 Aboubakr Haredi Abdelmonsef1*
Aboubakr Haredi Abdelmonsef1* Mahmoud Eldeeb Mohamed1Mohamed El-Naggar2Hussain Temairk1Ahmed Mohamed Mosallam1
Mahmoud Eldeeb Mohamed1Mohamed El-Naggar2Hussain Temairk1Ahmed Mohamed Mosallam1- 1Chemistry Department, Faculty of Science, South Valley University, Qena, Egypt
- 2Chemistry Department, Faculty of Sciences, University of Sharjah, Sharjah, United Arab Emirates
The research explores the synthesis of a series of novel hybrid quinazolin-2,4-dione analogs bearing acetyl/amide bridged-nitrogen heterocyclic moieties such as azetidinone, pyrrole, oxazole, oxadiazole, thiazole, pyrazole, and thiazolidine scaffolds 2-16. The newly synthesized compounds were structurally confirmed by means of IR, 1H-NMR, 13C-NMR, MS and elemental analysis. In addition, an in silico molecular docking analysis of new compounds and standard drug (Chloroquine) has been performed to analyze the binding modes of interaction to the putative active site of Plasmodium falciparum Dihydroorotate dehydrogenase (pfDHODH). Aiming to search for potentially better antimalarials, a modern approach has been undertaken to identify new quinazolin-2,4-dione derivatives targeting pfDHODH. The identification of antimalarial activity of the newly synthesized compounds by using experimental techniques is expensive and requires extensive pains and labor. The compound 11 showed the highest binding affinity against pfDHODH. Moreover, the electrostatic potential (ESP) of the docked molecules was also calculated. Further, the pharmacokinetic properties (ADMET) of the prepared compounds were predicted through in silico technique.
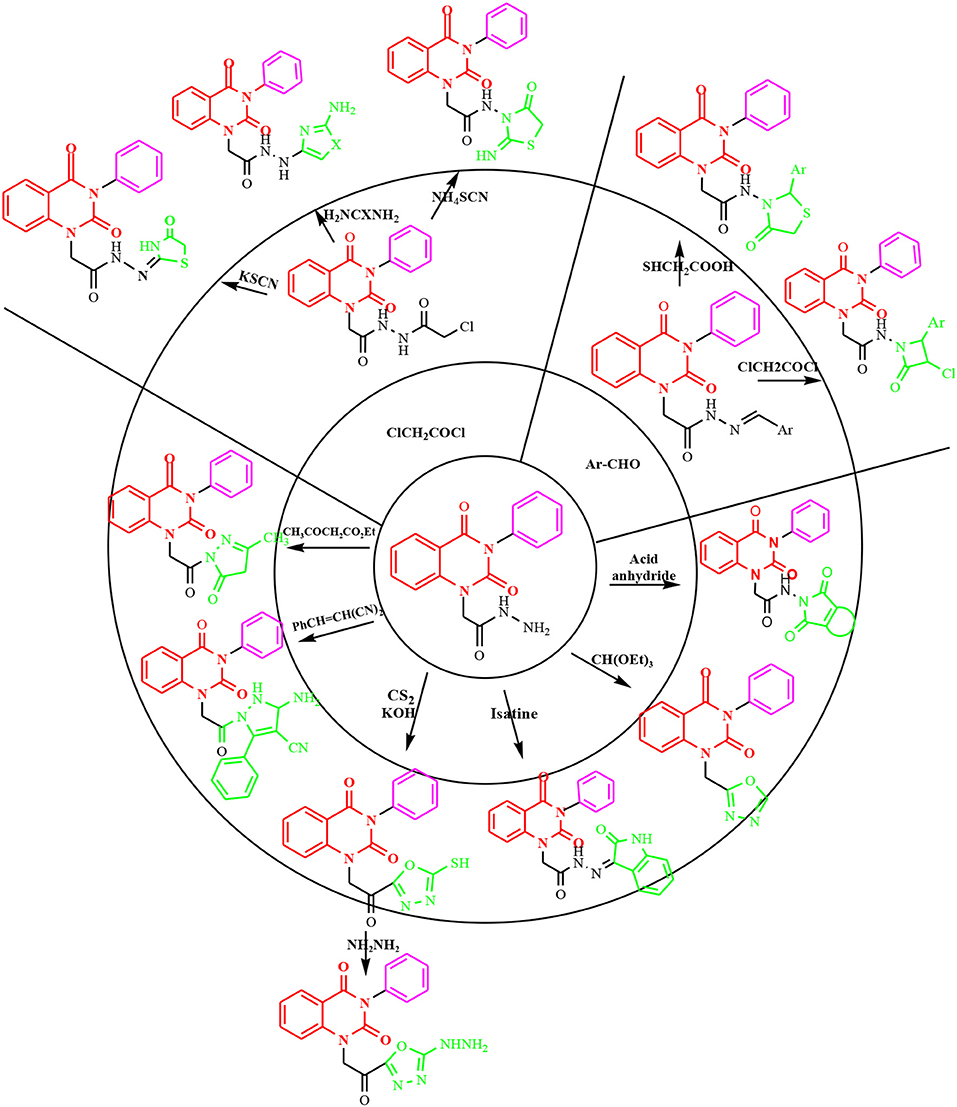
Graphical Abstract. Synthesis of new quinazolin-2,4-dione hybrid molecules.
Introduction
Malaria is a parasitic disease caused by single-celled microorganisms of Plasmodium group (Nivsarkar et al., 2005; Sahu et al., 2019). It is a widespread health problem in the tropical regions (Campbell, 1997; Autino et al., 2012). According to WHO, about 219 million cases and 430,000 related deaths worldwide of malaria were reported in the year 2017 (Talapko et al., 2019). Plasmodium falciparum Dihydroorotate dehydrogenase is an essential enzyme which is predominantly responsible for the growth of malaria parasite (Talapko et al., 2019; Vikram and Mishra, 2019). These parasites are transmitted to the human by the bites of infected female anopheles mosquitoes. Four species of plasmodia are commonly known to cause human malaria: P. falciparum, P. vivax, P. ovale, and P. malariae (Oguike et al., 2011). The 85% of malarial deaths are caused by the intracellular protozoan parasite Plasmodium falciparum (Blasco et al., 2017). Therefore, the targeting of the human enzyme pfDHODH is the main concern in combating malaria disease.
Molecular hybridization concept has gained much interest in last decades (Claudio et al., 2007). The synthesis of hybrid molecules by combination of two different biologically moieties gave promising information for medicinal chemistry, as they exhibited anti-fungal, anti-malarial, anti-cancer, and anti- inflammatory activities (Gómez and Kouznetsov, 2013). Quinazolin-2,4-diones are such interesting structural cores that found to have significant attention in medicinal and pharmaceutical chemistry (Hassan et al., 2013a,b; El-Sheshtawy et al., 2017; Abdelmonsef and Mosallam, 2020; El-Naggar et al., 2020). Heterocycles bearing nitrogen such as azetidinone, pyrrole, oxazole, oxadiazole, thiazole, pyrazole, and thiazolidine scaffolds are reported to possess a broad spectrum of applications in medicinal, biological as well as pharmaceutical activities as anti-fungal, anti-bacterial, anti-tumor, and anti-inflammatory drugs (Tanitame et al., 2004; Dua et al., 2011; Sokolova et al., 2017). Nowadays, the synthesis of nitrogen containing heterocyclic systems attached to quinazolin-2,4-dione ring have drawn considerable importance owing to their enormous applications in medicinal and pharmaceutical chemistry (Patel et al., 2018).
Guided by these findings, and as a part of our efforts to synthesis of novel dual hybrid molecules, our group planned to incorporate the quinazolin-2,4-dione moiety as a main scaffold attached with various N-heterocyclic moieties at nitrogen N-1 position through acetyl/amide linkage 2-16 which can be used as possible antimalarials targeting pfDHODH using structure-based drug discovery approach. Chloroquine is an aminoquinoline which belongs to a class of potent anti-malarial drugs (Bawa et al., 2010) and has been selected as a reference ligand in order to compare its binding affinity and their of the prepared compounds toward pfDHODH. The electrostatic potential of the molecules was performed through the determination of the electron charge density, to understand the intermolecular interactions to the target enzyme (Drissi et al., 2015). In addition, the pharmacological and pharmacokinetics properties of these compounds were also further calculated using in silico methods such as Mol inspiration and admetSAR web servers.
Results and Discussion
Chemistry
In continuation of our efforts, herein we reported the synthesis of 15 structural hybrid molecules of quinazolindione coupled to four and five membered N-heterocycles at N-1 position by using the synthetic protocols as depicted in Scheme 1. The acetyl/amide group serves as a connecting bridge fragment between both nucleolus. The carbohydrazide analog 1, was obtained using the method described by our group (El-Naggar et al., 2020) via the treatment of 2,4-Dioxo-3-phenyl-3,4-dihydroquinazolin-1(2H)-yl) acetate with hydrazine hydrate. The compound 1 is used as a reaction intermediate in construction of four and five membered heterocyclic systems such as azetidine, pyrrole, oxazole, oxadiazole, thiazole, and thiazolidine moieties. A series of Schiff bases 2a-e were synthesized by reaction of so formed 2-(2,4-dioxo-3-phenyl-3,4-dihydroquinazolin-1(2H)-yl)acetohydrazide 1 with an equimolar amount of different aromatic aldehydes namely, benzaldehyde, 4-nitrobenzaldehyde, 4-chlorobenzaldehyde, salicylaldehyde, and thiophene-2-carboxaldehyde, respectively. IR spectra of benzylidine hydrazide derivatives 2a-e provide valuable information regarding nature of functional group present in these compounds. The bands around 1,576–1,625 cm−1 suggested the presence of CH=N (azomethine linkage) in the compounds. In addition, the 1H-NMR spectra for compounds 2a-e revealed singlet signal at δ 4.37 ppm characteristic for N-CH2-CO, multiplet signals at the range of δ 7.25–8.06 ppm for aromatic protons, in addition, the important signal at δ 5.81 ppm equivalent to one hydrogen due to the CH=N group of the Schiff bases, plus singlet signals at 11.51 ppm assigned for NH's, respectively, which were exchangeable by D2O. The so formed Schiff base 2a was converted to azetidin-2-one 3a by [2+2] cycloaddition reaction with chloroacetyl chloride as shown in Scheme 2. The desired compound 3a has been approved by disappearance of absorption band of 1,576 cm−1 which is related to azomethine group (-N=CH), and appearance of the carbonyl azetidinone band at 1,660 cm−1. Furthermore, the appearance of new band at 673 cm−1 due to the stretching for C-Cl bond. Moreover, it can be confirmed on the basis of 1H-NMR, by disappearance of azomethine signal at δ 5.81 ppm and the appearance of two signals in the region δ 5.48–6.45 ppm due to aliphatic protons of new azetidinone ring, -N-CH-Ar and –CO-CH-Cl. Heterocyclization of compound 2a with thioglycolic acid yielded quinazolin-2,4-dione analog attached to 2-thiazolidine 4a, by nucleophilic addition to the carbonyl group of thioglycolic acid. The structure of the resulted compound 4a was efficiently characterized by IR and 1H-NMR. For instance, the FT-IR spectrum showed the appearance of stretching frequency of amidic carbonyl group (N-C=O) at 1,726 cm−1 due to the thiazolinone ring and this considered the most characteristic evidence for the success of cyclization step. Beside the disappearance of –N=C group at 1,576 cm−1 for Schiff base 2a. In addition, 1H-NMR spectrum of compound 4a exhibited new characteristic signal for CH2 group adjacent to S (CH2-S) appeared at δ 4.37 ppm; this again confirmed the formation of thiazolinone ring. The proposed mechanism of this reaction is declared in Scheme 3. Compound 5 was resulted from reaction of carbohydrazide analog 1 with β-ketoester like ethyl acetoacetate through Knorr pyrazole synthesis as shown in Scheme 4 (Knorr, 1883). The latter compound showed in its 1H-NMR spectrum characteristic peaks at δ 1.36, 4.38, and 7.25–8.07 ppm attributed to CH3, CH2 and aromatic protons, respectively. Moreover, 3-Amino-1-[2-(2,4-dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-5-phenyl-2,3-dihydro-1H-pyrazole-4-carbonitrile 6 was obtained employing the reaction of compound 1 with benzylidene malononitrile. The structure of compound 6 was corroborated by its IR, 1H-NMR and elemental analysis. The IR spectra of the resulted 6 showed absorption bands at 3,400 and 2,225 cm−1 for NH2 and C=N groups, respectively. In addition, the formation of compound 8 was prepared accordingly, by heating of carbohydrazide 1 with CS2 in presence of alc. KOH to yield compound 7, and then the latter compound was separated and treated with hydrazine hydrate. The 1H-NMR spectroscopic technique used to characterize the newly synthesized compound 8; it showed new characteristic signals for NH and NH2 groups at 11.53 and 8.73 ppm, respectively. Chloro-acetic acid N'-[2-(2,4-dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-hydrazide 9, which was synthesized by reaction of compound 1 with chloroacetyl chloride (El-Naggar et al., 2020), is used as starting material to synthesize of compounds 10-13. The reaction of compound 9 with potassium thiocyanate through Dimroth type rearrangement to yield the annealed compound 10 as declared in Scheme 5. The FT-IR spectrum of the compound 10 showed the appearance of stretching band of C=NH group at 1,580 cm−1. On the other hand, the compound 9 underwent heterocyclization with urea, and thiourea to afford compounds 11 and 12, respectively, as declared in Scheme 6. The 1H-NMR spectra of both compounds showed new characteristic signal at 8.72 ppm for NH2 group, in addition, appearance of the CH proton in interference with the aromatic protons in the same range for the newly synthesized compounds, which is considered the most characteristic evidence for the success of cyclization step and formation of oxazole and thiazole rings, respectively. Preparation of compound 13 was achieved by heterocyclization of compound 9 with ammonium thiocyanate. 1H-NMR spectrum of compound 13 shows new signal for CH2 group at 4.3 ppm of new thiazolinone ring. The formation compounds 14 and 15 were attempted through refluxing of carbohydrazide 1 with Isatine and Triethyl orthoformate, respectively. The 1H-NMR spectrum of compound 14 shows new signal for NH group at 8.74 ppm of new Isatine ring introduced to hydrazide 1. The 1H-NMR spectrum of compound 15 showed disappearance of signals related to NH and NH2 groups, and appearance of new signal related to CH proton of new oxadiazole ring, which is in interference with the aromatic protons. The FT-IR spectrum of the resulted compound showed the appearance of –N=C group at 1,600 cm−1 for oxadiazole derivative 15. Finally, reaction of carbohydrazide analog 1 with various acid anhydrides like maleic, phthalic, and tetrachlorophthalic anhydride through dehydrative condensation reaction, gives the corresponding imides 16a-c, respectively. The 1H-NMR/IR spectra of the resulted imides showed disappearance of signal/band related to NH2 group, with appearance of new signals/bands. The mass spectra of compounds 2-16 add additional confirmation for elucidation of the structures which showed molecular ion peaks corresponding to molecular formula of these compounds further confirmed the assigned structures. CHN analysis was confirmed the validity of the assumed structures of the newly synthesized compounds (see Experimental section).
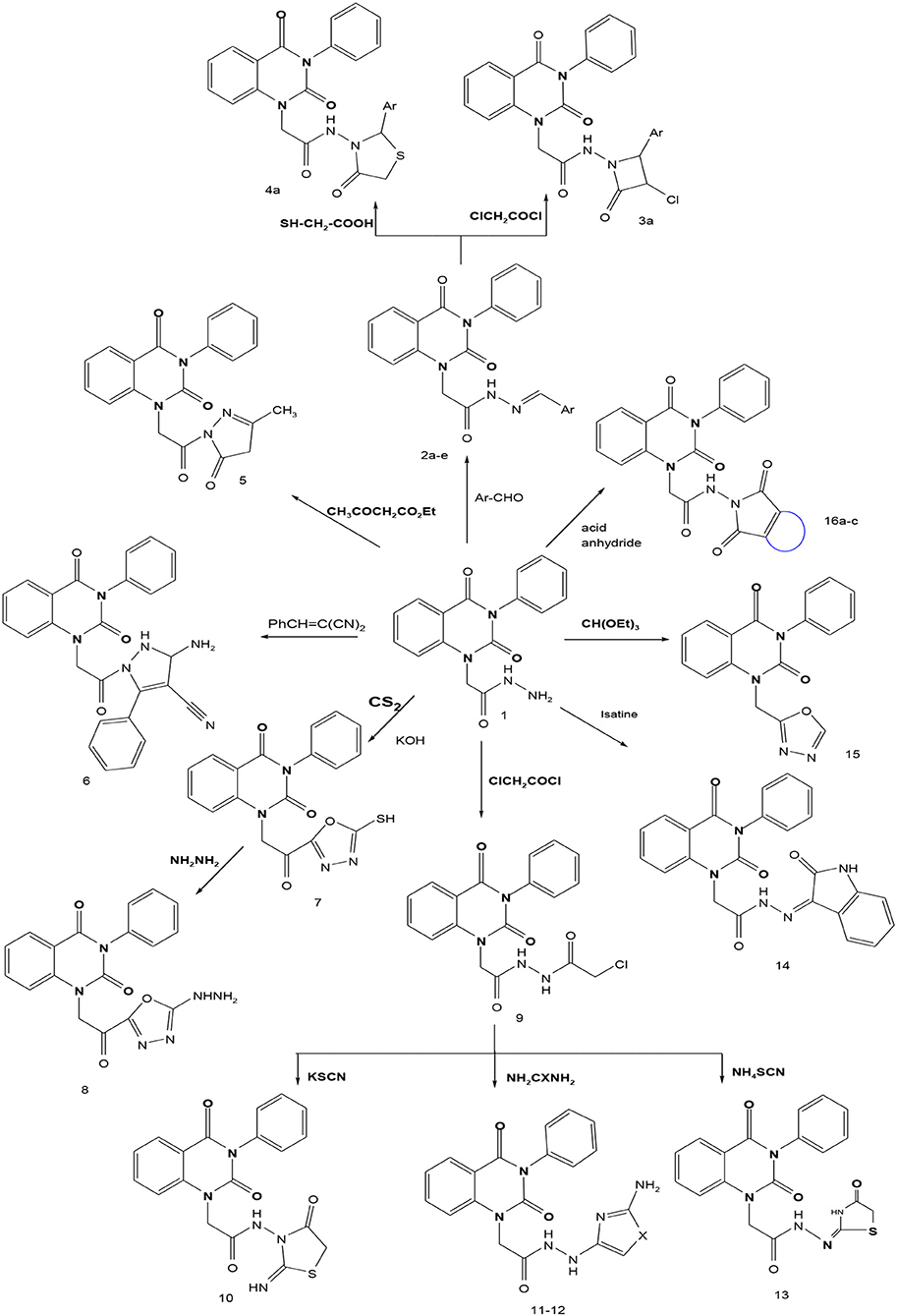
Scheme 1. Synthesis of quinazolin-2,4-dione analog bearing N-heterocyclic moieties 2-16. ArCHO (a- benzaldehyde, b- 4-nitrobenzaldehyde, c- 4-chloro benzaldehyde, d- salicylaldehyde, and e- thiophene-2-carboxaldehyde). X=O, and S. Acid anhydrides: a- maleic, b- phthalic, and c- tetrachlorophthalic anhydride.
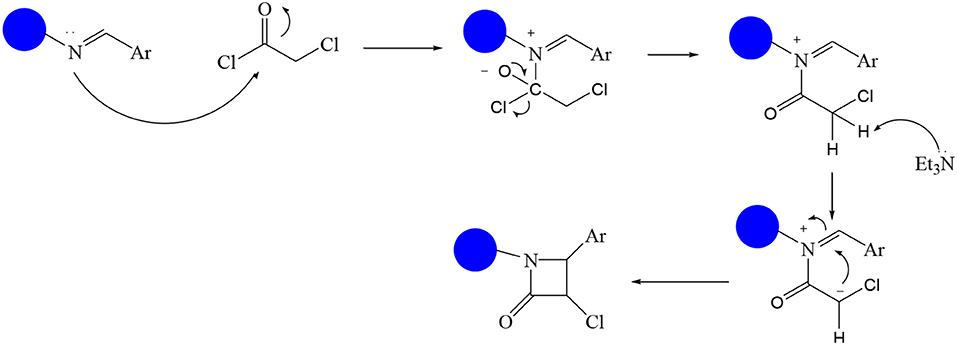
Scheme 2. Synthesis of azetidinone derivative 3a.
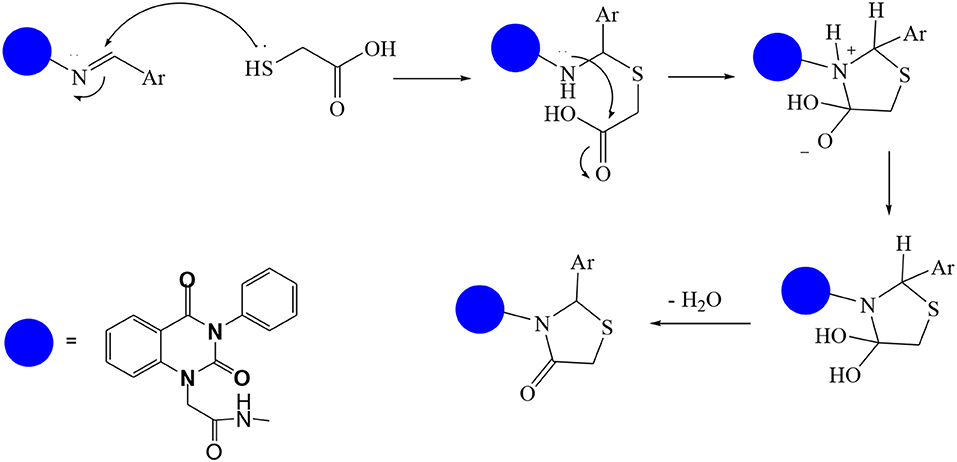
Scheme 3. Reaction protocol for the synthesis of compound 4a.
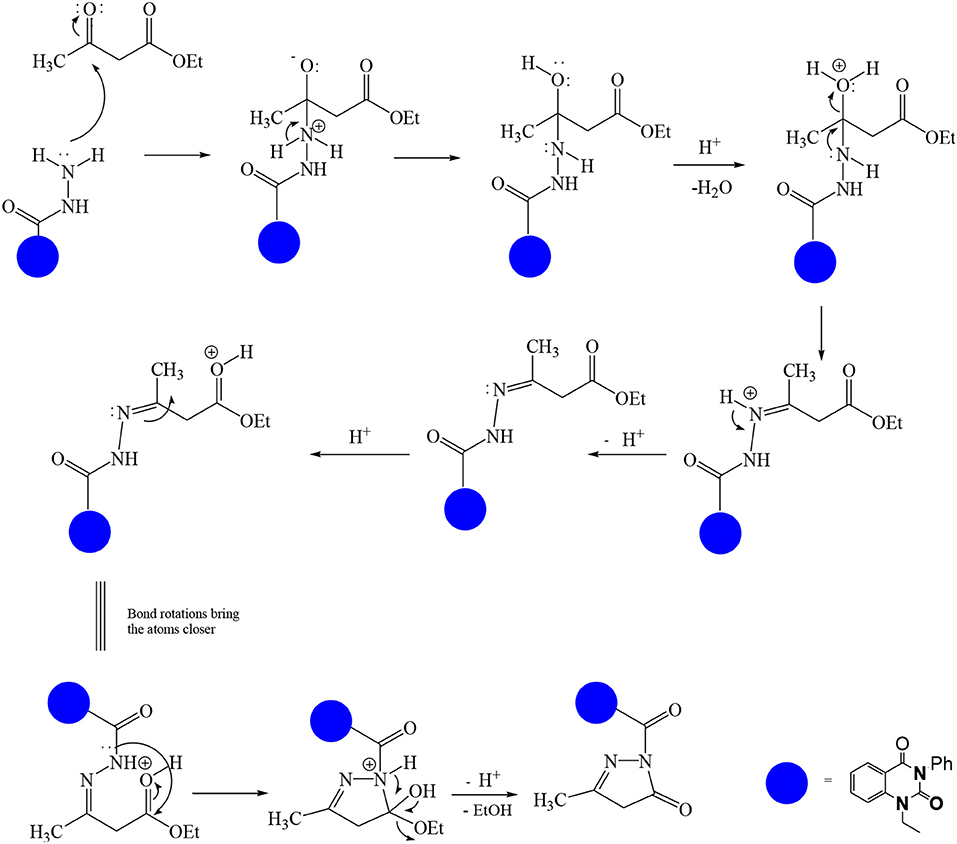
Scheme 4. Mechanism of formation of compound 5.
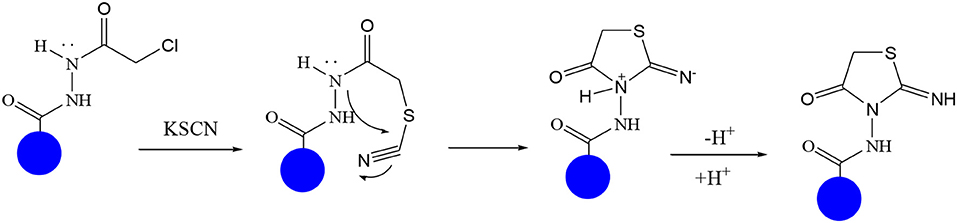
Scheme 5. Synthetic strategy of analog 10.
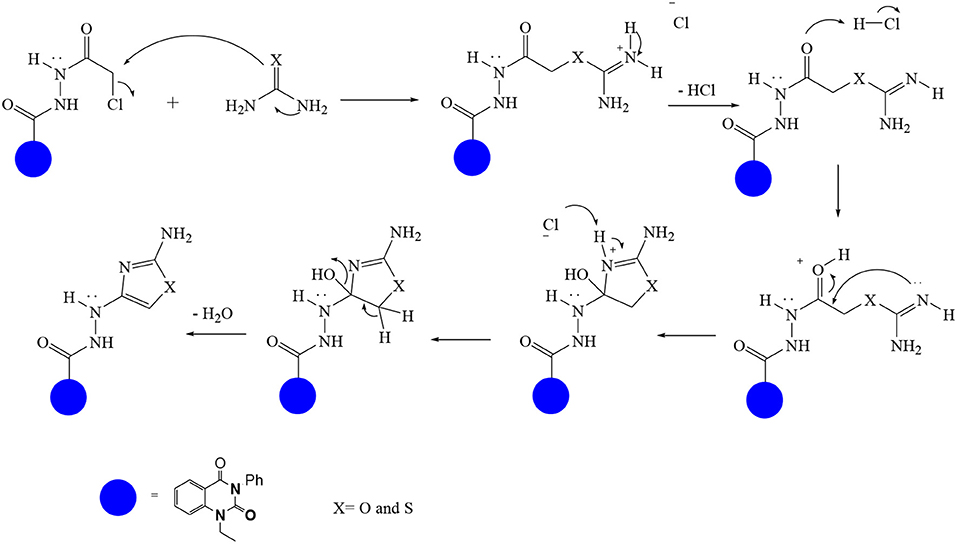
Scheme 6. Plausible mechanism for the synthesis of compounds 11-12.
In silico study
Three- dimensional crystal structure of pfDHODH (PDB ID: 1tv5) was retrieved from RCSB Protein Data Bank web server (Hurt et al., 2006) (Figure S1). The PDB file of the target enzyme was optimized by energy minimization and prepared prior to docking by removal of water molecules and atomic clashes. Moreover, H-atoms and gasteiger charges were added to the target enzyme for correct ionization and tautomeric states of residues. The protonation states of ionizable groups in receptor were theoretically calculated according to Propka (Table S1). In accordance with previously literatures, the putative binding site residues of the target enzyme pfDHODH are PHE171, LEU172, CYS175, GLY181, CYS184, HIS185, PHE188, LEU189, PHE227, ILE263, ARG265, TYR528, LEU531, VAL532, GLY535, and MET536 (Vikram and Mishra, 2019), which found to be playing a catalytic role by forming the intermolecular interactions with the ligand molecules. Grid was then allocated around the region of interest (binding site) to perform further structure-based screening approach (Abdelmonsef et al., 2016; Aboubakr et al., 2016). In-house set of 22 analogs of quinazolindione derivatives was used for study. In addition, ligand molecules must be prepared to create 3D geometries, assign proper bond orders, and generate accessible tautomerization and ionization sates prior for molecular docking studies (Figure S2). A library of 22 compounds was subjected to PyRx-Virtual screening tool version 8.0 through inbuilt Autodock 4.2 with the Lamarckian genetic algorithm (LGA) as scoring function (Morris et al., 1639), in order to carry the docking simulation with pfDHODH. Docking approach was performed on default parameters of number of generation and energy evaluation for 10 steps of run. Molecular docking study was accomplished to estimate the binding mode interactions between a novel series of prepared molecules and active site region of the target enzyme (Dasari et al., 2017; Rondla et al., 2017; Shehadi et al., 2020). Upon the completion of each docking approach, a maximum output of 9 distinct conformational clusters was calculated for each ligand. The molecule with lowest binding energy (i.e., more negative) indicates highest binding affinity to enzyme (Hussein et al., 2018; Abdelmonsef, 2019). The predicted binding energies were computed in units of kcal/mol. Generally, the target compounds fit well into the binding regions of pfDHODH with different binding energies. Docking of the target analogs to pfDHODH yielded scores between −12.2 and −5.7 kcal/mol, compared to −7.2 kcal/mol for reference drug (Chloroquine), and disclosed the derivative 11 as a lead compound. On close inspection of Table 1, Figure 1, and (Figure S3) potential interactions (hydrogen bonds, π-π stacking, π-cation, and π-sigma), bond lengths and atoms involved in these interactions were observed between the synthesized molecules and reference with enzyme. The analog 1 was successfully docked into pfDHODH with binding energy −9.6 kcal/mol. It exhibited hydrogen bond and π-π interactions (π-stacking) with the binding regions via four amino acid residues namely LYS429, SER477, SER505, and TYR528 at the distance of 2.91, 2.84, 2.31, 5.25, and 4.05 Å, respectively. In case of compounds 2a-e; all of them exhibited H-bond interactions, in addition they showed π-cation interactions except 2e with ARG544, ARG253, TYR207, and TYR528 at the distances of 6.47, 4.66, 5.93, 4.70, and 4.48 Å, respectively. Furthermore, they exhibited π-π interactions (edge to face) with TYR207 and TYR528, respectively, except derivatives 2b and 2e. Introduction of electron donating group (OH) on phenyl ring causing high activity as shown in compound 2d (best binding energy of them = −10.5 kcal/mol) (Metwally et al., 2018). On the other hand, the derivatives 2b (−7.1 kcal/mol) and 2c (−8.0 kcal/mol) with electron withdrawing groups such as –NO2 (strong) and –Cl (weak), respectively, exhibited less binding affinities in comparison to derivative 2d. Introducing of electron withdrawing groups (–NO2 and –Cl) on phenyl ring causing less activity of the compounds (Metwally et al., 2018). The compound 3a exhibited H-bond interactions with HIS185, ASN274, LYS429, SER477, and TYR528 at the distances of 2.95, 2.61, 2.96, 2.42, 2.97, and 2.95 Å, respectively. In addition to π-cation and π-sigma interactions with LYS429 and ILE263, respectively. The analog 4a formed π-π (edge to face, and face to face), and π-cation interactions with the residues ASN547, TYR207, and ARG544 at 2.93, 2.96, 5.12, 5.70 (edge to face), 3.70 Å (face to face), respectively. Compound 5 showed H-bond interactions with LYS260 and ARG262 at the distances of 3.12 and 2.92 Å. The product 6 of −8.5 kcal/mol exhibited three types of interactions like H-bond, π-π, and π-cation interactions with ASN547, TYR207, and ARG544 at 2.95, 4.28, 5.06, and 5.82 Å, respectively. In comparison between compounds 7 (−7.5 kcal/mol) and 8 (−8.4 kcal/mol); both of aromatic thiol and aromatic amine are electron donating groups, herein we noted that the presence of amino group enhanced the binding energy of compound 8 than compound 7 which contains thiol group. Compound 9 exhibited H-bond, π-π, π-cation interactions with the residues GLY507, SER529, TYR528, LYS229, and LYS429 at 3.01, 2.95, 3.69, 3.88, and 5.98 Å, respectively. Compound 10 showed H-bond and π-π interactions with the residues SER205, ASN547, and TYR207 at 3.09, 2.88, and 5.06 Å, respectively. In addition, the best binding energy was reported by compound 11 (−12.2 kcal/mol) formed different H-bonds with LYS429, SER477, SER505, and GLYS507 and π-π stacking with TYR528. The analog 12 exhibited H-bond and π-cation interactions with TYR168, SER160, ASN195, and PHE165 at the distances of 2.97, 2.22, 2.13, 2.15, and 6.71 Å, respectively. While the compound 13 of −7.7 kcal/mol docked into pfDHODH through one H-bond and two π-π interactions with TYR555 and TYR207. Moreover, the analog 14 exhibited H-bond and π-cation interactions with the active site pocket of the target enzyme through ARG253, THR318. The product 15 formed three types of interactions H-bond, π-cation and π-π through ASN274, LYS429, and TYR528 at the distances of 2.95, 2.64, 2.80, 2.85, 5.52, and 4.60 Å, respectively. Finally, the imide 16a formed H-bonds with SER477, 505, and 529, in addition to two π-π interactions with TYR528. In comparison between the phthalimides 16b-c, we noted that the presence of four chlorine groups in compound 16c caused the moderate binding energy (−8.6 kcal/mol) than compound 16b (−8.8 kcal/mol) which contains un-substituted benzene ring. To further understand the nature of flexible docking, tyrosine TYR207 and 528 formed unique binding mode via π-π stacking with some docked molecules, because it contains a phenyl ring on its side-chain that is involved in forming these interactions with the compounds. On the other hand, arginine ARG253, 262, and 544 contain a positively charged guanidinium group H2N-C(=NH)-NH-, that is involved in forming π-cation interaction with compounds 2a, 2b, 2c, 4a, 6, 7, and 14. In addition, lysine LYS229, 429 also contain a positively charged amino on its side-chain (H3N+) that is involved in forming π-cation interactions with compounds 3a, 8, 9, and 15. Meanwhile, the side chains of aromatic amino acids PHE165 and TYR528 in docked molecules 2d and 12 provide a surface of negative electrostatic potential that can bind to a wide range of cations (π-cation interaction) through electrostatic interactions. These interactions may explain the highest potency toward pfDHODH enzyme (Zhang et al., 2015). Interestingly, the tested compounds (with quinazoline moiety) reported in this work and the reference compound (with quinoline moiety) are of similar moieties and are of important N-based heterocyclic aromatic compounds with a broad range of bioactivities (Kumar et al., 2010). The activities of the newly synthesized compounds could be due to heterocyclic core structures with acetyl/amide linkage. Moreover, the electrostatic potential of the docked molecules was also calculated to describe the intermolecular interactions to the target (Drissi et al., 2015). Figure 2 shows the theoretical maps in 3D dimensions of the electrostatic potential of the best three docked molecules. The obtained results could be used to explain the interaction between the molecules and the target enzyme. For example, the best binding compounds 1, 2d, and 11 declared π-π stacking through their electronegative heterocyclic aromatic pyrimidine rings (Figure 2) and phenyl rings of TYR528. Further, an amidic carbonyl core -N-C=O of pyrimidine rings of the compounds exhibited three H-bonds with SER477 and LYS429, respectively. The electrostatic potential maps of the rest compounds are included as Figure S4. Moreover, the title analogs 1-16 were filtered depending on their Absorption, Distribution, Metabolism, Elimination and Toxicity (ADMET) (Table 2) and physicochemical significant descriptors (Table 3) using admetSAR and Mol inspiration online servers. The pharmacokinetic parameters (ADMET) reveal that newly synthesized compounds have better Human Intestinal Absorption (%HIA) score, and good Blood-Brain Barrier (BBB) values, which means that the compounds could be better absorbed by the human intestine. Furthermore, all prepared compounds displayed negative AMES toxicity and carcinogenicity test which means that the ligand molecules are non-mutagenic. In addition, the results of physiochemical properties predictions show interesting values for these compounds, whereas they have molecular weights in the range of 310–578 g/mol (<725), the partition coefficients of the compounds are <5. Also, the topological surface areas TPSA were found to be in the acceptable range (<140). In addition, the numbers of H-bond acceptors (HBA) and donors (HBD) in the tested compounds obey Lipinski's rule of five (Ro5), and were found to be in the range of 7–10 and 0–3, respectively. Finally, the synthesized compounds possess high numbers of rotatable bonds (3–6), which indicates that they are flexible. From all these results, we can conclude that all molecules exhibited good absorption, distribution and oral bioavailability within the body. These ligand molecules may be considered as potent drug candidates against pfDHODH and used as possible anti-malarial agents.
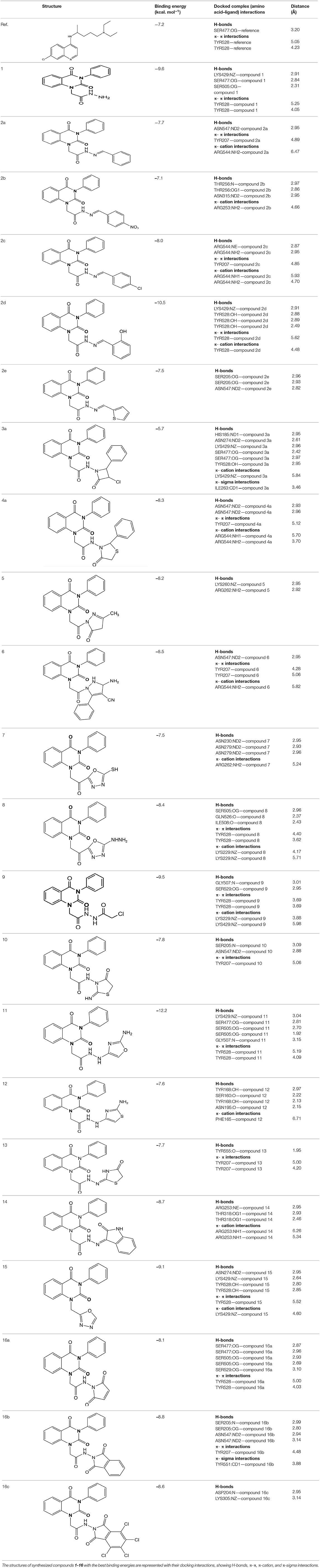
Table 1. Docking results and types of interactions between the title compounds 1–16 and active site region of pfDHODH enzyme.
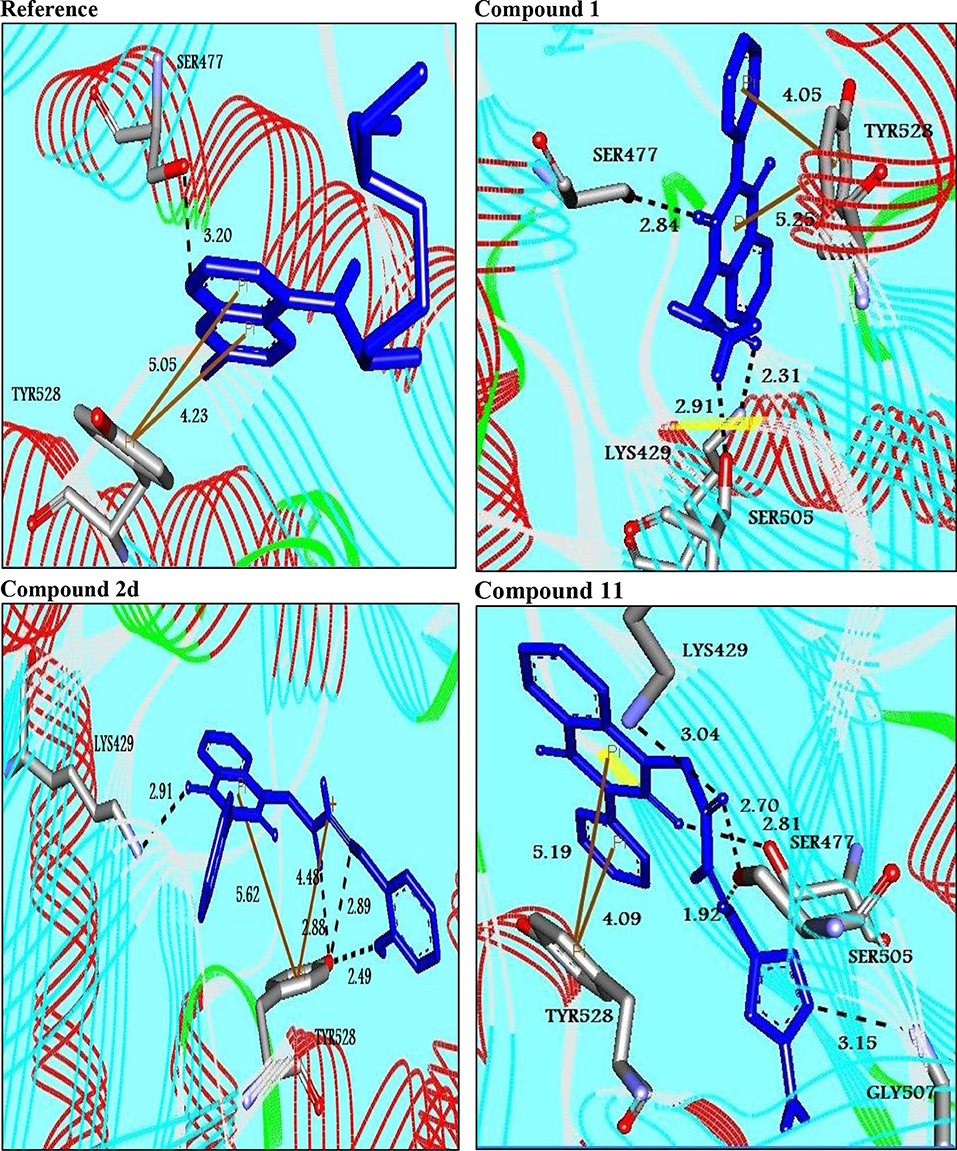
Figure 1. Molecular interactions between the best binders and pfDHODH. Three-dimensional of docking poses of the reference and three best binding compounds in the active pocket of pfDHODH. Blue stick models represented the docked compounds, and gray models represented the active site region. H-bond interactions are shown in black dotted lines. π- interactions are shown in orange lines.
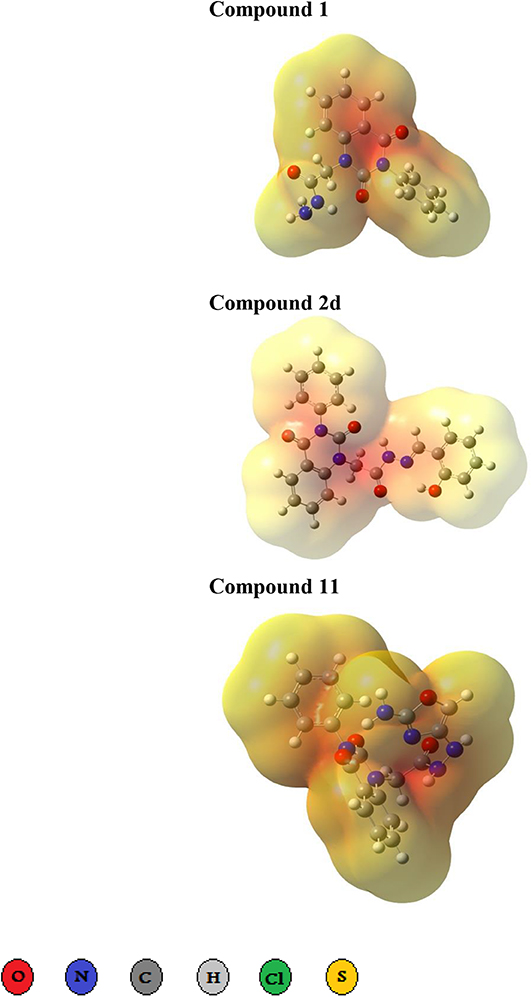
Figure 2. Three dimensional representation of electrostatic potential around the best three docked molecules. Electrostatic potential maps around the best binders 1, 2d, and 11. The red region represents highly electron density charges (electronegative), while pale yellow represents electropositive part.
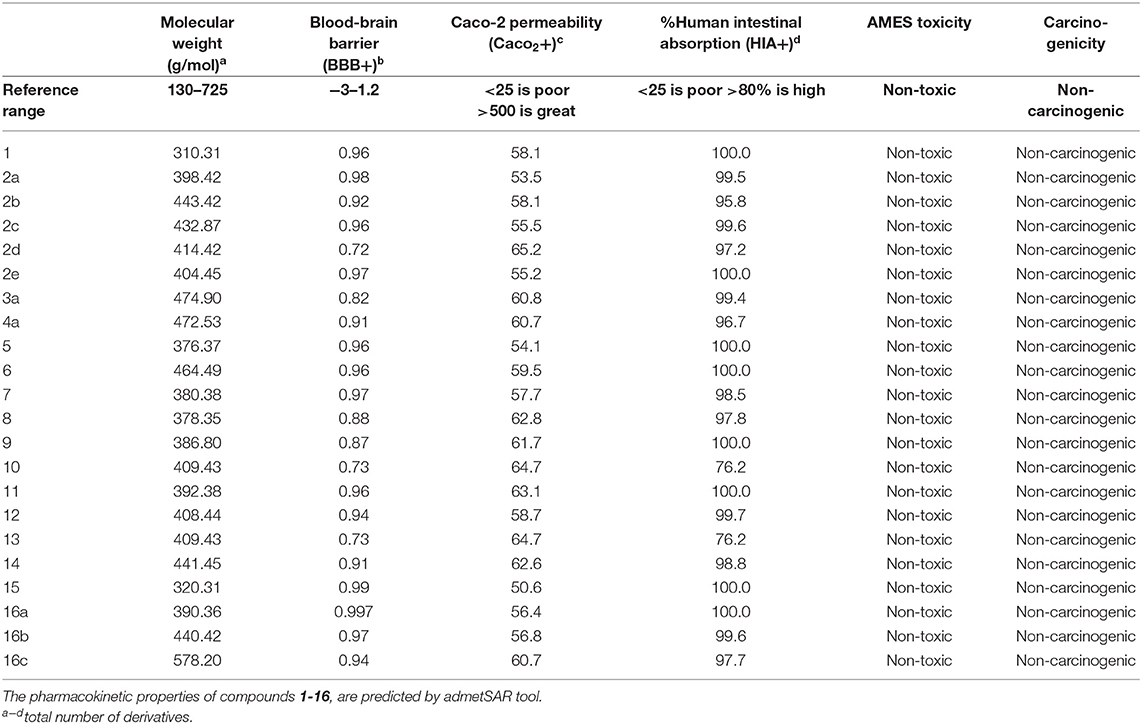
Table 2. List of ADMET properties of synthesized hybrid molecules 1-16.
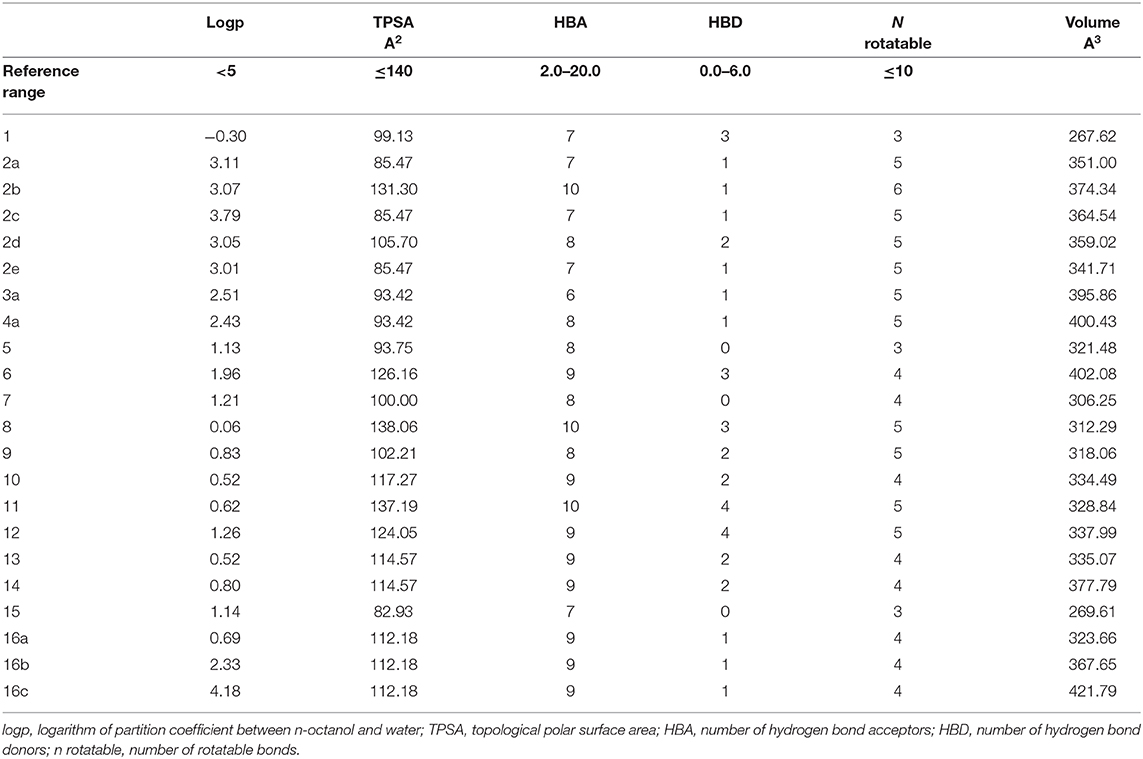
Table 3. Physicochemical properties of the title compounds 1–16.
Materials and Methods
The reagents used in this work were used without purification. The melting points of the prepared compounds were determined using Griffin apparatus and are uncorrected. The purity of the newly synthesized compounds 2-16 is mentioned by Thin Layer Chromatography TLC technique. IR spectra were recorded on Shimadzu 408 and Bruker Vect. 22. NMR spectra were run at 400 MHz using TMS as the internal standard. The chemical shifts were measured in ppm (δ) related to TMS (0.00 ppm). Finally, mass spectra were recorded on a HP model, Mass 5988 Mass spectrometer at 70 eV. All the prepared compounds were analyzed for C, H, and N at Cairo University, Egypt. Compounds 1 and 9 were already described (Hassan et al., 2013a).
General Procedures for Synthesis of Schiff Bases 2a-e
2-(2,4-dioxo-3-phenyl-3,4-dihydroquinazolin-1(2H)-yl)acetohydrazide 1 (1 mmol) was heated under reflux for 8–12 h with the appropriate aromatic aldehydes namely benzaldehyde, 4-nitrobenzaldehyde, 4-chlorobenzaldehyde, salicylaldehyde, and thiophene-2-carboxaldehyde (1 mmol) in absolute ethanol (20 mL) and drops of piperidine as a catalyst. After cooling, the reaction mixture was filtered off and crystallized from appropriate solvents to afford the Schiff bases 2a-e, respectively. 2a: Gray crystals; yield 86%; mp 210–211°C. FT-IR (KBr, υ, cm−1) = 3,333 (NH), 1,625, 1,655 (C=O's), 1,576 (CH=N). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) = 11.5 (s, 1H, NH), 7.2–8.06 (m, 14H, Ar-H), 5.8 (s, 1H, CH), 4.3 (s, 2H, CH2). 13C-NMR (CDCl3): δ 49.8, 114.4, 115.3, 124.7, 125.1, 126.5, 128.0, 128.2, 128.5, 128.7, 128.8, 128.9, 129.3, 129.5, 130.1, 131.8, 136.2, 136.6, 140.7, 142.5, 151.9, 161.3, 165.8. MS (EI): m/z (%) = 398.42 [M]+. Anal. Calcd for C23H18N4O3: C, 69.34; H, 4.55; N, 14.06%. Found C, 69.51; H, 4.72; N, 14.22%. 2b: White crystals; yield 78%; mp 170–171°C. FT-IR (KBr, υ, cm−1) = 3,416 (NH), 1,640, 1,655 (C=O's). MS (EI): m/z (%) = 443.42 [M]+. Anal. Calcd for C23H17N5O5: C, 62.30; H, 3.86; N, 15.79%. Found C, 62.55; H, 4.05; N, 15.99%. 2c: Yellow crystals; yield 69%; mp 230–231°C. FT-IR (KBr, υ, cm−1) = 3316 (NH), 1,640, 1,653 (C=O's). MS (EI): m/z (%) = 432.87 [M]+, 4.34.87 [M++2] due to the presence of chlorine atom. Anal. Calcd for C23H17N4O3Cl: C, 63.82; H, 3.96; N, 12.94, Cl, 8.19%. Found C, 64.02; H, 4.07; N, 13.12%. 2d: Faint yellow powder; yield 82%; mp 263–264°C. FT-IR (KBr, υ, cm−1) = 3,325 (NH), 1,640, 1,661 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.5 (s, 1H, NH), 7.2–8.06 (m, 13H, Ar-H), 6.4 (s, 1H, CH), 4.4 (s, 2H, CH2). MS (EI): m/z (%) = 414.42 [M]+. Anal. Calcd for C23H18N4O4: C, 66.66; H, 4.38; N, 13.52%. Found C, 66.81; H, 4.45; N, 13.72%. 2e: White crystals; yield 81%; mp 190–191°C. FT-IR (KBr, υ, cm−1) = 3,168 (NH), 1,625, 1,649 (C=O's). MS (EI): m/z (%) = 404.45 [M]+. Anal. Calcd for C21H16N4O3S: C, 62.36; H, 3.99; N, 13.85; S, 7.93%. Found C, 62.52; H, 4.09; N, 14.12%.
2-(2,4-Dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-N-(2-oxo-4-phenyl-azetidin-1-yl)-acetamide 3a
A mixture (2,4-Dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid benzylidene hydrazide 2a (1 mmol) and Chloroacetyl chloride (1 mmol) in absolute ethanol (20 mL) was heated under refluxed for 10 h. After cooling, the reaction mixture was filtered off, dried then recrystallized from ethanol to give compound 3a as gray crystals; yield 82%; mp 275–276°C. FT-IR (KBr, υ, cm−1) = 3,168 (NH), 1,555, 1,661 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) = 11.5 (s, 1H, NH), 7.3–8.06 (m, 14H, Ar-H), 6.4 (d, 1H, CH-Ph), 5.5 (d, 1H, CH-Cl), 4.3 (s, 2H, CH2). 13C-NMR (CDCl3): δ 49.8, 58.1, 59.5, 114.3, 115.2, 124.7, 125.0, 126.5, 126.7, 126.9, 127.1, 128.5, 128.6, 128.8, 128.9, 129.1, 129.3, 131.8, 136.2, 139.5, 140.7, 151.9, 161.2, 165.8, 167.2. MS (EI): m/z (%) = 474.91 [M]+, 476.91 [M++2] due to the presence of chlorine atom. Anal. Calcd for C25H19N4O4Cl: C, 63.23; H, 4.03; N, 11.80%. Found C, 63.54; H, 4.18; N, 12.08%.
2-(4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-N-(4-oxo-4-phenyl-thiazolidin-3-yl)-acetamide 4a
A mixture of Schiff base 2a (1 mmol) and thioglycolic acid (1 mmol) in ethanol was heated under refluxed for 7 h. After cooling, the solid formed was filtered and purified by recrystallization from ethanol to give pure compound 4a as yellow brownish crystals; yield 76%; mp 255–256°C. FT-IR (KBr, υ, cm−1) = 3,477 (NH), 1,653, 1,725 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) = 11.5 (s, 1H, NH), 7.2–8.06 (m, 14H, Ar-H), 6.4 (s, 1H, CH-Ph), 5.4 (s, 2H, CH2), 4.3 (s, 2H, CH2). 13C-NMR (CDCl3): δ 33.9, 49.8, 66.2, 114.4, 115.3, 124.7, 125.1, 126.5, 128.0, 128.1, 128.2, 128.4, 128.6, 128.8, 128.9, 129.3, 129.5, 131.8, 136.2, 137.0, 140.7, 151.9, 161.3, 165.8, 167.3. MS (EI): m/z (%) = 472.53 [M]+. Anal. Calcd for C25H20N4O4S: C, 63.55; H, 4.27; N, 11.86%. Found C, 63.77; H, 4.35; N, 12.12%.
4-Methylene-1-[2-(3-methyl-5-oxo-4,5-dihydro-pyrazol-1-yl)-2-oxo-ethyl]-3-phenyl-3,4-dihydro-1H-quinazolin-2-one 5
A solution of hydrazide hydrate 1 (1 mmol) and ethyl acetoacetate (1.1 mmol) in ethanol was refluxed for 8 h. After completion the reaction (monitored by TLC), the mixture was filtered off and then recrystallized from benzene to afford the product 5 as yellow crystals; yield 84%; mp 215–216°C. FT-IR (KBr, υ, cm−1) = 3,245 (NH), 1,605, 1,649 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) = 7.2–8.07 (m, 9H, Ar-H), 4.4 (s, 4H, 2CH2), 1.4 (s, 3H, CH3). MS (EI): m/z (%) = 376.80 [M]+. Anal. Calcd for C20H16N4O4: C, 63.83; H, 4.28; N, 14.89%. Found C, 64.03; H, 4.35; N, 15.17%.
3-Amino-1-[2-(2,4-dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-5-phenyl-2,3-dihydro-1H-pyrazole-4-carbonitrile 6
A mixture of hydrazide 1 (1 mmol) and benzylidene malononitrile (1 mmol) in ethanol was heated under reflux for 8 h. After cooling, the solid formed was filtered off and dried then recrystallized from benzene to yield compound 6 as white crystals; yield 83%; mp 287–288°C. FT-IR (KBr, υ, cm−1) = 3,416 (NH), 2,225 (CN), 1,649, 1,731 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.5 (s, 1H, NH), 7.2–8.06 (m, 14H, Ar-H), 8.7 (s, 2H, NH2), 4.3 (s, 2H, CH2). 13C-NMR (CDCl3): δ 49.8, 79.2, 114.4, 115.3, 115.9, 124.7, 125.1, 126.5, 126.7, 126.8, 128.4, 128.5, 128.6, 128.8, 128.9, 129.2, 129.3, 129.5, 131.8, 131.9, 136.2, 137.5, 140.7, 151.9, 161.3, 167.3. MS (EI): m/z (%) = 464.49 [M]+. Anal. Calcd for C26H20N6O3: C, 67.23; H, 4.34; N, 18.09%. Found C, 67.53; H, 4.55; N, 18.22%.
1-[2-(5-Mercapto-[1,3,4]oxadiazol-2-yl)-2-oxo-ethyl]-3-phenyl-dihydro-pyrimidine-2,4-dione 7
To an ethanolic solution of hydrazide 1 (1 mmol), a 20 mL of carbon disulfide was added then refluxed for 5 h. After completion the reaction (monitored by TLC), the reaction mixture was filtered off, dried then recrystallized from ethanol to afford pure product 7 as yellow crystals; yield 81%; mp 215–217°C. FT-IR (KBr, υ, cm−1) = 3,416 (NH), 1,623, 1,663 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) = 7.2–8.08 (m, 9H, Ar-H), 4.7 (s, 1H, SH), 4.3 (s, 2H, CH2). MS (EI): m/z (%) = 380.38 [M]+. Anal. Calcd for C18H12N4O4S: C, 56.84; H, 3.18; N, 14.73%. Found C, 57.13; H, 3.25; N, 14.92%.
1-[2-(5-Hydrazino-[1,3,4]oxadiazol-2-yl)-2-oxo-ethyl]-3-phenyl-1H-quinazoline-2,4-dione 8
Two gram of later product 7 was allowed to react with 4 ml hydrazine in ethanol under reflux for 7 h. After cooling, the solid formed was filtered off, dried then recrystallized from ethanol to give compound 8 as white crystals; yield 79%; mp 210–211°C. FT-IR (KBr, υ, cm−1) = 3,420 (NH), 1,623, 1,661 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.5 (s, 1H, NH), 8.7 (s, 2H, NH2), 7.2–8.06 (m, 9H, Ar-H), 4.3 (s, 2H, CH2). 13C-NMR (CDCl3): δ 54.4, 114.4, 115.3, 124.7, 125.1, 126.5, 128.6, 128.8, 129.3, 129.5, 131.8, 136.2, 140.7, 151.9, 161.3, 162.3, 161.8, 194.5. MS (EI): m/z (%) = 378.35 [M]+. Anal. Calcd for C18H14N6O4: C, 57.14; H, 3.73; N, 22.21%. Found C, 57.33; H, 4.05; N, 22.52%.
N-(2-Imino-4-oxo-thiazolidin-3-yl)-2-(2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetamide 10
To an ethanolic solution of chloro-acetic acid N'-[2-(2,4-dioxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetyl]-hydrazide 9 (1 mmol), potassium thiocyanate (1 mmol) was added then the reaction mixture was refluxed for 5 h. After cooling, the solid precipitated was filtered off and recrystallized from ethanol to yield compound 10 as white crystals; yield 80%; mp 255–257°C. FT-IR (KBr, υ, cm−1) = 3,483 (NH), 1,621, 1,675 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.5 (s, 2H, 2NH), 7.2–7.9 (m, 9H, Ar-H), 5.6 (s, 2H, CH2), 4.3 (s, 2H, CH2). 13C-NMR (CDCl3): δ 33.9, 49.8, 114.4, 115.3, 124.7, 125.1, 126.5, 128.6, 128.8, 129.3, 129.5, 131.8, 136.2, 140.7, 151.9, 160.3, 161.3, 165.8, 167.3. MS (EI): m/z (%) = 409.43 [M]+. Anal. Calcd for C19H15N5O4S: C, 55.74; H, 3.69; N, 17.11%. Found C, 56.03; H, 3.85; N, 17.32%.
(4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid N′-(2-amino-oxazol-4-yl)-hydrazide 11
A mixture of compound 9 (1 mmol) in dioxane and urea (1 mmol) was refluxed for 10 hr. Afterward, the formed precipitate was filtered and purified by recrystallization from benzene to give pure product 11 as yellow crystals; yield 86%; mp 245–246°C. FT-IR (KBr, υ, cm−1) = 3,378 (NH), 1,617, 1,645 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.5 (s, 1H, NH), 10.5 (s, 1H, NH), 8.7 (s, 2H, NH2), 7.2–8.06 (m, 9H, Ar-H), 4.3 (s, 4H, 2CH2). MS (EI): m/z (%) = 392.38 [M]+. Anal. Calcd for C19H16N6O4: C, 58.16; H, 4.11; N, 21.42%. Found C, 58.33; H, 4.32; N, 21.64%.
(4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid N′-(2-amino-thiazol-4-yl)-hydrazide 12
A mixture of compound 9 (1 mmol) in dioxane and thiourea (1 mmol) was heated under reflux for 10 h. Afterward, the formed precipitate was filtered and purified by recrystallization from benzene to give pure compound 12 as white crystals; yield 86%; mp 190–192°C. FT-IR (KBr, υ, cm−1) = 3,378 (NH), 1,617, 1,645 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.5 (s, 1H, NH), 10.5 (s, 1H, NH), 8.7 (s, 2H, NH2), 7.2–8.06 (m, 9H, Ar-H), 4.3 (s, 4H, 2CH2). MS (EI): m/z (%) = 408.44 [M]+. Anal. Calcd for C19H16N6O3S: C, 55.87; H, 3.95; N, 20.58%. Found C, 56.15; H, 4.08; N, 20.72%.
4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid (4-oxo-thiazolidin-2-ylidene)-hydrazide 13
A solution of compound 9 (1 mmol) and ammonium thiocyanate (1 mmol) in ethanol was refluxed for 12 h. After completion the reaction (monitored by TLC), the reaction mixture was filtered off, dried then recrystallized from ethanol to afford pure product 13 as white crystals; yield 82%; mp 290–291°C. FT-IR (KBr, υ, cm−1) = 3,316 (NH), 1,625, 1,674 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.3 (s, 2H, 2NH), 7.2–8.07 (m, 9H, Ar-H), 4.3 (s, 4H, 2CH2). MS (EI): m/z (%) = 409.43 [M]+. Anal. Calcd for C19H15N5O4S: C, 55.74; H, 3.69; N, 17.77%. Found C, 55.93; H, 3.91; N, 17.96%.
[3-(1-Methyl-buta-1,3-dienyl)-2,4-dioxo-3,4-dihydro-2H-quinazolin-1-yl]-acetic acid (2-oxo-1,2,3a,7a-tetrahydro-indol-3-ylidene)-hydrazide 14
To an ethanolic solution of compound 9 (1 mmol), Isatine (1 mmol) was added and the reaction mixture was heated under reflux for 8 h. After cooling; a solid product was filtered off and recrystallized from ethanol/benzene to give compound 14 as yellow crystals; yield 86%; mp 300–301°C. FT-IR (KBr, υ, cm−1) = 3,236 (NH), 1,643, 1,655 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.5 (s, 1H, NH), 8.7 (s, 1H, NH), 7.2–8.0 (m, 13H, Ar-H), 4.3 (s, 2H, CH2). 13C-NMR (CDCl3): δ 49.8, 110.6, 114.4, 115.3, 121.9, 123.1, 124.7, 125.1, 126.5, 128.6, 128.8, 129.3, 129.5, 129.6, 131.8, 132.0, 136.2, 140.7, 147.2, 151.9, 153.8, 161.3, 165.8, 167.3. MS (EI): m/z (%) = 441.45 [M]+. Anal. Calcd for C24H19N5O4: C, 65.30; H, 4.34; N, 15.86%. Found C, 65.51; H, 4.45; N, 16.02%.
4-Methylene-1-[1,3,4] oxadiazol-2-ylmethyl-3-phenyl-3,4-dihydro-1H-quinazolin-2-one 15
A mixture of hydrazide 1 (1 mmol) and triethyl orthoformate (1.1 mmol) in ethanol was heated reflux for 8 h. After cooling; the formed precipitate was filtered and purified by crystallization from benzene to give product 15 as yellow crystals; yield 80%; mp 200–201°C. FT-IR (KBr, υ, cm−1) = 3,236 (NH), 1,628, 1,685 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) = 7.2–8.06 (m, 10H, Ar-H+CH), 4.3 (s, 2H, CH2). MS (EI): m/z (%) = 320.31 [M]+. Anal. Calcd for C17H12N4O3: C, 63.75; H, 3.78; N, 17.49%. Found C, 63.93; H, 4.01; N, 17.62%.
General Procedure for Synthesis of Imide 16a-c
(4-Methylene-2-oxo-3-phenyl-3,4-dihydro-2H-quinazolin-1-yl)-acetic acid hydrazide 1 (1 mmol) was heated under reflux for 8–12 h with the appropriate anhydrides such as maleic, phthalic, and tetrachlorophthalic anhydrides, respectively (1 mmol) in glacial acetic acid (20 mL). After cooling, the reaction mixture was filtered off and crystallized from appropriate solvents to afford the imide 16a-c, respectively. 16a: yield 80%; mp 268–270°C. FT-IR (KBr, υ, cm−1) = 3,326 (NH), 1,610, 1,678 (C=O's). 13C-NMR (CDCl3): δ 49.8, 114.4, 115.3, 124.7, 125.1, 126.5, 128.6, 128.8, 129.3, 129.5, 131.8, 133.2, 133.4, 136.2, 140.7, 151.9, 161.3, 165.8, 168.2, 168.4. MS (EI): m/z (%) = 390.36 [M]+. Anal. Calcd for C20H14N4O5: C, 61.54; H, 3.62; N, 14.35%. Found C, 61.73; H, 3.85; N, 14.52%. 16b: yield 86%; mp 262–264°C. FT-IR (KBr, υ, cm−1) = 3,351 (NH), 1,645, 1,667 (C=O's). MS (EI): m/z (%) = 440.42 [M]+. Anal. Calcd for C24H16N4O5: C, 65.45; H, 3.66; N, 12.72%. Found C, 65.63; H, 3.81; N, 12.94%. 16c: yield 86%; mp 280–282°C. FT-IR (KBr, υ, cm−1) = 3,316 (NH), 1,663, 1,685 (C=O's). 1H-NMR (DMSO d6, 400 MHz): δ (ppm) =11.5 (s, 1H, NH), 7.2–8.06 (m, 9H, Ar-H), 4.3 (s, 2H, CH2). MS (EI): m/z (%) = 576.20 [M]+, 578.20 [M++2], 580.20 [M++4], 582.20 [M++6], due to the presence of four chlorine atoms. Anal. Calcd for C24H12N4O5Cl4: C, 49.86; H, 2.09; N, 9.69%. Found C, 50.09; H, 2.15; N, 9.82%.
In silico Docking
The chemical structures of the synthesized 22 molecules are drawn in cdx format using Chem Draw Ultra v.7.0.1, and the obtained were then converted to SDF format. In-house database of 22-small molecules is constructed. All the atomic coordinates were changed to pdbqt set-up using Open Babel GUI. The X-ray crystal structure of enzyme for this study was availed from RCSB Protein Data Bank (www.rscb.org/pdb/). The protonation states of the residues were theoretically calculated using PDB2PQR (Dolinsky et al., 2007) and Propka 3.1 (Olsson et al., 2011) servers. Furthermore, the three-dimensional 3D coordinates for the target enzyme and ligand molecules were energy minimized using CHARMm Force Field (Brooks et al., 2009) in Discovery Studio and AMBER Force Field (Salomon-Ferrer et al., 2013) in Open Babel, respectively (with a limit of 500 steps of steepest algorithm for each ligand), to get more stable confirmations then used for in silico screening approach. The grid size of dimensions 25 × 25 × 25 Å3 is set to cover all binding site region. Ligand-enzyme docking simulations are conducted using PyRx tool, Autodock 4.2 to find the best-fit orientation of a compound that bind to the target enzyme. The docking studies using Autodock 4.2 of PyRx 8.0 tool uses the binding free energy calculations to identify the best binding pattern of the ligand molecule to the active site region of the target. Understanding of the intermolecular interactions between ligand molecules and enzyme is a key for development of novel therapeutic agents. Three-dimensional 3D pose views of the ligands-enzyme complexes are visualized using Accelrys discovery studio 3.5 (Accelrys Discovery Studio Visualizer software 2010). The visualization of electrostatic potential of the molecules was obtained using Gaussian 03 program package, through PM6 semi empirical method (Stewart, 1989). The molecular parameters of the synthesized compounds were checked by Mol inspiration server https://molinspiration.com/ to fit into Lipinski rule of Five (Ro5), which is a key way to satisfy the rational drug design and to calculate the bioactivity score for drug meant for oral use. The drug-likeness was evaluated by helping of the following attributes: hydrogen bond donor HBD (not more than 5), hydrogen bond acceptor HBA (not more than 10), the topological surface areas TPSA (not more than 140 Å2), partition coefficient LogP (not more than 5), and rotatable bonds (not more than 10). Moreover, to further estimate the drug ability of the target compounds, we herein reported their ADMET properties by using admetSAR server http://lmmd.ecust.edu.cn/admetsar1/.
Conclusion
In summary, the efficient synthesis of novel series of quinazolin-2,4-dione analogs coupled to N-heterocyclic moieties via acetyl/amide linkage is described. In silico screening was performed for searching about potent pfDHODH inhibitors. Docking of the target analogs to pfDHODH yielded binding energy between −12.2 and −5.7 kcal/mol, compared to −7.2 kcal/mol for reference drug (Chloroquine), and disclosed the analog 11 as a possible lead compound. The obtained results revealed that the newly synthesized compounds represent promising starting points for development of novel drug candidates against malaria.
Data Availability Statement
All datasets generated for this study are included in the article/Supplementary Material.
Author Contributions
All authors designed the study, contributed to the revision of the drafts, and agreed on the final version to be submitted. AA, MM, and AM were responsible for synthesis of compounds. AA did the virtual screening approach and he wrote the preliminary draft of the manuscript. AM, ME-N, and HT were responsible for the spectral analysis section.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.00105/full#supplementary-material
Abbreviations
1H-NMR, Proton- Nuclear Magnetic Resonance; pfDHODH, Plasmodium falciparum Dihydroorotate dehydrogenase; ESP, Electrostatic potential; 3D, Three-dimensional; ADME, Adsorption, distribution, metabolic and excretion.
References
Abdelmonsef, A. H. (2019). Computer-aided identification of lung cancer inhibitors through homology modeling and virtual screening. Egypt. J. Med. Hum. Genet. 20:6. doi: 10.1186/s43042-019-0008-3
Abdelmonsef, A. H., Dulapalli, R., Dasari, T., Padmarao, L. S., Mukkera, T., and Vuruputuri, U. (2016). Identification of novel antagonists for rab38 protein by homology modeling and virtual screening. Comb. Chem. High Throughput Screen. 19, 875–892. doi: 10.2174/1386207319666161026153237
Abdelmonsef, A. H., and Mosallam, A. M. (2020). Synthesis, in vitro biological evaluation and in silico docking studies of new quinazolin-2,4-dione analogues as possible anticarcinoma agents. J. Heterocycl. Chem. 57, 1637–1654. doi: 10.1002/jhet.3889
Aboubakr, H. A., Lavanya, S. P., Thirupathi, M., Rohini, R., Sarita, R. P., and Uma, V. (2016). Human Rab8b protein as a cancer target - an in silico study. J. Comput. Sci. Syst. Biol. 9, 132–149. doi: 10.4172/jcsb.1000231
Autino, B., Noris, A., Russo, R., and Castelli, F. (2012). Epidemiology of malaria in endemic areas. Mediterr. J. Hematol. Infect. Dis. 4:e2012060. doi: 10.4084/mjhid.2012.060
Bawa, S., Kumar, S., Drabu, S., and Kumar, R. (2010). Structural modifications of quinoline-based antimalarial agents: recent developments. J. Pharm. Bioallied Sci. 2:64. doi: 10.4103/0975-7406.67002
Blasco, B., Leroy, D., and Fidock, D. A. (2017). Parasite biology to the clinic. Nat. Med. 23, 917–928. doi: 10.1038/nm.4381
Brooks, B. R., Brooks, C., Mackerell, A. D., Nilsson, L., Petrella, R. J., Roux, B., et al. (2009). CHARMM: molecular dynamics simulation package. J. Comput. Chem. 30, 1545–1614. doi: 10.1002/jcc.21287
Campbell, C. C. (1997). Malaria: an emerging and re-emerging global plague. FEMS Immunol. Med. Microbiol. 18, 325–331. doi: 10.1111/j.1574-695X.1997.tb01063.x
Claudio, V. J., Barreiro, J. E., and Manssour Fraga, C. A. (2007). Molecular hybridization: a useful tool in the design of new drug prototypes. Curr. Med. Chem. 14, 1829–1852. doi: 10.2174/092986707781058805
Dasari, T., Kondagari, B., Dulapalli, R., Abdelmonsef, A. H., Mukkera, T., Padmarao, L. S., et al. (2017). Design of novel lead molecules against RhoG protein as cancer target–a computational study. J. Biomol. Struct. Dyn. 35, 3119–3139. doi: 10.1080/07391102.2016.1244492
Dolinsky, T. J., Czodrowski, P., Li, H., Nielsen, J. E., Jensen, J. H., Klebe, G., et al. (2007). PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucl. Acids Res. 35, 522–525. doi: 10.1093/nar/gkm276
Drissi, M., Benhalima, N., Megrouss, Y., Rachida, R., Chouaih, A., and Hamzaoui, F. (2015). Theoretical and experimental electrostatic potential around the m-nitrophenol molecule. Molecules 20, 4042–4054. doi: 10.3390/molecules20034042
Dua, R., Shrivastava, S., Sonwane, S. K., and Srivastava, S. K. (2011). Pharmacological significance of synthetic heterocycles scaffold: a review. Adv. Biol. Res. 5, 120–144.
El-Naggar, M., Mohamed, M. E., Mosallam, A. M., Salem, W., Rashdan, H. R., and Abdelmonsef, A. H. (2020). Synthesis, characterization, antibacterial activity, and computer-aided design of novel quinazolin-2,4-dione derivatives as potential inhibitors against Vibrio Cholerae. Evol. Bioinform 16:117693431989759. doi: 10.1177/1176934319897596
El-Sheshtawy, H. S., Abdelmonsef, A. H., Abboudy, S. M., Younes, A. M. M., Taha, M. M., and Hassan, M. A. (2017). Synthesis, structural, and theoretical studies of quinazoline-2,4-dione derivatives. Polycycl. Aromat. Compd. 39, 279–286. doi: 10.1080/10406638.2017.1325747
Gómez, M. M. C., and Kouznetsov, V. V. (2013). “Recent developments on antimicrobial quinoline chemistry,” in Microbial Pathogens and Strategies for Combating Them: Science, Technology and Education, ed A. Méndez-Vilas (FORMATEX), 666–677.
Hassan, M. A., Seleem, M. A., Mohamed, A., Younes, M. M. A., Taha, M. M., and Abdel-Monsef, A. H. (2013a). Synthesis and spectral characterization of some heterocyclic nitrogen compounds. Eur. J. Chem. 4, 121–123. doi: 10.5155/eurjchem.4.2.121-123.740
Hassan, M. A., Seleem, M. A., Younes, A. M. M., Taha, M. M., and Abdel-Monsef, A. H. (2013b). Utility of (2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-acetic acid hydrazide in the synthesis of some heterocyclic nitrogen compounds. Eur. J. Chem. 4, 168–171. doi: 10.5155/eurjchem.4.2.168-171.762
Hurt, D. E., Widom, J., and Clardy, J. (2006). Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Crystallogr. Sect. D Biol. Crystallogr. 62, 312–323. doi: 10.1107/S0907444905042642
Hussein, M. A., Zyaan, O. H., Abdel Monsef, A. H., Rizk, S. A., Farag, S. M., Khaled, A. S., et al. (2018). Synthesis, molecular docking and insecticidal activity evaluation of chromones of date palm pits extract against Culex Pipiens (Diptera: Culicidae). Int. J. Mosq. Res. 5, 22–32.
Knorr, L. (1883). Knorr pyrazole synthesis. Ber. Dtsch. Chem. Ges. 16, 2597–2599. doi: 10.1002/cber.188301602194
Kumar, S., Bawa, S., and Gupta, H. (2010). Biological activities of quinoline derivatives. Mini Rev. Med. Chem. 9, 1648–1654. doi: 10.2174/138955709791012247
Metwally, N. H., Abdelrazek, F. M., and Eldaly, S. M. (2018). Synthesis, molecular docking, and biological evaluation of some novel bis-heterocyclic compounds based N,N′-([1,1′-biphenyl]-4,4′-diyl)bis(2-cyanoacetamide) as potential anticancer agents. J. Heterocycl. Chem. 55, 2668–2682. doi: 10.1002/jhet.3290
Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., et al. (1639). Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 19, 1639–1662. doi: 10.1002/(SICI)1096-987X(19981115)19:14<1639::AID-JCC10>3.0.CO;2-B
Nivsarkar, M., Thavaselvam, D., Prasanna, S., Sharma, M., and Kaushik, M. P. (2005). Design, synthesis and biological evaluation of novel bicyclic β-lactams as potential antimalarials. Bioorg. Med. Chem. Lett. 15, 1371–1373. doi: 10.1016/j.bmcl.2005.01.011
Oguike, M. C., Betson, M., Burke, M., Nolder, D., Stothard, J. R., Kleinschmidt, I., et al. (2011). Plasmodium ovale curtisi and Plasmodium ovale wallikeri circulate simultaneously in African communities. Int. J. Parasitol. 41, 677–683. doi: 10.1016/j.ijpara.2011.01.004
Olsson, H. M. M., SØndergaard, C. R., Rostkowski, M., and Jensen, J. H. (2011). PROPKA3: consistent treatment of internal and surface residues in empirical p K a predictions. J. Chem. Theory Comput. 7, 525–537. doi: 10.1021/ct100578z
Patel, K. D., Vekariya, R. H., Prajapati, N. P., Patel, D. B., Patel, H. D., Shaikh, T., et al. (2018). Synthesis of N′-(quinazolin-4-yl)isonicotinohydrazides and their biological screening, docking and ADME studies. Arab. J. Chem. 13, 1986–2000. doi: 10.1016/j.arabjc.2018.02.017
Rondla, R., Padmarao, L. S., Ramatenki, V., Haredi-Abdel-Monsef, A., Potlapally, S. R., and Vuruputuri, U. (2017). Selective ATP competitive leads of CDK4: discovery by 3D-QSAR pharmacophore mapping and molecular docking approach. Comput. Biol. Chem. 71, 224–229. doi: 10.1016/j.compbiolchem.2017.11.005
Sahu, S., Ghosh, S. K., Gahtori, P., Pratap Singh, U., Bhattacharyya, D. R., and Bhat, H. R. (2019). In silico ADMET study, docking, synthesis and antimalarial evaluation of thiazole-1,3,5-triazine derivatives as Pf-DHFR inhibitor. Pharmacol. Rep. 71, 762–767. doi: 10.1016/j.pharep.2019.04.006
Salomon-Ferrer, R., Case, D. A., and Walker, R. C. (2013). An overview of the amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3, 198–210. doi: 10.1002/wcms.1121
Shehadi, I. A., Rashdan, R. M. H., and Abdelmonsef, A. H. (2020). Homology modeling and virtual screening studies of antigen MLAA-42Protein: identification of noveldrugcandidates against leukemia-an in silico approach. Comput. Math. Methods Med. 2020, 1–12. doi: 10.1155/2020/8196147
Sokolova, A. S., Yarovaya, I. O., Shtro, A. A., Borisova, M. S., Morozova, E. A., Tolstikova, T. G., et al. (2017). Synthesis and biological activity of heterocyclic borneol derivatives. Chem. Heterocycl. Compd. 53, 371–377. doi: 10.1007/s10593-017-2063-3
Stewart, J. P. J. (1989). Optimization of parameters for semiempirical methods method I. J. Comput. Chem. 10, 209–220. doi: 10.1002/jcc.540100208
Talapko, J., Škrlec, I., Alebić, T., Jukić, M., and Včev, A. (2019). Malaria: the past and the present. Microorganisms 7:179. doi: 10.3390/microorganisms7060179
Tanitame, A., Oyamada, Y., Ofuji, K., Fujimoto, M., Iwai, N., Hiyama, Y., et al. (2004). Synthesis and antibacterial activity of a novel series of potent DNA gyrase inhibitors. Pyrazole derivatives. J. Med. Chem. 47, 3693–3696. doi: 10.1021/jm030394f
Vikram, S., and Mishra, S. (2019). Molecular docking studies of benzamide derivatives for pfdhodh inhibitor as potent antimalarial agent. Am. J. Biochem. Mol. Biol. 9, 1–6. doi: 10.3923/ajbmb.2019.1.6
Keywords: malaria, pfDHODH, hybrid molecules, quinazolin-2,4-dione, N-heterocyclic moieties, docking study
Citation: Haredi Abdelmonsef A, Eldeeb Mohamed M, El-Naggar M, Temairk H and Mohamed Mosallam A (2020) Novel Quinazolin-2,4-Dione Hybrid Molecules as Possible Inhibitors Against Malaria: Synthesis and in silico Molecular Docking Studies. Front. Mol. Biosci. 7:105. doi: 10.3389/fmolb.2020.00105
Received: 29 February 2020; Accepted: 06 May 2020;
Published: 05 June 2020.
Edited by:
Vojtech Spiwok, University of Chemistry and Technology in Prague, CzechiaReviewed by:
Stanislav Kozmon, Slovak Academy of Sciences (SAS), SlovakiaIvan Raich, University of Chemistry and Technology in Prague, Czechia
Copyright © 2020 Haredi Abdelmonsef, Eldeeb Mohamed, El-Naggar, Temairk and Mohamed Mosallam. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aboubakr Haredi Abdelmonsef, YWJvdWJha3IuYWhtZWRAc2NpLnN2dS5lZHUuZWc=