 Jinhang Zhang1
Jinhang Zhang1 Qinhui Liu
Qinhui Liu Jinhan He
Jinhan He Yanping Li
Yanping Li- 1Laboratory of Clinical Pharmacy and Adverse Drug Reaction, National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University, Sichuan, China
- 2Department of Pharmacy, State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Sichuan, China
Liver fibrosis is end-stage liver disease that can be rescued. If irritation continues due to viral infection, schistosomiasis and alcoholism, liver fibrosis can progress to liver cirrhosis and even cancer. The US Food and Drug Administration has not approved any drugs that act directly against liver fibrosis. The only treatments currently available are drugs that eliminate pathogenic factors, which show poor efficacy; and liver transplantation, which is expensive. This highlights the importance of clarifying the mechanism of liver fibrosis and searching for new treatments against it. This review summarizes how parenchymal, nonparenchymal cells, inflammatory cells and various processes (liver fibrosis, hepatic stellate cell activation, cell death and proliferation, deposition of extracellular matrix, cell metabolism, inflammation and epigenetics) contribute to liver fibrosis. We highlight discoveries of novel therapeutic targets, which may provide new insights into potential treatments for liver fibrosis.
Introduction
Liver Fibrosis
Liver fibrosis is a repair response to chronic liver injury caused by various pathogenic factors, and it is characterized mainly by the excessive accumulation of extracellular matrix (ECM), especially collagen fibers (Reeves and Friedman, 2002). If the pathogenic factor is not removed, liver fibrosis can progress to liver cirrhosis and even hepatocellular carcinoma, which elevates risk of mortality.
Liver fibrosis has become one of the most common liver diseases worldwide, and it has also become one of the leading indications for liver transplantation. The global prevalence of nonalcoholic fatty liver disease (NAFLD) is 25.24%, and its prevalence is particularly high in the Middle East, South America and Asia. Just over half of patients (59.1%) with NAFLD, and in particular 40.76% of patients with liver fibrosis, progress to nonalcoholic steatohepatitis (NASH) (Younossi et al., 2016). Liver disease accounts for approximately 2 million deaths per year worldwide, among which 1 million deaths occur due to complications from cirrhosis, which is currently the 11th most common cause of death globally (Asrani et al., 2019).
Alcohol abuse, chronic viral hepatitis, obesity, autoimmune hepatitis, metabolic syndrome and cholestasis are the most common causes of liver fibrosis (Bataller and Brenner, 2005). If the pathogenic factors are acute or self-limiting, wound-healing responses are transient, and the liver architecture can return to normal. When the factors persist, the inflammatory phase begins and hepatic stellate cells (HSCs) activate, leading to ECM deposition and destruction of the liver architecture. The pathogenesis of liver fibrosis is complicated and involves multiple types of liver cells and inflammatory reactions. Its main pathological features are collagen deposition and damage of liver structure. Numerous studies of anti-fibrosis targets have focused mainly on collagen deposition and various types of hepatic cells.
Hepatic Cells in Liver Fibrosis
Liver fibrosis is a complex process of liver self-repair that involves multiple types of hepatic cells. Intercellular crosstalk within the liver microenvironment is critical for the maintenance of normal hepatic functions and cell survival (Marrone et al., 2016). The chronic presence of external pathological factors can injury hepatocytes; activate inflammatory cells such as macrophages; promote infiltration of lymphocytes; trigger proliferation of sinusoidal endothelial cells and capillarization of sinusoidal endothelial cells, blocking perfusion between blood and liver cells and causing abdominal aortic hypertension; and activate bile duct cells (Boyer-Diaz et al., 2021). These changes ultimately lead to the activation of HSCs, the most important source of myofibroblasts, to synthesize excess ECM, resulting in liver fibrosis. HSC activation is considered central in liver fibrosis. However, other types of liver cells also play important roles in fibrosis. Indeed, HSCs activation depends on the interaction with other hepatic cells, including hepatocytes, liver sinusoidal endothelial cells, inflammatory cells and biliary cells (Figure 1). These cells interact with each other and promote or inhibit the activation of HSC through production of cytokines and other signalling molecules. Targeting the crosstalk between HSCs and other hepatic cells might be a novel option for liver fibrosis treatment.
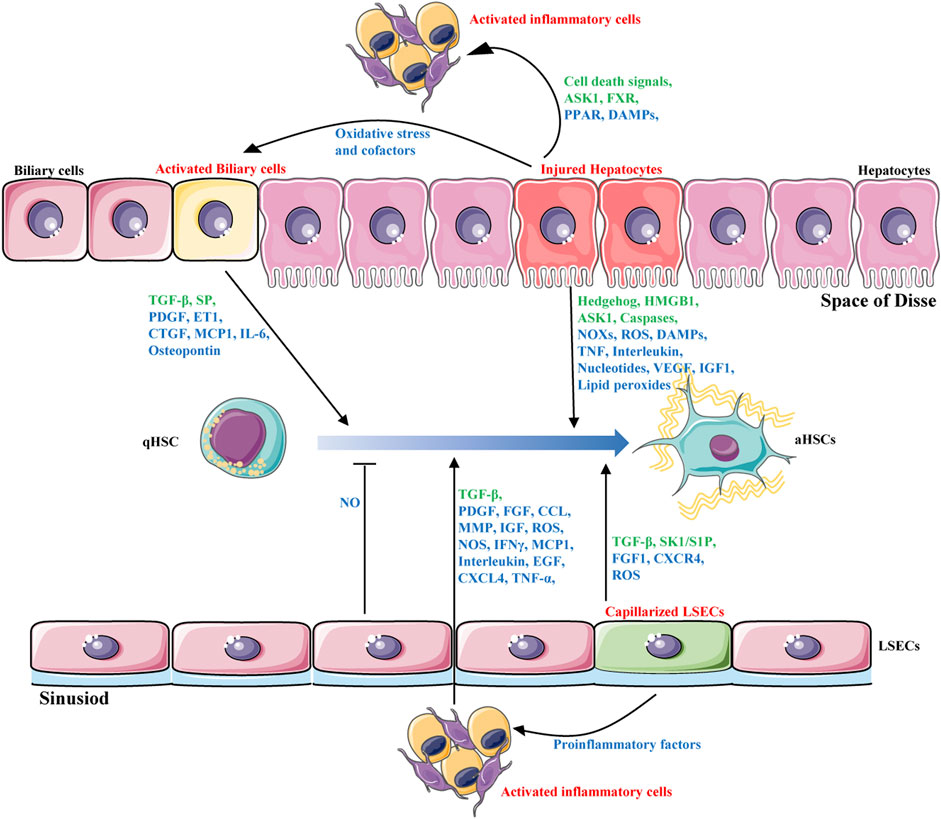
FIGURE 1. Intercellular crosstalk during liver fibrosis. HSCs activation is the major driver of liver fibrosis that depends on the interaction with other hepatic cells, including hepatocytes, biliary cells, liver sinusoidal endothelial cells and inflammatory cells. These cells interact with each other and promote or inhibit the activation of HSCs through production of hormones, cytokines(blue) and other signalling molecules(green).
Hepatic Stellate Cells
Activation of HSCs, often referred to as their “trans-differentiation”, is the major cellular source of matrix protein-secreting myofibroblasts, which are the major driver of liver fibrogenesis (Higashi et al., 2017; Tsuchida and Friedman, 2017; Cai et al., 2020). HSCs, which are also called vitamin A-storing cells, lipocytes, interstitial cells, fat-storing cells or Ito cells, exist in the space between parenchymal cells and liver sinusoidal endothelial cells of the hepatic lobule. They store 50–80% of the total vitamin A in the body; they store the vitamin A in the form of retinyl palmitate in lipid droplets in the cytoplasm (Senoo et al., 2010). HSCs in the space of Disse are also thought to contribute reversibly to portal hypertension (McConnell and Iwakiri, 2018).
Stimulating HSCs with external factors such as lipid peroxides or pro-fibrotic cytokines leads them to lose lipid droplets, proliferate, and transform into myofibroblasts. The cells then begin to produce ECM and they acquire contractile, pro-inflammatory, and fibrogenic properties (Bataller and Brenner, 2005). The disordered accumulation of ECM results in scar and liver fibrosis (Gandhi, 2017). Many current anti-fibrosis treatments aim to prevent HSCs from contributing to fibrosis, such as by blocking their activation by external factors, inhibiting their proliferation, promoting their apoptosis (Trivedi et al., 2021), and preventing their adoption of a high metabolic state (Du et al., 2018; Khomich et al., 2019).
The major signaling pathways involved in HSCs activation contains: growth factors and ligand-receptor signaling pathways (Wnt/β-catenin, Hedgehog, YAP/TAZ, FGF, cAMP-PKA-CREB), profibrogenic response pathways (TGF-β, PDGF/VEGF/CTGF, ROCKs, Axl/Gas6, Notch, renin angiotensin system), cell death signaling (autophagy, ER stress, oxidative stress), immune-related signaling (TLRs, LPS, DAMPs, interleukin), metabolic regulated pathways (Acc, Hedgehog, YAP, leptin), Nuclear receptors (FXR, LXR, PPARs, VDR) and epigenetic changes (miRNA, lncRNA, DNA methylation, histone modification) (Figure 2). Although some clinical drugs were found to be positive for liver fibrosis treatment in clinical trails including PPAR-γ agonist (pioglitazone), angiotensin receptor blockers (losartan, telmisartan, olmesartan and candesartan), and glucagon-like peptide-1 receptor agonists (liraglutide), the safety and effect of these drugs need to be further confirmed (Georgescu, 2008; Mantovani et al., 2021). Clinical existing drugs like angiotensin receptor blockers, PPAR-γ agonist may be accompanied by side effects due to their wide range of effects, and they may not be suitable for the treatment of simple liver fibrosis. It is necessary to find new therapeutic targets.
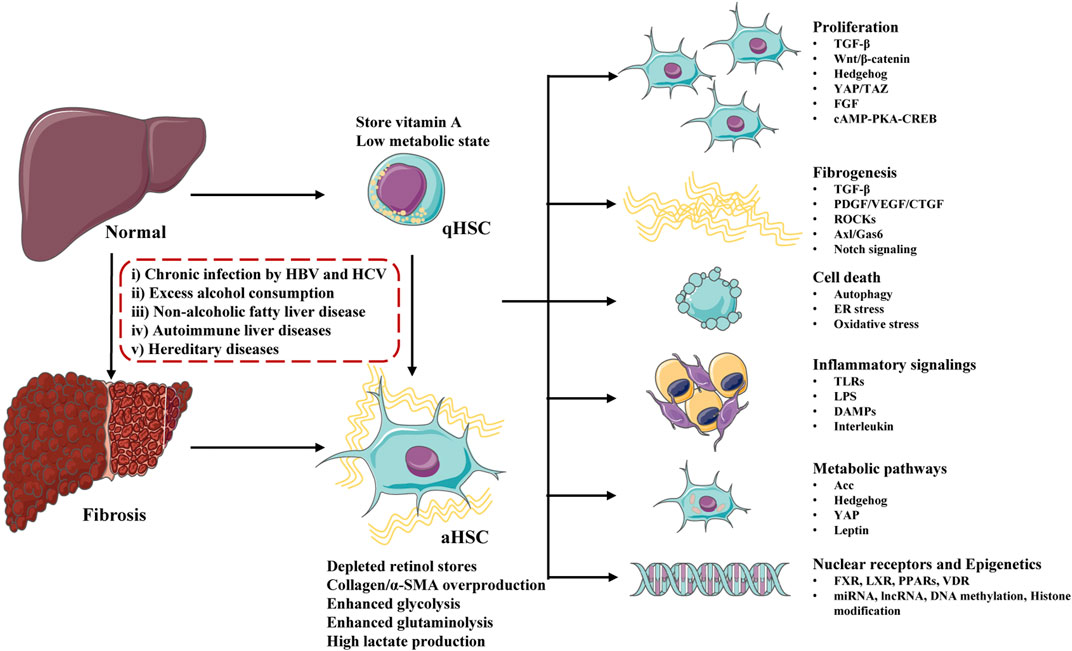
FIGURE 2. Major molecules and signaling pathways regulating hepatic stellate cells (HSCs) activation and liver fibrosis. Undergo physiological conditions, HSCs exist as a quiescent phenotype (qHSCs), which store vitamin A and stay in a low metabolic state. After stimulating by a serious of factors (including chronic infection, excess alcohol consumption, non-alcoholic fatty liver disease, autoimmune liver diseases and hereditary diseases), qHSCs activate into activated HSCs (aHSCs) and liver fibrosis develop. Activated HSCs lose stored retinol, produce excessive collagen/α-SMA and stay at a high metabolic state. HSCs activation is regulated by a number of signaling pathways and molecules, including proliferation, fibrogenesis, cell death, inflammatory signalings, metabolic pathways, nuclear receptors and epigenetics.
Myofibroblasts, which are not present in the healthy liver, are activated in response to liver injury (Friedman, 2008; Kisseleva and Brenner, 2021). They form from resident mesenchymal cells, epithelial cells (e.g. hepatocytes and cholangiocytes), endothelial cells, bone marrow stem cells, portal fibroblasts and HSCs (Wynn, 2008). The most important characteristic of liver fibrosis is the excessive deposition of ECM, in which myofibroblasts play the most important role (Karsdal et al., 2016). This makes ECM production a primary target for anti-fibrotic therapy. Inflammation often accompanied liver injury (Koyama and Brenner, 2017), which involves the production and release of such cytokines as CTGF, PDGF, and TGF-β. These cytokines activate myofibroblasts to produce abundant ECM (Wynn, 2008), inducing liver fibrosis.
Hepatocytes
Hepatocytes are the most important parenchymal cells in liver; they account for more than 80% of all liver cells. Together with cholangiocytes, hepatocytes help maintain liver homeostasis (Dai et al., 2020). Damage to hepatocytes, together with subsequent inflammatory and fibrogenic signaling cascades, is thought to trigger fibrosis (Tu et al., 2017). After damage by microenvironmental factors, hepatocytes secrete pro-inflammatory and pro-fibrotic factors, activating inflammatory cells and HSCs, in turn promoting fibrosis (Wang et al., 2016a; Zhang et al., 2019; An et al., 2020). The epithelial‐mesenchymal transition (EMT) in hepatocytes promotes the progression of liver fibrosis. Fructose induces hepatocytes to upregulate fibroblast‐specific protein1 and vimentin, while downregulating E‐cadherin, thereby promoting EMT (Cicchini et al., 2015; Song et al., 2019).
In early-stage liver disease, if liver cell damage can be reversed, then hepatocytes can be promoted and liver fibrosis reversed. This is the aim of hepatoprotective drugs currently used in the treatment of liver diseases. In late-stage liver disease, in contrast, it is often impossible to inhibit the death of hepatocytes. This may be because HSCs are in a highly activated state, and they can secrete pro-fibrotic factors. Drugs that fail to target liver cells selectively and that instead are taken up by HSCs can promote HSC proliferation and inhibit their apoptosis. The problem of this “two-way” action must be taken into account when designing treatments that promote apoptosis or inhibit proliferation.
Liver Endothelial Cells
LSECs act as permeable barriers and portal pressure regulators, they mediate the transcript of nutrients, they recruit lymphocytes from the blood, and they secrete cytokines and growth factors from their sinusoidal side (Asahara et al., 1999). LSECs have the highest endocytotic capacity of all human cells (Poisson et al., 2017). They also interact with HSCs and hepatocytes, and they are critical to maintain HSC quiescence and regenerate hepatocytes (Hu et al., 2014), thus inhibiting intrahepatic vasoconstriction and fibrosis. LSECs maintain HSC quiescence via a pathway that is stimulated by vascular endothelial growth factor (VEGF) and that depends on nitrous oxide (NO) (Asahara et al., 1999; Marrone et al., 2013; DeLeve, 2015). In chronic liver injury, LSECs undergo capillarization, they downregulate eNOS and NO synthesis, and they secrete profibrogenic and proinflammatory cytokines such as TGF-β1, PDGF, TNF-α and IL-6, thereby promoting liver fibrosis (Lafoz et al., 2020).
Cholangiocytes
Cholangiocytes are epithelial cells lining the intra- and extra-hepatic bile ducts; they are heterogeneous in size and function and mediate solute transport processes that determine the composition and flow of bile (Banales et al., 2019). Their dysfunction lies at the heart of cholangiopathies. During biliary disease, various pathological stimuli such as gastrointestinal hormones, bile acids, angiogenic factors, and nerve growth factor can activate cholangiocytes, leading to biliary proliferation, known as a ductular reaction. The result is an epigenetically-regulated transcriptional program involving secretion of TGF-β1, CTGF, p16, CCL2 and SA-β-gal, ultimately leading to a profibrogenic micro-environment, HSC activation, and enhanced liver fibrosis (Zhou et al., 2018; Elssner et al., 2019; Jalan-Sakrikar et al., 2019). The ductular reaction contributes to the initiation and progression of liver fibrosis (Glaser et al., 2009).
Many recent studies have explored the role of cholangiocytes in liver fibrosis. For example, non-canonical NF-κB can contribute to cholangiocyte proliferation and the ductular reaction, accelerating liver fibrosis (Elssner et al., 2019). The long non-coding RNA H19, present in exosomes from cholangiocytes, can activate HSCs and promote cholestatic liver fibrosis (Liu et al., 2019a). Knockout of the secretin receptor reduces biliary damage and liver fibrosis by slowing cholangiocyte senescence (Zhou et al., 2018). These findings suggest that targeting the activation of cholangiocytes and the ductular reaction can mitigate biliary fibrosis.
Inflammatory Cells
The acute inflammation that arises in response to liver injury is thought to help mitigate infection and promote liver repair and regeneration (Karin and Clevers, 2016). Chronic inflammation, in contrast, is detrimental and contributes to liver fibrosis through the involvement of multiple types of inflammatory cells, including Kupffer cells, recruited macrophages, neutrophils, Th17 cells and Tregs (Berumen et al., 2021; Wen et al., 2021). Macrophages play a dual role in the progress of fibrosis. M1 macrophages produce inflammatory cytokines, while M2 macrophages regulate inflammatory responses and secret matrix metalloproteases (MMPs), the main enzymes that degrade ECM, thereby reversing fibrosis (Pradere et al., 2013; Luo et al., 2019). Thus, the balance between M1 and M2 macrophages influences whether fibrosis progresses or not (Sica et al., 2014). Interestingly, recent studies have suggested that M1, but not M2 macrophages, inhibit liver fibrogenesis by recruiting endogenous macrophages and “polarizing” them into a restorative Ly6Clo phenotype, which secrets high levels of MMPs for collagen degradation, as well as high levels of hepatocyte growth factor for hepatocyte proliferation (Ramachandran et al., 2012; Ma et al., 2017).
Kupffer cells, the resident macrophages of the liver, play a central role in liver inflammation. They are resident macrophages that localize within the lumen of the liver sinusoids, and they account for about 30% of sinusoidal cells (Bouwens et al., 1986; Koyama and Brenner, 2017). In response to hepatocyte injury, Kupffer cells become active and secret pro-inflammatory and pro-fibrosis factors. TGF-β, which is secreted mainly by Kupffer cells and plays a key role in liver fibrosis (Xu et al., 2020), binds to a receptor on HSCs to activate them and induce production of collagen (Wang et al., 2019a). Hepatic macrophages also enhance liver fibrosis through the release of IL-1β, TNF-α, CCL2 and PDGF. During liver steatosis, neutrophils and Kupffer cells release reactive oxygen species (ROS), promoting HSC activation and liver fibrosis (Jiang et al., 2012; Dat et al., 2021). Th17 cells produce IL-17, which activates Kupffer cells and express the proinflammatory cytokines IL-6, IL-1β and TNF-α. IL-17 also directly activates HSCs and promotes collagen production via the STAT3 pathway (Meng et al., 2012). Th22 cells, for their part, produce IL-22, which drives TGF-β-dependent liver fibrosis (Fabre et al., 2018).
In this way, many types of inflammatory cells and complex molecular pathways are involved in liver fibrosis. Future studies aiming to treat liver fibrosis by targeting inflammatory cells should cautiously consider the potentially complex effects of such treatment.
New Therapeutic Targets
Liver fibrosis can be reversed in early stages if the pathological insult can be removed. Here we summarize recent reports on signaling pathways that contribute to liver fibrosis and on efforts to target such pathways as a therapeutic strategy.
TGF-β Signaling
TGF-β signaling is a core regulator of fibrosis, and it can induce fibrosis via canonical and non-canonical (non-Smad) pathways (Figure 3) (Finnson et al., 2020). In both cases, myofibroblasts are activated, leading to excessive ECM production and inhibition of ECM degradation (Meng et al., 2016). TGF-β binds to its cognate receptor TGF-β type II receptor, inducing the nuclear translocation of Smad2 and Smad3, which regulate the transcription of fibrotic target genes (Zhang et al., 2021a).
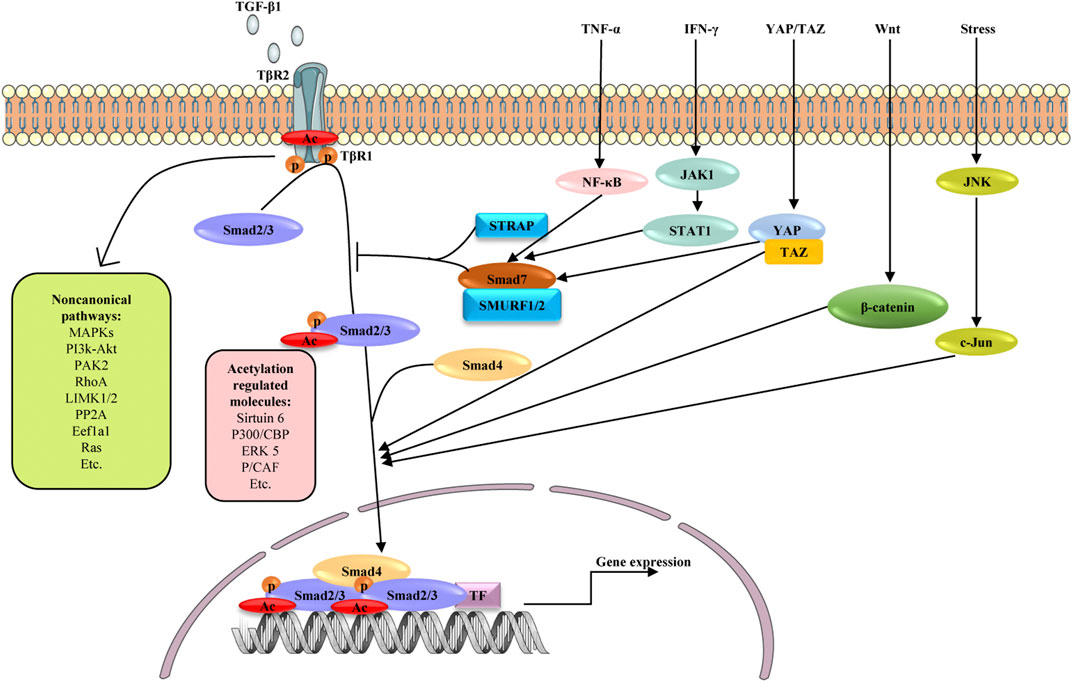
FIGURE 3. Schematic diagram depicting possible mechanisms involved in the fibrogenesis of TGF-β signaling and its crosstalk with other signaling pathways. Signaling starts with TGF-β binding to TGFβR2 (TβR2), which activates TGFβR1 (TβR1). The active TβR1 phosphorylates and acetylates Smad2/3, which complex with Smad4, translocate into nucleus and promote profibrotic genes expression. As another active form of Smad2/3, acetylated Smad2/3 are reported to be regulated by Sirtuin6, P300/CBP, ERK5 and P/CAF. Smad7 negatively regulates TGF-β signaling through competing with Smad2/3 for interaction with TβR1 in the presence of STRAP and SMURF1/2. TGF-β can also activate non-canonical TGF-β pathways, including MAPK, PI3K/AKT, PAK2, RhoA, LIMK1/2, pP2A, Eefla1and Ras pathways. In addition to canonical and non-canonical pathways, TGF-β/Smad signaling shares crosstalk with other signaling pathways in liver fibrosis, including TNF-α, IFN-γ, YAP/TAZ, Wnt and JNK signaling. These pathways influence TGF-β signaling by affecting the activation and nuclear translocation of Smad2/3. TF: transcription factors, p: phosphate group, Ac: acetylate group.
In the canonical pathway, Smad2/3 is activated by phosphorylation but potentially also by lysine acetylation to promote liver fibrosis (Bugyei-Twum et al., 2018; Wang et al., 2019a; Zhong et al., 2020; Zhang et al., 2021a). The chromatin deacylase Sirtuin 6 is also an important regulator of liver fibrosis through its influence on metabolism, DNA repair, gene expression, and mitochondrial biology (Andrew et al., 2020). Sirtuin6 deficiency induces aging-dependent fibrosis in liver and other organs in mice. Sirtuin6 may deacetylate Smad3 as well as Lys-9 and Lys-56 in histone 3 to repress the expression of key TGF-β signaling genes (Maity et al., 2019). By deacetylating lysines 333 and 378 of Smad3, sirtuin6 may inhibit Smad3 activity, protecting against liver fibrosis (Zhong et al., 2020). Like Smad3, Smad2 is also a major acetylated substrate of sirtuin6. Sirtuin6 deacetylates lysine 54 on Smad2, reducing TGF-β/Smad2 signaling in HSCs and thereby alleviating liver fibrosis. By deacetylating Smad2, sirtuin6 influences its phosphorylation and nuclear translocation (Zhang et al., 2021a). These findings suggest that TGF-β signaling is a master regulator of fibrosis and warrants multilayer control, and that sirtuin6 may regulate TGF-β signaling at multiple levels.
TGF-β also regulates other signaling pathways through non-Smad signaling pathways, such as pathways involving Wnt/β-catenin, MAPK, mTOR, IKK, PI3K/Akt, and Rho GTPase, thereby contributing to liver fibrosis (Zhang, 2017; Mi et al., 2019). TGF-β-mediated upregulation of FoxO3a and the DNA demethylase TET3 in HSCs facilitates hepatic fibrogenesis (Xu et al., 2020; Kim et al., 2021). TGF-β can also regulate proteasomal degradation of EZH2 in cholangiocytes, supporting biliary fibrosis (Jalan-Sakrikar et al., 2019). TGF-β upregulates hyaluronan (HA) synthase 2, leading to increased production of HA, a major extracellular matrix glycosaminoglycan and biomarker for cirrhosis. HA promotes the fibrogenic, proliferative, and invasive properties of HSCs via pathways involving the receptors CD44, Toll-like receptor 4 (TLR4), and Notch1 (Yang et al., 2019). TGF-β1 activates the p65/MAT2A pathway to decrease levels of S-adenosylmethionine, thereby facilitating liver fibrosis (Wang et al., 2019b).
Several pathways inhibit TGF-β signaling, making them interesting as therapeutic strategies. Transcriptional intermediary factor 1γ, a negative regulator of the TGF-β pathway, interacts with Smad2/3 and binds to the promoter of the α-smooth muscle gene (α-SMA), downregulating α-SMA and activating HSCs (Lee et al., 2020). ECM1 interacts with αv integrins to keep TGF-β in an inactive form, thereby preventing HSC activation and liver fibrosis (Fan et al., 2019). Recent studies have explored epigenetic regulation of Smad- and non-Smad-mediated pathways in TGF-β signaling, highlighting the complex role of such signaling in fibrosis.
Notch Signaling
Notch signaling is a conversed intercellular signaling pathway that regulates interactions between physically adjacent cells. Accumulating evidence suggests that Notch signaling participates in liver fibrosis by mediating myofibroblasts trans-differentiation and the EMT (Hu and Phan, 2016). When any one of five ligands (Delta-like1/3/4, Jagged-1/2) binds to the receptor for Notch1-4, the Notch intracellular domain (NICD) is released and translocates to the nucleus, where it binds to transcription factor CBF1/Suppressor of hairless/Lag1 (CSL) and modulates gene expression (Kovall and Blacklow, 2010). Notch activity in hepatocytes correlates with disease severity and treatment response in patients with NASH, and Notch is upregulated in a mouse model of diet-induced NASH and liver fibrosis. Forced activation of Notch in hepatocytes induces fibrosis by upregulating Sox9-dependent Osteopontin (Opn) secretion from hepatocytes, which activates resident HSCs (Zhu et al., 2018). Endothelial Notch1 overexpression results in LSEC dedifferentiation and accelerates liver fibrogenesis through eNOS-sGC signaling, and it alters the angiocrine profile of LSECs to compromise hepatocyte proliferation and liver regeneration (Duan et al., 2018). DLL4, a ligand of Notch signaling, is up-regulated in the LSECs of fibrotic liver of patients and of mice treated with CCl4, consistent with LSEC capillarization involving endothelin-1 (Chen et al., 2019a). Notch signaling is an attractive target for treating liver fibrosis; so far, Wnt/β-catenin signaling, miR-30c, liver fibrosis-associated lncRNA1 have been found to influence such signaling (Zhang et al., 2017; So et al., 2018; Gu et al., 2021). Notch signaling can also cross-talk with other signaling pathways involving TGF-β and Hedgehog, which together can regulate liver fibrosis (Xie et al., 2013; Wang et al., 2017; Fan et al., 2020).
Some compounds attenuate liver fibrosis by targeting Notch signaling and so may be novel potential therapeutic candidates for the treatment of liver fibrosis. For example, capsaicin shows liver fibrosis progression by regulating Notch signaling to reduce secretion of inflammatory cytokine TNF-α, which attenuates myofibroblast regeneration and fibrosis mediated by HSCs (Sheng et al., 2020). The natural sesquiterpene costunolide exerts potent antifibrotic effects by disrupting the WWP2/PPM1G complex, promoting Notch3 degradation and inhibiting the Notch3/HES1 pathway (Ge et al., 2020). The Notch inhibitor niclosamide exerts hepatoprotective effects against BDL-induced liver fibrosis (Esmail et al., 2021). Dibenzazepine, a bioavailable γ-secretase inhibitor and Notch antagonist, prevents activation of Notch receptors and is already in clinical trials as an anticancer treatment (Takebe et al., 2014). A nanoparticle system has been developed to deliver dibenzazepine to the liver for treatment of liver fibrosis and obesity-induced type 2 diabetes mellitus (Richter et al., 2020).
Wnt Signaling
During liver fibrosis, canonical(β-catenin-dependent) and non-canonical (β-catenin-independent) pathways of Wnt signaling are activated and some proteins in the pathways are upregulated (Wang et al., 2018; Hu et al., 2020; Yu et al., 2020). In β-catenin-dependent pathways, Wnt ligation to cell surface receptors induces downstream phosphorylation and stabilization of β-catenin, which then translocates to the nucleus, where it acts together with p300 or CBP as a transcriptional co-activator of the T cell factor/lymphoid enhancer-binding factor (TCF/LEF) promoter (Miao et al., 2013; Lien and Fuchs, 2014; Nusse and Clevers, 2017). Non-canonical pathways comprise the β-catenin-independent planar cell polarity pathway and the non-canonical Wnt/Ca2+ pathways (De, 2011). Better understanding of Wnt signaling may provide novel insights into the pathophysiology of liver fibrosis.
Wnt also interacts with other pathways to influence liver fibrosis. For example, it blocks the phosphorylation of Smad3 and ERK to inhibit TGF-β1-induced trans-differentiation of fibroblasts into myofibroblasts (Liu et al., 2020a). The Wnt/β-catenin pathway may interact with the Smo-independent Gli1 pathway to promote HSC contraction via TCF4-dependent transrepression of Sufu (Zhang et al., 2021b). The “protein regulator of cytokinesis 1” (PRC1), which regulates the Wnt/β-catenin signaling pathway, may induce Gli1-dependent osteopontin expression to contribute to liver fibrosis (Rao et al., 2019). Wnt signaling may play a dual role in liver repair and liver ECM deposition: it promotes liver fibrosis in the BDL mouse model of liver fibrosis, but it protects the liver in the MDR2 KO mouse model of cholestatic liver disease (Jarman and Boulter, 2020). Thus, efforts to target the Wnt signaling to alleviate liver fibrosis should consider how to reduce scarring without affecting repair.
Numerous molecules have been identified that can inhibit Wnt signaling, such as antagonists, short interfering RNA (siRNA), soluble receptors, and the transcription inhibitors DKK1, ICG-001, PRI-724, and honokiol (Miao et al., 2013; Akcora et al., 2018; Nishikawa et al., 2018; Hu et al., 2020; Lee et al., 2021). These molecules may be candidate drugs for fibrosis treatment.
YAP/TAZ Signaling
YAP/TAZ, a downstream effector of the alternative Wnt signaling pathway, is involved in liver fibrosis (Park et al., 2015). The YAP/TAZ-TEAD transcriptional complex plays an important role in the activity of the Hippo pathway (Bai et al., 2012; Crawford et al., 2018). YAP is activated in HSCs in patients with fibrotic livers, and inhibiting YAP using verteporfin impedes fibrogenesis in CCl4 mice (Mannaerts et al., 2015). Thus, inhibition of YAP may be a novel approach for treating fibrosis. Blockade of YAP reduces HSC activation and proliferation, while promoting their apoptosis. Loss of YAP also inhibits Wnt/β-catenin activity (Yu et al., 2019). Dynein-mediated interaction between YAP and acetylated microtubules may drive nuclear localization of YAP in the soft matrix, increasing TGF-β1-induced transcriptional activity of Smad for myofibroblast differentiation (You et al., 2020). Interestingly, activation of YAP attenuates hepatic damage and fibrosis in studies of liver ischemia-reperfusion injury, which may reflect the complex role of YAP in liver repair and fibrosis through processes such as Wnt signaling (Konishi et al., 2018; Liu et al., 2019b). The expression of YAP and TAZ in HSCs as well as hepatocytes promotes parenchymal inflammation and fibrosis (Mooring et al., 2020).
Verteporfin is the most commonly used small molecule inhibitor of YAP. Many other molecules alleviate fibrosis via inhibiting YAP/TAZ signaling, including magnesium isoglycyrrhizinate, acid ceramidease, dopamine receptor D1 agonist, and liquiritigenin (Li et al., 2018; Haak et al., 2019; Lee et al., 2019; Alsamman et al., 2020). Given its complex role in liver regeneration and HSC proliferation in different stages of NAFLD, balancing the activity of YAP in hepatocytes and HSCs during different disease stages is key for efficacy.
Hedgehog Signaling
Growing evidence indicates that the hedgehog pathway is a critical regulator of adult liver repair and, hence, a potential diagnostic and/or therapeutic target in cirrhosis (Figure 4) (Machado and Diehl, 2018). Gli1 is the downstream transcriptional activator of hedgehog signaling, and it is also a marker of mesenchymal cells. Previous studies have confirmed perivascular Gli1+ mesenchymal-like cells to be a major driver of organ fibrosis (Kramann et al., 2015). Peribiliary Gli1+ mesenchymal cells are a subset of stromal cells characterized by active hedgehog signaling; these cells proliferate, acquire a myofibroblast phenotype, and surround the biliary tree in response to cholestatic injury, promoting liver fibrosis (Gupta et al., 2020). In fact, aberrant activation of hedgehog signaling in not only mesenchymal cells but also HSCs is considered crucial in liver fibrosis (Li et al., 2020a).
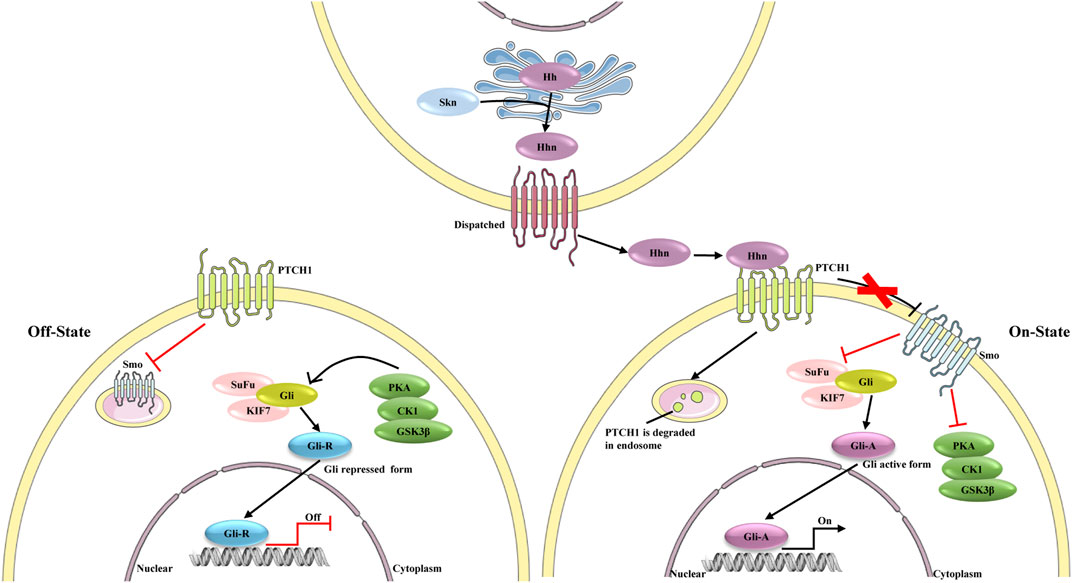
FIGURE 4. The different state of hedgehog signaling pathway. In the absence of ligands, the signaling is on “Off state”, PTCH1 inhibits the activity of Smo. Protein kinase (including PKA, CK1 and GSK3β), SuFu and KIF7 phosphorylate Gli, which leads proteolytic cleavage of Gli to Gli-repressor (Gli-R). Gli-R represses the expression of target genes. In Hhn secreting cells, the precursor of Hh is auto-cleaved and can be modified by a cholesterol at C-terminus to form Hhn on ER membrane. After this process, Hhn is secreted from the secreting cells and bind to PTCH1. PTCH1 is degraded in endosome, and consequently Smo repression is removed. Activated Smo inhibits the effect of PKA on Gli proteins, leading to the dissociation of SuFu, and Gli active form (Gli-A) is formed. Gli-A promotes the expression of target genes.
Some signaling factors and epigenetic modifications regulate hedgehog signaling and thereby influence HSC activation. These factors include lipopolysaccharide, palmitic acid, and the protein “predicted paired box 6” (Duan et al., 2017; Pan et al., 2017; Li et al., 2020a; Zhu et al., 2021), which may therefore be therapeutic targets in liver fibrosis. One study also suggested that miR-200a inhibits Gli3 expression and may function as a novel anti-fibrotic agent (Li et al., 2020b). Treating HSCs with the DNA methylation inhibitor 5-azadC prevents their proliferation and activation by restoring expression of Patched (PTCH1) (Yang et al., 2013). The metabolic state of HSCs affects their activation, and hedgehog signaling regulates metabolism (Chen et al., 2012; Du et al., 2018). Further work is needed to clarify exactly how hedgehog signaling regulates HSC activation and metabolism, thereby influencing liver.
Many chemical inhibitors of hedgehog inhibitors have been identified, including Gant61, GDC-0049, MD85, and vismodegib. These compounds have shown promise against liver fibrosis in vivo and in vitro (Li et al., 2019a; Kumar et al., 2019; Jiayuan et al., 2020). The naturally occurring iridoid glucoside geniposide, extracted from Gardenia jasminoides Ellis, inhibits hedgehog and thereby HSCs (Lin et al., 2019). These compounds are less effective against liver fibrosis in part because they cannot be delivered specifically to the liver. Two studies have achieved such delivery using nanoparticles, which improved drug efficacy. One group replaced the sulfonamide group of the hedgehog inhibitor GDC-0449 with two methylpyridine-2yl groups at the amide nitrogen, generating MDB5. This inhibitor was more potent at inhibiting hedgehog signaling and HSC proliferation in vitro. The research group also developed MDB5-loaded micelles, which enhanced systemic delivery of the drug and efficacy against liver fibrosis (Kumar et al., 2019). In another study, the hedgehog inhibitor vismodegib was loaded into cRGDyK-guided liposomes, which markedly inhibited the fibrogenic phenotype in vivo. The delivery system targeted the delivery of vismodegib to activated HSCs rather than quiescent HSCs, leading to preferential accumulation in fibrotic liver. These finding illustrate the promise of delivering therapeutic agents to activated HSCs to treat liver fibrosis (Li et al., 2019a).
Fibroblast Growth Factor Signaling
Fibroblast growth factor (FGF) signaling is a prerequisite for adequate would healing, repair and homeostasis in various tissues and organs (Seitz and Hellerbrand, 2021). Since liver fibrosis is a wound healing response to liver injury, FGFs play an important role in hepatic fibrosis by acting as paracrine, and endocrine mediators of hepatocyte regeneration and HSC migration, proliferation and trans-differentiation (Schumacher and Guo, 2016). The paracrine FGFs (FGF1-10, FGF16-18, FGF20 and FGF22) bind strongly to heparan sulphate proteoglycans, which limits FGF diffusion through ECM and restricts their action to the site of secretion (Ornitz and Itoh, 2015; Seitz and Hellerbrand, 2021).
The three endocrine FGFs, FGF19 (mouse homolog FGF15), FGF21 and FGF23 participate in phosphate, bile acid, carbohydrate and lipid metabolism and thereby affect liver homeostasis (Itoh, 2010; Itoh et al., 2016; Kuro-o, 2019). Recent studies illustrate how FGF15/19 and FGF21 affect hepatic fibrosis and HSC activation. Hepatic accumulation of bile acid is central to the pathogenesis of cholestasis-induced liver injury, and excessive levels of cytotoxic bile acids in the liver can lead to liver fibrosis (Schaap et al., 2014). Expression of FGF15/19 is strongly induced by farnesoid X receptor (FXR) in the ileum, and the protein is secreted into the portal blood and transported to the liver, where it represses the expression of CYP7a1, a rate-limiting enzyme in bile acid synthesis, thereby mitigating liver fibrosis (Inagaki et al., 2005; Liu et al., 2020b). While enterocytes of the terminal ileum likely produce most FGF15/19, HSCs express FGFR4, while HSCs secrete FGF19. Enhanced FGF19/FGFR4 signaling blocks HSC proliferation and activation, which may help explain the anti-fibrotic effects of FGF19 observed in previous studies (Zhou et al., 2017; Hirschfield et al., 2019; Tian et al., 2021). FGF15 deficiency inhibits the development of hepatic fibrosis in animal models of NASH or liver fibrosis (Uriarte et al., 2015; Schumacher et al., 2017; Schumacher et al., 2020).
These findings suggest that FGF15/19 exert hepatoprotective effects via a pathway independent of bile acids. In contrast to FGF15/19, FGF21 is expressed predominantly in hepatocytes and is released in response to high levels of glucose and free fatty acids as well as low levels of amino acids. In this way, FGF 21 can prevent fatty liver, hepatic steatosis and hepatotoxicity (Reinehr et al., 2012; Seitz and Hellerbrand, 2021). FGF21 also inhibits HSC activation via TGF-β and NF-κB pathways, and it can induce HSC apoptosis through caspase-3, which attenuates hepatic fibrogenesis (Xu et al., 2016). FGF21 may participate in metabolism-related liver disease.
Gas6 Signaling
Growth arrest-specific gene 6 (Gas6), a ligand of the TAM receptor (Tyro3, Axl, Mer), is a vitamin K-dependent protein expressed primarily by Kupffer cells, whereas Axl is found in both macrophages and quiescent HSCs in normal liver (Bárcena et al., 2015; Shrivastava et al., 2016). Serum levels of Gas6 correlate directly with liver stiffness and are significantly higher in patients with advanced fibrosis and primary biliary cholangitis (Bellan et al., 2016; Hayashi et al., 2020). In fact, the Gas6/TAM system has recently emerged as an important player in the progression of liver fibrosis and as a novel biomarker of liver fibrosis (Bellan et al., 2019; Smirne et al., 2019). CCl4-induced liver fibrosis activates the Gas6/Axl pathway, which in turn promotes HSC activation. Disrupting the pathway through Gas6 deficiency, Axl knockout or pharmacological inhibition attenuates hepatic fibrosis (Lafdil et al., 2006; Fourcot et al., 2011; Bárcena et al., 2015). Similarly, the rs4374383 polymorphism in the gene encoding Mer modulates HSC activation, affecting the severity of fibrosis in NAFLD (Petta et al., 2016). Gas6 also participates in both cardiac and pulmonary fibrosis by binding TAM (Espindola et al., 2018; Chen et al., 2019b; Li et al., 2019b). Targeting Gas6 signaling may be a potential treatment for liver fibrosis, so how Gas6 contributes to liver fibrosis should be further explored.
Ferroptosis Signaling
Ferroptosis is a recently recognized form of regulated cell death, characterized by the presence of unusually small mitochondria with quite dense mitochondrial membrane, loss of mitochondria crista, rupture of the outer mitochondrial membrane, and accumulation of iron-based lipid reactive oxygen species (Xie et al., 2016). Ferroptosis is a defensive mechanism against cancer, neurotoxicity and ischemia/reperfusion-induced injury (Liang et al., 2019; Li et al., 2020c; Song and Long, 2020). In mice, an MCD diet induces iron accumulation, cell death and hepatic ferroptosis. Conversely, ferroptosis inhibitors alleviate MCD-diet induced inflammation, fibrogenesis and liver injury, suggesting an important role of ferroptosis in NASH (Li et al., 2020d). Ferroptosis is also an iron-dependent form of regulated cell death triggered by toxic lipid peroxidation, which is inhibited by glutathione peroxidase 4 (GPX4) in steatohepatitic liver (Qi et al., 2020).
Ferroptosis is now considered a new strategy for inhibiting HSCs to alleviate liver fibrosis (Zhang et al., 2018; Zhang et al., 2020a; Zhang et al., 2020b). How ferroptosis is regulated in HSCs remains unclear, although the RNA-binding protein ELAVL1/HuR, ZFP36/TTP, iron regulatory protein2 and the BRD7-p53-SLC25A28 complex appear to be involved. Ferroptosis can be trigged by inhibiting GPX4 (e.g., altretamine), inhibiting system Xc- (e.g., sorafenib, erastin, and sulfasalazine), depleting glutathione (e.g., BSO), or applying certain environmental conditions (e.g., high extracellular glutamate, amino acid starvation, cystine deprivation) (Alim et al., 2019). Thus, these treatments may be effective against liver fibrosis.
cAMP-PKA-cAMP-Responsive Element-Binding Signaling
Cyclic cAMP (cAMP) is well-known as an antifibrogenic second messenger (Li et al., 2019c). The downstream cAMP-responsive element-binding (CREB) protein is a nuclear protein that binds to the cAMP-responsive element (CRE) in the promoter of the gene encoding neuropeptide, and CREB has been implicated in HSC activation and liver fibrosis (Cui et al., 2021). CREB-1, following its activation by phosphorylation, inhibits HSC proliferation and collagen expression in vitro (Deng et al., 2011), and it is involved in TGF-β3 auto-regulation in HSCs (Houglum et al., 1997). Phosphorylation or acetylation of CREB-1 in rat HSCs inhibits the TGF-β1 pathway, downregulating collagen I (Deng et al., 2016).
In contrast to these studies suggesting that CREB-1 can inhibit fibrosis, some studies indicate that it can promote hepatic fibrosis. One study, for example, suggested that phosphorylated CREB-1 promotes fibrosis by transactivating TGF-β1 expression (Wang et al., 2016b). Acetaldehyde can activate HSC-T6 cells, while caffeine can act via the adenosine A2A receptor to inhibit the cAMP/PKA pathway and thereby suppress such activation (Wang et al., 2015). Blocking the interaction between CREB and β-catenin using the selective inhibitor PRI-724 reduces liver fibrosis induced by CCl4 or bile duct ligation (Osawa et al., 2015).
The potentially opposite effects of CREB in different cellular contexts suggest its complex involvement in liver fibrosis, which requires further investigation. Many studies have detected interaction between cAMP-PKA-CREB signaling and pathways mediated by TGF-β and Wnt. This may be a fruitful direction for future research. The molecules currently known to interact with CREB and to show therapeutic potential against liver fibrosis inhibit cAMP-PKA-CREB signaling. These molecules include ICG-001, a selective inhibitor of the CBP/β-catenin interaction (Henderson et al., 2010); PRI-724, which is phosphorylated on C-82 and is rapidly hydrolyzed in vivo into its active form, which shows acceptable toxicity and efficacy in preclinical studies (Lenz and Kahn, 2014; Osawa et al., 2015), and caffeine (Wang et al., 2015).
Cellular Metabolism
Under normal circumstances, HSCs are in a resting state and show low metabolism. Their main function is to store small vitamin A fat droplets, which contain more than 70% of vitamin A in the body (Puche et al., 2013). Pro-inflammatory and pro-fibrotic cytokines can activate HSCs to release their stored vitamin A, proliferate, and produce ECM, with the cells adopting a high metabolic state (Chen et al., 2012; Higashi et al., 2017). In this way, HSCs undergo dramatic metabolic changes to meet the increased bioenergetic and biosynthetic demands of mitogenesis and ECM synthesis (Xie et al., 2015; Para et al., 2019; Zhao et al., 2020; Hewitson and Smith, 2021). These metabolic changes are often accompanied by increased glycolysis and mitochondrial respiration in order to optimize glucose consumption in HSCs and redirect them to support fibrogenic trans-differentiation (Lian et al., 2016).
Recent studies suggest that by changing the metabolism of activated HSCs, they can be converted into the resting type, offering opportunities for liver fibrosis treatment. In diseased livers of animals and patients, the number of glycolytic stromal cells is associated with the severity of fibrosis. Glycolysis is upregulated and lactate accumulates in quiescent HSCs that have been activated to become myofibroblasts. Hedgehog signaling regulates glycolysis to control the fate of HSCs (Chen et al., 2012). Increased aerobic glycolysis alone cannot meet the high metabolic demands of active HSCs: it works together with glutaminolysis (conversion of glutamine to α-ketoglutarate) to sustain energy metabolism and permit anabolism, and this is controlled by hedgehog signaling to YAP (Du et al., 2018). In vivo, glutaminolysis in HSCs is a marker of active fibrogenesis, and its cell-specific antagonism represents a potential therapeutic target by depriving the cells of glutamine (Du et al., 2020). Acetyl-CoA carboxylase (ACC), a regulator of fatty acid β-oxidation and de novo lipogenesis, has been implicated in metabolic reprogramming during HSC activation. ACC inhibitors prevent the de novo lipogenesis that is necessary for induction of glycolysis and oxidative phosphorylation during HSC activation, and such inhibitors thereby mitigate fibrosis (Bates et al., 2020). In addition to elevated levels of glutamine, elevated levels of fructose can increase risk of liver fibrosis (Song et al., 2019; Roeb and Weiskirchen, 2021). More studies are needed that explore metabolic regulation of HSCs, since this is a promising therapeutic strategy against liver fibrosis (Trivedi et al., 2021).
Epigenetics
With the rise of advanced molecular methods, studies have begun to describe the epigenetic landscape of liver fibrosis, involving changes in DNA methylation, histone modifications and levels of non-coding RNAs that control chromatin structure and DNA accessibility to the transcriptional machinery. DNA methylation is carried out by three enzyme: DNA methyltransferase 1 (DNMT1), DNMT3A and DNMT3B (Jin et al., 2011). MCP2 influences methylation of the gene encoding PPARγ, leading to its silencing, which in turn promotes HSC activation (Mann et al., 2010). DNMT1 and DNMT3B methylates the genes encoding regulator of calcineurin 1 (RCAN1), prostacyclin synthase (PTGIS), Septin9 and SAD1/UNC84 domain protein-2 (SUN2), promoting HSC activation and liver fibrosis (Wu et al., 2017; Chen et al., 2018; Pan et al., 2018; Pan et al., 2019). These findings suggest that gene methylation is important for HSC activation. In fact, DNA methylation affects other epigenetic process, including the expression and activity of long non-coding RNAs, which in turn influence HSC activation and fibrosis. One example is the DNMT1-LncRNA H19 epigenetic pathway, which is involved in HSC activation and liver fibrosis (Yang et al., 2018a). Hypermethylation of the gene encoding PSTPIP2 not only activates HSCs but also polarizes macrophages in mice with CCl4-induced hepatic fibrosis (Yang et al., 2018b).
It is not surprising, then, that the various histone modifications, which include methylation, acetylation, phosphorylation, ubiquitination, deamination, and sumoylation, are considered targets for fibrosis treatment (El Taghdouini and van Grunsven, 2016). For example, the enhancer of zeste homologue 2 (EZH2), which is responsible for the trimethylation of histone 3 at lysine 27(H3K27me3), is involved in TGF-β dependent fibrogenic pathways (Martin-Mateos et al., 2019). EZH2 and the demethylase JMJD3 regulate HSC activation and liver fibrosis (Jiang et al., 2021). Histone deacetylases 1/2 (HDAC1/2) may regulate liver fibrosis and may therefore be therapeutic targets (Liu et al., 2021; Zhu et al., 2021). Many genes are regulated through cross-talk between histone and DNA mehyltransferases such as G9a and DNMT1. CM272, a first-in-class reversible inhibitor of G9a and DNMT1, can halt fibrogenesis without causing toxic effects (Barcena-Varela et al., 2021).
Numerous non-coding RNAs such as microRNAs and long non-coding RNAs play important roles in liver fibrosis. For example, miR-199a, miR-200a/b, miR-122, miR-194/192, miR-223, miR-21, miR-155 and miR-29 are expressed or enriched in several types of hepatic cells or in the circulation specifically in the presence of liver disease, implying that they play important roles in pathogenesis (Murakami et al., 2011; Wang et al., 2021). Nanoparticle-based delivery of miR-30c to LSECs inhibits the DLL4/Notch pathway and angiogenesis, ameliorating liver fibrosis in vivo (Gu et al., 2021). The lncRNA-ATB is upregulated in fibrotic liver tissues and activated LX‐2 cells. Knockdown of lncRNA‐ATB downregulates β‐catenin by upregulating the endogenous miR‐200a and suppressing activation of LX‐2 cells (Fu et al., 2017). HOTAIR act as an endogenous “sponge” for miR‐148b to facilitate expression of DNMT1, which in turn promotes HSC proliferation and activation (Bian et al., 2017).
Several inhibitors of DNA methylation (e.g., 5-azadC, Sennoside A) and histone modifications (e.g., givinostat, DZNep, GSK-503, GSK-J4) as well as epigenetic inhibitors such as CM272 have shown promise for treating liver fibrosis (Yang et al., 2013; Martin-Mateos et al., 2019; Zhu et al., 2020; Barcena-Varela et al., 2021; Ding et al., 2021; Huang et al., 2021; Jiang et al., 2021). Epigenetic biomarkers may be useful not only as treatment targets but also for assaying in tissue and liquid biopsies in order to predict prognosis of patients with liver fibrosis. For example, the levels of H3K27ac in specific oncogenes and of TS, PPARγ-mediated DNA methylation have been suggested for this purpose (Hardy et al., 2017; Arechederra et al., 2021; Jühling et al., 2021).
Candidate Drugs During Clinical Trails for Liver Fibrosis
Recently, with the in-depth understanding of the pathogenesis of liver fibrosis, some new compounds with anti-fibrosis potential have emerged and are in clinical trial (Rotman and Sanyal, 2017; Lambrecht et al., 2020; Attia et al., 2021) (Table 1). The therapeutic targets of these compounds contain metabolism, gut-liver axis, inflammation and cell death, which share their effects among whole body. After fully confirming drug’s efficacy on liver fibrosis, finding a suitable targeted delivery system for drugs may help for its clinical use with better treatment efficacy and lower side effects. The future candidate drugs for liver fibrosis may develop from the novel targets or its combinatorial use.
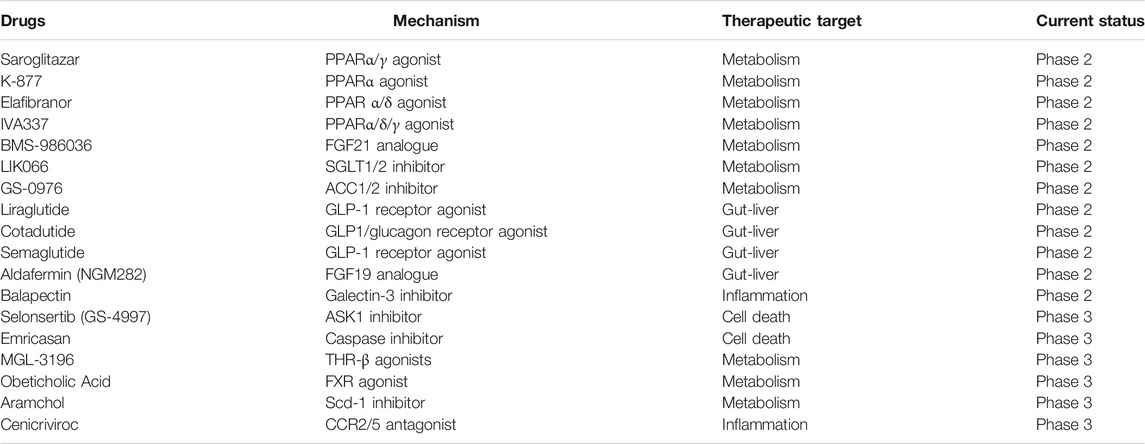
TABLE 1. New pharmacotherapeutics with antifibrotic effects currently in clinical trial.
Conclusion
In this review, we have outlined how major types of hepatic cells participate in liver fibrosis, and we have described several novel targets for fibrosis therapy. Our hope is to provide directions for future investigations.
Early research on the mechanism of fibrosis has focused mainly on HSC activation and collagen deposition. More recent research has focused on cellular state and processes, including metabolism, HSC proliferation and apoptosis, and epigenetic modifications. These studies have broadened our understanding of the pathogenesis of fibrosis, and have pointed out new directions for research into anti-fibrotic drugs. The many in-depth studies on the pathogenesis of liver fibrosis have identified novel signaling pathways as well as signaling crosstalk between TGF-β and Wnt/β-catenin, TGF-β and hedgehog, or YAP and hedgehog.
These studies will gradually build a complete picture of the pathogenesis of fibrosis and provide new ideas in the search for treatment targets. These studies highlight that exploiting crosstalk between signaling pathways may lead to the development of more effective drugs against liver fibrosis.
Author Contributions
JZ and YL wrote the manuscript.; QL and JH contributed to the conclusion and reviewed the manuscript. JH and YL obtained funding. JZ, JH and YL are the guarantors of this work and as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Funding
This study was supported by the National Natural Science Foundation of China (82025007, 81930020, 81873662 and 81870599).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Akcora, B. Ö., Storm, G., and Bansal, R. (2018). Inhibition of Canonical WNT Signaling Pathway by β-Catenin/CBP Inhibitor ICG-001 Ameliorates Liver Fibrosis In Vivo Through Suppression of Stromal CXCL12. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1864 (3), 804–818. doi:10.1016/j.bbadis.2017.12.001
Alim, I., Caulfield, J. T., Chen, Y., Swarup, V., Geschwind, D. H., Ivanova, E., et al. (2019). Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell. 177 (5), 1262–1279. doi:10.1016/j.cell.2019.03.032
Alsamman, S., Christenson, S. A., Yu, A., Ayad, N. M. E., Mooring, M. S., Segal, J. M., et al. (2020). Targeting Acid Ceramidase Inhibits YAP/TAZ Signaling to Reduce Fibrosis in Mice. Sci. Transl Med. 12 (557), eaay8798. doi:10.1126/scitranslmed.aay8798
An, P., Wei, L.-L., Zhao, S., Sverdlov, D. Y., Vaid, K. A., Miyamoto, M., et al. (2020). Hepatocyte Mitochondria-Derived Danger Signals Directly Activate Hepatic Stellate Cells and Drive Progression of Liver Fibrosis. Nat. Commun. 11 (1), 2362. doi:10.1038/s41467-020-16092-0
Andrew, R., Chang, C. M. F., and Mostoslavsky, R. (2020). SIRT6, a Mammalian Deacylase With Multitasking Abilities. Physiol. Rev. 100 (1), 145–169. doi:10.1152/physrev.00030.2018
Arechederra, M., Recalde, M., Gárate-Rascón, M., Fernández-Barrena, M. G., Ávila, M. A., and Berasain, C. (2021). Epigenetic Biomarkers for the Diagnosis and Treatment of Liver Disease. Cancers (Basel). 13 (6), 1265. doi:10.3390/cancers13061265
Asahara, T., Takahashi, T., Masuda, H., Kalka, C., Chen, D., Iwaguro, H., et al. (1999). VEGF Contributes to Postnatal Neovascularization by Mobilizing Bone Marrow-Derived Endothelial Progenitor Cells. EMBO J. 18 (14), 3964–3972. doi:10.1093/emboj/18.14.3964
Asrani, S. K., Devarbhavi, H., Eaton, J., and Kamath, P. S. (2019). Burden of Liver Diseases in the World. J. Hepatol. 70 (1), 151–171. doi:10.1016/j.jhep.2018.09.014
Attia, S. L., Softic, S., and Mouzaki, M. (2021). Evolving Role for Pharmacotherapy in NAFLD/NASH. Clin. Transl Sci. 14 (1), 11–19. doi:10.1111/cts.12839
Bai, H., Zhang, N., Xu, Y., Chen, Q., Khan, M., Potter, J. J., et al. (2012). Yes‐Associated Protein Regulates the Hepatic Response After Bile Duct Ligation. Hepatology. 56 (3), 1097–1107. doi:10.1002/hep.25769
Banales, J. M., Huebert, R. C., Karlsen, T., Strazzabosco, M., LaRusso, N. F., and Gores, G. J. (2019). Cholangiocyte Pathobiology. Nat. Rev. Gastroenterol. Hepatol. 16 (5), 269–281. doi:10.1038/s41575-019-0125-y
Bárcena, C., Stefanovic, M., Tutusaus, A., Joannas, L., Menéndez, A., García-Ruiz, C., et al. (2015). Gas6/Axl Pathway Is Activated in Chronic Liver Disease and its Targeting Reduces Fibrosis via Hepatic Stellate Cell Inactivation. J. Hepatol. 63 (3), 670–678. doi:10.1016/j.jhep.2015.04.013
Barcena-Varela, M., Paish, H., Alvarez, L., Uriarte, I., Latasa, M. U., Santamaria, E., et al. (2021). Epigenetic Mechanisms and Metabolic Reprogramming in Fibrogenesis: Dual Targeting of G9a and DNMT1 for the Inhibition of Liver Fibrosis. Gut. 70 (2), 388–400. doi:10.1136/gutjnl-2019-320205
Bataller, R., and Brenner, D. A. (2005). Liver Fibrosis. J. Clin. Invest. 115 (2), 209–218. doi:10.1172/jci24282
Bates, J., Vijayakumar, A., Ghoshal, S., Marchand, B., Yi, S., Kornyeyev, D., et al. (2020). Acetyl-CoA Carboxylase Inhibition Disrupts Metabolic Reprogramming During Hepatic Stellate Cell Activation. J. Hepatol. 73 (4), 896–905. doi:10.1016/j.jhep.2020.04.037
Bellan, M., Cittone, M. G., Tonello, S., Rigamonti, C., Castello, L. M., Gavelli, F., et al. (2019). Gas6/TAM System: A Key Modulator of the Interplay Between Inflammation and Fibrosis. Int. J. Mol. Sci. 20 (20), 5070. doi:10.3390/ijms20205070
Bellan, M., Pogliani, G., Marconi, C., Minisini, R., Franzosi, L., Alciato, F., et al. (2016). Gas6 as a Putative Noninvasive Biomarker of Hepatic Fibrosis. Biomarkers Med. 10 (12), 1241–1249. doi:10.2217/bmm-2016-0210
Berumen, J., Baglieri, J., Kisseleva, T., and Mekeel, K. (2021). Liver Fibrosis: Pathophysiology and Clinical Implications. Wiley Interdiscip. Rev. Syst. Biol. Med. 13 (1), e1499. doi:10.1002/wsbm.1499
Bian, E.-B., Wang, Y.-Y., Yang, Y., Wu, B.-M., Xu, T., Meng, X.-M., et al. (2017). Hotair Facilitates Hepatic Stellate Cells Activation and Fibrogenesis in the Liver. Biochim. Biophys. Acta (Bba) - Mol. Basis Dis. 1863 (3), 674–686. doi:10.1016/j.bbadis.2016.12.009
Bouwens, L., Baekeland, M., de Zanger, R., and Wisse, E. (1986). Quantitation, Tissue Distribution and Proliferation Kinetics of Kupffer Cells in Normal Rat Liver. Hepatology. 6 (4), 718–722. doi:10.1002/hep.1840060430
Boyer-Diaz, Z., Aristu-Zabalza, P., Andrés-Rozas, M., Robert, C., Ortega-Ribera, M., Fernández-Iglesias, A., et al. (2021). Pan-PPAR Agonist Lanifibranor Improves portal Hypertension and Hepatic Fibrosis in Experimental Advanced Chronic Liver Disease. J. Hepatol. 74 (5), 1188–1199. doi:10.1016/j.jhep.2020.11.045
Bugyei-Twum, A., Ford, C., Civitarese, R., Seegobin, J., Advani, S. L., Desjardins, J.-F., et al. (2018). Sirtuin 1 Activation Attenuates Cardiac Fibrosis in a Rodent Pressure Overload Model by Modifying Smad2/3 Transactivation. Cardiovasc. Res. 114 (12), 1629–1641. doi:10.1093/cvr/cvy131
Cai, X., Wang, J., Wang, J., Zhou, Q., Yang, B., He, Q., et al. (2020). Intercellular Crosstalk of Hepatic Stellate Cells in Liver Fibrosis: New Insights Into Therapy. Pharmacol. Res. 155, 104720. doi:10.1016/j.phrs.2020.104720
Chen, F.-F., Song, F.-Q., Chen, Y.-Q., Wang, Z.-H., Li, Y.-H., Liu, M.-H., et al. (2019a). Exogenous Testosterone Alleviates Cardiac Fibrosis and Apoptosis via Gas6/Axl Pathway in the Senescent Mice. Exp. Gerontol. 119, 128–137. doi:10.1016/j.exger.2019.01.029
Chen, L., Gu, T., Li, B., Li, F., Ma, Z., Zhang, Q., et al. (2019b). Delta-Like Ligand 4/DLL4 Regulates the Capillarization of Liver Sinusoidal Endothelial Cell and Liver Fibrogenesis. Biochim. Biophys. Acta (Bba) - Mol. Cel Res. 1866 (10), 1663–1675. doi:10.1016/j.bbamcr.2019.06.011
Chen, X., Li, W.-X., Chen, Y., Li, X.-F., Li, H.-D., Huang, H.-M., et al. (2018). Suppression of SUN2 by DNA Methylation Is Associated With HSCs Activation and Hepatic Fibrosis. Cell Death Dis. 9 (10), 1021. doi:10.1038/s41419-018-1032-9
Chen, Y., Choi, S. S., Michelotti, G. A., Chan, I. S., Swiderska-Syn, M., Karaca, G. F., et al. (2012). Hedgehog Controls Hepatic Stellate Cell Fate by Regulating Metabolism. Gastroenterology. 143 (5), 1319–1329. doi:10.1053/j.gastro.2012.07.115
Cicchini, C., Amicone, L., Alonzi, T., Marchetti, A., Mancone, C., and Tripodi, M. (2015). Molecular Mechanisms Controlling the Phenotype and the EMT/MET Dynamics of Hepatocyte. Liver Int. 35 (2), 302–310. doi:10.1111/liv.12577
Crawford, J. J., Bronner, S. M., and Zbieg, J. R. (2018). Hippo Pathway Inhibition by Blocking the YAP/TAZ-TEAD Interface: a Patent Review. Expert Opin. Ther. Patents. 28 (12), 867–873. doi:10.1080/13543776.2018.1549226
Cui, A., Ding, D., and Li, Y. (2021). Regulation of Hepatic Metabolism and Cell Growth by the ATF/CREB Family of Transcription Factors. Diabetes. 70 (3), 653–664. doi:10.2337/dbi20-0006
Dai, Z., Song, G., Balakrishnan, A., Yang, T., Yuan, Q., Möbus, S., et al. (2020). Growth Differentiation Factor 11 Attenuates Liver Fibrosis via Expansion of Liver Progenitor Cells. Gut. 69 (6), 1104–1115. doi:10.1136/gutjnl-2019-318812
Dat, N. Q., Thuy, L. T. H., Hieu, V. N., Hai, H., Hoang, D. V., Hai, N. T. T., et al. (2021). 6His-Tagged Recombinant Human Cytoglobin Deactivates Hepatic Stellate Cells and Inhibits Liver Fibrosis by Scavenging Reactive Oxygen Species. Hepatology. 73 (6), 2527–2545. doi:10.1002/hep.31752
De, A. (2011). Wnt/Ca2+ Signaling Pathway: a Brief Overview. Acta Biochim. Biophys. Sin (Shanghai). 43 (10), 745–756. doi:10.1093/abbs/gmr079
DeLeve, L. D. (2015). Liver Sinusoidal Endothelial Cells in Hepatic Fibrosis. Hepatology. 61 (5), 1740–1746. doi:10.1002/hep.27376
Deng, L., Li, Y., Huang, J. m., Zhou, G. y., Qian, W., and Xu, K. s. (2011). Effects of P-CREB-1 on Transforming Growth Factor-Β3 Auto-Regulation in Hepatic Stellate Cells. J. Cel. Biochem. 112 (4), 1046–1054. doi:10.1002/jcb.23017
Deng, X., Deng, L., Wang, P., Cheng, C., and Xu, K. (2016). Post-Translational Modification of CREB-1 Decreases Collagen I Expression by Inhibiting the TGF-Β1 Signaling Pathway in Rat Hepatic Stellate Cells. Mol. Med. Rep. 14 (6), 5751–5759. doi:10.3892/mmr.2016.5926
Ding, R., Zheng, J., Li, N., Cheng, Q., Zhu, M., Wang, Y., et al. (2021). DZNep, an Inhibitor of the Histone Methyltransferase EZH2, Suppresses Hepatic Fibrosis Through Regulating miR-199a-5p/SOCS7 Pathway. PeerJ. 9, e11374. doi:10.7717/peerj.11374
Du, K., Chitneni, S. K., Suzuki, A., Wang, Y., Henao, R., Hyun, J., et al. (2020). Increased Glutaminolysis Marks Active Scarring in Nonalcoholic Steatohepatitis Progression. Cell Mol. Gastroenterol. Hepatol. 10 (1), 1–21. doi:10.1016/j.jcmgh.2019.12.006
Du, K., Hyun, J., Premont, R. T., Choi, S. S., Michelotti, G. A., Swiderska-Syn, M., et al. (2018). Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology. 154 (5), 1465–1479. doi:10.1053/j.gastro.2017.12.022
Duan, J.-L., Ruan, B., Yan, X.-C., Liang, L., Song, P., Yang, Z.-Y., et al. (2018). Endothelial Notch Activation Reshapes the Angiocrine of Sinusoidal Endothelia to Aggravate Liver Fibrosis and Blunt Regeneration in Mice. Hepatology. 68 (2), 677–690. doi:10.1002/hep.29834
Duan, N.-N., Liu, X.-J., and Wu, J. (2017). Palmitic Acid Elicits Hepatic Stellate Cell Activation Through Inflammasomes and Hedgehog Signaling. Life Sci. 176, 42–53. doi:10.1016/j.lfs.2017.03.012
El Taghdouini, A., and van Grunsven, L. A. (2016). Epigenetic Regulation of Hepatic Stellate Cell Activation and Liver Fibrosis. Expert Rev. Gastroenterol. Hepatol. 10 (12), 1397–1408. doi:10.1080/17474124.2016.1251309
Elssner, C., Goeppert, B., Longerich, T., Scherr, A. L., Stindt, J., Nanduri, L. K., et al. (2019). Nuclear Translocation of RELB Is Increased in Diseased Human Liver and Promotes Ductular Reaction and Biliary Fibrosis in Mice. Gastroenterology. 156 (4), 1190–e14. doi:10.1053/j.gastro.2018.11.018
Esmail, M. M., Saeed, N. M., Michel, H. E., and El-Naga, R. N. (2021). The Ameliorative Effect of Niclosamide on Bile Duct Ligation Induced Liver Fibrosis via Suppression of NOTCH and Wnt Pathways. Toxicol. Lett. 347, 23–35. doi:10.1016/j.toxlet.2021.04.018
Espindola, M. S., Habiel, D. M., Narayanan, R., Jones, I., Coelho, A. L., Murray, L. A., et al. (2018). Targeting of TAM Receptors Ameliorates Fibrotic Mechanisms in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 197 (11), 1443–1456. doi:10.1164/rccm.201707-1519oc
Fabre, T., Malina, M. F., Soucy, G., Goulet, J.-P., Willems, B., Villeneuve, J.-P., et al. (2018). Type 3 Cytokines IL-17A and IL-22 Drive TGF-β-dependent Liver Fibrosis. Sci. Immunol. 3 (28), eaar7754. doi:10.1126/sciimmunol.aar7754
Fan, J., Shen, W., Lee, S.-R., Mathai, A. E., Zhang, R., Xu, G., et al. (2020). Targeting the Notch and TGF-β Signaling Pathways to Prevent Retinal Fibrosis In Vitro and In Vivo. Theranostics. 10 (18), 7956–7973. doi:10.7150/thno.45192
Fan, W., Liu, T., Chen, W., Hammad, S., Longerich, T., Hausser, I., et al. (2019). ECM1 Prevents Activation of Transforming Growth Factor β, Hepatic Stellate Cells, and Fibrogenesis in Mice. Gastroenterology. 157 (5), 1352–1367. doi:10.1053/j.gastro.2019.07.036
Finnson, K. W., Almadani, Y., and Philip, A. (2020). Non-Canonical (Non-Smad2/3) TGF-β Signaling in Fibrosis: Mechanisms and Targets. Semin. Cel Develop. Biol. 101, 115–122. doi:10.1016/j.semcdb.2019.11.013
Fourcot, A., Couchie, D., Chobert, M.-N., Zafrani, E.-S., Mavier, P., Laperche, Y., et al. (2011). Gas6 Deficiency Prevents Liver Inflammation, Steatohepatitis, and Fibrosis in Mice. Am. J. Physiology-Gastrointestinal Liver Physiol. 300 (6), G1043–G1053. doi:10.1152/ajpgi.00311.2010
Friedman, S. L. (2008). Mechanisms of Hepatic Fibrogenesis. Gastroenterology. 134 (6), 1655–1669. doi:10.1053/j.gastro.2008.03.003
Fu, N., Zhao, S.-X., Kong, L.-B., Du, J.-H., Ren, W.-G., Han, F., et al. (2017). LncRNA-ATB/microRNA-200a/β-Catenin Regulatory Axis Involved in the Progression of HCV-Related Hepatic Fibrosis. Gene. 618, 1–7. doi:10.1016/j.gene.2017.03.008
Gandhi, C. R. (2017). Hepatic Stellate Cell Activation and Pro-Fibrogenic Signals. J. Hepatol. 67 (5), 1104–1105. doi:10.1016/j.jhep.2017.06.001
Ge, M. x., Liu, H. t., Zhang, N., Niu, W. x., Lu, Z. n., Bao, Y. y., et al. (2020). Costunolide Represses Hepatic Fibrosis Through WW Domain‐Containing Protein 2‐Mediated Notch3 Degradation. Br. J. Pharmacol. 177 (2), 372–387. doi:10.1111/bph.14873
Georgescu, E. F. (2008). Angiotensin Receptor Blockers in the Treatment of NASH/NAFLD: Could They Be a First-Class Option? Adv. Ther. 25 (11), 1141–1174. doi:10.1007/s12325-008-0110-2
Glaser, S. S., Gaudio, E., Miller, T., Alvaro, D., and Alpini, G. (2009). Cholangiocyte Proliferation and Liver Fibrosis. Expert Rev. Mol. Med. 11, e7. doi:10.1017/s1462399409000994
Gu, T., Shen, B., Li, B., Guo, Y., Li, F., Ma, Z., et al. (2021). miR-30c Inhibits Angiogenesis by Targeting Delta-Like Ligand 4 in Liver Sinusoidal Endothelial Cell to Attenuate Liver Fibrosis. FASEB J. 35 (5), e21571. doi:10.1096/fj.202002694R
Gupta, V., Gupta, I., Park, J., Bram, Y., and Schwartz, R. E. (2020). Hedgehog Signaling Demarcates a Niche of Fibrogenic Peribiliary Mesenchymal Cells. Gastroenterology. 159 (2), 624–638. doi:10.1053/j.gastro.2020.03.075
Haak, A. J., Kostallari, E., Sicard, D., Ligresti, G., Choi, K. M., Caporarello, N., et al. (2019). Selective YAP/TAZ Inhibition in Fibroblasts via Dopamine Receptor D1 Agonism Reverses Fibrosis. Sci. Transl Med. 11 (516), eaau6296. doi:10.1126/scitranslmed.aau6296
Hardy, T., Zeybel, M., Day, C. P., Dipper, C., Masson, S., McPherson, S., et al. (2017). Plasma DNA Methylation: a Potential Biomarker for Stratification of Liver Fibrosis in Non-Alcoholic Fatty Liver Disease. Gut. 66 (7), 1321–1328. doi:10.1136/gutjnl-2016-311526
Hayashi, M., Abe, K., Fujita, M., Takahashi, A., Hashimoto, Y., and Ohira, H. (2020). Serum Gas6 and Axl as Non‐Invasive Biomarkers of Advanced Histological Stage in Primary Biliary Cholangitis. Hepatol. Res. 50 (12), 1337–1346. doi:10.1111/hepr.13568
Henderson, W. R., Chi, E. Y., Ye, X., Nguyen, C., Tien, Y.-t., Zhou, B., et al. (2010). Inhibition of Wnt/-Catenin/CREB Binding Protein (CBP) Signaling Reverses Pulmonary Fibrosis. Proc. Natl. Acad. Sci. 107 (32), 14309–14314. doi:10.1073/pnas.1001520107
Hewitson, T. D., and Smith, E. R. (2021). A Metabolic Reprogramming of Glycolysis and Glutamine Metabolism Is a Requisite for Renal Fibrogenesis-Why and How? Front. Physiol. 12, 645857. doi:10.3389/fphys.2021.645857
Higashi, T., Friedman, S. L., and Hoshida, Y. (2017). Hepatic Stellate Cells as Key Target in Liver Fibrosis. Adv. Drug Deliv. Rev. 121, 27–42. doi:10.1016/j.addr.2017.05.007
Hirschfield, G. M., Chazouillères, O., Drenth, J. P., Thorburn, D., Harrison, S. A., Landis, C. S., et al. (2019). Effect of NGM282, an FGF19 Analogue, in Primary Sclerosing Cholangitis: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Phase II Trial. J. Hepatol. 70 (3), 483–493. doi:10.1016/j.jhep.2018.10.035
Houglum, K., Lee, K. S., and Chojkier, M. (1997). Proliferation of Hepatic Stellate Cells Is Inhibited by Phosphorylation of CREB on Serine 133. J. Clin. Invest. 99 (6), 1322–1328. doi:10.1172/jci119291
Hu, B., and Phan, S. H. (2016). Notch in Fibrosis and as a Target of Anti-fibrotic Therapy. Pharmacol. Res. 108, 57–64. doi:10.1016/j.phrs.2016.04.010
Hu, H.-H., Cao, G., Wu, X.-Q., Vaziri, N. D., and Zhao, Y.-Y. (2020). Wnt Signaling Pathway in Aging-Related Tissue Fibrosis and Therapies. Ageing Res. Rev. 60, 101063. doi:10.1016/j.arr.2020.101063
Hu, J., Srivastava, K., Wieland, M., Runge, A., Mogler, C., Besemfelder, E., et al. (2014). Endothelial Cell-Derived Angiopoietin-2 Controls Liver Regeneration as a Spatiotemporal Rheostat. Science. 343 (6169), 416–419. doi:10.1126/science.1244880
Huang, H. M., Zhou, X. R., Liu, Y. J., Fan, S. J., Liao, L. P., Huang, J., et al. (2021). Histone Deacetylase Inhibitor Givinostat Alleviates Liver Fibrosis by Regulating Hepatic Stellate Cell Activation. Mol. Med. Rep. 23 (5), 305. doi:10.3892/mmr.2021.11944
Inagaki, T., Choi, M., Moschetta, A., Peng, L., Cummins, C. L., McDonald, J. G., et al. (2005). Fibroblast Growth Factor 15 Functions as an Enterohepatic Signal to Regulate Bile Acid Homeostasis. Cel Metab. 2 (4), 217–225. doi:10.1016/j.cmet.2005.09.001
Itoh, N. (2010). Hormone-like (Endocrine) Fgfs: Their Evolutionary History and Roles in Development, Metabolism, and Disease. Cell Tissue Res. 342 (1), 1–11. doi:10.1007/s00441-010-1024-2
Itoh, N., Nakayama, Y., and Konishi, M. (2016). Roles of FGFs as Paracrine or Endocrine Signals in Liver Development, Health, and Disease. Front. Cel Dev. Biol. 4, 30. doi:10.3389/fcell.2016.00030
Jalan-Sakrikar, N., De Assuncao, T. M., Shi, G., Aseem, S., Chi, C., Shah, V. H., et al. (2019). Proteasomal Degradation of Enhancer of Zeste Homologue 2 in Cholangiocytes Promotes Biliary Fibrosis. Hepatology. 70 (5), 1674–1689. doi:10.1002/hep.30706
Jarman, E. J., and Boulter, L. (2020). Targeting the Wnt Signaling Pathway: the challenge of Reducing Scarring Without Affecting Repair. Expert Opin. Investig. Drugs. 29 (2), 179–190. doi:10.1080/13543784.2020.1718105
Jiang, J. X., Chen, X., Serizawa, N., Szyndralewiez, C., Page, P., Schröder, K., et al. (2012). Liver Fibrosis and Hepatocyte Apoptosis Are Attenuated by GKT137831, a Novel NOX4/NOX1 Inhibitor In Vivo. Free Radic. Biol. Med. 53 (2), 289–296. doi:10.1016/j.freeradbiomed.2012.05.007
Jiang, Y., Xiang, C., Zhong, F., Zhang, Y., Wang, L., Zhao, Y., et al. (2021). Histone H3K27 Methyltransferase EZH2 and Demethylase JMJD3 Regulate Hepatic Stellate Cells Activation and Liver Fibrosis. Theranostics. 11 (1), 361–378. doi:10.7150/thno.46360
Jiayuan, S., Junyan, Y., Xiangzhen, W., Zuping, L., Jian, N., Baowei, H., et al. (2020). Gant61 Ameliorates CCl4-Induced Liver Fibrosis by Inhibition of Hedgehog Signaling Activity. Toxicol. Appl. Pharmacol. 387, 114853. doi:10.1016/j.taap.2019.114853
Jin, B., Li, Y., and Robertson, K. D. (2011). DNA Methylation: Superior or Subordinate in the Epigenetic Hierarchy? Genes & Cancer 2 (6), 607–617. doi:10.1177/1947601910393957
Jühling, F., Hamdane, N., Crouchet, E., Li, S., El Saghire, H., Mukherji, A., et al. (2021). Targeting Clinical Epigenetic Reprogramming for Chemoprevention of Metabolic and Viral Hepatocellular Carcinoma. Gut. 70 (1), 157–169. doi:10.1136/gutjnl-2019-318918
Karin, M., and Clevers, H. (2016). Reparative Inflammation Takes Charge of Tissue Regeneration. Nature. 529 (7586), 307–315. doi:10.1038/nature17039
Karsdal, M. A., Genovese, F., Madsen, E. A., Manon-Jensen, T., and Schuppan, D. (2016). Collagen and Tissue Turnover as a Function of Age: Implications for Fibrosis. J. Hepatol. 64 (1), 103–109. doi:10.1016/j.jhep.2015.08.014
Khomich, O., Ivanov, A. V., and Bartosch, B. (2019). Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells. 9 (1), 24. doi:10.3390/cells9010024
Kim, S. J., Kim, K. M., Yang, J. H., Cho, S. S., Jeong, E. H., Kim, J. H., et al. (2021). Transforming Growth Factor Beta-Induced Foxo3a Acts as a Profibrotic Mediator in Hepatic Stellate Cells. Toxicol. Sci. 179 (2), 241–250. doi:10.1093/toxsci/kfaa185
Kisseleva, T., and Brenner, D. (2021). Molecular and Cellular Mechanisms of Liver Fibrosis and its Regression. Nat. Rev. Gastroenterol. Hepatol. 18 (3), 151–166. doi:10.1038/s41575-020-00372-7
Konishi, T., Schuster, R. M., and Lentsch, A. B. (2018). Proliferation of Hepatic Stellate Cells, Mediated by YAP and TAZ, Contributes to Liver Repair and Regeneration After Liver Ischemia-Reperfusion Injury. Am. J. Physiology-Gastrointestinal Liver Physiol. 314 (4), G471–G482. doi:10.1152/ajpgi.00153.2017
Kovall, R. A., and Blacklow, S. C. (2010). Mechanistic Insights Into Notch Receptor Signaling From Structural and Biochemical Studies, in Notch Signaling. Curr. Top. Dev. Biol. 92, 31–71. doi:10.1016/s0070-2153(10)92002-4
Koyama, Y., and Brenner, D. A. (2017). Liver Inflammation and Fibrosis. J. Clin. Invest. 127 (1), 55–64. doi:10.1172/jci88881
Kramann, R., Schneider, R. K., DiRocco, D. P., Machado, F., Fleig, S., Bondzie, P. A., et al. (2015). Perivascular Gli1+ Progenitors Are Key Contributors to Injury-Induced Organ Fibrosis. Cell Stem Cell. 16 (1), 51–66. doi:10.1016/j.stem.2014.11.004
Kumar, V., Dong, Y., Kumar, V., Almawash, S., and Mahato, R. I. (2019). The Use of Micelles to Deliver Potential Hedgehog Pathway Inhibitor for the Treatment of Liver Fibrosis. Theranostics. 9 (25), 7537–7555. doi:10.7150/thno.38913
Kuro-o, M. (2019). The Klotho Proteins in Health and Disease. Nat. Rev. Nephrol. 15 (1), 27–44. doi:10.1038/s41581-018-0078-3
Lafdil, F., Chobert, M. N., Couchie, D., Brouillet, A., Zafrani, E. S., Mavier, P., et al. (2006). Induction of Gas6 Protein in CCl4-Induced Rat Liver Injury and Anti-Apoptotic Effect on Hepatic Stellate Cells. Hepatology. 44 (1), 228–239. doi:10.1002/hep.21237
Lafoz, E., Ruart, M., Anton, A., Oncins, A., and Hernández-Gea, V. (2020). The Endothelium as a Driver of Liver Fibrosis and Regeneration. Cells. 9 (4), 929. doi:10.3390/cells9040929
Lambrecht, J., van Grunsven, L. A., and Tacke, F. (2020). Current and Emerging Pharmacotherapeutic Interventions for the Treatment of Liver Fibrosis. Expert Opin. Pharmacother. 21 (13), 1637–1650. doi:10.1080/14656566.2020.1774553
Lee, E. H., Park, K.-I., Kim, K.-Y., Lee, J.-H., Jang, E. J., Ku, S. K., et al. (2019). Liquiritigenin Inhibits Hepatic Fibrogenesis and TGF-β1/Smad With Hippo/YAP Signal. Phytomedicine. 62, 152780. doi:10.1016/j.phymed.2018.12.003
Lee, E. J., Hwang, I., Lee, J. Y., Park, J. N., Kim, K. C., Kim, I., et al. (2020). Hepatic Stellate Cell-Specific Knockout of Transcriptional Intermediary Factor 1γ Aggravates Liver Fibrosis. J. Exp. Med. 217 (6), e20190402. doi:10.1084/jem.20190402
Lee, I. H., Im, E., Lee, H. J., Sim, D. Y., Lee, J. H., Jung, J. H., et al. (2021). Apoptotic and Antihepatofibrotic Effect of Honokiol via Activation ofGSK3βand Suppression of Wnt/β‐catenin Pathway in Hepatic Stellate Cells. Phytotherapy Res. 35 (1), 452–462. doi:10.1002/ptr.6824
Lenz, H. J., and Kahn, M. (2014). Safely Targeting Cancer Stem Cells via Selective Catenin Coactivator Antagonism. Cancer Sci. 105 (9), 1087–1092. doi:10.1111/cas.12471
Li, C., Tan, Y. H., Sun, J., Deng, F. M., and Liu, Y. L. (2020a). PAX6 Contributes to the Activation and Proliferation of Hepatic Stellate Cells via Activating Hedgehog/GLI1 Pathway. Biochem. Biophysical Res. Commun. 526 (2), 314–320. doi:10.1016/j.bbrc.2020.03.086
Li, L., Ran, J., Li, L., Chen, G., Zhang, S., and Wang, Y. (2020b). Gli3 Is a Novel Downstream Target of miR200a With an Anti-fibrotic Role for Progression of Liver Fibrosis In vivo and In Vitro. Mol. Med. Rep. 21 (4), 1861–1871. doi:10.3892/mmr.2020.10997
Li, Y., Cao, Y., Xiao, J., Shang, J., Tan, Q., Ping, F., et al. (2020c). Inhibitor of Apoptosis-Stimulating Protein of P53 Inhibits Ferroptosis and Alleviates Intestinal Ischemia/reperfusion-Induced Acute Lung Injury. Cell Death Differ. 27 (9), 2635–2650. doi:10.1038/s41418-020-0528-x
Li, X., Wang, T. X., Huang, X., Li, Y., Sun, T., Zang, S., et al. (2020d). Targeting Ferroptosis Alleviates Methionine‐Choline Deficient (MCD)‐diet Induced NASH by Suppressing Liver Lipotoxicity. Liver Int. 40 (6), 1378–1394. doi:10.1111/liv.14428
Li, L., Zhou, J., Li, Q., Xu, J., Qi, J., and Bian, H. (2018). The Inhibition of Hippo/Yap Signaling Pathway Is Required for Magnesium Isoglycyrrhizinate to Ameliorate Hepatic Stellate Cell Inflammation and Activation. Biomed. Pharmacother. 106, 83–91. doi:10.1016/j.biopha.2018.06.102
Li, Y., Pu, S., Liu, Q., Li, R., Zhang, J., Wu, T., et al. (2019a). An Integrin-Based Nanoparticle That Targets Activated Hepatic Stellate Cells and Alleviates Liver Fibrosis. J. Controlled Release. 303, 77–90. doi:10.1016/j.jconrel.2019.04.022
Li, W., Xie, L., Ma, J., Yang, M., Wang, B., Xu, Y., et al. (2019b). Genetic Loss of Gas6/Mer Pathway Attenuates Silica-Induced Lung Inflammation and Fibrosis in Mice. Toxicol. Lett. 313, 178–187. doi:10.1016/j.toxlet.2019.07.008
Li, G., Jiang, Q., and Xu, K. (2019c). CREB Family: A Significant Role in Liver Fibrosis. Biochimie. 163, 94–100. doi:10.1016/j.biochi.2019.05.014
Lian, N., Jin, H., Zhang, F., Wu, L., Shao, J., Lu, Y., et al. (2016). Curcumin Inhibits Aerobic Glycolysis in Hepatic Stellate Cells Associated With Activation of Adenosine Monophosphate-Activated Protein Kinase. IUBMB Life. 68 (7), 589–596. doi:10.1002/iub.1518
Liang, C., Zhang, X., Yang, M., and Dong, X. (2019). Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 31 (51), e1904197. doi:10.1002/adma.201904197
Lien, W.-H., and Fuchs, E. (2014). Wnt Some Lose Some: Transcriptional Governance of Stem Cells by Wnt/-Catenin Signaling. Genes Develop. 28 (14), 1517–1532. doi:10.1101/gad.244772.114
Lin, X., Li, J., and Xing, Y.-Q. (2019). Geniposide, a Sonic Hedgehog Signaling Inhibitor, Inhibits the Activation of Hepatic Stellate Cell. Int. Immunopharmacology. 72, 330–338. doi:10.1016/j.intimp.2019.04.016
Liu, J., Zhao, B., Zhu, H., Pan, Q., Cai, M., Bai, X., et al. (2020a). Wnt4 Negatively Regulates the TGF-Β1-Induced Human Dermal Fibroblast-To-Myofibroblast Transition via Targeting Smad3 and ERK. Cel Tissue Res. 379 (3), 537–548. doi:10.1007/s00441-019-03110-x
Liu, Y., Chen, K., Li, F., Gu, Z., Liu, Q., He, L., et al. (2020b). Probiotic Lactobacillus Rhamnosus GG Prevents Liver Fibrosis Through Inhibiting Hepatic Bile Acid Synthesis and Enhancing Bile Acid Excretion in Mice. Hepatology. 71 (6), 2050–2066. doi:10.1002/hep.30975
Liu, R., Li, X., Zhu, W., Wang, Y., Zhao, D., Wang, X., et al. (2019a). Cholangiocyte‐Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology. 70 (4), 1317–1335. doi:10.1002/hep.30662
Liu, Y., Lu, T., Zhang, C., Xu, J., Xue, Z., Busuttil, R. W., et al. (2019b). Activation of YAP Attenuates Hepatic Damage and Fibrosis in Liver Ischemia-Reperfusion Injury. J. Hepatol. 71 (4), 719–730. doi:10.1016/j.jhep.2019.05.029
Liu, Y.-R., Wang, J.-Q., Huang, Z.-G., Chen, R.-N., Cao, X., Zhu, D.-C., et al. (2021). Histone Deacetylase 2: A Potential Regulator and Therapeutic Target in Liver Disease (Review). Int. J. Mol. Med. 48 (1), 131. doi:10.3892/ijmm.2021.4964
Luo, X.-Y., Meng, X.-J., Cao, D.-C., Wang, W., Zhou, K., Li, L., et al. (2019). Transplantation of Bone Marrow Mesenchymal Stromal Cells Attenuates Liver Fibrosis in Mice by Regulating Macrophage Subtypes. Stem Cel Res Ther. 10 (1), 16. doi:10.1186/s13287-018-1122-8
Ma, P.-F., Gao, C.-C., Yi, J., Zhao, J.-L., Liang, S.-Q., Zhao, Y., et al. (2017). Cytotherapy with M1-Polarized Macrophages Ameliorates Liver Fibrosis by Modulating Immune Microenvironment in Mice. J. Hepatol. 67 (4), 770–779. doi:10.1016/j.jhep.2017.05.022
Machado, M. V., and Diehl, A. M. (2018). Hedgehog Signalling in Liver Pathophysiology. J. Hepatol. 68 (3), 550–562. doi:10.1016/j.jhep.2017.10.017
Maity, S., Muhamed, J., Sarikhani, M., Kumar, S., Ahamed, F., Spurthi, K. M., et al. (2019). Sirtuin 6 Deficiency Transcriptionally Up-Regulates TGF-β Signaling and Induces Fibrosis in Mice. J. Biol. Chem. 295 (2), 415–434. doi:10.1074/jbc.RA118.007212
Mann, J., Chu, D. C. K., Maxwell, A., Oakley, F., Zhu, N. L., Tsukamoto, H., et al. (2010). MeCP2 Controls an Epigenetic Pathway that Promotes Myofibroblast Transdifferentiation and Fibrosis. Gastroenterology. 138 (2), 705–714. doi:10.1053/j.gastro.2009.10.002
Mannaerts, I., Leite, S. B., Verhulst, S., Claerhout, S., Eysackers, N., Thoen, L. F. R., et al. (2015). The Hippo Pathway Effector YAP Controls Mouse Hepatic Stellate Cell Activation. J. Hepatol. 63 (3), 679–688. doi:10.1016/j.jhep.2015.04.011
Mantovani, A., Petracca, G., Beatrice, G., Csermely, A., Lonardo, A., and Targher, G. (2021). Glucagon-Like Peptide-1 Receptor Agonists for Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: An Updated Meta-Analysis of Randomized Controlled Trials. Metabolites. 11 (2), 73. doi:10.3390/metabo11020073
Marrone, G., Russo, L., Rosado, E., Hide, D., García-Cardeña, G., García-Pagán, J. C., et al. (2013). The Transcription Factor KLF2 Mediates Hepatic Endothelial protection and Paracrine Endothelial-Stellate Cell Deactivation Induced by Statins. J. Hepatol. 58 (1), 98–103. doi:10.1016/j.jhep.2012.08.026
Marrone, G., Shah, V. H., and Gracia-Sancho, J. (2016). Sinusoidal Communication in Liver Fibrosis and Regeneration. J. Hepatol. 65 (3), 608–617. doi:10.1016/j.jhep.2016.04.018
Martin-Mateos, R., De Assuncao, T. M., Arab, J. P., Jalan-Sakrikar, N., Yaqoob, U., Greuter, T., et al. (2019). Enhancer of Zeste Homologue 2 Inhibition Attenuates TGF-β Dependent Hepatic Stellate Cell Activation and Liver Fibrosis. Cell Mol. Gastroenterol. Hepatol. 7 (1), 197–209. doi:10.1016/j.jcmgh.2018.09.005
McConnell, M., and Iwakiri, Y. (2018). Biology of Portal Hypertension. Hepatol. Int. 12 (Suppl. 1), 11–23. doi:10.1007/s12072-017-9826-x
Meng, F., Wang, K., Aoyama, T., Grivennikov, S. I., Paik, Y., Scholten, D., et al. (2012). Interleukin-17 Signaling in Inflammatory, Kupffer Cells, and Hepatic Stellate Cells Exacerbates Liver Fibrosis in Mice. Gastroenterology. 143 (3), 765–776. doi:10.1053/j.gastro.2012.05.049
Meng, X.-M., Nikolic-Paterson, D. J., and Lan, H. Y. (2016). TGF-β: the Master Regulator of Fibrosis. Nat. Rev. Nephrol. 12 (6), 325–338. doi:10.1038/nrneph.2016.48
Mi, X.-J., Hou, J.-G., Jiang, S., Liu, Z., Tang, S., Liu, X.-X., et al. (2019). Maltol Mitigates Thioacetamide-Induced Liver Fibrosis Through TGF-Β1-Mediated Activation of PI3K/Akt Signaling Pathway. J. Agric. Food Chem. 67 (5), 1392–1401. doi:10.1021/acs.jafc.8b05943
Miao, C.-G., Yang, Y.-Y., He, X., Huang, C., Huang, Y., Zhang, L., et al. (2013). Wnt Signaling in Liver Fibrosis: Progress, Challenges and Potential Directions. Biochimie. 95 (12), 2326–2335. doi:10.1016/j.biochi.2013.09.003
Mooring, M., Fowl, B. H., Lum, S. Z. C., Liu, Y., Yao, K., Softic, S., et al. (2020). Hepatocyte Stress Increases Expression of Yes‐Associated Protein and Transcriptional Coactivator With PDZ‐Binding Motif in Hepatocytes to Promote Parenchymal Inflammation and Fibrosis. Hepatology. 71 (5), 1813–1830. doi:10.1002/hep.30928
Murakami, Y., Toyoda, H., Tanaka, M., Kuroda, M., Harada, Y., Matsuda, F., et al. (2011). The Progression of Liver Fibrosis Is Related with Overexpression of the miR-199 and 200 Families. PLoS One. 6 (1), e16081. doi:10.1371/journal.pone.0016081
Nishikawa, K., Osawa, Y., and Kimura, K. (2018). Wnt/β-Catenin Signaling as a Potential Target for the Treatment of Liver Cirrhosis Using Antifibrotic Drugs. Int. J. Mol. Sci. 19 (10), 3103. doi:10.3390/ijms19103103
Nusse, R., and Clevers, H. (2017). Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 169 (6), 985–999. doi:10.1016/j.cell.2017.05.016
Ornitz, D. M., and Itoh, N. (2015). The Fibroblast Growth Factor Signaling Pathway. Wires Dev. Biol. 4 (3), 215–266. doi:10.1002/wdev.176
Osawa, Y., Oboki, K., Imamura, J., Kojika, E., Hayashi, Y., Hishima, T., et al. (2015). Inhibition of Cyclic Adenosine Monophosphate (cAMP)-Response Element-Binding Protein (CREB)-Binding Protein (CBP)/β-Catenin Reduces Liver Fibrosis in Mice. EBioMedicine. 2 (11), 1751–1758. doi:10.1016/j.ebiom.2015.10.010
Pan, X.-R., Jing, Y.-Y., Liu, W.-T., Han, Z.-P., Li, R., Yang, Y., et al. (2017). Lipopolysaccharide Induces the Differentiation of Hepatic Progenitor Cells Into Myofibroblasts via Activation of the Hedgehog Signaling Pathway. Cell Cycle. 16 (14), 1357–1365. doi:10.1080/15384101.2017.1325976
Pan, X.-Y., Yang, Y., Meng, H.-W., Li, H.-D., Chen, X., Huang, H.-M., et al. (2018). DNA Methylation of PTGIS Enhances Hepatic Stellate Cells Activation and Liver Fibrogenesis. Front. Pharmacol. 9, 553. doi:10.3389/fphar.2018.00553
Pan, X.-Y., You, H.-M., Wang, L., Bi, Y.-H., Yang, Y., Meng, H.-W., et al. (2019). Methylation of RCAN1.4 Mediated by DNMT1 and DNMT3b Enhances Hepatic Stellate Cell Activation and Liver Fibrogenesis Through Calcineurin/NFAT3 Signaling. Theranostics. 9 (15), 4308–4323. doi:10.7150/thno.32710
Para, R., Romero, F., George, G., and Summer, R. (2019). Metabolic Reprogramming as a Driver of Fibroblast Activation in PulmonaryFibrosis. Am. J. Med. Sci. 357 (5), 394–398. doi:10.1016/j.amjms.2019.02.003
Park, H. W., Kim, Y. C., Yu, B., Moroishi, T., Mo, J.-S., Plouffe, S. W., et al. (2015). Alternative Wnt Signaling Activates YAP/TAZ. Cell. 162 (4), 780–794. doi:10.1016/j.cell.2015.07.013
Petta, S., Valenti, L., Marra, F., Grimaudo, S., Tripodo, C., Bugianesi, E., et al. (2016). MERTK Rs4374383 Polymorphism Affects the Severity of Fibrosis in Non-Alcoholic Fatty Liver Disease. J. Hepatol. 64 (3), 682–690. doi:10.1016/j.jhep.2015.10.016
Poisson, J., Lemoinne, S., Boulanger, C., Durand, F., Moreau, R., Valla, D., et al. (2017). Liver Sinusoidal Endothelial Cells: Physiology and Role in Liver Diseases. J. Hepatol. 66 (1), 212–227. doi:10.1016/j.jhep.2016.07.009
Pradere, J.-P., Kluwe, J., De Minicis, S., Jiao, J.-J., Gwak, G.-Y., Dapito, D. H., et al. (2013). Hepatic Macrophages but Not Dendritic Cells Contribute to Liver Fibrosis by Promoting the Survival of Activated Hepatic Stellate Cells in Mice. Hepatology. 58 (4), 1461–1473. doi:10.1002/hep.26429
Puche, J. E., Saiman, Y., and Friedman, S. L. (2013). Hepatic Stellate Cells and Liver Fibrosis. Compr. Physiol. 3 (4), 1473–1492. doi:10.1002/cphy.c120035
Qi, J., Kim, J.-W., Zhou, Z., Lim, C.-W., and Kim, B. (2020). Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation-Mediated Cell Death in Mice. Am. J. Pathol. 190 (1), 68–81. doi:10.1016/j.ajpath.2019.09.011
Ramachandran, P., Pellicoro, A., Vernon, M. A., Boulter, L., Aucott, R. L., Ali, A., et al. (2012). Differential Ly-6C Expression Identifies the Recruited Macrophage Phenotype, Which Orchestrates the Regression of Murine Liver Fibrosis. Proc. Natl. Acad. Sci. 109 (46), E3186–E3195. doi:10.1073/pnas.1119964109
Rao, S., Xiang, J., Huang, J., Zhang, S., Zhang, M., Sun, H., et al. (2019). PRC1 Promotes GLI1-Dependent Osteopontin Expression in Association With the Wnt/β-Catenin Signaling Pathway and Aggravates Liver Fibrosis. Cell Biosci. 9, 100. doi:10.1186/s13578-019-0363-2
Reeves, H. L., and Friedman, S. L. (2002). Activation of Hepatic Stellate Cells - a Key Issue in Liver Fibrosis. Front. Biosci. 7, d808–26. doi:10.2741/reeves
Reinehr, T., Woelfle, J., Wunsch, R., and Roth, C. L. (2012). Fibroblast Growth Factor 21 (FGF-21) and its Relation to Obesity, Metabolic Syndrome, and Nonalcoholic Fatty Liver in Children: a Longitudinal Analysis. J. Clin. Endocrinol. Metab. 97 (6), 2143–2150. doi:10.1210/jc.2012-1221
Richter, L. R., Wan, Q., Wen, D., Zhang, Y., Yu, J., Kang, J. k., et al. (2020). Targeted Delivery of Notch Inhibitor Attenuates Obesity-Induced Glucose Intolerance and Liver Fibrosis. ACS Nano. 14 (6), 6878–6886. doi:10.1021/acsnano.0c01007
Roeb, E., and Weiskirchen, R. (2021). Fructose and Non-Alcoholic Steatohepatitis. Front. Pharmacol. 12, 634344. doi:10.3389/fphar.2021.634344
Rotman, Y., and Sanyal, A. J. (2017). Current and Upcoming Pharmacotherapy for Non-Alcoholic Fatty Liver Disease. Gut. 66 (1), 180–190. doi:10.1136/gutjnl-2016-312431
Schaap, F. G., Trauner, M., and Jansen, P. L. M. (2014). Bile Acid Receptors as Targets for Drug Development. Nat. Rev. Gastroenterol. Hepatol. 11 (1), 55–67. doi:10.1038/nrgastro.2013.151
Schumacher, J. D., and Guo, G. L. (2016). Regulation of Hepatic Stellate Cells and Fibrogenesis by Fibroblast Growth Factors. Biomed. Res. Int. 2016, 8323747. doi:10.1155/2016/8323747
Schumacher, J. D., Kong, B., Pan, Y., Zhan, L., Sun, R., Aa, J., et al. (2017). The Effect of Fibroblast Growth Factor 15 Deficiency on the Development of High Fat Diet Induced Non-Alcoholic Steatohepatitis. Toxicol. Appl. Pharmacol. 330, 1–8. doi:10.1016/j.taap.2017.06.023
Schumacher, J. D., Kong, B., Wu, J., Rizzolo, D., Armstrong, L. E., Chow, M. D., et al. (2020). Direct and Indirect Effects of Fibroblast Growth Factor (FGF) 15 and FGF19 on Liver Fibrosis Development. Hepatology. 71 (2), 670–685. doi:10.1002/hep.30810
Seitz, T., and Hellerbrand, C. (2021). Role of Fibroblast Growth Factor Signalling in Hepatic Fibrosis. Liver Int. 41 (6), 1201–1215. doi:10.1111/liv.14863
Senoo, H., Yoshikawa, K., Morii, M., Miura, M., Imai, K., and Mezaki, Y. (2010). Hepatic Stellate Cell (Vitamin A-Storing Cell) and its Relative - Past, Present and Future. Cell. Biol. Int. 34 (12), 1247–1272. doi:10.1042/cbi20100321
Sheng, J., Zhang, B., Chen, Y., and Yu, F. (2020). Capsaicin Attenuates Liver Fibrosis by Targeting Notch Signaling to Inhibit TNF-α Secretion from M1 Macrophages. Immunopharmacology and Immunotoxicology. 42 (6), 556–563. doi:10.1080/08923973.2020.1811308
Shrivastava, A., Khan, A. A., Khurshid, M., Kalam, M. A., Jain, S. K., and Singhal, P. K. (2016). Recent Developments in L-Asparaginase Discovery and its Potential as Anticancer Agent. Crit. Rev. Oncology/Hematology. 100, 1–10. doi:10.1016/j.critrevonc.2015.01.002
Sica, A., Invernizzi, P., and Mantovani, A. (2014). Macrophage Plasticity and Polarization in Liver Homeostasis and Pathology. Hepatology. 59 (5), 2034–2042. doi:10.1002/hep.26754
Smirne, C., Rigamonti, C., De Benedittis, C., Sainaghi, P. P., Bellan, M., Burlone, M. E., et al. (2019). Gas6/TAM Signaling Components as Novel Biomarkers of Liver Fibrosis. Dis. Markers. 2019, 2304931. doi:10.1155/2019/2304931
So, J., Khaliq, M., Evason, K., Ninov, N., Martin, B. L., Stainier, D. Y. R., et al. (2018). Wnt/β-Catenin Signaling Controls Intrahepatic Biliary Network Formation in Zebrafish by Regulating Notch Activity. Hepatology. 67 (6), 2352–2366. doi:10.1002/hep.29752
Song, L., Chen, T. Y., Zhao, X. J., Xu, Q., Jiao, R. Q., Li, J. M., et al. (2019). Pterostilbene Prevents Hepatocyte Epithelial‐mesenchymal Transition in Fructose‐induced Liver Fibrosis through Suppressing miR‐34a/Sirt1/p53 and TGF‐β1/Smads Signalling. Br. J. Pharmacol. 176 (11), 1619–1634. doi:10.1111/bph.14573
Song, X., and Long, D. (2020). Nrf2 and Ferroptosis: A New Research Direction for Neurodegenerative Diseases. Front. Neurosci. 14, 267. doi:10.3389/fnins.2020.00267
Takebe, N., Nguyen, D., and Yang, S. X. (2014). Targeting Notch Signaling Pathway in Cancer: Clinical Development Advances and Challenges. Pharmacol. Ther. 141 (2), 140–149. doi:10.1016/j.pharmthera.2013.09.005
Tian, S., Chen, M., Wang, B., Han, Y., Shang, H., and Chen, J. (2021). Salvianolic Acid B Blocks Hepatic Stellate Cell Activation via FGF19/FGFR4 Signaling. Ann. Hepatol. 20, 100259. doi:10.1016/j.aohep.2020.07.013
Trivedi, P., Wang, S., and Friedman, S. L. (2021). The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cel Metab. 33 (2), 242–257. doi:10.1016/j.cmet.2020.10.026
Tsuchida, T., and Friedman, S. L. (2017). Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 14 (7), 397–411. doi:10.1038/nrgastro.2017.38
Tu, X., Zhang, Y., Zheng, X., Deng, J., Li, H., Kang, Z., et al. (2017). TGF-β-induced Hepatocyte lincRNA-P21 Contributes to Liver Fibrosis in Mice. Sci. Rep. 7 (1), 2957. doi:10.1038/s41598-017-03175-0
Uriarte, I., Latasa, M. U., Carotti, S., Fernandez-Barrena, M. G., Garcia-Irigoyen, O., Elizalde, M., et al. (2015). Ileal FGF15 Contributes to Fibrosis-Associated Hepatocellular Carcinoma Development. Int. J. Cancer. 136 (10), 2469–2475. doi:10.1002/ijc.29287
Wang, J.-N., Li, L., Li, L.-Y., Yan, Q., Li, J., and Xu, T. (2018). Emerging Role and Therapeutic Implication of Wnt Signaling Pathways in Liver Fibrosis. Gene. 674, 57–69. doi:10.1016/j.gene.2018.06.053
Wang, X., Zheng, Z., Caviglia, J. M., Corey, K. E., Herfel, T. M., Cai, B., et al. (2016a). Hepatocyte TAZ/WWTR1 Promotes Inflammation and Fibrosis in Nonalcoholic Steatohepatitis. Cel Metab. 24 (6), 848–862. doi:10.1016/j.cmet.2016.09.016
Wang, P., Deng, L., Zhuang, C., Cheng, C., and Xu, K. (2016b). p-CREB-1 Promotes Hepatic Fibrosis Through the Transactivation of Transforming Growth Factor-Β1 Expression in Rats. Int. J. Mol. Med. 38 (2), 521–528. doi:10.3892/ijmm.2016.2630
Wang, Q., Dai, X., Yang, W., Wang, H., Zhao, H., Yang, F., et al. (2015). Caffeine Protects against Alcohol-Induced Liver Fibrosis by Dampening the cAMP/PKA/CREB Pathway in Rat Hepatic Stellate Cells. Int. Immunopharmacology. 25 (2), 340–352. doi:10.1016/j.intimp.2015.02.012
Wang, X., He, Y., Mackowiak, B., and Gao, B. (2021). MicroRNAs as Regulators, Biomarkers and Therapeutic Targets in Liver Diseases. Gut. 70 (4), 784–795. doi:10.1136/gutjnl-2020-322526
Wang, Y., Shen, R.-W., Han, B., Li, Z., Xiong, L., Zhang, F.-Y., et al. (2017). Notch Signaling Mediated by TGF-β/Smad Pathway in Concanavalin A-Induced Liver Fibrosis in Rats. Wjg. 23 (13), 2330–2336. doi:10.3748/wjg.v23.i13.2330
Wang, Y., Tu, K., Liu, D., Guo, L., Chen, Y., Li, Q., et al. (2019a). p300 Acetyltransferase Is a Cytoplasm‐to‐Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor β-Stimulated Hepatic Stellate Cells. Hepatology. 70 (4), 1409–1423. doi:10.1002/hep.30668
Wang, K., Fang, S., Liu, Q., Gao, J., Wang, X., Zhu, H., et al. (2019b). TGF-β1/p65/MAT2A Pathway Regulates Liver Fibrogenesis via Intracellular SAM. EBioMedicine. 42, 458–469. doi:10.1016/j.ebiom.2019.03.058
Wen, Y., Lambrecht, J., Ju, C., and Tacke, F. (2021). Hepatic Macrophages in Liver Homeostasis and Diseases-Diversity, Plasticity and Therapeutic Opportunities. Cell Mol Immunol. 18 (1), 45–56. doi:10.1038/s41423-020-00558-8
Wu, Y., Bu, F., Yu, H., Li, W., Huang, C., Meng, X., et al. (2017). Methylation of Septin9 Mediated by DNMT3a Enhances Hepatic Stellate Cells Activation and Liver Fibrogenesis. Toxicol. Appl. Pharmacol. 315, 35–49. doi:10.1016/j.taap.2016.12.002
Wynn, T. (2008). Cellular and Molecular Mechanisms of Fibrosis. J. Pathol. 214 (2), 199–210. doi:10.1002/path.2277
Xie, G., Karaca, G., Swiderska-Syn, M., Michelotti, G. A., Krüger, L., Chen, Y., et al. (2013). Cross-Talk Between Notch and Hedgehog Regulates Hepatic Stellate Cell Fate in Mice. Hepatology. 58 (5), 1801–1813. doi:10.1002/hep.26511
Xie, N., Tan, Z., Banerjee, S., Cui, H., Ge, J., Liu, R.-M., et al. (2015). Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 192 (12), 1462–1474. doi:10.1164/rccm.201504-0780oc
Xie, Y., Hou, W., Song, X., Yu, Y., Huang, J., Sun, X., et al. (2016). Ferroptosis: Process and Function. Cel Death Differ. 23 (3), 369–379. doi:10.1038/cdd.2015.158
Xu, P., Zhang, Y., Liu, Y., Yuan, Q., Song, L., Liu, M., et al. (2016). Fibroblast Growth Factor 21 Attenuates Hepatic Fibrogenesis Through TGF-Β/smad2/3 and NF-Κb Signaling Pathways. Toxicol. Appl. Pharmacol. 290, 43–53. doi:10.1016/j.taap.2015.11.012
Xu, Y., Sun, X., Zhang, R., Cao, T., Cai, S.-Y., Boyer, J. L., et al. (2020). A Positive Feedback Loop of TET3 and TGF-Β1 Promotes Liver Fibrosis. Cel Rep. 30 (5), 1310–1318. doi:10.1016/j.celrep.2019.12.092
Yang, J.-J., She, Q., Yang, Y., Tao, H., and Li, J. (2018a). DNMT1 Controls LncRNA H19/ERK Signal Pathway in Hepatic Stellate Cell Activation and Fibrosis. Toxicol. Lett. 295, 325–334. doi:10.1016/j.toxlet.2018.07.013
Yang, Y., Wu, X.-Q., Li, W.-X., Huang, H.-M., Li, H.-D., Pan, X.-Y., et al. (2018b). PSTPIP2 Connects DNA Methylation to Macrophage Polarization in CCL4-Induced Mouse Model of Hepatic Fibrosis. Oncogene. 37 (47), 6119–6135. doi:10.1038/s41388-018-0383-0
Yang, J.-J., Tao, H., Huang, C., Shi, K.-H., Ma, T.-T., Bian, E.-B., et al. (2013). DNA Methylation and MeCP2 Regulation of PTCH1 Expression During Rats Hepatic Fibrosis. Cell Signal. 25 (5), 1202–1211. doi:10.1016/j.cellsig.2013.01.005
Yang, Y. M., Noureddin, M., Liu, C., Ohashi, K., Kim, S. Y., Ramnath, D., et al. (2019). Hyaluronan Synthase 2-Mediated Hyaluronan Production Mediates Notch1 Activation and Liver Fibrosis. Sci. Transl Med. 11 (496), eaat9284. doi:10.1126/scitranslmed.aat9284
You, E., Ko, P., Jeong, J., Keum, S., Kim, J.-W., Seo, Y.-J., et al. (2020). Dynein-Mediated Nuclear Translocation of Yes-Associated Protein Through Microtubule Acetylation Controls Fibroblast Activation. Cell. Mol. Life Sci. 77 (20), 4143–4161. doi:10.1007/s00018-019-03412-x
Younossi, Z. M., Koenig, A. B., Abdelatif, D., Fazel, Y., Henry, L., and Wymer, M. (2016). Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology. 64 (1), 73–84. doi:10.1002/hep.28431
Yu, H.-X., Yao, Y., Bu, F.-T., Chen, Y., Wu, Y.-T., Yang, Y., et al. (2019). Blockade of YAP Alleviates Hepatic Fibrosis through Accelerating Apoptosis and Reversion of Activated Hepatic Stellate Cells. Mol. Immunol. 107, 29–40. doi:10.1016/j.molimm.2019.01.004
Yu, L., Wang, L., Yi, H., and Wu, X. (2020). LRP6-CRISPR Prevents Activation of Hepatic Stellate Cells and Liver Fibrogenesis in Rats. Am. J. Transl Res. 12 (2), 397–408.
Zhang, F., Wang, F., He, J., Lian, N., Wang, Z., Shao, J., et al. (2021). Regulation of Hepatic Stellate Cell Contraction and Cirrhotic portal Hypertension by Wnt/β‐catenin Signalling via Interaction With Gli1. Br. J. Pharmacol. 178 (11), 2246–2265. doi:10.1111/bph.15289
Zhang, J., Li, Y., Liu, Q., Huang, Y., Li, R., Wu, T., et al. (2021). Sirt6 Alleviated Liver Fibrosis by Deacetylating Conserved Lysine 54 on Smad2 in Hepatic Stellate Cells. Hepatology. 73 (3), 1140–1157. doi:10.1002/hep.31418
Zhang, K., Han, X., Zhang, Z., Zheng, L., Hu, Z., Yao, Q., et al. (2017). The Liver-Enriched Lnc-LFAR1 Promotes Liver Fibrosis by Activating TGFβ and Notch Pathways. Nat. Commun. 8 (1), 144. doi:10.1038/s41467-017-00204-4
Zhang, K., Zhang, M., Yao, Q., Han, X., Zhao, Y., Zheng, L., et al. (2019). The Hepatocyte-Specifically Expressed Lnc-HSER Alleviates Hepatic Fibrosis by Inhibiting Hepatocyte Apoptosis and Epithelial-Mesenchymal Transition. Theranostics. 9 (25), 7566–7582. doi:10.7150/thno.36942
Zhang, Y. E. (2017). Non-Smad Signaling Pathways of the TGF-β Family. Cold Spring Harb Perspect. Biol. 9 (2), a022129. doi:10.1101/cshperspect.a022129
Zhang, Z., Guo, M., Li, Y., Shen, M., Kong, D., Shao, J., et al. (2020a). RNA-Binding Protein ZFP36/TTP Protects Against Ferroptosis by Regulating Autophagy Signaling Pathway in Hepatic Stellate Cells. Autophagy. 16 (8), 1482–1505. doi:10.1080/15548627.2019.1687985
Zhang, Z., Guo, M., Shen, M., Kong, D., Zhang, F., Shao, J., et al. (2020b). The BRD7-P53-Slc25a28 Axis Regulates Ferroptosis in Hepatic Stellate Cells. Redox Biol. 36, 101619. doi:10.1016/j.redox.2020.101619
Zhang, Z., Yao, Z., Wang, L., Ding, H., Shao, J., Chen, A., et al. (2018). Activation of Ferritinophagy Is Required for the RNA-Binding Protein ELAVL1/HuR to Regulate Ferroptosis in Hepatic Stellate Cells. Autophagy. 14 (12), 2083–2103. doi:10.1080/15548627.2018.1503146
Zhao, X., Kwan, J. Y. Y., Yip, K., Liu, P. P., and Liu, F.-F. (2020). Targeting Metabolic Dysregulation for Fibrosis Therapy. Nat. Rev. Drug Discov. 19 (1), 57–75. doi:10.1038/s41573-019-0040-5
Zhong, X., Huang, M., Kim, H.-G., Zhang, Y., Chowdhury, K., Cai, W., et al. (2020). SIRT6 Protects against Liver Fibrosis by Deacetylation and Suppression of SMAD3 in Hepatic Stellate Cells. Cell Mol Gastroenterol Hepatol. 10 (2), 341–364. doi:10.1016/j.jcmgh.2020.04.005
Zhou, M., Learned, R. M., Rossi, S. J., DePaoli, A. M., Tian, H., and Ling, L. (2017). Engineered FGF19 Eliminates Bile Acid Toxicity and Lipotoxicity Leading to Resolution of Steatohepatitis and Fibrosis in Mice. Hepatol. Commun. 1 (10), 1024–1042. doi:10.1002/hep4.1108
Zhou, T., Wu, N., Meng, F., Venter, J., Giang, T. K., Francis, H., et al. (2018). Knockout of Secretin Receptor Reduces Biliary Damage and Liver Fibrosis in Mdr2−/− Mice by Diminishing Senescence of Cholangiocytes. Lab. Invest. 98 (11), 1449–1464. doi:10.1038/s41374-018-0093-9
Zhu, C., Kim, K., Wang, X., Bartolome, A., Salomao, M., Dongiovanni, P., et al. (2018). Hepatocyte Notch Activation Induces Liver Fibrosis in Nonalcoholic Steatohepatitis. Sci. Transl Med. 10 (468), eaat0344. doi:10.1126/scitranslmed.aat0344
Zhu, H., He, C., Zhao, H., Jiang, W., Xu, S., Li, J., et al. (2020). Sennoside A Prevents Liver Fibrosis by Binding DNMT1 and Suppressing DNMT1‐Mediated PTEN Hypermethylation in HSC Activation and Proliferation. FASEB j. 34 (11), 14558–14571. doi:10.1096/fj.202000494rr
Keywords: liver fibrosis, hepatic stellate cells, therapeutic targets, molecular mechanism, non-alcocholic fatty liver disease
Citation: Zhang J, Liu Q, He J and Li Y (2021) Novel Therapeutic Targets in Liver Fibrosis. Front. Mol. Biosci. 8:766855. doi: 10.3389/fmolb.2021.766855
Received: 30 August 2021; Accepted: 18 October 2021;
Published: 05 November 2021.
Edited by:
Hua Wang, Anhui Medical University, ChinaReviewed by:
Bin Gao, National Institutes of Health (NIH), United StatesSabine Klein, University Hospital Frankfurt, Germany
Sandra Torres, Goethe University Frankfurt, Germany
Sung Hwan Ki, Chosun University, South Korea
Copyright © 2021 Zhang, Liu, He and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinhan He, amluaGFuaGVAc2N1LmVkdS5jbg==; Yanping Li, bGl5YW5waW5nXzUxMkAxNjMuY29t