 Carlos A. V. Barreto1,2†
Carlos A. V. Barreto1,2† Salete J. Baptista 1,3†
Salete J. Baptista 1,3† António J. Preto1,2
António J. Preto1,2 Daniel Silvério1
Daniel Silvério1 Rita Melo1,4
Rita Melo1,4 Irina S. Moreira4,5*
Irina S. Moreira4,5*- 1CNC - Center for Neuroscience and Cell Biology, University of Coimbra, Cantanhede, Portugal
- 2PhD Programme in Experimental Biology and Biomedicine, Institute for Interdisciplinary Research (IIIUC), University of Coimbra, Coimbra, Portugal
- 3Centro de Ciências e Tecnologias Nucleares, Instituto Superior Técnico, University of Coimbra, Coimbra, Portugal
- 4Department of Life Sciences, University of Coimbra, Coimbra, Portugal
- 5Center for Innovative Biomedicine and Biotechnology, Center for Neuroscience and Cell Biology, University of Coimbra, Coimbra, Portugal
This paper describes an exciting big data analysis compiled in a freely available database, which can be applied to characterize the coupling of different G-Protein coupled receptors (GPCRs) families with their intracellular partners. Opioid receptor (OR) family was used as case study in order to gain further insights into the physiological properties of these important drug targets, known to be associated with the opioid crisis, a huge socio-economic issue directly related to drug abuse. An extensive characterization of all members of the ORs family (μ (MOR), δ (DOR), κ (KOR), nociceptin (NOP)) and their corresponding binding partners (ARRs: Arr2, Arr3; G-protein: Gi1, Gi2, Gi3, Go, Gob, Gz, Gq, G11, G14, G15, G12, Gssh, Gslo) was carried out. A multi-step approach including models’ construction (multiple sequence alignment, homology modeling), complex assembling (protein complex refinement with HADDOCK and complex equilibration), and protein-protein interface characterization (including both structural and dynamics analysis) were performed. Our database can be easily applied to several GPCR sub-families, to determine the key structural and dynamical determinants involved in GPCR coupling selectivity.
1 Introduction
G-Protein coupled receptors (GPCRs), the biggest family of membrane receptors, share a common three-dimensional structure: seven transmembrane domains (TM1-7) connected by three intracellular loops (ICL1-3) and three extracellular loops (ECL1-3). GPCRs also have an α-helix that runs parallel to the membrane in the intracellular side, commonly named Helix-8 (H8).
GPCR function is determined by the coupling with two main binding partners: heterotrimeric guanine nucleotide-binding proteins (G-proteins) and arrestins (Arrs). These receptors can interact with all four Gα classes (Gαi/o, Gαs, Gαq/11 and Gα12/13) that upregulate/downregulate distinct effectors. For example, Gαi/o inhibits cAMP production while Gαs increases its production, or Gαq/11, which in turn activates protein phospholipase C and the phosphatidylinositol signaling pathway. Furthermore, Arrs can also activate G-protein independent signaling pathways or promote GPCRs internalization (Ma and Pei, 2007; Smith and Rajagopal, 2016). The experimentally determined three-dimensional (3D) structures helped decipher some GPCR-partner binding determinants. However, a detailed knowledge of the key interactions between GPCR and their partners has not yet attained. (Barreto et al., 2021 in press).
Arrestins consist of two retinal isoforms (visual arrestin (arrestin 1) and cone arrestin (arrestin 4), and two nonvisual arrestins, β-arrestin 1 (arrestin 2) and β-arrestin 2 (arrestin 3). The visual arrestins (arrestin 1 and 4) are confined to visual sensory tissue, β-arrestins are widely expressed. Therefore, arrestin 2 and 3 are the OR binding partners described in our study. For complexes with non-visual Arr, two different conformations were recently reported: one complexed with muscarinic two receptor (M2R) (PDB ID: 6U1N (Staus et al., 2020)), highly similar to the arrestin-rhodopsin complex (PDB ID:4ZWJ (Kang et al., 2015)), and another complexed with neurotensin receptor 1 (NTS1), in which Arr2 was shown to undergo a 90° rotation relative to the receptor. Therefore, and despite the lack of solved GPCR-Arr complexes, the data available so far suggests divergent bioactive Arr conformations. (Yin et al., 2019; Staus et al., 2020). Moreover, we now have new data that points to the possibility of major macromolecular complexes, involving the simultaneous binding to both Arrs and G-proteins (Nguyen and Lefkowitz, 2021). Although the number of available structures has quickly increased in the last decade, structures are often not available for an entire family of receptors and typically only cover one or two families of partners. Hence, computational approaches such as homology modeling, molecular dynamics and docking are still indispensable to generate reliable structural information of non-available receptors in both unbound and bound forms (Wang and Miao, 2019; Jaiteh et al., 2020).
Researchers in life science related disciplines (especially drug development) need to access, aggregate and analyze copious amounts of interdisciplinary data. The possibility to deliver digital infrastructures/platforms that can deliver such requirements is now a reality in the light of recent advances in big data analysis. Our in-house pipeline published in Preto et al. (2020), applied to the study of dopamine receptors, was further extended and its usefulness as a web-tool to generate a database that allows protein-protein interactions (PPIs) evaluation of key GPCRs sub-families was once again demonstrated by application to the characterization of the four opioid receptors (OR): δ (DOR), κ (KOR), μ (MOR), and nociceptin (NOP). This pipeline was created to circumvent the scarcity of experimental available GPCR-partner complexes structures and to generate extensive structural and dynamical data of these documented complexes (Preto et al., 2020). All generated data is available at http://www.moreiralab.com/resources/oxr.
2 Methods
2.1 Workflow
Our GPCR PPI database is re-configurable in order to facilitate dynamical orchestration of operational components and provide a faster development/innovation lifecycle. The overall user workflow is available through a self-contained front-end application, which displays input information from compatible sources, and allows exploration of results. The resulting knowledge and full datasets are made freely accessible to its users. Also, the code to reproduce the analysis of the complexes is freely available at https://github.com/MoreiraLAB/or. We expanded our platform (Preto et al., 2020), to the OR family, for which all documented partners are described in Supplementary Table S1. The main pipeline steps, represented in Figure 1, are described below:
• Protein sequence retrieval from adequate database (e.g., Uniprot (The UniProt Consortium et al., 2021));
• Multiple Sequence Alignment (e.g., using Basic Local Alignment Search Tool (Altschul et al., 1990)) and biological inspection to pick adequate structural template;
• Homology modeling with Modeller (Šali and Blundell, 1993; Webb and Sali, 2016) or SWISS-Model (Waterhouse et al., 2018);
• Protein complex refinement with High Ambiguity Driven protein-protein DOCKing (HADDOCK) (van Zundert et al., 2016);
• Interface information retrieved using in-house Python (Van Rossum and Drake, 1995) tools and selenium (Software Freedom Conservancy, 2020), based on COCOMAPS (Vangone et al., 2011) and InterProSurf (Negi et al., 2007) servers;
• Additional features (amino acid and amino acid group percentages; 8 Å α-carbon distances; Salt-bridges; interhelical distance) calculated with in-house developed Python scripts (Van Rossum and Drake, 1995);
• Normal Mode Analysis performed using R (R Core Team, 2017);
• Hosting and webserver displayed with R (R Core Team, 2017) and shiny (Chang et al., 2021). Plots developed and displayed with ggplot2 (Wickham, 2016) and plotly (Plotly Technologies Inc., 2015).
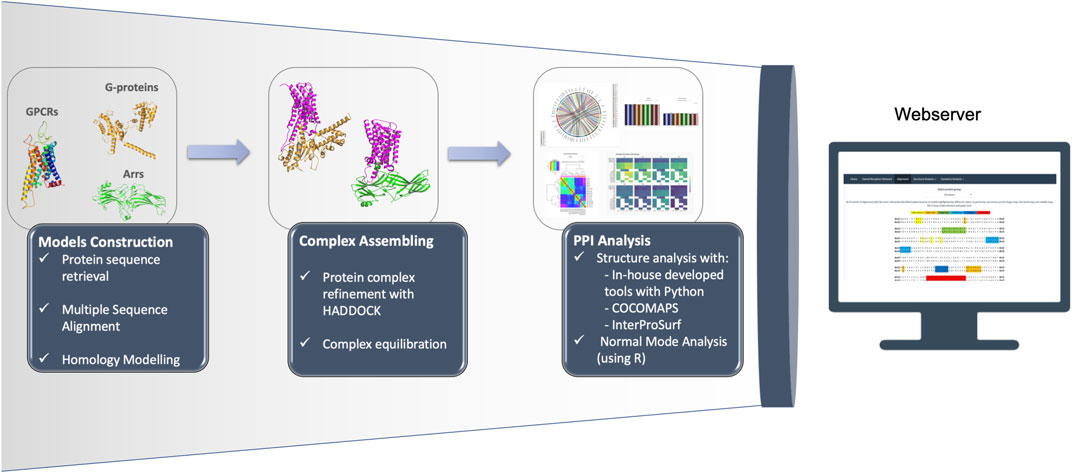
FIGURE 1. Representation of the OR pipeline.
2.1.1 Constructing Models of Receptors and Partners
MODELLER package (Šali and Blundell, 1993; Webb and Sali, 2016) was used to generate 3D models of OR. Three distinct structures were used as templates to model the studied the receptors:
• the MOR-Gi complex (PDB-ID: 6DDF (Koehl et al., 2018));
• the M2R-Arr2 complex (PDB-ID: 6U1N (Staus et al., 2020));
• the NTS1-Arr2 complex (PDB-ID: 6PWC (Yin et al., 2019)).
MOR structure was used to model G-protein coupled conformation, while M2R and NTS1 were used to model two different Arr coupled conformations. Target sequences were extracted from UniProt (The UniProt Consortium et al., 2021) sequence IDs P41143, P41145, P35372 and P41146 for DOR, KOR, MOR and NOP, respectively. The sequence alignment, model building and model selection were performed according to Preto et al. (2020). Receptors’ models were embedded in a POPC:CHL1 membrane and subjected to an equilibration protocol, as described in Preto et al. (2020). The Gi structure from Rhodopsin-Gi complex (PDB-ID: 6CMO (Kang et al., 2018)) was used as template for partner models of Gi/o, Gq/11 and G12 subfamilies members. The Gslo and Gssh model were built using as template the Gs structure retrieved from the β2AR-Gs complex (PDB-ID: 3SN6 (Rasmussen et al., 2011)). For Arr2 and Arr3 models, two distinct structure of Arr2 were used as template: one from NTS1-Arr2 complex (PDB-ID: 6PWC (Yin et al., 2019)) and the other from M2R-Arr2 complex (PDB-ID: 6U1N (Staus et al., 2020)). Human sequences of Gi1, Gi2, Gi3, Go, Gob, Gz, Gslo, Gssh, Gq, G11, G14, G15, G12, Arr2 and Arr3 were retrieved from UniProt (The UniProt Consortium et al., 2021) IDs P63096, P04899, P08754, P09471-1, P09471-2, P19086, P63092-1, P63092-2, P50148, P29992, O95837, P30679, Q03113, P49407-1 and P32121-1, respectively.
2.1.2 Assembling Complexes
Models of receptors and partners were superimposed to templates using PyMOL (Schrödinger, 2015) and then refined with HADDOCK (van Zundert et al., 2016). HADDOCK software enables optimization of backbone and side chains at the complex interface, and as such the determination of the best possible model for each OR-partner complex. The complexes with Gi/o, Gq/11 and G12/13 members were superimposed on MOR-Gi structure (PDB-ID: 6DDF (Koehl et al., 2018)). The OR-Gq/11 and OR-G12/13 complexes were also superimposed on M1R-G11 structure (PDB-ID: 6OIJ (Maeda et al., 2019)). The complexes with Gs members were superimposed with β2AR-Gs complex (PDB-ID: 3SN6 (Rasmussen et al., 2011)). Arrestin complexes were superimposed on NTS1-Arr2 complex (PDB-ID: 6PWC (Yin et al., 2019)) and M2R-Arr2 complex (PDB-ID: 6U1N (Staus et al., 2020)).
2.1.3 Analysis of the Complexes
As our previous work show, detailed PPI characterization is fundamental to shed light into complex biological mechanisms (Preto et al., 2020). Some of the key elements to understand these problems (e.g., GPCR-partner coupling), implies measurement of structural features like interacting residues, number of h-bonds (HB) and salt bridges (SB) and solvent accessible surface area (SASA) analysis, as well as an atomistic characterization of their dynamic behavior. As such, normal mode analysis (NMA), to investigate flexibility and fluctuation changes between receptor structure in monomer and in complex, as well as distance determination between TM domains, to characterize GPCR activation, were also implemented.
2.2 Server Architecture
The OR portal can be accessed by modern popular Web browsers, including Chrome, Internet Explorer, Safari, and Firefox, without installing any specialized software or browser plug-ins. The implementation was performed using shiny (Chang et al., 2021) within R (R Core Team, 2017). In the view tier, the front-end plots were developed using ggplot2 (Wickham, 2016), for static information, and embedded with plotly (Plotly Technologies Inc., 2015), when possible, for a more intuitive and dynamic analysis. The back-end deployment was performed and hosted in our own servers.
3 Case Study: Interface Characterization of OR-Partners
In our previous work, we have already detected significant signatures for the differential coupling of dopamine receptors, alongside a detailed characterization of all involved partners (Preto et al., 2020). Herein, we constructed a dedicated database focused on the OR family. Our pipeline was able to capture and reproduce the known data as well as infer new possibilities, that could be tested by the scientific community. In the next subsection, we detail the main biological information retrieved for the OR complexes. A summary of the interacting residues is provided in a heatmap-like figure (Figure 2) and by a 3D structural representation of receptor’s and partner’s interface (Figure 3). A more detailed discussion of the test case can be found at SI.
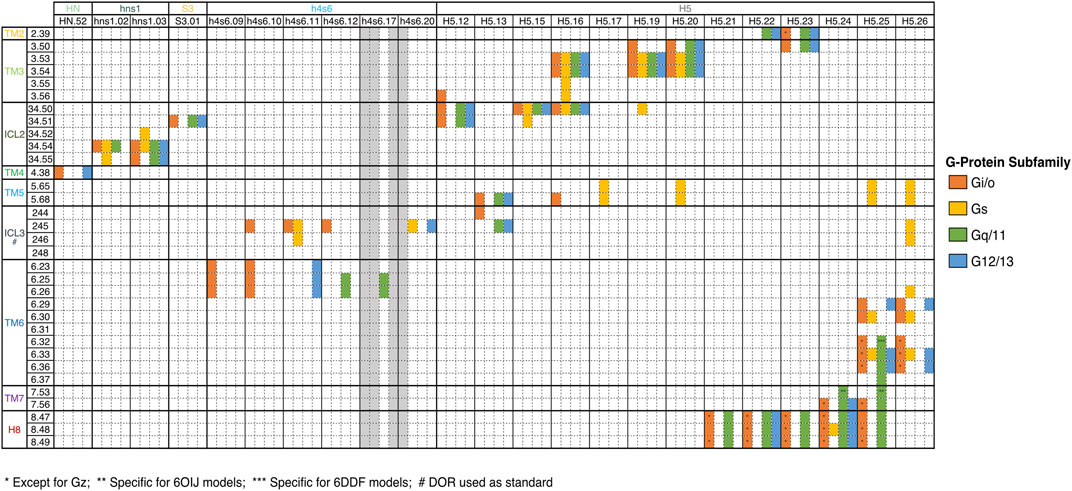
FIGURE 2. Key interactions between OR and G-Proteins. Only common interactions between the four receptors were depicted. The Ballesteros-Weinstein (BW) numbering scheme, which allows the comparison of any amino acid residue in the structurally conserved TM helices of GPCRs, was used. For ICL2, residues were labeled 34. X, indicating the positions between TM3 and TM4. X.50 was once again attributed to the most conserved residue. DOR residues numbers were used for ICL3 numbering, since this substructure is not conserved among OR.
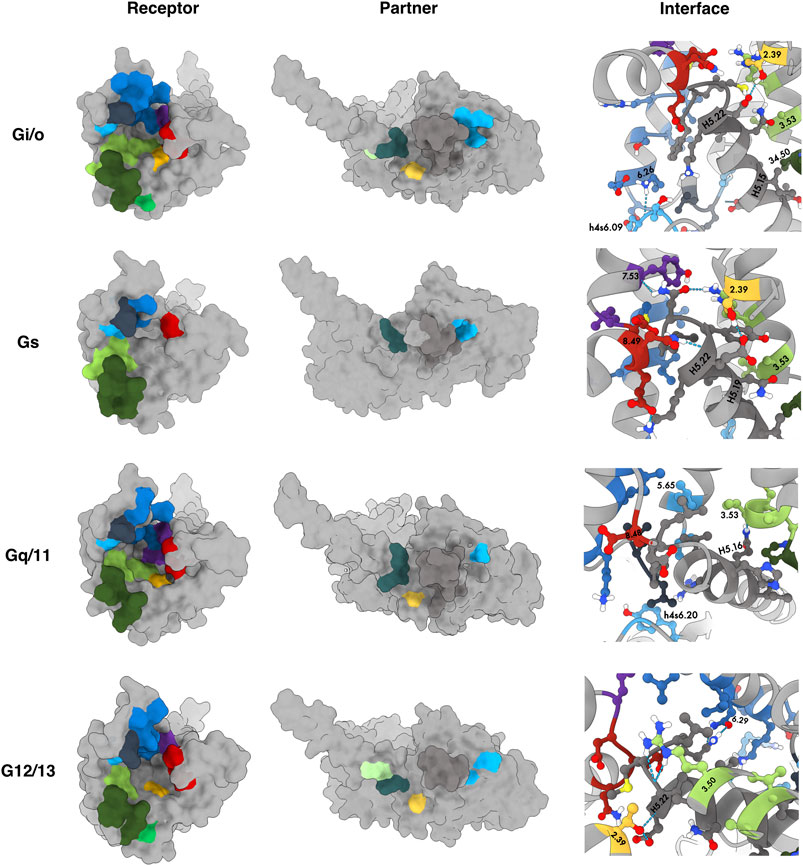
FIGURE 3. Common interaction binding motifs in the OR family. Receptors and partners are both represented as surface. 3D structures correspond to DOR-Gi1, KOR-Gssh, DOR-Gq and KOR-G12 (from top to bottom). Residues reported in Figure 2 were colored by their subdomain using the color scheme available at the sequence alignment on the webserver. Receptor: TM2 - yellow; TM3 - light green; ICL2 - dark green, TM4 - lime; TM5 - light blue; ICL3 - dark blue; TM6 - blue, TM7 - purple; H8 - red. Partner: HN - light green, hns1 - dark teal; S3 - yellow; h4s6 - light blue; H5 - dark grey.
Binding motifs were named using one-code amino acid nomenclature and in agreement with Ballesteros-Weinstein numbering scheme (Ballesteros and Weinstein, 1995), which defines X.50 as the most conserved residue in each TM domain or the residue number in case of belonging to an ICL or the binding partner in superscript. A wild-card amino acid was defined using “x”. The analysis of the main interactions established between OR and G-proteins pointed out some interesting interaction patterns, involving ICL2, TM3, TM5, ICL3 and TM6, which are in line with the MOR-Gi complex structure determined by Koehl et al. (2018). Particularly, P34.50–V/I from P34.50–V/I–x2LD34.55 pattern (ICL2) interacts with a hydrophobic patch of Gi/o and G12, which is constituted by T/KH5.12–x2I–I/LH5.16 pattern from H5 and L/IS3.01 from S3 (Figure 2). Although this hydrophobic pattern is not conserved for Gq/11 and Gs, ORs are still interacting through P34.50V with the corresponding residues at H5 and S3 positions. Moreover, the D34.55 (ICL2) interacts with R/K/G/Ahns1.03 of hns1, in all OR-G-proteins complexes (except for KOR-Gs). Even though D34.50 is a conserved residue among OR family, the same does not apply to other Gi-protein primary coupling GPCRs, such as D2R where the above-mentioned interaction is not observed (Koehl et al., 2018; Preto et al., 2020; Yin et al., 2020). Some hydrophobic residues from TM5 (V5.68) and ICL3 (L245/258/247/M266) were also involved in interactions with G-proteins’ H5, in a similar way to what was previously reported by Koehl et al. (2018). It is also worth mentioning that R244/257/265/246, an important residue in Gi signaling, was found to be engaged in interactions not only with Gi, but also with Gq/11 and G12 subfamilies, in most of OR-complexes, through H5 (DH5.13) and/or h4s6 (e.g., Eh4s6.12 (Gi/o), I/V/R/Ph4s6.20 (Gq/11 and G12 subfamilies)). This interaction is not observed for OR-Gs complexes, which agrees with previous data showing that an identical interaction was not present in the β2AR-Gs complex (Rasmussen et al., 2011; Koehl et al., 2018). Furthermore, the cytosolic ends of TM3 and TM6 were engaged in interactions with highly conserved residues at H5 C-terminal. Similarly to the information reported for MOR-Gi complex by Koehl et al., TM3 (mostly through the conserved R3.50), interacted with the Gi/o conserved CH5.23. This interaction mediated by TM3 was not observed for OR when complexed with Gz (IH5.23), or KOR-Gs complexes. Nevertheless, a similar interaction between R3.50 and Y/F/IH5.23 was also observed for OR-Gq/11/G12 or MOR-Gs complexes. The fully conserved LH5.25, from C-terminal H5, also interacts with a hydrophobic pocket formed by TM6 residues such as I6.33, M/L6.36, and V6.37, for OR upon coupling to Gi/o and Gq/11 subfamilies, as well as for MOR-Gs, data corroborated by literature (Koehl et al., 2018). An additional interaction between LH5.25 and R6.32, can further stabilize the interface of interaction in OR-Gi/o and OR-Gq_6DDF complexes, particularly for DOR, MOR and NOP ones. Through the interaction plot analysis, we also disclosed that T2.39 was relevant in all OR-G-protein complexes, with the exception of MOR-Go, Gs or Gz, where this residue (or any other TM2 residue) did not mediated any interaction. The interaction between T2.39 and the C/Y/F/IH5.23 (H5) was a characteristic one for Gi/o and Gq complexes. This fact agrees with our previous findings, in which both Gi/o and Gq complexed with DXR revealed this same interaction (Preto et al., 2020). On the contrary, no interaction involving TM2 was shown in OR-Gs or DXR-Gs. Another interesting sequence pattern highlighted in the present interface characterization study involves both TM7 and H8, through L7.56–D8.47EN8.49 interacting motif, and Gi/o, G12 and Gq/11 subfamilies. This interaction pattern was not, however, observed for either Gs or Gz complexes that did not interacted with OR TM7 subdomain. These results also follow previous reported data demonstrating a higher number of interactions involving the TM7-H8 boundary in Gi-coupled receptors, when compared with Gs-coupled ones (Draper-Joyce et al., 2018; Sandhu et al., 2019). This is also in line with our previous study, in which we identified the interacting pattern F7.56N–A/I–E8.45 for Gi/o and Gq couplings with D2-like Receptors (Preto et al., 2020). The recent solved D2R-Gi structure also suggested the interaction of TM7-H8 junction (namely F7.56, N8.43 and E8.45) with the C-terminal H5 interface of Gi (Yin et al., 2020).
Regarding OR-Arrs interactions analysis, some interesting features were also revealed. One of the most remarkable differences between OR-Arrs_6U1N and OR-Arrs_6PWC was the number of interactions established between OR and their intracellular effectors. Clearly, the latter complex group had a smaller number of interactions comparing with the former one. This is in accordance with dopamine complexes data (Preto et al., 2020), and could be linked to the 90° rotation of Arr2 in NTS1-Arr2 (PDB-ID: 6PWC (Yin et al., 2019)) structure when compared with M2R-Arr2 (PDB-ID: 6U1N (Staus et al., 2020)). The finger loop was the most interacting subdomain for both OR-Arrestins_6U1N and OR-Arrestins_6PWC groups. We observed that both Arrs interacted through a more embracing finger loop motif (Y63/64–G64/65x4DVLGL73/74) in OR-Arrs_6U1N complexes when compared with the OR-Arrestins_6PWC ones (D67/68x2VL71/72). This follows the differences found in both templates (NTS1-Arr2 and M2R-Arr2). In the M2R-Arr2, the finger loop had a higher number of interacting residues and contacts extensively with TM2, TM3, ICL2, TM5, and TM6. Like the template, two interactions appears to be conserved in all OR-Arrestins_6U1N and include the DRY motif (TM3) residues: the interaction between D69/70 and T2.39 or V70/71 and V3.50. The change of R3.50 (in NTS1) by a V3.50 (in OR) does not significantly affect the interaction profile (Staus et al., 2020). Moreover, M2R-Arr2 also reports a special positioning of ICL2 inside a hydrophobic cleft between the two Arrs domains. The results obtained for OR-Arrs_6U1N complexes, showed that ICL2 interacts with residues from both domains endorsing the authors conclusions (Staus et al., 2020). Concerning OR-Arrs_6PWC, it was observed that TM6, TM7 and H8 were always involved in interactions with finger loop, in line with the solved NTS1-Arr2 structure (Yin et al., 2019). Furthermore, TM3 and TM5 also contacted the finger loop at KOR-Arrs_6PWC complexes. The same was not observed for the NTS1-Arr2 complex (Yin et al., 2019).
Another interesting difference between OR-Arrs_6PWC and the original modeling template can be pointed out. While ICL1 was an interacting domain in NTS1-Arr2, particularly with lariat and bottom loops, in OR-Arrestins_6PWC, both Arrs only interacted with ICL1 in three complexes (MOR-Arr2_6PWC and NOP-Arrs_6PWC), all of them through the finger loop. On the other hand, the ICL2 formed contacts in all complex models, establishing interactions with finger loop and C-loop, instead of the lariat loop as reported in the template. This draws the hypothesis that ICL2 is not deeply inserted into the reported cleft (with middle loop, bottom loop and lariat loop), present in Arrs, interacting with finger loop at the intracellular cavity. It is also noteworthy that the ICL3 interaction pattern was in line with the NTS1-Arr2 structure, although only one interacting ICL3 residue was involved in establishing protein-protein contacts, for all OR-Arrs_6PWC complexes. This was a significant difference regarding arrestin recognition, when compared to other GPCRs with long ICL3, where multiple ICL3 residues interact with the receptor (Kang et al., 2015; Yin et al., 2019). On the other hand, some interactions previously described by Goddard et al.Mafi et al. (2020). were not observed in this study, namely the interaction between R34.57 (ICL2) and R65/66 (finger loop). R34.57 (ICL2) was reported as a key residue in Arr-2 binding regulation, mediating the creation of a polar network interactions which stabilizes the active-state of Arr2-pp-KOR complex bound to the full agonist DAMGO. Its interaction with R65/66 allowed the coordination of the later residue, which is thus able to establish a salt-bridge with the D67/68. This promote the reorientation of D67/68 in order to form a hydrogen bond with the TM2. (Mafi et al., 2020).
Since our published work on DxR complexes, experimentally determined complex structures of D1R-Gs (PDB-ID: 7JVP (Zhuang et al., 2021b), 7JVQ (Zhuang et al., 2021b), 7JV5 (Zhuang et al., 2021b), 7LJD (Zhuang et al., 2021a), 7LJC (Zhuang et al., 2021a), 7CKY (Xiao et al., 2021), 7CRH (Xiao et al., 2021), 7CKW (Xiao et al., 2021), 7CKX (Xiao et al., 2021), 7CKZ (Xiao et al., 2021)) and D2R-Gi (PDB-ID: 6VMS (Yin et al., 2020), 7JVR (Zhuang et al., 2021b)) have been released. The similarly between our complexes and the experimental reported is very high, with RMSD values ranging from 1 Å to 1.5 Å. Xiao et al. (2021) reported 16 different contacts between D1R-Gs structure and D2R-Gi structure. D1-like receptors have a Phe (F34.51) while D2-like have Met (M34.51) and Leu (L34.52) at the ICL2, which interacts with an hydrophobic pocket created by S1, S3 and H5 of Gs (Xiao et al., 2021). This interaction extends to other residues from ICL2 (P34.50, R34.52, E34.54 and R34.55). In Preto et al., where we used a similar protocol, we were able to correctly predict the same Phe interaction with Gs in D1-like receptors inside P34.50Fx2–E/K–R34.55 pattern (Preto et al., 2020). Another important interaction between D1R-Gs was predicted in Preto et al. and identified in Xiao et al. at TM5 (A5.65xxQ5.68-I5.69).
4 Conclusion
Our integrated end-to-end engine platform provides access to the putative protein-protein interfaces and key pairwise interactions involved in the coupling of OR receptors and G-proteins and/or Arrs partners. Although the modeling and web server can be assembled in a system independent way, we applied it to a particular relevant sub-family as we aim to link our platform to a careful analysis of all data in a consistent and integrative way.
Our pipeline was successful in predicting the interactions reported for previous solved GPCR-Partners complexes, disclosing the main differences engaged in the coupling of the distinct G-proteins subfamilies, as well as the different Arrs conformations involved. Our demonstrated success upon application to two different GPCR sub-families (DxR and OR), demonstrates the protocol scalability giving non-expert researcher a tool to quickly analyze and assess the interface between a receptor and its intracellular partners.
Data Availability Statement
The datasets generated and analyzed for this study can be found in http://www.moreiralab.com/resources/oxr. The platform is free and open to all. There is not no login requirement. The code and models used to generate the datasets are fully available at https://github.com/MoreiraLAB/or.
Author Contributions
CAVB, SJB, and DS performed model construction. CAVB and DS performed the molecular dynamics calculations. SJB, DS, and AJP performed model submission and selection for HADDOCK models. AJP constructed and deployed the web-tool implementation. CAVB, SJB, AJP, and DS performed formal analysis of results. CAVB and SJB authors wrote the original paper draft and all authors participated in writing the final article. RM and ISM conceptualized the experience, supervised the research, and acquired the necessary funding.
Funding
This work was funded by COMPETE 2020 - Operational Programme for Competitiveness and Internationalization and Portuguese national funds via FCT - Fundação para a Ciência e a Tecnologia, under projects LA/P/0058/2020, UIDB/04539/2020, UIDP/04539/2020 and PTDC/QUI-OUT/32243/2017. AJP and CAVB were also supported by FCT through Ph.D. scholarships SFRH/BD/144966/2019 and SFRH/BD/145457/2019, respectively. Authors would also like to acknowledge ERNEST–European research Network on Signal Transduction, CA18133.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We would like to acknowledge José Guilherme for his participation in the original platform and for contributing to elements of the main pipeline.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.715215/full#supplementary-material
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic Local Alignment Search Tool. J. Mol. Biol. 215, 403–410. doi:10.1016/S0022-2836(05)80360-2
Ballesteros, J. A., and Weinstein, H. (1995). “[19] Integrated Methods for the Construction of Three-Dimensional Models and Computational Probing of Structure-Function Relations in G Protein-Coupled Receptors,” in Methods in Neurosciences. Receptor Molecular Biology. Editor S. C. Sealfon (Academic Press), 25, 366–428. doi:10.1016/S1043-9471(05)80049-7
Barreto, C. A. V., Baptista, S. J., Bueschbell, B., Magalhães, P., Preto, A. J., Lemos, A., et al. (2021) “Arrestin and G Protein Interactions with GPCRs: a Structural Perspective,” in GPCRs as Therapeutic Targets. 1st edn. (John Wiley and Sons Ltd)). (in press).
Chang, W., Cheng, J., Allaire, J., Sievert, C., Schloerke, B., Xie, Y., et al. (2021). Shiny: Web Application Framework For R. R Package Version 1.6.0.
Draper-Joyce, C. J., Khoshouei, M., Thal, D. M., Liang, Y.-L., Nguyen, A. T. N., Furness, S. G. B., et al. (2018). Structure of the Adenosine-Bound Human Adenosine A1 Receptor-Gi Complex. Nature 558, 559–563. doi:10.1038/s41586-018-0236-6
Jaiteh, M., Rodríguez-Espigares, I., Selent, J., and Carlsson, J. (2020). Performance of Virtual Screening against GPCR Homology Models: Impact of Template Selection and Treatment of Binding Site Plasticity. Plos Comput. Biol. 16, e1007680. doi:10.1371/journal.pcbi.1007680
Kang, Y., Zhou, X. E., Gao, X., He, Y., Liu, W., Ishchenko, A., et al. (2015). Crystal Structure of Rhodopsin Bound to Arrestin by Femtosecond X-ray Laser. Nature 523, 561–567. doi:10.1038/nature14656
Kang, Y., Kuybeda, O., de Waal, P. W., Mukherjee, S., Van Eps, N., Dutka, P., et al. (2018). Cryo-EM Structure of Human Rhodopsin Bound to an Inhibitory G Protein. Nature 558, 553–558. doi:10.1038/s41586-018-0215-y
Koehl, A., Hu, H., Maeda, S., Zhang, Y., Qu, Q., Paggi, J. M., et al. (2018). Structure of the µ-Opioid Receptor-Gi Protein Complex. Nature 558, 547–552. doi:10.1038/s41586-018-0219-7
Ma, L., and Pei, G. (2007). β-Arrestin Signaling and Regulation of Transcription. J. Cel Sci. 120, 213–218. doi:10.1242/jcs.03338
Maeda, S., Qu, Q., Robertson, M. J., Skiniotis, G., and Kobilka, B. K. (2019). Structures of the M1 and M2 Muscarinic Acetylcholine receptor/G-Protein Complexes. Science 364, 552–557. doi:10.1126/science.aaw5188
Mafi, A., Kim, S.-K., and Goddard, W. A. (2020). Mechanism of β-arrestin Recruitment by the μ-opioid G Protein-Coupled Receptor. Proc. Natl. Acad. Sci. USA. 117, 16346–16355. doi:10.1073/pnas.1918264117
Negi, S. S., Schein, C. H., Oezguen, N., Power, T. D., and Braun, W. (2007). Interprosurf: a Web Server for Predicting Interacting Sites on Protein Surfaces. Bioinformatics 23, 3397–3399. doi:10.1093/bioinformatics/btm474
Nguyen, A. H., and Lefkowitz, R. J. (2021). Signaling at the Endosome: cryo‐EM Structure of a GPCR-G Protein-Beta‐arrestin Megacomplex. Febs J. 288, 2562–2569. doi:10.1111/febs.15773
Preto, A. J., Barreto, C. A. V., Baptista, S. J., Almeida, J. G. d., Lemos, A., Melo, A., et al. (2020). Understanding the Binding Specificity of G-Protein Coupled Receptors toward G-Proteins and Arrestins: Application to the Dopamine Receptor Family. J. Chem. Inf. Model. 60, 3969–3984. doi:10.1021/acs.jcim.0c00371
R Core Team (2017). R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing.
Rasmussen, S. G. F., DeVree, B. T., Zou, Y., Kruse, A. C., Chung, K. Y., Kobilka, T. S., et al. (2011). Crystal Structure of the β2 Adrenergic Receptor-Gs Protein Complex. Nature 477, 549–555. doi:10.1038/nature10361
Šali, A., and Blundell, T. L. (1993). Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 234, 779–815. doi:10.1006/jmbi.1993.1626
Sandhu, M., Touma, A. M., Dysthe, M., Sadler, F., Sivaramakrishnan, S., and Vaidehi, N. (2019). Conformational Plasticity of the Intracellular Cavity of GPCR-G-Protein Complexes Leads to G-Protein Promiscuity and Selectivity. Proc. Natl. Acad. Sci. U S A. 116, 11956–11965. doi:10.1073/pnas.1820944116
Smith, J. S., and Rajagopal, S. (2016). The β-Arrestins: Multifunctional Regulators of G Protein-Coupled Receptors. J. Biol. Chem. 291, 8969–8977. doi:10.1074/jbc.R115.713313
Staus, D. P., Hu, H., Robertson, M. J., Kleinhenz, A. L. W., Wingler, L. M., Capel, W. D., et al. (2020). Structure of the M2 Muscarinic Receptor-β-Arrestin Complex in a Lipid Nanodisc. Nature, 579, 297–302. doi:10.1038/s41586-020-1954-0
The UniProt Consortium Bateman, A., Martin, M.-J., Orchard, S., Magrane, M., Agivetova, R., et al. (2021). UniProt: the Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Van Rossum, G., and Drake, F. L. (1995). Python tutorial (Centrum voor Wiskunde en Informatica Amsterdam, The Netherlands).
van Zundert, G. C. P., Rodrigues, J. P. G. L. M., Trellet, M., Schmitz, C., Kastritis, P. L., Karaca, E., et al. (2016). The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 428, 720–725. doi:10.1016/j.jmb.2015.09.014
Vangone, A., Spinelli, R., Scarano, V., Cavallo, L., and Oliva, R. (2011). Cocomaps: a Web Application to Analyze and Visualize Contacts at the Interface of Biomolecular Complexes. Bioinformatics 27, 2915–2916. doi:10.1093/bioinformatics/btr484
Wang, J., and Miao, Y. (2019). “Recent Advances in Computational Studies of GPCR-G Protein Interactions,” in Advances in Protein Chemistry and Structural Biology (Elsevier), 116, 397–419. doi:10.1016/bs.apcsb.2018.11.011
Waterhouse, A., Bertoni, M., Bienert, S., Studer, G., Tauriello, G., Gumienny, R., et al. (2018). SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 46, W296–W303. doi:10.1093/nar/gky427
Webb, B., and Sali, A. (2016). Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 54, 5.6.1–5.6.37. doi:10.1002/cpbi.3
Xiao, P., Yan, W., Gou, L., Zhong, Y.-N., Kong, L., Wu, C., et al. (2021). Ligand Recognition and Allosteric Regulation of DRD1-Gs Signaling Complexes. Cell 184, 943–956.e18. doi:10.1016/j.cell.2021.01.028
Yin, W., Li, Z., Jin, M., Yin, Y.-L., de Waal, P. W., Pal, K., et al. (2019). A Complex Structure of Arrestin-2 Bound to a G Protein-Coupled Receptor. Cell Res. 29, 971–983. doi:10.1038/s41422-019-0256-2
Yin, J., Chen, K.-Y. M., Clark, M. J., Hijazi, M., Kumari, P., Bai, X.-c., et al. (2020). Structure of a D2 Dopamine Receptor-G-Protein Complex in a Lipid Membrane. Nature 584, 125–129. doi:10.1038/s41586-020-2379-5
Zhuang, Y., Krumm, B., Zhang, H., Zhou, X. E., Wang, Y., Huang, X.-P., et al. (2021a). Mechanism of Dopamine Binding and Allosteric Modulation of the Human D1 Dopamine Receptor. Cel Res. 31, 593–596. doi:10.1038/s41422-021-00482-0
Keywords: database, functional signature, GPCRs, opioid receptor, G-protein, arrestin
Citation: Barreto CAV, Baptista SJ, Preto AJ, Silvério D, Melo R and Moreira IS (2021) Decoding Partner Specificity of Opioid Receptor Family. Front. Mol. Biosci. 8:715215. doi: 10.3389/fmolb.2021.715215
Received: 26 May 2021; Accepted: 10 August 2021;
Published: 21 September 2021.
Edited by:
Jessica Andreani, UMR9198 Institut de Biologie Intégrative de la Cellule (I2BC), FranceReviewed by:
Yinglong Miao, University of Kansas, United StatesElodie Laine, Université Pierre et Marie Curie, France
Copyright © 2021 Barreto, Baptista , Preto, Silvério, Melo and Moreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Irina S. Moreira , aXJpbmEubW9yZWlyYUBjbmMudWMucHQ=
†These authors have contributed equally to this work and share first authorship