 Xiaopan Gao1†
Xiaopan Gao1† Xia Yu1,2†
Xia Yu1,2† Kaixiang Zhu1†
Kaixiang Zhu1† Bo Qin1†Wei Wang1Pu Han3Justyna Aleksandra Wojdyla4Meitian Wang4
Bo Qin1†Wei Wang1Pu Han3Justyna Aleksandra Wojdyla4Meitian Wang4 Sheng Cui1,5*
Sheng Cui1,5*- 1NHC Key Laboratory of Systems Biology of Pathogens, Institute of Pathogen Biology, And Center for Tuberculosis Research, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
- 2National Clinical Laboratory on Tuberculosis, Beijing Key Laboratory for Drug-resistant Tuberculosis Research Beijing Chest Hospital, Beijing Tuberculosis and Thoracic Tumor Institute, Capital Medical University, Beijing, China
- 3CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
- 4Swiss Light Source at the Paul Scherrer Institut, Villigen, Switzerland
- 5Sanming Project of Medicine in Shenzhen on Construction of Novel Systematic Network Against Tuberculosis, National Clinical Research Center for Infectious Diseases, Shenzhen Third People’s Hospital, Southern University of Science and Technology, Shenzhen, China
Mycobacterium tuberculosis (Mtb) caused an estimated 10 million cases of tuberculosis and 1.2 million deaths in 2019 globally. The increasing emergence of multidrug-resistant and extensively drug-resistant Mtb is becoming a public health threat worldwide and makes the identification of anti-Mtb drug targets urgent. Elongation factor G (EF-G) is involved in tRNA translocation on ribosomes during protein translation. Therefore, EF-G is a major focus of structural analysis and a valuable drug target of antibiotics. However, the crystal structure of Mtb EF-G1 is not yet available, and this has limited the design of inhibitors. Here, we report the crystal structure of Mtb EF-G1 in complex with GDP. The unique crystal form of the Mtb EF-G1-GDP complex provides an excellent platform for fragment-based screening using a crystallographic approach. Our findings provide a structure-based explanation for GDP recognition, and facilitate the identification of EF-G1 inhibitors with potential interest in the context of drug discovery.
Introduction
Mycobacterium tuberculosis (Mtb), one of the deadliest bacterial pathogens causing tuberculosis, is still threatening humanity in the 21st century. According to the World Health Organization (WHO) global tuberculosis report, this single pathogen claimed nearly 1.2 million lives worldwide in 2019 (https://www.who.int/tb/publications/global_report/en/). The emergence of multidrug-resistant tuberculosis (MDR-TB) and extensively drug-resistant tuberculosis (XDR-TB) undermine conventional treatments. For this reason, it is urgent to identify novel inhibitor targets and develop new and effective Mtb drugs.
Elongation factor G (EF-G) is a highly conserved and essential GTPase protein that catalyzes tRNA translocation on ribosomes at several stages of protein synthesis. In addition, EF-G is involved in ribosome recycling and suppression of ribosomal frameshifting (Rodnina et al., 2019). Therefore, EF-G represents a promising target for drug design. Fusidic acid (FA), a well-known drug, is widely used in the clinic against EF-G in Gram-positive bacteria (Fernandes, 2016). FA also exhibits antibacterial activity against Mtb in vitro (Cicek-Saydam et al., 2001). FA inhibits protein synthesis by binding to EF-G and preventing its release after GTP hydrolysis and translocation (Akinpelu et al., 2020). In addition, argyins exhibiting antibacterial activity against Gram-negative pathogens also target EF-G via a novel mechanism (Nyfeler et al., 2012; Jones et al., 2017).
Structural characterization of Mtb EF-G1 has been limited by the lack of a Mtb EF-G1 crystal, and this has hampered structure-based inhibitor design. Although FA is ineffective against Mtb in vivo owning to pharmacokinetic limitations, as demonstrated using a rodent model of Mtb characterized by rapid clearance and poor exposure (Kigondu et al., 2014; Njoroge et al., 2019), hence repurposing the idea that FA and its derivatives could represent a potential novel strategy for TB drug discovery (Njoroge et al., 2019). Indeed, various FA derivatives have been proposed as anti-TB agents (Kigondu et al., 2014; Dziwornu et al., 2019; Njoroge et al., 2019; Strydom et al., 2020). The structure determination of Mtb EF-G1 represents the key step for the structure-based design of inhibitors, in future activities, which will be investigated to assess their potential in the context of anti-TB drug development.
In this study, we purified recombinantly expressed Mtb EF-G1 and characterized its GDP binding activity. We further present the first crystal structure of Mtb EF-G1 in complex with GDP. The Mtb EF-G1●GDP crystal structure presents a complete description of GDP recognition and provides solid structural insight into the design and development inhibitors targeting Mtb EF-G1.
Materials and Methods
Plasmid Construction, Protein Expression, and Purification
The gene encoding Mtb EF-G1 was amplified from the genomic DNA of the Mtb H37RV strain by PCR. The PCR product was then cloned into the expression vector, pCOATexp, derived from pTYB1 vector (New England Biolabs) as previously described (Srivastava et al., 2016), expressing Mtb EF-G1 with a C-terminal His tag. The sequence of the constructed plasmid was verified by DNA sequencing. For protein expression and production, the plasmids encoding Mtb EF-G1 with a C-terminal His tag was transformed in to E. coli BL21 (DE3) competent cells. A single clone was randomly picked from a LB plate and inoculated in LB medium at 37°C and protein expression was induced by the addition of 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) when the culture OD600 reached approximately 1.0. the bacterial culture was cooled to 22°C in incubator and continued with shaking speed of 180rpm overnight after induction. Cells were harvested and washed three times with lysis buffer (20 mM Tris-HCl, pH 8.0,150 mM NaCl, 10 mM imidazole, 10 mM β-mercaptoethanol, 1 mM PMSF) by centrifugation and lysed sonication in lysis buffer. Cells debris was removed after centrifugation at 16000g for 1 h at 4°C and the supernatant was filtered through a 0.45 μm syringe filter, and applied to Ni-NTA resin (Qiagen) pre-equilibrated with lysis buffer. The resin was washed with 10 column volumes of washing buffer (20 mM Tris-HCl, pH 8.0,150 mM NaCl, 20 mM imidazole, 10 mM β-mercaptoethanol, 1 mM PMSF) to remove unbound and nonspecifically bound proteins. The targeted protein was eluted with elution buffer containing 300 mM imidazole. The his-tagged Mtb EF-G1 was loaded into a 5 ml anion exchange column Hitrap Q HP column (GE Healthcare) and eluted with a linear gradient of 75–1,000 mM NaCl. The eluted protein was collected and concentrated and was further purified using a Superdex 200 column pre-equilibrated with a buffer (20 mM Tris-HCl, pH 8.0,150 mM NaCl). The purified protein was collected, concentrated using AmiconUltra-15 with 10 kDa cut-off (Millipore) to 10 mg/ml before crystallization trials.
When preparing the selenomethionine-substituted Mtb EF-G1 for crystallization, the encoding gene was expressed in the E.coli methionine auxotrophic strain B834 (DE3) and cultured in media composed of SelenoMet Medium Base Mix (Molecular Dimensions) supplemented with L-selenomethionine (Sigma). The purification procedure for the SeMet derivative was the same as described above for the native protein, except that the buffers were supplemented with 2 mM DTT.
Crystallization
Crystallization trials of Mtb EF-G1 in complex with GDP were conducted in a hanging-drop vapor-diffusion system at 18◦C. Briefly, the Mtb EF-G1 (10 mg/ ml) was mixed with 5 mM GDP on ice for 30 min before crystallization. The Mtb EF-G1●GDP was crystallized by mixing 1 μL sample and reservoir buffer containing 20 mM magnesium chloride, 50 mM MOPS pH 7.0, 55%TacsimateTM pH 7.0 and 5 mM Hexammine cobalt (III) chloride. Crystals were cryoprotected by soaking crystals in reservoir buffer supplemented with 20% ethylene glycol and flash cooled in liquid nitrogen.
Data Collection, Phasing, and Structure Determination
Complete datasets were collected at beamline BL17U at the Shanghai Synchrotron Radiation Facility (SSRF), Shanghai, China, and at SLS beamline X06DA at the Swiss Light Source, Paul Scherrer Institut, Villigen, Switzerland. Highly redundant X-ray diffraction data for Mtb EF-G1●GDP crystal containing selenomethionine were collected at a wavelength of 0.97931 Å. The reflections were integrated and processed using XDS (Kabsch, 2010). The structure was determined by the single-wavelength anomalous diffraction (SAD) method. The heavy atom substructure with 18 Se atoms were firstly located by dual-space direct-methods program SHELXC/D from the difference of Friedel pairs in the Se-SAD data (Sheldrick, 2010). The initial phases had a figure of merit (FOM) of 0.444 in the resolution range 47.95–3.30 Å. The intrinsic phase ambiguity was broken by iterative direct-methods SAD phasing rooted in the pipeline Iterative Protein Crystal structure Automatic Solution (IPCAS) (Ding et al., 2020). The iteration control was set as OASIS-DM-AutoBuild/Buccaneer with 15 cycles. For each cycle of this dual-space iterative framework, phases were refined and improved in reciprocal space using OASIS and the real space constrains was applied to electron density map using DM (Cowtan and Main, 1996; Hao et al., 2000). The model was built alternatively between AutoBuild and Buccaneer within the iterations to avoid the premature convergence (Cowtan, 2006; Terwilliger et al., 2008). To further improve the quality of the model and phases, the iterative direct-methods model completion was also conducted through IPCAS under the iteration control as OASIS-DM-AutoBuild/Buccaneer with 10 cycles (He et al., 2007). A partial structure was automatically built with 94.6% of the total residues docked to the sequence after two direct-methods iterations.
Coot and Phenix.refine were subsequently used for final model building and refinement, respectively (Emsley et al., 2010; Echols et al., 2012). The final model was validated by MolProbity Chen V. B. et al. (2010) and showed excellent refinement statistics and stereochemical quality. The detailed statistics of data collection, reduction, and structure refinement are presented in Supplementary Table S1. All structural figures were generated by PyMOL.
Structure Alignment and Modeling
Structural alignment was performed with PyMOL (http://www.pymol.org) using the corresponding coordinates deposited in the PDB. PyMOL performs a sequence alignment followed by a structural superposition, and then carries out zero or more cycles of refinement in order to minimize the RMSD (Root Mean Square Deviation) between the aligned residues. All structure superposition was achieved by aligning domains I and II of the EF-G structures. The homology model for Mtb EF-G1-FA was carried out by superimposing Mtb EF-G1 onto the structure of EF-G bound to the T. thermophilus 70S ribosome with GDP and FA (PDB id: 4V5F) based on domains I and II in PyMOL.
Isothermal Titration calorimetry
All titration were performed using a MicroCalTM iTC200 calorimeter (MicroCal, United States) at 25◦C as previously described (Gao et al., 2018). Both the protein and the GDP were dissolved in the same buffer (20 mM Tris-HCl, pH = 8.0, 100 mM NaCl) to ensure a reasonable baseline. The concentrations of Mtb EF-G1 was 0.02 mM and GDP was 1 mM. For each titration, 18 injections of 2 μL of titrant were made at a 120-s interval using a stir rate of 600 rpm. Data were subtracted with dilution heat of the ligand from control assay, where GDP was titrated into the buffer. Raw data were integrated and analyzed by nonlinear curve fitting using Origin isothermal titration calorimetry analysis program equipped by Microcal.
Surface Plasmon Resonance Analysis
The interaction of FA with Mtb EF-G1was assessed by surface plasmon resonance (SPR) using a BiaCore T200 equipped with CM5 sensor chips (GE Healthcare) at 25°C. The surfaces of the sample and reference flow cells were activated as previously described (Chi et al., 2020). The Mtb EF-G1 was diluted in 10 mM sodium acetate buffer, pH 4.5, and immobilized on the chip at 19,000 resonance units (RU). For analysis of FA binding, a series of FA concentrations were passed sequentially over chips immobilized with Mtb EF-G1. The data were fitted with a 1:1 steady-state affinity model using BIA evaluation 1.0 software.
Size Exclusion Chromatography
Gel-filtration chromatography was conducted using a Superdex 200 Column (GE healthcare) .The column was pre-equilibrated with buffer containing 20 Mm Tris HCl (pH 8.0), 150 mM NaCl and calibrated using molecular weight standards. Purified Mtb EF-G1were fractionated at a flow rate of 0.15 ml/ min.
Results and Discussion
Biochemical Characterization of Mtb EF-G1
To provide solid structural insight into the Mtb EF-G1, we first expressed soluble Mtb EF-G1 by performing systematic optimization of the overexpression condition. This included varying the induction temperature (37°C, 22°C, 18°C), the concentration of the IPTG inducing reagent (0.1 mM, 0.5 mM), and the Escherichia coli host bacterial strain (BL21 [DE3], Rosetta [DE3]). Finally, we obtained stably expressed Mtb EF-G1 at high yield in E. coli BL21 [DE3] with an induction temperature of 22°C and 0.5 mM IPTG (Supplementary Figures S1A,B). To investigate the GDP binding ability of Mtb EF-G1, we first carried out isothermal titration calorimetry (ITC) experiments to verify the GDP affinity of Mtb EF-G1.The results showed that Mtb EF-G1 binds GDP with an apparent dissociation constant (Kd) of 11.5 ± 0.3 μM (Supplementary Figure S1C). This Kd value is comparable to the dissociation constant reported for other species EF-G (Martemyanov and Gudkov, 2000). These results indicated that Mtb EF-G1 was correctly folded and is suitable for subsequent crystallography.
Structure Determination of Mtb EF-G1
The crystals of Mtb EF-G1 diffracted the X-ray to 3 Å. The three-dimensional structure of Mtb EF-G1 was solved by the SAD method. The data collection, structure-refinement, and validation statistics are summarized in Supplementary Table S1. Comparing to several EF-G structures complexed with GDP that crystallized in the P212121 space group (PDB id: 1DAR, 4MYT, and 2BM0) in other species, the current crystal form of Mtb EF-G1 has unique space group C2221, larger cell dimensions and unique crystal packing (Supplementary Table S1).
Interestingly, Matthews coefficient calculations indicated the presence of either a dimer (VM = 3.58 Å3/Da, a solvent content of approximately 65.6%) or a trimer (VM = 2.38 Å3/Da,a solvent content of 48.39%) in the asymmetric unit (ASU). The highest probability (0.77) was for three molecules in the ASU based on the resolution of the data (Matthews, 1968; Kantardjieff and Rupp, 2003). However, further crystallographic analysis confirmed that the ASU contained two enzyme molecules with a correct electron density map and reasonable Rwork (20.6%)/Rfree (24.1%) values, instead of a crystallographic monomer in the ASU as in other EF-G structures (Supplementary Figures S2A,B).
The unique crystal packing, high solvent space (65.6%), and reasonable resolution (3 Å) of the Mtb EF-G1 crystal form provides several advantages for high-throughput fragment-based screening using a crystallographic approach targeting Mtb EF-G1: (1)Although the active site is occupied by GDP, which likely prevents the screening of inhibitors/fragments, it is still useful for identifying allosteric sites. For example, Pseudomonas aeruginosa EF-G1 in complex with argyrin B revealed a novel binding site at the interface of domains III and V that is away from the GDP binding site (PDB id: 4FN5), which supports our assertion; (2) The novel crystal form of Mtb EF-G1 can be produced easily with high repeatability, and has a higher solvent content and looser packing than previously reported crystal forms, which may facilitate rapid diffusion of compounds into crystals during soaking experiments.
Overall Structure of Mtb EF-G1
Mtb EF-G1 has five structural domains that fold into an elongated shape with dimensions of 118 Å × 60 Å × 50 Å (Figures 1A,B). The N-terminal G domains are composed of a classic GTPase domain (G domain) and an additional G′ subdomain. The other domains are numbered consecutively based on sequence and structural similarity (II-V; Figure 1A; Supplementary Figure S3). Alignment of the primary sequences of EF-G from different species indicated that Mtb EF-G1 shares higher identity with Mycobacterium smegmatis EF-G (85%) and Arthrobacter globiformis EF-G (72%) than with Staphylococcus aureus, P. aeruginosa, E. coli, and Thermococcus thermophilus EF-G (55–58%; Supplementary Figure S3). Comparison between EF-G1 and another EF-G-like protein in Mtb (FusA2 or EF-G2) revealed a low degree of homology (30%; Supplementary Figure S4). However, Domains I−V are conserved amongst the EF-G1 and EF-G2 sequences. The G domain (residues 2-292) shows great structural similarity with other GTPase family except with an insert of ∼90 residues (residues 160-256). The core of the G domain exhibits an α/ fold with six β-sheets (β1-β5, β11) surrounded by five α helices (α1-α4, α8). The G′ subdomain is comprised of a five-stranded mixed β-sheet (β6-β10) followed by three α-helices (α5-α7), and exhibits βββββααα topology (Figure 1C; Supplemenatry Figure S5A, colored wheat). The “effector loop” or “switch I” region (residues45-65) is largely disordered and is not visible in any of the reported EF-G structures. The G domain, including G1, G2, G3, G4, and G5 motifs, is highly conserved in various organisms (Supplementary Figure S3), and the GTP binding motifs (G1, G3, G4) are also conserved in Mtb EF-G2 (Supplementary Figure S4). The G1 motif (Walker A motif) forms a P-loop that interacts with the α- and β-phosphates of GTP or GDP. The G3 motif (Walker B motif) forms the switch II region that interacts with the γ-phosphates of GTP and binds a water-bridged Mg2+ ion. The G4 motif mainly engages in interactions with the guanine (Seshadri et al., 2009). However, the G2 motif forming the switch I region and the G5 motif are not well conserved in Mtb EF-G2. The conserved RGITI motif is replaced by QQRSV in the G2 motif and the GSAF motif is replaced by VCSS in the G5 motif in Mtb EF-G2 (Supplementary Figure S4). The additional G′ subdomain is not very well conserved amongst various EF-Gs and EF-G2 in Mtb (Supplementary Figures S3, S4). However, some of the functionally important residues (E221, E225, and E228) have been shown to interact with the L7/L12 stalk of the 50S subunit in the G′ subdomain, and are mostly conserved between EF-G in various organisms and EF-G2 in Mtb (Supplementary Figures S3, S4).(Nechifor et al., 2007; Nechifor and Wilson, 2007; Seshadri et al., 2009). Domain II is a β sandwich domain built up of 11 β sheets (β13-β23).Two strands (β18-β19)extend from the barrel to the G domain, very close to the invisible effector region (Figure 1C; Supplementary Figure S5B, colored cyan). Interestingly, domain II of Mtb EF-G2 contains an insert of 17 residues not present in Mtb EF-G1. Residue R334 in the consensus motif of domain II is highly conserved among EF-G1 and EF-G2 in Mtb, and it forms salt bridge with D103 in the G-domain (Supplementary Figure S4).
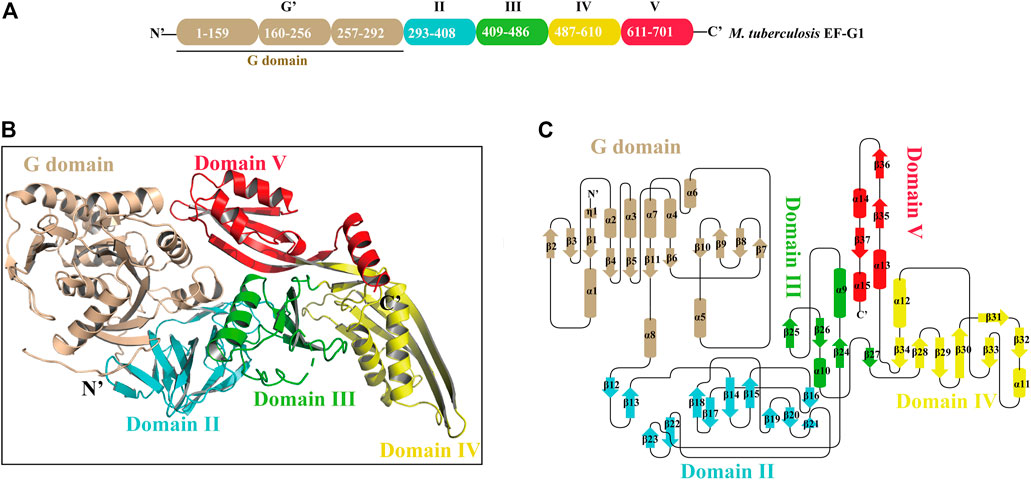
FIGURE 1. The overall structure of Mtb EF-G1. (A) Domain organization of Mtb EF-G1. (B) Ribbon model of the Mtb EF-G1 crystal structure. The individual domains are differently colored, with G domain in wheat, domain II in cyan, domain III in green, domain IV in yellow and domain V in red. (C) Topology diagram of Mtb EF-G1.The domains are colored the same as in panel (B).
Domain III comprises four antiparallel β-sheets (β24, β25, β26, β27) with two α helices on one side of the β-sheet (α9 and α10) and bridges between domains II and IV (Figure 1C; Supplementary Figure S5C, colored green). Domain III in different species is flexible, as is the case for the switch I and switch II regions, which indicates that domain III may be of functional importance. Domain IV is an elongated fold at the end of EF-G1. It contains two parallel β-sheets (β32, β33) connected by an α helix (α11) displaying an unusual left-handed structure (Figure 1C; Supplementary Figure S5D, colored yellow). Residue H580 of domain IV, which has been shown to play an important role in promoting tRNA movement, is conserved in Mtb EF-G1 and EF-G2 (Supplementary Figure S4) (Savelsbergh et al., 2000). The small β-sheets (β29, β34) connect domains III and V to domain IV, respectively. Finally, domain V has a double-split βαβ fold (Figure 1C; Supplementary Figure S5E, colored red).
Two interesting parts (switch I and switch II) of the Mtb EF-G1 G domain in molecule A are missing as a result of structural disorder. However, we could model switch II of Mtb EF-G1 in molecule B following refinement. This indicates that when GDP is bound, a large conformational change occurs in the two regions, or that these two regions are flexible and this may be important for EF-G function.
The Active Site for GDP Binding
We pre-incubated GDP and/or GTP with the Mtb EF-G1 protein in a molar ratio of 5:1 prior to crystallization trials. After extensive efforts, we obtained crystals of the EF-G1●GDP complex. However, we were unable to crystallize the EF-G1●GTP complex. As illustrated in Figure 2A, the GDP molecule is bound in the G domain. The electron density for the GDP is well defined in the final map (Figure 2B). The Walker A motif (23DAGKTT28) forms the A-loop (P-loop), which mainly binds the α- and β-phosphates through main chain atoms in the same way as it does in other EF-G proteins (Czworkowski et al., 1994; Al-Karadaghi et al., 1996) (Figures 2C,D). The O3B, O2B, and O1B atoms of the β-phosphate are specifically recognized by the backbone NH groups of T27, K26, and D23 through multiple hydrogen bonds. The O1A of the α-phosphate is specifically recognized by two hydrogen bonds donated by T28 (Figures 2C,D). The absence of the ε amino group of the K26-β phosphate in the GDP hydrogen bond but the presence of the ε amino group of the K26-hydroxyl group in the T85 hydrogen bond is also observed in Mtb EF-G1. Therefore, EF-G has a low affinity for GDP. Additionally, we did not observe the presence of Mg2+, even when MgCl2 was added during crystallization.
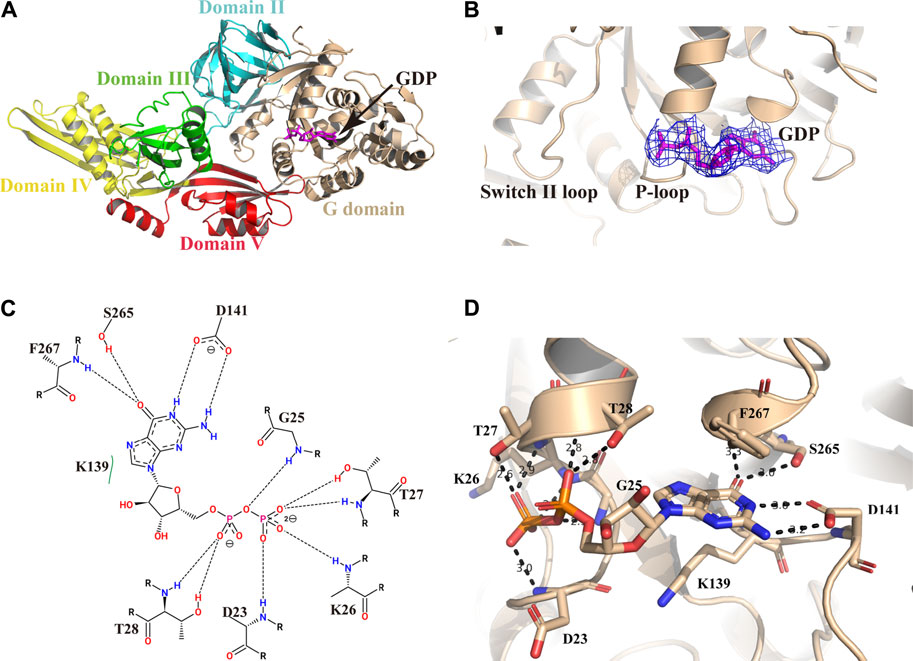
FIGURE 2. Structure of Mtb EF-G1 in complex with GDP. (A) Cartoon representation of Mtb EF-G1 colored by different domains. The docking of GDP in the active site is shown as a stick model in magenta. (B) A magnified view of GDP in the active site of Mtb EF-G1, with the final electron density map (2Fo-Fc, contoured at 0.8 σ) superimposed. (C) Detailed two-dimensional diagram of the active site of Mtb EF-G1 occupied by GDP. Directed bonds between Mtb EF-G1 and GDP are drawn as dashed lines. Image generated by PoseView (https://www.zbh.uni-hamburg.de/en/forschung/amd/server/poseview.html). (D) Ribbon model of the GDP binding pocket containing GDP. Residues recognizing GDP are shown in stick model. Hydrogen bonds between Mtb EF-G1 and GDP are indicated by dashed lines.
The ribose part of GDP is only recognized by K139 through hydrophobic contacts. The guanosine base is surrounded by several hydrogen bonds donated by S265, F267, and D141. The HN1 and HN2 of the guanosine base accept two hydrogen bonds from the side chain of D141. The O6 atom of the guanosine base accepts two hydrogen bonds from the S265 side chain and the backbone NH group of F267 (Figures 2C,D).
The switch I region of Mtb EF-G1, essential for coupling GTP hydrolysis, was found to be disordered, which is consistent with previous study indicating that the switch I region of EF-G1 has not been modeled (Czworkowski et al., 1994; Hansson et al., 2005; Gao et al., 2009), except those in the EF-G in GTP form bound to a ratcheted ribosome and in the S.aureus EF-G structure (Chen Y. et al., 2010; Chen et al., 2013). Therefore, Mtb EF-G1 is inactive, and upon ribosome and GTP binding, switch I of Mtb EF-G1 may be stabilized as the structure of a pretranslocational ribosome bound to EF-G trapped with a GTP analog (Chen et al., 2013). The switch II region of EF-G, which is important in coupling transition from the GDP-bound to the GTP-bound conformation, is ordered in the Mtb EF-G1 structure. It was previously reported that the largest conformational changes between GDP-bound and GDP-unbound EF-G1 occur in the switch II region (Al-Karadaghi et al., 1996). By superimposing the two EF-G without GDP fragment crystal structures (PDB id:5TY0 and 5VH6) with Mtb EF-G1●GDP, we could see that the conformations of the backbones and side chains between G domain and domain II are very similar (Supplementary Figure S6A). In addition, the side chains of key residues constituting the GDP binding site have the same orientations as those in Mtb EF-G1●GDP (Supplementary Figure S6B). The differences between these three EF-G structures are conformational changes of the switch II and P-loop regions (Supplementary Figure S6C). These conformational changes probably result from GDP binding.
Comparison of Mtb EF-G1 Structure With Previous EF-G Structures
To date, available data indicate that EF-G assumes different conformations that are mainly characterized by the movement of domains III, IV and V relative to domains I and II and no alternations in domains I and II. To gain insight into the structural differences between single domains of EF-G from different sources, we first compared Mtb EF-G1 with the previously isolated T. thermophilus EF-G bound to GDP, and the apo structure of S.aureus EF-G. The results showed that the individual domains (I-V) are highly similar. However, domains III, IV, and V display high variability relative to domains I and II (Figure 3A), and domains III-V are rotated relative to domains I-II, resulting in a movement of the tip of domain IV differing by 8 Å between our structure and apo S.aureus EF-G (Figure 3A), we examined the equivalent position displaying a movement by ∼23 Å between our structure and that of T.thermophilus EF-G bound to GDP (Figure 3A). Furthermore, comparison with the crystal structure of the ribosome with EF-G trapped in the post-translocational state reveals a significant structural rearrangement of domains III, IV, and V, resulting in a shift of the tip domain IV by up to ∼36 Å (Figure 3B). This distance is larger than that previously reported for T.thermophilus EF-G as shown by comparisons between apo EF-G and ribosome-bound EF-G (Chen Y. et al., 2010), these movements are consistent with previous results that EF-G undergoes interdomain rearrangement during the translocation cycle (Gao et al., 2009; Chen Y. et al., 2010; Brilot et al., 2013; Salsi et al., 2015; Ling and Ermolenko, 2016).
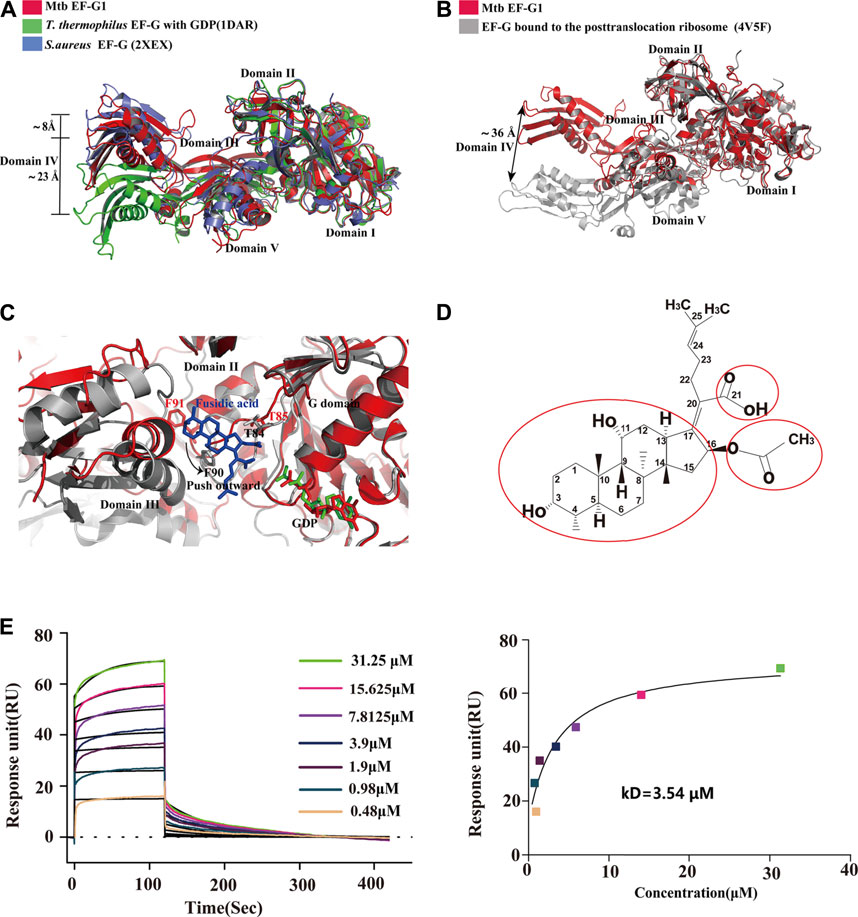
FIGURE 3. Comparison of the structures of Mtb EF-G1 with EF-G from other species and EF-G trapped in the post-translocational ribosome. (A) Comparison of Mtb EF-G1 with T.thermophiles EF-G (PDB id:1DAR) and S.aureus EF-G (PDB id:2XEX) revealing movement of the tip of domain IV between Mtb EF-G1 and T.thermophiles EF-G, and between Mtb EF-G1 and S.aureus EF-G. Superposition was achieved by aligning domains I and II of the two EF-G structures. Domains of EF-G are labeled with latin numbers. (B) Mtb EF-G1 (red)and EF-G bound to the post-translocational ribosome (gray, PDB id:4V5F) superposed by structural alignment of domains I and II. The arrow indicates the shift of the tip of domain IV between the two structures. (C) Superposition of domains I and II between the fusidic acid-bound structure (gray,PDB id:4V5F) with Mtb EF-G1 indicates movement outward or downward of the switch II region between the two structures. (D) Chemical structure of FA. The essential groups for activity are marked with red circles. (E) SPR analysis of FA binding to Mtb EF-G1(left)FA at various concentrations were injected over immobilized Mtb EF-G1. (right) Fitting curve for equilibrium binding that produced a kD of 3.54 μM.
Proposed FA Binding Site of Mtb EF-G1 and Future Chemical Modification of FA
FA is a natural steroid antibiotic that has been used clinically for the treatment of Gram-positive infections and methicillin-resistant S.aureus (Whitby, 1999; Sahm et al., 2013) . FA has also been shown to exhibit activity in vitro against Mtb (Cicek-Saydam et al., 2001). FA specially inhibits EF-G and locks it onto the ribosome after GTP hydrolysis and translocation. Because the EF-G-FA structure remains unavailable, we modeled binding of FA to Mtb EF-G1 by superimposing our structure onto the structure of EF-G bound to the T. thermophilus 70S ribosome with GDP and FA (PDB id: 4V5F) based on domains I and II. FA binds to a pocket surrounded by domains I, II, and III of Mtb EF-G1, similar to the T. thermophilus 70 S ribosome with GDP and FA (Figure 3C). Further comparison of the structure of Mtb EF-G1 and the EF-G–70S complex with GDP and FA indicates that binding of FA induces evident conformational changes in switch II, which includes movement of switch II outward or downward, relative to the corresponding switch II Mtb EF-G1 structure (Figure 3C). Two EF-G mutations at T85 and F91 in switch II caused a shift of 3–5 Å, resulting in a direct interaction with FA. In other words, FA binding prevents the switch II region from adopting its GDP conformation, and locks it in a conformation similar to that of the GTP form (Gao et al., 2009). Modeling of FA on Mtb EF-G1 indicated that the trans-syn-trans conformation of the tetracylic triterpene backbone, the carboxylic acid (C-21), and acetoxy (C-16) groups of FA occupy a hydrophobic cavity on EF-G, and all are indispensable for inhibitor design, consistent with the structure-activity relationship (SAR) (Figures 3C,D). In addition, the orientation of the lipophilic side chain and the carboxyl group around the Δ17,20 bond, rather than the double bond, are essential for anti-mycobacterial activity, and should be considered in future inhibitor design (von Daehne et al., 1979; Duvold et al., 2001). To validate our model, we employed SPR to investigate the binding affinity of FA to Mtb EF-G1. The binding affinity of Mtb EF-G1 with FA determined by our SPR experiments exhibited a kD of 3.54 μM (Figure 3E), which falls in with the range of kD values of 4.34–8.33 μM reported previously for E.coli EF-G using equilibrium dialysis (Okura et al., 1970; 1971) . This result indicates that FA can indeed bind to Mtb EF-G1, explaining why FA is effective against Mtb in vitro. However, it is still disputed whether FA can directly bind to EF-G in the absence of ribosomes (Willie et al., 1975). Further verification of FA binding to EF-G from different organisms using various biophysical techniques and structural studies should finally resolve this dispute. To prevent FA from being rapidly metabolized in vivo, one group repurposed FA via chemical modification and performed biological characterization, and found that the antimycobacterial activity of C-3 silicate esters was comparable to that of FA, and these compounds were stable in microsomes and plasma, revealing them to be attractive compounds for in vivo antimycobacterial activity evaluation (Njoroge et al., 2019). Finally, The structure of the Mtb ribosome in complex with EF-G and FA derivatives (C-3 silicates) will offer valuable insight into the design of FA analog inhibitors targeting EF-G.
Conclusion
In summary, we purified recombinantly expressed Mtb EF-G1 and characterized its GDP binding activity, and then provide the first crystal structure of Mtb EF-G1 in complex with GDP. Our unique crystal form provide an excellent platform for the fragment-based screening using crystallographic approach and will play an important role in design and development of enzyme inhibitors of potential interest for future studies in the context of TB drug discovery.
Data Availability Statement
The atomic coordinates and structure factors have been deposited in the Protein Data Bank (http://www.wwpdb.org) under the accession codes: 7CDW.
Author Contributions
SC and XG designed the study. SC, PH and XG solved the structure and wrote the paper. XG, XY, KZ, BQ and WW performed experiments. SC, XG, WW, JAW, BQ and MW analyzed the data. All authors reviewed the results and approved the final version of the manuscript.
Funding
National Natural Science Foundation of China (award No. 81971985, 82072291, 81772207, 11775308 and 81802057); Chinese Academy of Medical Science (CAMS) Innovation Fund for Medical Sciences (award No. 2016-I2M-1-013,2017-I2M-1-014); National Key Research and Development Program of China (2016YFD0500300 and 2019YFC0840602); Beijing Municipal Natural Science Foundation (award No.7182117, 7174288); Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (award No. 2018PT51009, 2017PT31049). The Guangdong Foundation for Basic and Applied Basic Research (No. 2019B1515120041).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the staff of PX III beamline at the Swiss Light Source, Paul Scherrer Institut (Villigen Switzerland) for assistance in data collection. We thank the staff of BL17B1/BL18U1/BL19U1/ BL19U2/ BL01B beamlines at National Center for Protein Science Shanghai and Shanghai Synchrotron Radiation Facility, Shanghai China, for help with data collection.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.667638/full#supplementary-material
References
Akinpelu, O. I., Lawal, M. M., Kumalo, H. M., and Mhlongo, N. N. (2020). Drug Repurposing: Fusidic Acid as a Potential Inhibitor of M. tuberculosis FtsZ Polymerization - Insight from DFT Calculations, Molecular Docking and Molecular Dynamics Simulations. Tuberculosis 121, 101920. doi:10.1016/j.tube.2020.101920
Al-Karadaghi, S., Ævarsson, A., Garber, M., Zheltonosova, J., and Liljas, A. (1996). The Structure of Elongation Factor G in Complex with GDP: Conformational Flexibility and Nucleotide Exchange. Structure 4 (5), 555–565. doi:10.1016/s0969-2126(96)00061-5
Brilot, A. F., Korostelev, A. A., Ermolenko, D. N., and Grigorieff, N. (2013). Structure of the Ribosome with Elongation Factor G Trapped in the Pretranslocation State. Proc. Natl. Acad. Sci. 110 (52), 20994–20999. doi:10.1073/pnas.1311423110
Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., et al. (2010a). MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr. D Biol. Cryst. 66 (Pt 1), 12–21. doi:10.1107/S0907444909042073
Chen, Y., Feng, S., Kumar, V., Ero, R., and Gao, Y.-G. (2013). Structure of EF-G-Ribosome Complex in a Pretranslocation State. Nat. Struct. Mol. Biol. 20 (9), 1077–1084. doi:10.1038/nsmb.2645
Chen, Y., Koripella, R. K., Sanyal, S., and Selmer, M. (2010b). Staphylococcus aureus Elongation Factor G - Structure and Analysis of a Target for Fusidic Acid. FEBS J. 277 (18), 3789–3803. doi:10.1111/j.1742-4658.2010.07780.x
Chi, X., Liu, X., Wang, C., Zhang, X., Li, X., Hou, J., et al. (2020). Humanized Single Domain Antibodies Neutralize SARS-CoV-2 by Targeting the Spike Receptor Binding Domain. Nat. Commun. 11 (1), 4528. doi:10.1038/s41467-020-18387-8
Cicek-Saydam, C., Cavusoglu, C., Burhanoglu, D., Hilmioglu, S., Ozkalay, N., and Bilgic, A. (2001). In Vitro susceptibility of Mycobacterium tuberculosis to Fusidic Acid. Clin. Microbiol. Infect. 7 (12), 700–702. doi:10.1046/j.1469-0691.2001.00341.x
Cowtan, K. D., and Main, P. (1996). Phase Combination and Cross Validation in Iterated Density-Modification Calculations. Acta Cryst. D 52 (1), 43–48. doi:10.1107/S090744499500761X
Cowtan, K. (2006). TheBuccaneersoftware for Automated Model Building. 1. Tracing Protein Chains. Acta Crystallogr. D Biol. Cryst. 62 (Pt 9), 1002–1011. doi:10.1107/S0907444906022116
Czworkowski, J., Wang, J., Steitz, T. A., and Moore, P. B. (1994). The crystal Structure of Elongation Factor G Complexed with GDP, at 2.7 A Resolution. EMBO J. 13 (16), 3661–3668. doi:10.1002/j.1460-2075.1994.tb06675.x
Ding, W., Zhang, T., He, Y., Wang, J., Wu, L., Han, P., et al. (2020). IPCAS: a Direct-Method-Based Pipeline from Phasing to Model Building and Refinement for Macromolecular Structure Determination. J. Appl. Cryst. 53 (1), 253–261. doi:10.1107/S1600576719015115
Duvold, T., Sørensen, M. D., Björkling, F., Henriksen, A. S., and Rastrup-Andersen, N. (2001). Synthesis and Conformational Analysis of Fusidic Acid Side Chain Derivatives in Relation to Antibacterial Activity. J. Med. Chem. 44 (19), 3125–3131. doi:10.1021/jm010899a
Dziwornu, G. A., Kamunya, S., Ntsabo, T., and Chibale, K. (2019). Novel Antimycobacterial C-21 Amide Derivatives of the Antibiotic Fusidic Acid: Synthesis, Pharmacological Evaluation and Rationalization of media-dependent Activity Using Molecular Docking Studies in the Binding Site of Human Serum Albumin. Med. Chem. Commun. 10 (6), 961–969. doi:10.1039/c9md00161a
Echols, N., Grosse-Kunstleve, R. W., Afonine, P. V., Bunkóczi, G., Chen, V. B., Headd, J. J., et al. (2012). Graphical Tools for Macromolecular Crystallography inPHENIX. J. Appl. Cryst. 45 (Pt 3), 581–586. doi:10.1107/S0021889812017293
Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010). Features and Development ofCoot. Acta Crystallogr. D Biol. Cryst. 66 (Pt 4), 486–501. doi:10.1107/S0907444910007493
Fernandes, P. (2016). Fusidic Acid: A Bacterial Elongation Factor Inhibitor for the Oral Treatment of Acute and Chronic Staphylococcal Infections. Cold Spring Harb Perspect. Med. 6 (1), a025437. doi:10.1101/cshperspect.a025437
Gao, X., Mu, Z., Yu, X., Qin, B., Wojdyla, J., Wang, M., et al. (2018). Structural Insight into Conformational Changes Induced by ATP Binding in a Type III Secretion-Associated ATPase from Shigella Flexneri. Front. Microbiol. 9, 1468. doi:10.3389/fmicb.2018.01468
Gao, Y.-G., Selmer, M., Dunham, C. M., Weixlbaumer, A., Kelley, A. C., and Ramakrishnan, V. (2009). The Structure of the Ribosome with Elongation Factor G Trapped in the Posttranslocational State. Science 326 (5953), 694–699. doi:10.1126/science.1179709
Hansson, S., Singh, R., Gudkov, A. T., Liljas, A., and Logan, D. T. (2005). Structural Insights into Fusidic Acid Resistance and Sensitivity in EF-G. J. Mol. Biol. 348 (4), 939–949. doi:10.1016/j.jmb.2005.02.066
Hao, Q., Gu, Y. X., Zheng, C. D., and Fan, H. F. (2000). OASIS: a Computer Program for Breaking Phase Ambiguity in One-Wavelength Anomalous Scattering or Single Isomorphous Substitution (Replacement) Data. J. Appl. Cryst. 33 (3 Part 2), 980–981. doi:10.1107/S0021889800001424
He, Y., Yao, D.-Q., Gu, Y.-X., Lin, Z.-J., Zheng, C.-D., and Fan, H.-F. (2007). OASISand Molecular-Replacement Model Completion. Acta Crystallogr. D Biol. Cryst. 63 (Pt 7), 793–799. doi:10.1107/S0907444907023451
Jones, A. K., Woods, A. L., Takeoka, K. T., Shen, X., Wei, J.-R., Caughlan, R. E., et al. (2017). Determinants of Antibacterial Spectrum and Resistance Potential of the Elongation Factor G Inhibitor Argyrin B in Key Gram-Negative Pathogens. Antimicrob. Agents Chemother. 61 (4), e02400. doi:10.1128/AAC.02400-16
Kabsch, W. (2010). Xds. Acta Crystallogr. D Biol. Cryst. 66 (Pt 2), 125–132. doi:10.1107/S0907444909047337
Kantardjieff, K. A., and Rupp, B. (2003). Matthews Coefficient Probabilities: Improved Estimates for Unit Cell Contents of Proteins, DNA, and Protein-Nucleic Acid Complex Crystals. Protein Sci. 12 (9), 1865–1871. doi:10.1110/ps.0350503
Kigondu, E. M., Wasuna, A., Warner, D. F., and Chibale, K. (2014). Pharmacologically Active Metabolites, Combination Screening and Target Identification-Driven Drug Repositioning in Antituberculosis Drug Discovery. Bioorg. Med. Chem. 22 (16), 4453–4461. doi:10.1016/j.bmc.2014.06.012
Ling, C., and Ermolenko, D. N. (2016). Structural Insights into Ribosome Translocation. Wiley Interdiscip. Rev. RNA 7 (5), 620–636. doi:10.1002/wrna.1354
Martemyanov, K. A., and Gudkov, A. T. (2000). Domain III of Elongation Factor G from Thermus Thermophilus Is Essential for Induction of GTP Hydrolysis on the Ribosome. J. Biol. Chem. 275 (46), 35820–35824. doi:10.1074/jbc.M002656200
Matthews, B. W. (1968). Solvent Content of Protein Crystals. J. Mol. Biol. 33 (2), 491–497. doi:10.1016/0022-2836(68)90205-2
Nechifor, R., Murataliev, M., and Wilson, K. S. (2007). Functional Interactions between the G′ Subdomain of Bacterial Translation Factor EF-G and Ribosomal Protein L7/L12. J. Biol. Chem. 282 (51), 36998–37005. doi:10.1074/jbc.M707179200
Nechifor, R., and Wilson, K. S. (2007). Crosslinking of Translation Factor EF-G to Proteins of the Bacterial Ribosome before and after Translocation. J. Mol. Biol. 368 (5), 1412–1425. doi:10.1016/j.jmb.2007.03.009
Njoroge, M., Kaur, G., Espinoza-Moraga, M., Wasuna, A., Dziwornu, G. A., Seldon, R., et al. (2019). Semisynthetic Antimycobacterial C-3 Silicate and C-3/C-21 Ester Derivatives of Fusidic Acid: Pharmacological Evaluation and Stability Studies in Liver Microsomes, Rat Plasma, and Mycobacterium tuberculosis Culture. ACS Infect. Dis. 5 (9), 1634–1644. doi:10.1021/acsinfecdis.9b00208
Nyfeler, B., Hoepfner, D., Palestrant, D., Kirby, C. A., Whitehead, L., Yu, R., et al. (2012). Identification of Elongation Factor G as the Conserved Cellular Target of Argyrin B. PLoS One 7 (9), e42657. doi:10.1371/journal.pone.0042657
Okura, A., Kinoshita, T., and Tanaka, N. (1970). Complex Formation of Fusidic Acid with G Factor, Ribosome and Guanosine Nucleotide. Biochem. Biophysical Res. Commun. 41 (6), 1545–1550. doi:10.1016/0006-291x(70)90563-2
Okura, A., Kinoshita, T., and Tanaka, N. (1971). Formation of Fusidic Acid-G Factor-GDP-Ribosome Complex and the Relationship to the Inhibition of GTP Hydrolysis. J. Antibiot. 24 (10), 655–661. doi:10.7164/antibiotics.24.655
Rodnina, M. V., Peske, F., Peng, B.-Z., Belardinelli, R., and Wintermeyer, W. (2019). Converting GTP Hydrolysis into Motion: Versatile Translational Elongation Factor G. Biol. Chem. 401 (1), 131–142. doi:10.1515/hsz-2019-0313
Sahm, D. F., Deane, J., Pillar, C. M., and Fernandes, P. (2013). In VitroActivity of CEM-102 (Fusidic Acid) against Prevalent Clones and Resistant Phenotypes of Staphylococcus aureus. Antimicrob. Agents Chemother. 57 (9), 4535–4536. doi:10.1128/AAC.00206-13
Salsi, E., Farah, E., Netter, Z., Dann, J., and Ermolenko, D. N. (2015). Movement of Elongation Factor G between Compact and Extended Conformations. J. Mol. Biol. 427 (2), 454–467. doi:10.1016/j.jmb.2014.11.010
Savelsbergh, A., Matassova, N. B., Rodnina, M. V., and Wintermeyer, W. (2000). Role of Domains 4 and 5 in Elongation Factor G Functions on the Ribosome. J. Mol. Biol. 300 (4), 951–961. doi:10.1006/jmbi.2000.3886
Seshadri, A., Samhita, L., Gaur, R., Malshetty, V., and Varshney, U. (2009). Analysis of the fusA2 Locus Encoding EFG2 in Mycobacterium Smegmatis. Tuberculosis 89 (6), 453–464. doi:10.1016/j.tube.2009.06.003
Sheldrick, G. M. (2010). Experimental Phasing withSHELXC/D/E: Combining Chain Tracing with Density Modification. Acta Crystallogr. D Biol. Cryst. 66 (Pt 4), 479–485. doi:10.1107/S0907444909038360
Srivastava, A., Asahara, H., Zhang, M., Zhang, W., Liu, H., Cui, S., et al. (2016). Reconstitution of Protein Translation of Mycobacterium Reveals Functional Conservation and Divergence with the Gram-Negative Bacterium Escherichia coli. PLoS One 11 (8), e0162020. doi:10.1371/journal.pone.0162020
Strydom, N., Kaur, G., Dziwornu, G. A., Okombo, J., Wiesner, L., and Chibale, K. (2020). Pharmacokinetics and Organ Distribution of C-3 Alkyl Esters as Potential Antimycobacterial Prodrugs of Fusidic Acid. ACS Infect. Dis. 6 (3), 459–466. doi:10.1021/acsinfecdis.9b00405
Terwilliger, T. C., Grosse-Kunstleve, R. W., Afonine, P. V., Moriarty, N. W., Zwart, P. H., Hung, L.-W., et al. (2008). Iterative Model Building, Structure Refinement and Density Modification with thePHENIX AutoBuildwizard. Acta Crystallogr. D Biol. Cryst. 64 (Pt 1), 61–69. doi:10.1107/S090744490705024X
von Daehne, W., Godtfredsen, W. O., and Rasmussen, P. R. (1979). Structure-activity Relationships in Fusidic Acid-type Antibiotics. Adv. Appl. Microbiol. 25, 95–146. doi:10.1016/s0065-2164(08)70148-5
Whitby, M. (1999). Fusidic Acid in the Treatment of Methicillin-Resistant Staphylococcus aureus. Int. J. Antimicrob. Agents 12 (Suppl. 2), S67–S71. doi:10.1016/s0924-8579(98)00075-2
Keywords: Mycobacteria tuberculosis, Elongatin factor G (EF-G), crystal structure, antituberculosis drug, ribosome-bound EF-G
Citation: Gao X, Yu X, Zhu K, Qin B, Wang W, Han P, Aleksandra Wojdyla J, Wang M and Cui S (2021) Crystal Structure of Mycobacterium tuberculosis Elongation Factor G1. Front. Mol. Biosci. 8:667638. doi: 10.3389/fmolb.2021.667638
Received: 14 February 2021; Accepted: 19 August 2021;
Published: 03 September 2021.
Edited by:
Menico Rizzi, University of Eastern Piedmont, ItalyReviewed by:
Marco Bellinzoni, Institut Pasteur, FranceMassimo Degano, San Raffaele Scientific Institute (IRCCS), Italy
Copyright © 2021 Gao, Yu, Zhu, Qin, Wang, Han, Aleksandra Wojdyla, Wang and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sheng Cui, Y3VpLnNoZW5nQGlwYi5wdW1jLmVkdS5jbg==
†These authors have contributed equally to this work