 Ranjeet Kumar
Ranjeet Kumar Pooja Singh
Pooja Singh Afsal Kolloli
Afsal Kolloli Lanbo Shi
Lanbo Shi Yuri Bushkin
Yuri Bushkin Sanjay Tyagi
Sanjay Tyagi Selvakumar Subbian
Selvakumar Subbian- Public Health Research Institute, New Jersey Medical School, Rutgers, The State University of New Jersey, Newark, NJ, United States
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb) remains as a leading killer among infectious diseases worldwide. The nature of the host immune response dictates whether the initial Mtb infection is cleared or progresses toward active disease, and is ultimately determined by intricate host-pathogen interactions that are yet to be fully understood. The early immune response to infection is mediated by innate immune cells, including macrophages and neutrophils that can phagocytose Mtb and mount an antimicrobial response. However, Mtb can exploit these innate immune cells for its survival and dissemination. Recently, it has become clear that the immune response and metabolic remodeling are interconnected, which is highlighted by the rapid evolution of the interdisciplinary field of immunometabolism. It has been proposed that the net outcome to Mtb infection—clearance or chronic disease—is likely a result of combined immunologic and metabolic activities of the immune cells. Indeed, host cells activated by Mtb infection have strikingly different metabolic requirements than naïve/non-infected cells. Macrophages activated by Mtb-derived molecules or upon phagocytosis acquire a phenotype similar to M1 with elevated production of pro-inflammatory molecules and rely on glycolysis and pentose phosphate pathway to meet their bioenergetic and metabolic requirements. In these macrophages, oxidative phosphorylation and fatty acid oxidation are dampened. However, the non-infected/naive, M2-type macrophages are anti-inflammatory and derive their energy from oxidative phosphorylation and fatty acid oxidation. Similar metabolic adaptations also occur in other phagocytes, including dendritic cells, neutrophils upon Mtb infection. This metabolic reprogramming of innate immune cells during Mtb infection can differentially regulate their effector functions, such as the production of cytokines and chemokines, and antimicrobial response, all of which can ultimately determine the outcome of Mtb-host interactions within the granulomas. In this review, we describe key immune cells bolstering host innate response and discuss the metabolic reprogramming in these phagocytes during Mtb infection. We focused on the major phagocytes, including macrophages, dendritic cells and neutrophils and the key regulators involved in metabolic reprogramming, such as hypoxia-inducible factor-1, mammalian target of rapamycin, the cellular myelocytomatosis, peroxisome proliferator-activator receptors, sirtuins, arginases, inducible nitric acid synthase and sphingolipids.
Introduction
Tuberculosis (TB) remains as significant cause of mortality and morbidity among infectious diseases around the world. In 2017, an estimated 10 million people developed active disease, while 1.6 million died from TB (WHO, 2018). About a third of the world's population is believed to be latently infected with Mycobacterium tuberculosis (Mtb) without obvious disease symptoms. Of these, 5–10% will develop active TB in their lifetime, if/when their host immunity wanes (WHO, 2018).
The host immune response during the various stages of Mtb infection is complex and not fully understood. Studies on human patients and experimental animal models of TB have shown that phagocytes such as macrophages and neutrophils are the primary immune effector cells against Mtb (Keane et al., 2001; North and Jung, 2004; Alcais et al., 2005). Engagement of surface receptors in these cells with mycobacterial cell wall molecules such as lipoarabinomannan and secreted proteins ultimately activates the immune cells to secrete a plethora of cytokines and chemokines, which aid in the recruitment of other leukocytes from circulation to the site of infection. A hallmark of successful initial Mtb infection is the formation of granuloma in the infected tissues, which is an organized cellular structure comprised of a variety of innate and adaptive immune cells (Ramakrishnan, 2012). Mature macrophages, characterized by an increased cytoplasm-to-nucleus ratio and larger number of organelles, are capable of developing into multinucleated giant cells as well as foam cells that accumulate lipid bodies. These cells are critical constituents of TB granulomas, which undergo structural changes over time (Russell et al., 2009; Guerrini et al., 2018). Initial development of granuloma is marked by its extensive vascularization through vascular endothelial growth factor (VEGF) mediated responses, leading to recruitment of macrophages, lymphocytes, and DCs, into the site of infection (Caceres et al., 2009). Further structural changes in granuloma are marked with the presence of different morphotypes of macrophages, including multinucleated giant cells, epithelioid cells, and foamy macrophages (Murphy, 2001; Ordway et al., 2005; D'Avila et al., 2006). The later cells are generated as a result of intracellular accumulation of lipid droplets consisting of neutral lipids, mainly cholesteryl esters (CE) and/or triglycerides (TAG) surrounded by a monolayer of phospholipids comprising structural proteins, cholesterol and enzymes (Martin and Parton, 2006; Saka and Valdivia, 2012; Guerrini et al., 2018). Accumulation of TAG, a significant constituent of foam cells within the granuloma, in Mtb-infected human primary macrophages requires tumor necrosis factor receptor (TNFR) signaling, activation of caspase cascade and mTORC1 (Russell et al., 2009). Rupture of foam cells due to exacerbated infection and/or inflammation and the release of their contents likely sustains the disease pathology and generation of caseum, which leads to progressive destruction of lung tissues (Russell et al., 2009). Foamy macrophages (FM) are also reported to provide Mtb their physiological niche similar to non-replicative vegetative state (Russell et al., 2009). In response to strong anti-mycobacterial response, the structure tends to become stratified, and a fibrous cuff is formed delineating central macrophages rich region from peripheral lymphocytic milieu (Russell et al., 2009). In majority of cases, granuloma maintains a dynamic state where Mtb is contained, and disease is resolved. However, in some cases there is increased accumulation of caseum in central region of granuloma, which leads to necrosis of cells and subsequent release of Mtb into the airways (Russell et al., 2009).
Other cell types that are present in the granulomas are neutrophils, dendritic cells (DCs), various subtypes of B and T lymphocytes, natural killer cells, fibroblasts, and epithelial cells (Ramakrishnan, 2012). Findings from rabbit and non-human primate models of Mtb infection, which are immunopathologically “relevant-to-human,” have shown that granulomas contain a macrophage-rich core with necrosis of infected cells surrounded by a lymphocytic cuff, consisting mainly of T and B cells (Capuano et al., 2003; Subbian et al., 2011, 2013; Mattila et al., 2013). However, findings in human pulmonary TB cases and relevant experimental animal models have revealed many different types of granulomas based on the presence and arrangement of various immune cell types. These divergent types include highly cellular, “solid” granulomas, caseating, necrotic and non-necrotic, fibrotic nodules, suppurative (comprising a surge of neutrophils), and closed and open cavitary lesions (Capuano et al., 2003; Subbian et al., 2011, 2013; Mattila et al., 2013). These granulomas vary in size, maturation state, and the quantity and quality of the resident immune cells. Partially mineralized and highly fibrotic (fibrocalcific) granulomas are primarily found in latent infection, while the number and cellular complexity of granulomas is of a higher order during active TB (Gideon and Flynn, 2011). However, different types of granulomas have been reported in the tissues of active TB and latent infection, making it difficult to assess the infection status of a host-based on the type of granulomas present in the organ (Mattila et al., 2013).
Although granulomas are thought to restrict bacterial growth and expansion by “walling off,” Mtb can exploit the immune cells within and outside of the granuloma for its persistence, proliferation, and dissemination (Davis and Ramakrishnan, 2009; Volkman et al., 2010). Based on the intricate interaction between the host immune response and bacterial determinants that are yet to be fully understood, the infecting Mtb is either contained and/or cleared or replicates and disseminates in the granuloma. The outcome of this interaction also results in a spectrum of clinical manifestations in the infected host, ranging from symptomatic active disease to sub-clinical, incipient, percolator, and intermittent disease to persistent, asymptomatic latent infection (Vynnycky and Fine, 2000).
The infection of host immune cells by Mtb causes several changes including metabolic reprogramming, which differentially regulates various cytokines and chemokines associated with clearance, containment, or progression of Mtb infection in host cells (Olakanmi et al., 2002; Gleeson et al., 2016; Qualls and Murray, 2016). In particular, a shift in glucose and lipid metabolism is critical for defining the fate of host cell function in the context of mycobacterial survival within the granuloma (Shi et al., 2016). Rapid advances in the field of immunology and metabolism have given rise to an interdisciplinary field termed immunometabolism (Mathis and Shoelson, 2011). Core metabolic processes such as glycolysis, Krebs cycle, fatty acid metabolism, and nitrogen metabolism are vital for the proper functioning of any cell (DeBerardinis and Thompson, 2012; Escoll and Buchrieser, 2018). In general, cells in a “resting” condition mostly rely on oxidative phosphorylation (OXPHOS) to generate ATP from NADH, and FADH made during the Krebs cycle. However, under oxygen-depleting or hypoxic conditions and during high-energy requirements, the cells switch to aerobic glycolysis to generate ATP (Escoll and Buchrieser, 2018). Although aerobic glycolysis produces ATP less efficiently than OXPHOS, it produces more ATP at a faster rate, which meets the high energy demands of the immune cells. Apart from energy needs, aerobic glycolysis also provides the precursors for chemical constituents, such as nucleotides, amino acids, and lipids (Lunt and Vander Heiden, 2011). Therefore, metabolic reprogramming fulfills both the bioenergetic and biosynthetic needs of cells for rapid proliferation, differentiation, and/or production of secretory components (DeBerardinis and Thompson, 2012; Escoll and Buchrieser, 2018).
Metabolic reprogramming in immune cells is controlled by host-derived transcriptional regulators such as hypoxia-inducible factor (HIF), mechanistic/mammalian target of rapamycin (mTOR), myelocytomatosis virus oncogene (Myc), and glycogen synthase kinase 3 (GSK3) (Wahl et al., 2012; O'Neill and Pearce, 2016; Jellusova and Rickert, 2017) (Table 1). Besides, immune cells can initiate metabolic reprogramming through signaling pathways elicited in response to host-pathogen interactions through receptors, such as pattern recognition receptors (PRR), cytokine receptors, and antigen receptors (Escoll and Buchrieser, 2018). In these cases, the onset of aerobic glycolysis is essential for immune cell activation and corresponding production of cytokines, chemokines, and antibacterial molecules. Therefore, the metabolic needs of an infected and/or activated immune cell are significantly different from a resting or non-infected cell. Taken together, recent advances focused on the immunometabolism during microbial infections have provided new insights into host immune regulation mechanisms, opening up new avenues for the development of host-directed therapies to enhance treatment outcomes (Eisenreich et al., 2019; Prusinkiewicz and Mymryk, 2019; Wang et al., 2019).

Table 1. Key metabolic regulators that modulate immune responses.
In this review, we describe the role of metabolic reprogramming in various phagocytic cells that bolster host innate immune response and their relevance to Mtb infection. Since a full report on the host immune response and the cells involved is beyond the scope of this article, we focus on major phagocytes and the key regulators that control metabolic reprogramming in these cells during Mtb infection.
Primary Phagocytes Involved in TB Pathogenesis
Macrophages
Macrophages shape the nature and course of host immunity against Mtb infection by playing a crucial early role, such as phagocytosis of Mtb and triggering an antimicrobial response (van Crevel et al., 2002; Gutierrez et al., 2004). Although macrophages elicit an early host protective immunity against Mtb, these innate cells are also thought to provide an intracellular niche where Mtb thrives (Schluger and Rom, 1998). Macrophages demonstrate a high level of heterogeneity in morphology and function (Austyn and Gordon, 1981; Kaplan and Gaudernack, 1982). Based on the heterogeneity observed mainly in in vitro cell culture systems, macrophages are categorized into four major groups: type I, type II, alternatively activated and deactivated macrophages (Figure 1) (Gordon, 2003; Gordon and Taylor, 2005). Type I macrophages are activated through lymphoid cell mediators such as IFN-γ and LPS, while type II is differentiated upon ligation of receptors by immune complexes (Guirado et al., 2013). Both of these macrophage types possess high microbicidal activity by producing pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6, and reactive oxygen species (ROS) and inducible nitric oxide synthase (iNOS), which leads to the production of nitric oxide (NO) (Bogdan, 2001). The third group, alternatively activated macrophages, is generated in response to Th-2 type cytokines such as IL-4 or IL-13 (Gordon, 2003). These macrophages secrete anti-inflammatory cytokines such as IL-10 and TGF-β and are mostly involved in tissue repair and humoral immunity (Gordon, 2003; Mosser, 2003). Finally, deactivated macrophages are produced in response to anti-inflammatory cytokines IL-10 or TGF-β, and are involved in the production of anti-inflammatory cytokines and prostaglandin E2, and have reduced MHC II expression (Guirado et al., 2013).
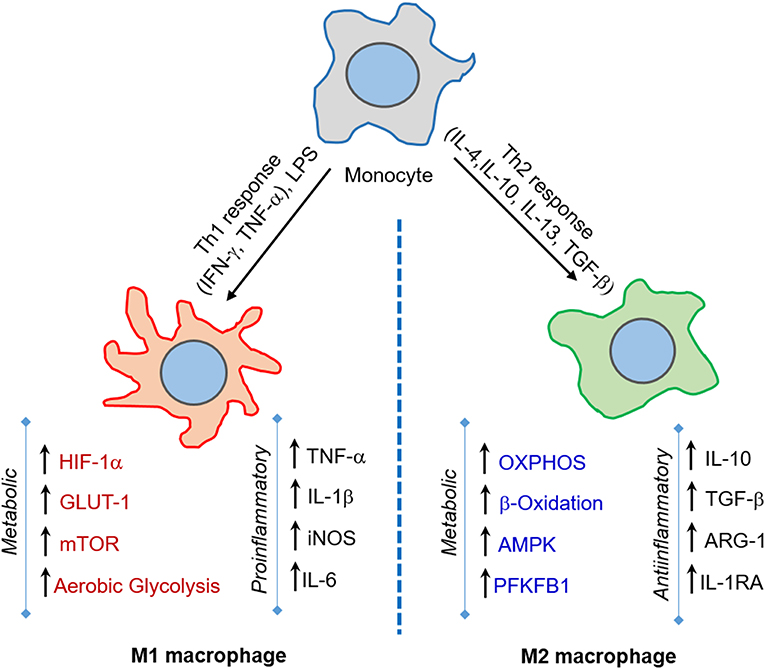
Figure 1. M1 and M2 macrophages orchestrate host immunity in response to a variety of stimuli. Macrophages are activated to M1-type upon challenge with pro-inflammatory IFN-γ/LPS or microbial infection, while M2-type macrophages are induced upon IL-4/IL-10/IL-13 or TGF-β exposure. The M1 state is marked by increase in aerobic glycolysis induced by HIF-1α, GLUT-1, and mTOR expression, and is involved in microbicidal activities. In contrast, the M2 state is marked by OXPHOS and enhanced lipid metabolic activities, such as fatty acid oxidation, and predominantly elicits an anti-inflammatory response and tissue repair.
One of the initial events following inhalation of Mtb is phagocytosis by alveolar macrophages that secrete IL-8 and facilitate the recruitment of neutrophils to the infection site (Schlesinger, 1996). Various host cell receptors, including complement receptors, C-type lectins, mannose receptors, surfactant proteins (SpA), and CD14, are involved in the uptake of Mtb by macrophages (Kleinnijenhuis et al., 2011). The association between the type of receptor involved in phagocytosis and the fate of engulfed Mtb within the macrophages remains poorly understood. The nature of the engulfing receptor can influence the downstream signaling and subsequent processing of the bacilli (Sanchez et al., 2010; Marakalala and Ndlovu, 2017; Queval et al., 2017). Thus, Mtb phagocytosed by specific cell surface receptors is thought to be processed differentially that can favor intracellular bacillary growth, while engagement and signaling by other receptors destroy the bacteria (Killick et al., 2013; Mortaz et al., 2015; Liu et al., 2017). Ultimately, the fusion of mycobacteria-containing phagosomes with lysosomes results in bacterial killing within the phagocyte. Similarly, host cell production of ROS and nitrogen (RNS) intermediates facilitate intracellular bacterial killing (MacMicking et al., 1997; Cooper et al., 2000; Ehrt and Schnappinger, 2009). Besides, macrophages phagocytose and eliminate Mtb through autophagy and apoptosis (Eruslanov et al., 2005; Wolf et al., 2007; Eum et al., 2010; Behar et al., 2011; Lowe et al., 2012). In general, these antibacterial mechanisms of phagocytes are successful in eliminating non-virulent bacillus; however, virulent Mtb strains have developed mechanisms to bypass these efforts, by preventing phagolysosome fusion, and surviving in the presence of ROS and RNS, thereby adapting to the hostile intracellular environment (Sturgill-Koszycki et al., 1994). Successful pathogenesis by Mtb further relies on subverting host cell death pathways; for example, Mtb inhibits host cell apoptosis and induces necrosis as a strategy to survive intracellularly and disseminate to other cells (Hinchey et al., 2007; Behar et al., 2011; Welin et al., 2011; Wong and Jacobs, 2011).
German Nobel laureate Otto Heinrich Warburg first described aerobic glycolysis for tumor cells in the 1920s (Warburg, 1956). Recent reports have revealed that Mtb can induce a Warburg-like metabolic reprogramming in infected cells both in vivo and in vitro (Shin et al., 2011; Somashekar et al., 2011; Appelberg et al., 2015; Shi et al., 2015; Gleeson et al., 2016; Lachmandas et al., 2016a,b; Billig et al., 2017). This effect is characterized by classical upregulation of glucose uptake and glycolysis-coupled deviation of glycolytic intermediates to the synthesis of large lipid bodies, which accumulate in the macrophages (Kelly and O'Neill, 2015). Fatty acids in these lipid bodies of infected macrophages provide a nutritional niche for the intracellular bacteria (Lee et al., 2013; Podinovskaia et al., 2013; VanderVen et al., 2015; Nazarova et al., 2017). It has also been reported that induction of aerobic glycolysis by Mtb in human monocyte-derived macrophages and primary human alveolar macrophages are required for the optimal production of IL-1β and suppression of the anti-inflammatory cytokine IL-10, leading to reduced bacterial load (Gleeson et al., 2016).
One of the critical virulent factors of pathogenic Mtb, ESAT-6, stimulates translocation of the glucose transporters Glut-1 and Glut-3 from the cytosol to the cell membrane, which correlates with enhanced uptake of glucose into human macrophages (Singh et al., 2015). Glucose uptake by THP-1 macrophages increases proportionately with the virulence of the Mtb strain, supporting a key metabolic function in Mtb progression (Singh et al., 2015). In addition, augmentation of aerobic glycolysis promoted activation of the cellular apoptotic response upon Mtb infection (Matta and Kumar, 2016). However, Mtb adapts to the host cell environment by switching its central carbon metabolism (CCM) toward catabolism of host lipid substrates, which includes the pathogen-induced lipid bodies accumulated within the macrophages (Lee et al., 2013). Recent studies also suggest that this lipid body accumulation in human macrophages relies on the induction of de novo cholesterol and fatty acid synthesis (FAS), and the generation of ketone body D-3-hydroxybutyrate by the host cell (Kim et al., 2010; Singh et al., 2012). Although Mtb ESAT-6 is involved in the initiation of lipid body formation, BCG, a non-pathogenic vaccine strain of M. bovis can also induce lipid bodies in infected macrophages (Melo and Dvorak, 2012). Therefore, lipid body formation may be a general consequence of mycobacterial infection rather than playing a key role in TB pathogenesis.
An important metabolic regulator, PPAR-γ, is highly upregulated in human and mouse macrophages upon mycobacterial infection (Almeida et al., 2009; Lagranderie et al., 2010; Rajaram et al., 2010). The increased expression of PPAR-γ leads to enhanced lipid droplet formation inside macrophages and down-modulation of immune responses against Mtb (Rajaram et al., 2010; Mahajan et al., 2012). This observation suggests that increased PPAR-γ expression is a strategy adopted by Mtb to establish infection in the host. Consistent with this, inhibition of PPAR-γ expression in macrophages resulted in enhanced mycobacterial killing (Almeida et al., 2012).
Different lineages of macrophages have differential responses to Mtb infection (Epelman et al., 2014; Gibbings et al., 2015; van de Laar et al., 2016), although macrophages derived from different ontogenies coexist in several tissues. A recent study in mice showed that alveolar macrophages are committed to fatty acid oxidation, which serves as a favorable niche for Mtb, while interstitial macrophages are glycolytically active, restricting bacterial growth (Huang et al., 2018). Further, studies using Mtb-infected mouse bone marrow-derived macrophages (BMDM) treated with 2-DG (2-D-Glucose, a glycolysis inhibitor) or etomoxir (ETO; a fatty acid oxidation inhibitor) showed that inhibition of glycolysis enhanced bacterial growth, while inhibition of fatty acid oxidation reduced bacterial count (Huang et al., 2018). Thus, it was proposed that restricting alveolar macrophages and increasing the number of interstitial macrophages might be an effective strategy to combat Mtb.
Activation of macrophages by signals emerging from Mtb infection leads to their polarization toward an M1 state. These cells undergo a metabolic shift toward aerobic glycolysis and arginine metabolism and express inflammatory cytokines such as TNF-α, IL-1β, IFN-γ, and IL-12 (McClean and Tobin, 2016) (Figure 1). During the advanced disease stage, the macrophages adapt to an M2- polarization state, whereby metabolic reprogramming leads to a dependence on oxidative phosphorylation to supply energy needs and the expression of anti-inflammatory cytokines such as IL-4, IL-10, and TGF-β (McClean and Tobin, 2016). The M2-polarized state is also partly required to maintain homeostasis and prevent self-destruction from an over-active immune response (O'Neill and Pearce, 2016; Escoll and Buchrieser, 2018). However, a study using computational genome-scale metabolic models predicted that Mtb decelerates OXPHOS and glycolysis in human alveolar macrophages, which results in decreased ATP production (Bordbar et al., 2010). This study also predicted that BCG and other non-pathogenic or dead mycobacteria could accelerate glycolysis and OXPHOS (Bordbar et al., 2010). In this study, a reduced glycolytic proton efflux rate was observed in Mtb-infected hMDMs and THP-1 macrophages. In contrast, an increased glycolytic proton efflux rate was noted in macrophages upon BCG infection. Therefore, reducing the glycolytic rate could be a strategy through which Mtb subverts the host immune response. It was also proposed that citrate is involved in this glycolysis reduction. Because citrate is an inhibitor of phosphofructokinase, an enzyme responsible for the conversion of fructose-6-phosphate to fructose-1, 6 biphosphate, and ADP, which is a rate-limiting step in glycolysis, predictions from this study challenge the model that Mtb infection induces the Warburg effect in macrophages (Bordbar et al., 2010).
In addition to glucose and fatty acid metabolism, amino acid metabolism has also been investigated in the context of Mtb pathogenesis. In a recent study, it was observed that in murine and macaque macrophages, as well as in their lungs, Mtb induced the expression of indoleamine 2, 3 dioxygenases (IDO), an enzyme involved in tryptophan catabolism (Mehra et al., 2013; Gautam et al., 2018). In these experimental models, suppression of IDO activity resulted in reduced bacterial load and increased host survival (Gautam et al., 2018). IDO catabolizes tryptophan (Trp) to Kynurenine (Kyn) and other metabolites. It also suppresses the host immune response, particularly IFN-γ production by CD4+ T cells (Mbongue et al., 2015). Since Mtb can synthesize its own Trp de novo in infected host phagocytes, the production of IDO by the host cells have less effect on intracellular Mtb metabolism; yet both Mtb-Trp and host-IDO impact the protective host immune response. Indeed, IDO is particularly enriched in the macrophage-rich, inner layer of granulomas in the lungs of Mtb-infected non-human primates (Mehra et al., 2013). Inhibition of IDO activity [e.g., using 1-methyltryptophan (1-MT, D-1MT)] was also associated with reorganization of the granuloma that allowed lymphocytic trafficking into the macrophage-tropic internal layers (Gautam et al., 2018). Taken together, macrophages, as the primary phagocytes of Mtb infection, can rewire their metabolic pathways upon infection and/or activation through multiple mechanisms, which are yet to be fully explored.
Dendritic Cells
Dendritic cells (DC) serve as a bridge between the host innate and adaptive immune response during Mtb infection (Mellman and Steinman, 2001; Kapsenberg, 2003). DCs carrying live Mtb and/or its antigens migrate to the regional lymph nodes where they prime T cells, leading to generation and proliferation of effector and memory T cells (Gallegos et al., 2008; Reiley et al., 2008; Wolf et al., 2008). Migration of DCs to lymph nodes is facilitated by IL-12-p40-dependent mechanisms and upregulation of CCR-7 (Khader et al., 2006). DCs are crucial for mounting CD4+ T cell response involving Th1 cytokines, such as IFN-γ and TNF-α (Tascon et al., 2000; Humphreys et al., 2006). The depletion of CD11C+ cells, which includes DCs, delays the CD4+T cell response in mice infected with Mtb (Tian et al., 2005). This shows that the early interaction of DCs with Mtb shapes the development of subsequent adaptive immune response (Figure 2). Since DCs are a heterogeneous cell population, the effectiveness of immune responses against Mtb is coordinated among the different subtypes. Lai et al. recently found in a mouse model that migratory CD11b+ DCs are involved in Th1 priming and activation following Mtb infection, while their counterpart, CD103+ DCs, plays a regulatory role through production of IL-10 and inhibiting priming of Th1 cells (Lai et al., 2018). It has also been reported that treatment of DCs with glutathione (GSH) precursor in vitro enhanced their ability to inhibit Mtb by innate immune mechanisms, and facilitated an efficient adaptive immune response (Morris et al., 2013a). However, several microbial factors, including cell wall components and secretion products, can interfere with DC-T cell signaling in vivo (Ting et al., 1999; Fortune et al., 2004; Reed et al., 2004; Banaiee et al., 2006; Pathak et al., 2007). Recently, Mtb Hip1 was identified as a critical factor that impairs DC function; infection of DCs with hip1-mutant Mtb resulted in significantly higher expression of pro-inflammatory cytokines such as IL-12 and enhanced surface expression of MHCII, CD4, and CD86, which are required for DC maturation (Madan-Lala et al., 2014).
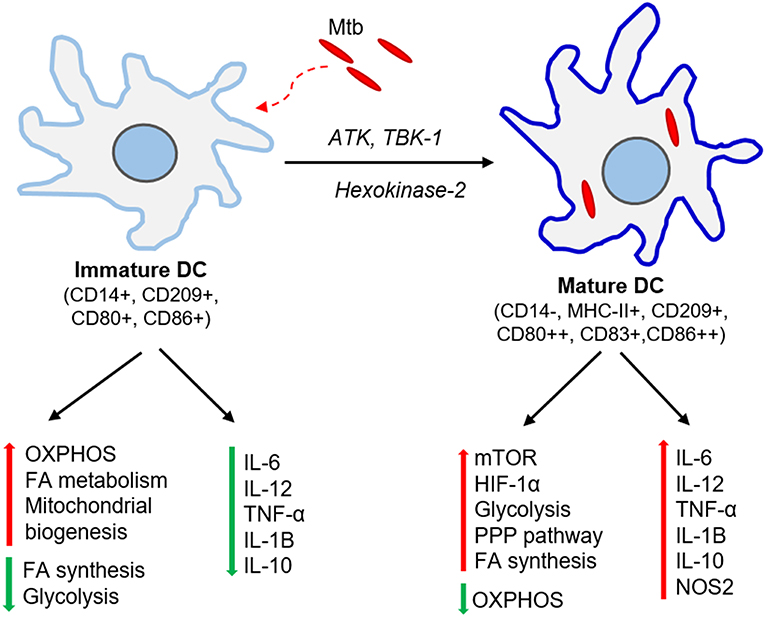
Figure 2. Immunometabolic changes associated with DC maturation. Immature DCs produce no or meager amount pro-inflammatory cytokines. These DCs have elevated oxidative phosphorylation (OXPHOS), fatty acid (FA) metabolism and mitochondrial biogenesis while dampening FA synthesis and glycolysis. Mtb infection activates ATK, TBK-1, and hexokinase-2 signaling pathways and induces DC maturation. The mature DCs express higher levels of major histocompatibility complex II (MHC-II) and co-stimulatory molecules (CD80, CD83, CD86). These DCs up-regulate molecules involved in mTOR and HIF-1α signaling pathways, elevate glycolysis, fatty acid synthesis and pentose phosphate pathway (PPP), and dampens OXPHOS. Mature DCs also produce significant amount pro-inflammatory cytokines such as IL-6, TNF-α, IL-1β.
The metabolic requirements for DCs are distinct among different subtypes, tissues, and stages of maturation (Figure 2). Bone marrow-derived dendritic cells (BMDCs) can adopt Warburg effect, i.e., aerobic glycolysis, upon stimulation with TLR agonists, leading to optimal maturation and effector functions in both human and mouse DCs (Krawczyk et al., 2010). This induction of glycolysis is regulated by PI3K/TBK1/IKKε/AKT signaling, which promotes rapid translocation of Hexokinase-2 to mitochondria, facilitating glucose catabolism required for DC maturation (Miyamoto et al., 2008; van der Windt et al., 2012). The by-products of glucose metabolism are used in pentose phosphate pathway (PPP) metabolism, lactate production, and fatty acid synthesis. This metabolic reprogramming regulates the breadth and depth of adaptive immune response induced by the DCs (Pearce and Everts, 2015). One of the essential regulators of DC metabolic remodeling, mTOR, is activated upon interaction of mycobacterial pathogen-associated molecular pattern (PAMP) molecules with DC surface receptors, which promotes protein synthesis and cell growth (Pearce and Everts, 2015).
Alteration in fatty acid metabolism of DCs in response to Mtb infection is another area of intense research. Differentiation of human monocytes to DCs in response to GM-CSF and IL-4 is accompanied by increased expression of the peroxisome proliferator-activated receptor-gamma (PPAR-γ), a transcription factor involved in lipid metabolism that is also upregulated in macrophages in response to Mtb infection (Le Naour et al., 2001; Ishikawa et al., 2007). Monocyte differentiation to DCs is dependent on phosphoinositol-3 kinase (PI3K)-mediated activation of the mTOR complex1 (mTORC1) that in turn activates its downstream target PPAR-γ, which affects cell maturation and function by controlling lipid metabolism (Nencioni et al., 2002; Szatmari et al., 2004, 2007; Gogolak et al., 2007). Further, inhibiting cytosolic fatty acid synthase (FASN) by blocking acetyl CoA-carboxylase (AAC1) reduced monocyte-derived dendritic cell (moDC) differentiation (Rehman et al., 2013). Differentiated moDCs contain higher numbers of mitochondria, show a higher oxygen consumption rate (OCR), and produce more ATPs than other non-differentiated monocytes (Del Prete et al., 2008; Zaccagnino et al., 2012). Consistent with this, moDC differentiation can be partially prevented by blocking the mitochondrial electron transport chain (ETC) with complex-I inhibitor, rotenone (Del Prete et al., 2008; Zaccagnino et al., 2012). Thus, differentiation of DCs from human monocytes is governed by OXPHOS and fatty acid metabolism. Glycolysis has also been shown to be essential for the development and proliferation of murine DCs (Kratchmarov et al., 2018). Similarly, DC-like cells differentiated from mouse BMDMs using GM-CSF also showed higher glucose uptake and high mitochondrial membrane potential and oxygen consumption (Krawczyk et al., 2010).
As well as promoting monocyte differentiation to DCs, fatty acid metabolism is also essential for immunogenicity and the antigen-presenting capacity of DCs in human and mouse liver (Ibrahim et al., 2012). And recently, it has been reported that, apart from glucose, DCs store glycogen and express enzymes required for its metabolism. Indeed, defective glycogen metabolism in DCs impaired their activation and effector functions (Thwe et al., 2017). Taken together, although DCs are phagocytes and antigen-presenting cells like macrophages, that have different metabolic requirements and utilize various signaling pathways to regulate their metabolic remodeling during cell growth and development vs. Mtb infection. While it is clear that glycolysis and fatty acid metabolism in particular play a vital role in DC function, the relevance of these metabolic pathways specifically during Mtb infection needs further investigation.
Neutrophils
Neutrophils or polymorphonuclear (PMN) cells are the most abundant constituent of white blood cells or granulocytes and are some of the first cells to arrive at the site of bacterial infections, including TB. However, among the various phagocytes, the specific role of neutrophils during Mtb infection is less well-characterized.
Neutrophils are efficient phagocytes that also facilitate the killing of pathogens by secreting human neutrophil peptides (HNP), which are cationic proteins of the α-defensin family that bind to anionic components of plasma membranes and disrupt their integrity (Fu, 2003). After phagocytosis of Mtb, neutrophils secrete several chemokines and cytokines that attract inflammatory monocytes from the circulation (Petrofsky and Bermudez, 1999; Seiler et al., 2003; Mantovani et al., 2011). Monocyte recruitment and maturation at the site of infection can redefine the microenvironment at both metabolic and immunologic levels (Blomgran and Ernst, 2011). Studies suggest that upon mycobacterial infection, neutrophils facilitate the activation of naïve antigen-specific CD4+ T cells, triggering an adaptive immune response (Blomgran and Ernst, 2011). However, there are also reports indicating that neutrophils can act as permissive hosts for Mtb (Abadie et al., 2005; Sutherland et al., 2009; Eum et al., 2010). Depending on host- and pathogen-associated factors, and their interactions, neutrophils can also contribute to disease progression. Specifically, recruitment of neutrophils during early stages of Mtb infection has been shown to halt infection (Lyadova, 2017). However, initial activation and presence of neutrophils in lungs infected by a hypervirulent strain of Mtb resulted in exacerbation of inflammation and disease progression (Koo et al., 2012). During mycobacterial infection, neutrophils also induce transcriptional changes in macrophages leading to the generation of pro-inflammatory cytokines (Andersson et al., 2014). Alternatively, Mtb-infected, necrotic macrophages can be engulfed by neutrophils, thereby clearing cellular debris during Mtb infection. Neutrophils affect T cell development and maturation during Mtb infection by releasing IL-12 (p40 and p70), IL-10, IP-10 (interferon γ induced protein 10), and MIP-1α (macrophage inflammatory protein) (Petrofsky and Bermudez, 1999; Seiler et al., 2003; Mantovani et al., 2011). Neutrophils also communicate with DCs to deliver signals and for antigen presentation (Hedlund et al., 2010). It has been shown that the enhanced levels of GSH inside neutrophils increase the fusion of phagosomes containing Mtb with lysosomes, which inhibits mycobacterial growth (Morris et al., 2013b).
Only a few studies have explored the metabolic changes associated with neutrophil maturation and activation. One reason for the lack in this area is the relatively short life span and rapid activation of neutrophils that limit technical manipulations for long term studies. In humans, Mtb infection-induced hypoxic conditions stabilize HIF-1α in neutrophils (Ong et al., 2018). This event leads to increased secretion of matrix metalloproteases (MMP)-8 and MMP-9, which contribute to the destruction of the matrix of type1 collagen, gelatin and elastin, which are the main structural proteins of the human lung (Ong et al., 2018). In summary, more research is needed to identify the metabolic changes occurring in neutrophils and to link them to the corresponding immune responses in the context of Mtb infection.
Mediators of Host Metabolism During Mtb Infection
Cellular signals sensed by host cells activate specific regulators that control immunometabolism (Figure 3). These signals can originate either from intrinsic stimuli of the host, such as starvation or from extrinsic factors such as microbes. Similarly, the downstream response to these stimulants shares many common molecules and pathways between homeostatic and disease conditions (Linke et al., 2017; Patsoukis et al., 2017; Krzywinska and Stockmann, 2018). The following are the key host transcriptional regulators involved in immunometabolic changes relevant for Mtb infection (Figure 4).
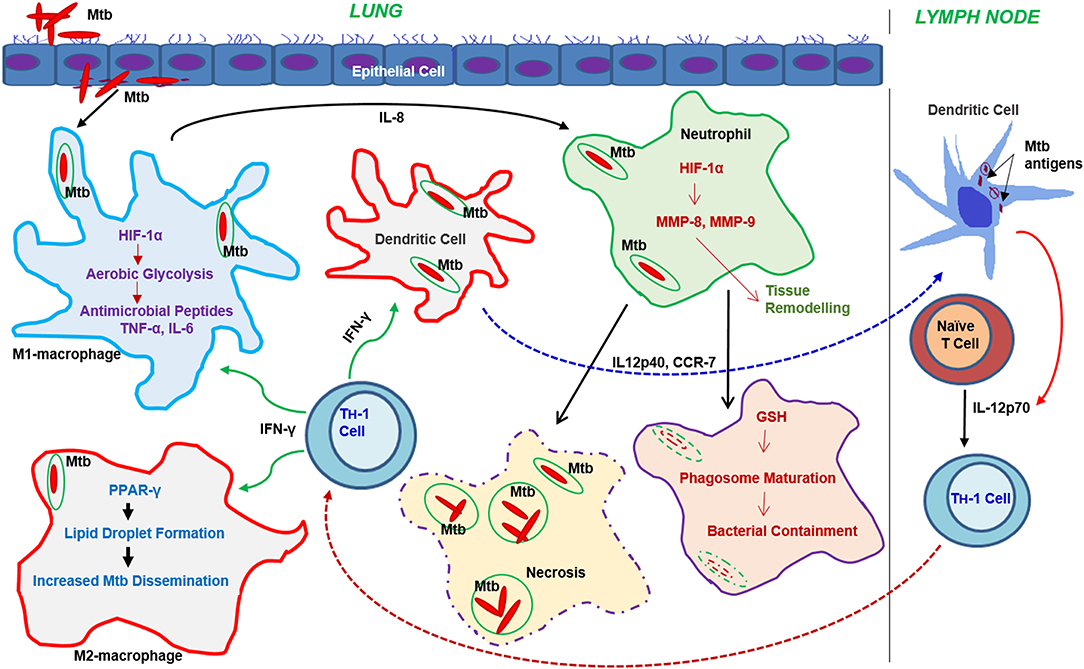
Figure 3. Progression of the immune response and metabolic changes in immune cells following Mtb infection. Following infection, Mtb reaches the lung where it encounters various immune cells. Macrophages and DCs are among the first immune cells to encounter Mtb. Activation of macrophages leads to the generation of antimicrobial peptides or dissemination of Mtb through the autophagy machinery. Neutrophil interaction with Mtb leads to either bacterial containment or dissemination through necrosis. DCs infected with Mtb migrate to the draining lymph nodes where they drive T cell differentiation toward a Th-1 phenotype. The activated Th-1 cells migrate back to the lungs, where they produce IFN-γ and TNF-α, which further activate macrophages leading to bacterial clearance. Metabolic reprogramming, mainly via the activation of aerobic glycolysis in macrophages, neutrophils, and dendritic cells, plays a significant role in effective functioning of these cells.
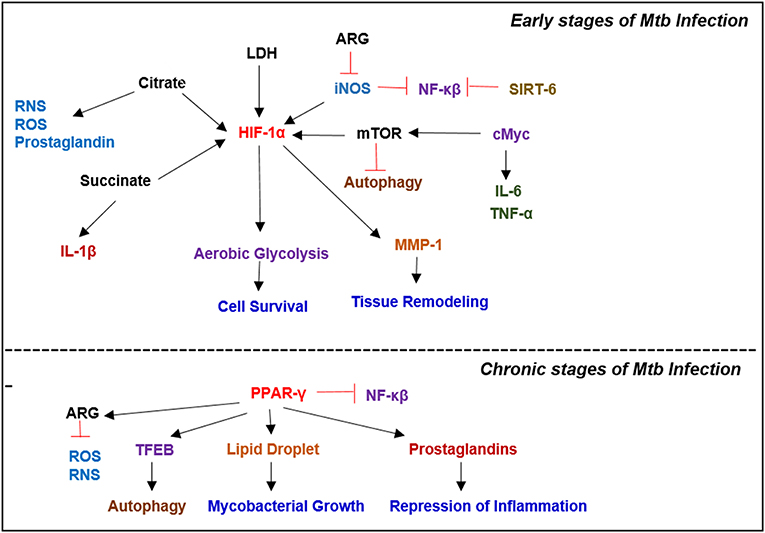
Figure 4. Key metabolic regulators involved in Mycobacterial infection. Infection with Mtb results in a plethora of metabolic reprogramming in immune cells leading to the induction of HIF-1α, which induces aerobic glycolysis. In the latter stages of the disease, when host immunity wanes, Mtb modulates the immune response by inducing PPAR-γ expression, which facilitates lipid droplet formation and prostaglandin synthesis, which augment bacterial survival.
HIF-1
The hypoxia-inducible factor-1 (HIF-1) is a heterodimeric transcription factor composed of two subunits: oxygen responsive HIF-1α, and constitutively expressed HIF-1β (Nizet and Johnson, 2009). In human colon and ovarian cancer cells, under non-hypoxic conditions, prolyl-1-4 hydroxylase proteins (PHDs) hydroxylate HIF-1α, which then binds von-Hippel-Lindau proteins, causing proteasomal degradation of HIF-1α (Huang et al., 1998; Semenza, 2000). Under hypoxic conditions, PHD activity is down-regulated resulting in HIF-1α accumulation and heterodimerization with HIF-1β (Semenza, 2001). This complex then binds to the promoter regions of several hypoxia-inducible genes and upregulates their expression (Corcoran and O'Neill, 2016). Hypoxia-inducible genes encode proteins involved in a myriad of cellular pathways that regulate cell survival, apoptosis, angiogenesis, erythropoiesis, glucose metabolism, and pH regulation (Semenza, 2000).
Various stimuli activate HIF-1α and, together with NF-κB, these signaling pathways regulate the immune response in hypoxic conditions (Rius et al., 2008). Under hypoxic conditions, HIF-1α fulfills the energy needs of cells by promoting aerobic glycolysis via inducing the expression of the glycolytic enzymes, such as hexokinase and phosphofructokinase (Riddle et al., 2000; Obach et al., 2004). Hypoxia and inflammation are inherently linked, as upon activation, immune cells undergo considerable metabolic reprogramming to sustain energy needs, thereby mostly switching to aerobic glycolysis. Thus, HIF-1α mediated pathways are essential in regulating immunity and inflammation. The role of HIF-1α in inducing aerobic glycolysis was elucidated in macrophages activated by LPS, and in Th-17 cells, which produce IL-17 (Tannahill et al., 2013; Gerriets et al., 2015). HIF-1α was also found to be upregulated in murine lungs upon Mtb infection inducing the Warburg effect (Shi et al., 2015).
HIF-1α is expressed in primary innate immune cells like macrophages, DCs, neutrophils, and Th-17 cells (Corcoran and O'Neill, 2016). In DCs, HIF-1α was shown to play a role in activation-induced by LPS, as blocking glycolysis using 2-DG inhibited DC maturation, measured through the reduced expression of costimulatory molecules CD80 and CD86 (Jantsch et al., 2008). A similar role for HIF-1α in macrophage activation by LPS was also demonstrated: inhibition of glycolysis using 2-DG caused a HIF-1α-mediated downregulation of IL-1β that blocked macrophage activation (Zhang et al., 2006; Tannahill et al., 2013). Additional roles for HIF-1α in macrophage differentiation and function have also been demonstrated. Specifically, HIF-1α-mediated metabolic reprogramming plays a significant role in polarization of macrophages toward the M1 or M2 phenotype (Corcoran and O'Neill, 2016). TCA cycle intermediates succinate and citrate accumulate in macrophages upon LPS stimulation, which stabilizes HIF-1α (Newsholme et al., 1986; Tannahill et al., 2013; Jha et al., 2015). Succinate increases the transcription of target gene IL-1β, whereas citrate accumulation leads to increased production of pro-inflammatory mediators- NO, ROS, and prostaglandins (Infantino et al., 2011; Tannahill et al., 2013). In neutrophils, HIF signaling leads to production of the PHD enzymes, especially PHD3, which prolong the survival of neutrophils under hypoxic conditions in both human and mice (Walmsley et al., 2011). Thus, HIF-1α plays a critical role in the homeostasis of immune cells through metabolic reprogramming during inadequate oxygen levels and also governs immune responses. In a recent study, it was shown that HIF-1α-dependent MMP-1 (collagenase) expression and secretion were synergistically upregulated by hypoxia during Mtb infection in MDMs and normal human bronchial epithelial cells (NHBFs) (Belton et al., 2016). They also showed that HIF-1α is stabilized by Mtb even in the absence of hypoxia (Belton et al., 2016). Consistent with this, in mice models of Mtb infection, IFNγ-activated macrophages exhibited increased HIF-1α levels (Braverman et al., 2016). In a zebrafish model of M. avium infection, increasing HIF-1α levels were associated with increased bacterial killing (Elks et al., 2013). Similarly, HIF-1α deficiency in the myeloid lineage blocked IFNγ-induced killing of Mtb (Braverman et al., 2016).
HIF-1α, which regulates half of the transcriptional responses to IFNγ, induces the cellular metabolic shift from OXPHOS to aerobic glycolysis in IFNγ-activated macrophages by inducing LDH (lactate dehydrogenase) activity to catalyze lactate production, and by limiting acetyl CoA for the TCA cycle (Nagao et al., 2019). HIF-1α also regulates the inflammatory response and NO production in IFNγ-activated macrophages against infection. Indeed, lack of HIF-1α reduces ATP levels and NO production at the inflammatory sites (Nagao et al., 2019). In contrast, HIF-2α reduces the levels of RNS in infected neutrophils and opposes control of bacterial burden (Elks et al., 2013). The IFNγ signaling cascade involves the induction of iNOS, which reduces the expression of RelA (NF-kB family), thereby negatively regulating the inflammatory response (Braverman and Stanley, 2017). This activity of iNOS has been suggested to stabilize the HIF-1α-induced immune response in IFNγ-activated macrophages during Mtb infection (Braverman and Stanley, 2017).
mTOR
The mammalian target of rapamycin (mTOR) belongs to the PI3K- related serine/threonine protein kinase family and comprises two distinct molecular complexes: mTOR complex1 (mTORC1) and mTOR complex 2 (mTORC2) (Laplante and Sabatini, 2012; Singh and Cuervo, 2012). mTOR is involved in balancing anabolic vs. catabolic reactions to maintain cellular homeostasis. mTOR is a master regulator of a myriad of cellular pathways and functions, including protein synthesis, metabolism, disease progression, and cell death (Saxton and Sabatini, 2017). mTOR phosphorylates the kinase p70S6 kinase1 (S6K1) (Holz et al., 2005). Upon activation, S6K1 phosphorylates and activates several downstream substrates including eIF4B (a positive regulator of the eIF4F complex) and PDCD4 (an inhibitor of eIF4B), which promotes translation (Dorrello et al., 2006). 4EBP is an essential cellular regulator of translation. It inhibits translation by binding and sequestering eIF4E, thereby preventing assembly of the eIF4E complex. Upon phosphorylation by mTORC1 at multiple sites, 4EBP dissociates from eIF4E, and 5′ cap-dependent mRNA translation is facilitated (Brunn et al., 1997; Gingras et al., 1999). mTOR is also involved in de novo lipid synthesis through the transcription factor, sterol responsive element binding protein (SERBP), which controls the expression of genes involved in fatty acid and cholesterol biosynthesis (Singh and Subbian, 2018). mTORC1 also promotes nucleotide biosynthesis by inducing ATF-4 dependent methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 2 (MTHFD2) expression, which is a vital component of the mitochondrial tetrahydrofolate cyclase that provides one-carbon units for purine biosynthesis (Ben-Sahra et al., 2016). mTORC1 also activates CAD (carbamoyl- phosphate synthetase), a key component required for pyrimidine synthesis (Ben-Sahra et al., 2013; Robitaille et al., 2013). mTOR also increases HIF-1α translation and thus is involved in the metabolic shift to aerobic glycolysis (Saxton and Sabatini, 2017). In addition, the Mtb-induced metabolic switch of the host cellular machinery toward aerobic glycolysis is mediated by a TLR-2-dependent pathway in human monocytes and macrophages, which requires the AKT-mTOR axis (Lachmandas et al., 2016a).
Together with positively regulating anabolic processes, mTOR also suppresses catabolic processes to promote cellular growth. One of the major cellular processes regulated by mTOR is autophagy. mTOR phosphorylates, and thereby inactivates ULK1, which is a kinase that triggers autophagy by forming a complex with ATG13, FIP2000, and ATG10, to drive autophagosome formation (Kim et al., 2011). One of the survival strategies adopted by Mtb in host cells is to subvert the autophagy pathway for its benefit (Zhai et al., 2019). In mice, prolonged exposure to rapamycin before Mtb aerosol infection inhibited T cell activation and production of pro-inflammatory cytokines (Lachmandas et al., 2016a). This was also shown in human PBMCs. Studies using inhibitors have demonstrated a role specifically for mTORC1 in Th1 and Th17 activation, and associated metabolic changes; however, no role for mTORC2 has been shown in immune cell metabolic shift (Delgoffe et al., 2011). One of the signaling pathways through which mTORC1 induces metabolic reprogramming involves HIF-1α (Yecies and Manning, 2011). Using immortalized human retinal pigment epithelial cells, mTORC1 activation was shown to upregulate glycolytic genes like Glut-1, which promotes cholesterol and fatty acid synthesis involving a pathway regulated by SREBPs and PPAR-γ (Porstmann et al., 2008). In contrast, mTORC2 activates metabolic reprogramming through Myc and AKT in the human breast cancer cell line MCF-7 (Sarbassov et al., 2005; Zou et al., 2015).
cMyc
The cellular myelocytomatosis (cMyc) is a member of a proto-oncogene family of transcription factors involved in regulating cell proliferation (Stine et al., 2015). Structurally, cMyc consists of an N-terminal transactivation domain and a C-terminal basic helix-loop-helix (HLH) leucine zipper (LZ) domain, which binds to a CACGTG E-box DNA sequence (Meyer and Penn, 2008). This binding facilitates the recruitment of histone acetyltransferase and elongation factors that can modify the transcriptional response of several genes. Deregulated and sustained Myc expression is a hallmark of cancer cells, and it constitutively activates a cell growth program in an mTOR-dependent manner (Dang, 2011). Myc controls metabolic reprogramming by binding to open chromatin of target genes involved in glycolysis and glutaminolysis, which promotes their efficient transcription. Myc dimerizes with Max, which is also a DNA-binding helix-loop-helix leucine zipper protein, to alter gene expression (Stine et al., 2015).
Upon mycobacterial infection of human PBMCs with different pathogenicity M. bovis (BCG), M. avium, M. kansasii, and M. chelonae, the Wnt/beta-catenin signaling pathway was found to stimulate cMyc through the MAPK/ERK pathway, resulting in the upregulation of key cytokines, including TNF-α and IL-6, which inhibit mycobacterial growth (Yim et al., 2011). In this context, Myc played a role in the anti-mycobacterial response without influencing cell proliferation or changing the G0/G1 cell cycle phase of the macrophages.
Sirtuins (SIRTs)
Sirtuins are a family of seven proteins with deacetylase activity that requires NAD+ for their function (Sauve and Youn, 2012). Generally, sirtuins modulate cellular processes by inhibiting NFκB signaling and associated pro-inflammatory responses in several clinical conditions associated with chronic and acute inflammation and other metabolic disorders such as diabetes and obesity (Kauppinen et al., 2013; Vachharajani et al., 2016). TLR4 stimulation in THP-1 macrophages leads to a sirtuin-mediated metabolic shift from enhanced glycolysis to increased fatty acid oxidation, which typically delays early inflammation (Liu et al., 2012). In addition, the role of sirtuins in modulating the host immune response toward a more anti-inflammatory response during Mtb infection has been reported (Cheng et al., 2017). Mtb infection down-regulates SIRT1 expression in THP-1 macrophages, as well as in mice lung, which downregulates the expression of the RelA/p65 units of NFκB and promotes inflammatory resolution (Cheng et al., 2017). Furthermore, in contrast to an earlier observation, it has been shown that inhibition of SIRT6 increases the expression of glucose transporters such as GLUT1 and GLUT4, which promote glucose absorption and glycolysis in a mouse model of type 2 diabetes (Sociali et al., 2017). Another study reported increased SIRT6 expression during the early stages of Mtb infection in murine macrophages, which suppresses both pro-inflammatory and other anti-microbial responses at this early stage of infection (Shi et al., 2019). This observation suggests that clinical intervention or host-directed therapeutic strategies that regulate availability of sirtuins might help control disease by promoting a pro-inflammatory immune response.
PPARs
PPAR-γ is a member of the ligand-activated transcription factor family (Theocharis et al., 2004). It forms a heterodimer with the retinoid X receptor (RXR) that binds to specific PPAR-response elements (PPREs) on the promoter regions of target genes (Kota et al., 2005). Expression of PPAR-γ can be activated by lipid metabolites; PPAR-γ also modulates lipid and glucose metabolism, and other cellular functions such as proliferation, differentiation, and inflammation (Almeida et al., 2012). Mtb infection increases PPAR-γ gene expression in host immune cells, which downregulates NF-κB signaling and enhances prostaglandin (PG) E2 production that represses pro-inflammatory cytokines and Th1 responses (Almeida et al., 2012; Mahajan et al., 2012). Increased PPAR-γ expression in Mtb-infected macrophages has also been associated with lipid droplet formation (Mahajan et al., 2012; Salamon et al., 2014). The accumulated lipids in these infected cells provide nutrients and promote bacterial growth. In addition, this PPAR-γ mediated lipid accumulation positively correlates with increased expression of scavenger receptors, including macrophage receptor with collagenous structure (MARCO), macrophage scavenger receptor (MRS), and CD36 in macrophages within human leprosy lesions (Cruz et al., 2008). These findings suggest that PPAR-γ promotes intracellular lipid accumulation by modulating the expression of several genes involved in lipid absorption as well as fatty acid synthesis during Mtb infection.
PPAR-α is another isoform of the PPAR family. It is a transcription factor that binds to cis-acting DNA elements and regulates the expression of several genes involved in lipid and glucose metabolism (Rakhshandehroo et al., 2010). PPAR-α regulates mitochondrial as well as peroxisome function and thus energy metabolism (Kersten, 2014). It enhances fatty acid oxidation and ketogenesis, whereas it inhibits fatty acid synthesis and glycolysis (Mandard et al., 2004). Thus, PPAR-α activation helps prevent lipid accumulation in Mtb-infected cells. PPAR-α activation also upregulates expression of transcription factor EB (TFEB) and promotes autophagy as well as host innate immunity during Mtb infection (Kim et al., 2017). The induction of TFEB also promotes lipid catabolism, which reduces intracellular Mtb growth in bone marrow-derived macrophages (Kim et al., 2017).
Arginases
Arginases are involved in the final steps of the urea cycle, where they convert arginine to ornithine and urea. Arginases exist in two isoforms; a cytosolic Arginase 1(ARG1) and a mitochondrial Arginase 2 (ARG2). Arginases compete with nitric oxide synthase (NOS) for a common substrate, arginine (Mori, 2007). While inducible NOS promotes a pro-inflammatory response, ARG is associated with the anti-inflammatory response in host immune cells (Yang and Ming, 2014). Thus, a shift in arginine metabolism is associated with the outcome of both innate and adaptive immune responses. ARG1 is predominantly present in M2 macrophages, which are mainly localized in the periphery of granulomas (Mattila et al., 2013). Increased expression of ARG1 in macrophages suppresses their antimicrobial activities by downregulating the production of nitric oxide (NO) and reactive RNS. In contrast, another study suggested a host-protective role for ARG1 in hypoxic TB granulomas: ARG1 regulated T cell proliferation and hyper-inflammation that controlled tissue necrosis and inflammatory lung pathology in murine TB model granulomas (Duque-Correa et al., 2014). This study underscored the crucial role of ARG1 in controlling inflammation and necrosis in hypoxic granulomas, where NO is ineffective. Similar to ARG1, ARG2 also has been shown to downregulate NO production and restrict an effective immune response during Helicobacter pylori infection (Lewis et al., 2011). This process involved the positive regulation of ARG2 by the liver X receptor, shifting arginine metabolism toward polyamine synthesis to exert an anti-inflammatory response in macrophages. In the same study, the authors also reported a decreased bacterial burden in Agr2−/− mice as compared to wild type, which was associated with increased NO production in the absence of ARG (Lewis et al., 2011). Thus, the metabolic shift mediated by ARG1 and 2 exerts an anti-inflammatory response in immune cells, which helps immune evasion by the pathogen.
Inducible Nitric Oxide Synthase (iNOS)
Inducible nitric oxide synthase (iNOS) catalyzes the production of NO, especially in macrophages activated by IFN-γ (Riquelme et al., 2013). NO is a key anti-mycobacterial molecule, and it can be converted into highly RNS such as NO3 and NO2 within Mtb-infected macrophages (Jamaati et al., 2017). In addition to its microbicidal activity, NO also acts as a secondary messenger and modulates the expression of genes involved in IFN-γ signaling pathways during Mtb infection (Lee and Kornfeld, 2010; Herbst et al., 2011). HIF-1α is an important factor for IFN-γ-mediated control of Mtb infection (Braverman et al., 2016). NO positively regulates HIF-1α expression and contributes to the protective macrophage response against Mtb infection (Braverman and Stanley, 2017). Furthermore, NO inhibits prolonged NFκB activation in macrophages, and limits excessive inflammation and tissue damage during Mtb infection (Braverman and Stanley, 2017). Thus, iNOS and its product NO exert direct antimicrobial responses and also modulate IFN-γ-mediated anti-Mtb activities and the inflammatory response during infection. In addition, increased NO leads to nitrosylation of iron-sulfur proteins present in the electron transport chain, inhibiting mitochondrial respiration (Brown, 2001). Nitrosylation of cysteine residues of enzymes involved in glycolysis, TCA cycle, and fatty acid oxidation, alters their activity and modulates both glucose and fatty acid metabolism in activated immune cells (Doulias et al., 2013; Kelly and O'Neill, 2015). It has been shown that NO production in LPS-activated macrophages and DCs is associated with mitochondrial dysfunction and a shift toward increased glycolysis (Everts et al., 2012). Overall, NO produced during infection causes nitrosylation of enzymes involved in glucose and fatty acid metabolism, which modulates the host immune response to infection.
Sphingolipids
The mucus secreted by lung alveolar epithelial cells acts as the first line of defense against microbial infections (Sharma and Prakash, 2017). One of the major components of these surfactants are sphingolipids, which play a crucial role in protecting the host against invasion of macrophages by Mtb (Garg et al., 2004). Sphingolipids are regulated by the de novo sphingolipid biosynthesis pathway and the sphingomyelin catabolic pathway (Gault et al., 2010). Specific sphingolipids associated with inflammatory and metabolic diseases are sphingomyelin (SM), ceramide (Cer), ceramide-1 phosphate (C1P), sphingosine (Sph), sphingosine-1 phosphate (S1P), and lactosylceramide (LacCer). All metabolites of sphingolipid biosynthesis are metabolically associated with each other to maintain a state of equilibrium within the innate immune cells and cope with the lipid overload produced during inflammatory disorders (Sharma and Prakash, 2017). The reversible/interconvertible nature of these metabolites establishes their roles as physiological (cell membrane integration) and functional (macrophage-activation) components of cells to maintain homeostasis (Ohanian and Ohanian, 2001). S1P, one of the most studied sphingolipids produced by the enzymatic activity of sphingosine-1 kinase (SK1), signals through five G-coupled receptors (S1PRs). Among these S1PRs, S1PR-1, 2, and 3 are immune regulators involved in pathogen control (Sanchez and Hla, 2004). Induction of S1PR2 has been shown to enhance the anti-mycobacterial activity of alveolar macrophages. SK1 activity is also important for phagosome maturation, innate activation of macrophages, and control of Mtb replication. Mtb is capable of inhibiting SK1 activity leading to a decrease in Ca2+ ion concentration that can block phagolysosome maturation (Malik et al., 2003). This process is facilitated by the increased levels of sialylated glycosphingolipid content in epithelial lung cells due to environmental or genetic factors. In pulmonary TB patients, the low serum level of S1P is suggested to be the result of reduced activity of SK1 by Mtb. Enhanced S1P levels are also shown to promote phagosome maturation and reduce the lung Mtb burden, thus improving histopathology in mouse models of infection (Anes et al., 2003; Garg et al., 2004). While S1P and C1P are known to promote cell repair, division, and survival, other sphingolipids such as ceramide are involved in cell death and inflammation (Sawai and Hannun, 1999). Ceramides are also suggested to induce drug resistance against rifampin in Mtb clinical isolates, and establish persistent survival of Mtb (Speer et al., 2015). This prolonged survival of Mtb is also linked with the S1P-pAKT-mediated upregulation of mTOR signaling in macrophages (Liu et al., 2009, 2010). During Mtb infection, FM formation indicates lipid overload in the host cells (Russell, 2011). To maintain sphingolipid balance in FM, saponin C (SapC) degrades glycosphingolipids to ceramide; SapC also mediates transfer of Mtb lipid antigen from intra-lysosomal membranes to CD1b to activate antigen-specific T cells (Kim et al., 2010). Upregulation of SapC seen in granulomas justifies the accumulation of LacCer, an important intermediate of sphingolipid metabolism found in abundance in the caseum compartment of granulomas, but only in trace amounts in other regions/cells (Kolter and Sandhoff, 2005, 2006). Since FM favors Mtb growth, accumulation of sphingolipid and its metabolites indicates altered phagolysosome biogenesis in Mtb-infected host cells. This lipid overload in granulomas also suggests a shift in lipid metabolism conferred by infection, which is crucial for Mtb survival.
Epigenetic Regulation of Immunometabolism
Epigenetic modifications can be induced by nutritional states of cells, which drive the inflammatory response accompanying metabolic disease (Barres et al., 2013). Mechanisms by which epigenetic modulations occur, include (I) Genomic DNA methylation by DNA methyltransferases including DNMT1, DNMT3A, and DNMT3B, which generally results in transcriptional repression, (II) histone modifications including acetylation, methylation, phosphorylation, ubiquitylation and sumoylation (Bannister and Kouzarides, 2011; Raghuraman et al., 2016). Immune regulation through methylation changes is reported in TNF-α, UBASH3B (Ubiquitin-associated and SH3 domain-containing protein B) and TRIM3 (Tripartite motif-containing-3) genes (Wang et al., 2010; Hermsdorff et al., 2013). Role of altered DNA methylation in regulating immune function of T cells and macrophages is well documented in metabolic disorders, including obesity and T2DM (Barres et al., 2013; Raghuraman et al., 2016). Macrophage differentiation from monocytes is associated with various histone-modifying components involving H3K4me1, H3K4me3, and H3K27ac at promoter and enhancer regions (Saeed et al., 2014). Besides, cytokine production by macrophages is induced by H3K4me3 and demethylation of H3K27 (Takeuch and Akira, 2011). Also, HDAC3 regulates inflammatory genes in macrophages, while HDAC2 resolves inflammation by suppressing IL-6 (Chen et al., 2014; Zhang et al., 2015). The role of H3 acetylation in regulating inflammatory genes in LPS stimulated monocytes has also been reported (Iglesias et al., 2012).
Increased glycolysis and β-oxidation lead to accumulation of acetyl CoA which is used as a group donor by histone acetyltransferases (HAT) leading to relaxation of chromatin structure resulting positive regulation of transcription (Laker and Ryall, 2016). Similarly, NAD+ was found to control epigenetic remodeling in monocytes through SIRT1 during acute systemic inflammation (Davis and Gallagher, 2019). The chromatin remodeling in BCG stimulated innate immune cells is primarily due to histone modification (H3K4me3 and H3K9me3) in the glycolysis genes leading to activation of AKT/mTOR/HIF1α pathway, which induces a metabolic shift in host cells from OXPHOS to aerobic glucose metabolism. This finding shows the rewiring of cellular metabolism to facilitate increased production of cytokines such as TNF-α and IL6 that promote mycobacterial killing (Kleinnijenhuis et al., 2011; Arts et al., 2016).
Mtb tends to prevent the expression of immune regulatory genes such as CIITA, CD64, and HLA DR through altering chromatin dynamics and modifying histones at specific promoter sites (Pattenden et al., 2002). In blood cells from patients with pulmonary tuberculosis, DNA hypermethylation of CpG sites in TLR2 promoter was observed (Chen et al., 2014). In another study, using global histone acetylation (Ac)/methylation (Me) in blood leukocytes of pulmonary tuberculosis patients showed H3K14 hypoacetylation and H3K27 hypermethylation played a role in developing active pulmonary tuberculosis (Chen et al., 2017). Mtb (Rv1988) can interfere with the host genetic remodeling also by inducing histone modifications (H3) of non-tail arginine of H3 to alter the antigen presentation of macrophages (Yaseen et al., 2015). In a recent study, it was observed that Mtb infection in DCs led to induction of stable DNA demethylation at enhancers elements across the genome (Pacis et al., 2019).
Concluding Remarks
Recent advances in TB research, including investigations into the role of metabolic reprogramming in different immune cells, have expanded our understanding of the pathogenesis and progression of the disease. It is now widely recognized that the immune response and metabolic remodeling are interconnected. Therefore, the net host response to Mtb infection is a result of combined immunologic and metabolic activities of the immune cells. However, much more work is needed to elucidate the intricate interactions among various metabolic programming pathways, which can tip the balance in the survival advantage toward the host or Mtb. Similarly, the causal link between immune response and metabolic remodeling needs further exploration, including investigations on how Mtb interferes with these host processes. A comprehensive understanding of the immunometabolic processes will not only help in our understanding of how pathogenic bacteria subvert immune response in their favor, but will also aid in uncovering new treatment/vaccination strategies to control TB more effectively. In this regard, the components of glycolysis and oxidative phosphorylation reactions associated with Mtb infection of immune cells could be harnessed for designing host-directed treatment strategies to enhance bacterial clearance and improve treatment outcomes.
Author Contributions
LS, SS, YB, and ST conceived the concept. RK, PS, AK, and SS wrote the manuscript. PS and AK contributed equally. All authors read, edited, and agreed to publish the manuscript.
Funding
The grant support to our immunometabolism research was provided by NIAID/NIH (Grant number: AI127844 to LS and SS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors like to acknowledge the NIAID/NIH for providing grant support to immunometabolism research (Grant number: AI127844 to LS and SS).
References
Abadie, V., Badell, E., Douillard, P., Ensergueix, D., Leenen, P. J., et al. (2005). Neutrophils rapidly migrate via lymphatics after Mycobacterium bovis BCG intradermal vaccination and shuttle live bacilli to the draining lymph nodes. Blood 106, 1843–1850. doi: 10.1182/blood-2005-03-1281
Alcais, A., Fieschi, C., Abel, L., and Casanova, J. L. (2005). Tuberculosis in children and adults: two distinct genetic diseases. J. Exp. Med. 202, 1617–1621. doi: 10.1084/jem.20052302
Almeida, P. E., Carneiro, A. B., Silva, A. R., Bozza, P., et al. (2012). PPARγ expression and function in mycobacterial infection: roles in lipid metabolism, immunity, and bacterial killing. PPAR Res. 2012:383829. doi: 10.1155/2012/383829
Almeida, P. E., Silva, A. R., Maya-Monteiro, C. M., Torocsik, D., et al. (2009). Mycobacterium bovis bacillus Calmette-Guerin infection induces TLR2-dependent peroxisome proliferator-activated receptor gamma expression and activation: functions in inflammation, lipid metabolism, and pathogenesis. J. Immunol. 183, 1337–1345. doi: 10.4049/jimmunol.0900365
Andersson, H., Andersson, B., Eklund, D., Ngoh, E., Persson, A., Svensson, K., et al. (2014). Apoptotic neutrophils augment the inflammatory response to Mycobacterium tuberculosis infection in human macrophages. PLoS ONE 9:e101514. doi: 10.1371/journal.pone.0101514
Anes, E., Kuhnel, M. P., Bos, E., Moniz-Pereira, J., Habermann, A., et al. (2003). Selected lipids activate phagosome actin assembly and maturation resulting in killing of pathogenic mycobacteria. Nat. Cell Biol. 5, 793–802. doi: 10.1038/ncb1036
Appelberg, R., Moreira, D., Barreira-Silva, P., Borges, M., Silva, L., Dinis-Oliveira, R., et al. (2015). The Warburg effect in mycobacterial granulomas is dependent on the recruitment and activation of macrophages by interferon- γ. Immunology 145, 498–507. doi: 10.1111/imm.12464
Arts, R. J. W., Joosten, L. A. B., and Netea, M. G. (2016). Immunometabolic circuits in trained immunity. Semin. Immunol. 28, 425–430. doi: 10.1016/j.smim.2016.09.002
Austyn, J. M., and Gordon, S. (1981). F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur. J. Immunol. 11, 805–815. doi: 10.1002/eji.1830111013
Banaiee, N., Kincaid, E. Z., Buchwald, U., Jacobs, W. R., et al. (2006). Potent inhibition of macrophage responses to IFN- γ by live virulent Mycobacterium tuberculosis is independent of mature mycobacterial lipoproteins but dependent on TLR2. J. Immunol. 176, 3019–3027. doi: 10.4049/jimmunol.176.5.3019
Bannister, A. J., and Kouzarides, T. (2011). Regulation of chromatin by histone modifications. Cell Res. 21, 381–395. doi: 10.1038/cr.2011.22
Barres, R., Kirchner, H., Rasmussen, M., Yan, J., Kantor, F. R., et al. (2013). Weight loss after gastric bypass surgery in human obesity remodels promoter methylation. Cell Rep. 3, 1020–1027. doi: 10.1016/j.celrep.2013.03.018
Behar, S. M., Martin, C. J., Booty, M. G., Nishimura, T., et al. (2011). Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol. 4, 279–287. doi: 10.1038/mi.2011.3
Belton, M., Brilha, S., Manavaki, R., Mauri, F., Nijran, K., Hong, Y., et al. (2016). Hypoxia and tissue destruction in pulmonary TB. Thorax 71, 1145–1153. doi: 10.1136/thoraxjnl-2015-207402
Ben-Sahra, I., Howell, J. J., Asara, J. M., Manning, B., et al. (2013). Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328. doi: 10.1126/science.1228792
Ben-Sahra, I., Hoxhaj, G., Ricoult, S. J. H., Asara, J. M., et al. (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. doi: 10.1126/science.aad0489
Billig, S., Schneefeld, M., Huber, C., Grassl, G. A., Eisenreich, W., et al. (2017). Lactate oxidation facilitates growth of Mycobacterium tuberculosis in human macrophages. Sci. Rep. 7:6484. doi: 10.1038/s41598-017-05916-7
Blomgran, R., and Ernst, J. D. (2011). Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J. Immunol. 186, 7110–7119. doi: 10.4049/jimmunol.1100001
Bogdan, C. (2001). Nitric oxide and the immune response. Nat. Immunol. 2, 907–916. doi: 10.1038/ni1001-907
Bordbar, A., Lewis, N. E., Schellenberger, J., Palsson, B. O., et al. (2010). Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol. Syst. Biol. 6:422. doi: 10.1038/msb.2010.68
Braverman, J., Sogi, K. M., Benjamin, D., Nomura, D. K., et al. (2016). HIF-1α is an essential mediator of IFN- γ -dependent immunity to Mycobacterium tuberculosis. J. Immunol. 197, 1287–1297. doi: 10.4049/jimmunol.1600266
Braverman, J., and Stanley, S. A. (2017). Nitric oxide modulates macrophage responses to Mycobacterium tuberculosis infection through activation of HIF-1α and repression of NF-κB. J. Immunol. 199, 1805–1816. doi: 10.4049/jimmunol.1700515
Brown, G. C. (2001). Regulation of mitochondrial respiration by nitric oxide inhibition of cytochrome c oxidase. Biochim. Biophys. Acta 1504, 46–57. doi: 10.1016/S0005-2728(00)00238-3
Brunn, G. J., Hudson, C. C., Sekulic, A., Williams, J. M., et al. (1997). Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 277, 99–101. doi: 10.1126/science.277.5322.99
Caceres, N., Tapia, G., Ojanguren, I., Altare, F., Gil, O., Pinto, S., et al. (2009). Evolution of foamy macrophages in the pulmonary granulomas of experimental tuberculosis models. Tuberculosis 89, 175–182. doi: 10.1016/j.tube.2008.11.001
Capuano, S. V. III., Croix, D. A., Pawar, S., Zinovik, A., Myers, A., Lin, P., et al. (2003). Experimental Mycobacterium tuberculosis infection of cynomolgus macaques closely resembles the various manifestations of human M. tuberculosis infection. Infect. Immun. 71, 5831–5844. doi: 10.1128/IAI.71.10.5831-5844.2003
Chen, Y. C., Chao, T. Y., Leung, S. Y., Chen, C., et al. (2017). Histone H3K14 hypoacetylation and H3K27 hypermethylation along with HDAC1 up-regulation and KDM6B down-regulation are associated with active pulmonary tuberculosis disease. Am. J. Transl. Res. 9, 1943–1955.
Chen, Y. C., Hsiao, C. C., Chen, C. J., Chao, T., et al. (2014). Aberrant Toll-like receptor 2 promoter methylation in blood cells from patients with pulmonary tuberculosis. J. Infect. 69, 546–557. doi: 10.1016/j.jinf.2014.08.014
Cheng, C. Y., Gutierrez, N. M., Marzuki, M. B., Lu, X., et al. (2017). Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci. Immunol. 2:eaaj1789. doi: 10.1126/sciimmunol.aaj1789
Cooper, A. M., Segal, B. H., Frank, A. A., Holland, S., et al. (2000). Transient loss of resistance to pulmonary tuberculosis in p47(phox-/-) mice. Infect. Immun. 68, 1231–1234. doi: 10.1128/IAI.68.3.1231-1234.2000
Corcoran, S. E., and O'Neill, L. A. (2016). HIF1α and metabolic reprogramming in inflammation. J. Clin. Invest. 126, 3699–3707. doi: 10.1172/JCI84431
Cruz, D., Watson, A. D., Miller, C. S., Montoya, D., et al. (2008). Host-derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J. Clin. Invest. 118, 2917–2928. doi: 10.1172/JCI34189
Dang, C. V. (2011). Therapeutic targeting of Myc-reprogrammed cancer cell metabolism. Cold Spring Harb. Symp. Quant. Biol. 76, 369–374. doi: 10.1101/sqb.2011.76.011296
D'Avila, H., Melo, R. C., Parreira, G. G., Werneck-Barroso, E., et al. (2006). Mycobacterium bovis bacillus Calmette-Guerin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J. Immunol. 176, 3087–3097. doi: 10.4049/jimmunol.176.5.3087
Davis, F. M., and Gallagher, K. A. (2019). Epigenetic mechanisms in monocytes/macrophages regulate inflammation in cardiometabolic and vascular disease. Arterioscler. Thromb. Vasc. Biol. 39, 623–634. doi: 10.1161/ATVBAHA.118.312135
Davis, J. M., and Ramakrishnan, L. (2009). The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell 136, 37–49. doi: 10.1016/j.cell.2008.11.014
DeBerardinis, R. J., and Thompson, C. B. (2012). Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148, 1132–1144. doi: 10.1016/j.cell.2012.02.032
Del Prete, A., Zaccagnino, P., Di Paola, M., Saltarella, M., Oliveros Celis, C., Nico, B., et al. (2008). Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free Radic. Biol. Med. 44, 1443–1451. doi: 10.1016/j.freeradbiomed.2007.12.037
Delgoffe, G. M., Pollizzi, K. N., Waickman, A. T., Heikamp, E., et al. (2011). The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 12, 295–303. doi: 10.1038/ni.2005
Diotallevi, M., Checconi, P., Palamara, A. T., Celestino, I., Coppo, L., et al. (2017). Glutathione fine-tunes the innate immune response toward antiviral pathways in a macrophage cell line independently of its antioxidant properties. Front. Immunol. 8:1239. doi: 10.3389/fimmu.2017.01239
Donnelly, R. P., Loftus, R. M., Keating, S. E., Liou, K., et al. (2014). mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 193, 4477–4484. doi: 10.4049/jimmunol.1401558
Dorrello, N. V., Peschiaroli, A., Guardavaccaro, D., Colburn, N. H., Sherman, N., et al. (2006). S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471. doi: 10.1126/science.1130276
Doulias, P. T., Tenopoulou, M., Greene, J. L., Raju, K., and Ischiropoulos, H. (2013). Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 6:rs1. doi: 10.1126/scisignal.2003252
Duque-Correa, M. A., Kuhl, A. A., Rodriguez, P. C., Zedler, U., et al. (2014). Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc. Natl. Acad. Sci. U.S.A. 111, E4024–E4032. doi: 10.1073/pnas.1408839111
Ehrt, S., and Schnappinger, D. (2009). Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell. Microbiol. 11, 1170–1178. doi: 10.1111/j.1462-5822.2009.01335.x
Eisenreich, W., Rudel, T., Heesemann, J., and Goebel, W. (2019). How viral and intracellular bacterial pathogens reprogram the metabolism of host cells to allow their intracellular replication. Front. Cell. Infect. Microbiol. 9:42. doi: 10.3389/fcimb.2019.00042
Elks, P. M., Brizee, S. M., van der Vaart, Walmsley, S. R., van Eeden, F., et al. (2013). Hypoxia inducible factor signaling modulates susceptibility to mycobacterial infection via a nitric oxide dependent mechanism. PLoS Pathog. 9:e1003789. doi: 10.1371/journal.ppat.1003789
Epelman, S., Lavine, K. J., Beaudin, A. E., Sojka, D., et al. (2014). Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40, 91–104. doi: 10.1016/j.immuni.2013.11.019
Eruslanov, E. B., Lyadova, I. V., Kondratieva, T. K., Majorov, K., et al. (2005). Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect. Immun. 73, 1744–1753. doi: 10.1128/IAI.73.3.1744-1753.2005
Escoll, P., and Buchrieser, C. (2018). Metabolic reprogramming of host cells upon bacterial infection: why shift to a Warburg-like metabolism? FEBS J. 285, 2146–2160. doi: 10.1111/febs.14446
Eum, S. Y., Kong, J. H., Hong, M. S., Lee, Y., et al. (2010). Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest 137, 122–128. doi: 10.1378/chest.09-0903
Everts, B., Amiel, E. G. J., van der Windt, Freitas, T. C., Chott, R., et al. (2012). Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431. doi: 10.1182/blood-2012-03-419747
Fortune, S. M., Solache, A., Jaeger, A., Hill, P. J., Belisle, J., et al. (2004). Mycobacterium tuberculosis inhibits macrophage responses to IFN- γ through myeloid differentiation factor 88-dependent and -independent mechanisms. J. Immunol. 172, 6272–6280. doi: 10.4049/jimmunol.172.10.6272
Fu, L. M. (2003). The potential of human neutrophil peptides in tuberculosis therapy. Int. J. Tuberc. Lung Dis. 7, 1027–1032.
Gallegos, A. M., Pamer, E. G., and Glickman, M. S. (2008). Delayed protection by ESAT-6-specific effector CD4+ T cells after airborne M. tuberculosis infection. J. Exp. Med. 205, 2359–2368. doi: 10.1084/jem.20080353
Garg, S. K., Volpe, E., Palmieri, G., Mattei, M., Galati, D., Martino, A., et al. (2004). Sphingosine 1-phosphate induces antimicrobial activity both in vitro and in vivo. J. Infect. Dis. 189, 2129–2138. doi: 10.1086/386286
Gault, C. R., Obeid, L. M., and Hannun, Y. A. (2010). An overview of sphingolipid metabolism: from synthesis to breakdown. Adv. Exp. Med. Biol. 688, 1–23. doi: 10.1007/978-1-4419-6741-1_1
Gautam, U. S., Foreman, T. W., Bucsan, A. N., Veatch, A., et al. (2018). In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 115, E62–E71. doi: 10.1073/pnas.1711373114
Gerriets, V. A., Kishton, R. J., Nichols, A. G., Macintyre, A., et al. (2015). Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Invest. 125, 194–207. doi: 10.1172/JCI76012
Gibbings, S. L., Goyal, R., Desch, A. N., Leach, S. M., et al. (2015). Transcriptome analysis highlights the conserved difference between embryonic and postnatal-derived alveolar macrophages. Blood 126, 1357–1366. doi: 10.1182/blood-2015-01-624809
Gideon, H. P., and Flynn, J. L. (2011). Latent tuberculosis: what the host sees? Immunol. Res. 50, 202–212. doi: 10.1007/s12026-011-8229-7
Gingras, A. C., Gygi, S. P., Raught, B., Polakiewicz, R. D., et al. (1999). Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev. 13, 1422–1437. doi: 10.1101/gad.13.11.1422
Gleeson, L. E., Sheedy, F. J. E. M., Palsson-McDermott, Triglia, D., O'Leary, S., et al. (2016). Cutting edge: Mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J. Immunol. 196, 2444–2449. doi: 10.4049/jimmunol.1501612
Gogolak, P., Rethi, B., Szatmari, I., Lanyi, A., Dezso, B., Nagy, L., et al. (2007). Differentiation of CD1a- and CD1a+ monocyte-derived dendritic cells is biased by lipid environment and PPAR γ. Blood 109, 643–652. doi: 10.1182/blood-2006-04-016840
Gordon, S. (2003). Alternative activation of macrophages. Nat. Rev. Immunol. 3, 23–35. doi: 10.1038/nri978
Gordon, S., and Taylor, P. R. (2005). Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964. doi: 10.1038/nri1733
Guerrini, V., Prideaux, B., Blanc, L., Bruiners, N., Arrigucci, R., Singh, S., et al. (2018). Storage lipid studies in tuberculosis reveal that foam cell biogenesis is disease-specific. PLoS Pathog. 14:e1007223. doi: 10.1371/journal.ppat.1007223
Guirado, E., Schlesinger, L. S., and Kaplan, G. (2013). Macrophages in tuberculosis: friend or foe. Semin. Immunopathol. 35, 563–583. doi: 10.1007/s00281-013-0388-2
Gutierrez, M. G., Master, S. S., Singh, S. B., Taylor, G., et al. (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766. doi: 10.1016/j.cell.2004.11.038
Hedlund, S., Persson, A., Vujic, A., Che, K. F., Stendahl, O., et al. (2010). Dendritic cell activation by sensing Mycobacterium tuberculosis-induced apoptotic neutrophils via DC-SIGN. Hum. Immunol. 71, 535–540. doi: 10.1016/j.humimm.2010.02.022
Herbst, S., Schaible, U. E., and Schneider, B. E. (2011). Interferon γ activated macrophages kill mycobacteria by nitric oxide induced apoptosis. PLoS ONE 6:e19105. doi: 10.1371/journal.pone.0019105
Hermsdorff, H. H., Mansego, M. L., Campion, J., Milagro, F. I., et al. (2013). TNF-α promoter methylation in peripheral white blood cells: relationship with circulating TNFα, truncal fat and n-6 PUFA intake in young women. Cytokine 64, 265–271. doi: 10.1016/j.cyto.2013.05.028
Hinchey, J., Lee, S., Jeon, B. Y., Basaraba, R. J., et al. (2007). Enhanced priming of adaptive immunity by a proapoptotic mutant of Mycobacterium tuberculosis. J. Clin. Invest. 117, 2279–2288. doi: 10.1172/JCI31947
Holz, M. K., Ballif, B. A., Gygi, S. P., and Blenis, J. (2005). mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123, 569–580. doi: 10.1016/j.cell.2005.10.024
Huang, L., Nazarova, E. V., Tan, S., Liu, Y., Russell, D., et al. (2018). Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med. 215, 1135–1152. doi: 10.1084/jem.20172020
Huang, L. E., Gu, J., Schau, M., and Bunn, H. F. (1998). Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. U.S.A. 95, 7987–7992. doi: 10.1073/pnas.95.14.7987
Humphreys, I. R., Stewart, G. R., Turner, D. J., Patel, J., et al. (2006). A role for dendritic cells in the dissemination of mycobacterial infection. Microbes Infect. 8, 1339–1346. doi: 10.1016/j.micinf.2005.12.023
Ibrahim, J., Nguyen, A. H., Rehman, A., Ochi, A., Jamal, M., et al. (2012). Dendritic cell populations with different concentrations of lipid regulate tolerance and immunity in mouse and human liver. Gastroenterology 143, 1061–1072. doi: 10.1053/j.gastro.2012.06.003
Iglesias, M. J., Reilly, S. J., Emanuelsson, O., Sennblad, B., Pirmoradian Najafabadi, M., et al. (2012). Combined chromatin and expression analysis reveals specific regulatory mechanisms within cytokine genes in the macrophage early immune response. PLoS ONE 7:e32306. doi: 10.1371/journal.pone.0032306
Infantino, V., Convertini, P., Cucci, L., Panaro, M. A., Di Noia, M., et al. (2011). The mitochondrial citrate carrier: a new player in inflammation. Biochem. J. 438, 433–436. doi: 10.1042/BJ20111275
Ishikawa, F., Niiro, H., Iino, T., Yoshida, S., Saito, N., Onohara, S., et al. (2007). The developmental program of human dendritic cells is operated independently of conventional myeloid and lymphoid pathways. Blood 110, 3591–3660. doi: 10.1182/blood-2007-02-071613
Jamaati, H., Mortaz, E., Pajouhi, Z., Folkerts, G., Movassaghi, M., Moloudizargari, M., et al. (2017). Nitric oxide in the pathogenesis and treatment of tuberculosis. Front. Microbiol. 8:2008. doi: 10.3389/fmicb.2017.02008
Jantsch, J., Chakravortty, D., Turza, N., Prechtel, A. T., Buchholz, B., et al. (2008). Hypoxia and hypoxia-inducible factor-1α modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 180, 4697–4705. doi: 10.4049/jimmunol.180.7.4697
Jellusova, J., and Rickert, R. C. (2017). A Brake for B Cell Proliferation: appropriate responses to metabolic stress are crucial to maintain B cell viability and prevent malignant outgrowth. Bioessays 39. doi: 10.1002/bies.201700079
Jha, A. K., Huang, S. C., Sergushichev, A., Lampropoulou, V., Ivanova, Y., et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. doi: 10.1016/j.immuni.2015.02.005
Kaplan, G., and Gaudernack, G. (1982). In vitro differentiation of human monocytes. Differences in monocyte phenotypes induced by cultivation on glass or on collagen. J. Exp. Med. 156, 1101–1114. doi: 10.1084/jem.156.4.1101
Kapsenberg, M. L. (2003). Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 3, 984–993. doi: 10.1038/nri1246
Kauppinen, A., Suuronen, T., Ojala, J., Kaarniranta, K., and Salminen, A. (2013). Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell. Signal. 25, 1939–1948. doi: 10.1016/j.cellsig.2013.06.007
Keane, J., Gershon, S., Wise, R. P., Mirabile-Levens, E., Kasznica, J., et al. (2001). Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N. Engl. J. Med. 345, 1098–1104. doi: 10.1056/NEJMoa011110
Kelly, B., and O'Neill, L. A. (2015). Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 25, 771–784. doi: 10.1038/cr.2015.68
Kersten, S. (2014). Integrated physiology and systems biology of PPARα. Mol. Metab. 3, 354–371. doi: 10.1016/j.molmet.2014.02.002
Khader, S. A., Partida-Sanchez, S., Bell, G., Jelley-Gibbs, D. M., Swain, S., et al. (2006). Interleukin 12p40 is required for dendritic cell migration and T cell priming after Mycobacterium tuberculosis infection. J. Exp. Med. 203, 1805–1815. doi: 10.1084/jem.20052545
Killick, K. E., Ni Cheallaigh, C., O'Farrelly, C., Hokamp, K., MacHugh, D. E., et al. (2013). Receptor-mediated recognition of mycobacterial pathogens. Cell. Microbiol. 15, 1484–1495. doi: 10.1111/cmi.12161
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132–141. doi: 10.1038/ncb2152
Kim, M. J., Wainwright, H. C., Locketz, M., Bekker, L. G., et al. (2010). Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol. Med. 2, 258–274. doi: 10.1002/emmm.201000079
Kim, Y. S., Lee, H. M., Kim, J. K., Yang, C., et al. (2017). PPAR-α Activation mediates innate host defense through induction of TFEB and lipid catabolism. J. Immunol. 198, 3283–3295. doi: 10.4049/jimmunol.1601920
Kleinnijenhuis, J., Oosting, M., Joosten, L. A., Netea, M. G., et al. (2011). Innate immune recognition of Mycobacterium tuberculosis. Clin. Dev. Immunol. 2011:405310. doi: 10.1155/2011/405310
Kolter, T., and Sandhoff, K. (2005). Principles of lysosomal membrane digestion: stimulation of sphingolipid degradation by sphingolipid activator proteins and anionic lysosomal lipids. Annu. Rev. Cell Dev. Biol. 21, 81–103. doi: 10.1146/annurev.cellbio.21.122303.120013
Kolter, T., and Sandhoff, K. (2006). Sphingolipid metabolism diseases. Biochim. Biophys. Acta 1758, 2057–2079. doi: 10.1016/j.bbamem.2006.05.027
Koo, M. S., Subbian, S., and Kaplan, G. (2012). Strain specific transcriptional response in Mycobacterium tuberculosis infected macrophages. Cell Commun. Signal. 10:2. doi: 10.1186/1478-811X-10-2
Kota, B. P., Huang, T. H., and Roufogalis, B. D. (2005). An overview on biological mechanisms of PPARs. Pharmacol. Res. 51, 85–94. doi: 10.1016/j.phrs.2004.07.012
Kratchmarov, R., Viragova, S., Kim, M. J., Rothman, N. J., et al. (2018). Metabolic control of cell fate bifurcations in a hematopoietic progenitor population. Immunol. Cell Biol. 96, 863–871. doi: 10.1111/imcb.12040
Krawczyk, C. M., Holowka, T., Sun, J., Blagih, J., Amiel, E., DeBerardinis, R., et al. (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. doi: 10.1182/blood-2009-10-249540
Krzywinska, E., and Stockmann, C. (2018). Hypoxia, metabolism and immune cell function. Biomedicines 6:56. doi: 10.3390/biomedicines6020056
Lachmandas, E., Beigier-Bompadre, M., Cheng, S. C., Kumar, V., van Laarhoven, A., et al. (2016a). Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur. J. Immunol. 46, 2574–2586. doi: 10.1002/eji.201546259
Lachmandas, E., Boutens, L., Ratter, J. M., Hijmans, A., Hooiveld, G., et al. (2016b). Microbial stimulation of different Toll-like receptor signalling pathways induces diverse metabolic programmes in human monocytes. Nat. Microbiol. 2:16246. doi: 10.1038/nmicrobiol.2016.246
Lagranderie, M., Abolhassani, M., Vanoirbeek, J. A., Lima, C., Balazuc, A., et al. (2010). Mycobacterium bovis bacillus Calmette-Guerin killed by extended freeze-drying targets plasmacytoid dendritic cells to regulate lung inflammation. J. Immunol. 184, 1062–1070. doi: 10.4049/jimmunol.0901822
Lai, R., Jeyanathan, M., Afkhami, S., Zganiacz, A., Hammill, J. A., et al. (2018). CD11b(+) Dendritic cell-mediated anti-Mycobacterium tuberculosis Th1 activation is counterregulated by CD103(+) dendritic cells via IL-10. J. Immunol. 200, 1746–1760. doi: 10.4049/jimmunol.1701109
Laker, R. C., and Ryall, J. G. (2016). DNA methylation in skeletal muscle stem cell specification, proliferation, and differentiation. Stem Cells Int. 2016:5725927. doi: 10.1155/2016/5725927
Laplante, M., and Sabatini, D. M. (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. doi: 10.1016/j.cell.2012.03.017
Le Naour, F., Hohenkirk, L., Grolleau, A., Misek, D. E., Lescure, P., et al. (2001). Profiling changes in gene expression during differentiation and maturation of monocyte-derived dendritic cells using both oligonucleotide microarrays and proteomics. J. Biol. Chem. 276, 17920–17931. doi: 10.1074/jbc.M100156200
Lee, J., and Kornfeld, H. (2010). Interferon-γ regulates the death of M. tuberculosis-infected macrophages. J Cell Death 3: 1–11. doi: 10.4137/JCD.S2822
Lee, W., VanderVen, B. C., Fahey, R. J., Russell, D., et al. (2013). Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. J. Biol. Chem. 288, 6788–6800. doi: 10.1074/jbc.M112.445056
Lewis, N. D., Asim, M., Barry, D. P., de Sablet, T., Singh, K., et al. (2011). Immune evasion by Helicobacter pylori is mediated by induction of macrophage arginase II. J. Immunol. 186, 3632–3641. doi: 10.4049/jimmunol.1003431
Linke, M., Fritsch, S. D., Sukhbaatar, N., Hengstschlager, M., and Weichhart, T. (2017). mTORC1 and mTORC2 as regulators of cell metabolism in immunity. FEBS Lett. 591, 3089–3103. doi: 10.1002/1873-3468.12711
Liu, C. H., Liu, H., and Ge, B. (2017). Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell. Mol. Immunol. 14, 963–975. doi: 10.1038/cmi.2017.88
Liu, G., Burns, S., Huang, G., Boyd, K., Proia, R. L., et al. (2009). The receptor S1P1 overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat. Immunol. 10, 769–777. doi: 10.1038/ni.1743
Liu, G., Yang, K., Burns, S., Shrestha, S., and Chi, H. (2010). The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nat. Immunol. 11, 1047–1056. doi: 10.1038/ni.1939
Liu, T. F., Vachharajani, V. T., Yoza, B. K., McCall, C., et al. (2012). NAD+-dependent sirtuin 1 and 6 proteins coordinate a switch from glucose to fatty acid oxidation during the acute inflammatory response. J. Biol. Chem. 287, 25758–25769. doi: 10.1074/jbc.M112.362343
Lowe, D. M., Redford, P. S., Wilkinson, R. J., O'Garra, A., et al. (2012). Neutrophils in tuberculosis: friend or foe? Trends Immunol. 33, 14–25. doi: 10.1016/j.it.2011.10.003
Lunt, S. Y., and Vander Heiden, M. G. (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464. doi: 10.1146/annurev-cellbio-092910-154237
Lyadova, I. V. (2017). Neutrophils in tuberculosis: heterogeneity shapes the Way? Mediators Inflamm. 2017:8619307. doi: 10.1155/2017/8619307
MacMicking, J. D., North, R. J., LaCourse, R., Mudgett, J. S., et al. (1997). Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA. 94, 5243–5248. doi: 10.1073/pnas.94.10.5243
Madan-Lala, R., Sia, J. K., King, R., Adekambi, T., Monin, L., et al. (2014). Mycobacterium tuberculosis impairs dendritic cell functions through the serine hydrolase Hip1. J. Immunol. 192, 4263–4272. doi: 10.4049/jimmunol.1303185
Mahajan, S., Dkhar, H. K., Chandra, V., Dave, S., Nanduri, R., et al. (2012). Mycobacterium tuberculosis modulates macrophage lipid-sensing nuclear receptors PPARγ and TR4 for survival. J. Immunol. 188, 5593–5603. doi: 10.4049/jimmunol.1103038
Mak, T. W., Grusdat, M., Duncan, G. S., Dostert, C., Nonnenmacher, Y., et al. (2017). Glutathione primes T cell metabolism for inflammation. Immunity 46, 675–689. doi: 10.1016/j.immuni.2017.03.019
Malik, Z. A., Thompson, C. R., Hashimi, S., Porter, B., Iyer, S., et al. (2003). Cutting edge: Mycobacterium tuberculosis blocks Ca2+ signaling and phagosome maturation in human macrophages via specific inhibition of sphingosine kinase. J. Immunol. 170, 2811–2815. doi: 10.4049/jimmunol.170.6.2811
Mandard, S., Muller, M., and Kersten, S. (2004). Peroxisome proliferator-activated receptor alpha target genes. Cell. Mol. Life Sci. 61, 393–416. doi: 10.1007/s00018-003-3216-3
Mantovani, A., Cassatella, M. A., Costantini, C., and Jaillon, S. (2011). Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 11, 519–531. doi: 10.1038/nri3024
Marakalala, M. J., and Ndlovu, H. (2017). Signaling C-type lectin receptors in antimycobacterial immunity. PLoS Pathog. 13:e1006333. doi: 10.1371/journal.ppat.1006333
Martin, S., and Parton, R. G. (2006). Lipid droplets: a unified view of a dynamic organelle. Nat. Rev. Mol. Cell Biol. 7, 373–378. doi: 10.1038/nrm1912
Mathis, D., and Shoelson, S. E. (2011). Immunometabolism: an emerging frontier. Nat. Rev. Immunol. 11:81. doi: 10.1038/nri2922
Matta, S. K., and Kumar, D. (2016). Hypoxia and classical activation limits Mycobacterium tuberculosis survival by Akt-dependent glycolytic shift in macrophages. Cell Death Discov. 2:16022. doi: 10.1038/cddiscovery.2016.22
Mattila, J. T., Ojo, O. O., Kepka-Lenhart, D., Marino, S., Kim, J., et al. (2013). Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. J. Immunol. 191, 773–784. doi: 10.4049/jimmunol.1300113
Mbongue, J. C., Nicholas, D. A., Torrez, T. W., Kim, N., et al. (2015). The role of indoleamine 2, 3-dioxygenase in immune suppression and autoimmunity. Vaccines (Basel) 3, 703–729. doi: 10.3390/vaccines3030703
McClean, C. M., and Tobin, D. M. (2016). Macrophage form, function, and phenotype in mycobacterial infection: lessons from tuberculosis and other diseases. Pathog. Dis. 74:ftw068. doi: 10.1093/femspd/ftw068
Mehra, S., Alvarez, X., Didier, P. J., Doyle, L. A., et al. (2013). Granuloma correlates of protection against tuberculosis and mechanisms of immune modulation by Mycobacterium tuberculosis. J. Infect. Dis. 207, 1115–1127. doi: 10.1093/infdis/jis778
Mellman, I., and Steinman, R. M. (2001). Dendritic cells: specialized and regulated antigen processing machines. Cell 106, 255–258. doi: 10.1016/S0092-8674(01)00449-4
Melo, R. C., and Dvorak, A. M. (2012). Lipid body-phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS Pathog. 8:e1002729. doi: 10.1371/journal.ppat.1002729
Meyer, N., and Penn, L. Z. (2008). Reflecting on 25 years with MYC. Nat. Rev. Cancer 8, 976–990. doi: 10.1038/nrc2231
Miyamoto, S., Murphy, A. N., and Brown, J. H. (2008). Akt mediates mitochondrial protection in cardiomyocytes through phosphorylation of mitochondrial hexokinase-II. Cell Death Differ. 15, 521–529. doi: 10.1038/sj.cdd.4402285
Mori, M. (2007). Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. J Nutr. 137, 1616S–1620S. doi: 10.1093/jn/137.6.1616S
Morris, D., Gonzalez, B., Khurasany, M., Kassissa, C., Luong, J., Kasko, S., et al. (2013a). Characterization of dendritic cell and regulatory T cell functions against Mycobacterium tuberculosis infection. Biomed Res. Int. 2013:402827. doi: 10.1155/2013/402827
Morris, D., Nguyen, T., Kim, J., Kassissa, C., Khurasany, M., Luong, J., et al. (2013b). An elucidation of neutrophil functions against Mycobacterium tuberculosis infection. Clin. Dev. Immunol. 2013:959650. doi: 10.1155/2013/959650
Mortaz, E., Adcock, I. M., Tabarsi, P., Masjedi, M. R., et al. (2015). Interaction of pattern recognition receptors with Mycobacterium Tuberculosis. J. Clin. Immunol. 35, 1–10. doi: 10.1007/s10875-014-0103-7
Mosser, D. M. (2003). The many faces of macrophage activation. J. Leukoc. Biol. 73, 209–212. doi: 10.1189/jlb.0602325
Murphy, D. J. (2001). The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog. Lipid Res. 40, 325–438. doi: 10.1016/S0163-7827(01)00013-3
Nagao, A., Kobayashi, M., Koyasu, S., Chow, C. C. T., et al. (2019). HIF-1-dependent reprogramming of glucose metabolic pathway of cancer cells and its therapeutic significance. Int. J. Mol. Sci. 20:238. doi: 10.3390/ijms20020238
Nazarova, E. V., Montague, C. R., La, T., Wilburn, K. M., et al. (2017). Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. Elife 6:e26969. doi: 10.7554/eLife.26969
Nencioni, A., Grunebach, F., Zobywlaski, A., Denzlinger, C., Brugger, W., and Brossart, P. (2002). Dendritic cell immunogenicity is regulated by peroxisome proliferator-activated receptor gamma. J. Immunol. 169, 1228–1235. doi: 10.4049/jimmunol.169.3.1228
Newsholme, P., Curi, R., Gordon, S., and Newsholme, E. A. (1986). Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem. J. 239, 121–125. doi: 10.1042/bj2390121
Nizet, V., and Johnson, R. S. (2009). Interdependence of hypoxic and innate immune responses. Nat. Rev. Immunol. 9, 609–617. doi: 10.1038/nri2607
North, R. J., and Jung, Y. J. (2004). Immunity to tuberculosis. Annu. Rev. Immunol. 22:599–623. doi: 10.1146/annurev.immunol.22.012703.104635
Obach, M., Navarro-Sabate, A., Caro, J., Kong, X., Duran, J., Gomez, M., et al. (2004). 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. J. Biol. Chem. 279, 53562–53570. doi: 10.1074/jbc.M406096200
O'Brien, A., Loftus, R. M., Pisarska, M. M., Tobin, L., et al. (2019). Obesity reduces mTORC1 activity in mucosal-associated invariant T cells, driving defective metabolic and functional responses. J. Immunol. 202, 3404–3411. doi: 10.4049/jimmunol.1801600
Ohanian, J., and Ohanian, V. (2001). Sphingolipids in mammalian cell signalling. Cell. Mol. Life Sci. 58, 2053–2068. doi: 10.1007/PL00000836
Olakanmi, O., Schlesinger, L. S., Ahmed, A., and Britigan, B. E. (2002). Intraphagosomal Mycobacterium tuberculosis acquires iron from both extracellular transferrin and intracellular iron pools. Impact of interferon-γ and hemochromatosis. J. Biol. Chem. 277, 49727–49734. doi: 10.1074/jbc.M209768200
O'Neill, L. A., and Pearce, E. J. (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213, 15–23. doi: 10.1084/jem.20151570
Ong, C. W. M., Fox, K., Ettorre, A., Elkington, P. T., Friedland, J., et al. (2018). Hypoxia increases neutrophil-driven matrix destruction after exposure to Mycobacterium tuberculosis. Sci. Rep. 8:11475. doi: 10.1038/s41598-018-29659-1
Ordway, D., Henao-Tamayo, M., Orme, I. M., and Gonzalez-Juarrero, M. (2005). Foamy macrophages within lung granulomas of mice infected with Mycobacterium tuberculosis express molecules characteristic of dendritic cells and antiapoptotic markers of the TNF receptor-associated factor family. J. Immunol. 175, 3873–3881. doi: 10.4049/jimmunol.175.6.3873
Pacis, A., Mailhot-Leonard, F., Tailleux, L., Randolph, H. E., Yotova, V., et al. (2019). Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 116, 6938–6943. doi: 10.1073/pnas.1814700116
Pathak, S. K., Basu, S., Basu, K. K., Banerjee, A., Pathak, S., et al. (2007). Direct extracellular interaction between the early secreted antigen ESAT-6 of Mycobacterium tuberculosis and TLR2 inhibits TLR signaling in macrophages. Nat. Immunol. 8, 610–618. doi: 10.1038/ni1468
Patsoukis, N., Weaver, J. D., Strauss, L., Herbel, C., Seth, P., et al. (2017). Immunometabolic regulations mediated by coinhibitory receptors and their impact on T cell immune responses. Front. Immunol. 8:330. doi: 10.3389/fimmu.2017.00330
Pattenden, S. G., Klose, R., Karaskov, E., and Bremner, R. (2002). Interferon-γ -induced chromatin remodeling at the CIITA locus is BRG1 dependent. EMBO J. 21, 1978–1986. doi: 10.1093/emboj/21.8.1978
Pearce, E. J., and Everts, B. (2015). Dendritic cell metabolism. Nat. Rev. Immunol. 15, 18–29. doi: 10.1038/nri3771
Petrofsky, M., and Bermudez, L. E. (1999). Neutrophils from Mycobacterium avium-infected mice produce TNF-α, IL-12, and IL-1β and have a putative role in early host response. Clin. Immunol. 91, 354–358. doi: 10.1006/clim.1999.4709
Podinovskaia, M., Lee, W., Caldwell, S., and Russell, D. G. (2013). Infection of macrophages with Mycobacterium tuberculosis induces global modifications to phagosomal function. Cell. Microbiol. 15, 843–859. doi: 10.1111/cmi.12092
Porstmann, T., Santos, C. R., Griffiths, B., Cully, M., Wu, M., et al. (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 8, 224–236. doi: 10.1016/j.cmet.2008.07.007
Prusinkiewicz, M. A., and Mymryk, J. S. (2019). Metabolic reprogramming of the host cell by human adenovirus infection. Viruses 11:E141. doi: 10.3390/v11020141
Qualls, J. E., and Murray, P. J. (2016). Immunometabolism within the tuberculosis granuloma: amino acids, hypoxia, and cellular respiration. Semin. Immunopathol. 38, 139–152. doi: 10.1007/s00281-015-0534-0
Queval, C. J., Brosch, R., and Simeone, R. (2017). The macrophage: a disputed fortress in the battle against Mycobacterium tuberculosis. Front. Microbiol. 8:2284. doi: 10.3389/fmicb.2017.02284
Raghuraman, S., Donkin, I., Versteyhe, S., Barres, R., and Simar, D. (2016). The emerging role of epigenetics in inflammation and immunometabolism. Trends Endocrinol. Metab. 27, 782–795. doi: 10.1016/j.tem.2016.06.008
Rajaram, M. V., Brooks, M. N., Morris, J. D., Torrelles, J., et al. (2010). Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor gamma linking mannose receptor recognition to regulation of immune responses. J. Immunol. 185, 929–942. doi: 10.4049/jimmunol.1000866
Rakhshandehroo, M., Knoch, B., Muller, M., and Kersten, S. (2010). Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010:612089. doi: 10.1155/2010/612089
Ramakrishnan, L. (2012). Revisiting the role of the granuloma in tuberculosis. Nat. Rev. Immunol. 12, 352–366. doi: 10.1038/nri3211
Reed, M. B., Domenech, P., Manca, C., Su, H., Barczak, A. K., et al. (2004). A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature 431, 84–87. doi: 10.1038/nature02837
Rehman, A., Hemmert, K. C., Ochi, A., Jamal, M., Henning, J., et al. (2013). Role of fatty-acid synthesis in dendritic cell generation and function. J. Immunol. 190, 4640–4649. doi: 10.4049/jimmunol.1202312
Reiley, W. W., Calayag, M. D., Wittmer, S. T., Huntington, J., et al. (2008). ESAT-6-specific CD4 T cell responses to aerosol Mycobacterium tuberculosis infection are initiated in the mediastinal lymph nodes. Proc. Natl. Acad. Sci. U.S.A. 105, 10961–10966. doi: 10.1073/pnas.0801496105
Riddle, S. R., Ahmad, A., Ahmad, S., Deeb, S. S., Malkki, M., et al. (2000). Hypoxia induces hexokinase II gene expression in human lung cell line A549. Am. J. Physiol. Lung Cell. Mol. Physiol. 278, L407–416. doi: 10.1152/ajplung.2000.278.2.L407
Riquelme, P., Tomiuk, S., Kammler, A., Fandrich, F., Schlitt, H. J., et al. (2013). IFN-γ-induced iNOS expression in mouse regulatory macrophages prolongs allograft survival in fully immunocompetent recipients. Mol. Ther. 21, 409–422. doi: 10.1038/mt.2012.168
Rius, J., Guma, M., Schachtrup, C., Akassoglou, K., Zinkernagel, A. S., et al. (2008). NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 453, 807–811. doi: 10.1038/nature06905
Robitaille, A. M., Christen, S., Shimobayashi, M., Cornu, M., Fava, L. L., et al. (2013). Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320–1323. doi: 10.1126/science.1228771
Russell, D. G. (2011). Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunol. Rev. 240, 252–268. doi: 10.1111/j.1600-065X.2010.00984.x
Russell, D. G., Cardona, P. J., Kim, M. J., Allain, S., et al. (2009). Foamy macrophages and the progression of the human tuberculosis granuloma. Nat. Immunol. 10, 943–948. doi: 10.1038/ni.1781
Saeed, S., Quintin, J., Kerstens, H. H., Rao, N. A., et al. (2014). Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 345:1251086. doi: 10.1126/science.1251086
Saka, H. A., and Valdivia, R. (2012). Emerging roles for lipid droplets in immunity and host-pathogen interactions. Annu. Rev. Cell Dev. Biol. 28, 411–437. doi: 10.1146/annurev-cellbio-092910-153958
Salamon, H., Bruiners, N., Lakehal, K., Shi, L., Ravi, J., Yamaguchi, K., et al. (2014). Cutting edge: vitamin D regulates lipid metabolism in Mycobacterium tuberculosis infection. J. Immunol. 193, 30–34. doi: 10.4049/jimmunol.1400736
Sanchez, D., Rojas, M., Hernandez, I., Radzioch, D., Garcia, L. F., et al. (2010). Role of TLR2- and TLR4-mediated signaling in Mycobacterium tuberculosis-induced macrophage death. Cell. Immunol. 260, 128–136. doi: 10.1016/j.cellimm.2009.10.007
Sanchez, T., and Hla, T. (2004). Structural and functional characteristics of S1P receptors. J. Cell. Biochem. 92, 913–922. doi: 10.1002/jcb.20127
Sarbassov, D. D., Guertin, D. A., Ali, S. M., Sabatini, D., et al. (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. doi: 10.1126/science.1106148
Sauve, A. A., and Youn, D. Y. (2012). Sirtuins: NAD(+)-dependent deacetylase mechanism and regulation. Curr. Opin. Chem. Biol. 16, 535–543. doi: 10.1016/j.cbpa.2012.10.003
Sawai, H., and Hannun, Y. A. (1999). Ceramide and sphingomyelinases in the regulation of stress responses. Chem. Phys. Lipids 102, 141–147. doi: 10.1016/S0009-3084(99)00082-1
Saxton, R. A., and Sabatini, D. M. (2017). mTOR signaling in growth, metabolism, and disease. Cell 168, 960–976. doi: 10.1016/j.cell.2017.02.004
Schlesinger, L. S. (1996). Entry of Mycobacterium tuberculosis into mononuclear phagocytes. Curr. Top. Microbiol. Immunol. 215, 71–96. doi: 10.1007/978-3-642-80166-2_4
Schluger, N. W., and Rom, W. N. (1998). The host immune response to tuberculosis. Am. J. Respir. Crit. Care Med. 157, 679–691. doi: 10.1164/ajrccm.157.3.9708002
Seiler, P., Aichele, P., Bandermann, S., Hauser, A. E., Lu, B., et al. (2003). Early granuloma formation after aerosol Mycobacterium tuberculosis infection is regulated by neutrophils via CXCR3-signaling chemokines. Eur. J. Immunol. 33, 2676–2686. doi: 10.1002/eji.200323956
Semenza, G. L. (2000). HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 88, 1474–1480. doi: 10.1152/jappl.2000.88.4.1474
Semenza, G. L. (2001). HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell 107, 1–3. doi: 10.1016/S0092-8674(01)00518-9
Sharma, L., and Prakash, H. (2017). Sphingolipids are dual specific drug targets for the management of pulmonary infections: perspective. Front. Immunol. 8:378. doi: 10.3389/fimmu.2017.00378
Shi, L., Eugenin, E. A., and Subbian, S. (2016). Immunometabolism in tuberculosis. Front. Immunol. 7:150. doi: 10.3389/fimmu.2016.00150
Shi, L., Jiang, Q., Bushkin, Y., Subbian, S., and Tyagi, S. (2019). Biphasic dynamics of macrophage immunometabolism during Mycobacterium tuberculosis infection. MBio 10:e02550–18. doi: 10.1128/mBio.02550-18
Shi, L., Salamon, H., Eugenin, E. A., Pine, R., Cooper, A., et al. (2015). Infection with Mycobacterium tuberculosis induces the Warburg effect in mouse lungs. Sci. Rep. 5:18176. doi: 10.1038/srep18176
Shin, J. H., Yang, J. Y., Jeon, B. Y., Yoon, Y., et al. (2011). (1)H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J. Proteome Res. 10, 2238–2247. doi: 10.1021/pr101054m
Singh, P., and Subbian, S. (2018). Harnessing the mTOR pathway for tuberculosis treatment. Front. Microbiol. 9:70. doi: 10.3389/fmicb.2018.00070
Singh, R., and Cuervo, A. M. (2012). Lipophagy: connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012:282041. doi: 10.1155/2012/282041
Singh, V., Jamwal, S., Jain, R., Verma, P., Gokhale, R., Rao, K., et al. (2012). Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 12, 669–681. doi: 10.1016/j.chom.2012.09.012
Singh, V., Kaur, C., Chaudhary, V. K., Rao, K. V., et al. (2015). M. tuberculosis secretory protein ESAT-6 induces metabolic flux perturbations to drive foamy macrophage differentiation. Sci. Rep. 5:12906. doi: 10.1038/srep12906
Sociali, G., Magnone, M., Ravera, S., Damonte, P., Vigliarolo, T., Von Holtey, M., et al. (2017). Pharmacological Sirt6 inhibition improves glucose tolerance in a type 2 diabetes mouse model. FASEB J. 31, 3138–3149. doi: 10.1096/fj.201601294R
Somashekar, B. S., Amin, A. G., Rithner, C. D., Troudt, J., et al. (2011). Metabolic profiling of lung granuloma in Mycobacterium tuberculosis infected guinea pigs: ex vivo 1H magic angle spinning NMR studies. J. Proteome Res. 10, 4186–4195. doi: 10.1021/pr2003352
Speer, A., Sun, J., Danilchanka, O., Meikle, V., Rowland, J. L., et al. (2015). Surface hydrolysis of sphingomyelin by the outer membrane protein Rv0888 supports replication of Mycobacterium tuberculosis in macrophages. Mol. Microbiol. 97, 881–897. doi: 10.1111/mmi.13073
Stine, Z. E., Walton, Z. E., Altman, B. J., Hsieh, A., et al. (2015). MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039. doi: 10.1158/2159-8290.CD-15-0507
Sturgill-Koszycki, S., Schlesinger, P. H., Chakraborty, P., Haddix, P. L., et al. (1994). Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 263, 678–681. doi: 10.1126/science.8303277
Subbian, S., O'Brien, P., Kushner, N. L., Yang, G., Tsenova, L., et al. (2013). Molecular immunologic correlates of spontaneous latency in a rabbit model of pulmonary tuberculosis. Cell Commun. Signal. 11:16. doi: 10.1186/1478-811X-11-16
Subbian, S., Tsenova, L., Yang, G., O'Brien, P., Parsons, S., Peixoto, B., et al. (2011). Chronic pulmonary cavitary tuberculosis in rabbits: a failed host immune response. Open Biol. 1:110016. doi: 10.1098/rsob.110016
Sutherland, J. S., Jeffries, D. J., Donkor, S., Walther, B., Hill, P., et al. (2009). High granulocyte/lymphocyte ratio and paucity of NKT cells defines TB disease in a TB-endemic setting. Tuberculosis 89, 398–404. doi: 10.1016/j.tube.2009.07.004
Szatmari, I., Gogolak, P., Im, J. S., Dezso, B., Rajnavolgyi, E., et al. (2004). Activation of PPARγ specifies a dendritic cell subtype capable of enhanced induction of iNKT cell expansion. Immunity 21, 95–106. doi: 10.1016/j.immuni.2004.06.003
Szatmari, I., Torocsik, D., Agostini, M., Nagy, T., Gurnell, M., Barta, E., et al. (2007). PPARγ regulates the function of human dendritic cells primarily by altering lipid metabolism. Blood 110, 3271–3280. doi: 10.1182/blood-2007-06-096222
Takeuch, O., and Akira, S. (2011). Epigenetic control of macrophage polarization. Eur. J. Immunol. 41, 2490–2493. doi: 10.1002/eji.201141792
Tannahill, G. M., Curtis, A. M., Adamik, J. E. M., Palsson-McDermott, McGettrick, A., et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242. doi: 10.1038/nature11986
Tascon, R. E., Soares, C. S., Ragno, S., Stavropoulos, E., Hirst, E., et al. (2000). Mycobacterium tuberculosis-activated dendritic cells induce protective immunity in mice. Immunology 99, 473–480. doi: 10.1046/j.1365-2567.2000.00963.x
Theocharis, S., Margeli, A., Vielh, P., and Kouraklis, G. (2004). Peroxisome proliferator-activated receptor-gamma ligands as cell-cycle modulators. Cancer Treat. Rev. 30, 545–554. doi: 10.1016/j.ctrv.2004.04.004
Thwe, P. M., Pelgrom, L. R., Cooper, R., Beauchamp, S., Reisz, J., et al. (2017). Cell-intrinsic glycogen metabolism supports early glycolytic reprogramming required for dendritic cell immune responses. Cell Metab. 26, 558–567 e555. doi: 10.1016/j.cmet.2017.08.012
Tian, T., Woodworth, J., Skold, M., and Behar, S. M. (2005). In vivo depletion of CD11c+ cells delays the CD4+ T cell response to Mycobacterium tuberculosis and exacerbates the outcome of infection. J. Immunol. 175, 3268–3272. doi: 10.4049/jimmunol.175.5.3268
Ting, L. M., Kim, A. C., Cattamanchi, A., and Ernst, J. D. (1999). Mycobacterium tuberculosis inhibits IFN-γ transcriptional responses without inhibiting activation of STAT1. J. Immunol. 163, 3898–3906.
Vachharajani, V. T., Liu, T., Wang, X., Hoth, J. J., Yoza, B., et al. (2016). Sirtuins link inflammation and metabolism. J. Immunol. Res. 2016:8167273. doi: 10.1155/2016/8167273
van Crevel, R., Ottenhoff, T. H., and van der Meer, J. W. (2002). Innate immunity to Mycobacterium tuberculosis. Clin. Microbiol. Rev. 15, 294–309. doi: 10.1128/CMR.15.2.294-309.2002
van de Laar, L., Saelens, W., De Prijck, S., Martens, L., Scott, C. L., et al. (2016). Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity 44, 755–768. doi: 10.1016/j.immuni.2016.02.017
van der Windt, G. J., Everts, B., Chang, C. H., Curtis, J. D., et al. (2012). Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78. doi: 10.1016/j.immuni.2011.12.007
VanderVen, B. C., Fahey, R. J., Lee, W., Liu, Y., Abramovitch, R., et al. (2015). Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium's metabolism is constrained by the intracellular environment. PLoS Pathog. 11:e1004679. doi: 10.1371/journal.ppat.1004679
Volkman, H. E., Pozos, T. C., Zheng, J., Davis, J. M., et al. (2010). Tuberculous granuloma induction via interaction of a bacterial secreted protein with host epithelium. Science 327, 466–469. doi: 10.1126/science.1179663
Vynnycky, E., and Fine, P. E. (2000). Lifetime risks, incubation period, and serial interval of tuberculosis. Am. J. Epidemiol. 152, 247–263. doi: 10.1093/aje/152.3.247
Wahl, D. R., Byersdorfer, C. A., Ferrara, J. L., Opipari, A., et al. (2012). Distinct metabolic programs in activated T cells: opportunities for selective immunomodulation. Immunol. Rev. 249, 104–115. doi: 10.1111/j.1600-065X.2012.01148.x
Walmsley, S. R., Chilvers, E. R., Thompson, A. A., Vaughan, K., et al. (2011). Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J. Clin. Invest. 121, 1053–1063. doi: 10.1172/JCI43273
Wang, S., Liu, R., Yu, Q., Dong, L., Bi, Y., and Liu, G. (2019). Metabolic reprogramming of macrophages during infections and cancer. Cancer Lett. 452, 14–22. doi: 10.1016/j.canlet.2019.03.015
Wang, X., Zhu, H., Snieder, H., Su, S., Munn, D., Harshfield, G., et al. (2010). Obesity related methylation changes in DNA of peripheral blood leukocytes. BMC Med. 8:87. doi: 10.1186/1741-7015-8-87
Warburg, O. (1956). On the origin of cancer cells. Science 123, 309–314. doi: 10.1126/science.123.3191.309
Welin, A., Eklund, D., Stendahl, O., and Lerm, M. (2011). Human macrophages infected with a high burden of ESAT-6-expressing M. tuberculosis undergo caspase-1- and cathepsin B-independent necrosis. PLoS ONE 6:e20302. doi: 10.1371/journal.pone.0020302
Wolf, A. J., Desvignes, L., Linas, B., Banaiee, N., Tamura, T., Takatsu, K., et al. (2008). Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 205, 105–115. doi: 10.1084/jem.20071367
Wolf, A. J., Linas, B., Trevejo-Nunez, G. J., Kincaid, E., Tamura, T., et al. (2007). Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo. J. Immunol. 179, 2509–2519. doi: 10.4049/jimmunol.179.4.2509
Wong, K. W., and Jacobs, W. R. Jr (2011). Critical role for NLRP3 in necrotic death triggered by Mycobacterium tuberculosis. Cell. Microbiol. 13, 1371–1384. doi: 10.1111/j.1462-5822.2011.01625.x
Yang, Z., and Ming, X. F. (2014). Functions of arginase isoforms in macrophage inflammatory responses: impact on cardiovascular diseases and metabolic disorders. Front. Immunol. 5:533. doi: 10.3389/fimmu.2014.00533
Yaseen, I., Kaur, P., Nandicoori, V. K., and Khosla, S. (2015). Mycobacteria modulate host epigenetic machinery by Rv1988 methylation of a non-tail arginine of histone H3. Nat. Commun. 6:8922. doi: 10.1038/ncomms9922
Yecies, J. L., and Manning, B. D. (2011). Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res. 71, 2815–2820. doi: 10.1158/0008-5472.CAN-10-4158
Yim, H. C., Li, J. C., Pong, J. C., Lau, A., et al. (2011). A role for c-Myc in regulating anti-mycobacterial responses. Proc. Natl. Acad. Sci. U.S.A. 108, 17749–17754. doi: 10.1073/pnas.1104892108
Zaccagnino, P., Saltarella, M., Maiorano, S., Gaballo, A., Santoro, G., Nico, B., et al. (2012). An active mitochondrial biogenesis occurs during dendritic cell differentiation. Int. J. Biochem. Cell Biol. 44, 1962–1969. doi: 10.1016/j.biocel.2012.07.024
Zhai, W., Wu, F., Zhang, Y., Fu, Y., and Liu, Z. (2019). The immune escape mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 20:340. doi: 10.3390/ijms20020340
Zhang, Q., Zhao, K., Shen, Q., Han, Y., Gu, Y., Li, X., et al. (2015). Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 525, 389–393. doi: 10.1038/nature15252
Zhang, W., Petrovic, J. M., Callaghan, D., Jones, A., Cui, H., et al. (2006). Evidence that hypoxia-inducible factor-1 (HIF-1) mediates transcriptional activation of interleukin-1β (IL-1β) in astrocyte cultures. J. Neuroimmunol. 174:63–73. doi: 10.1016/j.jneuroim.2006.01.014
Keywords: tuberculosis, Mycobacteria, innate immunity, immunometabolism, immune cells, epigenetics, metabolic regulators, infection
Citation: Kumar R, Singh P, Kolloli A, Shi L, Bushkin Y, Tyagi S and Subbian S (2019) Immunometabolism of Phagocytes During Mycobacterium tuberculosis Infection. Front. Mol. Biosci. 6:105. doi: 10.3389/fmolb.2019.00105
Received: 31 July 2019; Accepted: 26 September 2019;
Published: 14 October 2019.
Edited by:
Hannes Findeisen, University Hospital Münster, GermanyReviewed by:
Rob J. W. Arts, Radboud University Nijmegen Medical Centre, NetherlandsPrasun K. Datta, Temple University, United States
Copyright © 2019 Kumar, Singh, Kolloli, Shi, Bushkin, Tyagi and Subbian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Selvakumar Subbian, c3ViYmlhc2VAbmptcy5ydXRnZXJzLmVkdQ==
†These authors have contributed equally to this work