 Eleonora Napoli1
Eleonora Napoli1 Yingratana Amabel McLennan2
Yingratana Amabel McLennan2 Andrea Schneider2,3
Andrea Schneider2,3 Flora Tassone2,4
Flora Tassone2,4 Randi J. Hagerman2,3
Randi J. Hagerman2,3 Cecilia Giulivi1,2*
Cecilia Giulivi1,2*- 1Department of Molecular Biosciences, School of Veterinary Medicine, University of California, Davis, Davis, CA, United States
- 2MIND Institute, University of California Davis Medical Center, Sacramento, CA, United States
- 3Department of Pediatrics, University of California Davis Medical Center, Sacramento, CA, United States
- 4Department of Biochemistry and Molecular Medicine, School of Medicine, University of California, Davis, Davis, CA, United States
The X-linked FMR1 premutation (PM) is characterized by a 55–200 CGG triplet expansion in the 5′-untranslated region (UTR). Carriers of the PM were originally thought to be asymptomatic; however, they may present general neuropsychiatric manifestations including learning disabilities, depression and anxiety, among others. With age, both sexes may also develop the neurodegenerative disease fragile X-associated tremor/ataxia syndrome (FXTAS). Among carriers, females are at higher risk for developing immune disorders, hypertension, seizures, endocrine disorders and chronic pain, among others. Some female carriers younger than 40 years old may develop fragile X-associated primary ovarian insufficiency (FXPOI). To date, no studies have addressed the metabolic footprint – that includes mitochondrial metabolism – of female carriers and its link to clinical/cognitive manifestations. To this end, we performed a comprehensive biochemical assessment of 42 female carriers (24–70 years old) compared to sex-matched non-carriers. By applying a multivariable correlation matrix, a generalized bioenergetics impairment was correlated with diagnoses of the PM, FXTAS and its severity, FXPOI and anxiety. Intellectual deficits were strongly correlated with both mitochondrial dysfunction and with CGG repeat length. A combined multi-omics approach identified a down-regulation of RNA and mRNA metabolism, translation, carbon and protein metabolism, unfolded protein response, and up-regulation of glycolysis and antioxidant response. The suboptimal activation of the unfolded protein response (UPR) and endoplasmic-reticulum-associated protein degradation (ERAD) response challenges and further compromises the PM genetic background to withstand other, more severe forms of stress. Mechanistically, some of the deficits were linked to an altered protein expression due to decreased protein translation, but others seemed secondary to oxidative stress originated from the accumulation of either toxic mRNA or RAN-derived protein products or as a result of a direct toxicity of accumulated metabolites from deficiencies in critical enzymes.
Introduction
Carriers of the premutation (PM) are characterized by a moderate (55 to 200) expansion of the cytosine-guanine-guanine (CGG) nucleotide repeats in the first exon and promoter of the X-linked FMR1 gene (Verkerk et al., 1991; Bagni et al., 2012). Originally, PM carriers were assumed to be free of any apparent phenotypic traits. However, over the last decade, a growing number of neuropsychiatric manifestations (including depression, anxiety, and insomnia), visuospatial deficits, and immune dysregulation have been reported to occur at a greater frequency among adult PM carriers than in the general population (Hagerman and Hagerman, 2013). Generally, PM carriers show lower performance in neuropsychological testing including full-scale intellectual quotient (FSIQ) and working memory (WM) subtests on the Wechsler Adult Intelligence Scale (Lozano et al., 2016). In the case of children with the PM, they are often diagnosed with ADHD, autism, anxiety, and other psychopathologies (Napoli et al., 2018). The PM has also been associated with conditions beyond those involving the CNS, such as hypertension, hypothyroidism, high blood glucose, as well as a higher incidence of thyroid, prostate and other cancers (Lozano et al., 2016). With age, both female and male carriers of the PM are at a higher risk for developing the late-onset (usually appearing after age 50) neurodegenerative disorder fragile X-associated tremor/ataxia syndrome [FXTAS; OMIM:300623; (Hagerman et al., 2001, 2004; Hagerman and Hagerman, 2013)]. FXTAS-affected carriers may exhibit intention tremor and gait ataxia, accompanied by cerebral atrophy, white matter disease, parkinsonism, neuropathy, autonomic dysfunction, and cognitive deficits (Berry-Kravis et al., 2007a; Bourgeois et al., 2007; Lozano et al., 2016; Claeys et al., 2020).
As it is the case for many X-linked disorders, women have a lower absolute risk of developing FXTAS symptoms compared to men (Hagerman and Hagerman, 2016). However, women carrying FMR1 PM allele have a higher risk of developing premature or primary ovarian insufficiency (POI) [16% (Schwartz et al., 1994; Allingham-Hawkins et al., 1999; Rifé et al., 2004; Terracciano et al., 2004; Schuettler et al., 2011; Elizur et al., 2014; Pouresmaeili and Fazeli, 2014)] as compared to full mutation females (>200 CGG repeats), who carry the same risk for POI as the general population (1%). About 20 to 30% of female carriers experiencing irregular periods or amenorrhea due to ovarian insufficiency prior to age 40 are diagnosed with fragile X-associated primary ovarian insufficiency [FXPOI; (Allingham-Hawkins et al., 1999; Allen et al., 2007)]. Even PM carriers without signs of ovarian dysfunction have an earlier (on average by 5 years) age at menopause compared with non-carriers (Patsalis et al., 1999; Sullivan et al., 2005; Besterman et al., 2014). Women with alleles between 35–44 CGG repeats seem to present diminished ovarian function but regular menses and occult primary ovarian insufficiency (Streuli et al., 2009; Karimov et al., 2011; Pastore et al., 2012); however, other studies found no association between FMR1 intermediate alleles and POI (Bennett et al., 2010; Murray et al., 2014; Voorhuis et al., 2014). Other medical and psychological issues reported in females are hypothyroidism, hypertension, endocrine dysfunctions, chronic pain, fibromyalgia, autoimmune diseases, neuropathies, migraines, dementia, and psychiatric conditions, such as anxiety and depression (Allen et al., 2007, 2020; Bailey et al., 2008; Hunter et al., 2010; Winarni et al., 2012; Wheeler et al., 2014a,b; Lozano et al., 2016; Movaghar et al., 2019). Collectively included under the term FXAND [fragile X–associated neuropsychiatric disorders; (Hagerman et al., 2018)], such emotional and neuropsychiatric disorders, have been shown to be more common in female carriers compared to non-carriers. In a recent work by Dr. S. Sherman’s group, which investigated the association between the PM diagnosis and CGG repeat expansion in female carriers, the most common symptoms reported were anxiety and depression, migraine, headaches, and sleep problems (Allen et al., 2020).
Our team was the first to report mitochondrial dysfunction as a common feature in biological samples from PM carriers (Napoli et al., 2013, 2016a,b, 2018; Song et al., 2016) as well as in murine models of the PM (Napoli et al., 2016a). This decreased mitochondrial bioenergetics is present in PM carriers with and without FXTAS and even in some pediatric carriers (Napoli et al., 2018). However, to our knowledge, no study has to date characterized the metabolic footprint of the PM and related clinical and cognitive features in female carriers of the PM.
To bridge this gap in knowledge, we performed a comprehensive biochemical assessment (including metabolomics and proteomics profiling, and bioenergetics) in peripheral blood mononuclear cells (PBMC) and plasma samples obtained from 24- to 70-year-old female carriers. To elucidate peripheral bioenergetics markers that may function as surrogates for CNS function, we utilized a multivariable correlation matrix to identify correlations between mitochondrial outcomes and cognitive parameters (FSIQ), executive function (BDS-2), anxiety, tremor, and FXTAS (and its severity) and FXPOI diagnoses. As such, this study is ideally positioned to perform comprehensive deep metabolic and mitochondrial phenotyping by gathering complementary outcomes on genomics, proteomics, metabolomics, mitochondrial physiology and clinical information to systematically develop a metabolic profile from established female carriers by taking advantage of a substantial repository of patient samples. This multi-faceted approach, as opposed to a simple model based on the statistical differences of few (and sometimes unconnected) metabolites or proteins between diagnostic groups. The profile generated will, for the first time, allow researchers to fully assess the larger biological impact of the premutation on metabolic status of female carriers, thereby providing unprecedented insight into the biological consequences of metabolic deficits and mitochondrial dysfunction, aiding the discovery of disease mechanisms. Importantly, due to the depth of the phenotyping across multiple readouts, the integrated metabolic profiles generated will have the detail required to cluster patients according to their clinical pathology when more patients’ data will be available. This necessary step, in turn, will lead to truly novel and testable hypotheses regarding individualized pathogenesis and treatment.
Materials and Methods
Subjects
Blood samples were obtained from 42 female carriers of the FMR1 PM ranging from 24- to 70-year old, recruited through the Fragile X Treatment and Research Center at the MIND Institute at University of California, Davis. Blood samples were also obtained from 10 female non-carriers aged 25 to 60 years. The study was approved by the IRB ethics committee at University of California Davis Medical Center. Blood samples were obtained by venipuncture with informed consent. FXTAS was diagnosed utilizing criteria reported before (Jacquemont et al., 2003; Hagerman and Hagerman, 2016). For returning participants, outcomes evaluated at one single visit collected at the indicated age were included in the analysis.
Genotyping
CGG repeat expansion in all individuals included in this study were evaluated by both PCR and Southern Blot analysis, as previously described (Tassone et al., 2008; Filipovic-Sadic et al., 2010). The X-activation ratio (XAR), representing the percentage of cells with the normal allele on the active X chromosome, was calculated by the ratio of the densitometric intensity of the normal FMR1 unmethylated band over the sum of the intensities of the normal unmethylated and methylated bands (Tassone et al., 1999; Berry-Kravis et al., 2003). In a population of normal (Z) distribution, 1.65 × SD leads to a tail that gives the probability of 5% of the data to be excluded from normal. If this value is subtracted from the mean XAR, then anything below this value has <5% probability of being significant. Thus, XAR values <36% of non-carrier ones were considered unfavorable (<0.2).
Lymphocyte Preparation
Blood (5–7 ml) was collected in BD vacutainer CPT tubes (BD Biosciences, Franklin Lakes, NJ, United States) and lymphocytes were isolated as previously described (Napoli et al., 2016a). Upon collection, lymphocyte suspension was divided into 2 aliquots in Eppendorf tubes and pelleted by centrifugation 1 min 2,000 rpm in a microfuge at 4°C. The supernatant was removed, and the pellet was used immediately for mitochondrial outcomes. An aliquot of PBMC was suspended in 0.5 ml cold 10 mM HEPES, pH 7.4, frozen at −80°C overnight and subsequently transferred for extended storage, into liquid nitrogen.
Mitochondrial Outcomes
All chemicals and biochemicals were of analytical grade or higher. Enzymatic activities of Complexes I–V in digitonin-permeabilized lymphocytes determined by polarography essentially as described before (Giulivi et al., 2010; Napoli et al., 2016a). Briefly, an aliquot (0.5–1.0 × 106) of lymphocytes was added to the oxygen chamber in 0.3 ml of a buffer containing 0.22 M sucrose, 50 mM KCl, 1 mM EDTA, 10 mM KH2PO4, and 10 mM HEPES, pH 7.4. Oxygen consumption rates were evaluated in the presence of (i) 1 mM ADP plus 1 mM malate-10 mM glutamate followed by the addition of 5 μM rotenone; (ii) 10 mM succinate followed by the addition of 1 mM malonate; (iii) 1 mM α-glycerophosphate followed by the addition of 3.6 μM antimycin A; and (iv) 10 mM ascorbate and 0.2 mM N,N,N′,N′-tetramethyl-p-phenylenediamine followed by the addition of 1 mM KCN. Activities of individual electron transport chain (ETC) segments were evaluated as the difference of oxygen uptake recorded before and after the addition of specific inhibitors. Citrate synthase activity was evaluated spectrophotometrically with a Tecan Infinite M200 microplate reader equipped with the Magellan software (Austria) at 412 nm as described elsewhere (Napoli et al., 2016a). The respiratory control ratio (RCR) was calculated as the ratio between oxygen uptake rates of intact cells supplemented with 10 mM glucose (present in RPMI-1640) in State 3 μ (with 2 μM carbonylcyanide-p-trifluoromethoxyphenylhydrazone, or FCCP) and State 4 (with 0.2 μM oligomycin) (Giulivi et al., 2013). Mitochondrial ROS production was calculated from the oligomycin-resistant oxygen consumption rates and normalized by basal respiration in the presence of 10 mM glucose.
Plasma Metabolomics
Plasma samples were obtained from age-matched 8 non-carriers and 7 PM carriers as previously described (Napoli et al., 2016a), and metabolites were extracted and analyzed by mass spectrometry (Napoli et al., 2015). Briefly, 30-μl aliquots were extracted by 1 ml of degassed acetonitrile:isopropanol:water (3:3:2, V/V/V) at −20°C, centrifuged and decanted with subsequent evaporation of the solvent to complete dryness. A clean-up step with acetonitrile/water (1:1) removed membrane lipids and triglycerides. Details on the identification of metabolites and data analysis are reported in Napoli et al. (2015).
Proteomics
Peripheral blood mononuclear cells samples were hand-homogenized on ice in 20 mM HEPES pH 6.8, containing protease and phosphatase inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, United States). Protein concentration was determined by BCA protein assay kit (Thermo Scientific, Sunnyvale, CA, United States) and samples from 7 controls and 7 PM carriers were submitted to the UC Davis Mass Spectrometry Facility and analyzed as described in detail elsewhere (Napoli et al., 2016a).
Statistics
As the population assessed in this study did not follow a normal distribution, Spearman’s correlation coefficients and correspondent p values for each pair of variables tested were computed with a correlation matrix for multiple variables. Due to the limited information available for some subjects on some of the variables included in our analysis (Table 1), and to exclude the possibility of considering significant interactions without true biological relevance, we applied a more stringent cut-off conditions by setting p values at ≤0.01. We did not employ a Bonferroni correction to adjust the p values for the number of variables tested as (i) it ignores dependencies among the data and is therefore too conservative if the number of tests is large, (ii) it assumes that all null hypotheses are true simultaneously, which was not our assumption, (iii) it increases the likelihood of type II errors, so that truly important differences are considered non-significant (Perneger, 1998). A two-way ANOVA was carried out to test for the contribution of age, diagnosis and age x diagnosis on the mitochondrial outcomes’ variability. Graph Pad Prism v. 8.1.2 was used for all the statistical analyses. Metabolomics and proteomics data were analyzed as previously described (Giulivi et al., 2016; Napoli et al., 2016a,b).
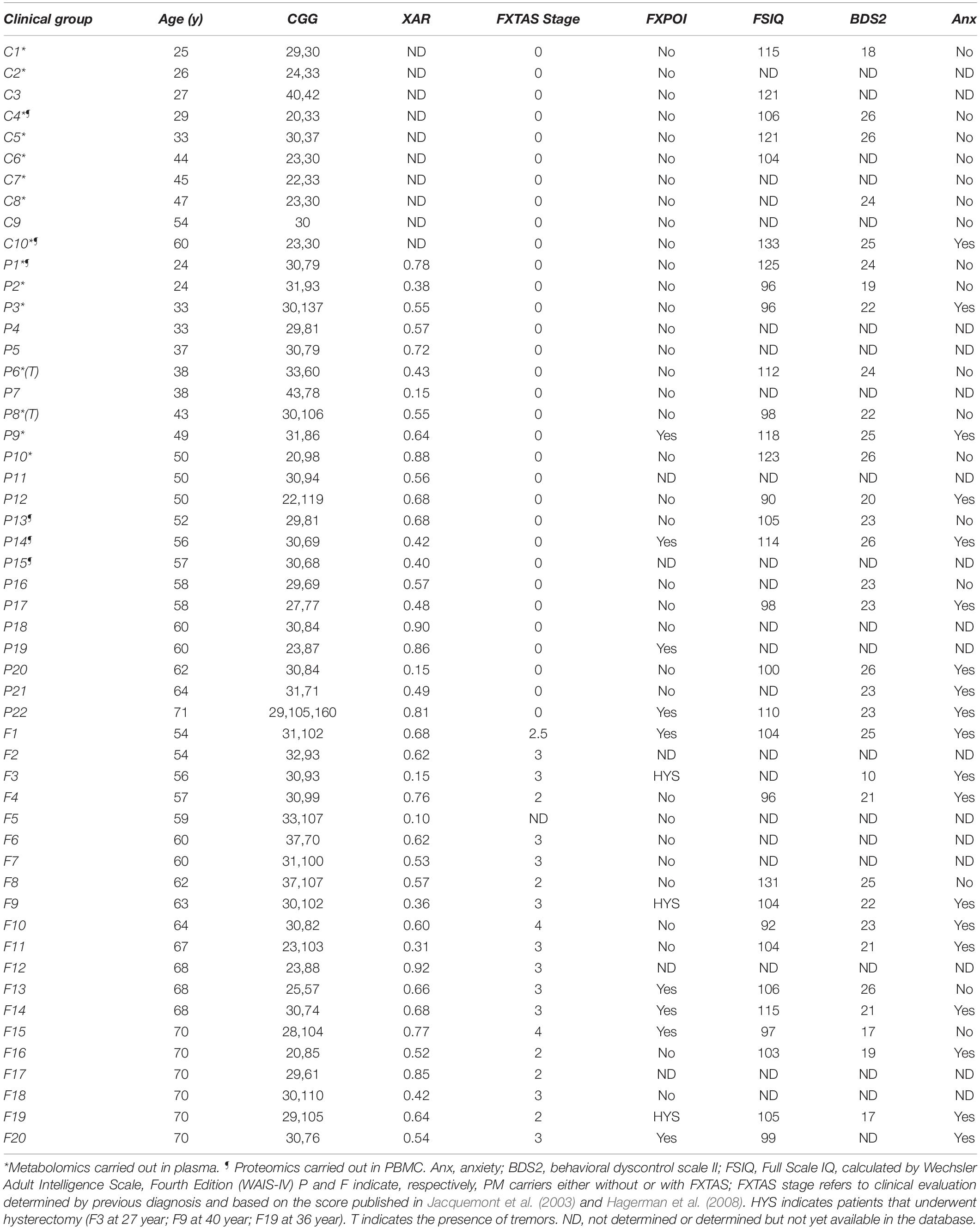
Table 1. Demographic and clinical data of the women included in this study.
Results
Demographics and Clinical Characteristics of the Cohorts
Our cohorts consisted of women with the PM with and without FXTAS, and sex-matched non-carriers (named hereafter controls; Table 1). The control study group consisted of 25–60 year old women with CGG repeats at the 5′- untranslated region (UTR) of FMR1 of 31 ± 1 (mean ± SEM; n = 10). The PM group included 24- to 70-year-old females (n = 42), from which about half (n = 20) were diagnosed with FXTAS. The FXTAS stages ranged from 1 to 4, with most subjects at stage 3. The CGG repeats of the mutant allele ranged from 57 to 137, with an average of 87 ± 4 for PM carriers without FXTAS and 90 ± 4 for FXTAS-affected carriers with no significant difference between these two groups (p = 0.156). Of the 42 carriers, 10 were diagnosed with FXPOI, and 3 had undergone hysterectomy between the age of 26 and 40 years. XAR ranged from 0.1 to 0.92 with an average of 0.57 ± 0.04 in PM without FXTAS and 0.57 ± 0.05 in FXTAS-affected with no significant difference between these two groups (p = 0.878), and not different from the expected random inactivation of the X-chromosome. Only 4 of 42 carriers had an unfavorable XAR.
Both cohorts were assessed for most of the neurological/neuropsychiatric outcomes. They included full-scale intellectual quotient (FSIQ; Table 1) assessed with the Wechsler Adult Intelligence Scale, WAIS-IV (Drozdick et al., 2012), and executive function [assessed by the Behavioral Dyscontrol Scale II or BDS-2; (Belanger et al., 2005)]. Subjects were also tested for generalized anxiety through the Structured Clinical Interview for Diagnosis of Mental Disorders (SCID)-1 [Diagnostic and Statistical Manual of Mental Disorders (DSM)-IV; Table 1].
Deficits in Peripheral Mitochondrial Energy Production Are Linked to Cognitive and Psychiatric Outcomes, and Diagnoses of FXPOI and FXTAS and Its Morbidity (FXTAS Stage)
A multivariable correlation matrix was built by utilizing as input data clinical, psychiatric and molecular outcomes from non-carriers and PM carriers (Figure 1). Continuous variables were used as such, whereas categorical ones (e.g., three diagnoses: non-carriers, PM, FXTAS; FXPOI diagnosis, and presence of anxiety) were assigned numerical values. Tremor, one of FXTAS hallmarks, was excluded from the correlation matrix as it was available for 23 of the 42 participants, of which only 2 showed tremors. Similarly, the XAR was not shown in Figure 1, as this outcome did not show any correlation with any of the other reported outcomes, due to the low frequency (9.5%) of unfavorable (<0.2) XAR in our cohort. The apparent discrepancy with the findings of other studies (Hagerman and Hagerman, 2004; Berry-Kravis et al., 2005, 2007b) could be bridged by considering that they utilized other neurological features (Berry-Kravis et al., 2007b); the use of an updated clinical diagnostic criteria for FXTAS (Hagerman and Hagerman, 2013).
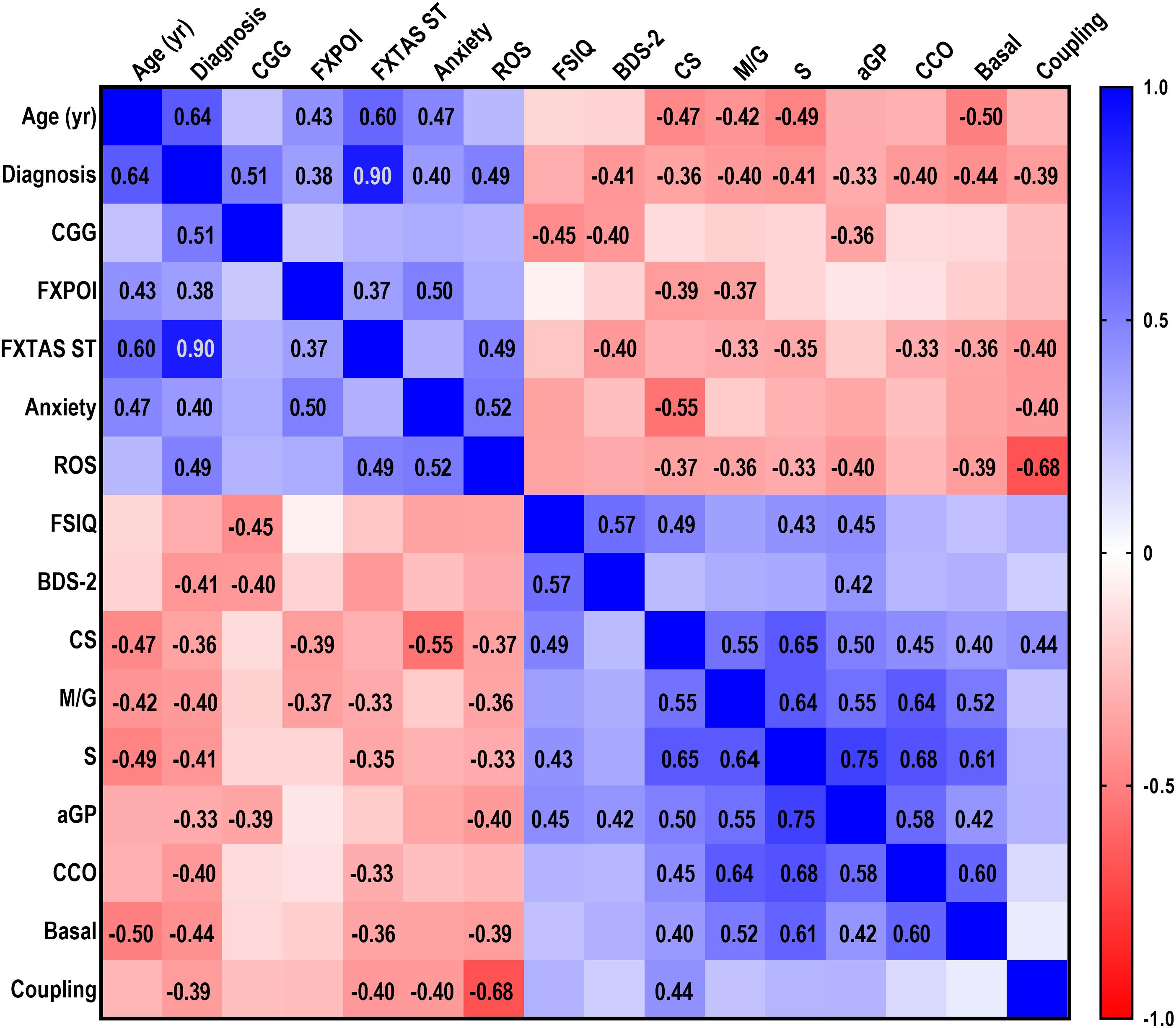
Figure 1. Correlations between demographic, clinical and mitochondrial outcomes. A multivariable correlation matrix was built with demographic, clinical and functional data relative to PM without (n = 22) and with (n = 20) FXTAS symptoms and control (n = 10) women age 24–70 year, along with biochemical mitochondrial outcomes measured in lymphocytes obtained from the same individuals. Outcomes analyzed were: age, diagnosis, CGG (all with n = 52), FXPOI (n = 47), FXTAS stage (n = 51), anxiety (n = 36), Full Scale IQ (FSIQ; n = 31), Behavioral Dyscontrol Scale-2 (BDS-2; n = 32), ROS (n = 46), citrate synthase activity (CS; n = 52), NADH-fueled ATP-linked O2 consumption (M/G; n = 52), succinate (S)-sustained FADH2-fueled ATP-linked linked O2 consumption (n = 52), α-glycerophosphate (αGP)-dependent ATP-linked O2 consumption (n = 50), cytochrome c oxidase activity (CCO; 51), basal respiration (n = 51), coupling (n = 50). Categorical variables (i.e., diagnosis, FXPOI, anxiety) were assigned a numerical value. Diagnosis: control = 0, PM without FXTAS = 1, PM with FXTAS = 2; FXPOI and anxiety: absence = 0, presence = 1. Due to the non-Gaussian distribution of the data, the non-parametric Spearman test was run. R values are shown for those correlations which were statistically significant at p ≤ 0.01. A scale showing the range of r values (from –1.0 for inversely correlated outcomes to 1.0 for positively correlated ones) is also shown.
Overall, demographic (age), clinical (diagnoses of PM and FXTAS, CGG repeats, diagnosis of FXPOI and FXTAS stage) and psychiatric (anxiety) variables correlated positively with each other. Notably, all of these outcomes were associated with increased oxidative stress as they were directly correlated with mitochondrial ROS production. As expected, these outcomes were inversely correlated with intellectual and functional capacities (FSIQ and BDS-2) and mitochondrial function (Figure 1).
In order to narrow down the significant associations among variables, the non-parametric Spearman’s correlation coefficients, and correspondent p values were calculated.
Diagnoses of PM, FXTAS and FXPOI
Using the above mentioned significance threshold, FXTAS and PM diagnoses significantly and inversely correlated with (i) activity of citrate synthase, marker of mitochondrial mass, (CS; Spearman’s r = −0.361, p = 0.004); (ii) mitochondrial ATP production supported by NADH- (r = −0.397, p = 0.001) and FADH2-linked substrates (r = −0.413, p = 0.001); (iii) α-glycerophosphate-fueled ATP production (αGP; r = −0.330, p = 0.01); (iv) cytochrome c oxidase activity (r = −0.402, p = 0.001); (v) glucose-sustained mitochondrial ATP production (r = −0.439, p < 0.0001); and (vi) coupling between electron transport and ATP production (r = −0.391, p = 0.002). In turn, diagnoses directly correlated with mitochondrial ROS production (r = 0.490, p < 0.0001). The same biochemical outcomes, except for mitochondrial mass (CS activity) and αGP-mediated respiration, correlated with FXTAS severity (i.e., stage; Figure 1) indicating that the morbidity of this neurodegenerative disease is also reflected as a peripheral deficit in bioenergetics.
Lower CS activity and NADH-linked ATP production characterized FXPOI, diagnosis strongly linked to the occurrence of anxiety (r = 0.497, p = 0.001; Figure 1). While PM and FXTAS diagnoses, morbidity of FXTAS and FXPOI were all inversely correlated with CS activity, it could be assumed that the lower bioenergetics was a result of lower mitochondrial mass. However, even after normalization of the rates of ATP production by mitochondrial mass, several outcomes were still correlated (some directly and others inversely) highlighting the delicate balance among Complexes within the electron transport chain that needs to be preserved for its adequate production of ATP while minimizing ROS production. Among them (normalized by mitochondrial mass), NADH-linked ATP production with anxiety (r = 0.469; p = 0.002); succinate-linked ATP production with anxiety and PRI (r = 0.370; p = 0.01); CCO activity with FXPOI diagnosis (r = 0.310; p = 0.02), anxiety (r = 0.407; p = 0.008), PRI (r = −0.565; p = 0.01) and FSIQ (r = −0.369; p = 0.02); and basal respiration with PRI (r = −0.653; p = 0.003) and PSI (r = −0.517; p = 0.02).
Anxiety
Anxiety was inversely correlated with CS activity (r = −0.553, p < 0.0001) and coupling (r = −0.398, p = 0.01), and directly with mitochondrial ROS production (r = 0.521, p = 0.001).
Cognition and Executive Function
FSIQ strongly and positively correlated with executive function (BDS-2; r = 0.566, p < 0.0001; Figure 1). Similarly, and considering that the brain represents the main site of energy consumption with over 20% of body’s total oxygen consumption (Watts et al., 2018), a strong and direct correlation was identified between FSIQ and overall mitochondrial bioenergetics (Figure 1), with statistically significant associations with mitochondrial mass (CS, r = 0.489, p = 0.002), FADH2-linked ATP production (r = 0.427, p = 0.009), and α-glycerophosphate-fueled ATP production (αGP; r = 0.446, p = 0.008). Overall, BDS-2 positively correlated with mitochondrial function, with the correlation with α-glycerophosphate-fueled ATP production resulting statistically significant (r = 0.417, p = 0.008). [NADH- and FADH2-linked ATP production were significantly correlated with BDS-2 at p < 0.05 with r = 0.318 and 0.337, respectively].
FMR1 Gene Structure
No correlation was obtained between CGG expansions and FXPOI diagnosis likely due to the significant number of carriers with >100 CGG repeats (28.6%) as reported by others (Sullivan et al., 2005; Ennis et al., 2006; Allen et al., 2007). However, CGG repeats showed a statistically significant inverse correlation with α-glycerophosphate-fueled ATP production (r = −0.391, p = 0.009), probably indicating issues with the redox state and regulation of cellular energy metabolism, as well as with FSIQ (r = −0.448, p = 0.008) and BDS-2 performance (r = −0.400, p = 0.01).
Age
A strong and inverse correlation was observed between age and mitochondrial function in both carriers and non-carriers (Figure 1) consistent with the age-dependent decline of OXPHOS capacity (Burch et al., 1963; Cardellach et al., 1989; Linnane et al., 1989; Yen et al., 1989; Byrne et al., 1991; Cooper et al., 1992; Müller-Höcker, 1992; Torii et al., 1992; Münscher et al., 1993; Boffoli et al., 1994, 1996; Lezza et al., 1994; Wallace et al., 1995; Capkova et al., 2002) being more evident in tissues with high OXPHOS demand (Wallace, 1992; Ojaimi et al., 1999; Kwong and Sohal, 2000; Quiles et al., 2002). Relevant to our study, the overall age-dependent decline in the OXPHOS capacity is more evident in women (Cooper et al., 1992; Papa, 1996). To discriminate between the contribution of age- and the PM-dependent decline in mitochondrial function, we carried out a two-way ANOVA analysis for each mitochondrial outcome evaluated in PM and non-carriers at two age ranges, i.e., younger (23–43 year) and older (44–60) (Table 2). Then the same analysis was done with PM and FXTAS-affected females at two age ranges, i.e., younger (50–60 year) and older (60–70 year), to test for the contribution of age and FXTAS diagnosis (Table 2). When comparing controls and PM carriers, a simple main effect analysis showed that NADH- and FADH2-linked ATP production along with the activity of CCO were solely attributed to the carrier status. Only CS activity showed a decline dependent with both age and PM status (Table 2). When the same analysis was performed to compare mitochondrial outcomes between PM and FXTAS-affected females, FXTAS diagnosis affected significantly both mtROS production and coupling, whereas all other outcomes were not influenced either by age or diagnosis. Of note, the observed correlation between age and FXPOI does not reflect the age at which these carriers first experienced ovarian insufficiency, but merely the age at which the PM carriers reported their medical history.

Table 2. Effect of diagnosis, age and diagnosis x age interaction on mitochondrial outcomes evaluated in controls, PM and FXTAS female carriers.
RNA and Protein Metabolism, Glycolysis and Cellular Response to Stress Is Differentially Regulated in Females With PM
Considering that impaired energy metabolism in the brain is integral to many CNS diseases, cells display complex proteomic responses to energy deficits, including activation of UPR and limited protein synthesis, among others. To gain a deeper insight on the molecular mechanisms of the responses originating from the impaired energy deficit in a PM genetic background, we utilized a proteomics approach on PBMCs from a subset of carriers (see demographics details of this subset under Table 1). Proteins significantly altered between PM and non-carriers were uploaded with their corresponding fold change values (Supplementary Dataset) to STRING (Szklarczyk et al., 2019). Interactomes were algorithmically generated based on direct associations (physical or functional) between eligible proteins. STRING generated a score for each interactome which is a putative measure of probability, with the shading of each node being positively correlated with the magnitude of the fold change. The main functional interactome showed upregulation of the following pathways: HIF-1 signaling, glycolysis, sugars and carbon metabolism, pentose phosphate pathway, pathways involved in cancer-related faulty transcriptional regulation, and pathways affected in Alzheimer’s disease (Table 3). Among the downregulated ones were ribosomal-, spliceosomal- (Supplementary Figure 1) and proteasomal-related pathways (Table 3; Supplementary Figure 2; Supplementary Dataset), pyruvate metabolism, amino acids degradation (in particular branched amino acids), RNA processing (Table 3; Supplementary Figure 2; Supplementary Dataset) and transport, fatty acid synthesis, and TCA (Table 3; Figure 2A; KEGG and REACTOME Pathway tabs of Supplementary Dataset). This interactome highlighted a shift in mitochondrial energy production (and consistent with the previous results on bioenergetics), enhanced endoplasmic reticulum (ER) stress and ribosomal dysfunction. A sub-interactome (named “Energy production interactome”) was linked to energy-producing pathways including mitochondrial bioenergetics and glycolysis (Figure 2B; Supplementary Figure 3; Supplementary Dataset, tabs highlighted in gray). The decrease in proteins involved in butanoate metabolism, fatty acid elongation and beta-oxidation along with BCAA metabolism (ECH1, HSD17B10, and ECHS1) indicates a shift toward glycolysis (higher HK1, ALDOA, but lower LDHB) as alternative energy source without incurring into increased ketone bodies’ metabolism.
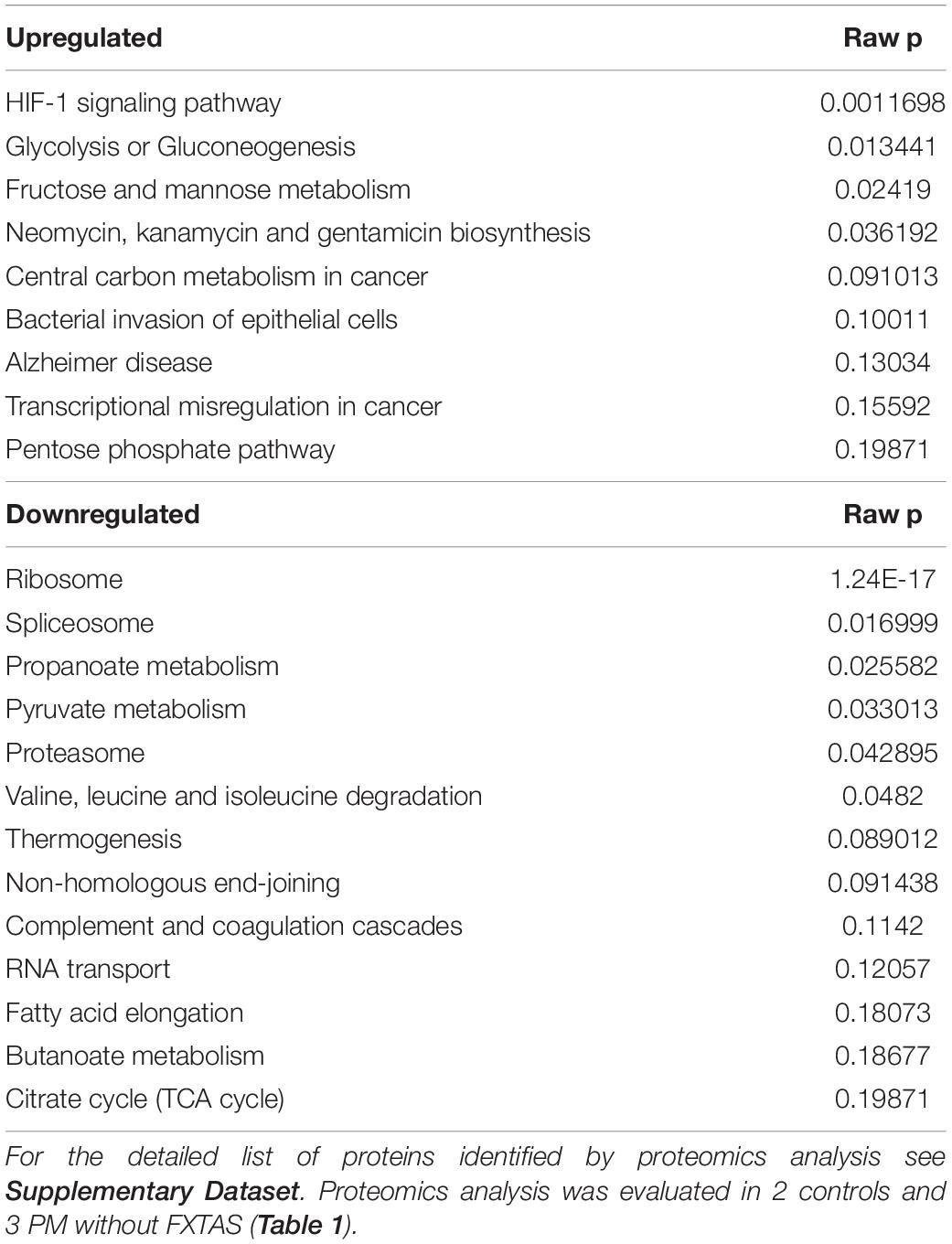
Table 3. Differential expression of pathways in PBMC from PM females.
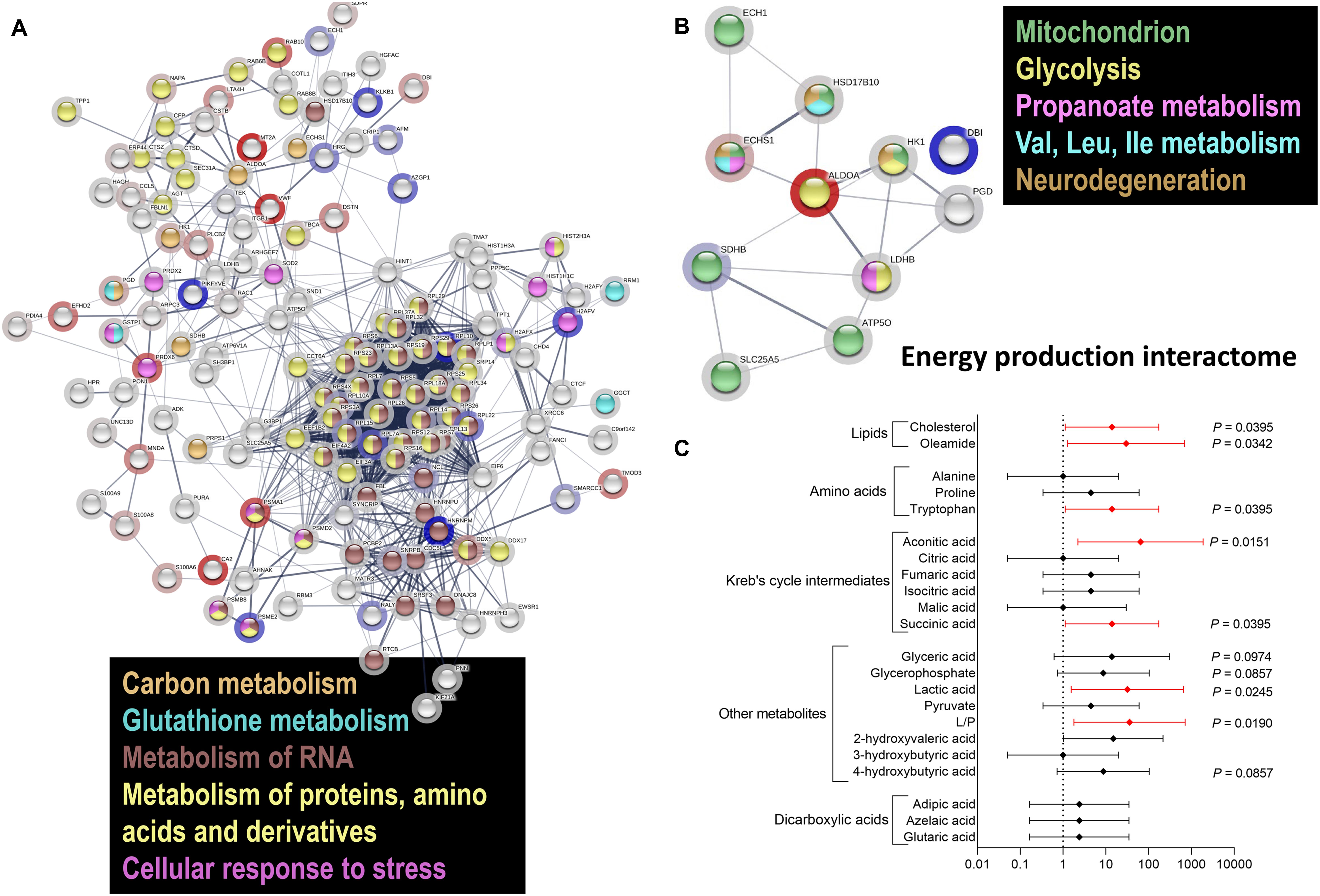
Figure 2. Differential protein and metabolite enrichment in PM females. (A) Proteomics analysis was carried out in PBMC from PM females 29–63 year old and age-matched controls. Proteins detected in both PM and non-carriers were uploaded with their corresponding fold change values (see Supplementary Dataset) to STRING. Interactomes were algorithmically generated based on direct associations (physical or functional) between eligible proteins. The interactomes are color coded with blue nodes representing proteins that were down-regulated, red upregulated in PM, and clear when there were detected but whose levels were not statistically different between diagnostic groups. The shading of each node is correlated with the magnitude of the fold change. Differentially regulated metabolic pathways included carbon metabolism, metabolism of RNA, proteins, amino acids, and cellular response to oxidative stress. (B) A subset of differentially regulated proteins (Supplementary Dataset, tabs highlighted in gray) had key roles in mitochondrial function, glycolysis, fatty acid and amino acid metabolism, as well as in RNA processing (pathways reported in Supplementary Figures 1–3) and pathways affected in neurodegeneration. (C) Metabolomics analysis was performed in plasma from 8 controls and 7 age-matched PM carriers, 24–52 year, and age-matched controls. Differentially enriched metabolites are shown, with their respective p values, in Supplementary Figure 4. A forest plot was built with the odds ratios (X axis) and the 95% CI (error bars) calculated with control values for each selected metabolite based on its role in glycolysis and mitochondrial metabolism. In red are metabolites with statistically significant OR, indicating a higher probability to be affected in the PM, and as such considered as putative biomarkers for female carriers.
Since the protein changes provided insight into the complex molecular alterations underpinning the cellular adaptation to the PM-mediated stress, we sought to evaluate circulating metabolites and biomarkers of mitochondrial dysfunction by performing untargeted plasma metabolomics. To this end, untargeted metabolomics was performed by using gas chromatography mass spectrometry in plasma from 7 carriers and 8 age-matched controls (see demographics details under Table 1). Similar to the metabolic disarray observed in some mitochondrial disorders, Krebs’ cycle intermediates, dicarboxylic acids, and “other metabolites” associated with glycolysis and leucine metabolism were mostly up-regulated (Supplementary Figure 4), with levels of aconitate, succinate, lactic acid, and lactate-to-pyruvate, being significantly higher in PM than controls (Supplementary Figure 4). These results confirm and expand findings of increased Krebs’ cycle intermediates, likely the result of an impaired TCA activity, already observed by our group in PM carriers of both sexes (Giulivi et al., 2016), with aconitate, and isocitrate being those that differentiated the most between diagnostic groups, along with oleamide (Giulivi et al., 2016). In this regard, levels of oleamide, along with tryptophan, were found significantly lower in female PM whereas those of cholesterol were significantly higher in PM (Supplementary Figure 4). The odds ratio (OR) for these metabolites to be altered in the PM (Figure 2C with p < 0.1) was significant for cholesterol, oleamide, tryptophan, aconitate, succinate, lactate, and lactate-to-pyruvate ratio, glycerate, alpha-glycerophosphate, 4-hydroxybutyrate and some of the dicarboxylic acids tested.
Discussion
Mounting evidence has shown that female carriers of the PM are at higher risk for developing several health-related issues compared to non-carrier females (Bailey et al., 2008; Hunter et al., 2010; Winarni et al., 2012; Wheeler et al., 2014b; Movaghar et al., 2019). Awareness of these risks and correlation of the clinical signs with the biochemical footprint of carriers could help to identify critical biomarkers in early diagnosis and likely prognosis. If ascertained, these associations could lead to an integrated approach between clinical specialties and basic science, which could be extremely beneficial in the management of symptoms and challenges that female PM carriers experience in their day-to-day life.
The brain’s high mitochondrial energy consumption makes neurons highly vulnerable to impaired glucose metabolism (Hyder et al., 2013). Then, it is not surprising that a decline in mitochondrial activity has been associated with memory loss and, particularly, with age-dependent cognitive impairment (Freeman and Young, 2000; Liu et al., 2002). Moreover, most mtDNA diseases are associated with brain disorders because adequate neuronal development (Atamna et al., 2002; Zini et al., 2002) and structure (Johnson and Byerly, 1993; Bristow et al., 2002; Liu et al., 2002) and axonal and synaptic activity (Freeman and Young, 2000; Zenisek and Matthews, 2000) all involve mitochondrial genes (Wallace, 1999; Schon, 2000). Mitochondrial dysfunction has been reported not only in neurodegeneration (Friedland et al., 1983; Minoshima et al., 1997; Chiaravalloti et al., 2015) but also in pre-symptomatic, genetically-susceptible individuals (Small et al., 1995; Reiman et al., 1996; Mosconi et al., 2006; Ossenkoppele et al., 2013). In line with these studies and our previous reports on the PM (Napoli et al., 2016a, 2018), our study strongly indicates that global impairment of bioenergetics, and likely the subsequent energy depletion, is one of the earliest functional changes prior to the onset of overt clinical symptoms. This is supported by the relatively milder mitochondrial dysfunction in PM carriers without FXTAS and FXPOI which is enhanced with the diagnosis and progression of FXTAS. While the decline in cognitive/intellectual (FSIQ) and executive (BDS-2) function was correlated with longer CGGs, intellectual decline (FSIQ) was significantly correlated with an overall mitochondrial deficit. This is highly suggestive that mitochondrial deficits may influence neuronal dysregulation and, over the years, degenerative mechanisms such as described for AD (Rice et al., 2014).
The metabolomics findings are particularly relevant in the context of several psychiatric and neurological symptoms experienced by some female carriers as oleamide has a critical role in mood and sleep disorders as well as depression due to its interaction with serotonergic and GABAergic neurotransmission (Mendelson and Basile, 2001). Similarly, tryptophan is not only an essential amino acid for protein synthesis but also a precursor of several biological mediators involved in stress response, antioxidant system, behavioral response, and immune system (Firk and Markus, 2009; Hoglund et al., 2017). Decreased tryptophan levels have been linked to lower serotonin levels, likely setting the basis for the establishment of neuropsychological symptoms including depression, anxiety, irritability, attention deficits, and insomnia (Jenkins et al., 2016). The higher cholesterol levels are of interest in the context of neurodegeneration, as mitochondrial cholesterol loading has recently emerged as a key player in the pathology of neurological disorders such as AD and Niemann-Pick Type C (NPC) disease (Elustondo et al., 2017; Torres et al., 2019).
In line with these findings, the lower fatty acid beta-oxidation resulting mainly from lower ABAD and ECHS1, deserves a separate discussion. Lower levels of ABAD may disrupt the ABAD-beta-amyloid interaction in mitochondria and suppresses apoptosis by increasing the levels of beta-amyloid as reported for AD patients and transgenic mouse models (Lustbader et al., 2004). Deficiency in ECHS1 results in metabolic acidosis with a combined respiratory chain deficiency (Sakai et al., 2015). Consistent with these findings, higher levels of lactic acid and a trend towards higher 2-hydroxyvaleric acid, proline and organic acids, such as adipic, azelaic and glutaric, were observed in plasma from female carriers (Figures 2C, 3) similar to the cases reported by others (Fitzsimons et al., 2018). Then, the significant lower expression of ABAD and ECHS1 in female carriers makes them as interesting candidates to investigate further in terms of its regulation in the context of beta-amyloid and proteostasis. Furthermore, since the OXPHOS decline in ECHS1 deficiency has been attributed to the accumulation of inhibitory fatty acid intermediates and to the disruption of ETC Complex biogenesis and/or stability (Burgin and McKenzie, 2020), it would not be unlikely that these mechanisms are contributing to the OXPHOS deficiencies observed in the PM.
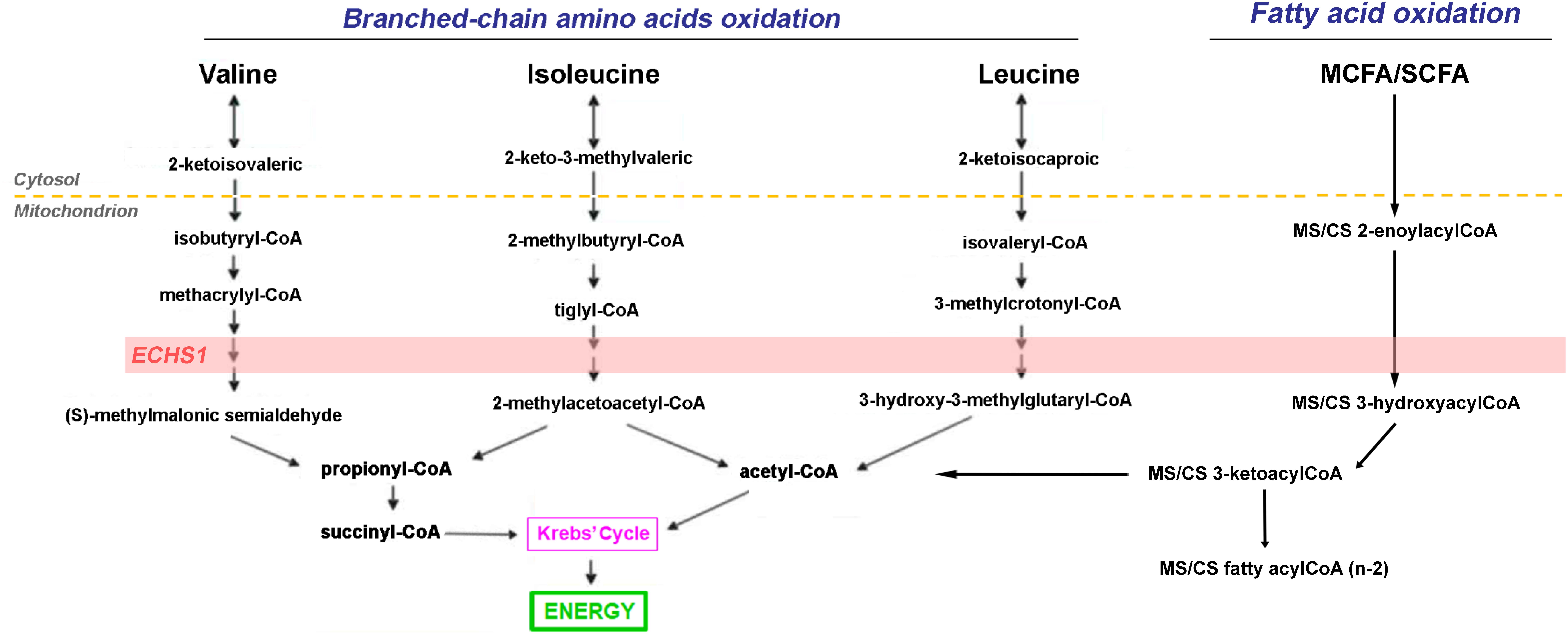
Figure 3. Role of ECSH1 in branched-chain amino acids and short- and medium-chain fatty acid catabolism. The short-chain enoyl-CoA hydratase ECHS1 has a critical role in branched chain amino acid (BCAA) catabolism as well as fatty acid catabolism generating succinyl-CoA and acetyl-CoA which are fed into the Krebs’ cycle for the generation of reducing equivalents.
Proteomics analysis not only confirmed and extended the functional and metabolomics results, but also shed light into the proteostasis status of the PM. The biological consequence of higher mitochondrial ROS was identified with the higher levels of aconitate likely the result of inactivation of aconitase, an enzyme highly sensitive to oxidative stress damage. The increase in oxidative stress may contribute to proteotoxicity and to the generation of misfolded proteins that accumulate upon ER stress, as a result, increase in the detoxification demands. However, an uncoordinated proteomic response in the ER was identified in the PM. Only one chaperone, HSPA5, was higher in PM vs. non-carriers, and a mixed response was observed for subunits of the proteasome. These results suggest that proteasomal degradation, a fundamental mechanism for degrading the misfolded proteins that accumulate in response to the metabolic challenge, is somehow not coordinated to adequately cope with the proteotoxicity. Indeed, it has been a long-standing hypothesis that protein aggregates in diseased brain impair the protein degradation function of the 26S proteasome (Taylor et al., 2002; Ciechanover and Brundin, 2003; Valera et al., 2005), then it would be reasonable to hypothesize that as the UPR is not fully coordinated in female carriers, any proteasomal response expected to ensue in response to a metabolic, environmental or pharmacological challenge might be adversely affected.
Our findings are particularly relevant for female carriers, as mitochondria have a critical role in oocyte developmental competence and function (Dumollard et al., 2007; Wang et al., 2009), and in the development and function of the reproductive system fertility (Reynier et al., 2001; El Shourbagy et al., 2006; Santos et al., 2006; Cagnone et al., 2016; Demain et al., 2017) with an irrefutable implication in primary ovarian insufficiency (POI) (Conca Dioguardi et al., 2016; Tiosano et al., 2019). The energy deficit along with the lower levels of ECHS1 in carriers, one of the genes identified as critical for oocyte developmental competence (Biase, 2017), set the basis for future research to identify more clearly the role of this protein in POI and FXPOI.
In conclusion, through a combined multi-omics approach in PBMC and plasma of PM females in association with thorough clinical and bioenergetics assessments, we found impaired metabolic pathways which can result from the direct action of toxic intermediates derived from the PM genetic background (either accumulation of mRNA or proteotoxicity of RAN-derived protein products). In this regard, we are adding the novel observation that the accumulation of aberrant metabolites resulting from deficiencies in critical metabolic steps may add to the altered interaction between fatty acid oxidation and the electron transport chain contributing to the overall OXPHOS decline as it has been described for ECHS1 deficiency (Burgin and McKenzie, 2020; Figure 3). In turn, either mechanism elicits a suboptimal activation of UPR and ERAD responses, setting a challenging scenario to withstand other, more severe forms of stress. Along these lines, the development of neurodegeneration or other clinical symptoms in older carriers could be linked to a lifetime accumulation of cellular damage, aggravated by the aging process.
Limitations
There are potential limitations to our study, starting with its partly retrospective design. Of all the participants included, neurocognitive testing and omics analyses were performed in a subset of individuals. The selection of the cohort was from a controlled sample of individuals enrolled in the study upon referral to the medical center clinic. For this reason, this cohort might not be representative of the full PM population range but rather the most affected ones. Similarly, a fraction of the controls included in our analysis were family members of probands with typical FMR1 repeat sizes and, while they may represent the non-carrier group, they may be affected by extra emotional burden by having a relative or partner with PM diagnosis. The challenging recruitment of healthy donors is reflected in their relatively lower number particularly in the 60–70 year age bracket. Information on anxiety was self-reported. The OXPHOS variability could be ascribed to the lack of a clinical diagnosis of a mitochondrial disease (i.e., borderline OXPHOS capacity without overt mitochondrial disease symptoms) which is manifested in front of a challenge (high-intensity exercise, a co-morbidity, or aging), the influence of different dietary habits and lifestyle, and the contribution of mtDNA polymorphisms to cognitive deficits (Skuder et al., 1995).
Financial Disclosure
RH has received funding from the Azrieli Foundation, Zynerba, Ovid and Neuren for treatment trials in fragile X syndrome, and consulted with Zynerba and Fulcrum regarding treatment for fragile X syndrome. FT has received funding from the Azrieli Foundation and Zynerba for studies on fragile X syndrome and has consulted with Zynerba. The other authors have no financial disclosures relevant to this article.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The studies involving human participants were reviewed and approved by IRB Ethics Committee at UC Davis Medical Center. The patients/participants provided their written informed consent to participate in this study.
Author Contributions
EN processed all samples, carried out all polarographic and spectrophotometric measurements, performed correspondent statistical analyses, wrote the manuscript, and revised and approved the final version as submitted. YM collected and provided demographic, clinical, and molecular data, revised the manuscript, and approved the final version as submitted. AS carried out the neuropsychological testing and also revised the manuscript and approved the final as submitted. FT provided CGG repeats and XAR, revised the manuscript, and approved the final version as submitted. RH carried out clinical assessment of the women enrolled in this study and wrote clinical findings, revised the manuscript, and approved the final manuscript as submitted. CG conceptualized and designed the study, analyzed the omics data, wrote the manuscript, and approved the final manuscript as submitted.
Funding
This study was funded by the National Institutes of Health (HD036071). Support was also obtained from the MIND Institute Intellectual and Developmental Disabilities Research Center (U54 HD079125) and the Health and Human Administration of Developmental Disabilities grants 90DD0596 and UL1 TR001860.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We wish to thank all women who provided these samples making this study possible.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2020.578640/full#supplementary-material
References
Allen, E. G., Charen, K., Hipp, H. S., Shubeck, L., Amin, A., He, W., et al. (2020). Clustering of comorbid conditions among women who carry an FMR1 premutation. Genet. Med. 22, 758–766. doi: 10.1038/s41436-019-0733-5
Allen, E. G., Sullivan, A. K., Marcus, M., Small, C., Dominguez, C., Epstein, M. P., et al. (2007). Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum. Reprod. 22, 2142–2152. doi: 10.1093/humrep/dem148
Allingham-Hawkins, D. J., Babul-Hirji, R., Chitayat, D., Holden, J. J. A., Yang, K. T., Lee, C., et al. (1999). Fragile X premutation is a significant risk factor for premature ovarian failure: the international collaborative POF in fragile X study - Preliminary data. Am. J. Med. Genet. 83, 322–325. doi: 10.1002/(SICI)1096-8628(19990402)83:4<322::AID-AJMG17>3.0.CO;2-B
Atamna, H., Killilea, D. W., Killilea, A. N., and Ames, B. N. (2002). Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc. Natl. Acad. Sci. U.S.A. 99, 14807–14812. doi: 10.1073/pnas.192585799
Bagni, C., Tassone, F., Neri, G., and Hagerman, R. (2012). Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J. Clin. Invest. 122, 4314–4322. doi: 10.1172/JCI63141
Bailey, D. B. Jr., Raspa, M., Olmsted, M., and Holiday, D. B. (2008). Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am. J. Med. Genet. A 146A, 2060–2069. doi: 10.1002/ajmg.a.32439
Belanger, H. G., Wilder-Willis, K., Malloy, P., Salloway, S., Hamman, R. F., and Grigsby, J. (2005). Assessing motor and cognitive regulation in AD, MCI, and controls using the behavioral dyscontrol scale. Arch. Clin. Neuropsychol. 20, 183–189. doi: 10.1016/j.acn.2004.05.003
Bennett, C. E., Conway, G. S., Macpherson, J. N., Jacobs, P. A., and Murray, A. (2010). Intermediate sized CGG repeats are not a common cause of idiopathic premature ovarian failure. Hum. Reprod. 25, 1335–1338. doi: 10.1093/humrep/deq058
Berry-Kravis, E., Abrams, L., Coffey, S. M., Hall, D. A., Greco, C., Gane, L. W., et al. (2007a). Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov. Disord. 22, 2018–2030. doi: 10.1002/mds.21493
Berry-Kravis, E., Goetz, C. G., Leehey, M. A., Hagerman, R. J., Zhang, L., Li, L., et al. (2007b). Neuropathic features in fragile X premutation carriers. Am. J. Med. Genet. A 143A, 19–26. doi: 10.1002/ajmg.a.31559
Berry-Kravis, E., Lewin, F., Wuu, J., Leehey, M., Hagerman, R., Hagerman, P., et al. (2003). Tremor and ataxia in fragile X premutation carriers: blinded videotape study. Ann. Neurol. 53, 616–623. doi: 10.1002/ana.10522
Berry-Kravis, E., Potanos, K., Weinberg, D., Zhou, L., and Goetz, C. G. (2005). Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann. Neurol. 57, 144–147. doi: 10.1002/ana.20360
Besterman, A. D., Wilke, S. A., Mulligan, T. E., Allison, S. C., Hagerman, R., Seritan, A. L., et al. (2014). Towards an understanding of neuropsychiatric manifestations in fragile X premutation carriers. Fut. Neurol. 9, 227–239. doi: 10.2217/fnl.14.11
Biase, F. H. (2017). Oocyte developmental competence: insights from cross-species differential gene expression and human oocyte-specific functional gene networks. OMICS 21, 156–168. doi: 10.1089/omi.2016.0177
Boffoli, D., Scacco, S. C., Vergari, R., Persio, M. T., Solarino, G., Laforgia, R., et al. (1996). Ageing is associated in females with a decline in the content and activity on the b-c1 complex in skeletal muscle mitochondria. Biochim. Biophys. Acta 1315, 66–72. doi: 10.1016/0925-4439(95)00107-7
Boffoli, D., Scacco, S. C., Vergari, R., Solarino, G., Santacroce, G., and Papa, S. (1994). Decline with age of the respiratory chain activity in human skeletal muscle. Biochim. Biophys. Acta 1226, 73–82. doi: 10.1016/0925-4439(94)90061-2
Bourgeois, J. A., Cogswell, J. B., Hessl, D., Zhang, L., Ono, M. Y., Tassone, F., et al. (2007). Cognitive, anxiety and mood disorders in the fragile X-associated tremor/ataxia syndrome. Gen. Hosp. Psychiatry 29, 349–356. doi: 10.1016/j.genhosppsych.2007.03.003
Bristow, E. A., Griffiths, P. G., Andrews, R. M., Johnson, M. A., and Turnbull, D. M. (2002). The distribution of mitochondrial activity in relation to optic nerve structure. Arch. Ophthalmol. 120, 791–796. doi: 10.1001/archopht.120.6.791
Burch, H. B., Lowry, O. H., Kuhlman, A. M., Skerjance, J., Diamant, E. J., Lowry, S. R., et al. (1963). Changes in patterns of enzymes of carbohydrate metabolism in the developing rat liver. J. Biol. Chem. 238, 2267–2273.
Burgin, H. J., and McKenzie, M. (2020). Understanding the role of OXPHOS dysfunction in the pathogenesis of ECHS1 deficiency. FEBS Lett. 594, 590–610. doi: 10.1002/1873-3468.13735
Byrne, E., Dennett, X., and Trounce, I. (1991). Oxidative energy failure in post-mitotic cells: a major factor in senescence. Rev. Neurol. 147, 6–7.
Cagnone, G. L., Tsai, T. S., Makanji, Y., Matthews, P., Gould, J., Bonkowski, M. S., et al. (2016). Restoration of normal embryogenesis by mitochondrial supplementation in pig oocytes exhibiting mitochondrial DNA deficiency. Sci. Rep. 6:23229. doi: 10.1038/srep23229
Capkova, M., Houstek, J., Hansikova, H., Hainer, V., Kunesova, M., and Zeman, J. (2002). Activities of cytochrome c oxidase and citrate synthase in lymphocytes of obese and normal-weight subjects. Int. J. Obes. Relat. Metab. Disord. 26, 1110–1117. doi: 10.1038/sj.ijo.0802055
Cardellach, F., Galofre, J., Cusso, R., and Urbano-Marquez, A. (1989). Decline in skeletal muscle mitochondrial respiration chain function with ageing. Lancet 334, 44–45. doi: 10.1016/S0140-6736(89)90282-1
Chiaravalloti, A., Martorana, A., Koch, G., Toniolo, S., Di Biagio, D., Di Pietro, B., et al. (2015). Functional correlates of t-Tau, p-Tau and Abeta(1)(-)(4)(2) amyloid cerebrospinal fluid levels in Alzheimer’s disease: a (1)(8)F-FDG PET/CT study. Nucl. Med. Commun. 36, 461–468. doi: 10.1097/MNM.0000000000000272
Ciechanover, A., and Brundin, P. (2003). The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 40, 427–446. doi: 10.1016/s0896-6273(03)00606-8
Claeys, T., Boogers, A., and Vanneste, D. (2020). MRI findings in fragile X-associated tremor/ataxia syndrome. Acta Neurol. Belg. 120, 181–183. doi: 10.1007/s13760-019-01237-w
Conca Dioguardi, C., Uslu, B., Haynes, M., Kurus, M., Gul, M., Miao, D. Q., et al. (2016). Granulosa cell and oocyte mitochondrial abnormalities in a mouse model of fragile X primary ovarian insufficiency. Mol. Hum. Reprod. 22, 384–396. doi: 10.1093/molehr/gaw023
Cooper, J. M., Mann, V. M., and Schapira, A. H. (1992). Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: effect of ageing. J. Neurol. Sci. 113, 91–98. doi: 10.1016/0022-510x(92)90270-u
Demain, L. A., Conway, G. S., and Newman, W. G. (2017). Genetics of mitochondrial dysfunction and infertility. Clin. Genet. 91, 199–207. doi: 10.1111/cge.12896
Drozdick, L. W., Wahlstrom, D., Zhu, J., and Weiss, L. G. (2012). “The Wechsler adult intelligence scale—fourth edition and the wechsler memory scale—fourth edition,” in Contemporary Intellectual Assessment: Theories, Tests, and Issues, 3rd Edn, eds D. P. Flanagan and P. L. Harrison (New York, NY: The Guilford Press), 197–223.
Dumollard, R., Duchen, M., and Carroll, J. (2007). The role of mitochondrial function in the oocyte and embryo. Curr. Top. Dev. Biol. 77, 21–49. doi: 10.1016/S0070-2153(06)77002-8
El Shourbagy, S. H., Spikings, E. C., Freitas, M., and St John, J. C. (2006). Mitochondria directly influence fertilisation outcome in the pig. Reproduction 131, 233–245. doi: 10.1530/rep.1.00551
Elizur, S. E., Lebovitz, O., Derech-Haim, S., Dratviman-Storobinsky, O., Feldman, B., Dor, J., et al. (2014). Elevated levels of FMR1 mRNA in granulosa cells are associated with low ovarian reserve in FMR1 premutation carriers. PLoS One 9:e0105121. doi: 10.1371/journal.pone.0105121
Elustondo, P., Martin, L. A., and Karten, B. (2017). Mitochondrial cholesterol import. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1862, 90–101. doi: 10.1016/j.bbalip.2016.08.012
Ennis, S., Ward, D., and Murray, A. (2006). Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Europ. J. Hum. Genet. 14, 253–255. doi: 10.1038/sj.ejhg.5201510
Filipovic-Sadic, S., Sah, S., Chen, L., Krosting, J., Sekinger, E., Zhang, W., et al. (2010). A novel FMR1 PCR method for the routine detection of low abundance expanded alleles and full mutations in fragile X syndrome. Clin. Chem. 56, 399–408. doi: 10.1373/clinchem.2009.136101
Firk, C., and Markus, C. R. (2009). Mood and cortisol responses following tryptophan-rich hydrolyzed protein and acute stress in healthy subjects with high and low cognitive reactivity to depression. Clin. Nutr. 28, 266–271. doi: 10.1016/j.clnu.2009.03.002
Fitzsimons, P. E., Alston, C. L., Bonnen, P. E., Hughes, J., Crushell, E., Geraghty, M. T., et al. (2018). Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Am. J. Med. Genet. A 176, 1115–1127. doi: 10.1002/ajmg.a.38658
Freeman, F. M., and Young, I. G. (2000). The mitochondrial benzodiazepine receptor and avoidance learning in the day-old chick. Pharmacol. Biochem. Behav. 67, 355–362. doi: 10.1016/S0091-3057(00)00373-7
Friedland, R. P., Budinger, T. F., Ganz, E., Yano, Y., Mathis, C. A., Koss, B., et al. (1983). Regional cerebral metabolic alterations in dementia of the Alzheimer type: positron emission tomography with [18F]fluorodeoxyglucose. J. Comput. Assist. Tomogr. 7, 590–598. doi: 10.1097/00004728-198308000-00003
Giulivi, C., Napoli, E., Schwartzer, J., Careaga, M., and Ashwood, P. (2013). Gestational exposure to a viral mimetic poly(i:C) results in long-lasting changes in mitochondrial function by leucocytes in the adult offspring. Mediators Inflamm. 2013:609602. doi: 10.1155/2013/609602
Giulivi, C., Napoli, E., Tassone, F., Halmai, J., and Hagerman, R. (2016). Plasma biomarkers for monitoring brain pathophysiology in FMR1 premutation carriers. Front. Mol. Neurosci. 9:71. doi: 10.3389/fnmol.2016.00071
Giulivi, C., Zhang, Y. F., Omanska-Klusek, A., Ross-Inta, C., Wong, S., Hertz-Picciotto, I., et al. (2010). Mitochondrial dysfunction in autism. JAMA 304, 2389–2396. doi: 10.1001/jama.2010.1706
Hagerman, P. J., and Hagerman, R. J. (2004). The fragile-X premutation: a maturing perspective. Am. J. Hum. Genet. 74, 805–816. doi: 10.1086/386296
Hagerman, R., and Hagerman, P. (2013). Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 12, 786–798. doi: 10.1016/S1474-4422(13)70125-X
Hagerman, R. J., and Hagerman, P. (2016). Fragile X-associated tremor/ataxia syndrome - features, mechanisms and management. Nat. Rev. Neurol. 12, 403–412. doi: 10.1038/nrneurol.2016.82
Hagerman, R. J., Hall, D. A., Coffey, S., Leehey, M., Bourgeois, J., Gould, J., et al. (2008). Treatment of fragile X-associated tremor ataxia syndrome (FXTAS) and related neurological problems. Clin. Interv. Aging 3, 251–262. doi: 10.2147/cia.s1794
Hagerman, R. J., Leavitt, B. R., Farzin, F., Jacquemont, S., Greco, C. M., Brunberg, J. A., et al. (2004). Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am. J. Hum. Genet. 74, 1051–1056. doi: 10.1086/420700
Hagerman, R. J., Leehey, M., Heinrichs, W., Tassone, F., Wilson, R., Hills, J., et al. (2001). Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 57, 127–130. doi: 10.1212/wnl.57.1.127
Hagerman, R. J., Protic, D., Rajaratnam, A., Salcedo-Arellano, M. J., Aydin, E. Y., and Schneider, A. (2018). Fragile X-associated neuropsychiatric disorders (FXAND). Front. Psychiatry 9:564. doi: 10.3389/fpsyt.2018.00564
Hoglund, E., Overli, O., Andersson, M. A., Silva, P., Laursen, D. C., Moltesen, M. M., et al. (2017). Dietary l-tryptophan leaves a lasting impression on the brain and the stress response. Br. J. Nutr. 117, 1351–1357. doi: 10.1017/S0007114517001428
Hunter, J. E., Rohr, J. K., and Sherman, S. L. (2010). Co-occurring diagnoses among FMR1 premutation allele carriers. Clin. Genet. 77, 374–381. doi: 10.1111/j.1399-0004.2009.01317.x
Hyder, F., Rothman, D. L., and Bennett, M. R. (2013). Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. U.S.A. 110, 3549–3554. doi: 10.1073/pnas.1214912110
Jacquemont, S., Hagerman, R. J., Leehey, M., Grigsby, J., Zhang, L., Brunberg, J. A., et al. (2003). Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet. 72, 869–878. doi: 10.1086/374321
Jenkins, T. A., Nguyen, J. C., Polglaze, K. E., and Bertrand, P. P. (2016). Influence of tryptophan and serotonin on mood and cognition with a possible role of the gut-brain axis. Nutrients 8:56. doi: 10.3390/nu8010056
Johnson, B. D., and Byerly, L. (1993). A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+. Neuron 10, 797–804. doi: 10.1016/0896-6273(93)90196-X
Karimov, C. B., Moragianni, V. A., Cronister, A., Srouji, S., Petrozza, J., Racowsky, C., et al. (2011). Increased frequency of occult fragile X-associated primary ovarian insufficiency in infertile women with evidence of impaired ovarian function. Hum. Reprod. 26, 2077–2083. doi: 10.1093/humrep/der168
Kwong, L. K., and Sohal, R. S. (2000). Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch. Biochem. Biophys. 373, 16–22. doi: 10.1006/abbi.1999.1495
Lezza, A. M. S., Boffoli, D., Scacco, S., Cantatore, P., and Gadaleta, M. N. (1994). Correlation between mitochondrial DNA 4977-bp deletion and respiratory chain enzyme activities in aging human skeletal muscles. Biochem. Biophys. Res. Commun. 205, 772–779. doi: 10.1006/bbrc.1994.2732
Linnane, A., Ozawa, T., Marzuki, S., and Tanaka, M. (1989). Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases Lancet. Lancet 333, 642–645. doi: 10.1016/S0140-6736(89)92145-4
Liu, J., Head, E., Gharib, A. M., Yuan, W., Ingersoll, R. T., Hagen, T. M., et al. (2002). Memory loss in old rats is associated with brain mitochondrial decay and RNA/DNA oxidation: Partial reversal by feeding acetyl-L-carnitine and/or R-α-lipoic acid. Proc. Natl. Acad. Sci. U.S.A. 99, 2356–2361. doi: 10.1073/pnas.261709299
Lozano, R., Saito, N., Reed, D., Eldeeb, M., Schneider, A., Hessl, D., et al. (2016). Aging in Fragile X Premutation carriers. Cerebellum 15, 587–594. doi: 10.1007/s12311-016-0805-x
Lustbader, J. W., Cirilli, M., Lin, C., Xu, H. W., Takuma, K., Wang, N., et al. (2004). ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. doi: 10.1126/science.1091230
Mendelson, W. B., and Basile, A. S. (2001). The hypnotic actions of the fatty acid amide, oleamide. Neuropsychopharmacol. 25, S36–S39. doi: 10.1016/S0893-133X(01)00341-4
Minoshima, S., Giordani, B., Berent, S., Frey, K. A., Foster, N. L., and Kuhl, D. E. (1997). Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 42, 85–94. doi: 10.1002/ana.410420114
Mosconi, L., Sorbi, S., De Leon, M. J., Li, Y., Nacmias, B., Myoung, P. S., et al. (2006). Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J. Nucl. Med. 47, 1778–1786.
Movaghar, A., Page, D., Brilliant, M., Baker, M. W., Greenberg, J., Hong, J., et al. (2019). Data-driven phenotype discovery of FMR1 premutation carriers in a population-based sample. Sci. Adv. 5:eaaw7195. doi: 10.1126/sciadv.aaw7195
Müller-Höcker, J. (1992). Mitochondria and ageing. Brain Pathol. 2, 149–158. doi: 10.1111/j.1750-3639.1992.tb00683.x
Münscher, C., Müller-Höcker, J., and Kadenbach, B. (1993). Human aging is associated with various point mutations in trna genes of mitochondrial DNA. Biol. Chem. Hoppe Seyler 374, 1099–1104. doi: 10.1515/bchm3.1993.374.7-12.1099
Murray, A., Schoemaker, M. J., Bennett, C. E., Ennis, S., Macpherson, J. N., Jones, M., et al. (2014). Population-based estimates of the prevalence of FMR1 expansion mutations in women with early menopause and primary ovarian insufficiency. Genet. Med. 16, 19–24. doi: 10.1038/gim.2013.64
Napoli, E., Schneider, A., Hagerman, R., Song, G., Wong, S., Tassone, F., et al. (2018). Impact of FMR1 premutation on neurobehavior and bioenergetics in young monozygotic twins. Front. Genet. 9:338. doi: 10.3389/fgene.2018.00338
Napoli, E., Song, G., Schneider, A., Hagerman, R., Eldeeb, M. A., Azarang, A., et al. (2016a). Warburg effect linked to cognitive-executive deficits in FMR1 premutation. FASEB J. 30, 3334–3351. doi: 10.1096/fj.201600315R
Napoli, E., Song, G., Wong, S., Hagerman, R., and Giulivi, C. (2016b). Altered bioenergetics in primary dermal fibroblasts from adult carriers of the FMR1 premutation before the onset of the neurodegenerative disease Fragile X-Associated Tremor/Ataxia Syndrome. Cerebellum 15, 552–564. doi: 10.1007/s12311-016-0779-8
Napoli, E., Tassone, F., Wong, S., Angkustsiri, K., Simon, T. J., Song, G., et al. (2015). Mitochondrial citrate transporter-dependent metabolic signature in the 22q11.2 deletion syndrome. J. Biol. Chem. 290, 23240–23253. doi: 10.1074/jbc.M115.672360
Napoli, E., Wong, S., Hung, C., Ross-Inta, C., Bomdica, P., and Giulivi, C. (2013). Defective mitochondrial disulfide relay system, altered mitochondrial morphology and function in Huntington’s disease. Hum. Mol. Genet. 22, 989–1004. doi: 10.1093/hmg/dds503
Ojaimi, J., Masters, C. L., Opeskin, K., Mckelvie, P., and Byrne, E. (1999). Mitochondrial respiratory chain activity in the human brain as a function of age. Mech. Ageing Dev. 111, 39–47. doi: 10.1016/s0047-6374(99)00071-8
Ossenkoppele, R., Van Der Flier, W. M., Zwan, M. D., Adriaanse, S. F., Boellaard, R., Windhorst, A. D., et al. (2013). Differential effect of APOE genotype on amyloid load and glucose metabolism in AD dementia. Neurology 80, 359–365. doi: 10.1212/WNL.0b013e31827f0889
Papa, S. (1996). Mitochondrial oxidative phosphorylation changes in the life span. Molecular aspects and physiopathological implications. Biochim. Biophys. Acta 1276, 87–105. doi: 10.1016/0005-2728(96)00077-1
Pastore, L. M., Young, S. L., Baker, V. L., Karns, L. B., Williams, C. D., and Silverman, L. M. (2012). Elevated prevalence of 35-44 FMR1 trinucleotide repeats in women with diminished ovarian reserve. Reprod. Sci. 19, 1226–1231. doi: 10.1177/1933719112446074
Patsalis, P. C., Sismani, C., Hettinger, J. A., Holden, J. J. A., Lawson, J. S., Chalifoux, M., et al. (1999). Frequencies of’ ‘grey-zone’ and premutation-size FMR1 CGG-repeat alleles in patients with developmental disability in Cyprus and Canada [1]. Am. J. Med. Genet. 84, 195–197. doi: 10.1002/(SICI)1096-8628(19990528)84:3<195::AID-AJMG4<3.0.CO;2-1
Perneger, T. V. (1998). What’s wrong with bonferroni adjustments. BMJ 316, 1236–1238. doi: 10.1136/bmj.316.7139.1236
Pouresmaeili, F., and Fazeli, Z. (2014). Premature ovarian failure: a critical condition in the reproductive potential with various genetic causes. Intl. J. Fert. Steril. 8, 1–12.
Quiles, J. L., Martinez, E., Ibanez, S., Ochoa, J. J., Martin, Y., Lopez-Frias, M., et al. (2002). Ageing-related tissue-specific alterations in mitochondrial composition and function are modulated by dietary fat type in the rat. J. Bioenerg. Biomembr. 34, 517–524. doi: 10.1023/a:1022530512096
Reiman, E. M., Caselli, R. J., Yun, L. S., Chen, K., Bandy, D., Minoshima, S., et al. (1996). Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N. Engl. J. Med. 334, 752–758. doi: 10.1056/NEJM199603213341202
Reynier, P., May-Panloup, P., Chretien, M. F., Morgan, C. J., Jean, M., Savagner, F., et al. (2001). Mitochondrial DNA content affects the fertilizability of human oocytes. Mol. Hum. Reprod. 7, 425–429. doi: 10.1093/molehr/7.5.425
Rice, A. C., Keeney, P. M., Algarzae, N. K., Ladd, A. C., Thomas, R. R., and Bennett, J. P. Jr. et al. (2014). Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer’s disease hippocampi. J. Alzheimers Dis. 40, 319–330. doi: 10.3233/JAD-131715
Rifé, M., Nadal, A., Milà, M., and Willemsen, R. (2004). Immunohistochemical FMRP studies in a full mutated female fetus. Am. J. Med. Genet. 124, 129–132. doi: 10.1002/ajmg.a.20342
Sakai, C., Yamaguchi, S., Sasaki, M., Miyamoto, Y., Matsushima, Y., and Goto, Y. (2015). ECHS1 mutations cause combined respiratory chain deficiency resulting in Leigh syndrome. Hum. Mutat. 36, 232–239. doi: 10.1002/humu.22730
Santos, T. A., El Shourbagy, S., and St John, J. C. (2006). Mitochondrial content reflects oocyte variability and fertilization outcome. Fertil. Steril. 85, 584–591. doi: 10.1016/j.fertnstert.2005.09.017
Schon, E. A. (2000). Mitochondrial genetics and disease. Trends Biochem. Sci. 25, 555–560. doi: 10.1016/S0968-0004(00)01688-1
Schuettler, J., Peng, Z., Zimmer, J., Sinn, P., Von Hagens, C., Strowitzki, T., et al. (2011). Variable expression of the Fragile X Mental Retardation 1 (FMR1) gene in patients with premature ovarian failure syndrome is not dependent on number of (CGG)n triplets in exon 1. Hum. Reprod. 26, 1241–1251. doi: 10.1093/humrep/der018
Schwartz, C. E., Dean, J., Howard-Peebles, P. N., Bugge, M., Mikkelsen, M., Tommerup, N., et al. (1994). Obstetrical and gynecological complications in fragile X carriers: a multicenter study. Am. J. Med. Genet. 51, 400–402. doi: 10.1002/ajmg.1320510419
Skuder, P., Plomin, R., Mcclearn, G. E., Smith, D. L., Vignetti, S., Chorney, M. J., et al. (1995). A polymorphism in mitochondrial DNA associated with IQ? Intelligence 21, 1–11. doi: 10.1016/0160-2896(95)90035-7
Small, G. W., Mazziotta, J. C., Collins, M. T., Baxter, L. R., Phelps, M. E., Mandelkern, M. A., et al. (1995). Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273, 942–947.
Song, G., Napoli, E., Wong, S., Hagerman, R., Liu, S., Tassone, F., et al. (2016). Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: use of antioxidants in precision medicine. Mol. Med. 22, 548–559. doi: 10.2119/molmed.2016.00122
Streuli, I., Fraisse, T., Ibecheole, V., Moix, I., Morris, M. A., and De Ziegler, D. (2009). Intermediate and premutation FMR1 alleles in women with occult primary ovarian insufficiency. Fertil. Steril. 92, 464–470. doi: 10.1016/j.fertnstert.2008.07.007
Sullivan, A. K., Marcus, M., Epstein, M. P., Allen, E. G., Anido, A. E., Paquin, J. J., et al. (2005). Association of FMR1 repeat size with ovarian dysfunction. Hum. Reprod. 20, 402–412. doi: 10.1093/humrep/deh635
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi: 10.1093/nar/gky1131
Tassone, F., Hagerman, R. J., Ikle, D. N., Dyer, P. N., Lampe, M., Willemsen, R., et al. (1999). FMRP expression as a potential prognostic indicator in fragile X syndrome. Am. J. Med. Genet. 84, 250–261. doi: 10.1002/(SICI)1096-8628(19990528)84:3<250::AID-AJMG17<3.0
Tassone, F., Pan, R., Amiri, K., Taylor, A. K., and Hagerman, P. J. (2008). A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J. Mol. Diagn. 10, 43–49. doi: 10.2353/jmoldx.2008.070073
Taylor, J. P., Hardy, J., and Fischbeck, K. H. (2002). Toxic proteins in neurodegenerative disease. Science 296, 1991–1995. doi: 10.1126/science.1067122
Terracciano, A., Pomponi, M. G., Marino, G. M. E., Chiurazzi, P., Rinaldi, M. M., Dobosz, M., et al. (2004). Expansion to full mutation of a FMR1 intermediate allele over two generations. Europ. J. Hum. Genet. 12, 333–336. doi: 10.1038/sj.ejhg.5201154
Tiosano, D., Mears, J. A., and Buchner, D. A. (2019). Mitochondrial dysfunction in primary ovarian insufficiency. Endocrinology 160, 2353–2366. doi: 10.1210/en.2019-00441
Torii, K., Sugiyama, S., Takagi, K., Satake, T., and Ozawa, T. (1992). Age-related decrease in respiratory muscle mitochondrial function in rats. Am. J. Resp. Cell. Mol. Biol. 6, 88–92. doi: 10.1165/ajrcmb/6.1.88
Torres, S., Garcia-Ruiz, C. M., and Fernandez-Checa, J. C. (2019). Mitochondrial cholesterol in Alzheimer’s disease and niemann-pick type C Disease. Front. Neurol. 10:1168. doi: 10.3389/fneur.2019.01168
Valera, A. G., Diaz-Hernandez, M., Hernandez, F., Ortega, Z., and Lucas, J. J. (2005). The ubiquitin-proteasome system in Huntington’s disease. Neuroscientist 11, 583–594. doi: 10.1177/1073858405280639
Verkerk, A. J. M. H., Pieretti, M., Sutcliffe, J. S., Fu, Y. H., Kuhl, D. P. A., Pizzuti, A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. doi: 10.1016/0092-8674(91)90397-H
Voorhuis, M., Onland-Moret, N. C., Janse, F., Ploos Van Amstel, H. K., Goverde, A. J., Lambalk, C. B., et al. (2014). The significance of fragile X mental retardation gene 1 CGG repeat sizes in the normal and intermediate range in women with primary ovarian insufficiency. Hum. Reprod. 29, 1585–1593. doi: 10.1093/humrep/deu095
Wallace, D. C. (1992). Diseases of the mitochondrial DNA. Annu. Rev. Biochem. 61, 1175–1212. doi: 10.1146/annurev.bi.61.070192.005523
Wallace, D. C. (1999). Mitochondrial diseases in man and mouse. Science 283, 1482–1488. doi: 10.1126/science.283.5407.1482
Wallace, D. C., Bohr, V. A., Cortopassi, G., Kadenbach, B., Linn, S., Linnane, A. W., et al. (1995). “Group report: the role of bioenergetics and mitochondrial DNA mutations in aging and age-related diseases,” in Molecular Aspects of Aging, eds K. Esser and G. M. Martin (England: Wiley), 199–226.
Wang, L. Y., Wang, D. H., Zou, X. Y., and Xu, C. M. (2009). Mitochondrial functions on oocytes and preimplantation embryos. J. Zhejiang Univ. Sci. B 10, 483–492. doi: 10.1631/jzus.B0820379
Watts, M. E., Pocock, R., and Claudianos, C. (2018). Brain energy and oxygen metabolism: emerging role in normal function and disease. Front. Mol. Neurosci. 11:216. doi: 10.3389/fnmol.2018.00216
Wheeler, A. C., Bailey, D. B. Jr., Berry-Kravis, E., Greenberg, J., Losh, M., Mailick, M., et al. (2014a). Associated features in females with an FMR1 premutation. J. Neurodev. Disord. 6:30. doi: 10.1186/1866-1955-6-30
Wheeler, A. C., Raspa, M., Green, A., Bishop, E., Bann, C., Edwards, A., et al. (2014b). Health and reproductive experiences of women with an FMR1 premutation with and without fragile X premature ovarian insufficiency. Front. Genet. 5:300. doi: 10.3389/fgene.2014.00300
Winarni, T. I., Chonchaiya, W., Sumekar, T. A., Ashwood, P., Morales, G. M., Tassone, F., et al. (2012). Immune-mediated disorders among women carriers of fragile X premutation alleles. Am. J. Med. Genet. A 158A, 2473–2481. doi: 10.1002/ajmg.a.35569
Yen, T. C., Chen, Y. S., King, K. L., Yeh, S. H., and Wei, Y. H. (1989). Liver mitochondrial respiratory functions decline with age. Biochem. Biophys. Res. Comm. 165, 994–1003. doi: 10.1016/0006-291X(89)92701-0
Zenisek, D., and Matthews, G. (2000). The role of mitochondria in presynaptic calcium handling at a ribbon synapse. Neuron 25, 229–237. doi: 10.1016/S0896-6273(00)80885-5
Keywords: mitochondrial dysfunction, omics, cellular response to stress, oxidative phosphorylation, glycolysis, fragile X-associated primary ovarian insufficiency, fragile X-associated tremor and ataxia syndrome
Citation: Napoli E, McLennan YA, Schneider A, Tassone F, Hagerman RJ and Giulivi C (2020) Characterization of the Metabolic, Clinical and Neuropsychological Phenotype of Female Carriers of the Premutation in the X-Linked FMR1 Gene. Front. Mol. Biosci. 7:578640. doi: 10.3389/fmolb.2020.578640
Received: 30 June 2020; Accepted: 24 September 2020;
Published: 22 October 2020.
Edited by:
Simona Paladino, University of Naples Federico II, ItalyReviewed by:
Shai E. Elizur, IVF Unit, Sheba Medical Center, IsraelEmily G. Allen, Emory University, United States
Copyright © 2020 Napoli, McLennan, Schneider, Tassone, Hagerman and Giulivi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cecilia Giulivi, Y2dpdWxpdmlAdWNkYXZpcy5lZHU=