 Otto Luiz Dutra Cerqueira1
Otto Luiz Dutra Cerqueira1 Fernanda Antunes1
Fernanda Antunes1 Nadine G Assis1
Nadine G Assis1 Elaine C Cardoso2
Elaine C Cardoso2 Maria A Clavijo-Salomón1
Maria A Clavijo-Salomón1 Ana C Domingues1
Ana C Domingues1 Nayara G Tessarollo1
Nayara G Tessarollo1 Bryan E Strauss1*
Bryan E Strauss1*- 1Centro de Investigação Translacional em Oncologia (CTO)/LIM, Instituto do Câncer do Estado de São Paulo (ICESP), Faculdade de Medicina da Universidade de São Paulo (FMUSP), São Paulo, Brazil
- 2Department of Pediatrics, Faculdade de Medicina da Universidade de São Paulo (FMUSP), São Paulo, Brazil
Melanoma is the deadliest type of skin cancer with steadily increasing incidence worldwide during the last few decades. In addition to its tumor associated antigens (TAAs), melanoma has a high mutation rate compared to other tumors, which promotes the appearance of tumor specific antigens (TSAs) as well as increased lymphocytic infiltration, inviting the use of therapeutic tools that evoke new or restore pre-existing immune responses. Innovative therapeutic proposals, such as immune checkpoint inhibitors (ICIs), have emerged as effective options for melanoma. However, a significant portion of these patients relapse and become refractory to treatment. Likewise, strategies using viral vectors, replicative or not, have garnered confidence and approval by different regulatory agencies around the world. It is possible that further success of immune therapies against melanoma will come from synergistic combinations of different approaches. In this review we outline molecular features inherent to melanoma and how this supports the use of viral oncolysis and immunotherapies when used as monotherapies or in combination.
Introduction
Immunotherapy has revolutionized cancer treatment in unprecedented ways (Mellman et al., 2011). It consists of mobilizing the immune system’s own defenses to recognize and eliminate neoplastic cells, thus reinstating cancer immunosurveillance (Zitvogel et al., 2013). The promising early results generated high expectations and gave hope to many patients to reach long-term remission (Cable et al., 2021). However, only a minority of patients with advanced cancer undergoing this therapeutic modality increase survival in a lasting way (Hegde and Chen, 2020). In this scenario, the use of rational combinations of immunotherapies has been increasing and may link different approaches and technologies in order to improve patient benefit of the treatment (Finck et al., 2020). Oncolytic viruses (OV) are therapeutic tools with the property of not only selectively inducing oncolysis, but also attracting cells of the immune system, activating them and thus mobilizing innate and adaptive antitumor responses (Vähä-Koskela et al., 2007; Russell et al., 2012). This property is suitable for combination with therapies aimed at cell-mediated cytotoxic effect, whether adoptive or naturally intrinsic to the immune system, acting synergistically at different stages of the cancer-immunity cycle (Chen and Mellman, 2013). Here we provide a historical and biological perspective regarding the advent and clinical implementation of immunotherapies, including oncolytic viruses, for the treatment of melanoma and we explore the possible combinations of oncolytic viruses with additional immunotherapy strategies.
Molecular Alterations in Melanoma That May Serve as Therapeutic Targets
Cancer is a multifactorial disease characterized by heterogeneous subpopulations of cells with different phenotypes and genetic properties leading to uncontrolled proliferation, migration, invasion as well as metastasis and drug resistance. Skin cancer can be classified as basal cell carcinoma, squamous carcinoma and melanoma. Non-melanoma skin cancers account for nearly 98% of skin cancer in the United States (Force et al., 2018). Although melanoma is the least frequent type of skin cancer, it is the deadliest with steadily increasing incidence worldwide during the last few decades, especially in Caucasian populations (Siegel et al., 2020). The latest data released by Global Cancer Observatory (GLOBOCAN) estimated more than 280,000 new cases of skin melanoma (1.6% of all cancers) with nearly 60,000 deaths in 2020 (Global Cancer Observatory–https://gco.iarc.fr/). Melanoma arises from malignant transformation of melanocytes, melanin-producing cells found in the epidermal skin layer, due to mutagenic damage that activates many oncogenes and inactivates tumor suppressor genes (Kobayashi et al., 1998; Cancer Genome Atlas, 2015; Hayward et al., 2017). Some studies demonstrated that incidence of skin cancer (non-melanoma and melanoma) is inversely related to skin pigmentation, with higher risk and incidence in individuals with fair skin along with the presence of nevi and freckles. Furthermore, a family history of melanoma, immunosuppression related to organ transplantation, and HIV or HPV infection also increase the predisposition to this neoplasm (Veierod et al., 2010; Force et al., 2018).
One of the main risk factors for the development of melanoma is intermittent excessive exposure to solar ultraviolet (UV) radiation, being particularly harmful when it occurs in childhood. In addition, there is growing evidence that exposure to artificial UV radiation through the use of artificial tanning chambers increases the propensity to develop melanomas (Veierod et al., 2010; Sun et al., 2020). UV radiation induces DNA damage directly (DNA photoproducts) or through ROS production that indirectly causes oxidative DNA damage, leading to DNA mutations and alterations in the transcriptional profile, resulting in dysregulation of several tumor suppressor genes and oncogenes (Kvam and Tyrrell, 1997; Anna et al., 2007). Moreover, UV radiation can also downregulate cutaneous immunity by apoptosis of epidermal immune cells (Langerhans cells) and inhibition of antigen presentation together with release of immunosuppressive cytokines, favoring tumor development and progression (Fisher and Kripke, 1982; Shreedhar et al., 1998). The UV-induced DNA damage response is modulated by the tumor suppressor gene TP53 that can be found downregulated, contributing to UV-mediated mutagenesis in non-melanoma and melanoma skin cancer (Smith et al., 2000; Decraene et al., 2001). In addition, UV exposure can alter TP53, resulting in cooperation with BRAF mutations to induce melanoma (Viros et al., 2014).
Sequencing studies revealed the genetic landscape of cutaneous melanoma and classified them into four subgroups: mutant BRAF, mutant NRAS, mutant NF1 and triple-wild type (Cancer Genome Atlas, 2015; Hayward et al., 2017; Zhou et al., 2019). In another study, the whole genome sequence analysis of melanoma samples also found mutations in other genes, such as TERT, TP53, CDKN2A and CDKN2B. Some of these mutations as BRAF, NRAS and TERT are also found in benign lesions whereas CDKN2A, TP73 and PTEN are observed only in invasive melanoma (Amaral et al., 2017; Consortium, 2020). Other mutations less frequently found, especially in melanomas missing heritability, are BPA1, POT1, ACD and TERF2IP (Potjer et al., 2019).
BRAF mutations are highly prevalent in melanoma and found in 40–60% of cultured primary melanoma cells but are not sufficient for melanoma progression and development since they are found in benign nevi (Pollock et al., 2003; Tschandl et al., 2013). The most frequent oncogenic mutation for BRAF in melanomas is the substitution of amino acid valine for glutamic acid at position 600 (V600 E), representing 70–90% of BRAF mutations. Other BRAF mutations, although less frequent, can be found in melanoma, including V600K, V600R, V600D for example (Rubinstein et al., 2010; Long et al., 2011; Lovly et al., 2012). Mutations in BRAF are not related to UV radiation exposition as 30–60% of patients without chronic sun-induced damage have been identified with somatic BRAF mutation (Curtin et al., 2005; Brash, 2015). These mutations have important clinical significance since mutated BRAF protein is active as a monomer instead of dimer and the monomer conformation is the target for the binding of BRAF inhibitors, such as vemurafenib, dabrafenib and encorafenib, used in melanoma therapy (Czarnecka et al., 2020). Moreover, the presence of BRAF mutations (BRAF (+)), despite not impacting recurrence-free survival from diagnosis of primary melanoma (stage I/II) to metastases development (stage IV) compared to BRAF WT patients, they do have a negative impact on median overall survival (OS) of patients who are newly diagnosed, untreated and with metastatic disease, since in BRAF (+) patients the OS is 5.7 months and for BRAF WT it is 8.5 months (Long et al., 2011). BRAFV600 E mutation resulted in altered BRAF protein conformation, increasing its kinase activity, leading to constitutive MAPK pathway activation, resulting in uncontrolled proliferation, cell survival and immune evasion which contribute to melanoma growth (Yang et al., 2019). The MAPK pathway is also activated by NRAS mutations that are frequently found in several tumor types and in 15–20% of melanoma patients but not concomitant with BRAF mutations (Wan et al., 2004; Chiappetta et al., 2015). Moreover, 15% of melanomas have NF1 mutations with loss of function that also result in MAPK hyperactivation (Wan et al., 2004; Krauthammer et al., 2015). Deregulation of RAS/MAPK/ERK pathway is found in nearly all melanomas (Hayward et al., 2017). The signaling pathway RAS/MAPK/ERK impacts more than 50 transcription factors involved in the regulation of genes that control cell growth, division, proliferation and differentiation (Molina and Adjei, 2006). The pathway is activated by cytokines, growth factors and hormones which interact with a membrane tyrosine kinase receptor, inducing its phosphorylation and leading to signal transduction by subsequent phosphorylation of a series of proteins from RAS, RAF (ARF, BRAF, CRAF), MEK (MEK1 and MEK2) and MAPK/ERK family. The activated ERK goes to the nucleus where it activates transcription factors such as cMyc and CREB by phosphorylation (Molina and Adjei, 2006). The activated MAPK pathway also has an immunosuppressive effect due to downregulation of tumor antigens and decreased recognition by immune cells together with upregulation and infiltration of immunosuppressive cells after cytokine secretion (Ott and Bhardwaj, 2013; Yang et al., 2019).
Another important pathway commonly upregulated in melanomas is that of PI3K/AKT/mTOR which regulates cell proliferation, cellular response during stress and quiescence, contributing to tumor growth, metastasis and angiogenesis induction in melanomas (Porta et al., 2014). The most common mutations contributing to this activation are found as upregulation of the oncogene NRAS (15–20%) and loss of function or expression of the tumor suppressor PTEN (20–30%), yet these are largely mutually exclusive events (Hocker and Tsao, 2007; Aguissa-Toure and Li, 2012). On the other hand, PTEN loss can occur concomitantly with BRAF mutations, resulting in activation of RAS/RAF/MAPK and PI3K/AKT pathways (Tsao et al., 2004; Goel et al., 2006). Activated AKT phosphorylates several proteins, including antiapoptotic proteins (XIAP, BAD, BIM), MDM2, p21 and many others, allowing survival and progression of melanoma cells together with apoptosis inhibition (Madhunapantula et al., 2011). Interestingly, PTEN loss is also related to immunosuppressive properties such as lower sensitivity to T cell mediated cell death and reduced infiltrations of T cell infiltration in the tumor site, contributing to melanoma immune resistance (Peng et al., 2016).
Melanomas bearing BRAFV600 E mutations commonly also have altered MITF expression and activity (Levy et al., 2006). The MITF gene encodes a central regulator of melanocyte differentiation, development and function, besides several biological processes such as DNA repair, senescence, cell metabolism, survival, differentiation, proliferation and metastases formation (Goding and Arnheiter, 2019). MITF can be employed as a diagnostic marker for tumors from melanocytic origin, however, with different levels of expression correlating with distinct behavior of malignant cells (Levy et al., 2006). High MITF expression is associated with highly proliferative and poorly invasive phenotype while low MITF expression correlates with a slowly proliferative and highly invasive profile. In vivo studies demonstrated that although different in MITF expression, both phenotypes can establish tumors when inoculated into nude mice but with the invasive phenotype requiring a longer period to develop palpable tumors (Hoek et al., 2008; Vachtenheim and Ondrusova, 2015). However, both invasive and proliferative phenotypes can be present simultaneously since melanoma progression is not associated just with differential gene expression (Harbst et al., 2012; Cancer Genome Atlas, 2015), but also with dynamic transcription signature plasticity which contributes to tumor metastization due to the adaptive response to the tumor microenvironment (Roesch et al., 2016) (Figure 1A) Also reported for melanoma is the P29 S mutation in the RAC1 gene that is found in approximately 3% of melanomas but in almost 20% of patients resistant to BRAF inhibitors (Watson et al., 2014). RAC1 is involved in cellular adhesion, motility and differentiation, and the consequences of mutation in this gene are melanocytic to mesenchymal phenotype, increased tumor size and presence of metastasis (Lionarons et al., 2019).
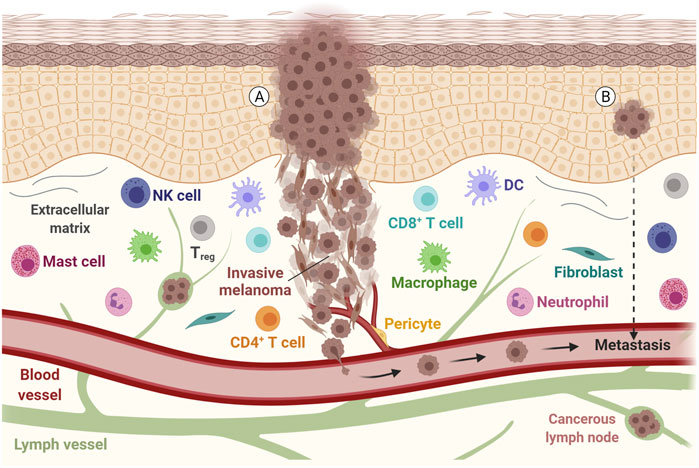
FIGURE 1. Representation of a melanoma tumor during invasion and metastasis. Tumor cells with epithelial and mesenchymal-like morphology are shown, along with other components of the tumor microenvironment, such as the extracellular matrix, immune cells and fibroblasts. The communication between these components, with lymph vessels, blood vessels and tumor cells may allow the tumor to spread. (A) Metastatic disease is found in patients with clinically identified proliferating melanoma, (B) but also in patients with an undetected source of tumor cells or evident primary lesion. DC, Dendritic cell; NK, Natural killer; Treg, Regulatory T cell. Adapted from “Melanoma Staging”, by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates.
The expression of immune-related genes also correlates with prognosis and response to melanoma immunotherapy as demonstrated after analysis of 45 patients submitted to anti-CTLA-4 therapy that had tumors with increased expression of genes related to immune signature (Ji et al., 2012). The interferon pathway is one of the key players in the response to immunotherapy and the type I and II interferons are mainly responsible for antitumor response due to increased immune recognition and apoptosis induction in tumor cells (Benci et al., 2016; Gao et al., 2016; Sharma et al., 2017). Many tumors have impaired interferon signaling resultant from alterations in regulatory genes such as loss of IFNGR1, IRF1, JAK2 and amplification of SOCS1 and PIAS4 (Gao et al., 2016). Analysis of the TCGA dataset and studies in vitro and in vivo showed that nearly 30% of melanoma samples present mutations in the interferon signaling pathway, which is associated with shorter overall survival (Gao et al., 2016). Moreover, increased expression of interferon-related genes (e.g., CXCL4, CXCL5, CXCL10, ID O 1, IRF1, STAT1 and others) was associated with benefit from anti-PD-1 and anti-CTLA-4 immunotherapy in melanoma patients (Ji et al., 2012; Gide et al., 2019). On the other hand, the suppression of the interferon pathway is associated with poor response to immunotherapy protocols due to immune evasion (Jerby-Arnon et al., 2018).
Melanoma patients’ deaths are mostly associated with distant metastasis development, showing a 5-years survival rate of around just 20% (Bomar et al., 2019). However, some melanoma patients have metastatic disease without evident primary lesion (Figure 1B) and in this case, the disease development is associated with immunoediting mechanisms together with loss of immunohistochemical melanocytic markers like S100 protein, HMB-45, Melan-A, SOX10 and MITF (Gyorki et al., 2013; Bankar et al., 2015). The exhaustion of the immune system and immune evasion are among the key factors that enable melanoma growth and metastasis formation (Passarelli et al., 2017; Motofei, 2019). Moreover, BRAF mutations are present in about 50–60% of metastatic melanoma cases (Zaman et al., 2019). Indeed, studies have revealed that cutaneous melanoma has a high mutation rate compared to other common tumors, with a mean tumor mutation burden (TMB) of over 20 mutations per megabase, one of the highest TMB among solid tumors (Cancer Genome Atlas, 2015; Zhang et al., 2016). Malignant melanoma is highly genetically heterogeneous, with prevalence of somatic mutations in primary tumors and metastatic lesions that also acquire numerous mutations during their formation (Hoek et al., 2006; Swick et al., 2012).
Melanoma Antigens and Immunogenicity
The immune system has the inherent property to distinguish self from non-self-antigens (Yarchoan et al., 2017). Despite the fact that tumor cells arise from healthy tissues, hence self, the ability of the immune system to recognize them is based on an important concept: neoantigens (also referred to as neoepitopes), which arise from tumor-specific mutations (Alexandrov et al., 2013). Since melanomas have a high mutational burden, which is reflected in the higher levels of neoantigens, they are more likely to promote immune response and be recognized by the immune system (Maleki Vareki, 2018). Described immunologically as “hot”, these tumors offer a huge repertoire of potential targets for T cells that, in principle, reflect a greater inflammatory infiltrate (Maleki Vareki, 2018). This point has been extensively explored in several approaches in cancer treatment, such as cancer vaccines against neoantigens and adoptive T cell transfer, which can be combined with immunotherapy targeting T cell inhibitory receptors, including cytotoxic T-lymphocyte associated antigen (CTLA)-4 and programmed cell death (PD)-1 (Peng et al., 2019). The clinical benefits of immune checkpoint inhibitors are often observed in high mutational load tumors, which may be related to the presence of tumor associated antigen-specific T cells (Banchereau and Palucka, 2018).
The antitumor immune response is mainly mediated by the adaptive immune system, especially the tumor-infiltrating lymphocytes (TILs) (Lugowska et al., 2018) that can recognize through the T cell receptors (TCRs) antigenic peptides presented via major histocompatibility complex molecules (Durgeau et al., 2018). In the case of human melanomas, the high degree of TILs and, more specifically, cytotoxic T cell infiltration, together with elevated expression of checkpoint receptors make melanoma patients more likely to respond successfully to immunotherapy (Galon and Bruni, 2019).
Antigen targets of immunotherapy can be divided into tumor associated antigens (TAAs), which include the cancer testis antigens (CTAs), and tumor specific antigens (TSAs) (Aurisicchio et al., 2018). TAAs include proteins encoded in the normal genome, usually expressed at low levels, and might be over-expressed in malignant cells. CTAs are normally expressed in testis, fetal ovaries, and trophoblasts, but can also be expressed in cancer cells. Because TAAs and CTAs are found in normal cells, their antigenicity depends on abnormal expression levels and, frequently, their presence in the tumor microenvironment can lead to immunological tolerance. The third class comprises antigens that are not encoded in the normal host genome and are originated by somatic mutations in the coding sequence, creating a unique peptide sequence (Gubin et al., 2015), or by insertion of oncogenic viral genes, such as E6 and E7 encoded by human papillomavirus type 16 that drive oral and cervical tumors (Walboomers et al., 1999).
Many TAAs have been used for years to assist clinical practice. For example, the human epidermal growth factor receptor (HER2) is routinely used for breast cancer prediction and prognosis (Patani et al., 2013). Melanoma TAAs include the type 1 melanoma antigen recognized by T cells (MART-1, also known as Melan-A) and the melanoma-associated antigen (MAGE). In a phase 1/2 clinical trial, a three-dose vaccine strategy using autologous DCs transduced with an adenoviral vector encoding the MART-1 antigen for metastatic melanoma patients showed that at least half of the treated patients had significant MART-1–specific T cell responses (Butterfield et al., 2008). Similarly, in a phase II study DCs were pulsed with a cocktail of melanoma-associated antigens, including MART-1 or MAGE-A1, MAGE-A2, MAGE-A3, gp100 and tyrosinase, and were subcutaneously injected in metastatic melanoma patients, for which 75% had an antigen-specific CTL response. Notably, patients in the vaccinated group with two or more peptide-specific responses had a significantly longer mean survival time (21.9 months) compared to treated patients who had less than two peptide-specific responses (8.1 months) (Oshita et al., 2012). Recently identified potential melanoma biomarkers, in addition to the more than 45 already studied (Belter et al., 2017), include metabolic components, for instance aminomalonic acid and phosphatidylinositol (PI) (Kim et al., 2017), as well as immune-related genes and TCRs (Charles et al., 2020; Huang et al., 2020). Although the clinical trials using TAA have shown initial immune response, most of them have failed to demonstrate durable beneficial effects. The main reasons are the lower TCR affinity to TAA and peripheral tolerance of TAA-reactive T cells (Melero et al., 2014).
Beyond TAAs, TSAs are attractive targets for immunotherapy. These neoantigens are expressed only in cancer cells and can be recognized by TCR with high affinity. Neoantigen-specific T cells are not subject to central and peripheral tolerance and, consequently, their activation leads to a lower induction of autoimmunity (Yarchoan et al., 2017). As a result, the antitumor immune responses to TSAs are more robust as compared to TAAs. An important advance in the understanding of TSAs and immune response was published in 2005 by Wölfel and colleagues. The authors found that the T cells of a patient were reactive against five mutated epitopes and the immunoreactivity against melanoma neoantigens predominated over the response to TAAs (Lennerz et al., 2005). In addition, Rosenberg’s group showed that the adoptive transfer of ex vivo–expanded TILs reactive against two neoantigens into a melanoma patient promoted complete tumor regression. All these studies support the role of neoantigens in the natural antitumoral T cell response (Zhou et al., 2005).
Besides its high TMB (Lawrence et al., 2013) and despite most human melanomas having a mutational load above 10 somatic mutations per megabase of coding DNA, which are generally sufficient to lead to the formation of neoantigens, T cell reactivity is not always observed (Linnemann et al., 2015). Recent studies revealed that both TMB and PD-L1 are not effective biomarkers for identifying patients who will have clinical benefit from checkpoint inhibitor therapy. An important point that may be considered is the clonality of neoantigens. Some evidence suggested that a minimum quantity of cells is required to generate T cell-mediated immune rejection (Gejman and Chang, 2018) and subclonal neoantigens are not presented by every cancer cell, so they are less effective in immune control of disease (Mcgranahan and Swanton, 2019). Furthermore, the majority of neoantigens are considered passenger events and, usually, their loss during tumor progression may be tolerated. However, when the mutations occur in genes required for tumor cell survival (such as cancer driver genes and genes required for cancer cell viability) and these genes are retained despite the events of copy number loss or transcriptional repression through methylation, the neoantigens are considered as essential neoantigens. Due to positive selection, these high-quality neoantigens cannot be repressed or deleted during tumor progression. Thus, both the quantity and the quality of neoantigens, more emphatically the quality, may explain why some patients are good responders to immunotherapy and others are not (Mcgranahan and Swanton, 2019).
However, the study of neoantigens has encountered barriers due to the lack of effective tools for their identification. In 2012, using a combination of next generation sequencing and algorithms for predicting the binding of peptides to MHC class I and class II molecules, Castle and coworkers identified TSAs in B16-F10 mouse melanoma cells (Castle et al., 2012). In parallel, exome sequencing and high-throughput MHC tetramer screening showed higher expansion of pre-existing T cells specific for tumor neoantigens in a human melanoma patient after treatment with checkpoint blockade immunotherapy (Van Rooij et al., 2013). To improve the adoptive transfer of T cells, Lu et al. genetically engineered autologous T cells to have neoantigen-specific TCRs. Isolated TILs were cultured and screened for the identification of neoantigen-reactive T cells to be further co-cultured with peptide-pulsed APCs. The single-cell RNA-sequencing allowed the identification of different neoantigen-specific TCRs, for instance a mutated KRAS-specific TCR, which could be successfully transduced into autologous T cells and recognize the specific neoantigens presented by the donor APCs (Lu and Robbins, 2016). The use of genomics and bioinformatics approaches in both mouse and human studies supported the rapid identification of mutant proteins expressed exclusively in cancer cells that act as neoantigens compared to conventional antigen-cloning approaches (Gubin et al., 2015) and highlight the potential of personalized cancer vaccines targeting neoantigens.
In a phase I study, 10 patients with stage IIIB/C or IVM1a/b melanoma were vaccinated with 13–20 personalized neoantigens peptides synthesized from sequencing of the tumors. After 20–32 months from vaccination, four patients with stage III disease were recurrence-free. Two patients with lung metastases had a complete response with the anti-PD-1 antibody, indicating the expansion of neoantigens specific T cells (Ott et al., 2017). Similar results were found by Sahin and colleagues who used personalized RNA-based ‘poly-epitope’ vaccine in 13 patients with stage III or IV melanoma. Each patient developed an immune response against at least three mutations. One patient with relapse and progressive disease at the time of vaccination presented a complete response after administration of anti-PD-1 antibody and eight continued disease-free 12–23 months later (Sahin et al., 2017). Both studies revealed that immune response was generated by CD4+ T cells and the vaccination provided the expansion of the neoantigen-specific T cells (Li L. et al., 2017). These studies confirm the potential of the immunogenic melanoma neoantigens and open novel possibilities for approaches using neoantigens as vaccinogenic agents associated with diverse delivery vehicles such as synthetic and biological nanoparticles and adenoviral vectors.
Although neoantigens are a promising strategy, the non-synonymous mutations that will originate the mutated protein depend on several factors that need to be present; the sequence with the mutation must be translated into protein, the mutated protein must be processed, and the peptides must be presented by MHC molecules. At the end of the process, the affinity between the mutated peptide and the patient’s MHC molecules will determine recognition by the TCR (Schumacher and Schreiber, 2015). All of these processes are susceptible to complications that can alter the TCR-MHC binding, contributing to tumor escape from the immune system and also confounding in silico approaches for the prediction of the most effective neoantigens.
Current Immunotherapeutic Strategies and Challenges
More than 10 different drug types have been approved by the FDA for the treatment of melanoma, including dacarbazine chemotherapy, BRAF and MEK-targeted therapy, recombinant interferon alpha-2b and IL-2, immune checkpoint inhibitors (ICIs), and oncolytic viral therapy (T-VEC). These strategies, together with radiation therapy and surgery comprise the clinical arsenal against primary and metastatic melanoma (Garbe et al., 2020; Jenkins and Fisher, 2020). During the past 20 years, ICIs (commonly referred to as immunotherapy) have taken on a leading role and occupied center stage in the melanoma treatment scene. Extraordinary results were achieved with anti-CTLA-4, anti-PD-1 and anti-PD-L1 in a time when targeted therapy only offered reasonable short-term, but poor long-term, overall survival (Ribas et al., 2012; Spagnolo et al., 2016; Li X. et al., 2017; Karlsson and Saleh, 2017). Their success turned ICIs into the standard of care for advanced melanoma, after surgery, demonstrating that when the immune system is activated properly, it may lead to durable long-term responses.
In terms of improved efficacy and reduced toxicity, studies have found that anti-PD-1 (Nivolumab or Pembrolizumab) is superior to anti-CTLA-4 (Ipilimumab) (Li X. et al., 2017; Karlsson and Saleh, 2017). No statistically significant differences have been found between Nivolumab and Pembrolizumab, although Nivolumab presents a slight improvement in terms of median overall survival (Moser et al., 2020). Attempts to reduce toxicity through the combination of Nivolumab and Ipilimumab in reduced doses resulted in greater efficacy, but less tolerability than monotherapy (Karlsson and Saleh, 2017; Turajlic et al., 2018; Moser et al., 2020). Nevertheless, most patients still fall in the non-responder-to-ICIs category and many experience immune-related adverse events (irAE), suggesting that despite their potential, resistance and toxicity continue to be their major hurdles.
The lack of ICI response in melanoma patients may be due to different immunological reasons, such as the absence or exclusion of T cells within the tumor microenvironment (TME) or insufficient antigen presentation and priming. Combinations of strategies that turn ICI resistant tumors into responders are being pursued, but, in addition to immunotherapies that directly aim to activate the immune system, all therapeutic strategies would potentially offer some degree of immune activation as well, either by inducing immunogenic cell death of tumor cells and providing antigenic supplies to antigen-presenting cells (APCs), or by triggering inflammatory pathways that will set the tone of the microenvironment towards a possible antitumor response (Chen and Mellman, 2013). Thus, the most obvious way to tackle resistance and perhaps reduce toxicity is through the combination of currently approved therapies.
Recently, studies in melanoma have focused on combining targeted therapies such as BRAF and MEK inhibitors with ICIs, aiming to increase efficacy by tackling resistance; the biology behind this strategy is to promote immune changes within the TME, particularly the release of cancer cell antigens and antigen-presentation by APCs to T cells in a context of checkpoint inhibition, which in turn lead to the activation of effector, tumor-specific T cell clones (Chen and Mellman, 2013). The idea of the strategy is to combine the rapid and deep response of targeted therapy with the durable response of ICIs (Kim et al., 2014; Dummer et al., 2020). In vivo melanoma models have shown tumor growth delay, reduced tumor size and prolonged overall survival with the combination of anti-PD-1/anti-PD-L1 and BRAF and MEK inhibitors (Dummer et al., 2020). Currently, there are two studies in clinical phase III, IMspire150 to assess the combination of Atezolimumab (anti-PD-L1) and vemurafenib (BRAF inhibitor), and COMBI-I part 3, assessing the combination of Spartalizaumab (anti-PD-1) plus dabrafenib (BRAF inhibitor) and trametinib (MEK inhibitor); both studies are ongoing. Still, in terms of timing of administration, it is debatable if targeted therapy should be administered before, at the same time or after ICIs; although, the changes induced in the microenvironment by BRAF inhibitors including reducing immunosuppressive cytokines, increasing availability of tumor antigens and infiltration of tumors by immune cells may sensitize tumors to ICIs (Dummer et al., 2020).
Due to the important role that checkpoint molecules play in the homeostasis of the immune system, the intravenous administration and systemic action of ICIs are known to induce undesired irAE. A variety of inflammatory and autoimmune events, ranging from grade 1 to 4 toxicities, have been observed as a result of ICI usage; the most prevalent are the dermatologic toxicities (from grade 1 to 2 rash, pruritus, vitiligo, dermatitis to grade 3 to 4 Stevens-Johnsons syndrome and epidermal necrolysis), present in 50% of melanoma patients treated with anti-CTLA-4 and up to 40% for anti-PD-1 and anti-PD-L1. Other less frequent, but not less relevant, irAEs include gastrointestinal toxicities, from diarrhea to severe colitis, hepatitis with or without elevation of transaminases or bilirubin and fulminant hepatitis; endocrinopathies that include a range of thyroid and pituitary toxicities, adrenal insufficiency and type I diabetes; neurologic toxicities such as autoimmune encephalitis, myasthenia gravis and Guillain-Barré syndrome; renal toxicities include hematuria and acute interstitial nephritis and lupus-like nephritis; ocular toxicity such as uveitis, ulcerative keratitis and retinopathy; cardiac toxicities such as myocarditis, pericarditis, fibrosis, arrhythmias and heart failure and finally, hematological toxicities including hemolytic anemia, thrombocytopenia and eosinophilia (El Osta et al., 2017; Calvo, 2019; Kennedy and Salama, 2019). Despite the increased efficacy, the occurrence of irAEs is indeed more frequent with combinations of different ICIs (Li X. et al., 2017; El Osta et al., 2017; Karlsson and Saleh, 2017; Turajlic et al., 2018). As expected, irAE clinical manifestations are mostly managed with steroids or immunosuppressants, that when used carefully, ameliorate irAE without compromising ICIs efficacy (Karlsson and Saleh, 2017; Calvo, 2019). Even though the possibility of the occurrence of a grade 3 to 4 or chronic irAE is rare, future studies must consider the importance of segregating, through biomarkers, which patients will actually benefit from ICI therapy. Unfortunately, to date, the ideal biomarker for the indication of immunotherapy has not yet been identified (Jessurun et al., 2017). Recent studies in murine melanoma models have suggested a pivotal role of the gut microbiome for ICI efficacy, showing that the presence of some bacterial populations may be associated with increased response to ICIs, while others may be associated with the lack of response (Sivan et al., 2015; Vétizou et al., 2015). Theoretically, through the modulation of microbial populations, non-responders could be turned into responders and perhaps even become less susceptible to ICI toxic effects. In addition, microbial populations may comprise potential biomarkers of ICIs response (Vetizou and Trinchieri, 2018).
Still, there is a clear need for more effective and less toxic strategies. Other checkpoint molecules such as LAG-3, TIM-3, TIGIT, VISTA and B7-H3 and other TME molecules such as IDO have been demonstrated to be promising targets for melanoma and other advanced solid tumors in preclinical studies, granting their passage into clinical trials, although all of them are currently ongoing (Kwiatkowska et al., 2019; Qin et al., 2019). The most studied target, with more than 60 open clinical trials, is the LAG-3 molecule; after binding MHC-II molecules or fibrinogen-like protein 1 (FGL1), LAG-3 restrains the activation, proliferation and cytokine production capacity of Th1 cells while contributing to the suppression activity of Tregs (Qin et al., 2019; Murciano-Goroff and Warner, 2020). In an ongoing phase 1/2 study, anti-LAG-3 antibody (Relatlimab) as monotherapy or in combination with anti-PD-1 is being tested for melanoma patients who were resistant to classical ICIs; early results suggest that the combination is safe and can even increase the antitumor activity of anti-PD-1 alone in ICI-resistant melanoma patients (Kwiatkowska et al., 2019).
Stage IV metastatic melanomas have also been subjected to a personalized strategy developed by Steven Rosenberg at the NIH, resulting in outstanding outcomes and durable complete responses in a few melanoma patients (Prickett et al., 2016). From the lessons learned with IL-2 immunotherapy, Dr. Rosenberg recognized the potential of expanding functional T cells while circumventing the toxicity induced by IL-2 systemic administration, through the ex vivo activation with IL-2 of tumor infiltrating lymphocytes which, upon reinfusion, can then target tumor neoantigens (Rosenberg, 2011; Lu et al., 2014; Rosenberg, 2014; Prickett et al., 2016). Despite the great potential of this kind of strategy, known as adoptive cell therapy (ACT), the sophisticated methods required could hamper their inclusion in the clinical routine.
Other strategies that trigger important pathways of the innate immune response have been recognized in preclinical models for their importance in setting the tone of tumors for antitumor responses. Different agonists that trigger innate receptors and sensors such as TLR, STING, RIG-1 and NLR are currently being developed and some, such as STING agonists, have been considered in clinical trials as ICI adjuvants for advanced melanoma and other solid tumors (Hu and Li, 2020); the activation of these receptors is intended to mimic the immune response against viruses, that ultimately trigger cytokines and chemokines that will break the suppressive TME and allow the infiltration of immune cells inside tumors (Clavijo-Salomon et al., 2017). Consistent with the idea of harnessing innate receptor agonists and antiviral responses to fight tumors, oncolytic viruses have demonstrated tremendous potential for the treatment of melanoma, since in addition to awakening antiviral immunity, they can also directly kill tumor cells.
Using Engineered Viruses for Cancer Immunotherapy
The continued study of viruses has brought countless advances not only to the understanding of the molecular basis of diseases, such as cancer itself, but also perspectives for its use as a genetic and therapeutic tool (see Figure 2 and Supplementary Table S1 for key publications in the development of gene therapy), including in Brazil, the first Latin American country with a defined regulatory process for the registration of advanced therapy products. Intriguingly, on one hand viruses that trigger cancer have been discovered (Rous, 1910), yet on the other it has been suggested that some viral infections could improve clinical outcomes for some patients with different types of cancer (Hoster et al., 1949; Newman and Southam, 1954). Though unthinkable today, in 1949 Herman A. Hoster and coworkers used, deliberately, wild type hepatitis B virus in clinical trials of patients with Hodgkin’s disease. In this study, 21 patients were intentionally exposed to the hepatitis virus and 4 of them had improvement in the clinical course, at least with regard to Hodgkin’s. (Hoster et al., 1949). This study was one of the first to intentionally use viral activity to alter the progression of cancer. As presented below, current approaches use a deeper understanding of viral properties, the molecular basis of cancer as well as recombinant DNA techniques in order to use viruses as anti-cancer agents, an approach known as virotherapy or oncolytic viruses (Figure 3). The term “oncolytic viruses” (OV) is typically used to describe genetically modified viruses that selectively infect cancer cells inducing their death, theoretically, without affecting non-malignant tissues (Vähä-Koskela et al., 2007; Russell et al., 2012). Some viruses offer oncolytic properties naturally and do not require modification, for example, Vesicular stomatitis virus, Myxoma, Reovirus, and Newcastle disease virus (Roberts et al., 2006; Kelly and Russell, 2007; Jhawar et al., 2017). Although oncolytic viruses may enter normal cells, progression of the viral life cycle should be inhibited due to molecular components that block viral replication. In tumor cells, many of these mechanisms are dysfunctional or have been suppressed during tumor progression and thus provide a selective advantage for replication and dissemination of viral progeny (Kaufman et al., 2015). For example, resistance to apoptotic cell death, a critical hallmark of cancer, implies that tumor cells are lacking in a fundamental anti-viral defense, a point that may be exploited in order to promote viral replication (Russell et al., 2012). The interferon pathway was originally identified due to its anti-viral properties and, as mentioned above, plays an essential role in inducing innate and adaptive immune responses. While normal cells can defend themselves from viruses using the interferon pathway, tumor cells frequently present deficiencies in interferon response. Thus, this characteristic of tumor cells can be deliberately exploited for the development of OV (Murira and Lamarre, 2016; Gessani and Belardelli, 2021). Tumor cell killing in response to virotherapy occurs due to virus replication and induction of anti-viral responses. As we will detail below, the anti-viral response, which includes activation of innate and adaptive immunity, may be just as important, if not more so, than viral replication.
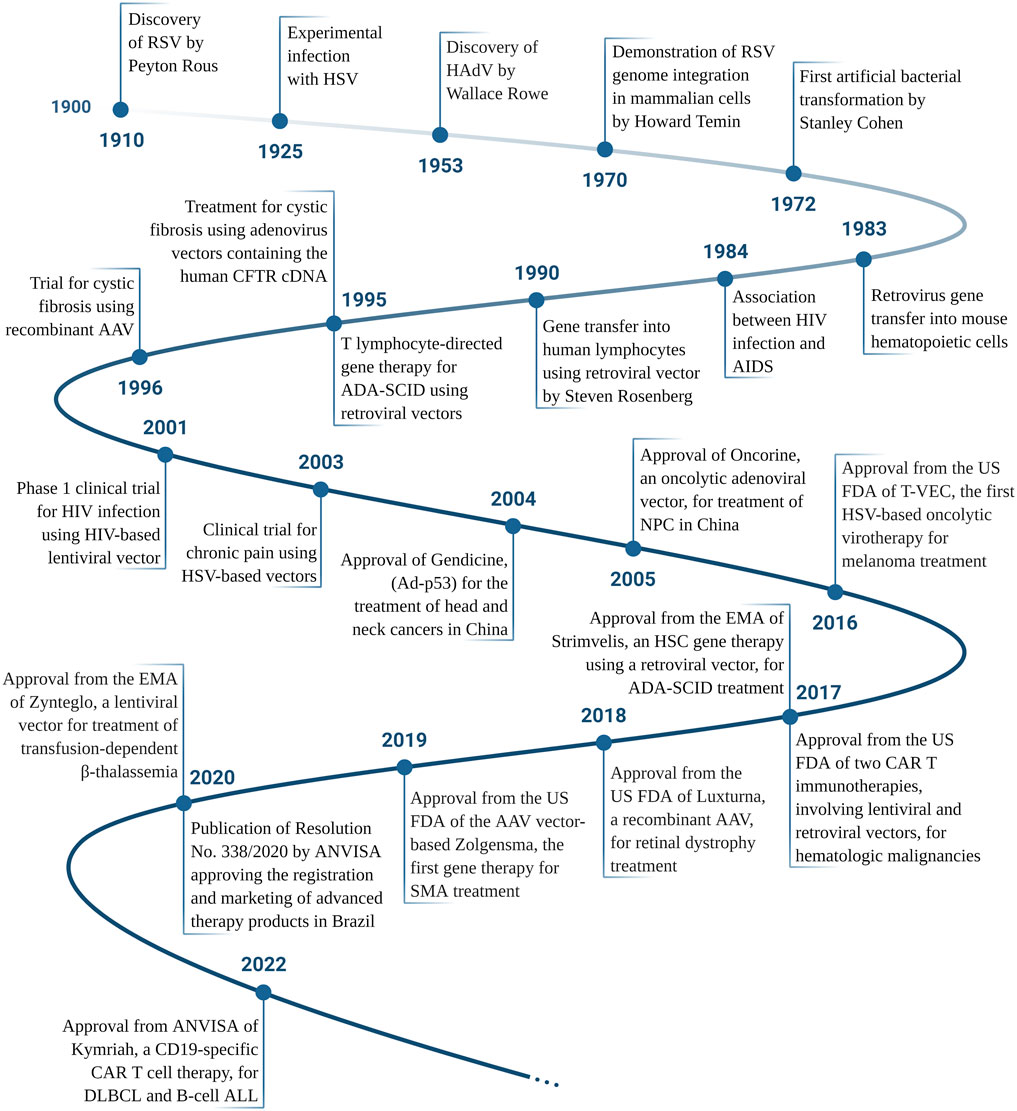
FIGURE 2. Timeline of the main achievements in the gene therapy and virotherapy fields, according to the date of publication. RSV, Rous Sarcoma Virus; HSV, Herpes Simplex Virus; HAdV, Human adenovirus; HIV, Human Immunodeficiency Virus; AIDS, Acquired Immunodeficiency Syndrome; ADA-SCID, Adenosine Deaminase Severe Combined Immunodeficiency; CFTR, Cystic Fibrosis Transmembrane Conductance Regulator; AAV, Adeno-Associated Virus; NPC, nasopharyngeal carcinoma; US FDA, The United States Food and Drug Administration; EMA, European Medicines Agency; CAR, Chimeric Antigen Receptor; SMA, Spinal Muscular Atrophy; ANVISA, Agência Nacional de Vigilância Sanitária (National Agency for Sanitary Vigilance, the federal body in Brazil that regulates new drugs, among other health related items); DLBCL, Diffuse Large B-Cell Lymphoma; ALL, Acute Lymphoblastic Leukemia. Please see Supplementary Table S1 for references. Created with BioRender.com.
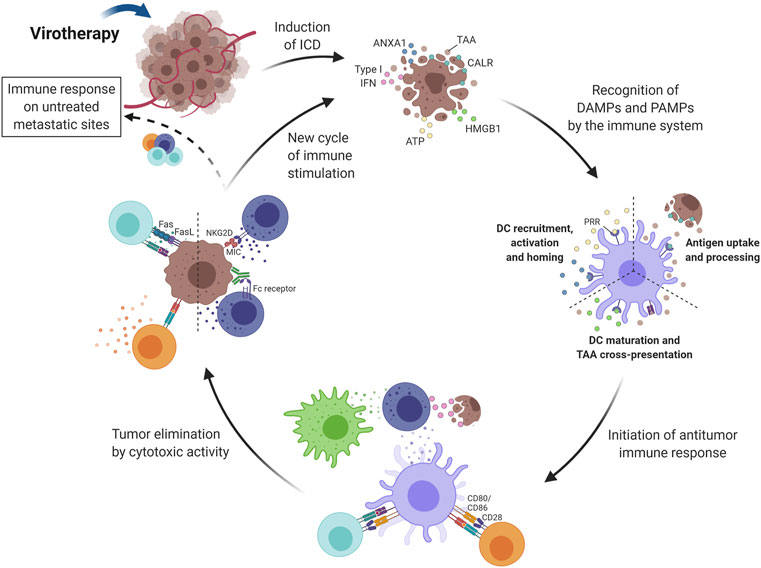
FIGURE 3. Induction of immune activity in response to virotherapy. Virotherapy can induce ICD, which is characterized by the release of DAMPs (i.e., CALR, HMGB1, ANXA1 and type I IFN) during cell death. These danger signals, along with TAAs and PAMPs also released by OV-treated tumor cells, promote the initiation of an immune response after recognition by antigen-presenting cells (mainly DCs). ATP and ANXA1 are responsible for DC recruitment, activation and homing; CALR increases antigen uptake and processing; and HMGB1 promotes DC maturation and antigen cross-presentation. Next, TAAs are presented to T lymphocytes that can differentiate into both helper and cytotoxic cells since presentation occurs by class I and II MHC proteins. The cytotoxic activity of NK cells is also important for tumor elimination and type I IFNs play an important role in their stimulation together with signals provided by activated DCs and macrophages. Activated effector cells are then capable of recognizing and eliminating tumor cells by different mechanisms, for example, Fas-FasL interaction with CD8+ T lymphocytes and MHC recognition by the NKG2D receptor on NK cells. Another key advantage of activating the immune system relies on the possibility of reaching untreated metastatic sites through circulating immune cells. The cycle restarts as dying tumor cells release more antigens and intracellular molecules that keep on activating the immune system. DC, Dendritic cell; NK, Natural killer; Treg, Regulatory T cell; ICD, Immunogenic Cell Death; TAA, Tumor-Associated Antigen; CALR, Calreticulin; HMGB1, High Mobility Group Box-1; ATP, Adenosine Triphosphate; IFN, Interferon; ANXA1, Annexin A1; DAMPs, Damage-Associated Molecular Pattern; PAMPs, Pathogen-Associated Molecular Pattern; PRR, Pattern Recognition Receptor; NKG2D, Natural Killer Group 2D; OV, Oncolytic Virus; MHC, Major Histocompatibility Complex. Created with BioRender.com.
Our understanding of anti-viral and immunostimulatory properties of both normal cells and neoplasms advanced in the late 1980 s with the discovery of Toll like receptors (TLRs), and later families of Nod (nucleotide-binding and oligomerization domain) -like receptors (NLRs) (Takeda and Akira, 2004; Hansson and Edfeldt, 2005; Inohara et al., 2005). TLRs are present in antigen-presenting cells, such as DCs, macrophages and B cells, as well as T cells, NK cells, and non-immune cells (epithelial and endothelial cells, and fibroblasts), and recognize pathogen-associated molecular patterns (PAMPs), present in both viruses and bacteria, which attract and activate other cells that mediate adaptive immune responses (Fitzgerald and Kagan, 2020). These receptors provided evidence for understanding Coley’s strategy (Coley’s Toxin) (Coley, 1991) and the successful application of BCG in cases of bladder cancer (Lamm et al., 1980), both containing bacterial PAMPs, as well initial works that explored adjuvant effects of viral preparations or natural infections (Dock, 1904; Hoster et al., 1949; Newman and Southam, 1954). In the case of OV, the vector itself provides PAMPs in the form of viral proteins, DNA and RNA that are detected by the cell and initiate the anti-viral cascade through TLRs and NLRs.
In addition, viral infections naturally trigger danger-associated molecular patterns (DAMPs), stress-signaling proteins and inflammatory cytokines (Matzinger, 2002; Tang et al., 2012). As a consequence, strategies employing viruses can induce immunogenic cell death (ICD) of cancer cells, generating chemo-attractants for cells of the immune system (Naik and Russell, 2009). Considering the characteristics of the tumor microenvironment, the use of these oncolytic strategies has the possibility of reversing the immunosuppressive profile and promoting the presentation of the repertoire of tumor antigens in an immunostimulatory context (Bartlett et al., 2013). Besides that, oncolysis triggered by viral particles, replicative or not, has the potential to aggregate immune responses against viral proteins and subvert this for antitumor immunity. When viral systems with replicative capacity are used, tumor selectivity gives these cells new viral epitopes, in addition to the TAAs and/or TSAs. This increases the exposure of these cells to both innate and adaptive immune responses, which may break the vicious cycle of tumor immunoediting. This is the main difference between oncolysis triggered by viral vectors and the approaches outlined above, and for this reason oncolytic virotherapy provides additional advantages over existing therapies that trigger ICD.
As shown in Table 1, several clinical trials have been performed using OV for the treatment of melanoma and novel approaches are being developed. Melanoma was the first neoplasm for which an oncolytic virus therapy was registered by the U.S. Food and Drug Administration (FDA). Approved in 2015, T-VEC, also known as Imlygic (OncoVex, talimogene laherparepvec) is prescribed for patients with advanced melanoma (Stage IIIB, IIIC or IV) that cannot be completely removed with surgery (Fukuhara et al., 2016). The history of this virotherapy exemplifies the path of a new biotechnological tool, from its conception in basic science to clinical trials aimed at proving its safety and efficiency for use in humans. Based on a modified herpes simplex virus (HSV-1), T-VEC was engineered deleting ICP34.5 and ICP47 viral genes. ICP34.5 blocks a cellular stress response to viral infection promoted by IFN-γ and ICP47 impairs the immune system’s CD8 T-cell response against infected cells, thus these viral components render normal cells susceptible to HSV replication. In T-VEC, the deletion of ICP34.5 and ICP47 prevents replication in normal cells, but tumor cells support viral replication due to defects in specific cellular pathways. The deletion of ICP47 also leads to upregulation of the viral protein US11, which further propels virus replication (Goldsmith et al., 1998; Liu et al., 2003). In addition, a constitutive expression cassette was inserted to provide granulocyte-macrophage colony-stimulating factor (GM-CSF) that, in conjunction with other cytokines, contributes to the attraction, differentiation and activation of APCs, such as DCs and macrophages, in the treated areas. Moving forward to phase I clinical trials, T-VEC was well tolerated and caused only mild adverse events such as local erythema and fever (Hu et al., 2006). Next, phase II clinical trials were realized in 50 patients with melanoma, stages III and IV, revealing a 26% response rate, including 8 with complete remission and another 5 with positive partial responses (RECIST) (Senzer et al., 2009). Finally, approval by the FDA and EMA was granted after an open-label phase III study that demonstrated the higher durable response rate (DRR) with a positive impact on overall survival compared to appropriate controls (Andtbacka et al., 2015). Kaufman and collaborators demonstrated that treatment with T-VEC induced a weakening of T cells responsive to MART-1 (melanoma-associated antigen) and, concomitantly, there was a decrease in regulatory T lymphocytes (Kaufman et al., 2010). Intriguingly, T-VEC is administered intratumorally, virus spread is only local, but immune response can mediate tumor regression in non-treated foci. This leads us to question the importance of viral replication itself vs. the induction of antitumor immunity for the success of the modality. Another point to be debated is if viral epitopes would indeed be needed for the effectiveness of these immune responses to contain and eliminate the primary tumor, as well metastases.
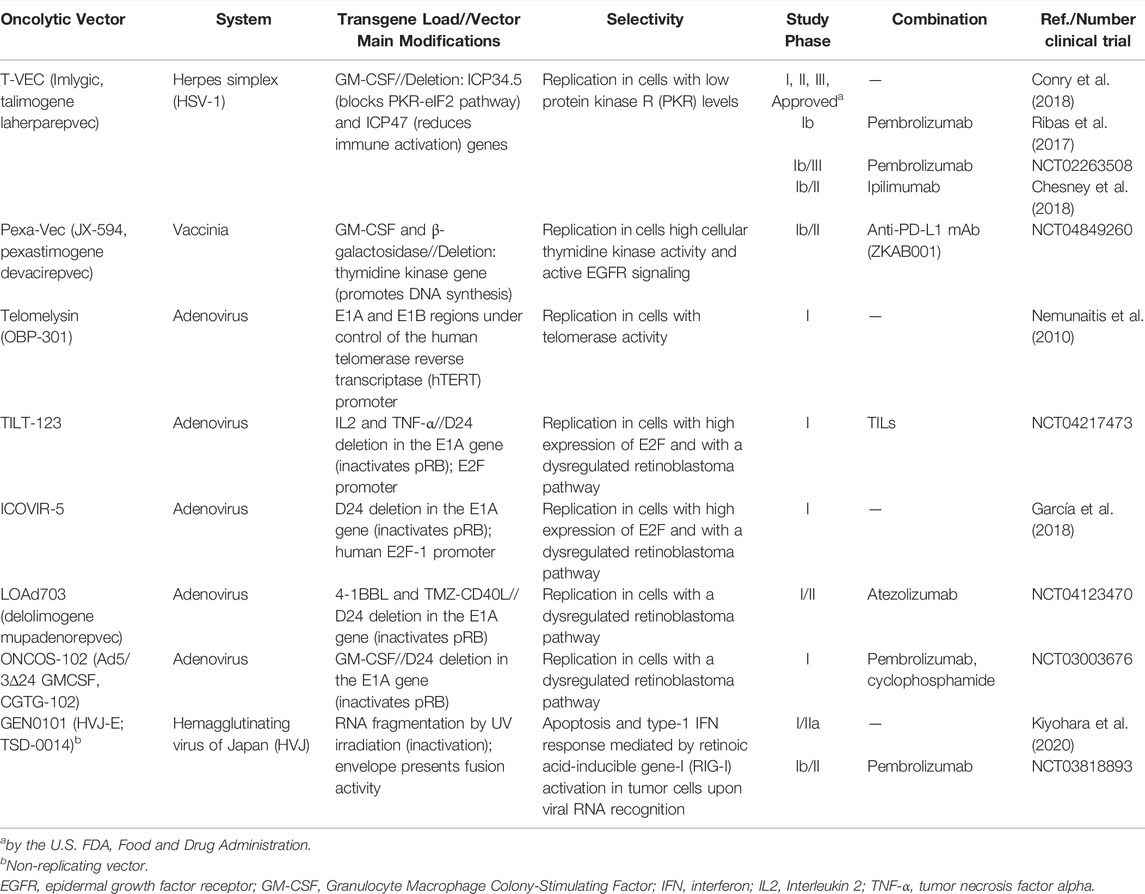
TABLE 1. Clinical trials for treatment of melanoma with oncolytic viruses.
Oncolytic viruses have also been developed based on other viral systems. Pexa-Vec (JX-594) is derived from vaccinia virus inactivated by the deletion of the thymidine kinase gene, and modified for the expression of GM-CSF and β-galactosidase transgenes, is in the clinical testing phase for colorectal cancer and hepatocellular carcinoma (Breitbach et al., 2015; Park et al., 2015). A phase I/II clinical trial suggests that intratumoral injection of Pexa-vec is safe and with promising results being effective in treating both injected and distant disease in patients with surgically incurable metastatic melanoma (NCT00429312) (Mastrangelo et al., 1999). Some clinical trials testing Pexa-vec for melanoma are in progress (NCT04849260, NCT02977156). These findings reinforce interest in the use of OV as an immunotherapeutic for melanoma.
Adenovirus is another viral system widely used in immunotherapy for cancer. Oncorine (Onyx-015, H101), for example, is an oncolytic adenovirus-based used for the treatment of head and neck squamous cell carcinoma (Liang, 2018). It was designed to be replicated only in cells that have lost p53 activity, which would cover a large percentage of human neoplasms (Wei et al., 2018). Oncorine was approved in 2005 by State Food and Drug Administration, China (SFDA) (Wei et al., 2018). Although clinical trials of Oncorine for human melanoma have not yet been performed, Hu and colleagues found evidence that the use of ZD55-IL-24 (similar to Oncorine) in an animal model of melanoma prevents tumor growth and induced systemic antitumor immunity (Hu et al., 2020).
Telomelysin (OBP-301) is an oncolytic adenovirus utilizing the human telomerase reverse transcriptase (hTERT) promoter to control the expression of E1A and E1B, key genes that regulate adenoviral replication. In normal cells, the hTERT promoter should not be active, thus the lack of E1A/E1B prevents viral replication. Since hTERT is generally over active in tumor cells, E1A/E1B will be expressed, thus providing selectivity of virus replication (Trager et al., 2020). A phase I clinical trial showed good tolerability, with patients presenting only mild symptoms (grades 1 and 2), such as pain, induration, fever, and chill, and none of them had severe symptoms (grades 3 and 4). Despite having a small cohort, the results were promising, with seven of the twelve patients fulfilling RECIST criteria for stable disease at 56 days after the treatment (Nemunaitis et al., 2010; Trager et al., 2020). In 2016, a phase II clinical trial was initiated testing Telomelysin in patients with unresectable stage III and IV melanoma, though results are not yet available (NCT03190824). In addition, several phase I clinical trials involving replicative adenoviral vectors for different types of cancer have already been carried out, such as TILT-123, ICOVIR-5, LOAd703, ONCOS-102, as shown in Table 1.
As mentioned above, the induction of ICD is essential for the success of oncolytic approaches, bringing into question the importance of virus replication to achieve this goal. Many approaches are being developed for the induction of oncolysis even when the virus, such as adenovirus, does not replicate (Tessarollo et al., 2021). Our research has focused on the use of non-replicating adenoviral vectors for the transfer of genes intended to induce both cell death and immune activation including reversal of the immunosuppressive TME. That is to say, our approach induces oncolysis without the need for a replicating vector. In the first instance, the objective of the gene transfer is to induce immunogenic cell death in cancer cells, and subsequently, a second wave of death due to cytotoxicity mediated by cells of the immune system, mainly T and NK cells. Evidence from our studies indicates that the combined use of interferon-β (IFNβ) and p19Arf (alternate reading frame, p19Arf in mice and p14ARF in humans) induces melanoma cell death by necroptosis and is associated with an anti-viral response and the release of immunogenic factors (such as HMGB1, ATP and calreticulin) (Merkel et al., 2013; Ribeiro et al., 2017; Cerqueira et al., 2020). In our pre-clinical models, this therapy was well tolerated in animals where no side effects, such as liver transaminase induction, were observed and showed promising results in inhibiting tumor growth in s.c. tumors after in situ gene therapy, as well as prolonging the survival of treated animals (Cerqueira et al., 2020; David et al., 2020). We have also confirmed the induction of an antitumor immune response in vaccine and immunotherapy settings, with critical involvement of NK cells, CD4+ and CD8+ T cells, when our vector is used in immunocompetent C57BL/6 mice and B16-F10 mouse melanoma cells (Medrano et al., 2016). When using the human melanoma cell line SK-MEL 147 we demonstrated that transduction with adenoviral vectors encoding p14ARF and IFNβ resulted in activation of monocyte-derived DCs (Cerqueira et al., 2020). In turn, these promoted the activation and priming of T cells, as well as the pro-inflammatory cytokine profile (Cerqueira et al., 2020). Together, these studies show that our gene transfer approach is a promising immunotherapy for melanoma (Hunger et al., 2017; Medrano et al., 2017; Cerqueira et al., 2020). The use of non-replicating vectors may be an advantage for our approach since the delivery of IFNβ can be counterproductive for replicating OVs (Cerqueira et al., 2020; Geoffroy and Bourgeois-Daigneault, 2020; Tessarollo et al., 2021). The results to date are encouraging and research will continue, with critical development using clinically relevant models, such as testing with patient-derived tumor samples, including PDO and immunological ex vivo models (Strauss et al., 2018).
Combining Virotherapy With Different Immunotherapeutic Interventions
Since the immune system consists of multiple components that act in an ordered and coordinated manner, immunotherapies that target a single step may not promote the entire cascade of events. Instead, combined approaches, especially those that facilitate different steps in the immune response, may provide an improved clinical outcome. As described above, the role of OV is to induce ICD, but this does not guarantee the effectiveness of the steps that follow, including antigen presentation, T cell priming and cytolytic activity. With the success of immunotherapies that target these points, especially ICI, a wide range of combination therapies is possible, as discussed here and summarized in Figures 4, 5.
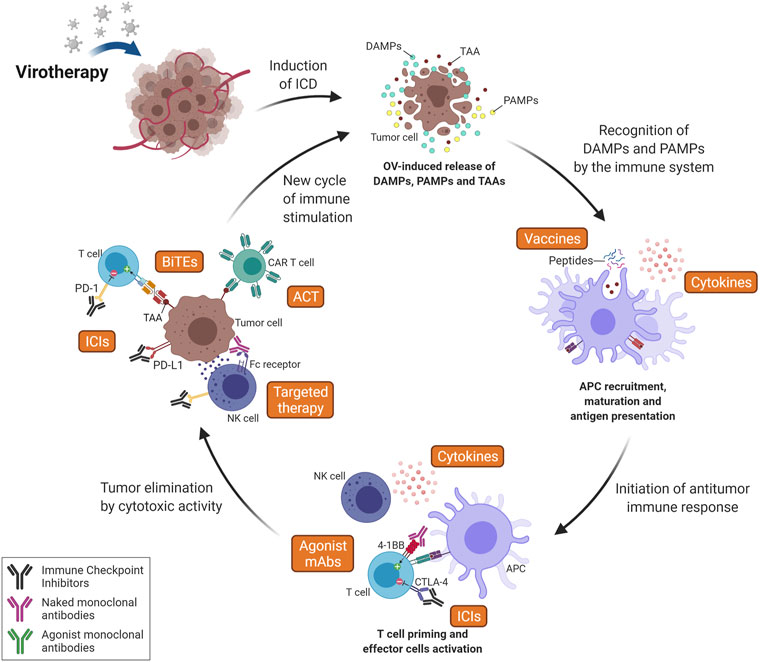
FIGURE 4. Combining Oncolytic Virus (OV) therapy with other immunotherapy strategies. Since OV acts at the first step of the cancer-immunity cycle, releasing DAMPs, PAMPs and TAAs, its association with additional interventions aiming to establish an antitumor response is favored. The induction of ICD leads to recruitment and activation of antigen-presenting cells (APCs), which can increase the efficiency of vaccine-based approaches (especially peptide vaccines) and generate a stronger T cell response. Immune Checkpoint Inhibitors (ICIs) can increase activation of T cells during priming (here represented by anti-CTLA-4 mAb) and also increase T-cell effector activity in the tumor (i.e., anti-PD-(L)1 mAbs) following OV-induced immune cell infiltration. Agonist monoclonal antibodies (mAbs) are also an interesting intervention to increase T cell activation against TAAs (released after oncolytic therapy) by recognizing and activating costimulatory T cell receptors (i.e., 4-1 BB). Targeted therapy can also potentiate the immune responses to target tumor cells by increasing the effector activity of the innate immune system, including NK cell-mediated ADCC, macrophage-mediated ADCP and complement-mediated CDC. It is important to note that the cytotoxic activity of innate immune cells increases antigen release and, consequently, T cell recruitment. Virotherapy can also increase efficiency of Adoptive Cell Therapy (ACT), here represented by CAR T cells, by promoting a pro-inflammatory microenvironment, enabling T cells enhanced functions and leading to a higher recognition and elimination of tumor cells even in solid tumors. Therapy using bi-specific antibodies, such as BiTEs, can also be enhanced by oncolytic therapy as OVs precondition tumors in terms of T cell recruitment and activation. Finally, cytokines can act at many key steps of the process, such as antigen presentation and T cell priming, activation and recruitment. Furthermore, these factors have an important role at maintaining a favorable microenvironment for the survival of activated immune cells and the sustainment of the immune response. ICD, Immunogenic Cell Death; TAA, Tumor-Associated Antigen; DAMPs, Damage-Associated Molecular Pattern; PAMPs, Pathogen-Associated Molecular Pattern; NK, Natural killer; CTLA-4, Cytotoxic T-Lymphocyte-Associated protein 4; PD-1, Programmed cell Death protein 1; PD-L1, Programmed cell Death Ligand 1; CAR, Chimeric Antigen Receptor; BiTEs, Bi-specific T-cell Engagers; ADCC, Antibody-Dependent Cell-mediated Cytotoxicity; ADCP, Antibody-Dependent Cellular Phagocytosis; CDC, Complement-Dependent Cytotoxicity. Created with BioRender.com.
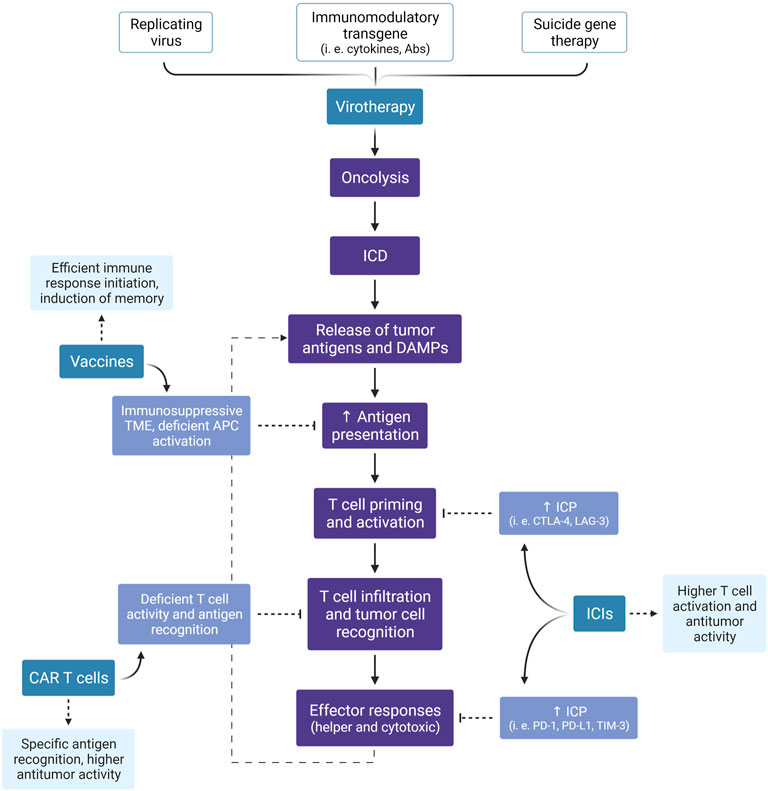
FIGURE 5. Expected benefits of combined immunotherapies. Besides the direct antitumor effect of virotherapy, especially by the lytic effect of oncolytic viruses, this treatment modality can induce an anti-cancer immune response through the promotion of ICD. Here, we summarize the events illustrated in Figures 3, 4 and highlight the expected advantage when combining virotherapy with other immunotherapy strategies. ICD, Immunogenic Cell Death; DAMPs, Damage-Associated Molecular Patterns; TME, Tumor Microenvironment; APC, Antigen-Presenting Cell; ICP, Immune Checkpoint; ICI, Immune Checkpoint Inhibitor; CTLA-4, Cytotoxic T-Lymphocyte-Associated protein 4; LAG-3, Lymphocyte-Activation Gene 3; PD-1, Programmed cell Death protein 1; PD-L1, Programmed cell Death Ligand 1; TIM-3, T cell Immunoglobulin and Mucin domain-containing protein 3; CAR, Chimeric Antigen Receptor. Created with BioRender.com.
In the same way that combinations can enhance therapeutic efficacy, they can also be counterproductive or even intensify unwanted side effects. Therefore, clinical trials are essential to provide critical correlative data that can support the combined use of these new therapeutic options. Despite the importance of pre-clinical assays for the initial proof of concept, the animal models used in this stage are limited, since the main potential is precisely the performance of the human immune system (Bareham et al., 2021; Patton et al., 2021). Macedo and collaborators published a review that provides a current overview of virotherapy clinical trials, as well as the resulting combinations (Macedo et al., 2020). According to what they observed, the majority of clinical trials (62.9%) published between 2000 and 2020 investigated only the action of oncolytic viruses used as monotherapy, while 37.1% where OV was administered in combination with at least one other anti-cancer treatment or medication. Of these combinations, the most frequent were cytotoxic chemotherapy agents, chemotherapy prodrugs and radiotherapy. Only a small fraction (5%) of all clinical tests with oncolytic viruses investigated the combination with other immunotherapies such as ICIs or cytokines (Macedo et al., 2020).
Inhibitors of Immune Checkpoints
As previously mentioned, melanomas have a high mutational load, which contributes to the generation of neoantigens which can be targeted by the patients’ T cells, but their function is often impeded due to upregulation of PD-1. There is also evidence that OV-based therapies increase infiltration of T cells in the tumor (Ribas et al., 2017). Thus, the combination of these immunotherapies is an interesting option (Chiu et al., 2020; Hwang et al., 2020). In randomized, open-label phase I and II studies, T-VEC combined with ipilimumab (anti-CTLA4) showed significantly greater efficacy compared to ipilimumab alone (Puzanov et al., 2016; Chesney et al., 2018; Trager et al., 2020). Likewise, another phase 1b study using T VEC plus pembrolizumab (anti-PD-1) in advanced melanoma showed that this combination was well tolerated, although some patients had mild side effects such as chills, fatigue and pyrexia. Phase III clinical study is already being conducted to expand clinical information regarding efficacy (Long et al., 2016). The patients who responded to the combination therapy showed an increase in the infiltrated CD8+ T cells, as well as an increase in the expression of PD-L1 protein, and thus providing mechanistic evidence for the improvement in the effectiveness of pembrolizumab therapy. In addition, greater expression of IFN-γ was detected in the tumor microenvironment in different cell subpopulations, contributing to a less immunosuppressive context (Ribas et al., 2017).
Several approaches using a variety of OVs and ICIs are in pre-clinical and clinical development. For example using a mouse model of metastatic pulmonary melanoma, it was demonstrated that virotherapy using influenza A viruses (IAVs) combined with ICI resulted in a sustained antitumor efficacy caused by the significant increase in the oncolytic effect (Sitnik et al., 2020). In other lines of evidence, Vijayakumar et al., applied Newcastle disease virus (NDV) in combination with radiotherapy plus checkpoint inhibitors (PD1 or CTLA4 targeted mAbs) induces an abscopal effect in immunocompetent B16-F10 murine melanoma model. These authors also show that recombinant NDV a single-chain variable fragment (scFv) anti-CTLA4 plus radiation was as effective as virus, radiation and systemic anti-CTLA4 in terms of survival benefit (Vijayakumar et al., 2019). An important limitation of the use of ICI therapy is that the majority of patients still fail to respond over time (Sharma et al., 2017; Grote et al., 2020). In this context, Liu and collaborators used therapy with oncolytic virus derived from a strain of alphavirus, M1, and were able to demonstrate refractory tumors were sensitized to subsequent checkpoint blockade by boosting T-cell recruitment and upregulating the expression of PD-L1 (Liu et al., 2020). Another important factor to be considered in cancer therapy is whether the effect of local treatment may also include untreated distant metastases. Often referred to as the abscopal effect, this could be an indication that systemic antitumor immune responses are being activated. Evidence in the literature indicates that this effect was observed experimentally in animal models when Kuryk et al. used oncolytic adenoviruses carrying GM-CSF (ONCOS-102) plus ICI therapy (Kuryk et al., 2019). In this scenario of cooperation and synergy, we could also imagine different combinations with other immune players.
Cytokines
A significant obstacle to successful immunotherapeutic interventions is the modulation of the TME, which, in addition to supporting tumor growth and dissemination, favors the evasion of antitumor immune responses. Dysfunctional interaction of tumor and stromal cellular components leads to a predominantly anti-inflammatory cytokine profile with interleukin-10 (IL-10) (Seo et al., 2001), transforming growth factor (TGF)-beta and other cytokines (Strauss et al., 2007), produced by immunosuppressive cells, for example, regulatory T cells (Tregs) (Knol et al., 2011). The administration of IL-2 was one of the first reproducible effective human cancer immunotherapies against metastatic melanoma (Rosenberg, 2014). Likewise, IFN-α also showed antitumor activity in animal models, in addition to its antiviral activity initially described (Gresser and Bourali, 1970). After clinical trials, both IL-2 and IFN-α demonstrated only mild clinical benefit when used as monotherapy and are approved by the FDA for use in melanoma (Atkins et al., 1999). However, due to the short half-life of most cytokines, high-dose IL-2 and IFN-α administration may be necessary, a situation that increases the incidence of adverse effects, which can make continued treatment unfeasible (Komenaka et al., 2004; Berraondo et al., 2019).
Viruses can be loco-regional adjuvants if applied intratumorally. In addition to their immunostimulatory properties associated with the release of DAMPs and PAMPs, OVs act by inducing acute localized inflammation, they can disturb the tumor niche through the production of inflammatory cytokines in infected/transduced cells. This implies a change in the pattern of cytokines present in the tumor microenvironment in a way that favors the breakdown of immunological tolerance. Few clinical trials have explored the combination of oncologic viruses and cytokine, IL-2 (Voit et al., 2003) and IFN-α (Macedo et al., 2020). However, instead of administering soluble cytokines directly, several approaches function for the design of recombinant virus oncolytic armed with immune modulators, such as cytokines and chemokines. Once the target cell is transduced, it starts to express the carried genes locally, reducing the systemic adverse effects (De Graaf et al., 2018). A classic example is the T-Vec, which locally induces the expression of GM-CSF (Andtbacka et al., 2015). For this purpose, several approaches, with different viral vectors, were used to express cytokines such as IL-2 (Carew et al., 2001; Bai et al., 2014), IL-12 (Varghese et al., 2006), IL-15 (Niu et al., 2015), IFN-γ (Vigil et al., 2007), IFN-β (Durham et al., 2017; Cerqueira et al., 2020; David et al., 2020), reinforces the potential of combining OV and cytokines for immunotherapy for melanomas (De Graaf et al., 2018).
Oncolytic Vaccines Use TSA/TAA in Combination With OV
Assuming that TSA/TAA are the main targets of the adaptive immune system, some strategies seek to incorporate these tumor antigens in OVs, referred to as “oncolytic vaccines”, designed to potentiate antitumor immune responses, especially cytotoxic T lymphocytes (Elsedawy and Russell, 2013; Holay et al., 2017). Mulryan and coworkers used engineered vaccinia virus expressing TAA 5T4 (an oncofetal antigen), in animal models of melanoma and showed significant melanoma tumor retardation compared with mice vaccinated with respective controls. Although it is a self-antigen, in this work no autoimmune effects inherent to the treatment were detected (Mulryan et al., 2002). In another work, using the B16-ova mosuse melanoma model, Diaz and coworkers verified an increase in the activation of ova-specific T cells after treatment with vesicular stomatitis virus (VSV) delivering ovalbumin (ova) (Diaz et al., 2007). Other studies went further and studied melanoma cDNA library delivery by the oncolytic viral vector VSV. After using these constructs to treat pre-established melanomas in animal models, remission was observed, which is associated with the ability of mouse lymphoid cells to mount a tumor-specific CD4+ interleukin (IL)-17 dependent response (Pulido et al., 2012). Collectively, these findings corroborate the principles of personalization of cancer treatment, since the gamut of potential TAA/TSA epitopes will be inherent in the evolutionary history of tumors (Holay et al., 2017). This strategy will be quite valuable in the not-too-distant future, where tumor genome and transcriptome sequencing data will be increasingly available, and thus, likely to be coupled with viral therapies (Finck et al., 2020).
Perspectives for Combining OV and CAR-T Cell Therapy for Melanoma
Adoptive transfer of chimeric antigen receptor (CAR)-modified T cells has demonstrated remarkable rates of long-lasting complete remission in patients with hematological tumors (Guedan and Alemany, 2018). The approval by the US Food and Drug Administration (FDA) of Kymriah (tisagenlecleucel) for acute lymphoblastic leukemia (ALL) (Maude et al., 2018) and Yescarta (axicabtagene ciloleucel) designed to treat large B-cell lymphoma (Neelapu et al., 2017), opened unprecedented perspectives for cancer treatment. In general, the CAR-T cell approach involves the ex vivo modification of the patients’ own T cells using lentiviral and retroviral vectors to deliver the CAR sequence, followed by expansion and reinfusion in the patient (Simon and Uslu, 2018; Chicaybam et al., 2020). Other applications for the use of CAR T have been approved by the FDA as Abecma (idecabtagene vicleucel) for the treatment of multiple myeloma and its use for autoimmune diseases is already being discussed (Hong et al., 2020).
Despite impressive results reported for hematological malignancies, CAR-T cell therapy in solid tumors has failed to meet expectations (Newick et al., 2017; Dana et al., 2021; Marofi et al., 2021). Unlike hematological malignancies, melanomas, like many solid tumors, do not have well-defined targets for the design of a CAR since the available antigens are often expressed in normal cells, thus promoting off-target cell killing. Adding to that difficulty, expression of possible candidate antigens can vary as tumor immunoediting is a continuous process and may give the targeted cells a selective advantage that results in their escape from the CAR-T cells (Dunn et al., 2002; Poggi et al., 2014). Even so, many studies are underway to use the CAR-T cell approach in melanoma (Table 2), a topic that has been reviewed recently (Soltantoyeh et al., 2021; Uslu, 2021). Currently, data from these clinical trials are not available. The immunosuppressive TME is another barrier that must be overcome if CAR-T cell therapies are to be successful. Melanoma, like other solid tumors, is composed of a complex network containing different cell types, such as fibroblasts, endothelial cells, adipocytes and several cells of the immune system immersed in the extracellular matrix (ECM) (Villanueva and Herlyn, 2008; Simiczyjew et al., 2020). Acting together, they comprise an immunosuppressive microenvironment that promotes evasion of antitumor responses, especially of T effector/cytotoxic lymphocytes (Ruiter et al., 2002; Maibach et al., 2020; Simiczyjew et al., 2020). This also directly affects the recruitment and activity of CAR-T cells that may have reached the tumor sites, suggesting that therapy with CAR-T cells alone will not be sufficient to induce complete responses in melanoma. From this perspective, virotherapy, with its ability to revert immunosuppression and promote infiltration of T cells, is expected to fill in some necessary gaps for the effectiveness of CAR-T cell therapy in solid tumors.
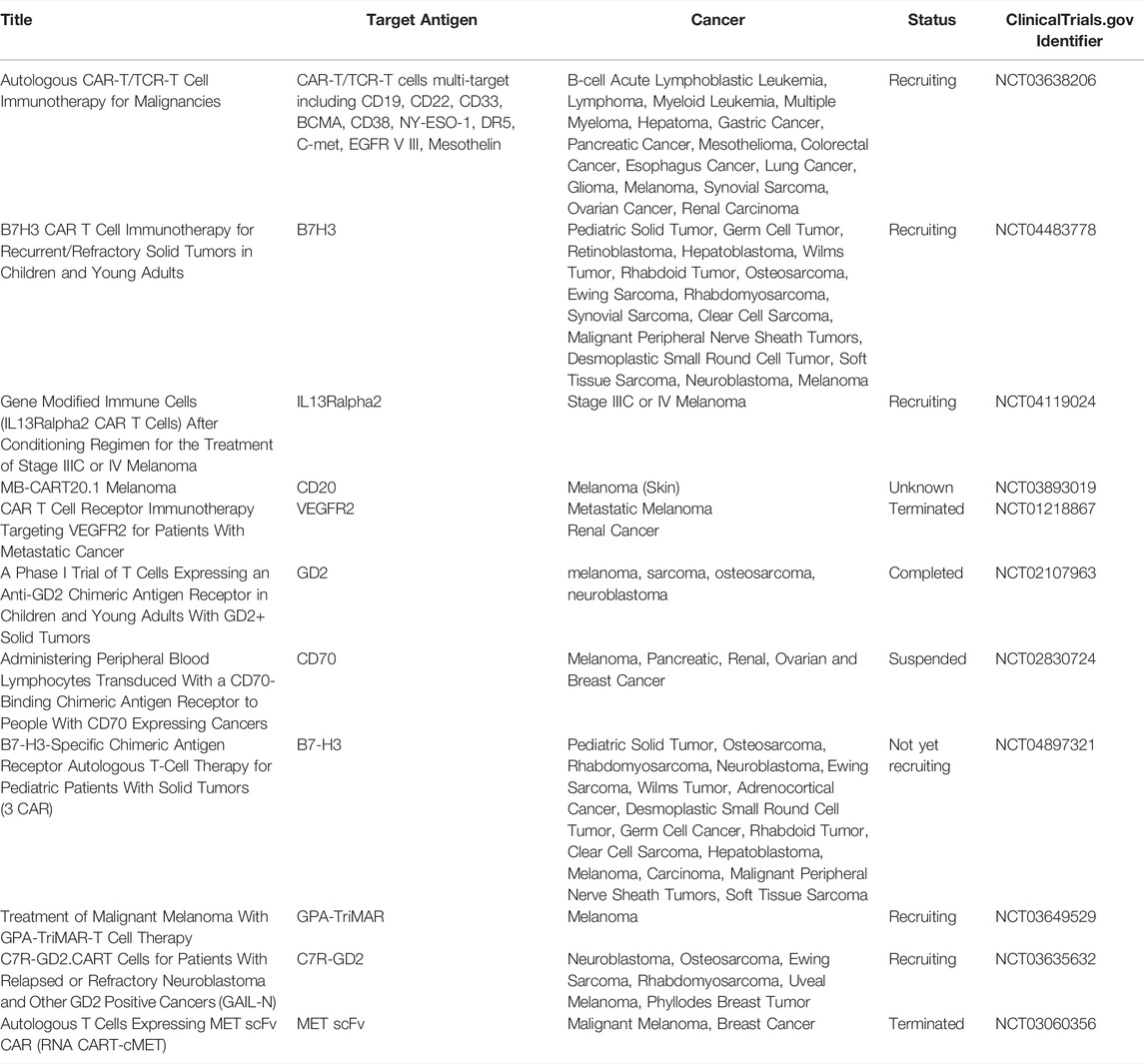
TABLE 2. Clinical trials using CAR T-cells for the treatment of melanoma and other solid tumors.
In this context, Wing and coworkers used an oncolytic adenovirus to deliver a Bispecific T-cell Engager (BiTE) targeting a second tumor antigen in order to augment tumor cell recognition by CAR-T cells. Surprisingly, this combination was able to activate new populations of antitumor T cells in addition to the CAR-T cells (Wing et al., 2018). Since these assays were conducted in immunocompromised mice (NSG, NOD/SCID/IL2rγ−/-), we hypothesize that in immunocompetent individuals, the chance of generating new cytotoxic T cell clones against neoantigens during oncolysis will be increased. Clinical trials will be needed to evaluate this hypothesis, as well as safety. Even though melanoma was not studied, this work opened perspectives for a strategy for other solid tumors. Recently, Jong and collaborators used the same strategy with different targets, sialylated CD43 × CD3 bispecific T cell engager, and demonstrated that it was not only able to bind to cultured patient-derived melanoma samples, but also reduced tumor outgrowth in grafted mice (De Jong et al., 2021). Other studies have pointed out that the use of OV armed with PD-L1 blocking mini-antibody (Tanoue et al., 2017) or IL12p70 and PD-L1 (Rosewell Shaw et al., 2017), combined with CAR-T cell therapy is more effective for tumor control and prolonged survival when compared to each agent as monotherapy. Another interesting evidence in the literature points to the use of oncolytic viral vectors armed with IL-2 and TNF-α to curtail the progression of the primary tumor, but not its metastases. Intriguingly, combining these viral vectors with CAR-T was able to control the primary tumor, as well as its metastases (Guedan and Alemany, 2018; Watanabe et al., 2018).
An important limitation of the use of OV is the incidence of immunity against viral components, including cells that present these viral antigens. Thus, antibodies may neutralize and inactivate viral particles before they reach their target, a limitation of particular concern for repeated administration of the OV. In an innovative approach, VanSeggelen et al. utilized CAR-T cells to protect the oncolytic virus from the immune system, delivering it only to the tumor niche. Thus, viral oncolysis could attract not only more CAR-T cells in a positive feedback loop, but also other immune cells contributing to antitumor responses (Vanseggelen et al., 2015). Collectively these studies point to the potential of these combinations, which need appropriate clinical trials (Guedan and Alemany, 2018).
Conclusion
Despite numerous advances in therapies for metastatic melanoma, many patients become refractory and succumb to the disease since they are left without treatment options (Keller and Bell, 2016). In this scenario, the search for innovative therapeutic interventions is urgent. Therapies employing oncolytic viruses, replicative or not, are gaining attention. Some successful examples include orphan drug designation for Pexa-Vec (Breitbach et al., 2015; Park et al., 2015) and Telomelysin (Trager et al., 2020) as well as the approval of T-VEC (Fukuhara et al., 2016) and Oncorine (Liang, 2018) by national regulatory agencies. At a time when millions of people are receiving anti-SARS-CoV2 vaccines based on recombinant viral vectors with few/low side effects reported (Yadav et al., 2021), it reinforces safety and reliability with regard to viral vectors as viewed by regulatory agencies around the world.
The use of virotherapy, including gene transfer with non-replicating viral vectors, has been shown to change the profile of TME acting as an adjuvant (Keller and Bell, 2016). As we can see in the diagram in Figure 3, the use of virotherapy induces oncolysis, at this moment by the direct action of the viral particles. In this first round of cell death, due to the release of DAMPs, such as ATP, HMGB1, type I IFNs and exposure of calreticulin, it is characterized as ICD. At the same time, the release of tumor antigens (TAA and TSA) also occurs along with PAMPs associated with the virus. This is a favorable scenario for the recruitment of APCs that capture these tumor antigens and prime T cells, stimulating the formation of adaptive cellular responses against tumor cells. Likewise, the cytokine profile in the tumor microenvironment tends to change from an immunosuppressive to an inflammatory profile, favoring the recruitment of effector T cells. Thereafter, a second wave of oncolysis begins, but this time due to cytotoxicity of T cell antitumor clones, which can even act in distant metastatic sites with the application of viral therapy.
As novel therapies emerge, rational combinations will need to be overcome tumor resistance and adaptations of tumor cells and their cellular partners in the tumor microenvironment (Raja et al., 2018). Taking advantage of the fact that virotherapy can attract T cells to the tumor niche, ICIs can have their effectiveness enhanced if used together (Figures 4, 5). Clinical trial has already been carried out to envision this combination (Long et al., 2016; Puzanov et al., 2016; Chesney et al., 2018; Trager et al., 2020). By the same reasoning, the use of CAR T cells against solid tumors such as melanoma is expected to be more effective when combined with virotherapy. However, as there is still no registered CAR-T cell therapy for melanoma, clinical trials will be necessary to verify this hypothesis.
Since the tumor microenvironment is abundant in anti-inflammatory cytokines such as IL-10 and TGF-β, the use of armed viral vectors can reverse this profile and restrict the expression of appropriately chosen cytokines, to favor a less immunosuppressive context. Unlike the systemic application of cytokines, which brings together a series of collateral effects, intratumoral expression by viral vectors tends to increase their availability in this microenvironment with a reduction in side effects. In summary, improvements in the design, delivery and targeting of oncolytic viral vectors will provide increasing potential as immunotherapies against melanoma. Allied to this, is the fact that combinations with different immunotherapy modalities can cooperate to increase therapeutic efficacy.
Author Contributions
OLDC: Organized, wrote and edited the text, tables and figures; NGA and ACD: Wrote and edited the text, tables, designed and elaborated figures; FA, ECC, MACS, and NGT: wrote and edited the text; BES: Edited the text, secured funding.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful for the financial support received from the Sao Paulo Research Foundation (FAPESP) for providing a grant (2015/26580-9; BES) and fellowships (2017/25284-2, OLDC; 2018/04800-5, FA; 2017/2590-2, NGT; 2018/25555-0, ACD; 2019/03055-7, NGA). Funding was also provided by the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), fellowship 302888/2017/9 (BES).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.777775/full#supplementary-material
References
Aguissa-Touré, A.-H., and Li, G. (2012). Genetic Alterations of PTEN in Human Melanoma. Cell. Mol. Life Sci. 69, 1475–1491. doi:10.1007/s00018-011-0878-0
Alexandrov, L. B., Nik-Zainal, S., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A. J. R., Behjati, S., et al. (2013). Signatures of Mutational Processes in Human Cancer. Nature 500, 415–421. doi:10.1038/nature12477
Amaral, T., Sinnberg, T., Meier, F., Krepler, C., Levesque, M., Niessner, H., et al. (2017). The Mitogen-Activated Protein Kinase Pathway in Melanoma Part I - Activation and Primary Resistance Mechanisms to BRAF Inhibition. Eur. J. Cancer 73, 85–92. doi:10.1016/j.ejca.2016.12.010
Andtbacka, R. H. I., Kaufman, H. L., Collichio, F., Amatruda, T., Senzer, N., Chesney, J., et al. (2015). Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. Jco 33, 2780–2788. doi:10.1200/jco.2014.58.3377
Anna, B., Blazej, Z., Jacqueline, G., Andrew, C. J., Jeffrey, R., and Andrzej, S. (2007). Mechanism of UV-Related Carcinogenesis and its Contribution to Nevi/melanoma. Expert Rev. Dermatol. 2, 451–469. doi:10.1586/17469872.2.4.451
Atkins, M. B., Lotze, M. T., Dutcher, J. P., Fisher, R. I., Weiss, G., Margolin, K., et al. (1999). High-dose Recombinant Interleukin 2 Therapy for Patients with Metastatic Melanoma: Analysis of 270 Patients Treated between 1985 and 1993. Jco 17, 2105. doi:10.1200/jco.1999.17.7.2105
Aurisicchio, L., Pallocca, M., Ciliberto, G., and Palombo, F. (2018). The Perfect Personalized Cancer Therapy: Cancer Vaccines against Neoantigens. J. Exp. Clin. Cancer Res. 37, 86. doi:10.1186/s13046-018-0751-1
Bai, F., Niu, Z., Tian, H., Li, S., Lv, Z., Zhang, T., et al. (2014). Genetically Engineered Newcastle Disease Virus Expressing Interleukin 2 Is a Potential Drug Candidate for Cancer Immunotherapy. Immunol. Lett. 159, 36–46. doi:10.1016/j.imlet.2014.02.009
Banchereau, J., and Palucka, K. (2018). Cancer Vaccines on the Move. Nat. Rev. Clin. Oncol. 15, 9–10. doi:10.1038/nrclinonc.2017.149
Bankar, S., Patkar, S., Desai, S., and Shrikhande, S. V. (2015). Unusual Presentation of Melanoma of Unknown Primary Origin: A Case Report and Review of Literature. J. Cancer Res. Ther. 11, 1025. doi:10.4103/0973-1482.148680
Bareham, B., Georgakopoulos, N., Matas-Cespedes, A., Curran, M., and Saeb-Parsy, K. (2021). Modeling Human Tumor-Immune Environments In Vivo for the Preclinical Assessment of Immunotherapies. Cancer Immunol. Immunother. 70, 2737–2750. doi:10.1007/s00262-021-02897-5
Bartlett, D. L., Liu, Z., Sathaiah, M., Ravindranathan, R., Guo, Z., He, Y., et al. (2013). Oncolytic Viruses as Therapeutic Cancer Vaccines. Mol. Cancer 12, 103. doi:10.1186/1476-4598-12-103
Belter, B., Haase-Kohn, C., and Pietzsch, J. (2017). “Biomarkers in Malignant Melanoma: Recent Trends and Critical Perspective,” in Cutaneous Melanoma: Etiology and Therapy. Editors W. H. Ward, and J. M. Farma (Brisbane (AU): Codon Publications). doi:10.15586/codon.cutaneousmelanoma.2017.ch3
Benci, J. L., Xu, B., Qiu, Y., Wu, T. J., Dada, H., Twyman-Saint Victor, C., et al. (2016). Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 167, 1540–1554. doi:10.1016/j.cell.2016.11.022
Berraondo, P., Sanmamed, M. F., Ochoa, M. C., Etxeberria, I., Aznar, M. A., Pérez-Gracia, J. L., et al. (2019). Cytokines in Clinical Cancer Immunotherapy. Br. J. Cancer 120, 6–15. doi:10.1038/s41416-018-0328-y
Bomar, L., Senithilnathan, A., and Ahn, C. (2019). Systemic Therapies for Advanced Melanoma. Dermatol. Clin. 37, 409–423. doi:10.1016/j.det.2019.05.001
Breitbach, C. J., Moon, A., Burke, J., Hwang, T.-H., and Kirn, D. H. (2015). A Phase 2, Open-Label, Randomized Study of Pexa-Vec (JX-594) Administered by Intratumoral Injection in Patients with Unresectable Primary Hepatocellular Carcinoma. Methods Mol. Biol. 1317, 343–357. doi:10.1007/978-1-4939-2727-2_19
Butterfield, L. H., Comin-Anduix, B., Vujanovic, L., Lee, Y., Dissette, V. B., Yang, J.-Q., et al. (2008). Adenovirus MART-1-engineered Autologous Dendritic Cell Vaccine for Metastatic Melanoma. J. Immunother. 31, 294–309. doi:10.1097/cji.0b013e31816a8910
Cable, J., Greenbaum, B., Pe'er, D., Bollard, C. M., Bruni, S., Griffin, M. E., et al. (2021). Frontiers in Cancer Immunotherapy-A Symposium Report. Ann. N.Y. Acad. Sci. 1489, 30–47. doi:10.1111/nyas.14526
Calvo, R. (2019). Hematological Side Effects of Immune Checkpoint Inhibitors: The Example of Immune-Related Thrombocytopenia. Front. Pharmacol. 10, 454. doi:10.3389/fphar.2019.00454
Cancer Genome Atlas, N. (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. doi:10.1016/j.cell.2015.05.044
Carew, J. F., Kooby, D. A., Halterman, M. W., Kim, S.-H., Federoff, H. J., and Fong, Y. (2001). A Novel Approach to Cancer Therapy Using an Oncolytic Herpes Virus to Package Amplicons Containing Cytokine Genes. Mol. Ther. 4, 250–256. doi:10.1006/mthe.2001.0448
Castle, J. C., Kreiter, S., Diekmann, J., Löwer, M., Van De Roemer, N., De Graaf, J., et al. (2012). Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 72, 1081–1091. doi:10.1158/0008-5472.can-11-3722
Cerqueira, O. L. D., Clavijo-Salomon, M. A., Cardoso, E. C., Citrangulo Tortelli Junior, T., Mendonça, S. A., Barbuto, J. A. M., et al. (2020). Combined p14ARF and Interferon-β Gene Transfer to the Human Melanoma Cell Line SK-MEL-147 Promotes Oncolysis and Immune Activation. Front. Immunol. 11, 576658. doi:10.3389/fimmu.2020.576658
Charles, J., Mouret, S., Challende, I., Leccia, M. T., De Fraipont, F., Perez, S., et al. (2020). T‐cell Receptor Diversity as a Prognostic Biomarker in Melanoma Patients. Pigment Cel Melanoma Res 33, 612–624. doi:10.1111/pcmr.12866
Chen, D. S., and Mellman, I. (2013). Oncology Meets Immunology: the Cancer-Immunity Cycle. Immunity 39, 1–10. doi:10.1016/j.immuni.2013.07.012
Chesney, J., Puzanov, I., Collichio, F., Singh, P., Milhem, M. M., Glaspy, J., et al. (2018). Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination with Ipilimumab versus Ipilimumab Alone in Patients with Advanced, Unresectable Melanoma. Jco 36, 1658–1667. doi:10.1200/jco.2017.73.7379
Chiappetta, C., Proietti, I., Soccodato, V., Puggioni, C., Zaralli, R., Pacini, L., et al. (2015). BRAF and NRAS Mutations Are Heterogeneous and Not Mutually Exclusive in Nodular Melanoma. Appl. Immunohistochem. Mol. Morphol. 23, 172–177. doi:10.1097/pai.0000000000000071
Chicaybam, L., Bonamino, M. H., Luckow Invitti, A., Bortman Rozenchan, P., De Luna Vieira, I., and Strauss, B. E. (2020). Overhauling CAR T Cells to Improve Efficacy, Safety and Cost. Cancers (Basel) 12. doi:10.3390/cancers12092360
Chiu, M., Armstrong, E. J. L., Jennings, V., Foo, S., Crespo-Rodriguez, E., Bozhanova, G., et al. (2020). Combination Therapy with Oncolytic Viruses and Immune Checkpoint Inhibitors. Expert Opin. Biol. Ther. 20, 635–652. doi:10.1080/14712598.2020.1729351
Clavijo-Salomon, M. A., Azevedo, R. A., Jorge, S. D., and Ferreira, A. K. (2017). Overcoming the Tumor Microenvironment with STING Agonism: Lessons Lerned from Viruses. J. Immuno Virol. 1. doi:10.19080/JOJIV.2017.01.555569
Coley, W. B. (19911893). The Classic. Clin. Orthopaedics Relat. Res. 262, 3–11. doi:10.1097/00003086-199101000-00002
Conry, R. M., Westbrook, B., Mckee, S., and Norwood, T. G. (2018). Talimogene Laherparepvec: First in Class Oncolytic Virotherapy. Hum. Vaccin. Immunother. 14, 839–846. doi:10.1080/21645515.2017.1412896
Consortium, I. T. P.-C. a. O. W. G. (2020). Pan-cancer Analysis of Whole Genomes. Nature 578, 82–93. doi:10.1038/s41586-020-1969-6
Curtin, J. A., Fridlyand, J., Kageshita, T., Patel, H. N., Busam, K. J., Kutzner, H., et al. (2005). Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 353, 2135–2147. doi:10.1056/nejmoa050092
Czarnecka, A. M., Bartnik, E., Fiedorowicz, M., and Rutkowski, P. (2020). Targeted Therapy in Melanoma and Mechanisms of Resistance. Int. J. Mol. Sci. 21. doi:10.3390/ijms21134576
Dana, H., Chalbatani, G. M., Jalali, S. A., Mirzaei, H. R., Grupp, S. A., Suarez, E. R., et al. (2021). CAR-T Cells: Early Successes in Blood Cancer and Challenges in Solid Tumors. Acta Pharmaceutica Sinica B 11, 1129–1147. doi:10.1016/j.apsb.2020.10.020
David, T. I. P., Cerqueira, O. L. D., Lana, M. G., Medrano, R. F. V., Hunger, A., and Strauss, B. E. (2020). Response of Human Melanoma Cell Lines to Interferon-Beta Gene Transfer Mediated by a Modified Adenoviral Vector. Sci. Rep. 10, 17893. doi:10.1038/s41598-020-74826-y
De Graaf, J. F., De Vor, L., Fouchier, R. A. M., and Van Den Hoogen, B. G. (2018). Armed Oncolytic Viruses: A Kick-Start for Anti-tumor Immunity. Cytokine Growth Factor. Rev. 41, 28–39. doi:10.1016/j.cytogfr.2018.03.006
De Jong, G., Bartels, L., Kedde, M., Verdegaal, E. M. E., Gillissen, M. A., Levie, S. E., et al. (2021). Melanoma Cells Can Be Eliminated by Sialylated CD43 × CD3 Bispecific T Cell Engager Formats In Vitro and In Vivo. Cancer Immunol. Immunother. 70, 1569–1581. doi:10.1007/s00262-020-02780-9
Decraene, D., Agostinis, P., Pupe, A., De Haes, P., and Garmyn, M. (2001). Acute Response of Human Skin to Solar Radiation: Regulation and Function of the P53 Protein. J. Photochem. Photobiol. B: Biol. 63, 78–83. doi:10.1016/s1011-1344(01)00204-4
Diaz, R. M., Galivo, F., Kottke, T., Wongthida, P., Qiao, J., Thompson, J., et al. (2007). Oncolytic Immunovirotherapy for Melanoma Using Vesicular Stomatitis Virus. Cancer Res. 67, 2840–2848. doi:10.1158/0008-5472.can-06-3974
Dock, G. (1904). The Influence of Complicating Diseases upon Leukæmia.*. Am. J. Med. Sci. 127, 563–592. doi:10.1097/00000441-190412740-00001
Dummer, R., Ascierto, P. A., Nathan, P., Robert, C., and Schadendorf, D. (2020). Rationale for Immune Checkpoint Inhibitors Plus Targeted Therapy in Metastatic Melanoma: A Review. JAMA Oncol. 6, 1957–1996. doi:10.1001/jamaoncol.2020.4401
Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J., and Schreiber, R. D. (2002). Cancer Immunoediting: from Immunosurveillance to Tumor Escape. Nat. Immunol. 3, 991–998. doi:10.1038/ni1102-991
Durgeau, A., Virk, Y., Corgnac, S., and Mami-Chouaib, F. (2018). Recent Advances in Targeting CD8 T-Cell Immunity for More Effective Cancer Immunotherapy. Front. Immunol. 9, 14. doi:10.3389/fimmu.2018.00014
Durham, N. M., Mulgrew, K., Mcglinchey, K., Monks, N. R., Ji, H., Herbst, R., et al. (2017). Oncolytic VSV Primes Differential Responses to Immuno-Oncology Therapy. Mol. Ther. 25, 1917–1932. doi:10.1016/j.ymthe.2017.05.006
El Osta, B., Hu, F., Sadek, R., Chintalapally, R., and Tang, S.-C. (2017). Not all Immune-Checkpoint Inhibitors Are Created Equal: Meta-Analysis and Systematic Review of Immune-Related Adverse Events in Cancer Trials. Crit. Rev. Oncology/Hematology 119, 1–12. doi:10.1016/j.critrevonc.2017.09.002
Elsedawy, N. B., and Russell, S. J. (2013). Oncolytic Vaccines. Expert Rev. Vaccin. 12, 1155–1172. doi:10.1586/14760584.2013.836912
Finck, A., Gill, S. I., and June, C. H. (2020). Cancer Immunotherapy Comes of Age and Looks for Maturity. Nat. Commun. 11, 3325. doi:10.1038/s41467-020-17140-5
Fisher, M. S., and Kripke, M. L. (1982). Suppressor T Lymphocytes Control the Development of Primary Skin Cancers in Ultraviolet-Irradiated Mice. Science 216, 1133–1134. doi:10.1126/science.6210958
Fitzgerald, K. A., and Kagan, J. C. (2020). Toll-like Receptors and the Control of Immunity. Cell 180, 1044–1066. doi:10.1016/j.cell.2020.02.041
Force, U. S. P. S. T., Grossman, D. C., Curry, S. J., Owens, D. K., Barry, M. J., Caughey, A. B., et al. (2018). Behavioral Counseling to Prevent Skin Cancer: US Preventive Services Task Force Recommendation Statement. JAMA 319, 1134–1142. doi:10.1001/jama.2018.1623
Fukuhara, H., Ino, Y., and Todo, T. (2016). Oncolytic Virus Therapy: A new era of Cancer Treatment at Dawn. Cancer Sci. 107, 1373–1379. doi:10.1111/cas.13027
Galon, J., and Bruni, D. (2019). Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 18, 197–218. doi:10.1038/s41573-018-0007-y
Gao, J., Shi, L. Z., Zhao, H., Chen, J., Xiong, L., He, Q., et al. (2016). Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404. doi:10.1016/j.cell.2016.08.069
Garbe, C., Amaral, T., Peris, K., Hauschild, A., Arenberger, P., Bastholt, L., et al. (2020). European Consensus-Based Interdisciplinary Guideline for Melanoma. Part 2: Treatment - Update 2019. Eur. J. Cancer 126, 159–177. doi:10.1016/j.ejca.2019.11.015
García, M., Moreno, R., Gil-Martin, M., Cascallò, M., De Olza, M. O., Cuadra, C., et al. (2018). A Phase 1 Trial of Oncolytic Adenovirus ICOVIR-5 Administered Intravenously to Cutaneous and Uveal Melanoma Patients. Hum. Gene Ther. 30, 352–364.
Gejman, R. S., and Chang, A. Y. (2018). Rejection of Immunogenic Tumor Clones Is Limited by Clonal Fraction. Elife 7, 41090. doi:10.7554/elife.41090
Geoffroy, K., and Bourgeois-Daigneault, M.-C. (2020). The Pros and Cons of Interferons for Oncolytic Virotherapy. Cytokine Growth Factor. Rev. 56, 49–58. doi:10.1016/j.cytogfr.2020.07.002
Gessani, S., and Belardelli, F. (2021). Type I Interferons as Joint Regulators of Tumor Growth and Obesity. Cancers 13, 196. doi:10.3390/cancers13020196
Gide, T. N., Quek, C., Menzies, A. M., Tasker, A. T., Shang, P., Holst, J., et al. (2019). Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 35, 238–255. e236. doi:10.1016/j.ccell.2019.01.003
Goding, C. R., and Arnheiter, H. (2019). MITF-the First 25 Years. Genes Dev. 33, 983–1007. doi:10.1101/gad.324657.119
Goel, V. K., Lazar, A. J. F., Warneke, C. L., Redston, M. S., and Haluska, F. G. (2006). Examination of Mutations in BRAF, NRAS, and PTEN in Primary Cutaneous Melanoma. J. Invest. Dermatol. 126, 154–160. doi:10.1038/sj.jid.5700026
Goldsmith, K., Chen, W., Johnson, D. C., and Hendricks, R. L. (1998). Infected Cell Protein (ICP)47 Enhances Herpes Simplex Virus Neurovirulence by Blocking the CD8+ T Cell Response. J. Exp. Med. 187, 341–348. doi:10.1084/jem.187.3.341
Gresser, I., and Bourali, C. (1970). Antitumor Effects of Interferon Preparations in Mice. J. Natl. Cancer Inst. 45, 365–376.
Grote, H. J., Feng, Z., Schlichting, M., Helwig, C., Ruisi, M., Jin, H., et al. (2020). Programmed Death-Ligand 1 Immunohistochemistry Assay Comparison Studies in NSCLC: Characterization of the 73-10 Assay. J. Thorac. Oncol. 15, 1306–1316. doi:10.1016/j.jtho.2020.04.013
Gubin, M. M., Artyomov, M. N., Mardis, E. R., and Schreiber, R. D. (2015). Tumor Neoantigens: Building a Framework for Personalized Cancer Immunotherapy. J. Clin. Invest. 125, 3413–3421. doi:10.1172/jci80008
Guedan, S., and Alemany, R. (2018). CAR-T Cells and Oncolytic Viruses: Joining Forces to Overcome the Solid Tumor Challenge. Front. Immunol. 9, 2460. doi:10.3389/fimmu.2018.02460
Gyorki, D. E., Callahan, M., Wolchok, J. D., and Ariyan, C. E. (2013). The Delicate Balance of Melanoma Immunotherapy. Clin. Trans. Immunol. 2, e5. doi:10.1038/cti.2013.5
Hansson, G. K., and Edfeldt, K. (2005). Toll to Be Paid at the Gateway to the Vessel wall. Atvb 25, 1085–1087. doi:10.1161/01.atv.0000168894.43759.47
Harbst, K., Staaf, J., Lauss, M., Karlsson, A., Måsbäck, A., Johansson, I., et al. (2012). Molecular Profiling Reveals Low- and High-Grade Forms of Primary Melanoma. Clin. Cancer Res. 18, 4026–4036. doi:10.1158/1078-0432.ccr-12-0343
Hayward, N. K., Wilmott, J. S., Waddell, N., Johansson, P. A., Field, M. A., Nones, K., et al. (2017). Whole-genome Landscapes of Major Melanoma Subtypes. Nature 545, 175–180. doi:10.1038/nature22071
Hegde, P. S., and Chen, D. S. (2020). Top 10 Challenges in Cancer Immunotherapy. Immunity 52, 17–35. doi:10.1016/j.immuni.2019.12.011
Hocker, T., and Tsao, H. (2007). Ultraviolet Radiation and Melanoma: a Systematic Review and Analysis of Reported Sequence Variants. Hum. Mutat. 28, 578–588. doi:10.1002/humu.20481
Hoek, K. S., Eichhoff, O. M., Schlegel, N. C., Döbbeling, U., Kobert, N., Schaerer, L., et al. (2008). In Vivo switching of Human Melanoma Cells between Proliferative and Invasive States. Cancer Res. 68, 650–656. doi:10.1158/0008-5472.can-07-2491
Hoek, K. S., Schlegel, N. C., Brafford, P., Sucker, A., Ugurel, S., Kumar, R., et al. (2006). Metastatic Potential of Melanomas Defined by Specific Gene Expression Profiles with No BRAF Signature. Pigment Cel. Res. 19, 290–302. doi:10.1111/j.1600-0749.2006.00322.x
Holay, N., Kim, Y., Lee, P., and Gujar, S. (2017). Sharpening the Edge for Precision Cancer Immunotherapy: Targeting Tumor Antigens through Oncolytic Vaccines. Front. Immunol. 8, 800. doi:10.3389/fimmu.2017.00800
Hong, M., Clubb, J. D., and Chen, Y. Y. (2020). Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 38, 473–488. doi:10.1016/j.ccell.2020.07.005
Hoster, H. A., Zanes, R. P., and Von Haam, E. (1949). Studies in Hodgkin's Syndrome; the Association of Viral Hepatitis and Hodgkin's Disease; a Preliminary Report. Cancer Res. 9, 473–480.
Hu, H.-G., and Li, Y.-M. (2020). Emerging Adjuvants for Cancer Immunotherapy. Front. Chem. 8, 601. doi:10.3389/fchem.2020.00601
Hu, H.-J., Liang, X., Li, H.-L., Du, C.-M., Hao, J.-L., Wang, H.-Y., et al. (2020). The Armed Oncolytic Adenovirus ZD55-IL-24 Eradicates Melanoma by Turning the Tumor Cells from the Self-State into the Nonself-State besides Direct Killing. Cell Death Dis. 11, 1022. doi:10.1038/s41419-020-03223-0
Hu, J. C. C., Coffin, R. S., Davis, C. J., Graham, N. J., Groves, N., Guest, P. J., et al. (2006). A Phase I Study of OncoVEXGM-CSF, a Second-Generation Oncolytic Herpes Simplex Virus Expressing Granulocyte Macrophage colony-stimulating Factor. Clin. Cancer Res. 12, 6737–6747. doi:10.1158/1078-0432.ccr-06-0759
Huang, R., Mao, M., Lu, Y., Yu, Q., and Liao, L. (2020). A Novel Immune-Related Genes Prognosis Biomarker for Melanoma: Associated with Tumor Microenvironment. Aging 12, 6966–6980. doi:10.18632/aging.103054
Hunger, A., Medrano, R. F., and Strauss, B. E. (2017). Harnessing Combined p19Arf and Interferon-Beta Gene Transfer as an Inducer of Immunogenic Cell Death and Mediator of Cancer Immunotherapy. Cel. Death Dis. 8, e2784. doi:10.1038/cddis.2017.201
Hwang, J. K., Hong, J., and Yun, C. O. (2020). Oncolytic Viruses and Immune Checkpoint Inhibitors: Preclinical Developments to Clinical Trials. Int. J. Mol. Sci. 21. doi:10.3390/ijms21228627
Inohara, N., Chamaillard, M., McDonald, C., and Nuñez, G. (2005). NOD-LRR Proteins: Role in Host-Microbial Interactions and Inflammatory Disease. Annu. Rev. Biochem. 74, 355–383. doi:10.1146/annurev.biochem.74.082803.133347
Jenkins, R. W., and Fisher, D. E. (2020). Treatment of Advanced Melanoma in 2020 and Beyond. J. Invest. Dermatol. 141, 23–31. doi:10.1016/j.jid.2020.03.943
Jerby-Arnon, L., Shah, P., Cuoco, M. S., Rodman, C., Su, M.-J., Melms, J. C., et al. (2018). A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 175, 984–997. doi:10.1016/j.cell.2018.09.006
Jessurun, C. A. C., Vos, J. A. M., Limpens, J., and Luiten, R. M. (2017). Biomarkers for Response of Melanoma Patients to Immune Checkpoint Inhibitors: a Systematic Review. Front. Oncol. 7, 233. doi:10.3389/fonc.2017.00233
Jhawar, S. R., Thandoni, A., Bommareddy, P. K., Hassan, S., Kohlhapp, F. J., Goyal, S., et al. (2017). Oncolytic Viruses-Natural and Genetically Engineered Cancer Immunotherapies. Front. Oncol. 7, 202. doi:10.3389/fonc.2017.00202
Ji, R.-R., Chasalow, S. D., Wang, L., Hamid, O., Schmidt, H., Cogswell, J., et al. (2012). An Immune-Active Tumor Microenvironment Favors Clinical Response to Ipilimumab. Cancer Immunol. Immunother. 61, 1019–1031. doi:10.1007/s00262-011-1172-6
Karlsson, A., and Saleh, S. (2017). Checkpoint Inhibitors for Malignant Melanoma: a Systematic Review and Meta-Analysis. Ccid 10, 325–339. doi:10.2147/ccid.s120877
Kaufman, H. L., Kim, D. W., Deraffele, G., Mitcham, J., Coffin, R. S., and Kim-Schulze, S. (2010). Local and Distant Immunity Induced by Intralesional Vaccination with an Oncolytic Herpes Virus Encoding GM-CSF in Patients with Stage IIIc and IV Melanoma. Ann. Surg. Oncol. 17, 718–730. doi:10.1245/s10434-009-0809-6
Kaufman, H. L., Kohlhapp, F. J., and Zloza, A. (2015). Oncolytic Viruses: a New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 14, 642–662. doi:10.1038/nrd4663
Keller, B. A., and Bell, J. C. (2016). Oncolytic Viruses-Immunotherapeutics on the Rise. J. Mol. Med. 94, 979–991. doi:10.1007/s00109-016-1453-9
Kelly, E., and Russell, S. J. (2007). History of Oncolytic Viruses: Genesis to Genetic Engineering. Mol. Ther. 15, 651–659. doi:10.1038/sj.mt.6300108
Kennedy, L. B., and Salama, A. K. S. (2019). A Review of Immune-Mediated Adverse Events in Melanoma. Oncol. Ther. 7, 101–120. doi:10.1007/s40487-019-0096-8
Kim, H.-Y., Lee, H., Kim, S.-H., Jin, H., Bae, J., and Choi, H.-K. (2017). Discovery of Potential Biomarkers in Human Melanoma Cells with Different Metastatic Potential by Metabolic and Lipidomic Profiling. Sci. Rep. 7, 8864. doi:10.1038/s41598-017-08433-9
Kim, T., Amaria, R. N., Spencer, C., Reuben, A., Cooper, Z. A., and Wargo, J. A. (2014). Combining Targeted Therapy and Immune Checkpoint Inhibitors in the Treatment of Metastatic Melanoma. Cancer Biol. Med. 11, 237–246. doi:10.7497/j.issn.2095-3941.2014.04.002
Kiyohara, E., Tanemura, A., Nishioka, M., Yamada, M., Tanaka, A., Yokomi, A., et al. (2020). Intratumoral Injection of Hemagglutinating Virus of Japan-envelope Vector Yielded an Antitumor Effect for Advanced Melanoma: a Phase I/IIa Clinical Study. Cancer Immunol. Immunother. 69, 1131–1140. doi:10.1007/s00262-020-02509-8
Knol, A. C., Nguyen, J. M., Quéreux, G., Brocard, A., Khammari, A., and Dréno, B. (2011). Prognostic Value of Tumor-Infiltrating Foxp3+ T-Cell Subpopulations in Metastatic Melanoma. Exp. Dermatol. 20, 430–434. doi:10.1111/j.1600-0625.2011.01260.x
Kobayashi, N., Nakagawa, A., Muramatsu, T., Yamashina, Y., Shirai, T., Hashimoto, M. W., et al. (1998). Supranuclear Melanin Caps Reduce Ultraviolet Induced DNA Photoproducts in Human Epidermis. J. Invest. Dermatol. 110, 806–810. doi:10.1046/j.1523-1747.1998.00178.x
Komenaka, I., Hoerig, H., and Kaufman, H. L. (2004). Immunotherapy for Melanoma. Clin. Dermatol. 22, 251–265. doi:10.1016/j.clindermatol.2003.12.001
Krauthammer, M., Kong, Y., Bacchiocchi, A., Evans, P., Pornputtapong, N., Wu, C., et al. (2015). Exome Sequencing Identifies Recurrent Mutations in NF1 and RASopathy Genes in Sun-Exposed Melanomas. Nat. Genet. 47, 996–1002. doi:10.1038/ng.3361
Kuryk, L., Møller, A. S. W., and Jaderberg, M. (2019). Abscopal Effect when Combining Oncolytic Adenovirus and Checkpoint Inhibitor in a Humanized NOG Mouse Model of Melanoma. J. Med. Virol. 91, 1702–1706. doi:10.1002/jmv.25501
Kvam, E., and Tyrrell, R. M. (1997). Induction of Oxidative DNA Base Damage in Human Skin Cells by UV and Near Visible Radiation. Carcinogenesis 18, 2379–2384. doi:10.1093/carcin/18.12.2379
Kwiatkowska, D., Kluska, P., and Reich, A. (2019). Beyond PD-1 Immunotherapy in Malignant Melanoma. Dermatol. Ther. (Heidelb) 9, 243–257. doi:10.1007/s13555-019-0292-3
Lamm, D. L., Thor, D. E., Harris, S. C., Reyna, J. A., Stogdill, V. D., and Radwin, H. M. (1980). Bacillus Calmette-Guerin Immunotherapy of Superficial Bladder Cancer. J. Urol. 124, 38–42. doi:10.1016/s0022-5347(17)55282-9
Lawrence, M. S., Stojanov, P., Polak, P., Kryukov, G. V., Cibulskis, K., Sivachenko, A., et al. (2013). Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 499, 214–218. doi:10.1038/nature12213
Lennerz, V., Fatho, M., Gentilini, C., Frye, R. A., Lifke, A., Ferel, D., et al. (2005). The Response of Autologous T Cells to a Human Melanoma Is Dominated by Mutated Neoantigens. Proc. Natl. Acad. Sci. U.S.A. 102, 16013–16018. doi:10.1073/pnas.0500090102
Levy, C., Khaled, M., and Fisher, D. E. (2006). MITF: Master Regulator of Melanocyte Development and Melanoma Oncogene. Trends Mol. Med. 12, 406–414. doi:10.1016/j.molmed.2006.07.008
Li, L., Goedegebuure, S. P., and Gillanders, W. E. (2017a). Preclinical and Clinical Development of Neoantigen Vaccines. Ann. Oncol. 28, xii11–xii17. doi:10.1093/annonc/mdx681
Li, X., Wang, J., Yao, Y., Yang, L., Li, Z., Yu, C., et al. (2017b). Comparative Efficacy and Safety of Immune Checkpoint Inhibitor-Related Therapies for Advanced Melanoma: a Bayesian Network Analysis. Oncotarget 8, 83637–83649. doi:10.18632/oncotarget.18906
Liang, M. (2018). Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Ccdt 18, 171–176. doi:10.2174/1568009618666171129221503
Linnemann, C., Van Buuren, M. M., Bies, L., Verdegaal, E. M. E., Schotte, R., Calis, J. J. A., et al. (2015). High-throughput Epitope Discovery Reveals Frequent Recognition of Neo-Antigens by CD4+ T Cells in Human Melanoma. Nat. Med. 21, 81–85. doi:10.1038/nm.3773
Lionarons, D. A., Hancock, D. C., Rana, S., East, P., Moore, C., Murillo, M. M., et al. (2019). RAC1P29S Induces a Mesenchymal Phenotypic Switch via Serum Response Factor to Promote Melanoma Development and Therapy Resistance. Cancer Cell 36, 68–83. doi:10.1016/j.ccell.2019.05.015
Liu, B. L., Robinson, M., Han, Z.-Q., Branston, R. H., English, C., Reay, P., et al. (2003). ICP34.5 Deleted Herpes Simplex Virus with Enhanced Oncolytic, Immune Stimulating, and Anti-tumour Properties. Gene Ther. 10, 292–303. doi:10.1038/sj.gt.3301885
Liu, Y., Cai, J., Liu, W., Lin, Y., Guo, L., Liu, X., et al. (2020). Intravenous Injection of the Oncolytic Virus M1 Awakens Antitumor T Cells and Overcomes Resistance to Checkpoint Blockade. Cel. Death Dis. 11, 1062. doi:10.1038/s41419-020-03285-0
Long, G. V., Dummer, R., Ribas, A., Puzanov, I., Vanderwalde, A., Andtbacka, R. H. I., et al. (2016). Efficacy Analysis of MASTERKEY-265 Phase 1b Study of Talimogene Laherparepvec (T-VEC) and Pembrolizumab (Pembro) for Unresectable Stage IIIB-IV Melanoma. Jco 34, 9568. doi:10.1200/jco.2016.34.15_suppl.9568
Long, G. V., Menzies, A. M., Nagrial, A. M., Haydu, L. E., Hamilton, A. L., Mann, G. J., et al. (2011). Prognostic and Clinicopathologic Associations of Oncogenic BRAF in Metastatic Melanoma. Jco 29, 1239–1246. doi:10.1200/jco.2010.32.4327
Lovly, C. M., Dahlman, K. B., Fohn, L. E., Su, Z., Dias-Santagata, D., Hicks, D. J., et al. (2012). Routine Multiplex Mutational Profiling of Melanomas Enables Enrollment in Genotype-Driven Therapeutic Trials. PLoS One 7, e35309. doi:10.1371/journal.pone.0035309
Lu, Y.-C., and Robbins, P. F. (2016). Cancer Immunotherapy Targeting Neoantigens. Semin. Immunol. 28, 22–27. doi:10.1016/j.smim.2015.11.002
Lu, Y.-C., Yao, X., Crystal, J. S., Li, Y. F., El-Gamil, M., Gross, C., et al. (2014). Efficient Identification of Mutated Cancer Antigens Recognized by T Cells Associated with Durable Tumor Regressions. Clin. Cancer Res. 20, 3401–3410. doi:10.1158/1078-0432.ccr-14-0433
Lugowska, I., Teterycz, P., and Rutkowski, P. (2018). Immunotherapy of Melanoma. wo 2018, 61–67. doi:10.5114/wo.2018.73889
Macedo, N., Miller, D. M., Haq, R., and Kaufman, H. L. (2020). Clinical Landscape of Oncolytic Virus Research in 2020. J. Immunother. Cancer 8. doi:10.1136/jitc-2020-001486
Madhunapantula, S. V., Mosca, P. J., and Robertson, G. P. (2011). The Akt Signaling Pathway. Cancer Biol. Ther. 12, 1032–1049. doi:10.4161/cbt.12.12.18442
Maibach, F., Sadozai, H., Seyed Jafari, S. M., Hunger, R. E., and Schenk, M. (2020). Tumor-Infiltrating Lymphocytes and Their Prognostic Value in Cutaneous Melanoma. Front. Immunol. 11, 2105. doi:10.3389/fimmu.2020.02105
Maleki Vareki, S. (2018). High and Low Mutational burden Tumors versus Immunologically Hot and Cold Tumors and Response to Immune Checkpoint Inhibitors. J. Immunotherapy Cancer 6, 157. doi:10.1186/s40425-018-0479-7
Marofi, F., Motavalli, R., Safonov, V. A., Thangavelu, L., Yumashev, A. V., Alexander, M., et al. (2021). CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cel. Res. Ther. 12, 81. doi:10.1186/s13287-020-02128-1
Mastrangelo, M. J., Maguire, H. C., Eisenlohr, L. C., Laughlin, C. E., Monken, C. E., Mccue, P. A., et al. (1999). Intratumoral Recombinant GM-CSF-Encoding Virus as Gene Therapy in Patients with Cutaneous Melanoma. Cancer Gene Ther. 6, 409–422. doi:10.1038/sj.cgt.7700066
Matzinger, P. (2002). The Danger Model: a Renewed Sense of Self. Science 296, 301–305. doi:10.1126/science.1071059
Maude, S. L., Laetsch, T. W., Buechner, J., Rives, S., Boyer, M., Bittencourt, H., et al. (2018). Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 378, 439–448. doi:10.1056/nejmoa1709866
Mcgranahan, N., and Swanton, C. (2019). Neoantigen Quality, Not Quantity. Sci. Transl. Med. 11, eaax7918. doi:10.1126/scitranslmed.aax7918
Medrano, R. F. V., Catani, J. P. P., Ribeiro, A. H., Tomaz, S. L., Merkel, C. A., Costanzi-Strauss, E., et al. (2016). Vaccination Using Melanoma Cells Treated with P19arf and Interferon Beta Gene Transfer in a Mouse Model: a Novel Combination for Cancer Immunotherapy. Cancer Immunol. Immunother. 65, 371–382. doi:10.1007/s00262-016-1807-8
Medrano, R. F. V., Hunger, A., Catani, J. P. P., and Strauss, B. E. (2017). Uncovering the Immunotherapeutic Cycle Initiated by p19Arf and Interferon-β Gene Transfer to Cancer Cells: An Inducer of Immunogenic Cell Death. Oncoimmunology 6, e1329072. doi:10.1080/2162402x.2017.1329072
Melero, I., Gaudernack, G., Gerritsen, W., Huber, C., Parmiani, G., Scholl, S., et al. (2014). Therapeutic Vaccines for Cancer: an Overview of Clinical Trials. Nat. Rev. Clin. Oncol. 11, 509–524. doi:10.1038/nrclinonc.2014.111
Mellman, I., Coukos, G., and Dranoff, G. (2011). Cancer Immunotherapy Comes of Age. Nature 480, 480–489. doi:10.1038/nature10673
Merkel, C. A., Medrano, R. F. V., Barauna, V. G., and Strauss, B. E. (2013). Combined p19Arf and Interferon-Beta Gene Transfer Enhances Cell Death of B16 Melanoma In Vitro and In Vivo. Cancer Gene Ther. 20, 317–325. doi:10.1038/cgt.2013.23
Molina, J. R., and Adjei, A. A. (2006). The Ras/Raf/MAPK Pathway. J. Thorac. Oncol. 1, 7–9. doi:10.1016/s1556-0864(15)31506-9
Moser, J. C., Wei, G., Colonna, S. V., Grossmann, K. F., Patel, S., and Hyngstrom, J. R. (2020). Comparative-effectiveness of Pembrolizumab vs. Nivolumab for Patients with Metastatic Melanoma. Acta Oncologica 59, 434–437. doi:10.1080/0284186x.2020.1712473
Motofei, I. G. (2019). Melanoma and Autoimmunity: Spontaneous Regressions as a Possible Model for New Therapeutic Approaches. Melanoma Res. 29, 231–236. doi:10.1097/cmr.0000000000000573
Mulryan, K., Ryan, M. G., Myers, K. A., Shaw, D., Wang, W., Kingsman, S. M., et al. (2002). Attenuated Recombinant Vaccinia Virus Expressing Oncofetal Antigen (Tumor-associated Antigen) 5T4 Induces Active Therapy of Established Tumors. Mol. Cancer Ther. 1, 1129–1137.
Murciano-Goroff, Y. R., Warner, A. B., and Wolchok, J. D. (2020). The Future of Cancer Immunotherapy: Microenvironment-Targeting Combinations. Cell Res. 30, 507–519. doi:10.1038/s41422-020-0337-2
Murira, A., and Lamarre, A. (2016). Type-I Interferon Responses: From Friend to Foe in the Battle against Chronic Viral Infection. Front. Immunol. 7, 609. doi:10.3389/fimmu.2016.00609
Naik, S., and Russell, S. J. (2009). Engineering Oncolytic Viruses to Exploit Tumor Specific Defects in Innate Immune Signaling Pathways. Expert Opin. Biol. Ther. 9, 1163–1176. doi:10.1517/14712590903170653
Neelapu, S. S., Locke, F. L., Bartlett, N. L., Lekakis, L. J., Miklos, D. B., Jacobson, C. A., et al. (2017). Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 377, 2531–2544. doi:10.1056/nejmoa1707447
Nemunaitis, J., Tong, A. W., Nemunaitis, M., Senzer, N., Phadke, A. P., Bedell, C., et al. (2010). A Phase I Study of Telomerase-specific Replication Competent Oncolytic Adenovirus (Telomelysin) for Various Solid Tumors. Mol. Ther. 18, 429–434. doi:10.1038/mt.2009.262
Newick, K., O'brien, S., Moon, E., and Albelda, S. M. (2017). CAR T Cell Therapy for Solid Tumors. Annu. Rev. Med. 68, 139–152. doi:10.1146/annurev-med-062315-120245
Newman, W., and Southam, C. M. (1954). Virus Treatment in Advanced cancer.A Pathological Study of Fifty-Seven Cases. Cancer 7, 106–118. doi:10.1002/1097-0142(195401)7:1<106::aid-cncr2820070112>3.0.co;2-l
Niu, Z., Bai, F., Sun, T., Tian, H., Yu, D., Yin, J., et al. (2015). Recombinant Newcastle Disease Virus Expressing IL15 Demonstrates Promising Antitumor Efficiency in Melanoma Model. Technol. Cancer Res. Treat. 14, 607–615. doi:10.7785/tcrt.2012.500414
Oshita, C., Takikawa, M., Kume, A., Miyata, H., Ashizawa, T., Iizuka, A., et al. (2012). Dendritic Cell-Based Vaccination in Metastatic Melanoma Patients: Phase II Clinical Trial. Oncol. Rep. 28, 1131–1138. doi:10.3892/or.2012.1956
Ott, P. A., and Bhardwaj, N. (2013). Impact of MAPK Pathway Activation in BRAFV600 Melanoma on T Cell and Dendritic Cell Function. Front. Immunol. 4, 346. doi:10.3389/fimmu.2013.00346
Ott, P. A., Hu, Z., Keskin, D. B., Shukla, S. A., Sun, J., Bozym, D. J., et al. (2017). An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 547, 217–221. doi:10.1038/nature22991
Park, S. H., Breitbach, C. J., Lee, J., Park, J. O., Lim, H. Y., Kang, W. K., et al. (2015). Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. 23, 1532–1540. doi:10.1038/mt.2015.109
Passarelli, A., Mannavola, F., Stucci, L. S., Tucci, M., and Silvestris, F. (2017). Immune System and Melanoma Biology: a Balance between Immunosurveillance and Immune Escape. Oncotarget 8, 106132–106142. doi:10.18632/oncotarget.22190
Patani, N., Martin, L.-A., and Dowsett, M. (2013). Biomarkers for the Clinical Management of Breast Cancer: International Perspective. Int. J. Cancer 133, 1–13. doi:10.1002/ijc.27997
Patton, E. E., Mueller, K. L., Adams, D. J., Anandasabapathy, N., Aplin, A. E., Bertolotto, C., et al. (2021). Melanoma Models for the Next Generation of Therapies. Cancer Cell 39, 610–631. doi:10.1016/j.ccell.2021.01.011
Peng, M., Mo, Y., Wang, Y., Wu, P., Zhang, Y., Xiong, F., et al. (2019). Neoantigen Vaccine: an Emerging Tumor Immunotherapy. Mol. Cancer 18, 128. doi:10.1186/s12943-019-1055-6
Peng, W., Chen, J. Q., Liu, C., Malu, S., Creasy, C., Tetzlaff, M. T., et al. (2016). Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 6, 202–216. doi:10.1158/2159-8290.cd-15-0283
Poggi, A., Musso, A., Dapino, I., and Zocchi, M. R. (2014). Mechanisms of Tumor Escape from Immune System: Role of Mesenchymal Stromal Cells. Immunol. Lett. 159, 55–72. doi:10.1016/j.imlet.2014.03.001
Pollock, P. M., Harper, U. L., Hansen, K. S., Yudt, L. M., Stark, M., Robbins, C. M., et al. (2003). High Frequency of BRAF Mutations in Nevi. Nat. Genet. 33, 19–20. doi:10.1038/ng1054
Porta, C., Paglino, C., and Mosca, A. (2014). Targeting PI3K/Akt/mTOR Signaling in Cancer. Front. Oncol. 4, 64. doi:10.3389/fonc.2014.00064
Potjer, T. P., Bollen, S., Grimbergen, A. J. E. M., Doorn, R., Gruis, N. A., Asperen, C. J., et al. (2019). Multigene Panel Sequencing of Established and Candidate Melanoma Susceptibility Genes in a Large Cohort of Dutch Non-cdkn2a/cdk4 Melanoma Families. Int. J. Cancer 144, 2453–2464. doi:10.1002/ijc.31984
Prickett, T. D., Crystal, J. S., Cohen, C. J., Pasetto, A., Parkhurst, M. R., Gartner, J. J., et al. (2016). Durable Complete Response from Metastatic Melanoma after Transfer of Autologous T Cells Recognizing 10 Mutated Tumor Antigens. Cancer Immunol. Res. 4, 669–678. doi:10.1158/2326-6066.cir-15-0215
Pulido, J., Kottke, T., Thompson, J., Galivo, F., Wongthida, P., Diaz, R. M., et al. (2012). Using Virally Expressed Melanoma cDNA Libraries to Identify Tumor-Associated Antigens that Cure Melanoma. Nat. Biotechnol. 30, 337–343. doi:10.1038/nbt.2157
Puzanov, I., Milhem, M. M., Minor, D., Hamid, O., Li, A., Chen, L., et al. (2016). Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. Jco 34, 2619–2626. doi:10.1200/jco.2016.67.1529
Qin, S., Xu, L., Yi, M., Yu, S., Wu, K., and Luo, S. (2019). Novel Immune Checkpoint Targets: Moving beyond PD-1 and CTLA-4. Mol. Cancer 18, 155. doi:10.1186/s12943-019-1091-2
Raja, J., Ludwig, J. M., Gettinger, S. N., Schalper, K. A., and Kim, H. S. (2018). Oncolytic Virus Immunotherapy: Future Prospects for Oncology. J. Immunotherapy Cancer 6, 140. doi:10.1186/s40425-018-0458-z
Ribas, A., Dummer, R., Puzanov, I., Vanderwalde, A., Andtbacka, R. H. I., Michielin, O., et al. (2017). Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 170, 1109–1119. e1110. doi:10.1016/j.cell.2017.08.027
Ribas, A., Hersey, P., Middleton, M. R., Gogas, H., Flaherty, K. T., Sondak, V. K., et al. (2012). New Challenges in Endpoints for Drug Development in Advanced Melanoma. Clin. Cancer Res. 18, 336–341. doi:10.1158/1078-0432.ccr-11-2323
Ribeiro, A., Medrano, R., Bertolini Zanatta, D., Del Valle, P., Merkel, C., Salles, T., et al. (2017). Reestablishment of p53/Arf and Interferon-β Pathways Mediated by a Novel Adenoviral Vector Potentiates Antiviral Response and Immunogenic Cell Death. Cel Death Discov. 3, 17017.
Roberts, M. S., Groene, W. S., Lorence, R. M., and Bamat, M. K. (2006). Naturally Occurring Viruses for the Treatment of Cancer. Discov. Med. 6, 217–222.
Roesch, A., Paschen, A., Landsberg, J., Helfrich, I., Becker, J. C., and Schadendorf, D. (2016). Phenotypic Tumour Cell Plasticity as a Resistance Mechanism and Therapeutic Target in Melanoma. Eur. J. Cancer 59, 109–112. doi:10.1016/j.ejca.2016.02.023
Rosenberg, S. A. (2011). Cell Transfer Immunotherapy for Metastatic Solid Cancer-What Clinicians Need to Know. Nat. Rev. Clin. Oncol. 8, 577–585. doi:10.1038/nrclinonc.2011.116
Rosenberg, S. A. (2014). IL-2: the First Effective Immunotherapy for Human Cancer. J.I. 192, 5451–5458. doi:10.4049/jimmunol.1490019
Rosewell Shaw, A., Porter, C. E., Watanabe, N., Tanoue, K., Sikora, A., Gottschalk, S., et al. (2017). Adenovirotherapy Delivering Cytokine and Checkpoint Inhibitor Augments CAR T Cells against Metastatic Head and Neck Cancer. Mol. Ther. 25, 2440–2451. doi:10.1016/j.ymthe.2017.09.010
Rous, P. (1910). A Transmissible Avian Neoplasm. (Sarcoma of the Common Fowl.). J. Exp. Med. 12, 696–705. doi:10.1084/jem.12.5.696
Rubinstein, J. C., Sznol, M., Pavlick, A. C., Ariyan, S., Cheng, E., Bacchiocchi, A., et al. (2010). Incidence of the V600K Mutation Among Melanoma Patients with BRAF Mutations, and Potential Therapeutic Response to the Specific BRAF Inhibitor PLX4032. J. Transl Med. 8, 67. doi:10.1186/1479-5876-8-67
Ruiter, D., Bogenrieder, T., Elder, D., and Herlyn, M. (2002). Melanoma-stroma Interactions: Structural and Functional Aspects. Lancet Oncol. 3, 35–43. doi:10.1016/s1470-2045(01)00620-9
Russell, S. J., Peng, K.-W., and Bell, J. C. (2012). Oncolytic Virotherapy. Nat. Biotechnol. 30, 658–670. doi:10.1038/nbt.2287
Sahin, U., Derhovanessian, E., Miller, M., Kloke, B.-P., Simon, P., Löwer, M., et al. (2017). Personalized RNA Mutanome Vaccines Mobilize Poly-specific Therapeutic Immunity against Cancer. Nature 547, 222–226. doi:10.1038/nature23003
Schumacher, T. N., and Schreiber, R. D. (2015). Neoantigens in Cancer Immunotherapy. Science 348, 69–74. doi:10.1126/science.aaa4971
Senzer, N. N., Kaufman, H. L., Amatruda, T., Nemunaitis, M., Reid, T., Daniels, G., et al. (2009). Phase II Clinical Trial of a Granulocyte-Macrophage colony-stimulating Factor-Encoding, Second-Generation Oncolytic Herpesvirus in Patients with Unresectable Metastatic Melanoma. Jco 27, 5763–5771. doi:10.1200/jco.2009.24.3675
Seo, N., Hayakawa, S., Takigawa, M., and Tokura, Y. (2001). Interleukin-10 Expressed at Early Tumour Sites Induces Subsequent Generation of CD4+ T-Regulatory Cells and Systemic Collapse of Antitumour Immunity. Immunology 103, 449–457. doi:10.1046/j.1365-2567.2001.01279.x
Sharma, P., Hu-Lieskovan, S., Wargo, J. A., and Ribas, A. (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 168, 707–723. doi:10.1016/j.cell.2017.01.017
Shreedhar, V. K., Pride, M. W., Sun, Y., Kripke, M. L., and Strickland, F. M. (1998). Origin and Characteristics of Ultraviolet-B Radiation-Induced Suppressor T Lymphocytes. J. Immunol. 161, 1327–1335.
Siegel, R. L., Miller, K. D., and Jemal, A. (2020). Cancer Statistics, 2020. CA A. Cancer J. Clin. 70, 7–30. doi:10.3322/caac.21590
Simiczyjew, A., Dratkiewicz, E., Mazurkiewicz, J., Ziętek, M., Matkowski, R., and Nowak, D. (2020). The Influence of Tumor Microenvironment on Immune Escape of Melanoma. Int. J. Mol. Sci. 21. doi:10.3390/ijms21218359
Simon, B., and Uslu, U. (2018). CAR ‐T Cell Therapy in Melanoma: A Future success story? Exp. Dermatol. 27, 1315–1321. doi:10.1111/exd.13792
Sitnik, S., Masemann, D., Leite Dantas, R., Wixler, V., and Ludwig, S. (2020). PD-1 IC Inhibition Synergistically Improves Influenza A Virus-Mediated Oncolysis of Metastatic Pulmonary Melanoma. Mol. Ther. - Oncolytics 17, 190–204. doi:10.1016/j.omto.2020.03.023
Sivan, A., Corrales, L., Hubert, N., Williams, J. B., Aquino-Michaels, K., Earley, Z. M., et al. (2015). Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates Anti-PD-L1 Efficacy. Science 350, 1084–1089. doi:10.1126/science.aac4255
Smith, M. L., Ford, J. M., Hollander, M. C., Bortnick, R. A., Amundson, S. A., Seo, Y. R., et al. (2000). p53-Mediated DNA Repair Responses to UV Radiation: Studies of Mouse Cells Lacking P53 , P21 , And/or Gadd45 Genes. Mol. Cel Biol 20, 3705–3714. doi:10.1128/mcb.20.10.3705-3714.2000
Soltantoyeh, T., Akbari, B., Karimi, A., Mahmoodi Chalbatani, G., Ghahri-Saremi, N., Hadjati, J., et al. (2021). Chimeric Antigen Receptor (CAR) T Cell Therapy for Metastatic Melanoma: Challenges and Road Ahead. Cells 10. doi:10.3390/cells10061450
Spagnolo, F., Picasso, V., Lambertini, M., Ottaviano, V., Dozin, B., and Queirolo, P. (2016). Survival of Patients with Metastatic Melanoma and Brain Metastases in the Era of MAP-Kinase Inhibitors and Immunologic Checkpoint Blockade Antibodies: A Systematic Review. Cancer Treat. Rev. 45, 38–45. doi:10.1016/j.ctrv.2016.03.003
Strauss, B. E., Silva, G. R. O., De Luna Vieira, I., Cerqueira, O. L. D., Del Valle, P. R., Medrano, R. F. V., et al. (2018). Perspectives for Cancer Immunotherapy Mediated by p19Arf Plus Interferon-Beta Gene Transfer. Clinics (Sao Paulo) 73, e479s. doi:10.6061/clinics/2018/e479s
Strauss, L., Bergmann, C., Szczepanski, M., Gooding, W., Johnson, J. T., and Whiteside, T. L. (2007). A Unique Subset of CD4+CD25highFoxp3+ T Cells Secreting Interleukin-10 and Transforming Growth Factor-Β1 Mediates Suppression in the Tumor Microenvironment. Clin. Cancer Res. 13, 4345–4354. doi:10.1158/1078-0432.ccr-07-0472
Sun, X., Zhang, N., Yin, C., Zhu, B., and Li, X. (2020). Ultraviolet Radiation and Melanomagenesis: From Mechanism to Immunotherapy. Front. Oncol. 10, 951. doi:10.3389/fonc.2020.00951
Swick, J. M., Maize, J. C., and Sr, (2012). Molecular Biology of Melanoma. J. Am. Acad. Dermatol. 67, 1049–1054. doi:10.1016/j.jaad.2011.06.047
Takeda, K., and Akira, S. (2004). TLR Signaling Pathways. Semin. Immunol. 16, 3–9. doi:10.1016/j.smim.2003.10.003
Tang, D., Kang, R., Coyne, C. B., Zeh, H. J., and Lotze, M. T. (2012). PAMPs and DAMPs: Signal 0s that spur Autophagy and Immunity. Immunol. Rev. 249, 158–175. doi:10.1111/j.1600-065x.2012.01146.x
Tanoue, K., Rosewell Shaw, A., Watanabe, N., Porter, C., Rana, B., Gottschalk, S., et al. (2017). Armed Oncolytic Adenovirus-Expressing PD-L1 Mini-Body Enhances Antitumor Effects of Chimeric Antigen Receptor T Cells in Solid Tumors. Cancer Res. 77, 2040–2051. doi:10.1158/0008-5472.can-16-1577
Tessarollo, N. G., Domingues, A. C. M., Antunes, F., Luz, J. C. D. S. D., Rodrigues, O. A., Cerqueira, O. L. D., et al. (2021). Nonreplicating Adenoviral Vectors: Improving Tropism and Delivery of Cancer Gene Therapy. Cancers (Basel) 13. doi:10.3390/cancers13081863
Trager, M. H., Geskin, L. J., and Saenger, Y. M. (2020). Oncolytic Viruses for the Treatment of Metastatic Melanoma. Curr. Treat. Options. Oncol. 21, 26. doi:10.1007/s11864-020-0718-2
Tsao, H., Yang, G., Goel, V., Wu, H., and Haluska, F. G. (2004). Genetic Interaction between NRAS and BRAF Mutations and PTEN/MMAC1 Inactivation in Melanoma. J. Invest. Dermatol. 122, 337–341. doi:10.1046/j.0022-202x.2004.22243.x
Tschandl, P., Berghoff, A. S., Preusser, M., Burgstaller-Muehlbacher, S., Pehamberger, H., Okamoto, I., et al. (2013). NRAS and BRAF Mutations in Melanoma-Associated Nevi and Uninvolved Nevi. PLoS One 8, e69639. doi:10.1371/journal.pone.0069639
Turajlic, S., Gore, M., and Larkin, J. (2018). First Report of Overall Survival for Ipilimumab Plus Nivolumab from the Phase III Checkmate 067 Study in Advanced Melanoma. Ann. Oncol. 29, 542–543. doi:10.1093/annonc/mdy020
Uslu, U. (2021). Driving CAR T Cells towards Dermatologic Oncology. JDDG: J. der Deutschen Dermatologischen Gesellschaft 19, 359–362. doi:10.1111/ddg.14402
Vachtenheim, J., and Ondrušová, L. (2015). Microphthalmia-associated Transcription Factor Expression Levels in Melanoma Cells Contribute to Cell Invasion and Proliferation. Exp. Dermatol. 24, 481–484. doi:10.1111/exd.12724
Vähä-Koskela, M. J., Heikkilä, J. E., and Hinkkanen, A. E. (2007). Oncolytic Viruses in Cancer Therapy. Cancer Lett. 254, 178–216.
Van Rooij, N., Van Buuren, M. M., Philips, D., Velds, A., Toebes, M., Heemskerk, B., et al. (2013). Tumor Exome Analysis Reveals Neoantigen-specific T-Cell Reactivity in an Ipilimumab-Responsive Melanoma. Jco 31, e439–e442. doi:10.1200/jco.2012.47.7521
Vanseggelen, H., Tantalo, D. G., Afsahi, A., Hammill, J. A., and Bramson, J. L. (2015). Chimeric Antigen Receptor-Engineered T Cells as Oncolytic Virus Carriers. Mol. Ther. - Oncolytics 2, 15014. doi:10.1038/mto.2015.14
Varghese, S., Rabkin, S. D., Liu, R., Nielsen, P. G., Ipe, T., and Martuza, R. L. (2006). Enhanced Therapeutic Efficacy of IL-12, but Not GM-CSF, Expressing Oncolytic Herpes Simplex Virus for Transgenic Mouse Derived Prostate Cancers. Cancer Gene Ther. 13, 253–265. doi:10.1038/sj.cgt.7700900
Veierød, M. B., Adami, H. O., Lund, E., Armstrong, B. K., and Weiderpass, E. (2010). Sun and Solarium Exposure and Melanoma Risk: Effects of Age, Pigmentary Characteristics, and Nevi. Cancer Epidemiol. Biomarkers Prev. 19, 111–120. doi:10.1158/1055-9965.EPI-09-0567
Vétizou, M., Pitt, J. M., Daillère, R., Lepage, P., Waldschmitt, N., Flament, C., et al. (2015). Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science 350, 1079–1084.
Vetizou, M., and Trinchieri, G. (2018). Anti-PD1 in the Wonder-Gut-Land. Cel. Res. 28, 263–264. doi:10.1038/cr.2018.12
Vigil, A., Park, M.-S., Martinez, O., Chua, M. A., Xiao, S., Cros, J. F., et al. (2007). Use of Reverse Genetics to Enhance the Oncolytic Properties of Newcastle Disease Virus. Cancer Res. 67, 8285–8292. doi:10.1158/0008-5472.can-07-1025
Vijayakumar, G., Palese, P., and Goff, P. H. (2019). Oncolytic Newcastle Disease Virus Expressing a Checkpoint Inhibitor as a Radioenhancing Agent for Murine Melanoma. EBioMedicine 49, 96–105. doi:10.1016/j.ebiom.2019.10.032
Villanueva, J., and Herlyn, M. (2008). Melanoma and the Tumor Microenvironment. Curr. Oncol. Rep. 10, 439–446. doi:10.1007/s11912-008-0067-y
Viros, A., Sanchez-Laorden, B., Pedersen, M., Furney, S. J., Rae, J., Hogan, K., et al. (2014). Ultraviolet Radiation Accelerates BRAF-Driven Melanomagenesis by Targeting TP53. Nature 511, 478–482. doi:10.1038/nature13298
Voit1, C., Kron2, M., Schwurzer-Voit1, M., and Sterry1, W. (2003). Intradermal injection of Newcastle disease virus-modified autologous melanoma cell lysate and interleukin-2 for adjuvant treatment of melanoma patients with resectable stage III disease. Adjuvante Therapie von Melanompatienten im Stadium III mit einer Kombination aus Newcastle-disease-Virus-modifiziertem Tumorzelllysat und Interleukin-2. J. Deut Dermatol. Gesell 1, 120–125. doi:10.1046/j.1610-0387.2003.02014.x
Walboomers, J. M. M., Jacobs, M. V., Manos, M. M., Bosch, F. X., Kummer, J. A., Shah, K. V., et al. (1999). Human Papillomavirus Is a Necessary Cause of Invasive Cervical Cancer Worldwide. J. Pathol. 189, 12–19. doi:10.1002/(sici)1096-9896(199909)189:1<12::aid-path431>3.0.co;2-f
Wan, P. T. C., Garnett, M. J., Roe, S. M., Lee, S., Niculescu-Duvaz, D., Good, V. M., et al. (2004). Mechanism of Activation of the RAF-ERK Signaling Pathway by Oncogenic Mutations of B-RAF. Cell 116, 855–867. doi:10.1016/s0092-8674(04)00215-6
Watanabe, K., Luo, Y., Da, T., Guedan, S., Ruella, M., Scholler, J., et al. (2018). Pancreatic Cancer Therapy with Combined Mesothelin-Redirected Chimeric Antigen Receptor T Cells and Cytokine-Armed Oncolytic Adenoviruses. JCI Insight 3. doi:10.1172/jci.insight.99573
Watson, I. R., Li, L., Cabeceiras, P. K., Mahdavi, M., Gutschner, T., Genovese, G., et al. (2014). The RAC1 P29S Hotspot Mutation in Melanoma Confers Resistance to Pharmacological Inhibition of RAF. Cancer Res. 74, 4845–4852. doi:10.1158/0008-5472.can-14-1232-t
Wei, D., Xu, J., Liu, X.-Y., Chen, Z.-N., and Bian, H. (2018). Fighting Cancer with Viruses: Oncolytic Virus Therapy in China. Hum. Gene Ther. 29, 151–159. doi:10.1089/hum.2017.212
Wing, A., Fajardo, C. A., Posey, A. D., Shaw, C., Da, T., Young, R. M., et al. (2018). Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-Cell Engager. Cancer Immunol. Res. 6, 605–616. doi:10.1158/2326-6066.cir-17-0314
Yadav, R., Bajpai, P. K., Srivastava, D. K., and Kumar, R. (2021). Epidemiological Characteristics, Reinfection Possibilities and Vaccine Development of SARS CoV2: A Global Review. J. Fam. Med. Prim. Care 10, 1095–1101. doi:10.4103/jfmpc.jfmpc_2151_20
Yang, L., Li, A., Lei, Q., and Zhang, Y. (2019). Tumor-intrinsic Signaling Pathways: Key Roles in the Regulation of the Immunosuppressive Tumor Microenvironment. J. Hematol. Oncol. 12, 125. doi:10.1186/s13045-019-0804-8
Yarchoan, M., Johnson, B. A., Lutz, E. R., Laheru, D. A., and Jaffee, E. M. (2017). Targeting Neoantigens to Augment Antitumour Immunity. Nat. Rev. Cancer 17, 209–222. doi:10.1038/nrc.2016.154
Zaman, A., Wu, W., and Bivona, T. G. (2019). Targeting Oncogenic BRAF: Past, Present, and Future. Cancers (Basel) 11. doi:10.3390/cancers11081197
Zhang, T., Dutton-Regester, K., Brown, K. M., and Hayward, N. K. (2016). The Genomic Landscape of Cutaneous Melanoma. Pigment Cel. Melanoma Res. 29, 266–283. doi:10.1111/pcmr.12459
Zhou, J., Dudley, M. E., Rosenberg, S. A., and Robbins, P. F. (2005). Persistence of Multiple Tumor-specific T-Cell Clones Is Associated with Complete Tumor Regression in a Melanoma Patient Receiving Adoptive Cell Transfer Therapy. J. Immunother. 28, 53–62. doi:10.1097/00002371-200501000-00007
Zhou, R., Shi, C., Tao, W., Li, J., Wu, J., Han, Y., et al. (2019). Analysis of Mucosal Melanoma Whole-Genome Landscapes Reveals Clinically Relevant Genomic Aberrations. Clin. Cancer Res. 25, 3548–3560. doi:10.1158/1078-0432.ccr-18-3442
Keywords: immunotherapy, gene therapy, adenovirus, melanoma, checkpoint inhibitors, CAR-T cells, p53, interferon
Citation: Cerqueira OLD, Antunes F, Assis NG, Cardoso EC, Clavijo-Salomón MA, Domingues AC, Tessarollo NG and Strauss BE (2022) Perspectives for Combining Viral Oncolysis With Additional Immunotherapies for the Treatment of Melanoma. Front. Mol. Biosci. 9:777775. doi: 10.3389/fmolb.2022.777775
Received: 15 September 2021; Accepted: 22 March 2022;
Published: 14 April 2022.
Edited by:
Majid Jabir, University of Technology, IraqReviewed by:
Wolfgang Walther, Charité Universitätsmedizin Berlin, GermanyKonstantin Kousoulas, Louisiana State University, United States
Kayhan Azadmanesh, Pasteur Institute of Iran, Iran
Copyright © 2022 Cerqueira, Antunes, Assis, Cardoso, Clavijo-Salomón, Domingues, Tessarollo and Strauss. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bryan E Strauss, YnN0cmF1c3NAdXNwLmJy