 Liliana Florea
Liliana Florea Lindsay Payer1,3
Lindsay Payer1,3- 1McKusick-Nathans Department of Genetic Medicine, Johns Hopkins School of Medicine, Baltimore, MD, United States
- 2Department of Computer Science, Johns Hopkins University, Baltimore, MD, United States
- 3Department of Pathology, Johns Hopkins School of Medicine, Baltimore, MD, United States
- 4Department of Pathology, Dana-Farber Cancer Institute, Boston, MA, United States
- 5Harvard Medical School, Boston, MA, United States
Alu exonization events functionally diversify the transcriptome, creating alternative mRNA isoforms and accounting for an estimated 5% of the alternatively spliced (skipped) exons in the human genome. We developed computational methods, implemented into a software called Alubaster, for detecting incorporation of Alu sequences in mRNA transcripts from large scale RNA-seq data sets. The approach detects Alu sequences derived from both fixed and polymorphic Alu elements, including Alu insertions missing from the reference genome. We applied our methods to 117 GTEx human frontal cortex samples to build and characterize a collection of Alu-containing mRNAs. In particular, we detected and characterized Alu exonizations occurring at 870 fixed Alu loci, of which 237 were novel, as well as hundreds of putative events involving Alu elements that are polymorphic variants or rare alleles not present in the reference genome. These methods and annotations represent a unique and valuable resource that can be used to understand the characteristics of Alu-containing mRNAs and their tissue-specific expression patterns.
Introduction
Alu elements are ∼300 bp sequences belonging to an order of retrotransposons termed Short Interspersed Elements (SINEs) that have expanded in primates (Batzer and Deininger, 2002; Hormozdiari et al., 2013). Alu elements represent 11% of the human genome, with nearly one million copies located primarily in introns and intergenic space proximal to genes (Lander et al., 2001; Venter et al., 2001). They have contributed to genetic and functional diversity during evolution in multiple ways. Alu element insertions can influence gene regulation and affect RNA polyadenylation, splicing, and editing (Chen et al., 2008; Chen et al., 2009; Vorechovsky, 2010; Deininger, 2011; Shen et al., 2011; Payer and Burns, 2019). Alu elements can be deleterious, interrupting key gene regulatory elements, and serving as substrates for non-allelic recombination leading to copy number variations (CNVs).
Alu exonization, or recruitment of an intronic Alu element into a gene transcript can alter protein sequence (Lev-Maor et al., 2003) and function or, alternatively, can introduce a premature termination codon (PTC) and trigger nonsense mediated decay (Attig et al., 2016) (NMD) surveillance mechanisms to degrade transcripts. Examples of Alu exonizations causing disease include a mutation in an antisense Alu element, which created a splice donor and activated a cryptic splice acceptor in the ornithine-delta-aminotransferase mRNA leading to a loss-of-function (Mitchell et al., 1991). Other examples of mutations that lead to Alu exonization include a collagen, type IV, alpha 3 (Goodpasture antigen) (COL4A3) allele causing Alport syndrome (Knebelmann et al., 1995); a survivin allele (baculoviral IAP repeat containing 5 (BIRC5)) causing Sly syndrome (Mola et al., 2007); a fibroblast growth factor receptor 2 (FGFR2) allele causing Apert syndrome (Oldridge et al., 1999); and a 6-pyruvoyltetrahydropterin synthase (PTS) allele resulting in tetrahydrobiopterin deficiency (Meili et al., 2009).
Alu elements have contributed directly and in a significant way to the creation of new gene content. Approximately 5% of alternatively spliced exons internal to the human genes are estimated to have derived from exonizations of intronic Alu sequences (Sorek et al., 2002). The most common mechanism is via changes in the Alu sequence leading to the formation of new and typically weak 5′ splice sites (ss) (Lev-Maor et al., 2003). There are multiple potential 5’ ss, and the selection of a specific site is determined by a complex interplay between the relative strength of the candidate splice sites, coupled with splicing regulatory elements (enhancers) within the Alu exon sequence (Ram et al., 2008). There is a proclivity for Alu exonization when an intronic Alu is oriented antisense to the gene (Lev-Maor et al., 2003; Sela et al., 2007; Vorechovsky, 2010). The primary path to exonization is via alternative splicing, with the transcript incorporating the new Alu exon starting off as the minor isoform. From an evolutionary perspective, this scenario may allow a locus to “experiment” with new function while preserving its primary function. Over time, mutations in the Alu exon and surrounding intron sequence, and the selective pressures acting on them, may lead to relative permanence or the acquisition of new function, and to the transcript being promoted to the major isoform. Indeed, a significant number of the exonized Alu elements are unique to the human genome, and similarly for other primates, and thought to have played a part in the formation of species-specific traits (Shen et al., 2011).
With the ascent and democratization of deep RNA sequencing (RNA-seq), there are now vast collections of detailed gene expression data from large numbers of individuals, species and developmental or cellular conditions. Such resources present tremendous opportunities to identify instances of Alu element recruitment into transcripts under a wide variety of conditions. In particular, cataloguing exonization of fixed Alu elements may increase our understanding of tissue, organ and cell type specificity, and help uncover niche functions evolved through Alu exonizations. Such a wholesale discovery and curation effort has not yet been undertaken for Alu exons, and they remain poorly represented in the gene annotation databases.
While significantly improved in its representation of the Alu content and haplotype variation, the current reference genome is incomplete as a representation of individuals. Alu elements continue to insert into the human genome and to create structural variants, at a rate of about one new Alu insert per 20 human births (Xing et al., 2009; Deininger, 2011). This process leads to genetic diversity (Comas et al., 2004), but also to about one in 1,000 new human genetic (Deininger and Batzer, 1999; Vorechovsky, 2010) diseases. Understanding the preponderance and impact of polymorphic and rare Alu insertion variants in the population and in disease is a yet untapped reservoir.
Detection of Alu exonization from short read, high-throughput RNA-seq data, however, is challenging for multiple reasons. When the Alu is incorporated in the reference genome, Alu-containing RNA-seq reads may map to multiple locations on the genome or even within the same gene locus, leading to their exclusion or making it difficult to unambiguously determine the source. Further, unspliced, pre-mRNA sequences are present in RNA-seq experiments (Ameur et al., 2011) and Alu-containing reads from pre-mRNA can be difficult to distinguish from fully processed mRNAs containing Alu-derived exons. Furthermore, when the Alu element is not part of the reference assembly (i.e., is a polymorphic or de novo element), the location of the insert in the genome may be unknown. Most of these Alu insertions are members of a subfamily of AluY elements (Batzer and Deininger, 1991). The AluY subfamily is one of several that account for almost all recently integrated human Alu elements, including rare and polymorphic events not represented in the reference human genome (Batzer and Deininger, 1991; Batzer and Deininger, 2002). High similarity among AluY elements coupled with sequencing errors make it difficult to distinguish reads as derived from a non-reference Alu as opposed to from another AluY residing in the same intron. Adding to the complexity, unlike with DNA sequences where Alu insertions can be detected as local structural variations (Qian et al., 2015) that appear as breaks in the expected co-localization of paired end reads, the interrupted structure of mRNAs allows for reads in the same pair to be arbitrarily distanced, increasing the likelihood of a local Alu element (‘shadow’) confounding the prediction and creating a false positive. The precise site of a genomic Alu may be impossible to pinpoint even when an exonization event is evident. Lastly, most of these elements may be expressed at very low levels, limiting their deleterious effects but providing little read evidence to allow detection. The challenges inherent to identifying good candidate sites and distinguishing these from a large potential number of false signals make the task of predicting novel Alu exonizations particularly daunting.
We describe two methods for detecting Alu insertions in mRNA sequences, at elements already included in the reference genome, and at novel loci not encoded in the reference genome and representing likely polymorphic or rare variations, respectively. We then apply the tools to a collection of 117 RNA-seq data sets from human frontal cortex tissue, generated by the GTEx project (Consortium, 2015). The collection of sequences and annotations can be used as a starting point for validation experiments, and incorporated into functional and disease studies targeted at the feature level. Lastly, our study is a model for creating a comprehensive Alu mRNA feature repertoire in other tissues, across developmental stages and for a wide variety of disease and normal cellular conditions.
We implemented the algorithm into a software called Alubaster, available from https://github.com/splicebox/Alubaster.
Results
Detection of Exonization Events at Alu Elements in the Reference Genome
When the DNA sequence of an Alu insertion variant is included in the reference genome, traditional approaches to read alignment and RNA-seq analysis can be used to distinguish between Alu exonization (‘signal’) and ‘noise’ generated by unprocessed intronic RNA and from multi-mappings reads. Transcript assembly algorithms incorporate intronic read filters, using statistical modeling to distinguish intronic read levels in mRNA resulted from intron retention and alternatively spliced exons, including exonized Alu elements, and unprocessed RNA from incomplete co-transcriptional splicing (Song et al., 2016). Additionally, assemblers can detect and remove splicing patterns that have very low likelihood, for instance with low read support or creating uncharacteristic mRNA features, such as unexpectedly long introns and exons. Further, rather than assessing each read to determine its origin among multiple mappings on the genome, assemblers collectively analyze clusters of reads to assess the relative contribution of unique and multi-mapping sequences at the same location and to filter out potential paralogs.
We used the transcript assembler CLASS2, which implements all of the above filters and has been shown to capture alternative splicing variation, especially within the body of the gene, with very high accuracy (Song et al., 2016). Candidate Alu-containing exons that are internal to a gene, agree in size (<400), and are in antisense orientation to the annotated Alu are further retained (Figure 1). The exons are then collected across all samples to create a comprehensive list.
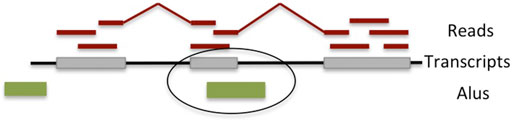
FIGURE 1. Overview of the detection algorithm for fixed (in the reference genome) Alu exonizations. RNA-seq reads are mapped to the reference genome and assembled into transcripts, some of which contain the Alu exons. Exons overlapping an Alu element located on the opposite strand and that are between 40 and 400 bp long are deemed to have occurred through Alu exonization.
Alu Exonization Events in the Human Frontal Cortex
We analyzed 117 human frontal cortex RNA-seq samples obtained from the GTEx (Consortium, 2015) repository, with 16,495,334-172,877,906 reads per sample (92,601,615 sample average). When assembled, the reads produced between 26,164 and 66,638 transcripts per sample. In the first pass, applying the exon filters above detected 1,019 Alu exons (45-343 per sample), at 870 reference Alu elements (loci) in 861 genes (Figure 2A and Supplementary Table S1). Of these, 725 events were found in 2 or more samples. Because the transcript assembly process may be too stringent to capture low expression exons, including some Alu exons, we relax the criteria to allow counting an exon in a sample if its flanking introns have read support. After expanding the search, 947 Alu exons, representing 92.9% of the total, had concomitant evidence for both splice sites in more than 2 samples. Therefore, the collection of Alu exonization events generated with our assembly-based method appeared robust and reliable.
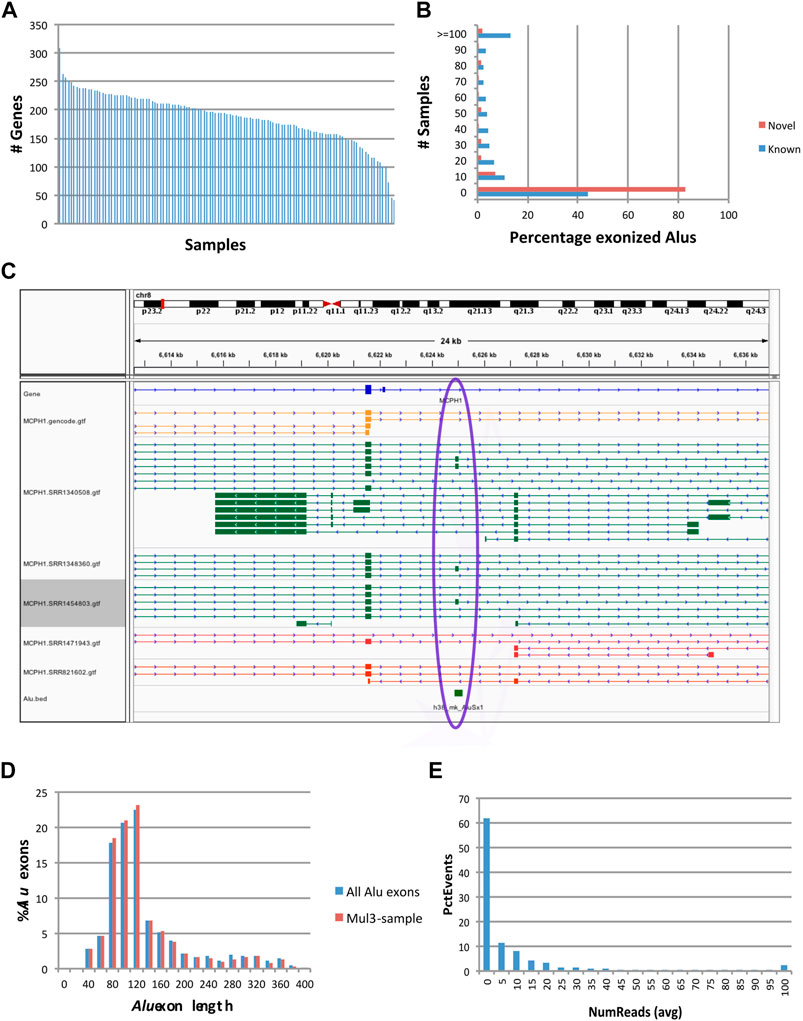
FIGURE 2. Characteristics of exonization events at fixed (reference) Alu loci. Events occurred at 870 reference Alu loci, of which 633 occurred at GENCODE annotated exons (‘known’) and 237 had not been previously known (‘novel’). (A) Numbers of genes with Alu exonization events, by sample. (B) Prevalence of Alu exonization events in the 117 samples. (C) Example of a fixed AluSx exonization event at the microcephalin 1 (MCPH1) gene. Gold: GENCODE gene annotations. Green and Red: samples with and without evidence of Alu exonization, respectively. This visualization was generated using the Integrated Genomics Viewer (IGV) (Robinson et al., 2011). (D) Histogram of Alu exon lengths. (E) Read counts of Alu exonization events. Total numbers of reads supporting the two flanking exons of the Alu exon were computed per sample, then averaged across samples and plotted into a histogram.
Alu exonization events occurred across multiple subfamilies of Alu (467 AluJ, 357 AluS, 40 AluY and 6 generic Alu instances). 633 of the exonized Alu elements were already annotated as exons in the GENCODE v.36 reference database, whereas 237 were not known to be exonized. A larger proportion of Alu elements known to undergo exonization had support in multiple samples compared to novel (unannotated) exonization events (Figure 2B). An example of a previously uncharacterized Alu exonization event at the Microcephalin 1 (MCPH1) gene is shown in Figure 2C, in which the event presents as an alternatively spliced 142 bp exon (chr8:6624896-6625037). The 142 exon inserts between exon 13 and the 3’ terminal exon 14, introducing a frame shift in the coding sequence of ENST00000344683.8 and creating a longer reading frame (from 2,508 bp/836 aa to 2,628 bp/875 aa), by reading through the new exon and 11 new aa into the terminal exon. While the new Alu exon is included in transcript predictions from 3 samples (SRR1340508, SRR1454803 and SRR1348360), the upstream and downstream flanking introns are present at very low levels (<3 reads) in 67 samples and 7 samples, respectively, suggesting that the exon may be expressed more broadly.
Features of Alu Exonization Events
Alu exon lengths were preponderantly between 80 and 140 bp (61.1%), with an average length of 144 bp and median 122 bp (Figure 2D). They were relatively uniformly distributed among in frame, frame +1 and frame +2 (342, 354, 323), and the distribution did not change when only the 947 exons present in multiple samples were considered (321, 323, 303). The above length distribution suggests that a majority of these Alu exonizations would likely be deleterious in a coding context, and hence may occur in non-coding RNAs or in untranslated regions of coding transcripts, or may be expressed at a low level that does not affect the overall output of the gene. Indeed, most events appear to show low read support in samples (Figure 2E).
Alu Exonization Events Are Alternatively Spliced
The primary mechanism for Alu inclusion into mRNA structure is via alternative splicing. We compared the exon-intron structure of the predicted transcripts across the 117 samples to determine alternative splicing events involving our Alu exons, especially cassette (skipped) exons. We imposed a stringent requirement to only consider simple events, where the exon had to be excised or included into the transcript without any changes to the flanking exons, to isolate the effects of the exon sequence from those of the context. 651 (63.9%) of Alu exons were found to undergo exon skipping, in 785 alternative splicing patterns (ASP) formed by alternative flanking introns (Supplementary Table S2).
We further analyzed the expression level of the 785 ASPs across all samples, as reflected by the read coverage, and the relative isoform expression levels. The Percent Splice In (PSI) value is defined as the ratio of expression levels of the exon-including isoform and the isoforms containing the locus, and is used to measure the relative isoforms’ contribution (Wang et al., 2008). After correcting for samples with low numbers of reads (< 10), which could not render a reliable PSI estimate, 68.6% of the remaining 303 events had the Alu exon included in the minor isoform (PSI<0.35), 11.2% had relatively equal contributions of the two forms (0.36<=PSI<0.65), and in 20.1% of ASPs the exon was included in the major isoform (PSI>=0.65) (Supplementary Figure S1 and Supplementary Table S2). This distribution is in sharp contrast to that observed when all of the exon skipping events in the 117 samples data set were considered (39,859 events), where the vast majority of the skipped exons, 77.9%, were expressed as part of the major isoform (PSI>=0.65).
Tissue Specificity of Alu Exonization Events
Lastly, we assessed the tissue specificity of 2,771 introns flanking the Alu exons by interrogating a comprehensive human introns database with the tool Snaptron (Wilks et al., 2018). Snaptron uses a collection of exon-exon junctions (introns), along with supporting read counts per sample, extracted from ∼50,000 publicly available RNA-seq samples, including those from the GTEx project. Read support for the 2,771 introns was extracted for all RNA-seq samples represented in GTEx, and a custom statistical test was designed to assess tissue specificity (see Methods). A total of 2,745 introns were found by Snaptron (Wilks et al., 2018), of which 45 introns were detected as brain specific using our stringent criteria (min 10 reads per sample, p-val<0.001). Roughly half of these introns occur in long non-coding RNAs (Supplementary Table S3).
Examples of tissue specific events are illustrated in Figure 3 and Supplementary Figure S2. All (coding) host genes were confirmed to be expressed solely in brain using the ProteinAtlas resource (https://www.proteinatlas.org/), except for Ubiquitin-Associated Protein 1-Like (UBAP1L), also expressed in retina, which nevertheless leaves the possibility that the Alu exonization may take place preferentially in brain. At the PDZ Domain Containing 7 (PDZD7) gene, an annotated Alu-containing terminal 3′ exon is converted into an internal Alu exon (chr10:101017253-101017615) with the introduction of a novel, not present in the GENCODE annotation, brain specific intron, chr10:101016831,101017253. The resulting exon is 363 bp long. Additional novel exons and splicing patterns were also revealed by the RNA-seq data (Figure 3). In another example, transcripts assembled for the gene UBAP1L show a partially exonized AluSc element, to generate a 118 bp exon (chr15:65107209-65107326) not present in the GENCODE annotation, along with an additional potential Alu recruitment event within the upstream exon (Supplementary Figure S2A). The downstream intron, chr15:65106359,65107209 is tissue specific and not found in the reference database. Further, exonization of an AluJo element creates a new exon chr11:45528970-45529061 within the long non-coding RNA AC103855.2. Here, both of the flanking introns (chr11:45528443,45528970 and chr11:45529061,45531525) were identified as tissue specific and are not annotated in GENCODE. The combination of new exons and new complex splicing patterns internal to and/or at the 3’ end of the gene suggest the presence of multiple previously unknown splice isoforms that may perform brain specific functions (Supplementary Figure S2B). Lastly, and serving as a control, an annotated Alu exon (chr5:162102554-162102673) at the GABRG2 gene is flanked by the brain specific intron chr5:162101317,162102554 (Supplementary Figure S2C). As expected, in all of these cases the exonized Alu is present uniquely in primates, as shown in the UCSC Genome Browser plots.
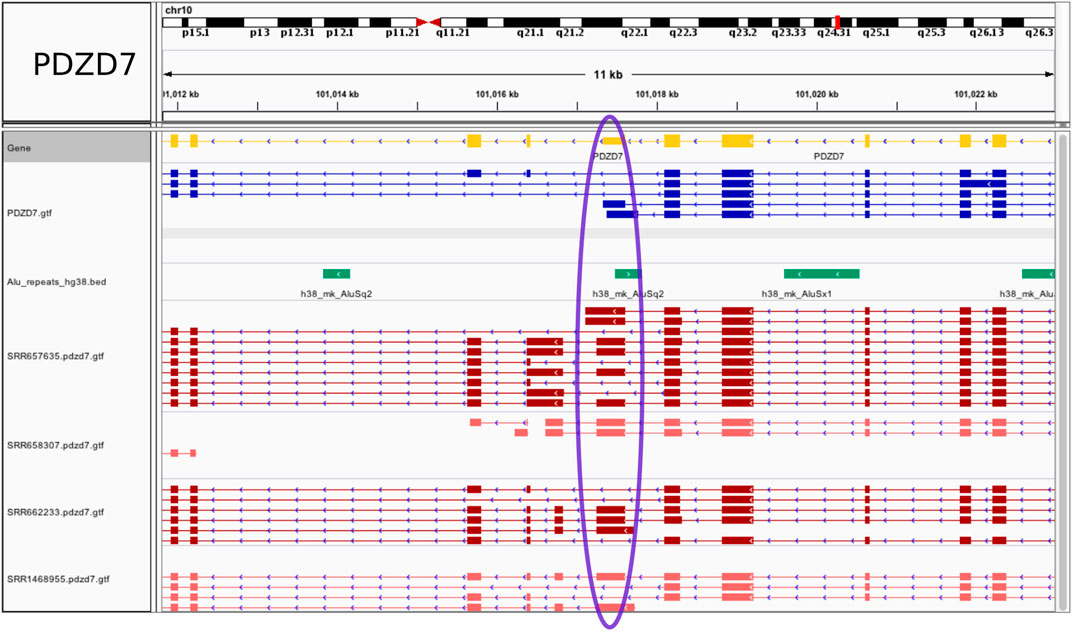
FIGURE 3. Fixed Alu exonization event in human frontal cortex at the PDZD7 gene. A gain of 5’ss at a 3′ UTR terminal exon at the PDZD7 gene creates a 363 bp internal Alu exon. The exon region is marked in blue on the display. The visualization was generated using the Integrated Genomics Viewer.
Prediction of Exons From Novel (Non-reference) Alu Insertions
When the DNA sequence of an Alu insertion variant is not part of the reference genome, standard RNA-seq analysis will discard or misplace informative reads. However, if reads or read pairs exist that span portions of the Alu element and of the adjacent exons, the unique exonic sequences flanking the exonization event can be used to ‘anchor’ the insertion and narrow down candidate reads for assembly.
We developed a pipeline to detect candidate Alu insertion loci in the genes, either within existing exons or as novel exons created by Alu exonizations. Given the interrupted structure of the gene and RNA-seq read alignments, it is not possible to a priori distinguish between these two cases; rather, the length of the predicted insertion interval can be used to infer the type of event from the predictions. To identify candidate insertion loci, we located mapped reads (‘anchors’) whose mates in the read pair contain a portion of an Alu sequence and could not be mapped on the genome (‘floats’) (Figure 4). To reduce the potentially large number of spurious matches, we only considered anchor reads overlapping annotated exons and therefore associated with known genes. A candidate read must then show evidence that it has originated from an exonized Alu sequence (‘signal’ test) and that is not likely to be sourced from a local reference Alu (‘shadow’ test). More specifically, a read is labeled as a ‘signal’ if it matches both a portion of the adjacent exonic sequences and a portion of the consensus Alu sequence. A read is deemed to be a ‘shadow’ if it aligns nearly exactly to the genomic sequence of a known Alu at the locus (see Methods). Further, to allow for small inaccuracies in the classification, the relative proportion of ‘signal’ to ‘shadow’ matches for a candidate insertion locus is used to call a putative novel Alu insertion event. Lastly, anchor reads and their ‘floats’ at that locus are assembled using a transcript assembly algorithm, which allows for multiple assembled sequences, potentially corresponding to different haplotypes. The assembled contigs are finally searched against the reference DNA sequence to eliminate any false positives missed in the previous steps, and to select a high confidence set of inserts for future curation and validation studies. The algorithm and calibration are described in detail in the Methods.
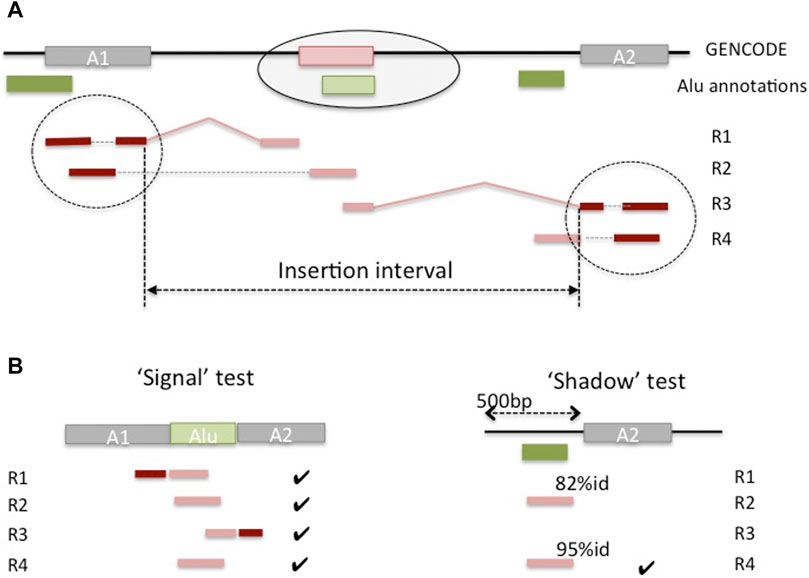
FIGURE 4. Outline of prediction of non-reference Alu insertions in genes. (A) A novel Alu insertion (light green) in a gene creates a novel exon (pink) that is not present in the genome, and for which there is evidence from RNA-seq reads (R1, R2, R3 and R4). Read pairs R1, R4 are all non-concordant and illustrate different scenarios. R1 has the first read mapped to (unique) sequence in anchor exon A1, and its mate is spliced between A1 and the new exon and missed by the aligner; similarly for R3, at the anchor exon A2. R2 has the first read mapping to A1, and its mate maps entirely inside the new exon. R4 shows a case of a ‘shadow’ read, from unprocessed intronic RNA, and was discarded by the aligner because it exceeded the maximum number of hits on the genome (e.g., 10 hits). (B) The ‘signal’ test matches the unmapped Alu-containing mate against the concatenation of the two anchor exons and the consensus Alu sequence. The ‘shadow’ test searches the same mate against the sequence adjacent to the anchor exon (shown), and against the genomic interval spanning the anchor exon and its neighbor (not shown).
Non-Reference Alu Insertion Events in the Human Frontal Cortex
We analyzed the 117 human frontal cortex samples above to determine candidate sites of Alu insertions. We detected putative polymorphic Alu insertions in 1,816 genes (4-387 per sample), of which 1,070 genes were reported to have undergone Alu insertions in multiple samples (Figure 5A). Many of the predicted Alu insertions were detected in repeat-rich regions, which makes them difficult to analyze and curate. Events at 1,353 genes (651 genes reported in multiple samples) occurred in non-repeat rich context (Figure 5B). The assembly process generated 11-1,756 contigs per sample; 17,585 contigs across all samples passed the alignment filter, at 1,363 genes.
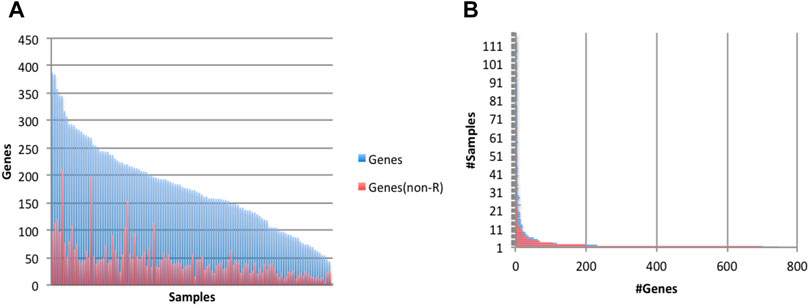
FIGURE 5. Characteristics of predicted (non-reference) Alu insertion events. Events were predicted to occur in 1,816 gene (1,353 genes when considering only events in a non-repeat rich context, ‘non-R’). (A) Numbers of genes (total and ‘non-R’) with putative Alu insertion events, by sample. (B) Prevalence of Alu-acquiring genes in the 117 samples.
We present several examples in Figure 6. For instance, at the Carboxymethylenebutenolidase-Like (Pseudomonas) (CMBL) gene, our algorithm predicts an Alu insertion within the interval chr5:10279825-10282219 in the gene’s 3′ most intron, in one sample only, SRR1432123 (Figure 6A). Of the four contigs assembled, three have high-quality (> 95% sequence identity) matches to the region, covering the full length of the contig sequences. The fourth, a 91 bp contig, has only partial paralogous matches to two separate local AluSz and AluSx1 sequences, at low 88 and 92% identity. Therefore, contig three is a likely novel Alu insertion. In a second example at the NOP2/Sun RNA methyltransferase 5 (NSUN5) gene (Figure 6B), the algorithm predicts an Alu exonization between positions 73,305,031 and 73,307,446 on chromosome 7, in 39 samples. In the sample SRR1435293, the 71 bp contig 3 has a high quality (91% sequence identity) partial match to an anchor exon, covering positions 25-71. The first 32 bases match in reverse orientation to a local intronic Alu, at 88% sequence identity, indicating a paralogous match. Therefore, contig three presents evidence of the insertion sequence spanning the junction between the Alu exon and the downstream anchor exon. Lastly, a novel Alu exonization is predicted at the PQ Loop Repeat Containing one/Solute Carrier Family 66 Member 2 (PQLC1/SLC66A2) gene locus, between 79,934,021 and 79,943,460 on chromosome 18. In sample SRR1474553, bases 1-68 of the 77 bp contig two have a low 87% identity match to the local Alu element which appears paralogous, and therefore is likely to represent a portion of the novel Alu insertion (Figure 6C).
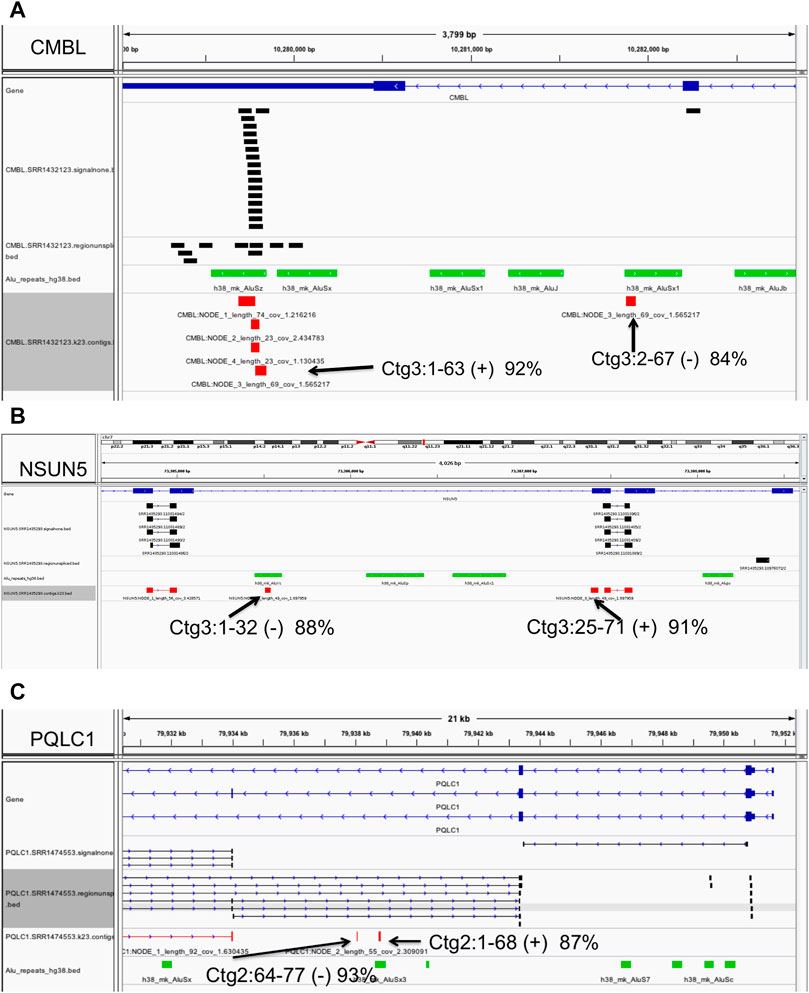
FIGURE 6. Example of polymorphic (non-reference) Alu insertions. Predicted Alu element insertions at the CMBL (A), NSUN5 (B) and PQLC1 (C) genes. Annotations: black - clusters of ‘signalnone’ and ‘regionunspliced’ matches flanking the insertion site (top), green - local Alu elements, and red - alignments of assembled contigs. Full descriptions of the predicted Alu insertion events are presented in the text and Methods. Displays were generated with the Integrative Genomics Viewer.
Methods
Detection of Fixed Alu Exonization
When an exonization event occurs at an Alu annotated in the genome, standard RNA-seq analysis approaches can be used to identify the isoforms containing it. Each of the 117 samples were analyzed as follows. RNA-seq reads were assessed for quality and mapped to the GRCh38 genome (chromosomes 1-22, X, Y and M; https://ensembl.org/Homo_sapiens/) with the program TopHat2 v.2.1.1 (Kim et al., 2013). Reads were then assembled into transcripts with the transcript assembler CLASS2 v.2.1.6 with the sensitive setting, ‘–f 0.01’. With this option, CLASS2 reports all isoforms that are present at 1% or more of the expression level of the most abundant isoform for that gene. CLASS2 with the ‘-f 0.01’ option was previously shown to detect more splicing variation, in particular events at internal exons such as exon skipping, and more accurately than any of the other tools tested (Song et al., 2016). Exons overlapping annotated Alu repeats in the human genome were identified with BEDtools (Quinlan and Hall, 2010). Lastly, candidate Alu exons were selected from among the overlaps with the criteria: i) the exon was internal to the transcript structure, ii) the Alu was in antisense to the gene orientation, and iii) the exon was shorter than 400 bp and longer than 40 bp.
Characterization of Fixed Alu Exonization Events
Alternative Splicing
We used the tool ASprofile (Florea et al., 2010) to determine alternative splicing events, in particular exon skipping, in the 117 samples. ASprofile compares the exon-intron structures of assembled transcripts to detect patterns indicative of alternative splicing events. Read counts for the introns, representing the number of spliced alignments that contain the intron, were calculated with the tool junc included in the CLASS2 package. Percent Splice In (PSI) values for the intron in each sample were then calculated as PSI = (read_count_iLeft + read_count_iRight)/(read_count_iLeft + read_count_iRight+2*read_count_iSpan)), where iLeft and iRight are the two exons flanking the Alu exon in the exon skipping event and iSpan is the exon-skipping intron. Lastly, the median or average of the samples’ PSI values were used in the analysis.
Tissue Specific Introns
To determine tissue specificity, we employed the following procedure. We used the Snaptron database and query tool to extract supporting read counts for all introns in the 19,081 samples from 31 tissues in the GTEx database. We used a combination of two tests, to assess: i) the ‘presence’ of the intron in the tissue, and ii) its ‘absence’ from all other tissues. For the ‘presence’ test, an intron is deemed to be ‘present’ in a given tissue if it is present (≥10 reads) in 15% or more of the samples for that ‘tissue’. For the ‘absence’ test, a 2 × n matrix (n is the number of tissues) is built representing, for each tissue, the number of samples in which the intron has >=10 reads. The first row represents the numbers of samples, for each tissue, from which the intron is expected to be absent, defined as 0.85 * the total number of samples for that tissue in GTEx. The second row contains the observed (O) numbers of samples from which the intron was absent. To perform the test, the matrix is restricted to only those tissues (columns) where O ≥ E, and analyzed with a χ2-square test. This test, therefore, excludes any introns that are ‘absent’ at the margin of statistical error. Lastly, introns that were ‘present’ in exactly one tissue (here, brain) and that passed the statistical ‘absence’ test (p-value≤0.001) are deemed as tissue specific.
Detection of Novel (Non-reference) Alu Exonization Events
Our approach uses non-concordant paired-end RNA-seq reads since the Alu-containing reads will be misaligned in the mapping step. A ‘concordant’ alignment pair is defined as one where the two paired-end reads are mapped to the reference with the correct relative orientations and distance between them. A ‘non-concordant’ read pair is one that is not ‘concordant’, i.e. for which there is no concordant pair of alignments anywhere in the genome. When a polymorphic Alu is not included in the reference genome, the mapped reads containing the unique exonic sequences flanking the exonization event can be used to ‘anchor’ the insertion site. The algorithm follows the following steps: 1) pre-process the data to determine non-concordant reads whose mates match the consensus Alu sequence; 2) ‘anchor’ the reads to annotated exons of known genes; 3) determine candidate reads for Alu insertions, with the ‘signal’ and ‘shadow’ tests; 4) apply context filters to clusters of reads and determine the likely insertion interval; 5) assemble the reads into contigs; and 6) align the contigs to the gene region to filter likely false positives and select a high confidence set for future validation.
We implemented the algorithm into a software called Alubaster, available without charge under the GPL license from http://github.com/splicebox/Alubaster.
Pre-Process the Reads
We classify each read as an Alu read or non-Alu read using the tool Kraken (Wood and Salzberg, 2014), a fast metagenomic classification tool that matches short sequencing reads to a database of sequences based on their k-mer profiles, and a custom database of Alu sequences extracted from the genome-wide Alu element annotations. We map all reads to the human genome GRCh38 with Tophat2 v.2.1.1 and extract all non-concordant read pairs in which one mate (say m2) is identified as an Alu read. The unique sequence of its mate (m1) will be used to ‘anchor’ the insertion event.
Anchor the Reads to Annotated Exons
Determine exons in the GENCODE v.22 annotation overlapping the ‘anchors’, with the program BEDtools (Quinlan and Hall, 2010). Group and then process ‘anchor’ reads by their co-located exons.
Determine Candidate Reads for Alu Insertions
One ‘anchor’ exon at a time, process each Alu-containing mate to determine whether it is a) a likely ‘signal’, and b) a likely ‘shadow’ match. To gauge potential ‘signals’, the reads are searched against the concatenated sequence of the exon, consensus Alu sequence, and the adjacent exon, with a traditional spliced alignment algorithm, sim4db (Walenz and Florea, 2011). The program allows for multiple gaps (insertions, deletions) and substitutions, including longer indels, which may arise when matching the particular instance of the inserted Alu against the consensus sequence, and when the insertion site within the Alu sequence is unknown. As determined by our studies, a match that explains≥80% of the read sequence, has >80% sequence identity, and covers 10 or more Alu bases is deemed a ‘signal’. To determine if the read is likely from a ‘shadow’ local Alu element in the genome, it is first searched against a 500 bp intronic region adjacent to the anchor exon (‘unspliced’ test), to eliminate matches due to unprocessed intronic RNA, and then to the genomic region between and including the anchor exon and the farthest adjacent exon (‘region’ test). A match that covers 80% of the read length at 93% sequence identity or higher is deemed real and determined to be a ‘shadow’. Alu reads that pass the ‘signal’ filter and are not classified as ‘shadow’ are used to initiate candidate insertion sites at the next step.
Apply Context Filters and Infer the Insertion Site
Within each gene, we create a vicinity, or context, around a putative insertion area by clustering overlapping ‘anchor’ reads on the genome. Reads are clustered separately by match orientation and by category, based on the classification of the Alu mate, as follows. Specifically, reads (and clusters) that passed the ‘signal’ test above and were not ‘shadows’ are deemed to be strongly indicative of an insertion and classified as S (‘signal’). Reads that tested as ‘shadows’ regardless of the ‘signal’ test are marked with RU (‘regionunspliced’), and deemed likely false positives. To recruit additional potentially informative reads, a hybrid category SN (‘signalnone’) jointly includes S reads and non-‘shadow’ reads that did not pass the ‘signal’ test. Lastly, we created a category SRU (‘signalregionunspliced’) for reads that passed the ‘signal’ test but also tested as ‘shadows’. For each vicinity, we apply additional context based filtering criteria. Let s, sn, ru and sru be the number of reads in the S, SN, RU and SRU clusters. The following criteria are applied to determine an insertion site: i) s >= MIN_SIGNALS; ii) sn >= MIN_SIGNALNONE; iii) the ratio s/sn>=MIN_S2SN; iv) s/sru >=MIN_S2SRU; and v) s/ru >=MIN_S2RU, where MIN_SIGNALS, MIN_SIGNALNONE, MIN_S2SN, MIN_S2SRU and MIN_S2RU are cutoffs that are optimized in an extensive calibration scheme (see below). Conditions i) - iii) are intended to determine that sufficient reads exist to provide evidence for a candidate insertion site, whereas conditions iv) and v) relax the criterion to exclude a site based on the binary presence/absence of ‘shadow’ reads, instead allowing for a proportion of spurious ‘shadows’. To determine suitable parameter cutoffs, we performed an exhaustive calibration and optimization on a simulated data set, as explained below.
Lastly, candidate insertion intervals are determined for each vicinity that passed the context filter above. For each S cluster in the vicinity, an insertion interval is defined as comprised between the initiating S cluster and the closest cluster in the opposing orientation, or the end of the gene if no such cluster exists.
Assemble the Reads Into Contigs
We assembled all reads (‘anchors’ and their Alu mates) in the clusters bounding the insertion intervals, using the transcript assembler Oases/Velvet (Schulz et al., 2012) with k = 23.
Prioritize Contigs
To further select a high-confidence set of candidate inserts, we align all contigs to the full genomic region for the gene, and select those contigs that do not have a high-quality alignment, defined as >=90% sequence identity and >=80% contig coverage. This subset will serve as a starting point for future curation and validation studies.
Simulation Study and Program Calibration
To calibrate the program and assess its performance, we generated a semi-control data set in which we excised expressed Alu elements from the reference genome, and used our method to infer them. We first used HEK293T cell line expression data to infer expressed Alu elements, as follows. Reads from one HEK293T RNA-seq sample (SRA Accession: SRR1284895) were mapped to the reference genome GRCh38 and assembled into transcript using CLASS2 v.2.1.6. Exons of the assembled transcripts were then intersected with Alu annotations, and 1,000 randomly selected (954 without repetitions) expressed Alu elements were excised from the GRCh38 genome to create a new reference genome, GRCh38sim. We also adjusted the coordinates of the GENCODE v.22 gene annotations accordingly. To measure the performance, we ran our algorithm with the SRR1284895 data on the GRCh38sim genome and predicted insertion sites in genes. Note that the data set may contain other real Alu element insertions characteristic of the HEK293T cell line, which will be counted as false positives. However, the genome and data set present a realistic and suitable scenario for our goals. For evaluation, we employed standard performance measures, sensitivity Sn = TP/(TP + FN) and precision Pr = TP/(TP + FP), and we used the set of genes harboring deleted Alu elements as the gold standard against which to compare the predicted genes.
Calibration
To determine suitable cutoff parameters for the context-based filter, in a comprehensive calibration scheme we varied parameters linearly: MIN_SIGNALS = {1, 2}; MIN_SIGNALNONE = {1, .., 10}; MIN_S2SN = {0, 0.1, .., 0.5}; MIN_S2SRU = {0, 0.5, .., 2}, and MIN_S2RU = {0, 0.05, 0.1, 0.15, 0.2, 0.25, 0.5} (see example in Supplementary Figure S2). The maximum Sn that could be achieved was 0.671 and the maximum Pr was 0.8. The best overall performance based on the F-value = 2*Sn*Pr/(Sn + Pr) was obtained for the parameter combination (2,5,0,2,0.5), namely Sn = 291/954 = 0.305, Pr = 291/522 = 0.557, F-value = 0.394, and accuracy Acc=(Sn + Pr)/2 = 0.431, for a run that reported 522 genes, of which 291 were among those listed, i.e. true positives (TP), and 231 were ‘false positives’ (FP). We used this set of parameters for our analyses.
Discussion and Conclusion
Alu interspersed repeats represent a large portion of human and other primate genomes, and have played an important part in evolution and potentially the acquisition of species and tissue-specific traits. Alu insertions, especially those in gene bodies, have impacted gene structure and function in a variety of ways and at all steps in the regulation of gene expression, including transcription, RNA splicing, RNA editing and translation. Alu exonizations, or the recruitment of intronic Alu sequences into coding regions by activation of Alu-encoded cryptic splice sites, can directly alter gene function and contribute to functional diversification through the formation of alternative gene isoforms. The acquisition of new functions through Alu exonization, however, has been a gradual process. The new Alu is incorporated in a minority of the gene’s transcripts, where it is able to evolve without significantly impacting the gene’s primary function and therefore under reduced selective pressure (Zarnack et al., 2013; Payer and Burns, 2019). Currently, Alu exonizations are estimated to account for ∼5% of alternatively spliced (skipped) exons in the human genome (Sorek et al., 2002). In time, the Alu isoform may evolve entirely new function and even become the major isoform for the gene. Hence, the coupling of Alu exonization with alternative splicing has provided an elegant mechanism to create functional diversity while safeguarding the primary gene function.
As Alu elements continue to insert into the human genome and create structural variants, the process leads to genetic diversity but also potentially to disease. Understanding the prevalence and impact of polymorphic and rare Alu insertion variants on the population and in disease at large scale is an important and as of yet little charted endeavor. In particular, when an Alu insertion interrupts a gene exon or is very close to an annotated exon, its functional consequence for protein coding capacity of the locus or for mRNA splicing are more likely to be recognized. However, currently there are no adequate tools for characterizing the effects of Alu insertions farther from exons in deep intronic space, even when these become incorporated in mRNA transcripts.
Over the past decade, a tremendous volume of publicly available RNA-seq data has been generated and become available, a rich resource that can now be mined to characterize Alu exonization events in different tissues, developmental stages and disease conditions. This study taps this potential, as we developed methods to identify exonizations of Alu elements with or without a priori knowledge of the Alu insertion site in the genome, from RNA-seq data. We applied these tools to analyze 117 human frontal cortex RNA-seq samples obtained from the GTEx repository.
Our study detected exonization events at 870 annotated Alu elements. In many instances, an Alu element was included in multiple Alu exons with varying coordinates, to create 1,019 distinct Alu-containing exons, and for each Alu exon there were potentially multiple flanking introns. Therefore, Alu exonization is strongly coupled with alternative splicing. Only 651 of the 1,019 Alu exons were uncovered as part of an exon skipping event. There are multiple explanations for this relatively small number. First, exon skipping is defined strictly to refer to exons that are included or excluded alone from gene transcripts, with no changes to the surrounding exons. In multiple examples, including at the UBAP1L and AC103855.2 genes (Supplementary Figure S3), however, we observed multiple exons and/or exon segments being skipped in a single complex alternative splicing event that involves multiple exons or portions of exons, which sometimes include novel unannotated exons (see example at the AC103855.2 gene). Further, some isoforms, particularly those with low expression, may have been missed by the assembler or misassembled. Indeed, a majority of the events had low read counts, with 73% having fewer than 10 reads supporting the flanking introns.
Exonized Alu elements belonged to different subfamilies (AluS, AluJ and AluY), suggesting that Alu insertions occurring at different times throughout primate and human evolution share a potential to be exonized. Our analyses based on our data set in a single tissue suggest that expression levels measured in read support for the flanking introns are similarly distributed across the three classes, but AluY exons tend to be included in a lower fraction of a gene’s transcripts compared to AluS and AluJ exons, as measured by the PSI value, and are less likely to be in-frame (24.4% compared to 36.2% and 32.8%) (Supplementary Figure S4 and Supplementary Table S4). Collectively these analyses, albeit limited, are consistent with the hypothesis of Alu exon birth through exonization and alternative splicing, and evolution to function acquisition. Overall, a majority (∼75%) of all skipped Alu exons were present in the minor isoform (PSI<=0.5). When considered alongside the generally low read counts and lack of a preference for in frame exon lengths, these observations suggest that most of the Alu exons discovered may play a niche or relatively low impact role in the gene’s function.
Alu exons are hypothesized to contribute to species, tissue or condition specific function. Our analyses of 2,771 flanking introns identified 1,260 (45.5%) that were novel, not present in the GENCODE v.36 annotation database. Further, 45 introns (29 novel) were determined to be tissue specific using a strict classification criterion, and a large portion (>21) were in non-coding RNA genes.
In contrast to fixed Alu exonizations, which are common in the population and may have been evolutionary selected, polymorphic or rare Alu insertions occur in a small number of samples and are more likely to be present at a low expression level in a carrying individual. These characteristics alone make them difficult to detect from the transcriptomic data. Further, there are significant computational challenges to detecting gene Alu insertions at uncharted non-reference locations and from whole-transcriptome RNA sequencing data, where an Alu read may match to thousands of genomic locations. While paired-end reads can help localize the search, local Alu elements located in the gene’s introns and exons present additional sources for the exon, leading to false positives. Consequently, despite the great interest in their potential to point to the mechanism and cause of uncharacterized genetic diseases, polymorphic Alu insertions giving rise to exonizations may be underreported by our pipeline and even more so in disease databases.
In conclusion, our novel software Alubaster detected putative Alu-containing exons in hundreds of genes expressed in the human cortex, with over half seen in multiple samples. These events can be leveraged on their own, or can be incorporated into gene annotations and other feature (exon, intron) databases that can be queried or included in differential splicing analyses, to allow the discovery of novel markers of disease. While further manual curation and experimental validation will be needed to confirm and characterize each event individually, we believe that our collection of tools, sites and sequences represents a valuable resource that can be employed to understand the characteristics of Alu (m)RNA insertions.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
LP, KB, and LF conceived the project and the experiment design. LF and LP developed the algorithm, and LF, LP, GY, and CA wrote and engineered the software and performed the analyses. LF and KB drafted, and all co-authors read and approved the manuscript.
Funding
This work was supported in part by grants R01-GM124531 and R01-GM129085 from the National Institutes of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
Calculations were performed partly on the Center for Advanced Research Computing at Hopkins (ARCH).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2021.727537/full#supplementary-material
References
Ameur, A., Zaghlool, A., Halvardson, J., Wetterbom, A., Gyllensten, U., Cavelier, L., et al. (2011). Total RNA Sequencing Reveals Nascent Transcription and Widespread Co-transcriptional Splicing in the Human Brain. Nat. Struct. Mol. Biol. 18, 1435–1440. doi:10.1038/nsmb.2143
Attig, J., Ruiz de Los Mozos, I., Haberman, N., Wang, Z., Emmett, W., Zarnack, K., et al. (2016). Splicing Repression Allows the Gradual Emergence of New Alu-Exons in Primate Evolution. Elife 5. doi:10.7554/eLife.19545
Batzer, M. A., and Deininger, P. L. (1991). A Human-specific Subfamily of Alu Sequences. Genomics 9, 481–487. doi:10.1016/0888-7543(91)90414-a
Batzer, M. A., and Deininger, P. L. (2002). Alu Repeats and Human Genomic Diversity. Nat. Rev. Genet. 3, 370–379. doi:10.1038/nrg798
Chen, C., Ara, T., and Gautheret, D. (2009). Using Alu Elements as Polyadenylation Sites: A Case of Retroposon Exaptation. Mol. Biol. Evol. 26, 327–334. doi:10.1093/molbev/msn249
Chen, L.-L., DeCerbo, J. N., and Carmichael, G. G. (2008). Alu Element-Mediated Gene Silencing. EMBO J. 27, 1694–1705. doi:10.1038/emboj.2008.94
Comas, D., Schmid, H., Braeuer, S., Flaiz, C., Busquets, A., Calafell, F., et al. (2004). Alu Insertion Polymorphisms in the Balkans and the Origins of the Aromuns. Ann. Hum. Genet 68, 120–127. doi:10.1046/j.1529-8817.2003.00080.x
Consortium, G. T. (2015). Human Genomics. The Genotype-Tissue Expression (GTEx) Pilot Analysis: Multitissue Gene Regulation in Humans. Science 348, 648–660. doi:10.1126/science.1262110
Deininger, P. (2011). Alu Elements: Know the SINEs. Genome Biol. 12, 236. doi:10.1186/gb-2011-12-12-236
Deininger, P. L., and Batzer, M. A. (1999). Alu Repeats and Human Disease. Mol. Genet. Metab. 67, 183–193. doi:10.1006/mgme.1999.2864
Florea, L., Souvorov, A., Kalbfleisch, T. S., and Salzberg, S. L. (2010). Genome Assembly Has a Major Impact on Gene Content: a Comparison of Annotation in Two Bos taurus Assemblies. PLoS ONE 6, e21400. doi:10.1371/journal.pone.0021400
Hormozdiari, F., Konkel, M. K., Prado-Martinez, J., Chiatante, G., Herraez, I. H., Walker, J. A., et al. (2013). Rates and Patterns of Great Ape Retrotransposition. Proc. Natl. Acad. Sci. 110, 13457–13462. doi:10.1073/pnas.1310914110
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. L. (2013). TopHat2: Accurate Alignment of Transcriptomes in the Presence of Insertions, Deletions and Gene Fusions. Genome Biol. 14, R36. doi:10.1186/gb-2013-14-4-r36
Knebelmann, B., Forestier, L., Drouot, L., Quinones, S., Chuet, C., Benessy, F., et al. (1995). Splice-mediated Insertion of an Alu Sequence in the COL4A3 mRNA Causing Autosomal Recessive Alport Syndrome. Hum. Mol. Genet. 4, 675–679. doi:10.1093/hmg/4.4.675
Lander, E. S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., et al. (2001). Initial Sequencing and Analysis of the Human Genome. Nature 409, 860–921. doi:10.1038/35057062
Lev-Maor, G., Sorek, R., Shomron, N., and Ast, G. (2003). The Birth of an Alternatively Spliced Exon: 3' Splice-Site Selection in Alu Exons. Science 300, 1288–1291. doi:10.1126/science.1082588
Meili, D., Kralovicova, J., Zagalak, J., Bonafé, L., Fiori, L., Blau, N., et al. (2009). Disease-causing Mutations Improving the branch Site and Polypyrimidine Tract: Pseudoexon Activation of LINE-2 and antisenseAlulacking the Poly(T)-tail. Hum. Mutat. 30, 823–831. doi:10.1002/humu.20969
Mitchell, G. A., Labuda, D., Fontaine, G., Saudubray, J. M., Bonnefont, J. P., Lyonnet, S., et al. (1991). Splice-mediated Insertion of an Alu Sequence Inactivates Ornithine delta-aminotransferase: a Role for Alu Elements in Human Mutation. Proc. Natl. Acad. Sci. 88, 815–819. doi:10.1073/pnas.88.3.815
Mola, G., Vela, E., Fernández-Figueras, M. T., Isamat, M., and Muñoz-Mármol, A. M. (2007). Exonization of Alu-Generated Splice Variants in the Survivin Gene of Human and Non-human Primates. J. Mol. Biol. 366, 1055–1063. doi:10.1016/j.jmb.2006.11.089
Oldridge, M., Zackai, E. H., McDonald-McGinn, D. M., Iseki, S., Morriss-Kay, G. M., Twigg, S. R. F., et al. (1999). De Novo Alu-element Insertions in FGFR2 Identify a Distinct Pathological Basis for Apert Syndrome. Am. J. Hum. Genet. 64, 446–461. doi:10.1086/302245
Payer, L. M., and Burns, K. H. (2019). Transposable Elements in Human Genetic Disease. Nat. Rev. Genet. 20, 760–772. doi:10.1038/s41576-019-0165-8
Qian, Y., Kehr, B., and Halldórsson, B. V. (2015). PopAlu: Population-Scale Detection of Alu Polymorphisms. PeerJ 3, e1269. doi:10.7717/peerj.1269
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 26, 841–842. doi:10.1093/bioinformatics/btq033
Ram, O., Schwartz, S., and Ast, G. (2008). Multifactorial Interplay Controls the Splicing Profile of Alu-Derived Exons. Mol. Cell Biol. 28, 3513–3525. doi:10.1128/mcb.02279-07
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative Genomics Viewer. Nat. Biotechnol. 29, 24–26. doi:10.1038/nbt.1754
Schulz, M. H., Zerbino, D. R., Vingron, M., and Birney, E. (2012). Oases: Robust De Novo RNA-Seq Assembly across the Dynamic Range of Expression Levels. Bioinformatics 28, 1086–1092. doi:10.1093/bioinformatics/bts094
Sela, N., Mersch, B., Gal-Mark, N., Lev-Maor, G., Hotz-Wagenblatt, A., and Ast, G. (2007). Comparative Analysis of Transposed Element Insertion within Human and Mouse Genomes Reveals Alu's Unique Role in Shaping the Human Transcriptome. Genome Biol. 8, R127. doi:10.1186/gb-2007-8-6-r127
Shen, S., Lin, L., Cai, J. J., Jiang, P., Kenkel, E. J., Stroik, M. R., et al. (2011). Widespread Establishment and Regulatory Impact of Alu Exons in Human Genes. Proc. Natl. Acad. Sci. 108, 2837–2842. doi:10.1073/pnas.1012834108
Song, L., Sabunciyan, S., and Florea, L. (2016). CLASS2: Accurate and Efficient Splice Variant Annotation from RNA-Seq Reads. Nucleic Acids Res. 44, e98. doi:10.1093/nar/gkw158
Sorek, R., Ast, G., and Graur, D. (2002). Alu-containing Exons Are Alternatively Spliced. Genome Res. 12, 1060–1067. doi:10.1101/gr.229302
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., et al. (2001). The Sequence of the Human Genome. Science 291, 1304–1351. doi:10.1126/science.1058040
Vorechovsky, I. (2010). Transposable Elements in Disease-Associated Cryptic Exons. Hum. Genet. 127, 135–154. doi:10.1007/s00439-009-0752-4
Walenz, B., and Florea, L. (2011). Sim4db and Leaff: Utilities for Fast Batch Spliced Alignment and Sequence Indexing. Bioinformatics 27, 1869–1870. doi:10.1093/bioinformatics/btr285
Wang, E. T., Sandberg, R., Luo, S., Khrebtukova, I., Zhang, L., Mayr, C., et al. (2008). Alternative Isoform Regulation in Human Tissue Transcriptomes. Nature 456, 470–476. doi:10.1038/nature07509
Wilks, C., Gaddipati, P., Nellore, A., and Langmead, B. (2018). Snaptron: Querying Splicing Patterns across Tens of Thousands of RNA-Seq Samples. Bioinformatics 34, 114–116. doi:10.1093/bioinformatics/btx547
Wood, D. E., and Salzberg, S. L. (2014). Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 15, R46. doi:10.1186/gb-2014-15-3-r46
Xing, J., Zhang, Y., Han, K., Salem, A. H., Sen, S. K., Huff, C. D., et al. (2009). Mobile Elements Create Structural Variation: Analysis of a Complete Human Genome. Genome Res. 19, 1516–1526. doi:10.1101/gr.091827.109
Keywords: Alu exonization, alternative splicing, RNA sequencing, computational prediction, frontal cortex
Citation: Florea L, Payer L, Antonescu C, Yang G and Burns K (2021) Detection of Alu Exonization Events in Human Frontal Cortex From RNA-Seq Data. Front. Mol. Biosci. 8:727537. doi: 10.3389/fmolb.2021.727537
Received: 18 June 2021; Accepted: 30 August 2021;
Published: 10 September 2021.
Edited by:
Abdullah Kahraman, University Hospital Zürich, SwitzerlandReviewed by:
Alexander Zelikovsky, Georgia State University, United StatesSarath Chandra Janga, Indiana University, Purdue University Indianapolis, United States
Copyright © 2021 Florea, Payer, Antonescu, Yang and Burns. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Liliana Florea, ZmxvcmVhQGpodS5lZHU=