Abstract
Interferon-induced protein with tetratricopeptide repeats (IFIT) genes are prominent interferon-stimulated genes (ISGs). The human IFIT gene family consists of four genes named IFIT1, IFIT2, IFIT3, and IFIT5. The expression of IFIT genes is very low in most cell types, whereas their expression is greatly enhanced by interferon treatment, viral infection, and pathogen-associated molecular patterns (PAMPs). The proteins encoded by IFIT genes have multiple tetratricopeptide repeat (TPR) motifs. IFIT proteins do not have any known enzymatic roles. However, they execute a variety of cellular functions by mediating protein-protein interactions and forming multiprotein complexes with cellular and viral proteins through their multiple TPR motifs. The versatile tertiary structure of TPR motifs in IFIT proteins enables them to be involved in distinct biological functions, including host innate immunity, antiviral immune response, virus-induced translation initiation, replication, double-stranded RNA signaling, and PAMP recognition. The current understanding of the IFIT proteins and their role in cellular signaling mechanisms is limited to the antiviral immune response and innate immunity. However, recent studies on IFIT protein functions and their involvement in various molecular signaling mechanisms have implicated them in cancer progression and metastasis. In this article, we focused on critical molecular, biological and oncogenic functions of human IFIT proteins by reviewing their prognostic significance in health and cancer. Research suggests that IFIT proteins could be novel therapeutic targets for cancer therapy.
Introduction
The interferon-induced protein with tetratricopeptide repeats (IFIT) gene family is well-studied interferon-stimulating genes (ISGs) for their antiviral properties (Fensterl and Sen, 2015). IFIT gene products are cytoplasmic proteins with no known enzymatic activity, but all of them share special structural motifs known as tetratricopeptide repeats (TPRs). The TPR motifs consist of helix-turn-helix structures that can make multiple protein complexes in the cells (Allan and Ratajczak, 2011). IFIT proteins are involved in a variety of biological processes, such as cell proliferation, migration, virus-induced translation initiation, replication and double-stranded RNA signaling (Fensterl and Sen, 2011). Most cell types without stimulation do not express IFIT genes at high levels, whereas the transcription of IFIT genes is rapidly induced by interferon (IFN) treatment and viral infection. The antiviral functions of human IFIT proteins have been extensively demonstrated in many research studies (Fensterl and Sen, 2011, 2015; Diamond and Farzan, 2013). Recent studies have implicated IFIT proteins as prognostic markers to determine the clinical outcome of many cancers, such as glioblastoma, hepatocellular carcinoma, breast cancer and pancreatic cancer (Danish et al., 2013; Zhang et al., 2016; Yang et al., 2017; Zhao et al., 2017). During the past several years, we explored the biological and clinical functions of IFIT proteins in oral squamous cell carcinoma (OSCC) (Blot et al., 1988; Lai et al., 2008, 2013; Pidugu et al., 2019b). In this review, we focus on the molecular and clinical significance of IFIT proteins in cancer and discuss their emerging oncogenic roles, with a special emphasis on the progression of human OSCC.
IFIT Expression in OSCC
Oral cancer is one of the most familiar malignant cancers worldwide, and it is estimated that 3.6% of cancer deaths were due to oral cancer in 2012 (Shield et al., 2017). It is also one of the fastest-increasing malignancies and the fourth leading cause of death in male cancer patients (Liu et al., 2010). OSCC accounts for more than 90% of oral cancers. Betel quid chewing, tobacco use and alcohol consumption are the major risk factors for OSCC (Su et al., 2007). In addition, human papillomavirus (HPV) infection has been reported as an aetiological agent of OSCC (Gillison et al., 2008; Husain and Neyaz, 2017). However, HPV-16-positive patients had better clinical outcomes than patients with HPV-negative tumors (Schwartz et al., 2001). Because of the synergistic tumor-promoting effect of cigarette smoking and alcohol consumption, people with heavy smoking and alcohol consumption habits are more prone to OSCC than those who only smoke or drink (Blot et al., 1988; Haddad and Shin, 2008; Jha, 2009). A high incidence of OSCC has been reported in Asian countries due to the cultural practice of chewing betel nuts (Su et al., 2007; Liu et al., 2010; Krishna Rao et al., 2013). Several studies have reported the relationship between betel nut chewing and increased mortality rate from OSCC (Jeng et al., 2001; Wen et al., 2010). OSCC is usually diagnosed in advanced stages due to ignorance of the patients or inaccurate diagnosis (Scott et al., 2006). OSCC patients with advanced-stage (III and IV) tumors are usually treated with extensive surgery combined with radiation and chemotherapies (Kirita et al., 2012; Noguti et al., 2012). The overall 5 year survival rate of OSCC patients usually varies from 40 to 50% (Markopoulos, 2012). Disappointingly, the 5 year survival rates are low in OSCC patients, and no significant improvement has been seen (Wang et al., 2013). The development of drug resistance by intrinsic molecular mechanisms causes treatment failure. Therefore, it is of fundamental importance to identify the tumor-intrinsic pathways involved in drug resistance and metastasis for the development of effective therapies for the treatment of OSCC patients.
During the course of our research, we found that IFIT protein levels were altered in OSCC patient tissues. However, how and why their expression was induced in OSCC is still unknown. That warrants further investigation. It has been reported that the increased incidence of OSCC in Asian countries is caused by chewing betel quid's (Krishna Rao et al., 2013). Many research studies have documented the association between OSCC progression and betel nut (BN) or betel quid (BQ) chewing (Jeng et al., 2001). The carcinogenic components of the areca nut cause malignant transformation of cells that leads to increased risk for the development of OSCC. Hence, chewing BNs/BQs is considered an independent risk factor for OSCC (Warnakulasuriya et al., 2002; Sharan et al., 2012). As shown in previous studies, areca nut extract treatment resulted in the differential expression of various cellular genes, including IFIT2, in human keratinocytes (Lai and Lee, 2006; Lai et al., 2008). Therefore, we speculated that BQ chewing may influence IFIT expression in tumor tissues of OSCC patients. We preliminary found that high IFIT1, IFIT3, and IFIT5 expression levels are significantly associated with betel quid chewing in patients. However, further prospective cohort studies are required to determine the influence of betel quid chewing on IFIT expression in patients with OSCC. The distinct expression of individual IFIT genes in the same cell or tissue is believed to result in non-redundant functions (Terenzi et al., 2007; Wacher et al., 2007). Hence, we hypothesized that individual IFIT proteins may exhibit unique functions depending on the cell type.
Gene Structure and Transcriptional Regulation of Human IFITs
The human IFIT gene family is composed of four genes: IFIT1, IFIT2, IFIT3, and IFIT5. They are clustered on human chromosome 10q23.31 (Table 1) (Varela et al., 2014). Besides, an untranscribed IFIT1 pseudogene, IFIT1P has been identified on human chromosome 13 (Wathelet et al., 1988). The IFIT genes have a simple gene architecture with 2 exons and a promoter. The two exons are separated by an intronic region that extends a few kilobases in length. The first exon is small and is next to interferon-stimulated response elements (ISREs), regulatory elements responsible for IFN treatment. The second exon encodes the protein-coding mRNA sequence (Fensterl and Sen, 2011, 2015; Liu et al., 2013) (Figure 1A). The presence of the ISREs in the promoter regions of IFIT genes could be the reason for their low basal expression and rapid IFN-mediated transcriptional induction (Sarkar and Sen, 2004; Pichlmair et al., 2011). INFs are broadly divided into two groups, type I and type II; type I contains IFN-α, IFN-β, IFN-δ, IFN-ε, IFN-κ, IFN-τ, and IFN-ω, and type II IFNs include IFN-γ (Pestka et al., 1987, 2004; Platanias, 2005). IFN-α, -β, or -γ treatment activates the transcription of a multitude of ISGs, including IFIT genes; it was roughly estimated that these number 500–1000 genes, depending upon cell or tissue type (Der et al., 1998; de Veer et al., 2001). It was noted that IFIT proteins are extensively induced by IFN-α compared to IFN-γ treatment (Der et al., 1998; de Veer et al., 2001). In addition to IFN stimuli, the expression of IFITs can also be triggered by several signaling pathways, such as those of Toll-like receptor 3 (TLR3), retinoic acid-inducible gene-I/melanoma differentiation-associated gene-5 (RIG-I/MDA-5) and pathogen-associated molecular patterns (PAMPs) (Fensterl and Sen, 2011; Diamond and Farzan, 2013) (Figure 1B).
Table 1
| Gene name | Entrez gene ID | HGNC ID | Ensemble ID | OMIM ID | Alternate names |
|---|---|---|---|---|---|
| IFIT1 | 3434 | 5407 | ENSG00000185745 | 147690 | IFI-56K, IFNAI1, IFIT-1, G10P1, IFI56, ISG56, P56, IFI56, RNM561, C56, Hup56 |
| IFIT2 | 3433 | 5409 | ENSG00000119922 | 147040 | ISG54K, IFI-54K, IFIT-2, G10P2, IFI54, P54, GARG-39, ISG-54K, IFI-54, CIG-42, Cig42 |
| IFIT3 | 3437 | 5411 | ENSG00000119917 | 604650 | IFI-60K, CIG-49, IFIT-3, IFIT-4, ISG-60, IFIT4, CIG49, IFI60, RIG-G, P60, GARG-49, Cig41, IRG2 |
| IFIT5 | 24138 | 13328 | ENSG00000152778 | 616135 | IFIT-5, ISG58, RI58, P58 |
Synopsis of all available gene database information and synonyms for Human IFIT genes.
Figure 1

Human IFIT gene family location, structure, and transcriptional regulation. (A) The genetic architecture of human IFITs. The human IFIT genes are located on human chromosome 10q23.31. The exons are depicted as black boxes, the introns as dark lines, and ISREs in the promoter regions as blue asterisks. The direction of the open reading frames (ORFs) is designated by white arrowheads. (B) Signaling pathways of IFIT gene transcription. The ISREs in the promoter regions of IFIT genes are induced by stimulation of cell surface receptors such as IFNAR-α/β, TLR-3, RIG-I/MDA-5, TLR-7, and TLR-9 by IFN-α/β, viral infection, and PAMPs. Transcriptional induction is mediated by various transcription factors, such as IRF-3, IRF-5, IRF-7, and IRF-9. The encoded proteins (IFIT1, IFIT2, IFIT3, and IFIT5) regulate OSCC progression. IFIT1/IFIT3 promote OSCC invasion and metastasis by EGFR activation. IFIT2 inhibits OSCC migration via CK-18. IFIT5 expression is correlated with HPV E6 protein in OSCC. However, its mechanism in OSCC is not yet understood. IFN-α/β, interferon alpha/beta; IFNAR-α/β, interferon alpha/beta receptors; STAT1/2, signal transducer and activator of transcription 1/2; TLRs, Toll-like receptors; RIG-I, retinoic acid-inducible gene-I; MDA-5, melanoma differentiation-associated gene-5; IRFs, interferon regulatory factors; ISRE, interferon-stimulated responsive elements; IFIT, interferon-induced proteins with tetratricopeptide repeats; EGFR, epidermal growth factor receptor, CK-18, cytokeratin-18; HPV E6, human papilloma virus early protein-6; OSCC, oral squamous cell carcinoma.
The transcription kinetics of individual IFIT genes vary depending on the stimulus, exposure time, and cell or tissue type. For instance, human IFIT1 expression is high at 6 h after IFN treatment and decreases rapidly within 12 to 24 h of treatment (Kusari and Sen, 1986). IFIT1 mRNA levels are high in HT1080 fibrosarcoma cells 24 h after treatment with IFN-β, whereas IFIT2 mRNA levels are substantially decreased (Terenzi et al., 2006). In HEK293 cells, the mRNA levels of both IFIT1 and IFIT2 remain high even after 24 h of treatment with IFN (Terenzi et al., 2006). However, anti-inflammatory therapies with glucocorticoids such as dexamethasone negatively regulate the transcription of IFIT genes due to the competitive binding of the glucocorticoid receptor with glucocorticoid receptor-interacting protein-1 (GRIP1), thereby inhibiting IRF-3 activation (Smith and Herschman, 1996; Reily et al., 2006). IFIT genes have been shown to undergo mutations. However, the mutation frequencies were relatively low compared to other cancer driver genes (Figure 2) (Cerami et al., 2012; Gao et al., 2013). In head and neck cancer, the mutation frequencies are lower than 1%. Since mutations in IFIT genes can alter the structure of encoded proteins and significantly affect protein stability and their interactions with other cellular proteins (Johnson et al., 2018), prior knowledge of mutations of IFIT genes may be crucial for investigating the cause of cancers.
Figure 2
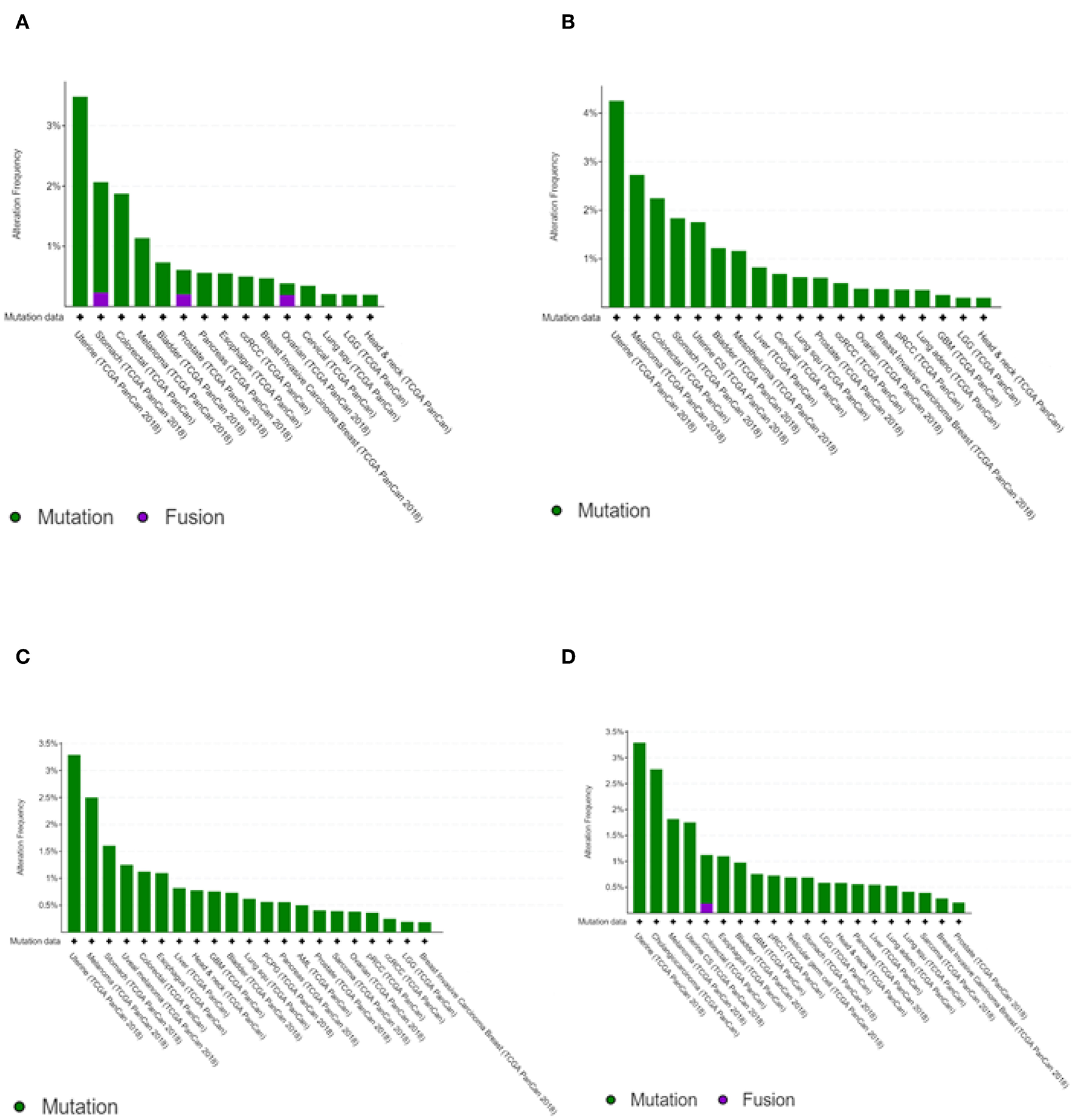
Cancer type summary of mutation frequencies in IFIT genes across the cancer studies. Pictures shows the histograms of alteration of mutation frequencies in IFIT1(A), IFIT2(B), IFIT3(C), and IFIT5(D) across the cancer studies. Data obtained from TCGA PanCancer Atlas studies. Picture courtesy of cBioPortal.
IFITs With Versatile TPR Motifs in Their Protein Structure
The molecular weights of the human IFIT proteins range from 54 to 60 kDa. Hence, IFIT1, IFIT2, IFIT3, and IFIT5 proteins are denoted as p56, p54, p60, and p58, respectively; the number represents the molecular weight of the proteins (Table 2). The unique characteristic feature of IFIT proteins is that all of them contain many TPR motifs distributed across the whole sequence. IFIT proteins have different numbers of TPR motifs in their structure. The TPR is a structural unit of IFIT proteins comprised of 3 to 16 degenerate tandem repeats of 34 amino acids that form helix-turn-helix arrangements enabling them to be involved in protein-protein interactions and are frequently found in scaffold proteins (Allan and Ratajczak, 2011). The antiparallel helices of multiple TPR motifs impart a unique folding nature to the IFIT proteins that allow for the binding of distinct cellular proteins (Blatch and Lassle, 1999; D'Andrea and Regan, 2003). The proposed consensus amino acid sequence conventionally found in TPR motifs is W4G8Y11G15Y17A20Y24A27P32 (D'Andrea and Regan, 2003). However, TPRs are degenerate in amino acid sequence, and the tandem repeats of each amino acid residue are variable and can be replaced by other classes of amino acids. Therefore, the prediction of TPR motifs in a protein is difficult. According to the UniProtKB database, the predicted number of TPR motifs in human IFIT1 (P09914), IFIT2 (P09913), IFIT3 (O14879), and IFIT5 (Q13325) proteins is 10, 9, 8, and 8, respectively (Figure 3A). Although all human IFIT proteins share TPR motifs in their structure, the amino acid sequence similarity among IFIT1, IFIT2, IFIT3, and IFIT5 is merely 25% (Figure 3B). Phylogenetic analysis of human IFIT proteins reveals evolutionary relationships between IFIT1 and IFIT5 and between IFIT2 and IFIT3 (Liu et al., 2013) (Figure 3C), whereas the sequence identity is 54% between IFIT1 and IFIT5 and 52% between IFIT2 and IFIT3. This implies that each IFIT protein may have unique biological functions, which is indeed witnessed in the cellular and molecular functions of different IFIT-family proteins. IFIT proteins such as IFIT1, IFIT2, and IFIT3 can form homo-oligomers in solutions (Pichlmair et al., 2011), except for IFIT5, which is solely monomeric. The crystal structure of human IFIT2 has shown that it makes homo- or heterodimers with other IFIT family members (Stawowczyk et al., 2011; Sen and Fensterl, 2012). Although the complete crystal structure of IFIT3 is not available, IFIT3 may be similar to IFIT2 in forming a homodimer because the N-terminal domain of IFIT3 has a 70% sequence similarity with IFIT2 (Yang et al., 2012). The first protein complex of IFIT1, IFIT2, and IFIT3 was found in the IFN-treated HeLa cell lysates (Stawowczyk et al., 2011). Subsequently, protein pulldown assays followed by mass spectrometry analysis have shown that IFIT proteins could associate with other members of the IFIT family except IFIT5 (Pichlmair et al., 2011; Habjan et al., 2013). In addition, we observed the protein complex of IFIT1, IFIT2, and IFIT3 in OSCC cells by co-immunoprecipitation assay (Pidugu et al., 2019b). These findings suggest that IFIT1, IFIT2, and IFIT3 can form protein complexes in cells. Recent research revealed that IFIT1, IFIT2, and IFIT3 proteins interact with each other via a conserved YxxxL motif in the C-terminus of each protein. In the functional complexity of these three proteins, IFIT3 acts as a central scaffold and regulates the functions of IFIT1 and IFIT2 (Kumar et al., 2014; Fleith et al., 2018; Mears and Sweeney, 2018). Recent protein crystallographic studies of IFIT1 and IFIT5 have also revealed that IFITs can interact with viral RNA containing 5' triphosphate group (PPP-RNA) (Abbas et al., 2013; Feng et al., 2013; Katibah et al., 2013). Furthermore, the crystal structure reveals that IFIT2 specifically binds with AU-rich RNAs (Sen and Fensterl, 2012; Yang et al., 2012).
Table 2
| Gene name | Protein name | UniprotKB ID | No. of amino acids | No. of predicted TPR motifs |
|---|---|---|---|---|
| IFIT1 | P56 | P09914 | 478 | 10 |
| IFIT2 | P54 | P09913 | 472 | 9 |
| IFIT3 | P60 | O14879 | 490 | 8 |
| IFIT5 | P58 | Q13325 | 482 | 8 |
Summary of Human IFIT gene encoded proteins.
Figure 3

The organization of tetratricopeptide repeat (TPR) motifs and phylogenetic analyses of human IFIT family proteins. (A) The schematic illustration shows the predicted number of TPR motifs in IFIT1 (P09914), IFIT2 (P09913), IFIT3 (O14879) and IFIT5 (Q13325) according to the UniProtKB database. The numbers are UniProtKB accession numbers. (B) Sequence alignments of human IFIT proteins with Clustal Omega. The multiple-sequence alignment shows that IFIT1, IFIT2, IFIT3, and IFIT5 share only 25% sequence similarity. (C) The cartoon depicts the evolutionary relationship among human IFIT proteins. The multiple-sequence alignment using Clustal Omega reveals that IFIT1 and IFIT5 have 54% sequence similarity, whereas IFIT2 and IFIT3 have 52%.
Interdependence Between IFIT1 and IFIT3
Protein sequence analysis revealed that IFIT1 and IFIT3 share only 37% sequence similarity. Intriguingly, the expression of IFIT1 was strongly correlated with IFIT3 in both OSCC cell lines and tissues derived from OSCC patients (Pidugu et al., 2019b). Ectopic expression of IFIT3 also induced endogenous IFIT1 expression in a dose-dependent manner (Johnson et al., 2018). These data indicate that IFIT1 and IFIT3 are interdependent. This was further supported by the studies where deletion or mutation in the C-terminal YxxxL motif of IFIT3 resulted in decreased IFIT1 expression (Fleith et al., 2018). This could be due to the enhanced IFIT1 protein stability or decreased degradation by IFIT3 in the complex. Alternatively, IFIT3 may promote IFIT1 activity by enhancing the IFIT1 concentration in the cell by averting its turnover by locking it in a stable complex (IFIT1: IFIT3) (Fleith et al., 2018; Johnson et al., 2018). Formation of the complex with IFIT3 also seems to increase the IFIT1 specificity, since IFIT3 binds with IFIT1 and facilitates the binding of IFIT1 with Cap0 RNAs, and hence functional studies demonstrated that IFIT3 is required for the stabilization of IFIT1 expression and its antiviral functions in the cell (Johnson et al., 2018). IFIT1 is phosphorylated at 24 h after Sendai virus infection in HEK293 cells, yet the phosphorylation site and its subsequent effect of phosphorylation remain elusive (Li et al., 2009). Since the tyrosine in the IFIT3 C-terminus YxxxL motif is crucial for the interaction between IFIT1 and IFIT3 (Fleith et al., 2018), it would be noteworthy to determine whether this tyrosine residue is prone to phosphorylation and how this impacts IFIT1: IFIT3 complex formation. Moreover, it was found that a small proportion of IFIT1 was ISGylated by human ISG15 protein in IFN-β-treated HeLa cells; however, the consequences of this modification remain unclear, and further studies are warranted to understand the mechanistic insights into IFIT1: IFIT3 complex formation (Zhao et al., 2005).
IFITs in Malignant Progression
Although IFITs are involved in the host immune response and antiviral defense, emerging research studies have shown their involvement in malignant progression (Lai et al., 2008, 2013; Niess et al., 2015; Zhang et al., 2016; Ohsugi et al., 2017; Yang et al., 2017; Zhao et al., 2017; Chen et al., 2018; Lo et al., 2018, 2019; Nushtaeva et al., 2018; Shen et al., 2018; Huang et al., 2019). The epithelial-mesenchymal transition (EMT) is a biological mechanism in which epithelial cells convert into a mesenchymal phenotype to acquire increased cell invasion and migration capacity as well as resistance to apoptosis (Kalluri and Weinberg, 2009), which lead to metastasis (Yilmaz and Christofori, 2009). Accumulated evidence has shown that EMT is linked to an increased risk of cancer invasion and decreased the survival rate of OSCC patients (Yilmaz and Christofori, 2009; Natarajan et al., 2014). In our previous studies, we found an inverse association of IFIT2 with the migration activity of OSCC cells (Lai et al., 2008) and showed reduced IFIT2 expression level was correlated with an increased rate of metastasis in OSCC patients (Lai et al., 2013). IFIT2 exhibited distinct cytoskeletal staining patterns in squamous cells and surrounding normal cells in OSCC patient tissues and interacted with cytokeratins such as CK8 and CK18. Knockdown of endogenous IFIT2 expression using IFIT2-specific siRNA enhanced OSCC cell migration rates. Strikingly, high IFIT2 expression in tumor tissues was correlated with better patient survival (Lai et al., 2008). The enhanced malignancy of IFIT2-depleted OSCC cells was attributed to increased expression and secretion of tumor necrosis factor-alpha (TNF-α), and hence blocking TNF-α abolished the angiogenic activity of IFIT2-silenced metastatic cells (Li et al., 2012). We also demonstrated that knockdown of IFIT2 induced EMT in OSCC by activating the atypical PKC signaling pathway (Lai et al., 2008, 2013).
We recently found that ectopic overexpression of IFIT1 and IFIT3 enhanced EGFR and AKT activation and subsequently promoted OSCC invasion through EMT (Pidugu et al., 2017). IFIT1 expression was strongly correlated with IFIT3 expression in OSCC cell lines and patient tissues. Additionally, the expression of both IFIT1 and IFIT3 was associated with phospho-EGFR in OSCC specimens. Clinicopathological and survival analyses showed that elevated IFIT1 and IFIT3 expression correlated with poor survival in OSCC patients. Furthermore, we demonstrated that IFIT1 and IFIT3 enhanced the EGFR endocytic recycling process by interacting with Annexin-2 (ANX2) (Pidugu et al., 2019b). However, IFIT3 has been shown to have antiproliferative activity by enhancing the expression of cell cycle negative regulators such as p27 and p21 in monocytic U937 cells (Xiao et al., 2006). IFIT3 increased p21 protein level by downregulating c-Myc, a repressor of p21 in the cell (Xiao et al., 2006). IFIT proteins execute multiple complex cellular functions based on cell type and tissue types. Therefore, the functions of IFIT proteins could be altered depending on the cell system. Hence, the controversial functions of IFIT3 warrant further investigation. IFIT5 has been implicated exclusively in an innate immune response. However, a newly identified mechanism of IFIT5 is regulation of the turnover of tumor suppressor microRNAs (miRNAs), including miR-363 and miR-128, resulting in increased expression of transcription factors of EMT such as slug and ZEB1, thereby enhancing invasion in renal cell carcinoma (RCC) (Lo et al., 2019). Moreover, the oncogenic role of IFIT5 has been identified in bladder cancer. IFIT5 promoted cell invasion and migration by inducing EMT by downregulating miR-99a in bladder cancer (Huang et al., 2019). IFIT5 expression was also inversely correlated with miR-363 expression in prostate cancer. IFIT5 is involved in the degradation of miR-363 and can form a complex with miR-101 and miR-128 to promote prostate cancer progression by inducing EMT (Lo et al., 2018).
IFITs in Apoptosis
Intriguingly, IFIT-family proteins display distinct functions in apoptosis. The negative association of IFIT2 with tumor malignancy is likely due to its pro-apoptotic activity (Stawowczyk et al., 2011; Chen et al., 2017). Ectopic IFIT2 overexpression induced the activation of caspase-3 and disturbed the plasma membrane asymmetry and permeability, which is a basic characteristic feature of apoptosis (Stawowczyk et al., 2011; Lai et al., 2013; Feng et al., 2014). In addition, IFIT2 was associated with the mediator of IRF3 activation (MITA) and regulated apoptotic cell death via the mitochondrial pathway (Stawowczyk et al., 2011). Inhibition of proteasome-mediated degradation of IFIT2 led to the aggregation of IFIT2 in the perinuclear region and promoted apoptosis (Chen et al., 2017). Derepression of IFIT2 made the cells prone to apoptotic death induced by external stimuli such as chemotherapeutic drugs and serum starvation (Feng et al., 2014; Wang et al., 2016). IFIT2-mediated apoptosis was not dependent on the DNA damage response (Chen et al., 2017; Ohsugi et al., 2017). Rather, IFIT2 induced apoptosis by regulating the balance between pro- and anti-apoptotic Bcl-2 family proteins, which altered the permeability of the mitochondrial membrane (Tait and Green, 2010; Stawowczyk et al., 2011). Several research studies have confirmed the ability of IFIT2 to promote the apoptotic death of cancer cells, including OSCC (Lai et al., 2013; Feng et al., 2014), colorectal cancer (Jia et al., 2017; Ohsugi et al., 2017), leukemia (Zhang et al., 2017), osteosarcoma (Wang et al., 2016), and hepatocellular carcinoma (Tang et al., 2017).
IFIT3 acts as a bridging molecule between the mitochondrial antiviral signaling (MAVS) complex and the TNFR-associated factor family member-associated NF-κB activator binding kinase 1 (TBK1) to regulate the activation of IRF3 and NF-κB (Liu et al., 2011). Overexpression of IFIT3 had a protective role in human lung epithelial cells, whereas depletion of IFIT3 expression induced apoptotic cell death (Hsu et al., 2013). Studies have shown that co-expression of IFIT3 inhibited IFIT2-dependent apoptotic cell death (Stawowczyk et al., 2011). Since IFIT2 and IFIT3 form a protein complex in the cell, the apoptotic effects of IFIT2 can be negatively regulated by IFIT3 (Reich, 2013). However, further studies are needed to understand whether the pro-survival signal of IFIT3 alone can protect the cell or whether the IFIT2: IFIT3 association is required to modulate IFIT2-mediated apoptosis.
IFIT1 negatively regulated NF-κB and IRF3 activation by interrupting the MITA-MAVS-TBK1 interaction in the cell (Li et al., 2009). Our recent study has shown that IFIT1 interacted with IFIT3 and that IFIT1 and IFIT3 expression promoted OSCC cell proliferation and metastasis (Pidugu et al., 2019b). However, the anti-apoptotic roles of IFIT1/IFIT3 have to be investigated in OSCC. Ectopic expression of IFIT5 induced IRF-3- and NF-κB-mediated gene expression. Furthermore, IFIT5 colocalized with MAVS (Zhang et al., 2013; Zheng et al., 2015). Recent research has shown that IFIT5 promoted the progression of various cancers, including renal cancer, prostate cancer, and bladder cancer (Lo et al., 2018, 2019; Huang et al., 2019). Hence, it would be interesting to investigate whether IFIT5 may also have anti-apoptotic functions.
IFITs in Drug Resistance
The failure of chemotherapy or targeted therapy has often been associated with intrinsic or acquired drug resistance in cancer cells (Mansoori et al., 2017). Anticancer drug resistance can be caused by various intrinsic cellular mechanisms, including alterations of intracellular drug distribution, changes in drug metabolism, decreased apoptosis, enhanced drug efflux by ATP-binding cassette transporters, enhanced DNA damage repair, cell cycle dysregulation, and reduced drug-target interactions (Larsen et al., 2000). Increased expression of IFIT1 and IFIT3 genes was observed in DNA damage-resistant sublines compared to parental cell lines in various cancers, including OSCC cells (Weichselbaum et al., 2008). IFIT1 and IFIT3 were also upregulated in estrogen-negative breast cancer cells in post-chemotherapy residual tumors (Legrier et al., 2016). Overexpression of IFIT1 or IFIT3 increased OSCC resistance to various chemotherapeutic drugs, including cisplatin, carboplatin, oxaliplatin, 5FU, and ganetespib. Interestingly, IFIT1 and IFIT3 expression made OSCC cells susceptible to gefitinib (EGFR-TKI) (Yen et al., 2014; Pidugu et al., 2018, 2019a). IFIT1 and IFIT3 promoted EGFR activation in OSCC cells and enhanced the tumor-preventive activity of gefitinib. In addition, the combination treatment of IFN-α and gefitinib showed synergistic anti-tumor activity in OSCC cells (Bruzzese et al., 2006; Pidugu et al., 2019b). These studies demonstrated that IFIT1 and IFIT3 modulate the drug response via EGFR signaling. The efficacy of the therapies with various drugs depends on the cellular mechanisms active in the cancer cells. Thus, advanced metastatic OSCC must be treated with multifaceted therapies to improve the clinical outcome in patients. Therefore, targeting specific IFITs can be a good clinical approach for OSCC treatment. We have also shown that IFIT2 knockdown enhances atypical PKC signaling in OSCC cells (Lai et al., 2013). Activation of PKC signaling is involved in the development of multidrug resistance (MDR) phenotype by phosphorylation of p-glycoprotein in cancer cells (Rumsby et al., 1998). On the other hand, inhibition of atypical PKC has improved clinical outcomes in advanced basal cell carcinoma (BCCs) (Mirza et al., 2017). Therefore, IFIT2 depletion may promote drug resistance. Hence, additional studies are warranted to determine the IFIT2-mediated drug resistance in OSCC.
IFITs in HVP Infection
The HPV infection is one of the aetiological factors of OSCC pathogenesis (Gillison et al., 2008; Husain and Neyaz, 2017). Studies have shown that OSCC patients with HPV infection had a better prognosis compared to patients with no infection (Schwartz et al., 2001). However, the biological mechanism that leads to better clinical outcomes in OSCC patients is not clear. IFIT1 has been shown to act against HPV by direct binding with key E1 replication protein, which is crucial for the synthesis of viral progeny DNA. The HPV E1 protein binds at the viral origin of replication DNA elements and makes a hexameric helicase complex with the help of HPV E2 protein. This protein complex interacts with several host cell proteins such as DNA polymerase alpha and recruits replication protein A (RPA) to initiate viral DNA replication (Wilson et al., 2002). The binding of the N-terminus of human IFIT1 with HPV E1 inhibits its helicase function and stops viral DNA replication by uncoupling it from the viral DNA. It has been demonstrated that IFIT1 binding activity with HPV E1 is not virus strain-specific, as it binds with E1 of HPV11, HPV18, and HPV31. HPV replication is inhibited to a great extent by the treatment with interferons, and this effect is weakened by the shRNA-mediated knockdown of IFIT1. Thus, human IFIT1 is considered the prime antiviral protein against HPV (Terenzi et al., 2008).
Human IFIT5 plays a crucial role in the host innate immune response during viral infection (Zhang et al., 2013; Zheng et al., 2015). Interestingly, we found that the expression of human IFIT5 protein is highly correlated with HPV E6 protein in tissues of OSCC patients (Yen et al., 2014). However, the underlying molecular mechanism involved in the association between IFIT5 and HPV E6 proteins is not yet understood. HPV E6 is one of the major oncoproteins that contribute to the malignant progression of OSCC. One of the major functions of HPV E6 is to enhance the proteasome-mediated degradation of tumor suppressor protein p53 via the interaction with E6-associated protein (E6AP). p53 regulates cell growth and apoptosis immediately after DNA damage. HPV E6 also impedes pro-apoptotic proteins, such as procaspase 8 and Bak, to block apoptosis (Mantovani and Banks, 2001; Gupta and Gupta, 2015). Over the past decade, an increasing number of HPV E6-associated proteins have contributed to cellular malignant transformation, with human telomerase as one of the best examples (Narisawa-Saito and Kiyono, 2007). Recently, human IFIT5 has been implicated in cancer progression (Lo et al., 2018, 2019; Huang et al., 2019). Therefore, the correlation between human IFIT5 and HVP E6 might contribute to OSCC malignancy, and further investigation is warranted to explore the reason for their association.
IFITs as Putative Co-chaperones
Heat shock protein 90 (Hsp90) is a ubiquitously expressed ATPase-directed molecular chaperone in the cell, which facilitates posttranslational protein homeostasis by regulating a variety of molecular processes such as stabilization, maturation, degradation of client proteins that are involved in various signal transduction pathways (Caplan, 1999; Whitesell and Lindquist, 2005; Pearl and Prodromou, 2006). Hsp90 regulates the folding and activation of more than 200 client proteins (https://www.picard.ch/downloads/Hsp90interactors.pdf), including EGFR (Ahsan et al., 2012), AKT (Basso et al., 2002), SAPK (Tatebe and Shiozaki, 2003), p38 (Ota et al., 2010), PKC (Gould et al., 2009), FAK (Xiong et al., 2014), and DNA-PK (Solier et al., 2012). Hsp90 controls the maturation and intracellular trafficking of ErbB2-family proteins, including EGFR (Xu et al., 2001, 2002; Yang et al., 2016). The biological function of Hsp90 relies on an inherent ATPase activity that is modulated by many of its co-chaperones. Emerging evidence has indicated that many TPR proteins, such as Hsp90-organizing protein (HOP), carboxy-terminus of Hsc70–interacting protein (CHIP), Cdc37, PP5, Unc45, Fkbp51, Fkbp51, Fkbp52, Sti1, and Cyp40, act as co-chaperones and play crucial roles in stabilizing the interaction between chaperones, such as Hsp90 or Hsp70, and their client proteins (Bose et al., 1996; Frydman and Hohfeld, 1997; Chen et al., 1998; Blatch and Lassle, 1999; Caplan, 1999; Scheufler et al., 2000; Li et al., 2012). HOP interacts with Hsp70 and Hsp90 with its TPR1 and TPR2A domains, respectively. Both TPR1 and TPR2A domains of HOP bind with EEVD motifs of Hsp70 and Hsp90 through electrostatic interactions between C-terminal aspartates and hydrophobic interactions of amino acid residues upstream of the EEVD motifs (Chen and Smith, 1998; Scheufler et al., 2000; Brinker et al., 2002; Travers and Fares, 2007). CHIP, a quality-control E3 ligase containing a co-chaperone protein, regulates the degradation of ubiquitinylated client proteins, which is crucial for maintaining protein turnover and cellular homeostasis. CHIP has three N-terminal tandem repeats of TPR motifs by which it interacts with and regulates Hsp70 and Hsp90 (Ballinger et al., 1999; Paul and Ghosh, 2014). Intriguingly, the balance between client protein folding and degradation is regulated by the binding of HOP or CHIP with Hsp90 and Hsp70. Emerging evidence has shown that the proteins with TPR motifs bind a C-terminal highly conserved motif, the EEVD-COOH tail, of Hsp70/Hsp90 to control protein folding and maturation (Demand et al., 1998; Scheufler et al., 2000; Brinker et al., 2002). Furthermore, the binding of these co-chaperones is determined by the C-terminal phosphorylation status of Hsp70 and Hsp90 (Muller et al., 2013). Since IFIT proteins contain multiple TPR motifs in their structures, we speculated that they may exert co-chaperone functions. Hsp90 has two isoforms, Hsp90α and Hsp90β, but the functional difference between these isoforms is not well-understood (Subbarao Sreedhar et al., 2004). Interestingly, co-immunoprecipitation followed by LC-MS/MS analysis showed 16 peptides with 21% sequence coverage and 17 peptides with 16% sequence coverage of Hsp90α; 59 peptides with 50% sequence coverage and 57 peptides with 44% sequence coverage of Hsp90β were identified with immunoprecipitates of IFIT1 and IFIT3, respectively. Protein-protein interaction network analysis using the STRING 10.5 database showed that heat shock protein 90 s (Hsp90α/β) directly bound IFIT1 and IFIT3 (Pidugu et al., 2019a,b). Increased C-terminal phosphorylation of Hsp90α was observed in IFIT1- or IFIT3-overexpressing OSCC cells. Overexpression of IFIT1 or IFIT3 in OSCC cells enhanced the phosphorylation of Hsp90 client proteins, including EGFR, AKT, p38, and SAPK/JNK. Alternatively, inhibition of Hsp90 activity by ganetespib led to decreased C-terminal phosphorylation of Hsp90α and expression and activation of its downstream client proteins (Pidugu et al., 2019a). These Hsp90-downstream signaling regulators have been widely shown to play crucial roles in cell survival, migration, invasion, and metastasis (Tsutsumi and Neckers, 2007; Pashtan et al., 2008; Tsutsumi et al., 2009). Since IFIT1 and IFIT3 contain TRP motifs, we may hypothesize that they function as co-chaperones. Further investigation is warranted to elucidate the TPR motifs of IFIT1 and IFIT3 that mediate protein-protein interactions in OSCC. A recent study has shown that IFIT2 is co-precipitated with p67phox and mitochondria-associated heat shock protein Hsc70 (Stawowczyk et al., 2018). As IFIT2 contains many TPR domains in its structure, it binds with p67phox and Hsc70 through TPR motif-mediated interactions; however, the exact TPR motifs involved in the interaction have not yet been mapped. Since increased Hsp90 activity directly influences client kinase activation and stabilization, Hsp90 is regarded as a promising therapeutic target for cancer treatment (Caplan et al., 2007; Trepel et al., 2010). Further studies are needed to delineate the co-chaperone functions of IFITs in the future.
The Clinical Relevance of IFITs in OSCC and Other Cancers
The ISGs and their signaling pathways play vital roles in the malignant transformation of cells in the tumor microenvironment. Although IFNs have been used as exogenous pharmaceuticals for the treatment of cancers, paradoxical findings revealed that constitutive expression of aberrantly regulated ISGs promotes neoplastic disease development and progression (Cheon et al., 2014). Abnormal expression of ISGs promotes tumor invasion and progression in many cancers, including skin cancer, breast cancer and head and neck cancers (Perou et al., 1999; Suomela et al., 2004; Andersen and Hassel, 2006; Hatano et al., 2008). Increased expression of ISGs has been reported in metastatic cancer cells compared to non-metastatic cells (Cai et al., 2009; Khodarev et al., 2009). ISGs can modulate the tumor cell response to therapeutic drugs by altering the tumor microenvironment. Defects in interferon signaling cause resistance to immunotherapies such as anti-CTLA-4 and PD1 blockade (Shin et al., 2015; Gao et al., 2016; Zaretsky et al., 2016). Constant exposure to low levels of IFNs induces the transcription of a subset of ISGs called interferon-related DNA damage-resistant signature (IRDS) genes that contribute to tumor growth, metastasis, resistance to radiation therapy and chemotherapeutic drugs (Rickardson et al., 2005; Duarte et al., 2012; Cheon et al., 2013). High expression of IRDS genes in cancer tissues promotes tumor growth, invasion, and metastasis (Wallace et al., 2011). IFIT1 and IFIT3 genes are classified as IRDS subset genes and are considered as predictive biomarkers for chemotherapy and radiation therapy in many primary human cancers, such as breast cancer, head and neck cancer, prostate cancer, lung cancer, and glioma (Weichselbaum et al., 2008). Therefore, cancer patients with IRDS expression are suggested to undergo therapies combined with adjuvant treatments. Accordingly, high IFIT1 and IFIT3 expression have been correlated with a better therapeutic response to IFNs along with chemotherapeutics and immunostimulating agents in patients with breast cancer, glioblastoma, and hepatocellular carcinoma (Zhang et al., 2016; Yang et al., 2017; Nushtaeva et al., 2018). However, the enhanced expression of IFIT3 in pancreatic cancer has been shown to cause pseudo-inflammation and result in cancer progression (Niess et al., 2015; Zhao et al., 2017). Our recent investigations also demonstrated that high IFIT1 or IFIT3 expression correlated with T-stage, lymph node metastasis, lymphovascular, perineural invasion, and poor overall survival in OSCC patients. Ectopic expression of IFIT1 or IFIT3 in OSCC cells promoted tumor growth and metastasis by activating EGFR signaling (Pidugu et al., 2019b). Strikingly, we also found that enhanced IFIT1 or IFIT3 expression in OSCC cells promoted sensitivity to gefitinib (EGFR-TKI). Moreover, the combination treatment of gefitinib and IFN-α resulted in a synergistic tumor-inhibitory effect in OSCC (Bruzzese et al., 2006; Pidugu et al., 2018). This could be due to the enhanced EGFR tyrosine kinase activity in OSCC cells caused by IFIT1 and IFIT3 expression. These data suggest that IFIT1 and IFIT3 expression can be used as prognostic biomarkers to predict the EGFR-TKI and IFN-α therapeutic response in OSCC patients and probably in patients with other cancer types.
Interestingly, we also observed that high IFIT1 or IFIT3 expression in OSCC cells increased resistance to various therapeutics, including cisplatin, oxaliplatin, carboplatin, 5-FU, and ganetespib. IFIT1 or IFIT3 expression enhanced C-terminal phosphorylation of Hsp90 and its client proteins, including PKC, EGFR, AKT, and p38 (Pidugu et al., 2017, 2019a). Protein-protein interaction analysis revealed that IFIT1 and IFIT3 colocalize with both Hsp90α and Hsp90β isoforms in OSCC cells. Collectively, these data suggest that IFIT1 and IFIT3 regulate the drug response and might function as co-chaperones in OSCC. Thus, IFIT1 and IFIT3 proteins can be considered potential therapeutic targets for OSCC. In contrast, IFIT2 has been shown to have tumor suppressor function in many cancers. Decreased expression of IFIT2 is associated with increased cell proliferation and metastasis and predicts poor clinical outcome in gastric cancer patients (Chen et al., 2018). Downregulation of IFIT2 expression was observed in colorectal cancer (CRC) tissues compared to normal tissues. Overexpression of IFIT2 in CRC cells suppressed cell growth and increased apoptosis (Ohsugi et al., 2017). Our findings agree with previous findings that IFIT2 depletion significantly increased OSCC metastasis both in vitro and in vivo (Lai et al., 2013). Besides, the high expression of IFIT2 in OSCC tumor tissues negatively correlated with the nodal stage. The positive association of IFIT2 expression with better overall patient survival suggests that IFIT2 can act as a novel prognostic biomarker to predict OSCC progression (Lai et al., 2008). Although IFIT2 belongs to the same family, it executes tumor suppressor functions, whereas IFIT1 and IFIT3 have tumor-promoting properties in OSCC. However, how IFITs regulate contradictory functions in OSCC cells is not yet known. Further investigations are required to understand the possible molecular mechanisms that regulate opposing functions of IFITs in OSCC.
Discussion
IFITs are quickly induced by multiple stimuli, such as IFN-dependent or IFN-independent signaling pathways. Over the past decade, the anti-viral functions of IFITs have been extensively studied. Many structural and functional studies propose that IFITs are also involved in the regulation of cell-intrinsic and cell-extrinsic immune responses via new pathways that must be corroborated. Although IFITs have a simple gene architecture and promoter structure, they execute multiple complex functions based on stimulus, cell type, and tissue type. The variable amino acid sequence in TPR motifs could be the reason for the broad range of non-redundant functions mediated by IFITs in the cell. Even though IFITs are renowned antiviral proteins, recent studies hint at their complex biological and molecular roles in cancer.
In this review, we hypothesize that IFIT protein may serve as co-chaperones of Hsp90 (Figure 4). Many studies have demonstrated that TPR proteins interact with the C-terminus of Hsp90 to facilitate the stabilization of the chaperone complex (Scheufler et al., 2000; Brinker et al., 2002; Muller et al., 2013). Hsp90 is a ubiquitously expressed molecular chaperone in the cells that regulates a multitude of cellular processes, such as client kinase folding, maturation, and degradation (Whitesell and Lindquist, 2005). The direct association of IFIT1 or IFIT3 with heat shock proteins (Hsp90α/β) suggests a unique mechanism of action of IFIT1 and IFIT3 that may be involved in activating Hsp90 and several of its downstream signaling regulators, which are crucial for OSCC tumor progression and drug resistance. Thus, IFIT1 and IFIT3 may likely function as co-chaperones and serve as potentially important therapeutic targets for OSCC.
Figure 4
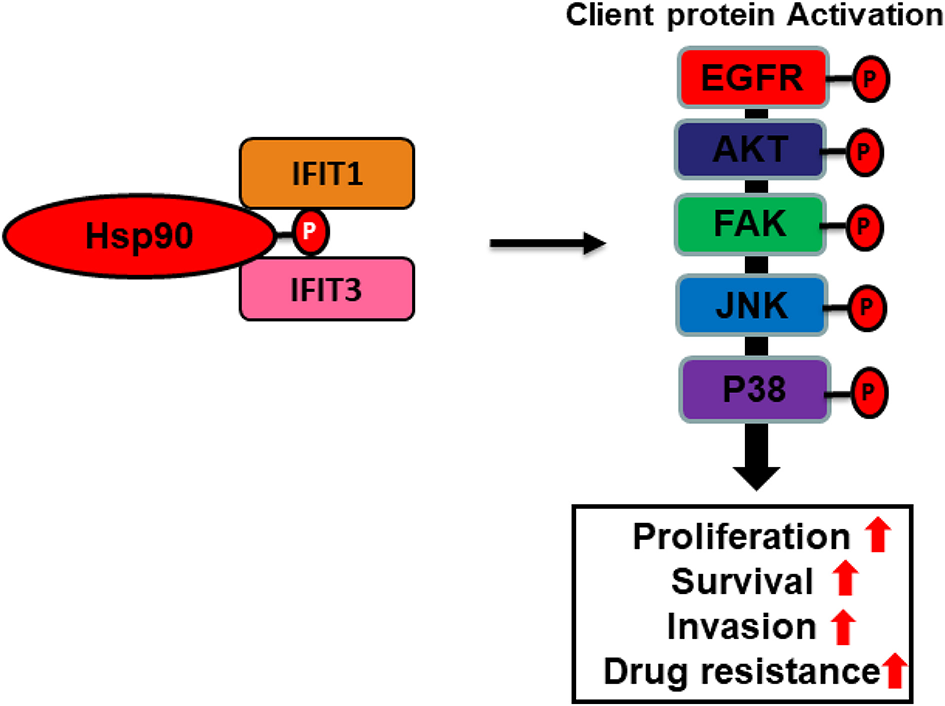
Cartoon illustration depicting putative co-chaperone functions of IFIT1 and IFIT3. A hypothetic scheme shows that IFIT1/IFIT3 may regulate C-terminal phosphorylation of Hsp90 and hence enhance proper folding and maturation of its downstream signaling regulators in OSCC.
Alternatively, our recent studies have shown that ectopic overexpression of IFIT1 and IFIT3 enhanced the EGFR endocytic recycling process by interacting with Annexin-2 (ANX2) recycling and thus activated several downstream pathways involving in cell proliferation, survival, and drug resistance. Activated EGFR subsequently promoted OSCC invasion through EMT (Figure 5). This hypothesis was supported by the clinical observations showing a positive association of phospho-EGFR levels with the expression of both IFIT1 and IFIT3 in OSCC specimens. In addition, elevated IFIT1 and IFIT3 expression indeed correlated with poor survival in OSCC patients. In contrast, decreasing IFIT2 expression activated PKC pathway and promoted the progression of tumor malignancy. However, how PKC is activated by depleting IFIT2 protein is still unknown. It cannot be neglected that IFIT proteins may execute controversial activity in different cell types. However, we may infer that IFITs play certain roles on modulation of survival signaling pathways.
Figure 5

Schematic model represents IFIT1 and IFIT3 interaction with ANXA2 promote EGFR recycling. Diagram illustrating the association of IFIT1 and IFIT3 with ANXA2 and enhance p-EGFRY1068 endocytic recycling, which subsequently leads to EGFR, AKT activation, and enhance the expression of downstream epithelial-mesenchymal transition (EMT) transcription factor slug. Activated EGFR and its downstream signaling regulators promote OSCC invasion and metastasis.
Additionally, recent studies have shown that IFIT1 undergoes ISGylation (Zhao et al., 2005) and phosphorylation (Li et al., 2009), indicating that IFIT protein stability and function can be regulated by posttranslational modification. The association between IFIT5 and HPV E6 indicates that IFIT5 may be involved in the malignant transformation of oral squamous cells during disease progression. Additional studies are warranted to ascertain the role of IFIT5 in HPV-positive OSCC.
In overall, the involvement of IFITs in the progression of cancer is an emerging question that warrants our concern. Apparently, these non-enzymatic proteins may exert their biologic activities via protein-protein interaction. Interruption of protein-protein interaction is a new clue for anti-cancer drug development. We prospect that in vivo studies with individual IFIT gene knockout mice can contribute to unraveling the pathophysiological roles of the respective IFIT proteins, and they could also open a new avenue for research into multiple disease systems and drug development in the future.
Statements
Author contributions
VP contributed to conception and manuscript writing. HP and M-MW contributed to the literature review. T-CL and C-JL defined the scope of the review and edited the manuscript. VP, HP, and T-CL revised the manuscript. All authors have read and approved the final manuscript.
Funding
This work was supported by grants (MOST 107-2320-B-001-007 and 108-2320-B-001-004) from the Ministry of Science and Technology, Taiwan (T-CL and C-JL).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AbbasY. M.PichlmairA.GórnaM. W.Superti-FurgaG.NagarB. (2013). Structural basis for viral 5'-PPP-RNA recognition by human IFIT proteins. Nature494, 60–64. 10.1038/nature11783
2
AhsanA.RamanandS. G.WhiteheadC.HinikerS. M.RehemtullaA.PrattW. B.et al. (2012). Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia14, 670–677. 10.1593/neo.12986
3
AllanR. K.RatajczakT. (2011). Versatile TPR domains accommodate different modes of target protein recognition and function. Cell Stress Chaperones16, 353–367. 10.1007/s12192-010-0248-0
4
AndersenJ. B.HasselB. A. (2006). The interferon regulated ubiquitin-like protein, ISG15, in tumorigenesis: friend or foe?Cytokine Growth Factor Rev.17, 411–21. 10.1016/j.cytogfr.2006.10.001
5
BallingerC. A.ConnellP.WuY.HuZ.ThompsonL. J.YinL. Y.et al. (1999). Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol. Cell. Biol.19, 4535–4545. 10.1128/MCB.19.6.4535
6
BassoA. D.SolitD. B.ChiosisG.GiriB.TsichlisP.RosenN. (2002). Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem.277, 39858–3966. 10.1074/jbc.M206322200
7
BlatchG. L.LassleM. (1999). The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. Bioessays21, 932–939.
8
BlotW. J.McLaughlinJ. K.WinnD. M.AustinD. F.GreenbergR. S.Preston-MartinS.Jr.et al. (1988). Smoking and drinking in relation to oral and pharyngeal cancer. Cancer Res.48, 3282–3287
9
BoseS.WeiklT.BuglH.BuchnerJ. (1996). Chaperone function of Hsp90-associated proteins. Science274, 1715–1717. 10.1126/science.274.5293.1715
10
BrinkerA.ScheuflerC. FVon Der MulbeFleckensteinB.HerrmannC.JungG.et al. (2002). Ligand discrimination by TPR domains. Relevance and selectivity of EEVD-recognition in Hsp70 x Hop x Hsp90 complexes. J Biol Chem. 277, 19265–75. 10.1074/jbc.M109002200
11
BruzzeseF.Di GennaroE.AvalloneA.PepeS.ArraC.CaragliaM.et al. (2006). Synergistic antitumor activity of epidermal growth factor receptor tyrosine kinase inhibitor gefitinib and IFN-alpha in head and neck cancer cells in vitro and in vivo. Clin. Cancer Res.12, 617–25. 10.1158/1078-0432.CCR-05-1671
12
CaiD.CaoJ.LiZ.ZhengX.YaoY.LiW.et al. (2009). Up-regulation of bone marrow stromal protein 2 (BST2) in breast cancer with bone metastasis. BMC Cancer9:102. 10.1186/1471-2407-9-102
13
CaplanA. J. (1999). Hsp90's secrets unfold: new insights from structural and functional studies. Trends Cell Biol.9, 262–268. 10.1016/S0962-8924(99)01580-9
14
CaplanA. J.MandalA. K.TheodorakiM. A. (2007). Molecular chaperones and protein kinase quality control. Trends Cell Biol.17, 87–92. 10.1016/j.tcb.2006.12.002
15
CeramiE.GaoJ.DogrusozU.GrossB. E.SumerS. O.AksoyB. A.et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov.2, 401–404. 10.1158/2159-8290.CD-12-0095
16
ChenL.LiuS.XuF.KongY.WanL.ZhangY.et al. (2017). Inhibition of proteasome activity induces aggregation of IFIT2 in the centrosome and enhances IFIT2-induced cell apoptosis. Int. J. Biol. Sci.13, 383–390. 10.7150/ijbs.17236
17
ChenL.ZhaiW.ZhengX.XieQ.ZhouQ.TaoM.et al. (2018). Decreased IFIT2 expression promotes gastric cancer progression and predicts poor prognosis of the patients. Cell. Physiol. Biochem. 45, 15–25. 10.1159/000486219
18
ChenS.SmithD. F. (1998). Hop as an adaptor in the heat shock protein 70 (Hsp70) and hsp90 chaperone machinery. J. Biol. Chem.273, 35194–35200. 10.1074/jbc.273.52.35194
19
ChenS.SullivanW. P.ToftD. O.SmithD. F. (1998). Differential interactions of p23 and the TPR-containing proteins Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaperones3, 118–129.
20
CheonH.BordenE. C.StarkG. R. (2014). Interferons and their stimulated genes in the tumor microenvironment. Semin. Oncol.41, 156–173. 10.1053/j.seminoncol.2014.02.002
21
CheonH.Holvey-BatesE. G.SchogginsJ. W.ForsterS.HertzogP.ImanakaN.et al. (2013). IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J.32, 2751–2763. 10.1038/emboj.2013.203
22
D'AndreaL. D.ReganL. (2003). TPR proteins: the versatile helix. Trends Biochem. Sci.28, 655–662. 10.1016/j.tibs.2003.10.007
23
DanishH. H.GoyalS.TaunkN. K.WuH.MoranM. S.HafftyB. G. (2013). Interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) as a prognostic marker for local control in T1-2 N0 breast cancer treated with breast-conserving surgery and radiation therapy (BCS + RT). Breast J.19, 231–239. 10.1111/tbj.12097
24
de VeerM. J.HolkoM.FrevelM.WalkerE.DerS.ParanjapeJ. M.et al. (2001). Functional classification of interferon-stimulated genes identified using microarrays. J. Leukoc. Biol.69, 912–920. 10.1189/jlb.69.6.912
25
DemandJ.LudersJ.HohfeldJ. (1998). The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol. Cell. Biol.18, 2023–2028. 10.1128/MCB.18.4.2023
26
DerS. D.ZhouA.WilliamsB. R.SilvermanR. H. (1998). Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. U.S.A.95, 15623–15628. 10.1073/pnas.95.26.15623
27
DiamondM. S.FarzanM. (2013). The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol.13, 46–57. 10.1038/nri3344
28
DuarteC. W.WilleyC. D.ZhiD.CuiX.HarrisJ. J.VaughanL. K.et al. (2012). Expression signature of IFN/STAT1 signaling genes predicts poor survival outcome in glioblastoma multiforme in a subtype-specific manner. PLoS ONE7:e29653. 10.1371/journal.pone.0029653
29
FengF.YuanL.WangY. E.CrowleyC.LvZ.LiJ.et al. (2013). Crystal structure and nucleotide selectivity of human IFIT5/ISG58. Cell Res.23, 1055–1058. 10.1038/cr.2013.80
30
FengX.WangY.MaZ.YangR.LiangS.ZhangM.et al. (2014). MicroRNA-645, up-regulated in human adencarcinoma of gastric esophageal junction, inhibits apoptosis by targeting tumor suppressor IFIT2. BMC Cancer14, 633–633. 10.1186/1471-2407-14-633
31
FensterlV.SenG. C. (2011). The ISG56/IFIT1 gene family. J. Interferon Cytokine Res.31, 71–78. 10.1089/jir.2010.0101
32
FensterlV.SenG. C. (2015). Interferon-induced Ifit proteins: their role in viral pathogenesis. J. Virol.89, 2462–2468. 10.1128/JVI.02744-14
33
FleithR. C.MearsH. V.LeongX. Y.SanfordT. J.EmmottE.GrahamS. C.et al. (2018). IFIT3 and IFIT2/3 promote IFIT1-mediated translation inhibition by enhancing binding to non-self RNA. Nucleic Acids Res.46, 5269–5285. 10.1093/nar/gky191
34
FrydmanJ.HohfeldJ. (1997). Chaperones get in touch: the Hip-Hop connection. Trends Biochem. Sci.22, 87–92. 10.1016/S0968-0004(97)01005-0
35
GaoJ.AksoyB. A.DogrusozU.DresdnerG.GrossB.SumerS. O.et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal.6:pl1. 10.1126/scisignal.2004088
36
GaoJ.ShiL. Z.ZhaoH.ChenJ.XiongL.HeQ.et al. (2016). Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell167, 397–404.e9. 10.1016/j.cell.2016.08.069
37
GillisonM. L.D'SouzaG.WestraW.SugarE.XiaoW.BegumS.et al. (2008). Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J. Natl. Cancer Inst.100, 407–420. 10.1093/jnci/djn025
38
GouldC. M.KannanN.TaylorS. S.NewtonA. C. (2009). The chaperones Hsp90 and Cdc37 mediate the maturation and stabilization of protein kinase C through a conserved PXXP motif in the C-terminal tail. J. Biol. Chem.284, 4921–4935. 10.1074/jbc.M808436200
39
GuptaS.GuptaS. (2015). Role of human papillomavirus in oral squamous cell carcinoma and oral potentially malignant disorders: a review of the literature. Indian J. Dent. 6, 91–98. 10.4103/0975-962X.155877
40
HabjanM.HubelP.LacerdaL.BendaC.HolzeC.EberlC. H.et al. (2013). Sequestration by IFIT1 impairs translation of 2'O-unmethylated capped RNA. PLoS Pathog.9:e1003663. 10.1371/journal.ppat.1003663
41
HaddadR. I.ShinD. M. (2008). Recent advances in head and neck cancer. N. Engl. J. Med. 359, 1143–1154. 10.1056/NEJMra0707975
42
HatanoH.KudoY.OgawaI.TsunematsuT.KikuchiA.AbikoY.et al. (2008). IFN-induced transmembrane protein 1 promotes invasion at early stage of head and neck cancer progression. Clin. Cancer Res.14, 6097–6105. 10.1158/1078-0432.CCR-07-4761
43
HsuY. L.ShiS. F.WuW. L.HoL. J.LaiJ. H. (2013). Protective roles of interferon-induced protein with tetratricopeptide repeats 3 (IFIT3) in dengue virus infection of human lung epithelial cells. PLoS ONE8:e79518. 10.1371/journal.pone.0079518
44
HuangJ.LoU. G.WuS.WangB.PongR.-C.LaiC.-H.et al. (2019). The roles and mechanism of IFIT5 in bladder cancer epithelial–mesenchymal transition and progression. Cell Death Dis.10:437. 10.1038/s41419-019-1669-z
45
HusainN.NeyazA. (2017). Human papillomavirus associated head and neck squamous cell carcinoma: Controversies and new concepts. J. Oral Biol. Craniofacial Res. 7, 198–205. 10.1016/j.jobcr.2017.08.003
46
JengJ. H.ChangM. C.HahnL. J. (2001). Role of areca nut in betel quid-associated chemical carcinogenesis: current awareness and future perspectives. Oral Oncol.37, 477–492. 10.1016/S1368-8375(01)00003-3
47
JhaP. (2009). Avoidable global cancer deaths and total deaths from smoking. Nat. Rev. Cancer9, 655–664. 10.1038/nrc2703
48
JiaH.SongL.CongQ.WangJ.XuH.ChuY.et al. (2017). The LIM protein AJUBA promotes colorectal cancer cell survival through suppression of JAK1/STAT1/IFIT2 network. Oncogene36, 2655–2666. 10.1038/onc.2016.418
49
JohnsonB.VanBlarganL. A.XuW.WhiteJ. P.ShanC.ShiP. Y.et al. (2018). Human IFIT3 modulates IFIT1 RNA binding specificity and protein stability. Immunity48, 487–499.e5. 10.1016/j.immuni.2018.01.014
50
KalluriR.WeinbergR. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest.119, 1420–1428. 10.1172/JCI39104
51
KatibahG. E.LeeH. J.HuizarJ. P.VoganJ. M.AlberT.CollinsK. (2013). tRNA binding, structure, and localization of the human interferon-induced protein IFIT5. Mol. Cell. 49, 743–750. 10.1016/j.molcel.2012.12.015
52
KhodarevN. N.RoachP.PitrodaS. P.GoldenD. W.BhayaniM.ShaoM. Y.et al. (2009). STAT1 pathway mediates amplification of metastatic potential and resistance to therapy. PLoS ONE4:e5821. 10.1371/journal.pone.0005821
53
KiritaT.YamanakaY.ImaiY.YamakawaN.AokiK.NakagawaY.et al. (2012). Preoperative concurrent chemoradiotherapy for stages II-IV oral squamous cell carcinoma: a retrospective analysis and the future possibility of this treatment strategy. Int. J. Oral Maxillofac. Surg.41, 421–428. 10.1016/j.ijom.2011.12.003
54
Krishna RaoS. V.MejiaG.Roberts-ThomsonK.LoganR. (2013). Epidemiology of oral cancer in Asia in the past decade–an update (2000-2012). Asian Pac. J. Cancer Prev.14, 5567–5577. 10.7314/APJCP.2013.14.10.5567
55
KumarP.SweeneyT. R.SkabkinM. A.SkabkinaO. V.HellenC. U. T.PestovaT. V. (2014). Inhibition of translation by IFIT family members is determined by their ability to interact selectively with the 5′-terminal regions of cap0-, cap1- and 5′ppp- mRNAs. Nucleic Acids Res.42, 3228–3245. 10.1093/nar/gkt1321
56
KusariJ.SenG. C. (1986). Regulation of synthesis and turnover of an interferon-inducible mRNA. Mol. Cell. Biol.6, 2062–2067. 10.1128/MCB.6.6.2062
57
LaiK. C.ChangK. W.LiuC. J.KaoS. Y.LeeT. C. (2008). IFN-induced protein with tetratricopeptide repeats 2 inhibits migration activity and increases survival of oral squamous cell carcinoma. Mol. Cancer Res.6, 1431–1439. 10.1158/1541-7786.MCR-08-0141
58
LaiK. C.LeeT. C. (2006). Genetic damage in cultured human keratinocytes stressed by long-term exposure to areca nut extracts. Mutat. Res.599, 66–75. 10.1016/j.mrfmmm.2006.01.005
59
LaiK. C.LiuC. J.ChangK. W.LeeT. C. (2013). Depleting IFIT2 mediates atypical PKC signaling to enhance the migration and metastatic activity of oral squamous cell carcinoma cells. Oncogene32, 3686–3697. 10.1038/onc.2012.384
60
LarsenA. K.EscargueilA. E.SkladanowskiA. (2000). Resistance mechanisms associated with altered intracellular distribution of anticancer agents. Pharmacol. Ther.85, 217–229. 10.1016/S0163-7258(99)00073-X
61
LegrierM. E.BiecheI.GastonJ.BeurdeleyA.YvonnetV.DeasO.et al. (2016). Activation of IFN/STAT1 signalling predicts response to chemotherapy in oestrogen receptor-negative breast cancer. Br. J. Cancer114, 177–187. 10.1038/bjc.2015.398
62
LiJ.SorokaJ.BuchnerJ. (2012). The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim. Biophys. Acta1823, 624–635. 10.1016/j.bbamcr.2011.09.003
63
LiY.LiC.XueP.ZhongB.MaoA. P.RanY.et al. (2009). ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. U.S.A.106, 7945–7950. 10.1073/pnas.0900818106
64
LiuS. Y.LuC. L.ChiouC. T.YenC. Y.LiawG. A.ChenY. C.et al. (2010). Surgical outcomes and prognostic factors of oral cancer associated with betel quid chewing and tobacco smoking in Taiwan. Oral Oncol.46, 276–282. 10.1016/j.oraloncology.2010.01.008
65
LiuX. Y.ChenW.WeiB.ShanY. F.WangC. (2011). IFN-induced TPR protein IFIT3 potentiates antiviral signaling by bridging MAVS and TBK1. J. Immunol.187, 2559–2568. 10.4049/jimmunol.1100963
66
LiuY.ZhangY.-B.LiuT.-K.GuiJ.-F. (2013). Lineage-specific expansion of IFIT gene family: an insight into coevolution with IFN gene family. PLoS ONE8:e66859. 10.1371/journal.pone.0066859
67
LoU.-G.PongR.-C.YangD.GandeeL.HernandezE.DangA.et al. (2018). IFN-r-induced IFIT5 promotes epithelial-to-mesenchymal transition in prostate cancer via microRNA processing. Cancer Res.79, 1098–1112. 10.1158/0008-5472.CAN-18-2207
68
LoU. G.BaoJ.CenJ.H.-,YehC.LuoJ.TanW.et al. (2019). Interferon-induced IFIT5 promotes epithelial-to-mesenchymal transition leading to renal cancer invasion. Am. J. Clin. Exp. Urol. 7, 31–45
69
MansooriB.MohammadiA.DavudianS.ShirjangS.BaradaranB. (2017). The different mechanisms of cancer drug resistance: a brief review. Adv. Pharmaceut. Bull. 7, 339–348. 10.15171/apb.2017.041
70
MantovaniF.BanksL. (2001). The Human Papillomavirus E6 protein and its contribution to malignant progression. Oncogene20, 7874–7887. 10.1038/sj.onc.1204869
71
MarkopoulosA. K. (2012). Current aspects on oral squamous cell carcinoma. Open Dent. J.6, 126–130. 10.2174/1874210601206010126
72
MearsH. V.SweeneyT. R. (2018). Better together: the role of IFIT protein-protein interactions in the antiviral response. J Gen Virol.99, 1463–1477. 10.1099/jgv.0.001149
73
MirzaA. N.FryM. A.UrmanN. M.AtwoodS. X.RoffeyJ.OttG. R.et al. (2017). Combined inhibition of atypical PKC and histone deacetylase 1 is cooperative in basal cell carcinoma treatment. JCI Insight2:97071. 10.1172/jci.insight.97071
74
MullerP.RuckovaE.HaladaP.CoatesP. J.HrstkaR.LaneD. P.et al. (2013). C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene32, 3101–3110. 10.1038/onc.2012.314
75
Narisawa-SaitoM.KiyonoT. (2007). Basic mechanisms of high-risk human papillomavirus-induced carcinogenesis: roles of E6 and E7 proteins. Cancer Sci.98, 1505–1511. 10.1111/j.1349-7006.2007.00546.x
76
NatarajanJ.ChandrashekarC.RadhakrishnanR. (2014). Critical biomarkers of epithelial-mesenchymal transition in the head and neck cancers. J. Cancer Res. Ther.10, 512–518. 10.4103/0973-1482.137926
77
NiessH.CamajP.MairR.RennerA.ZhaoY.JackelC.et al. (2015). Overexpression of IFN-induced protein with tetratricopeptide repeats 3 (IFIT3) in pancreatic cancer: cellular “pseudoinflammation” contributing to an aggressive phenotype. Oncotarget6, 3306–3318. 10.18632/oncotarget.2494
78
NogutiJ.De MouraC. F.De JesusG. P.Da SilvaV. H.HossakaT. A.OshimaC. T.et al. (2012). Metastasis from oral cancer: an overview. Cancer Genomics Proteomics. 9, 329–335. Available online at: http://cgp.iiarjournals.org/content/9/5/329.long
79
NushtaevaA. A.StepanovG. A.SemenovD. V.JuravlevE. S.BalahonovaE. A.GerasimovA. V.et al. (2018). Characterization of primary normal and malignant breast cancer cell and their response to chemotherapy and immunostimulatory agents. BMC Cancer18:728. 10.1186/s12885-018-4635-8
80
OhsugiT.YamaguchiK.ZhuC.IkenoueT.FurukawaY. (2017). Decreased expression of interferon-induced protein 2 (IFIT2) by Wnt/beta-catenin signaling confers anti-apoptotic properties to colorectal cancer cells. Oncotarget8, 100176–100186. 10.18632/oncotarget.22122
81
OtaA.ZhangJ.PingP.HanJ.WangY. (2010). Specific regulation of noncanonical p38alpha activation by Hsp90-Cdc37 chaperone complex in cardiomyocyte. Circ. Res.106, 1404–1412. 10.1161/CIRCRESAHA.109.213769
82
PashtanI.TsutsumiS.WangS.XuW.NeckersL. (2008). Targeting Hsp90 prevents escape of breast cancer cells from tyrosine kinase inhibition. Cell Cycle7, 2936–2941. 10.4161/cc.7.18.6701
83
PaulI.GhoshM. K. (2014). The E3 ligase CHIP: insights into its structure and regulation. Biomed Res. Int.2014:918183. 10.1155/2014/918183
84
PearlL. H.ProdromouC. (2006). Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem.75, 271–294. 10.1146/annurev.biochem.75.103004.142738
85
PerouC. M.JeffreyS. S. Mvan de RijnReesC. A.EisenM. B.RossD. T.et al. (1999). Distinctive gene expression patterns in human mammary epithelial cells and breast cancers. Proc. Natl. Acad. Sci. U.S.A.96, 9212–9217. 10.1073/pnas.96.16.9212
86
PestkaS.KrauseC. D.WalterM. R. (2004). Interferons, interferon-like cytokines, and their receptors. Immunol. Rev.202, 8–32. 10.1111/j.0105-2896.2004.00204.x
87
PestkaS.LangerJ. A.ZoonK. C.SamuelC. E. (1987). Interferons and their actions. Annu. Rev. Biochem.56, 727–777. 10.1146/annurev.bi.56.070187.003455
88
PichlmairA.LassnigC.EberleC. A.GornaM. W.BaumannC. L.BurkardT. R.et al. (2011). IFIT1 is an antiviral protein that recognizes 5'-triphosphate RNA. Nat. Immunol. 12, 624–30. 10.1038/ni.2048
89
PiduguV. K.WuM.-M.LiuC.-J.LeeT.-C. (2018). Abstract A135: IFIT1 and 3 modulate the drug sensitivity in human oral squamous cell carcinoma cells. Mol. Cancer Therapeut.17(Suppl. 1):A135. 10.1158/1535-7163.TARG-17-A135
90
PiduguV. K.WuM.-M.PiduguH. B.LeeT.-C. (2019a). Abstract 2098: IFIT1 and IFIT3 modulate the drug response in human oral squamous cell carcinoma through interaction and activation of Hsp90. Cancer Res.79(Suppl. 13):2098. 10.1158/1538-7445.A.~M.2019-2098
91
PiduguV. K.WuM. M.YenA. H.PiduguH. B.ChangK. W.LiuC. J.et al. (2019b). IFIT1 and IFIT3 promote oral squamous cell carcinoma metastasis and contribute to the anti-tumor effect of gefitinib via enhancing p-EGFR recycling. Oncogene38, 3232–3247. 10.1038/s41388-018-0662-9
92
PiduguV. K.YenA.-H.ChenY.-C.WuM.-M.LiuC.-J.LeeT.-C. (2017). Abstract 3933: characterization of oncogenic activity of interferon-induced protein with tetratricopeptide repeats 1 and 3 in human oral squamous cell carcinoma progression. Cancer Res.77(Suppl. 13):3933. 10.1158/1538-7445.AM2017-3933
93
PlataniasL. C. (2005). Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol.5:375. 10.1038/nri1604
94
ReichN. C. (2013). A death-promoting role for ISG54/IFIT2. J. Interferon Cytokine Res.33, 199–205. 10.1089/jir.2012.0159
95
ReilyM. M.PantojaC.HuX.ChinenovY.RogatskyI. (2006). The GRIP1:IRF3 interaction as a target for glucocorticoid receptor-mediated immunosuppression. EMBO J.25, 108–117. 10.1038/sj.emboj.7600919
96
RickardsonL.FryknasM.DharS.LovborgH.GullboJ.RydakerM.et al. (2005). Identification of molecular mechanisms for cellular drug resistance by combining drug activity and gene expression profiles. Br. J. Cancer. 93, 483–492. 10.1038/sj.bjc.6602699
97
RumsbyM. G.DrewL.WarrJ. R. (1998). Protein kinases and multidrug resistance. Cytotechnology27, 203–224. 10.1023/A:1008073006495
98
SarkarS. N.SenG. C. (2004). Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol. Ther.103, 245–259. 10.1016/j.pharmthera.2004.07.007
99
ScheuflerC.BrinkerA.BourenkovG.PegoraroS.MoroderL.BartunikH.et al. (2000). Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell101, 199–210. 10.1016/S0092-8674(00)80830-2
100
SchwartzS. R.YuehB.McDougallJ. K.DalingJ. R.SchwartzS. M. (2001). Human papillomavirus infection and survival in oral squamous cell cancer: a population-based study. Otolaryngol. Head Neck Surg.125, 1–9. 10.1067/mhn.2001.116979
101
ScottS. E.GrunfeldE. A.MainJ.McGurkM. (2006). Patient delay in oral cancer: a qualitative study of patients' experiences. Psychooncology15, 474–485. 10.1002/pon.976
102
SenG. C.FensterlV. (2012). Crystal structure of IFIT2 (ISG54) predicts functional properties of IFITs. Cell Res.22, 1407–1409. 10.1038/cr.2012.130
103
SharanR. N.MehrotraR.ChoudhuryY.AsotraK. (2012). Association of betel nut with carcinogenesis: revisit with a clinical perspective. PLoS ONE7:e42759. 10.1371/journal.pone.0042759
104
ShenH.ZhanM.ZhangY.HuangS.XuS.HuangX.et al. (2018). PLZF inhibits proliferation and metastasis of gallbladder cancer by regulating IFIT2. Cell Death Dis.9:71. 10.1038/s41419-017-0107-3
105
ShieldK. D.FerlayJ.JemalA.SankaranarayananR.ChaturvediA. K.BrayF.et al. (2017). The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. CA Cancer J. Clin.67, 51–64. 10.3322/caac.21384
106
ShinD.Garcia-DiazA.ZaretskyJ.Escuin-OrdinasH.Hu-LieskovanS.PalaskasN. J.et al. (2015). Innate resistance of PD-1 blockade through loss of function mutations in JAK resulting in inability to express PD-L1 upon interferon exposure. J. Immunother. Cancer3:P311. 10.1186/2051-1426-3-S2-P311
107
SmithJ. B.HerschmanH. R. (1996). The glucocorticoid attenuated response genes GARG-16, GARG-39, and GARG-49/IRG2 encode inducible proteins containing multiple tetratricopeptide repeat domains. Arch. Biochem. Biophys.330, 290–300. 10.1006/abbi.1996.0256
108
SolierS.KohnK. W.ScrogginsB.XuW.TrepelJ.NeckersL.et al. (2012). Heat shock protein 90alpha (HSP90alpha), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc. Natl. Acad. Sci. U. S. A.109, 12866–12872. 10.1073/pnas.1203617109
109
StawowczykM.NaseemS.MontoyaV.BakerD. P.KonopkaJ.ReichN. C. (2018). Pathogenic effects of IFIT2 and interferon-beta during fatal systemic Candida albicans infection. MBio9:e00365–e00418. 10.1128/mBio.00365-18
110
StawowczykM.Van ScoyS.KumarK. P.ReichN. C. (2011). The interferon stimulated gene 54 promotes apoptosis. J. Biol. Chem.286, 7257–7266. 10.1074/jbc.M110.207068
111
SuC. C.YangH. F.HuangS. J.Lian IeB. (2007). Distinctive features of oral cancer in Changhua County: high incidence, buccal mucosa preponderance, and a close relation to betel quid chewing habit. J. Formos. Med. Assoc.106, 225–233. 10.1016/S0929-6646(09)60244-8
112
Subbarao SreedharA.KalmárÉ.CsermelyP.ShenY.-F. (2004). Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett.562, 11–15. 10.1016/S0014-5793(04)00229-7
113
SuomelaS.CaoL.BowcockA.Saarialho-KereU. (2004). Interferon alpha-inducible protein 27 (IFI27) is upregulated in psoriatic skin and certain epithelial cancers. J. Invest. Dermatol.122, 717–721. 10.1111/j.0022-202X.2004.22322.x
114
TaitS. W.GreenD. R. (2010). Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol.11, 621–632. 10.1038/nrm2952
115
TangW. G.HuB.SunH. X.SunQ. M.SunC.FuP. Y.et al. (2017). Long non-coding RNA00364 represses hepatocellular carcinoma cell proliferation via modulating p-STAT3-IFIT2 signaling axis. Oncotarget8, 102006–102019. 10.18632/oncotarget.22039
116
TatebeH.ShiozakiK. (2003). Identification of Cdc37 as a novel regulator of the stress-responsive mitogen-activated protein kinase. Mol. Cell. Biol.23, 5132–542. 10.1128/MCB.23.15.5132-5142.2003
117
TerenziF.HuiD. J.MerrickW. C.SenG. C. (2006). Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J. Biol. Chem.281, 34064–34071. 10.1074/jbc.M605771200
118
TerenziF.SaikiaP.SenG. C. (2008). Interferon-inducible protein, P56, inhibits HPV DNA replication by binding to the viral protein E1. EMBO J.27, 3311–3321. 10.1038/emboj.2008.241
119
TerenziF.WhiteC.PalS.WilliamsB. R.SenG. C. (2007). Tissue-specific and inducer-specific differential induction of ISG56 and ISG54 in mice. J. Virol.81, 8656–8665. 10.1128/JVI.00322-07
120
TraversS. A.FaresM. A. (2007). Functional coevolutionary networks of the Hsp70-Hop-Hsp90 system revealed through computational analyses. Mol. Biol. Evol.24, 1032–1044. 10.1093/molbev/msm022
121
TrepelJ.MollapourM.GiacconeG.NeckersL. (2010). Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer10, 537–549. 10.1038/nrc2887
122
TsutsumiS.BeebeK.NeckersL. (2009). Impact of heat-shock protein 90 on cancer metastasis. Fut. Oncol.5, 679–688. 10.2217/fon.09.30
123
TsutsumiS.NeckersL. (2007). Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci.98, 1536–1539. 10.1111/j.1349-7006.2007.00561.x
124
VarelaM.Diaz-RosalesP.PereiroP.Forn-CuniG.CostaM. M.DiosS.et al. (2014). Interferon-induced genes of the expanded IFIT family show conserved antiviral activities in non-mammalian species. PLoS ONE9:e100015. 10.1371/journal.pone.0100015
125
WacherC.MullerM.HoferM. J.GettsD. R.ZabarasR.OusmanS. S.et al. (2007). Coordinated regulation and widespread cellular expression of interferon-stimulated genes (ISG) ISG-49, ISG-54, and ISG-56 in the central nervous system after infection with distinct viruses. J. Virol.81, 860–871. 10.1128/JVI.01167-06
126
WallaceT. A.MartinD. N.AmbsS. (2011). Interactions among genes, tumor biology and the environment in cancer health disparities: examining the evidence on a national and global scale. Carcinogenesis32, 1107–1121. 10.1093/carcin/bgr066
127
WangB.ZhangS.YueK.WangX.-D. (2013). The recurrence and survival of oral squamous cell carcinoma: a report of 275 cases. Chin. J. Cancer32, 614–618. 10.5732/cjc.012.10219
128
WangY.ZhangL.ZhengX.ZhongW.TianX.YinB.et al. (2016). Long non-coding RNA LINC00161 sensitises osteosarcoma cells to cisplatin-induced apoptosis by regulating the miR-645-IFIT2 axis. Cancer Lett.382, 137–146. 10.1016/j.canlet.2016.08.024
129
WarnakulasuriyaS.TrivedyC.PetersT. J. (2002). Areca nut use: an independent risk factor for oral cancer. BMJ324:799. 10.1136/bmj.324.7341.799
130
WatheletM. G.ClaussI. M.ContentJ.HuezG. A. (1988). The IFI-56K and IFI-54K interferon-inducible human genes belong to the same gene family. FEBS Lett. 231, 164–171. 10.1016/0014-5793(88)80724-5
131
WeichselbaumR. R.IshwaranH.YoonT.NuytenD. S. A.BakerS. W.KhodarevN.et al. (2008). An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc. Natl. Acad. Sci. U.S.A. 105, 18490–18495. 10.1073/pnas.0809242105
132
WenC. P.TsaiM. K.ChungW. S.HsuH. L.ChangY. C.ChanH. T.et al. (2010). Cancer risks from betel quid chewing beyond oral cancer: a multiple-site carcinogen when acting with smoking. Cancer Causes Control. 21, 1427–1435. 10.1007/s10552-010-9570-1
133
WhitesellL.LindquistS. L. (2005). HSP90 and the chaperoning of cancer. Nat. Rev. Cancer5, 761–772. 10.1038/nrc1716
134
WilsonV. G.WestM.WoytekK.RangasamyD. (2002). Papillomavirus E1 proteins: form, function, and features. Virus Genes24, 275–290. 10.1023/A:1015336817836
135
XiaoS.LiD.ZhuH. Q.SongM. G.PanX. R.JiaP. M.et al. (2006). RIG-G as a key mediator of the antiproliferative activity of interferon-related pathways through enhancing p21 and p27 proteins. Proc. Natl. Acad. Sci. U.S.A.103, 16448–16453. 10.1073/pnas.0607830103
136
XiongX.WangY.LiuC.LuQ.LiuT.ChenG.et al. (2014). Heat shock protein 90beta stabilizes focal adhesion kinase and enhances cell migration and invasion in breast cancer cells. Exp. Cell Res.326, 78–89. 10.1016/j.yexcr.2014.05.018
137
XuW.MimnaughE.RosserM. F.NicchittaC.MarcuM.YardenY.et al. (2001). Sensitivity of mature Erbb2 to geldanamycin is conferred by its kinase domain and is mediated by the chaperone protein Hsp90. J. Biol. Chem.276, 3702–3708. 10.1074/jbc.M006864200
138
XuW.MimnaughE. G.J.-,KimS.TrepelJ. B.NeckersL. M. (2002). Hsp90, not Grp94, regulates the intracellular trafficking and stability of nascent ErbB2. Cell Stress Chaperones7, 91–96. 10.1379/1466-1268(2002)007<0091:HNGRTI>2.0.CO;2
139
YangY.ZhouY.HouJ.BaiC.LiZ.FanJ.et al. (2017). Hepatic IFIT3 predicts interferon-alpha therapeutic response in patients of hepatocellular carcinoma. Hepatology66, 152–166. 10.1002/hep.29156
140
YangY. C.ChengT. Y.HuangS. M.SuC. Y.YangP. W.LeeJ. M.et al. (2016). Cytosolic PKM2 stabilizes mutant EGFR protein expression through regulating HSP90-EGFR association. Oncogene35, 3387–3398. 10.1038/onc.2015.397
141
YangZ.LiangH.ZhouQ.LiY.ChenH.YeW.et al. (2012). Crystal structure of ISG54 reveals a novel RNA binding structure and potential functional mechanisms. Cell Res.22, 1328–1338. 10.1038/cr.2012.111
142
YenA.-H.ChenY.-C.PiduguV. K.LiuC.-J.LeeT.-C. (2014). Abstract 40: Association of Interferon-induced protein with tetratricopeptide repeats 1 and 3 expression with regional metastasis and drug resistance in oral squamous cell carcinoma. Cancer Res.74(Suppl. 19):40. 10.1158/1538-7445.AM2014-40
143
YilmazM.ChristoforiG. (2009). EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev.28, 15–33. 10.1007/s10555-008-9169-0
144
ZaretskyJ. M.Garcia-DiazA.ShinD. S.Escuin-OrdinasH.HugoW.Hu-LieskovanS.et al. (2016). Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med.375, 819–29. 10.1056/NEJMoa1604958
145
ZhangB.LiuX.ChenW.ChenL. (2013). IFIT5 potentiates anti-viral response through enhancing innate immune signaling pathways. Acta Biochim. Biophys. Sin.45, 867–874. 10.1093/abbs/gmt088
146
ZhangJ. F.ChenY.LinG. S.ZhangJ. D.TangW. L.HuangJ. H.et al. (2016). High IFIT1 expression predicts improved clinical outcome, and IFIT1 along with MGMT more accurately predicts prognosis in newly diagnosed glioblastoma. Hum. Pathol.52, 136–144. 10.1016/j.humpath.2016.01.013
147
ZhangY.KongY.LiuS.ZengL.WanL.ZhangZ. (2017). Curcumin induces apoptosis in human leukemic cell lines through an IFIT2-dependent pathway. Cancer Biol. Ther.18, 43–50. 10.1080/15384047.2016.1276129
148
ZhaoC.DenisonC.HuibregtseJ. M.GygiS.KrugR. M. (2005). Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl. Acad. Sci. U. S. A.102, 10200–10205. 10.1073/pnas.0504754102
149
ZhaoY.Altendorf-HofmannA.PoziosI.CamajP.DaberitzT.WangX.et al. (2017). Elevated interferon-induced protein with tetratricopeptide repeats 3 (IFIT3) is a poor prognostic marker in pancreatic ductal adenocarcinoma. J. Cancer Res. Clin. Oncol.143, 1061–1068. 10.1007/s00432-017-2351-4
150
ZhengC.ZhengZ.ZhangZ.MengJ.LiuY.KeX.et al. (2015). IFIT5 positively regulates NF-kappaB signaling through synergizing the recruitment of IkappaB kinase (IKK) to TGF-beta-activated kinase 1 (TAK1). Cell. Signal.27, 2343–2354. 10.1016/j.cellsig.2015.08.018
Summary
Keywords
IFIT, cancer, TPRs, OSCC, progression, metastasis, drug resistance
Citation
Pidugu VK, Pidugu HB, Wu M-M, Liu C-J and Lee T-C (2019) Emerging Functions of Human IFIT Proteins in Cancer. Front. Mol. Biosci. 6:148. doi: 10.3389/fmolb.2019.00148
Received
25 September 2019
Accepted
04 December 2019
Published
19 December 2019
Volume
6 - 2019
Edited by
Robert M. Brosh, National Institute on Aging (NIA), United States
Reviewed by
Jennifer L. Hyde, School of Medicine, University of Washington, United States; Chinnadurai Mani, Texas Tech University Health Sciences Center, United States
Updates
Copyright
© 2019 Pidugu, Pidugu, Wu, Liu and Lee.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Te-Chang Lee bmtcl@ibms.sinica.edu.tw
This article was submitted to Molecular Diagnostics and Therapeutics, a section of the journal Frontiers in Molecular Biosciences
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.