2011.02.019
Shahnawaz, M., Park, K. W., Mukherjee, A., Diaz-Espinoza, R., and Soto, C. (2017). Prion-like characteristics of the bacterial protein Microcin E492. Sci. Rep. 7:45720. doi: 10.1038/srep45720
Shattuck, J. E., Waechter, A. C., and Ross, E. D. (2017). The effects of glutamine/asparagine content on aggregation and heterologous prion induction by yeast prion-like domains. Prion 11, 249–264. doi: 10.1080/19336896.2017.1344806
Shorter, J., and Lindquist, S. (2005). Prions as adaptive conduits of memory and inheritance. Nat. Rev. Genet. 6, 435–450. doi: 10.1038/nrg1616
Si, K., Choi, Y. B., White-Grindley, E., Majumdar, A., and Kandel, E. R. (2010). Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell 140, 421–435. doi: 10.1016/j.cell.2010.01.008
Si, K., Lindquist, S., and Kandel, E. R. (2003). A neuronal isoform of the aplysia CPEB has prion-like properties. Cell 115, 879–891. doi: 10.1016/S0092-8674(03)01020-1
Sideri, T., Yashiroda, Y., Ellis, D. A., Rodríguez-López, M., Yoshida, M., Tuite, M. F., et al. (2017). The copper transport-associated protein Ctr4 can form prion-like epigenetic determinants in Schizosaccharomyces pombe. Microb. Cell 4, 16–28. doi: 10.15698/mic2017.01.552
Sun, Z., Diaz, Z., Fang, X., Hart, M. P., Chesi, A., Shorter, J., et al. (2011). Molecular determinants and genetic modifiers of aggregation andtoxicity for the ALS disease protein FUS/TLS. PLoS Biol. 9:e1000614. doi: 10.1371/journal.pbio.1000614
Suzuki, G., Shimazu, N., and Tanaka, M. (2012). A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress. Science 336, 355–359. doi: 10.1126/science.1219491
Tetz, G., and Tetz, V. (2017). Prion-like domains in phagobiota. Front. Microbiol. 8:2239. doi: 10.3389/fmicb.2017.02239
Tetz, G., and Tetz, V. (2018). Prion-like domains in eukaryotic viruses. Sci. Rep. 8:8931. doi: 10.1038/s41598-018-27256-w
Toombs, J. A., McCarty, B. R., and Ross, E. D. (2010). Compositional determinants of prion formation in yeast. Mol. Cell. Biol. 30, 319–332. doi: 10.1128/MCB.01140-09
Toombs, J. A., Petri, M., Paul, K. R., Kan, G. Y., Ben-Hur, A., and Ross, E. D. (2012). De novo design of synthetic prion domains. Proc. Natl. Acad. Sci. U.S.A. 109, 6519–6524. doi: 10.1073/pnas.1119366109
True, H., Berlin, I., and Lindquist, S. (2004). Epigenetic regulation of translation reveals hidden genetic variation to produce comlex traits. Nature 431, 184–187. doi: 10.1038/nature02885
True, H., and Lindquist, S. (2000). A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature 407, 477–483. doi: 10.1038/35035005
Westaway, D., Daude, N., Wohlgemuth, S., and Harrison, P. (2011). The PrP-like proteins Shadoo and Doppel. Top. Curr. Chem. 305, 225–256. doi: 10.1007/128_2011_190
Wickner, R. (1994). [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science 264, 528–530.
Wickner, R., Edskes, H., Roberts, B., Baxa, U., Pierce, M., Ross, E., et al. (2004). Prions: proteins as genes and infectious entities. Genes Dev. 18, 470–485. doi: 10.1101/gad.1177104
Yuan, A. H., Garrity, S. J., Nako, E., and Hochschild, A. (2014). Prion propagation can occur in a prokaryote and requires the ClpB chaperone. Elife 3:e02949. doi: 10.7554/eLife.02949
Yuan, A. H., and Hochschild, A. (2017). A bacterial global regulator forms a prion. Science 355, 198–201. doi: 10.1126/science.aai7776
Keywords: prion, evolution, sequence analysis, fungi, disease, glutamine, asparagine
Citation: Su T-Y and Harrison PM (2019) Conservation of Prion-Like Composition and Sequence in Prion-Formers and Prion-Like Proteins of Saccharomyces cerevisiae. Front. Mol. Biosci. 6:54. doi: 10.3389/fmolb.2019.00054
Received: 21 May 2019; Accepted: 26 June 2019;
Published: 11 July 2019.
Edited by:
Walid A. Houry, University of Toronto, CanadaReviewed by:
Eric D. Ross, Colorado State University, United StatesLeonid Breydo, St. Jude Children's Research Hospital, United States
Copyright © 2019 Su and Harrison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paul M. Harrison, cGF1bC5oYXJyaXNvbkBtY2dpbGwuY2E=