 Weria Pezeshkian
Weria Pezeshkian Melanie König
Melanie König Siewert J. Marrink
Siewert J. Marrink John H. Ipsen
John H. Ipsen- 1Groningen Biomolecular Sciences and Biotechnology Institute and Zernike Institute for Advanced Materials, University of Groningen, Groningen, Netherlands
- 2Department of Physics, Chemistry and Pharmacy, Center for Biomembrane Physics (MEMPHYS), University of Southern Denmark, Odense, Denmark
We present a multi-scale simulation procedure to describe membrane-related biological processes that span over a wide range of length scales. At macroscopic length-scale, a membrane is described as a flexible thin film modeled by a dynamic triangulated surface with its spatial conformations governed by an elastic energy containing only a few model parameters. An implicit protein model allows us to include complex effects of membrane-protein interactions in the macroscopic description. The gist of this multi-scale approach is a scheme to calibrate the implicit protein model using finer scale simulation techniques e.g., all atom and coarse grain molecular dynamics. We previously used this approach and properly described the formation of membrane tubular invaginations upon binding of B-subunit of Shiga toxin. Here, we provide a perspective of our multi-scale approach, summarizing its main features and sketching possible routes for future development.
Introduction
Many biological processes involve large scale changes in lateral chemical organization and geometrical shapes of biological membranes (McMahon and Boucrot, 2015; Chavent et al., 2018). The modeling of these processes, by computer simulation, is a challenging task since they typically involve a wide range of length and time scales that cannot be captured in full by any single current simulation technique (Enkavi et al., 2019; Marrink et al., 2019). At large length scales, computational, and analytical techniques based on continuum models have played a great role in our understanding of these processes and has revealed many important generic phenomena (Seifert et al., 1991; Bozic et al., 1992; Ramakrishnan et al., 2013, 2015). Nevertheless, these predictions are often obscured by the simplicity of the model and by the approximations needed to make them mathematically tractable. In addition, such phenomenological models contain few model parameters that are typically hard to relate to their molecular origin. At small length scales, particle-based computer simulations techniques e.g., molecular dynamics (MD) and dissipative particle dynamics (DPD), are robust techniques to elucidate complex membrane behaviors but with a limited capacity to predict large length scale cooperative phenomena (Gao et al., 2007; Li et al., 2016; Enkavi et al., 2019; Marrink et al., 2019). To overcome these limitations, we have used a multi-scale simulation procedure that bridges the gap between the particle and continuum based models and allows the simulation of large biological membrane patches while retaining details from the atomistic length scale (Pezeshkian et al., 2016). Here, we summarize the main features of the method, extend its capacity to describe a wider range of processes and sketch possible routes for further development.
Methods
In our multi-scale approach, the large-scale physical properties of a membrane are described by a coarse-grained model which captures the elastic energy of membrane conformations and the energetics of the lateral organization of its chemical constituents. Such a model only contains a few model parameters which are calibrated using atomistic and mesoscopic simulations (Marrink et al., 2007).
Simulation of Continuum Model
A continuous membrane is discretized by a dynamical triangulated surface (DTS) containing Nυ vertices, NT triangles, NL links which together form an irregular planer triangulated network (Figure 1A). The difference between dynamical and static triangulation is that the mutual link between two neighboring triangles can flip (Alexander moves). This allows to sample through all possible triangulations for a given Nυ, NT, NL. Link flipping and positional updates of the vertices gives the fluid character with full translational invariance in the plane of the surface (Figure 1B). In this representation, a vertex can be visualized as a segment of a bilayer containing hundreds of lipids, this means that the resolution of the model is limited to the length-scales above few nanometers. To ensure self-avoidance of the surface each vertex is equipped with a spherical bead. Using a set of discretized geometrical operations, each vertex is furthermore assigned with a normal vector , surface area Aυ (one third of the area of its neighboring triangles), principal curvatures (c1υ, c2υ) and principal directions (X1(υ), X2(υ)) (Ramakrishnan et al., 2010) (Figure 1A). This suffices to construct an elastic energy function associated with membrane bending that allows us to obtain the surface equilibrium configurations using numerical update algorithms. In this work, we have employed the Metropolis Monte Carlo algorithm (Ramakrishnan et al., 2010; Bahrami et al., 2012; van der Wel et al., 2016), but many other updating schemes are possible (Noguchi and Takasu, 2001; Cooke et al., 2005; Noguchi and Gompper, 2006; Peng et al., 2013; Mauer et al., 2018).
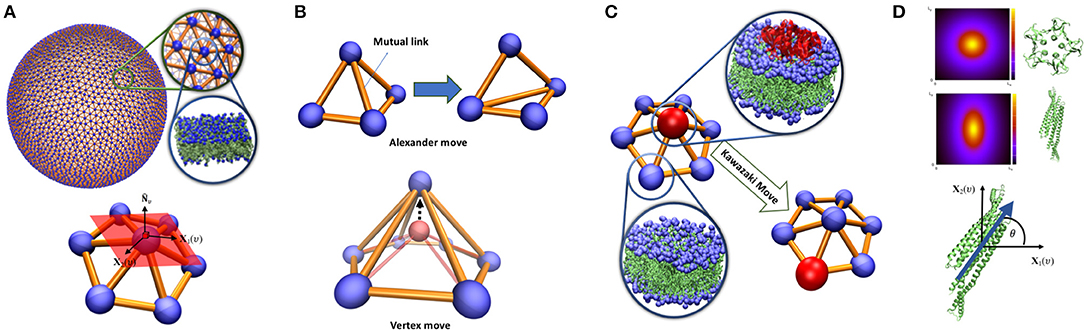
Figure 1. (A) Triangulated surface representation of a vesicle. Each vertex represents a segment of a bilayer containing hundreds of lipids. On each vertex, surface normal (), principal directions X1(υ), X2(υ) and their associated principal curvature (c1υ, c2υ) can be determined. (B) Top: Alexander move, mutual link between neighboring triangles is flipped and two new triangles are generated. Bottom: vertex move, a chosen vertex (red bead) is moved in a random direction (C) Proteins are modeled as an inclusion that can lay on a vertex and can jump to the neighboring vertex via Kawazaki moves. (D) Vertex based model of curvature map induced by proteins with lateral symmetry higher than 2 (top) and π-symmetric proteins (middle). The inclusion associated with these proteins should have a character of a two-dimensional vector in the plane of a vertex (bottom). The angle between the protein direction and the membrane main principal direction.
Elastic Energy
The Helfrich Hamiltonian (Helfrich, 1973) is the classic approach to describe membrane shape phenomena. The membrane elastic energy Eb can be expressed in the terms of two surface invariants, the mean curvature H = 0.5(c1 + c2), and Gaussian curvature, K = c1c2. A discretized form of the Helfrich Hamiltonian can be written as:
The second term of this equation only depends on the surface topology and does not change by continuous membrane deformation (Gauss-Bonnet theorem). The mean curvature elastic constant κ is called the bending elasticity, which carries the dimension of energy. The constant is called the spontaneous curvature, which represents a possible asymmetry between the two monolayers, e.g., differing solvent conditions. for a symmetric membrane. Equation (1) can be expanded in numerous ways depending on the membrane process at play. For example, for processes where a significant part of the total membrane surface undergoes deformations much faster than the flip flop rate of any monolayers chemical component, a monolayer area difference elastic term must be included (Seifert et al., 1991; Bozic et al., 1992). The difference in the area of the monolayers can be obtained as
Where h is the membrane thickness. Up to second order, the area-difference elastic energy is expressed as , with kr denoting the area compression modulus (Svetina and Žekš, 2014). Another relevant energy term that can be included is the elastic energy associated with change in the volume (V) of a closed surface (vesicle), where both the volume compression modulus KV and the equilibrium volume V0 are set by the osmotic conditions of the solvents in an experiment. For a triangulated surface, the volume can be easily obtained as
Here, is the position of any point on the triangle T, and AT are the normal vector and area of the oriented triangle T, respectively. For analysis of bounded membrane patches or semi-flat membranes in a periodic boundary box, a contribution τAp to the energy in Equation (1) becomes important. Ap and τ are the projected area and frame tension of the membrane, respectively.
When we are dealing with membranes with highly curved regions, e.g., formation of narrow necks prior to scission during a fission process, Equation (1) requires modification. In these regions, the curvatures of different monolayers can be significantly different. A practical approach to include this mismatch is to treat the bending energy associated with each monolayer separately. Using mid-plane principal curvatures, the mean curvature of each of the monolayers can be determined as Safran (1994):
Implicit Protein Model
Membrane proteins can locally influence bilayer shape through direct and indirect couplings. Direct impacts include local rigidification (Zhang et al., 2015), local membrane curvature imprint (Pezeshkian et al., 2017b; Corradi et al., 2018; Wang et al., 2018), local change in membrane thickness (Corradi et al., 2018) etc. Indirect effects arise from their interactions with other proteins that have the capacity to affect the membrane shape through cooperative phenomena. In our multi-scale simulation approach, these couplings are identified and quantified through atomistic and mesoscopic simulations and they are included in the system energy as new terms added to Equation (1). In the modeling, a protein or nanoparticle (an inclusion) is assigned to a vertex in the triangulation. Each vertex can at most occupy one inclusion, which naturally handles the in-plane excluded volume effect between inclusions. It also introduces a natural length scale to the model since we can associate the smallest possible area of a vertex with the projected area of the inclusion in question. Inclusions can move laterally through updates of the triangulation or by jumps between the neighboring vertices via Kawazaki moves (Figure 1C).
When an inclusion is situated in a vertex, it may change the elastic energy contribution from the vertex. For membrane proteins, the simplest model is to locally increase membrane bending rigidity (Frolov and Zimmerberg, 2008; Schweitzer et al., 2015). The most important effect of membrane proteins, that greatly influences the large-scale membrane shape, is to induce a local membrane curvature (Kozlov et al., 2014). This induced curvature can be in-plane rotationally symmetric or asymmetric. As a consequence of Eulers curvature formula, vertex-based inclusions, except π-symmetric inclusions (symmetric upon rotation by 180 degrees in the plane of the membrane, Figure 1D), can only induce symmetric curvature (Peliti and Prost, 1989). It may seem that this is a shortcoming of the model. Nevertheless, highly asymmetric curvature imprints decays quickly in the membrane plane (Dasgupta et al., 2017; Corradi et al., 2018) and does not appear in a macroscopic membrane model. The impact of these inclusions can be modeled by adding a local energy contribution eυ = −κHC0Aυ to the bending energy per vertex, where C0 is the local curvature imprint of the protein and needs calibration from finer scale simulations. Notice that C0 can only be identified with in Equation (1) for a fully covered membrane. π-symmetric inclusions can locally bend the membrane differently in different directions (Frolov and Zimmerberg, 2008). Such inclusions can thus be given an orientation in the plane in the direction with maximal directional curvature imprint while the perpendicular direction in the plane gives the lowest directional curvature imprint (Figure 1D). The membrane curvature in these directions can easily be obtained by Eulers curvature formula and where θ is the angle between the orientation of the inclusion and the direction of the main principal curvature of the membrane. Such inclusion will give rise to an additional local contribution to the total elastic energy in Equation (1), , where κ1 and κ2 are the directional bending rigidities imposed by the inclusion on the membrane. To complete the modeling, we need to include interactions between the inclusions. Here, we will only focus on the pair interactions but nevertheless the method can be extended to multi-body interactions. The pair-interactions between inclusions can be divided into two types: (i) as a function of distance between the proteins in the 3-dimensional space, e.g., electrostatic and van der Waals forces, (ii) as a function of a distance alongside the geodesic direction between two inclusions in the membrane, e.g., membrane mediated interactions (Haselwandter and Wingreen, 2014; Johannes et al., 2018). The former type of interactions can be modeled simply by a constant interaction energy when two inclusions are in proximity in the 3D space. This is a practical and valid choice, since the resolution of the model is well-below a range to capture the protein specific interactions. The second type of interactions is more challenging since it depends on the local curvature of the membrane. A particular consequence of this is that interactions between two neighboring non-isotropic inclusions can first be calculated after parallel transport between them, where the in-plane orientations of the inclusion is kept fixed along their mutual geodesic curve (Ramakrishnan et al., 2010). The interaction between two inclusions on the neighboring vertices is only a function of angle between their in-plane orientations alongside geodesic direction: is the orientation of inclusion residing on vertex i, represents the orientation of inclusion residing on vertex j after parallel transport to vertex i. This energy function can be written in term of Fourier series as
The first term (−ε0) models the isotropic part of the interaction between two inclusions while the second term is to model anisotropic interactions e.g., caused by steric factors and the distribution of the peptide groups in a protein (Domanski et al., 2017). M is a constant integer and its value depends on the chosen degree of coarse graining. Larger M allows to include more structural details of the protein shape in the interactions with other proteins. Ξk are the phase shift and μ0ak/M are amplitude of the Fourier modes and both need fitting from finer simulation techniques. By setting , μ0 can be defined as the lowest energy level of the anisotropic part of the interaction. Qij is the least common multiple of the degree of the i,j proteins symmetry in the plane of the membrane (N). Note that the interaction energy in Equation (5) can also be used to model lipid domain formations in multicomponent membranes (Ramakrishnan et al., 2010; Hansen et al., 2017).
Different approaches can be used to model proteins on triangulated surfaces e.g., introducing a curvature field and additional length scale to the model (Tourdot et al., 2014), however we prefer our procedure since it allows the calibration of all parameters solely through a bottom up approach. This increases the predication power of the model without need to tune the inputs parameters to reach the excepted outcome.
Calibration
To start a DTS simulation for a membrane containing different lipids and proteins, all the mentioned model parameters need to be calibrated using results from experiments or simulations of finer scales. Below we discuss several of these parameters .
Bending rigidity κ: Bending rigidity is known for many one component lipid bilayers from both experiment and simulations. However, for new lipid bilayers, fluctuation spectrum analysis is a powerful technique to extract this parameter. Both, coarse grained and all-atom MD simulation can be used to calibrate this parameter (Brandt et al., 2011; Watson et al., 2012; Venable et al., 2015).
Local curvature imprint : All-atom MD simulation has proven successful for calibration of these model parameters (Pezeshkian et al., 2016, 2017b; Kociurzynski et al., 2019). From an MD simulation trajectory, membrane curvature can be measured using different approaches. An accurate method is to use the first moment of the lateral membrane pressure profile, κC0 = ∫zΠ(z)dz (Safran, 1994). However, this approach has several problems. First, a converged lateral pressure profile requires very long simulations even for pure membrane systems. Secondly, it only provides the mean value of the induced curvature () unless the protein orientation is restricted (Bruhn et al., 2016; Ali Doosti et al., 2017). The second method is a geometrical approach and consists of fitting the upper and lower monolayer of the membrane to an analytical function and calculating the time-average curvature map on the surface of the bilayer. Note, since the typical radius of the curvature induced by proteins is much larger than a feasible MD simulation box size, the total average curvature of the fitted surface is zero. Therefore, one should only average the curvature of the surface up to a distance, in which the presence of the protein changes the lipid density, from the center of the proteins (Pezeshkian et al., 2016, 2017b; Corradi et al., 2018).
Protein-protein interaction parameters (ε0, μ0, N, ak, Ξk): An efficient approach to calibrate these parameters is to use coarse grained MD or DPD simulations. Typically, large simulation boxes are needed because the system size should be large enough so that the proteins do not interact (including membrane-mediated interactions) with their periodic image. Secondly a long simulation is required to disentangle the diffusive approach from the systematic interaction. In addition, mesoscale simulations allow us to derive a potential of mean force (PMF) profile that can be used to calibrate (ε0, μ0) (de Meyer et al., 2008; Periole et al., 2012; Domanski et al., 2017). In-plane symmetry of the protein structure (N) can be found from the crystal structure. ak and Ξk can be calibrated from both the density map or from free energy profile as a function of angle between the proteins.
Example: Shiga Toxin Induced Tubular Membrane Invaginations
The bacterial Shiga toxin is a member of the AB5 protein family that is composed of an enzymatically active A-subunit, and a receptor-binding B-subunit. STxB is homopentameric and mediates intracellular toxin trafficking via binding to the glycolipid globotriaosylceramide (Gb3) at the plasma membrane of target cells. Shiga toxin can enter the cell by both clathrin-dependent and independent endocytosis. The formation of tubular membrane invaginations is the first step in the clathrin-independent STxB uptake (Römer et al., 2007). Previously, we have used this multi-scale simulation approach to describe formation of membrane tubular invaginations upon STxB binding. Here we shortly discuss the scheme and results.
• Using the STxB crystal structure, we measured the projected area of STxB to be 38.5 nm2 (we approximated the lateral shape as a circle). The smallest area of a vertex is equal to (l is the minimum length of a link, or a vertex size). Therefore, l ≈ 6.7 nm.
• Local curvature induced by STxB was measured using all atom MD simulations (Pezeshkian et al., 2016). The radius of curvature is found to be R ≈ 29.4 nm = 4.39 l.
• DPD simulation was used to find STxB-STxB interactions. Using this method and experiments on unilamellar vesicles, we provide evidence that thermal Casimir-like force arising from membrane surface fluctuations are responsible for STxB clustering (Pezeshkian et al., 2017a). The computed PMF profile shows that the potential depth, taken to represent the isotropic strength of the pair interaction, is around 2.5 kBT (Figure 2A). STxB is a pentamer (2π/5-symmetric), therefore N = 5. Based on these numbers, we defined the simplest form of the interaction energy as in units of kBT.
• Using the above input parameters, we performed a Monte Carlo simulation of DTS in the constant frame tension ensemble (τ = 0) and could reproduce the behavior as seen in the experimental setups, namely formation of a tubular membrane invagination (Römer et al., 2007). We also found the minimum requirements for the formation of tubular membrane invaginations, i.e., (1) capacity of the individual proteins to induce local membrane curvature (2) their ability to cluster, by any mean, upon binding to the membrane (Figure 2A) (Pezeshkian et al., 2016).
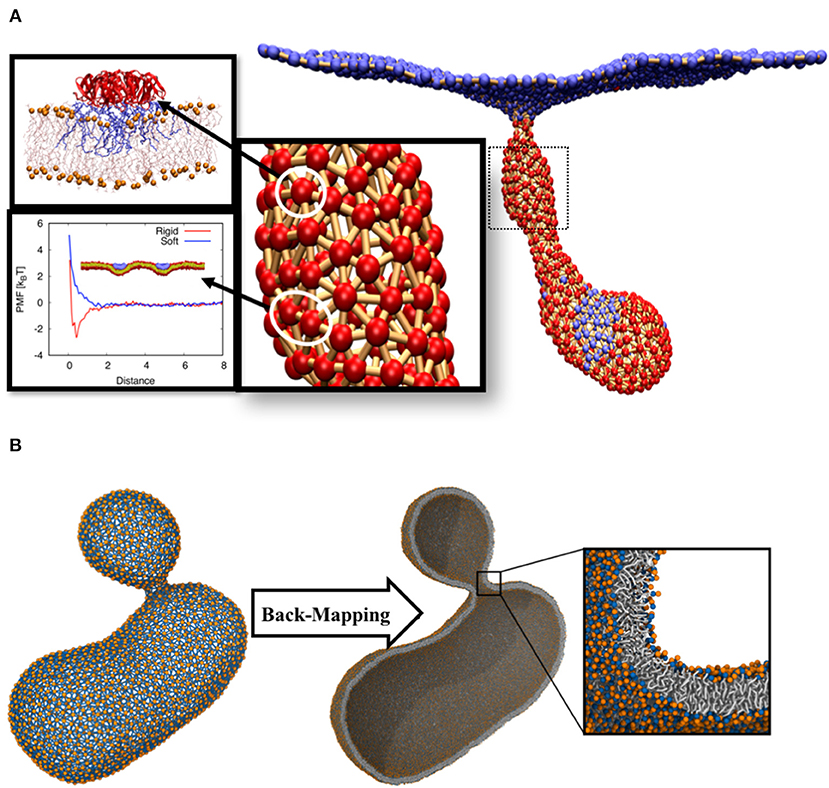
Figure 2. (A) Tubular membrane invaginations induced by Shiga toxin. The color code for the beads is the same as in Figure 1B. All atom molecular dynamic simulations were used to obtain local curvature induced by the protein (Pezeshkian et al., 2016). Protein-protein interaction was calibrated using DPD simulations. (B) The final structure of a budding vesicle after a DTS simulation is back-mapped to the particle based CG Martini model.
Back-Mapping to CG Model
The main assumption of this multi-scale simulation approach is that local properties of the membrane do not strongly get affected by large-scale membrane configurational changes. However, local lateral organizations of complex membranes chemical constituents can change upon large scale membrane deformations (Baoukina et al., 2018). In order to overcome this limitation, we have developed an algorithm that back-maps a DTS structure to its corresponding Martini CG model (Marrink et al., 2007; Marrink and Tieleman, 2013). This algorithm makes it possible to use DTS to equilibrate the slow large-scale membrane conformational change and exploit the Martini model to equilibrate the local lipid distributions. As a first attempt to explore this procedure, we performed a DTS simulation on a vesicle with a smaller volume/surface ratio of a perfect sphere (0.7) and a spontaneous curvature of 0.025 nm−1. Under this condition, the DTS simulation predicted the formation of a vesicular bud (Figure 2B) (Seifert et al., 1991; Markvoort et al., 2009; Bahrami et al., 2017). We then back-mapped the DTS structure to its corresponding Martini model and after a short energy minimization, it was ready for an MD simulation. The detail of this procedure is out of the scope of this article and will be published elsewhere.
Summary and Perspectives
We described an extended version of our multi-scale simulation procedure that uses a bottom up scheme to calibrate DTS model parameters (Pezeshkian et al., 2016). The approach is well-suited for investigating membrane involved biological processes that take place at a large-range of time and length scales that cannot be captured by any single current simulation techniques.
One of the clear advantages of exploiting DTS at macroscopic length scales is the speed. DTS allows us to simulate micron size vesicles, decorated with membrane proteins, on a single CPU core. This length-scale is hardly reachable (using much more computational power) by any particle-based computer simulation techniques (Cooke et al., 2005; Ayton and Voth, 2009). Nevertheless, the approach still suffers from several limitations that need to be resolved. For example, DTS simulations with dynamic topology has been only developed for several special purposes (Jeppesen and Ipsen, 1993; Shillcock and Boal, 1996; Gompper and Kroll, 1998; Shillcock and Seifert, 1998) that limits its applications, as a generic method, to describe processes that involve membrane topological changes, e.g., membrane scission and poration (Boye et al., 2017). Another limitation is the current implicit protein model that is only applicable for membrane proteins. One possibility is to adopt one protein to few beads strategy e.g., essential dynamics coarse-graining (Zhang et al., 2008) to extend the range of the DTS protein mapping. Another route to increase the molecular level detail is through dynamic coupling of macroscale and CG models. We shortly described a back-mapping algorithm that converts a DTS topology to a Martini structure. This algorithm opens up a new perspective to perform a dual resolution Martini/DTS simulation, so that DTS performs the large-scale moves while local moves of the chemical components is handled by the CG Martini model.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was funded by The Netherlands Organization for Scientific Research (NWO) within the framework of BaSyC—Building a Synthetic Cell Gravitation project.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank T. A. Wassenaar for constructive discussions and comments.
References
Ali Doosti, B., Pezeshkian, W., Bruhn, D. S., Ipsen, J. H., Khandelia, H., Jeffries, G. D. M., et al. (2017). Membrane tubulation in lipid vesicles triggered by the local application of calcium ions. Langmuir 33, 11010–11017. doi: 10.1021/acs.langmuir.7b01461
Ayton, G. S., and Voth, G. A. (2009). Hybrid coarse-graining approach for lipid bilayers at large length and time scales. J. Phys. Chem. B 113, 4413–4424. doi: 10.1021/jp8087868
Bahrami, A. H., Lin, M. G., Ren, X., Hurley, J. H., and Hummer, G. (2017). Scaffolding the cup-shaped double membrane in autophagy. PLoS Comput. Biol. 13:e1005817. doi: 10.1371/journal.pcbi.1005817
Bahrami, A. H., Lipowsky, R., and Weikl, T. R. (2012). Tubulation and aggregation of spherical nanoparticles adsorbed on vesicles. Phys. Rev. Lett. 109:188102. doi: 10.1103/PhysRevLett.109.188102
Baoukina, S., Ingólfsson, H. I., Marrink, S. J., and Tieleman, D. P. (2018). Curvature-induced sorting of lipids in plasma membrane tethers. Adv. Theory Simul. 1:1800034. doi: 10.1002/adts.201800034
Boye, T. L., Maeda, K., Pezeshkian, W., Sønder, S. L., Haeger, S. C., Gerke, V., et al. (2017). Annexin A4 and A6 induce membrane curvature and constriction during cell membrane repair. Nat. Commun. 8:1623. doi: 10.1038/s41467-017-01743-6
Bozic, B., Svetina, S., Zeks, B., and Waugh, R. E. (1992). Role of lamellar membrane structure in tether formation from bilayer vesicles. Biophys. J. 61, 963–973. doi: 10.1016/S0006-3495(92)81903-3
Brandt, E. G., Braun, A. R., Sachs, J. N., Nagle, J. F., and Edholm, O. (2011). Interpretation of fluctuation spectra in lipid bilayer simulations. Biophys. J. 100, 2104–2111. doi: 10.1016/j.bpj.2011.03.010
Bruhn, D. S., Lomholt, M. A., and Khandelia, H. (2016). Quantifying the relationship between curvature and electric potential in lipid bilayers. J. Phys. Chem. B 120, 4812–4817. doi: 10.1021/acs.jpcb.6b03439
Chavent, M., Duncan, A. L., Rassam, P., Birkholz, O., Hélie, J., Reddy, T., et al. (2018). How nanoscale protein interactions determine the mesoscale dynamic organisation of bacterial outer membrane proteins. Nat. Commun. 9:2846. doi: 10.1038/s41467-018-05255-9
Cooke, I. R., Kremer, K., and Deserno, M. (2005). Tunable generic model for fluid bilayer membranes. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 72:011506. doi: 10.1103/PhysRevE.72.011506
Corradi, V., Mendez-Villuendas, E., Ingólfsson, H. I., Gu, R. X., Siuda, I., Melo, M. N., et al. (2018). Lipid-protein interactions are unique fingerprints for membrane proteins. ACS Cent Sci. 4, 709–717. doi: 10.1021/acscentsci.8b00143
Dasgupta, S., Auth, T., and Gompper, G. (2017). Nano- and microparticles at fluid and biological interfaces. J. Phys. Condens. Matter 29:373003. doi: 10.1088/1361-648X/aa7933
de Meyer, F. J., Venturoli, M., and Smit, B. (2008). Molecular simulations of lipid-mediated protein-protein interactions. Biophys. J. 95, 1851–1865. doi: 10.1529/biophysj.107.124164
Domanski, J., Hedger, G., Best, R. B., Stansfeld, P. J., and Sansom, M. S. P. (2017). Convergence and sampling in determining free energy landscapes for membrane protein association. J. Phys. Chem. B 121, 3364–3375. doi: 10.1021/acs.jpcb.6b08445
Enkavi, G., Javanainen, M., Kulig, W., Róg, T., and Vattulainen, I. (2019). Multiscale simulations of biological membranes: the challenge to understand biological phenomena in a living substance. Chem. Rev. 119, 5607–5774. doi: 10.1021/acs.chemrev.8b00538
Frolov, V. A., and Zimmerberg, J. (2008). Flexible scaffolding made of rigid BARs. Cell 132, 727–729. doi: 10.1016/j.cell.2008.02.025
Gao, L., Shillcock, J., and Lipowsky, R. (2007). Improved dissipative particle dynamics simulations of lipid bilayers. J. Chem. Phys. 126:015101. doi: 10.1063/1.2424698
Gompper, G., and Kroll, D. M. (1998). Membranes with fluctuating topology: monte carlo simulations. Phys. Rev. Lett. 81, 2284–2287. doi: 10.1103/PhysRevLett.81.2284
Hansen, A. G., Ramakrishnan, N., Sunil Kumar, P. B., and Ipsen, J. H. (2017). Numerical insights into the phase diagram of p-atic membranes with spherical topology. Eur. Phys. J. E 40:32. doi: 10.1140/epje/i2017-11515-7
Haselwandter, C. A., and Wingreen, N. S. (2014). The role of membrane-mediated interactions in the assembly and architecture of chemoreceptor lattices. PLoS Comput. Biol. 10:e1003932. doi: 10.1371/journal.pcbi.1003932
Helfrich, W. (1973). Elastic properties of lipid bilayers: theory and possible experiments. Z Naturforsch. C 28, 693–703. doi: 10.1515/znc-1973-11-1209
Jeppesen, C., and Ipsen, J. H. (1993). Scaling properties of self-avoiding surfaces with free topology. Europhys. Lett. 22, 713–716. doi: 10.1209/0295-5075/22/9/013
Johannes, L., Pezeshkian, W., Ipsen, J. H., and Shillcock, J. C. (2018). Clustering on membranes: fluctuations and more. Trends Cell Biol. 28, 405–415. doi: 10.1016/j.tcb.2018.01.009
Kociurzynski, R., Beck, S. D., Bouhon, J. B., Römer, W., and Knecht, V. (2019). Binding of SV40's viral capsid protein VP1 to its glycosphingolipid receptor gm1 induces negative membrane curvature: a molecular dynamics study. Langmuir 35, 3534–3544. doi: 10.1021/acs.langmuir.8b03765
Kozlov, M. M., Campelo, F., Liska, N., Chernomordik, L. V., Marrink, S. J., and McMahon, H. T. (2014). Mechanisms shaping cell membranes. Curr. Opin. Cell Biol. 29, 53–60. doi: 10.1016/j.ceb.2014.03.006
Li, X., Gao, L., and Fang, W. (2016). Dissipative particle dynamics simulations for phospholipid membranes based on a four-to-one coarse-grained mapping scheme. PLoS ONE 11:e0154568. doi: 10.1371/journal.pone.0154568
Markvoort, A. J., Spijker, P., Smeijers, A. F., Pieterse, K., van Santen, R. A., and Hilbers, P. A. (2009). Vesicle deformation by draining: geometrical and topological shape changes. J. Phys. Chem. B. 113, 8731–8737. doi: 10.1021/jp901277h
Marrink, S. J., Corradi, V., Souza, P. C. T., Ingólfsson, H. I., Tieleman, D. P., and Sansom, M. S. P. (2019). Computational modeling of realistic cell membranes. Chem. Rev. 119, 6184–6226. doi: 10.1021/acs.chemrev.8b00460
Marrink, S. J., Risselada, H. J., Yefimov, S., Tieleman, D. P., and de Vries, A. H. (2007). The MARTINI force field: coarse grained model for biomolecular simulations. J. Phys. Chem. B 111, 7812–7824. doi: 10.1021/jp071097f
Marrink, S. J., and Tieleman, D. P. (2013). Perspective on the Martini model. Chem. Soc. Rev. 42, 6801–6822. doi: 10.1039/c3cs60093a
Mauer, J., Mendez, S., Lanotte, L., Nicoud, F., Abkarian, M., Gompper, G., et al. (2018). Flow-induced transitions of red blood cell shapes under shear. Phys. Rev. Lett. 121:118103. doi: 10.1103/PhysRevLett.121.118103
McMahon, H. T., and Boucrot, E. (2015). Membrane curvature at a glance. J. Cell. Sci. 128, 1065–1070. doi: 10.1242/jcs.114454
Noguchi, H., and Gompper, G. (2006). Meshless membrane model based on the moving least-squares method. Phys. Rev. E Stat. Nonlin Soft Matter Phys. 73:021903. doi: 10.1103/PhysRevE.73.021903
Noguchi, H., and Takasu, M. (2001). Self-assembly of amphiphiles into vesicles: a Brownian dynamics simulation. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 64:041913. doi: 10.1103/PhysRevE.64.041913
Peliti, L., and Prost, J. (1989). Fluctuations in membranes with reduced symmetry. J. Phys. 50, 1557–1571. doi: 10.1051/jphys:0198900500120155700
Peng, Z., Li, X., Pivkin, I. V., Dao, M., Karniadakis, G. E., and Suresh, S. (2013). Lipid bilayer and cytoskeletal interactions in a red blood cell. Proc. Natl. Acad. Sci. U. S. A. 110, 13356–13361. doi: 10.1073/pnas.1311827110
Periole, X., Knepp, A. M., Sakmar, T. P., Marrink, S. J., and Huber, T. (2012). Structural determinants of the supramolecular organization of G protein-coupled receptors in bilayers. J. Am. Chem. Soc. 134, 10959–10965. doi: 10.1021/ja303286e
Pezeshkian, W., Gao, H., Arumugam, S., Becken, U., Bassereau, P., Florent, J. C., et al. (2017a). Mechanism of Shiga toxin clustering on membranes. ACS Nano 11, 314–324. doi: 10.1021/acsnano.6b05706
Pezeshkian, W., Hansen, A. G., Johannes, L., Khandelia, H., Shillcock, J. C., Kumar, P. B., et al. (2016). Membrane invagination induced by Shiga toxin B-subunit: from molecular structure to tube formation. Soft Matter 12, 5164–5171. doi: 10.1039/C6SM00464D
Pezeshkian, W., Nåbo, L. J., and Ipsen, J. H. (2017b). Cholera toxin B subunit induces local curvature on lipid bilayers. FEBS Open Bio. 7, 1638–1645. doi: 10.1002/2211-5463.12321
Ramakrishnan, N., Ipsen, J. H., Rao, M., and Kumar, P. B. (2015). Organelle morphogenesis by active membrane remodeling. Soft Matter. 11, 2387–2393. doi: 10.1039/C4SM02311K
Ramakrishnan, N., Sunil Kumar, P. B., and Ipsen, J. H. (2010). Monte Carlo simulations of fluid vesicles with in-plane orientational ordering. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 81:041922. doi: 10.1103/PhysRevE.81.041922
Ramakrishnan, N., Sunil Kumar, P. B., and Ipsen, J. H. (2013). Membrane-mediated aggregation of curvature-inducing nematogens and membrane tubulation. Biophys. J. 104, 1018–1028. doi: 10.1016/j.bpj.2012.12.045
Römer, W., Berland, L., Chambon, V., Gaus, K., Windschiegl, B., Tenza, D., et al. (2007). Shiga toxin induces tubular membrane invaginations for its uptake into cells. Nature 450, 670–675. doi: 10.1038/nature05996
Safran, S. A. (1994). Statistical Thermodynamics of Surfaces, Interfaces, and Membranes. Reading, MA: Addison-Wesley Pub.
Schweitzer, Y., Shemesh, T., and Kozlov, M. M. (2015). A model for shaping membrane sheets by protein scaffolds. Biophys. J. 109, 564–573. doi: 10.1016/j.bpj.2015.06.001
Seifert, U., Berndl, K., and Lipowsky, R. (1991). Shape transformations of vesicles: phase diagram for spontaneous- curvature and bilayer-coupling models. Phys. Rev. A 44, 1182–1202. doi: 10.1103/PhysRevA.44.1182
Shillcock, J. C., and Boal, D. H. (1996). Entropy-driven instability and rupture of fluid membranes. Biophys. J. 71, 317–326. doi: 10.1016/S0006-3495(96)79227-5
Shillcock, J. C., and Seifert, U. (1998). Thermally induced proliferation of pores in a model fluid membrane. Biophys. J. 74, 1754–1766. doi: 10.1016/S0006-3495(98)77886-5
Svetina, S., and Žekš, B. (2014). Nonlocal membrane bending: a reflection, the facts and its relevance. Adv. Colloid Interface Sci. 208, 189–196. doi: 10.1016/j.cis.2014.01.010
Tourdot, R. W., Bradley, R. P., Ramakrishnan, N., and Radhakrishnan, R. (2014). Multiscale computational models in physical systems biology of intracellular trafficking. IET Syst. Biol. 8, 198–213. doi: 10.1049/iet-syb.2013.0057
van der Wel, C., Vahid, A., Saric, A., Idema, T., Heinrich, D., and Kraft, D. J. (2016). Lipid membrane-mediated attraction between curvature inducing objects. Sci. Rep. 6:32825. doi: 10.1038/srep37382
Venable, R. M., Brown, F. L. H., and Pastor, R. W. (2015). Mechanical properties of lipid bilayers from molecular dynamics simulation. Chem. Phys. Lipids 192, 60–74. doi: 10.1016/j.chemphyslip.2015.07.014
Wang, Z., Jumper, J. M., Wang, S., Freed, K. F., and Sosnick, T. R. (2018). A membrane burial potential with H-bonds and applications to curved membranes and fast simulations. Biophys. J. 115, 1872–1884. doi: 10.1016/j.bpj.2018.10.012
Watson, M. C., Brandt, E. G., Welch, P. M., and Brown, F. L. (2012). Determining biomembrane bending rigidities from simulations of modest size. Phys. Rev. Lett. 109:028102. doi: 10.1103/PhysRevLett.109.028102
Zhang, Y., Huang, C., Kim, S., Golkaram, M., Dixon, M. W., Tilley, L., et al. (2015). Multiple stiffening effects of nanoscale knobs on human red blood cells infected with Plasmodium falciparum malaria parasite. Proc. Natl. Acad. Sci. U. S. A. 112, 6068–6073. doi: 10.1073/pnas.1505584112
Keywords: dynamic triangulated surfaces, Martini coarse-grain simulation, Shiga toxin, simulation of continuum model, membrane remodeling, implicit protein model
Citation: Pezeshkian W, König M, Marrink SJ and Ipsen JH (2019) A Multi-Scale Approach to Membrane Remodeling Processes. Front. Mol. Biosci. 6:59. doi: 10.3389/fmolb.2019.00059
Received: 13 May 2019; Accepted: 08 July 2019;
Published: 23 July 2019.
Edited by:
Valentina Tozzini, Nanosciences Institute, National Research Council, ItalyReviewed by:
Riccardo Nifosì, Italian National Research Council (CNR), ItalyNicola Maria Pugno, University of Trento, Italy
Jeffery Klauda, University of Maryland, College Park, United States
Copyright © 2019 Pezeshkian, König, Marrink and Ipsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: John H. Ipsen, aXBzZW5AbWVtcGh5cy5zZHUuZGs=