 Xin-Yi Xu1
Xin-Yi Xu1 Yan Geng
Yan Geng Yilin Ren
Yilin Ren- 1Institute of Cancer, Affiliated Hospital of Jiangnan University, Wuxi, China
- 2School of Life Science and Health Engineering, Jiangnan University, Wuxi, China
- 3Department of Urology, Affiliated Wuxi No. 2 Hospital of Nanjing Medical University, Wuxi, China
- 4Department of Gastroenterology, Affiliated Hospital of Jiangnan University, Wuxi, China
- 5Department of Oncology, Affiliated Hospital of Jiangnan University, Wuxi, China
Hepatic stellate cells (HSCs) play an essential role in the development of liver fibrosis. Antrodia camphorata (A. camphorata) is a medicinal fungus with hepatoprotective effect. This study investigated whether Antrodin C, an A. camphorata-fermented metabolite, could exert a protective role on liver fibrosis both in vitro and in vivo. The anti-fibrotic effect of Antrodin C was investigated in CFSC-8B cell (hepatic stellate cell) stimulated by transforming growth factor-β1 (TGF-β1) or platelet-derived growth factor-BB (PDGF-BB) in vitro and in CCl4 induced liver fibrosis in mice. Antrodin C (50 μM) inhibited TGF-β1 or PDGF-BB stimulated CFSC-8B cell activation, migration and extracellular matrix (ECM) accumulation (all p < 0.05). Antrodin C (3, 6 mg/kg/d) oral administration reduced the degree of liver fibrosis induced by CCl4 in mice. Antrodin C down-regulated the expression of α-smooth muscle actin (α-SMA) and collagen I in fibrotic livers. Furthermore, Antrodin C ameliorated alanine aminotransferase (ALT) and aspartate aminotransferase (AST) elevation in serum (all p < 0.05). Mechanistically, Antrodin C executes its anti-fibrotic activity through negatively modulate TGF-β1 downstream SMAD Family Member 2 (Smad2), AKT Serine/Threonine Kinase 1 (AKT), extracellular signal-regulated kinase (ERK), and P38 MAP Kinase (P38), as well as PDGF-BB downstream AKT and ERK signaling pathways. Antrodin C ameliorates the activation, migration, ECM production in HSCs and CCl4-induced liver fibrosis in mice, suggesting that Antrodin C could serve as a protective molecule against liver fibrosis.
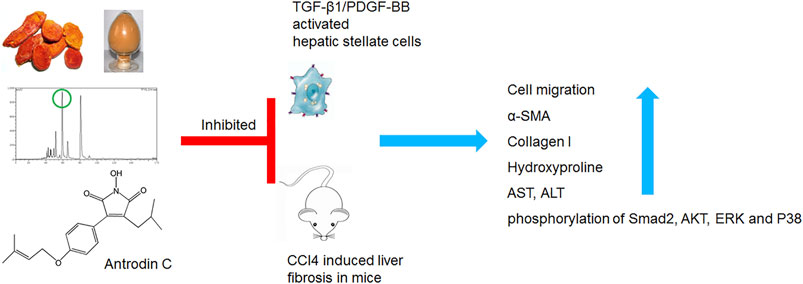
GRAPHICAL ABSTRACT
Introduction
Organ fibrosis refers to the excessive deposition of extracellular matrix (ECM) in response to chronic tissue injury (Caligiuri et al., 2021). Hepatic fibrosis is caused by various factors, such as genomic mutations, toxic chemicals, hepatitis B/C, excessive alcohol consumption, and nonalcoholic steatohepatitis (Guerra et al., 2016; George et al., 2019). Liver fibrosis are wound-healing responses generated against an insult to the liver that cause liver injury (Aydin and Akcali, 2018). Liver fibrosis has the potential to progress to cirrhosis, liver cancer, and liver failure and its complications represent a massive health care burden worldwide (Deng et al., 2020). Aging has been considered as a risk factor for progression of fibrosis in hepatitis C and for poor outcome in alcoholic hepatitis (Goldstein et al., 2005; David and George, 2008). Therefore, it has been suggested that aging increases the susceptibility of liver fibrosis. Recent studies suggest that hepatic fibrosis could be reversible, however, its underlying mechanism remains uncertain and efficient anti-fibrotic drugs are urgently needed (Tan et al., 2021).
Hepatic stellate cells (HSCs) constitute the major mesenchymal cell type of the liver and play pivotal roles in a hepatic injury response (Dhar et al., 2020). Upon chronic liver injury, the quiescent HSCs (qHSCs) receive secreted signals and become activated HSCs (aHSCs) which express α-smooth muscle actin (α-SMA) and produce excessive extracellular matrix (ECM), including collagens and fibronectin (Jin et al., 2020; Yoon et al., 2020; Trivedi et al., 2021). Among many aberrant signaling molecules, transforming growth factor β1 (TGF-β1) and platelet-derived growth factor (PDGF) mediated signaling plays prominent roles in the transition of qHSCs into aHSCs (Kang et al., 2013).
Antrodia camphorata (also known as Taiwanofungus camphoratus, Antrodia cinnamomea) has been used as a traditional medicine or functional food for a long history in China for treating diarrhea, viral infection, diabetes mellitus liver cirrhosis, hepatoma and more (Zhenwei et al., 2021). Fermented mycelium or mycelial extract from A. camphorata has been found to inhibit HSCs activation in vitro and CCl4 or thioacetamide induced liver fibrosis in vivo (Schyman et al., 2019). Two maleimide derivatives Antrodin B and Antrodin C isolated from the mycelia of A. camphorata inhibited the growth of Lewis lung carcinoma cells in vitro (Huang et al., 2019). Antrodin C inhibited breast cancer cell migration and invasion by suppressing Smad2/3 and β-Catenin signaling pathways (Kumar et al., 2015). We previously identified Antrodin B from A. camphorata as a novel anti-fibrotic compounds through a bioassay-guided fractionation approach (Geng et al., 2016). Antrodin C also suppressed lipopolysaccharide-induced inflammation (Lee et al., 2014). However, whether Antrodin C could inhibit HSCs activation and liver fibrosis, and the underlying mechanism remains unclear.
In this study, we isolated Antrodin C from mycelial extract of A. camphorata and investigated the effect and potential mechanism of Antrodin C on the aHSCs in vitro and CCl4 induced liver fibrosis in mice. Together, we unraveled Antrodin C as an active compound in A. camphorata against liver fibrosis.
Materials and Methods
Chemicals
3- (4, 5-dimethyl-2-thiazolyl)-2, 5-diphenyl-2H-tetrazolium bromide (MTT) (88417), silybin (S0292), SB431542 (616464), and other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, United States). Recombinant Human TGF-β1 (AF-100-21C), and PDGF-BB (AF-100-14B) were purchased from PeproTech (Rocky Hill, NJ, United States). RPMI 1640 medium, fetal bovine serum (FBS), trypsin and antibiotics (penicillin and streptomycin) were purchased from Gibco (Thermofisher, USA).
Isolation and Identification of Antrodin C
The extraction and fractionation procedures were described previously (Geng et al., 2016). Briefly, the dried mycelium of A. camphorata was extracted in methanol and then partitioned with n-hexane (319902, Sigma-Aldrich), chloroform (288306, Sigma-Aldrich) and ethyl acetate (319902, Sigma-Aldrich). The n-hexane-soluble fraction was chromatographed on a silica gel column eluted with a gradient of n-hexane and ethyl acetate. The fraction eluted by 16% of EtOAc was further separated by a semipreparative HPLC column (Waters XBridge C18 column, Ф19 × 250 mm, 5 μm) (Supplementary Figure S1). The mobile phase consisted of distilled water H2O (0.5% acetic acid) and acetonitrile (34851, Sigma-Aldrich) at 10 ml/min to obtain Antrodin C (purity > 95%). The structures of Antrodin C was analyzed by comparing their LC-MS, 1H, 13C NMR spectroscopic data (Supplementary Figures S2–S4) and compared with published data (Nakamura et al., 2004).
Cell Culture
Rat hepatic stellate cell line (CFSC-8B cells) were obtained from the cell bank of Xiangya Central Experiment Laboratory of Central South University (Changsha, China). CFSC-8B cells were cultured at 37°C in a humidified 5% CO2 incubator and RPMI 1640 medium, supplemented with 10% FBS, 100 U/mL penicillin and 100 mg/ml streptomycin.
Cell Viability
The viability of CFSC-8B cells was quantified by the ability of living cells to reduce the yellow dye MTT to a blue formazan product. CFSC-8B cells (8 × 104 cells/mL) were seeded in a 96-well cell culture plate (Corning Incorporated, USA) and grew to 80%–90% confluence. Then the cells were incubated with Antrodin C (10–200 μM) for 24 h. The viability (% of the control) of cells treated with Antrodin C was calculated as 100% × (absorbance of treated cells/absorbance of control cells).
Cell Migration Assay
CFSC-8B cells (3 × 106 cells/mL) were seeded onto the upper chambers (8 μm pore size, Milipore, Billerica, Massachusetts, United States) with 100 μL of 0.5% FBS medium and 0.5 ml normal growth medium was added to the lower chambers as a chemoattractant in 24 well plate for 24 h. The cells left on the upper chambers were removed using a cotton swab. Then the chambers were fixed using 4% paraformaldehyde for 30 min, washed and stained 0.5% crystal violet for 30 min at 37°C. Five random views were photographed under a microscope (Nikon, Tokyo, Japan). The positive cells were counted and quantified using ImageJ. Silybin (25 μM) and SB431542 (2 μM) were used as positive controls in the assay.
Animals and CCl4 Induced Liver Fibrosis
All animal experiments were approved by the Animal Research Committee of Jiangnan University. Male 6–8 weeks old BALB/c mice were obtained from Shanghai SLAC Laboratory Animal Co., Ltd. The animals were housed under standard conditions and fed with a normal chow diet (M01-F25-20150922034, Shanghai SLAC Laboratory Animal Co., Shanghai, China). The mice were randomly and equally divided into the CTL group, Silymarin group, CCl4 group and two Antrodin C treatment groups (n = 6 per group). In the Antrodin C treatment groups, mice were orally administered with Antrodin C (3 or 6 mg/kg/d, formulated in 0.5% Carboxymethylcellulose sodium) daily for 2 weeks before CCl4 injection. Silymarin was used as a positive drug control at the dose of 100 mg/kg/d daily for 2 weeks before CCl4 injection. Then the CCl4, Silymarin and Antrodin C groups were intraperitoneal injection CCl4 (0.5 ml/kg, 25% solution in olive oil) twice per week, and the control group was given the same dose of olive oil. The Silymarin or Antrodin C groups were orally administered with Antrodin C or Silymarin for 4 weeks together with CCl4 injection.
Histology Analysis
Liver tissues were embedded in paraffin and 4-μm-thick slices were cut, and placed on glass slides. Slides were stained with hematoxylin-eosin (H&E) or Sirius-red, and examined under a light microscope (Nikon, Tokyo, Japan). HE staining was performed to assess pathological changes in the liver. Sirius-red staining was performed to detect collagen deposition and was analyzed by ImageJ software (NIH, Bethsda, MD).
Measurement of Serum Aminotransferase Activities and Hydroxyproline Contents
The activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST) in serum, and hydroxyproline contents in liver tissues were estimated spectrophotometrically using commercial diagnostic kits (Jiancheng Institute of Biotechnology, Nanjing, China).
RNA Isolation and qRT-PCR Analysis
Total RNA was extracted from mouse liver tissue or cells with Trizol reagent (Thermofisher, CA, United States). Gene expressions were measured relative to the endogenous control gene Gapdh using the comparative CT method and the sequences of specific primer pairs for α-SMA, Col1, and Col3 were described previously (Geng et al., 2016).
Western Blotting Analysis
Protein extracted from cells was resolved by SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes. Antibodies against α-SMA, GAPDH were from Sigma-Aldrich (St. Louis, MO, United States). Antibodies against Smad2, phospho-Smad2, P38, phospho-P38, ERK, phospho-ERK, AKT, phospho-AKT were purchased from Cell Signaling Technology (Danvers, MA, United States). The bands were visualized using ECL reagents (Pierce, Thermofisher Scientific, USA). Band intensity was quantified using Image lab software (Bio-Rad Laboratories, Inc. USA) and expressed as relative intensity compared with control. GAPDH level served as an internal control.
Statistical Analysis
Data are expressed as means ± SD. Differences in measured variables among groups were assessed by using One-way analysis of variance (ANOVA), and the Tukey test was used for determining the significance (Graphpad, San Diego, CA, United States). Results were considered statistically significant at p < 0.05.
Results
Effect of Antrodin C on the Survival of CFSC-8B Cells
We examined the cytotoxic effects of Antrodin C (Figure 1A) on hepatic stellate CSFC-8B cells using an MTT assay. CFSC-8B cells were treated with 10–200 μM Antrodin C for 24 h (Figure 1B). When the concentration of Antrodin C was above 60 μM, the cell survival rate was less than 80%. Antrodin C at the concentration of 200 μM significantly inhibited CFSC-8B cell proliferation with 82% inhibition. The median inhibitory concentration (IC50) of Antrodin C for CFSC-8B cells was 147.91 μM. The data showed that Antrodin C is able to reduce the proliferation of HSCs, and because we found that the concentrations below 50 μM of Antrodin C do not have significant cytotoxic effects on CFSC-8B cells, we used these concentrations (12, 25, 50 μM) in the following experiments.
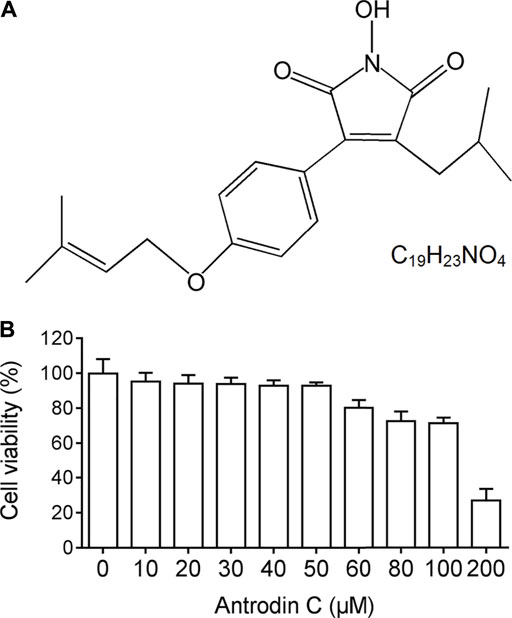
FIGURE 1. The chemical structure and cell viability of CSFC-8B cells treated with Antrodin C (A) The chemical structure of Antrodin C. (B) Effect of Antrodin C on cell viability of CFSC-8B cells by MTT assay.
Antrodin C Inhibits TGF-β1 Induced Cell Migration in CFSC-8B Cells
A previous study suggested that the cell migration of aHSCs is involved in the initial pathological development of liver fibrosis (Selenina et al., 2019). As anticipated, TGF-β1 (10 ng/ml) up-regulated CFSC-8B cell migration, while TGF-β receptor inhibitor SB431542 (Geng et al., 2016) dramatically decreased TGF-β1 induced cell migration (Figure 2A). Antrodin C (12–50 μM) inhibited cell migration stimulated by TGF-β1 in a dose-dependent manner in CFSC-8B cells (Figure 2A). These results suggest that Antrodin C might play an inhibitory role in TGF-β1-mediated HSCs activation.
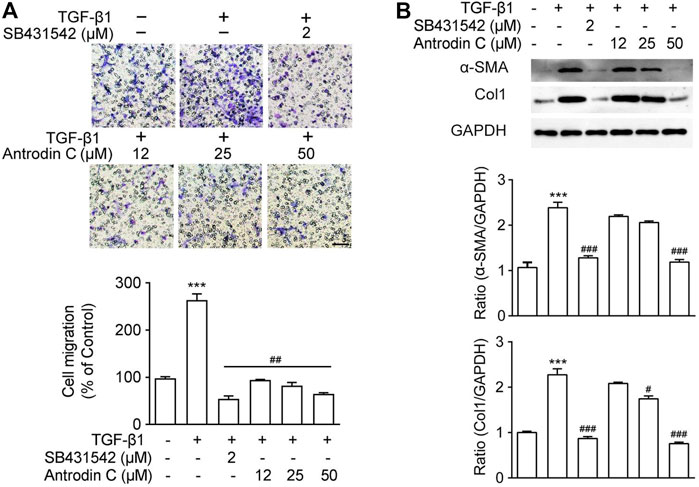
FIGURE 2. Effects of Antrodin C on TGF-β1 induced cell migration as well as α-SMA and Col1 production in CFSC-8B cells. (A) Representative images of migrated CSFC-8B cells treated with or without Antrodin C (12–50 μM) and SB431542 in the presence or absence of TGF-β1. The data were normalized to % of migrated control cells. Scale bar, 100 μm. (B) Western blot analysis of α-SMA and Col1 expression. GAPDH was used as an internal control. Data represent mean ± SD (n = 3), ***p < 0.001 compared with control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with cells treated with TGF-β1 only.
Antrodin C Reduces Activation and ECM Accumulation Induced by TGF-β1 in CFSC-8B Cells
To reveal the role of Antrodin C in HSCs activation, we assessed the effect of Antrodin C intervention on TGF-β1 induced expression of α-SMA and collagen I, that were the markers of HSCs activation (Tsuchida and Friedman, 2017). As shown in Figure 2B, TGF-β1 significantly increased a-SMA and Col1 expression in CFSC-8B cells. Antrodin C at a concentration of 50 μM suppressed a-SMA protein expression. The production of Col1 was also suppressed by the addition of Antrodin C (12–50 μM) in a dose-dependent manner (Figure 2B). Similar results were observed at the gene expression of a-SMA and Col1 by qRT-PCR analysis (Supplementary Figure S5A). Furthermore, Antrodin C (25–50 μM) inhibited TGF-β1 induced Col3 and Fibronectin (Fn) transcription in CFSC-8B cells (Supplementary Figure S5A). These data indicate that Antrodin C can inhibit the TGF-β1 induced HSCs activation and ECM production.
Antrodin C Suppresses TGF-β1-Stimulated Phosphorylation of Smad2, AKT, ERK, and P38 in CFSC-8B Cells
TGF-β1 is a well-known fibrogenic cytokine that activates HSCs and induces ECM production. Classical TGF-β1 signaling is initiated with ligand-induced oligomerization of serine/threonine receptor kinases, then through phosphorylation of the cytoplasmic signaling molecules Smad2 and Smad3 (Zhao et al., 2020). As expected, TGF-β1 stimulated p-Smad2 expression in CFSC-8B cells, which was blocked by TGF-β1 receptor inhibitor SB431542 (Figure 3A). Within the safe doses (25–50 μM), treatment with Antrodin C reversed TGF-β1 induced p-Smad2 (Figure 3). TGF-β signaling can also modulate Smad-independent pathways, including PI3K/AKT, ERK, JNK, and p38 MAPK pathways (Suwanabol et al., 2012). We found that TGF-β1 significantly up regulated the phosphorylation of AKT, ERK and P38 in CFSC-8B cells (Figure 3). Antrodin C treatment dose dependently inhibit TGF-β1 induced phosphorylation of AKT, ERK, and P38 (Figure 3). These results suggest that Antrodin C not only inhibits the classical TGF-β1 pathway, but also suppresses non-classical TGF-β1 signaling in HSCs activation, which is characterized by decreased TGF-β1 induced phosphorylation level of Smad2, AKT, ERK, and P38.
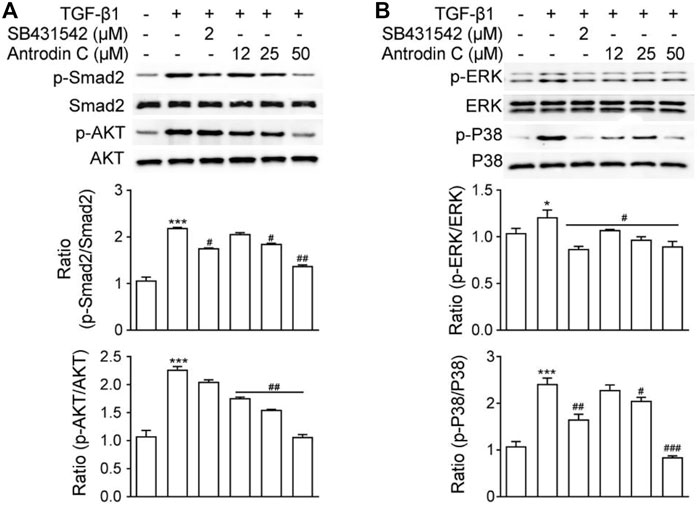
FIGURE 3. Effects of Antrodin C on the TGF-β1-induced signaling pathway. (A,B) CFSC-8B cells were treated with Antrodin C (12–50 μM) for 2 h and then induced by TGF-β1 for 1 h, and total cellular extracts were prepared and subjected to Western blot analysis to measure the levels of phosphorylated Smad2, AKT, ERK, and P38. Total Smad, AKT, ERK, and P38 were used for normalization. Data represent mean ± SD (n = 3), *p < 0.05, ***p < 0.001 compared with control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with TGF-β1 treated only.
Antrodin C Inhibits PDGF-BB-Induced Cell Migration in CFSC-8B Cells
The presence of PDGF-BB accelerated HSCs migration by chemoattractant mechanism (Ikeda et al., 2010). We found that PDGF-BB (10 ng/ml) induced cell migration (Figure 4A), while the positive control Silybin (25 μM) which is the major active constituent of hepatoprotective and anti-fibrotic drug, inhibited this effect (Brinda et al., 2012). Antrodin C (12–50 μM) suppressed the migration of CFSC-8B cells stimulated with PDGF-BB (10 ng/ml) in a dose-dependent manner (Figure 4A).
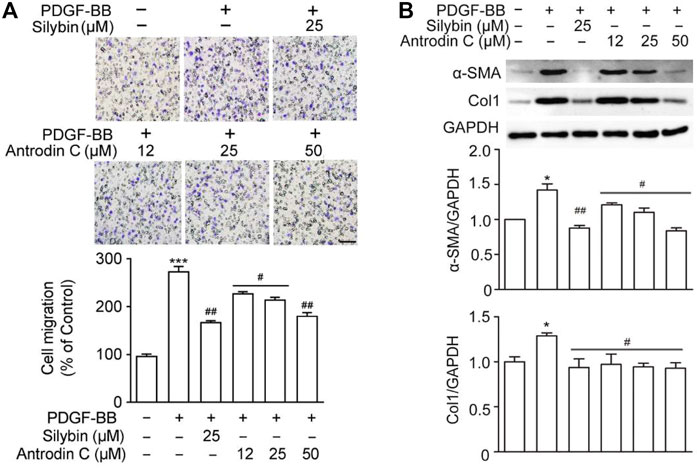
FIGURE 4. Effects of Antrodin C on PDGF-BB induced cell migration, α-SMA and Col1 production in CFSC-8B cells. (A) Representative images of migrated CSFC-8B cells treated with or without Antrodin C (12–50 μM) and Silybin in the presence or absence of PDGF-BB. The data were normalized to % of migrated control cells. Scale bar, 100 μm. (B) Western blot analysis of α-SMA and Col1 expression. GAPDH was used as an internal control. Data represent mean ± SD (n = 3), ***p < 0.001 compared with control; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with PDGF-BB treated only.
Antrodin C Reduces HSC Activation and ECM Accumulation Induced by PDGF-BB in CFSC-8B Cells
We then examined whether Antrodin C was able to inhibit the HSCs activation and ECM production induced by PDGF-BB at the molecular level. The remarkable makers of HSCs activation α-SMA and Col1 were both significantly induced by PDGF-BB treatment (Figure 4B). These inductions were suppressed by the addition of Silybin (25 μM) as well as Antrodin C (12–50 μM) (Figure 4B). Furthermore, α-SMA, Col1, Col3, and Fn transcription levels were significantly decreased in PDGF-BB stimulated CFSC-8B cells after 50 μM Antrodin C treatment (Supplementary Figure S5B). Thus, Antrodin C inhibits PDGF-BB induced HSCs activation and ECM production.
Antrodin C Treatment Downregulates PDGF-BB-Induced ERK and Akt Phosphorylation in CFSC-8B Cells
PDGF-BB is one of the most potent HSC mitogen, which binds to PDGFR-β, then sequentially activates Raf-1, MEK and extracellular-signal regulated kinase (ERK). PDGF also induces PI3K/Akt signaling pathway, which is necessary for both mitogenesis and chemotaxis and involved in activating the Ras-ERK pathway (Tricarico et al., 2002). As shown in Figure 5A, Antrodin C (25, 50 μM) decreased the phosphorylation of ERK compared to that of PDGF-BB-induced cells. Antrodin C (25, 50 μM) also significantly blocked PDGF-BB induced Akt phosphorylation (Figure 5B). These results demonstrate that the inhibitory effect of Antrodin C on liver fibrosis might be through inactivation of PDGF-BB induced p-ERK and p-Akt.
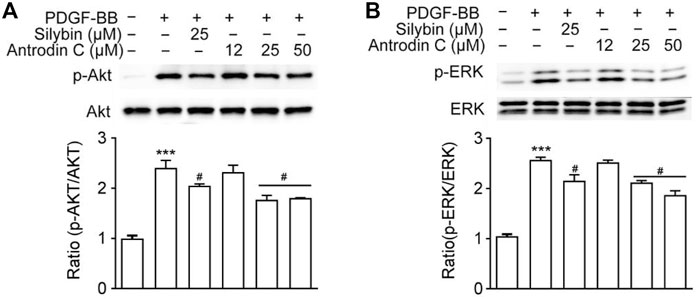
FIGURE 5. Effects of Antrodin C on the PDGF-BB-induced signaling pathway. (A,B) CFSC-8B cells were treated with Antrodin C (12–50 μM) for 2 h and then induced by PDGF-BB for 1 h, and total cellular extracts were prepared and subjected to Western blot analysis to measure the levels of phosphorylated AKT and ERK. Total AKT and ERK were used for normalization. Data represent means ± SD (n = 3), ***p < 0.001 compared with control; #p < 0.05 compared with PDGF-BB treated only.
Antrodin C Protects CCl4 Induced Liver Fibrosis in Mice
To determine the role of Antrodin C in vivo, we used the well-established model of CCl4 induced liver fibrosis in mice. Histopathological analysis by H&E and Sirius-red staining showed a considerable increase in the extent of liver fibrosis after repeated injection of CCl4 (Figure 6A). The oral administration of Antrodin C or positive control Silymarin reduced the degree of liver fibrosis as determined by histopathological analysis as well as the quantification of Sirus-red staining (Figures 6A,B). Hydroxyproline content further confirmed that Antrodin C inhibited CCl4 induced collagen production in mice (Figure 6C). ALT (alanine transaminase) and AST (aspartate aminotransferase) have been recognized as indicators of liver function (Odiba et al., 2014). The elevated serum ALT and AST revealed that CCl4 treatment cause hepatotoxicity, while Antrodin C treatment dramatically inhibited this effect (Figures 6D,E). HSCs are mainly responsible in the progression of liver fibrosis which produces massive ECM including collagens, and α-SMA is a marker of the activated HSCs (Tsuchida and Friedman, 2017). Repeated CCl4 injection resulted in the liver fibrosis with increased α-SMA and Col1 protein expression (Figure 6F). Immunoblotting analysis revealed that α-SMA and Col1 were less expressed in the liver tissues from the Antrodin C treated group than the CCl4 group (Figure 6F). Therefore, Antrodin C showed a protective effect on liver injury and fibrosis.
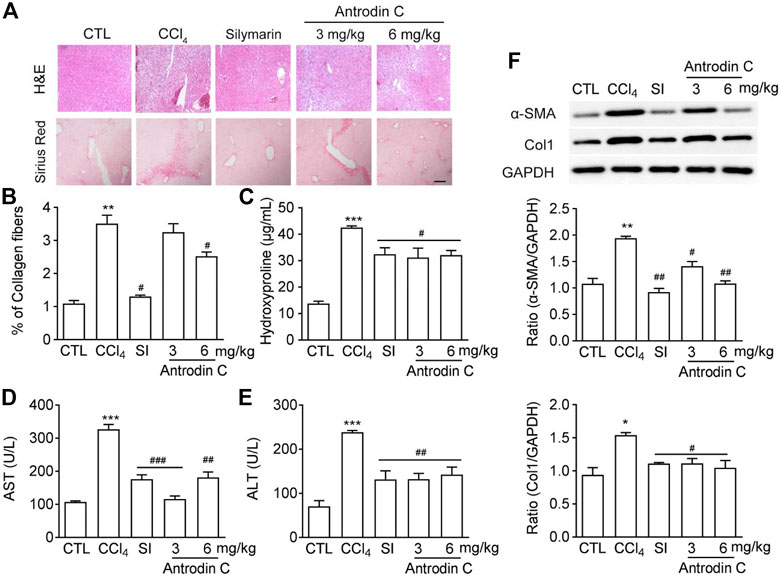
FIGURE 6. Effects of Antrodin C on liver fibrosis induced by CCl4 in vivo. (A) Representative images of H&E and Sirius Red staining of liver tissue sections from the different treatment groups. Scale bar, 100 μm. (B) The Sirius red staining was normalized to % of collagen fibers of the control group. (C) Hydroxyproline contents, (D) ALT and (E) AST activity in serum of CCl4-induced mice or control mice. (F) Western blot analysis of α-SMA and Col1 expression in livers tissues. GAPDH was used as an internal control. Data represent means ± SD (n = 6), *p < 0.05, **p < 0.01, ***p < 0.001 compared with control group; #p < 0.05, ##p < 0.01, ###p < 0.001 compared with CCl4-treated group. CTL: the control group; CCl4: the CCl4 treated group; SI: the Silymarin treated group.
Antrodin C Inhibits CCl4 Induced Phosphorylation of Smad2, -Akt, -ERK and -P38 MAPK in Mice
To explore a potential signaling pathways affected by Antrodin C treatments, we determined the levels of p-Smad2, p-Akt, p-ERK, and p-P38 MAPK, which belong to the TGF-β1 or PDGF-BB mediated signaling pathways. The results showed that the p-Smad2, p-Akt, p-ERK, and p-P38 were increased in mice treated with CCl4 compared with the control group (Figures 7A,B). Antrodin C treatment significantly inhibited the phosphorylation of Smad2, Akt, ERK, JNK and P38 (Figures 7A,B). These results demonstrate that Antrodin C offers a promising therapeutic strategy for patients with liver fibrosis through inhibiting p-Smad2, p-Akt, p-ERK, and p-P38.
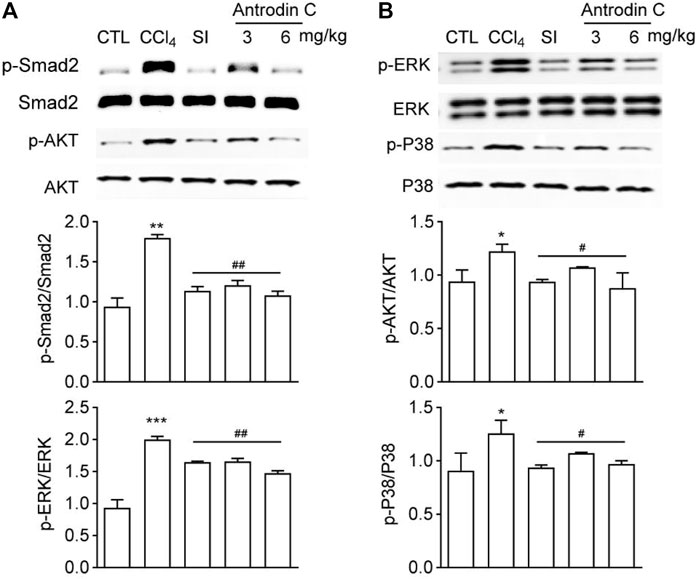
FIGURE 7. Effects of Antrodin C on phosphorylation of Smad2, AKT, ERK, and P38 in the livers of CCl4-treated mice. (A,B) Western blot analysis of phosphorylated and total Smad2, AKT, ERK, and P38 in livers tissues. Total Smad2, AKT, ERK, and P38 were used for normalization, respectively. Data represent mean ± SD (n = 6), *p < 0.05, **p < 0.01, ***p < 0.001 compared with control group; #p < 0.05, ##p < 0.01 compared with CCl4-treated group. CTL: the control group; CCl4: the CCl4-treated group; SI: the Silymarin treated group.
Discussion
Many bioactive components/compounds were identified in A. camphorata, including triterpenoids, polysaccharides, benzenoids, lignans, steroids, succinic, and maleic derivatives (Geethangili and Tzeng, 2010). However, little attention has been focused on why A. camphorate can inhibit liver fibrosis and what ingredients play a key role. In our study, we found that Antrodin C isolated from A. camphorata demonstrate significant anti-liver fibrosis properties. Antrodin C significantly inhibited cell migration, ECM production and HSCs activation induced by TGF-β1 or PDGF-BB in a dosage-dependent manner. In terms of mechanism, Antrodin C suppressed TGF-β1 induced p-Smad2, p-AKT, p-ERK, p-P38, as well as PDGF-BB induced p-AKT and p-ERK. We also showed that Antrodin C could attenuate CCl4-induced liver fibrosis in mice through blocking phosphorylation of Smad2/Akt/ERK/P38.
HSCs are believed to be a key player of liver fibrosis. Growth factors (TGF-β and PDGF) can provoke the activation of the HSCs, and activated HSCs migrate to the sites of damaged tissue and contribute to the expansion of fibrotic lesions in liver. We used CFSC-8B cells which are an immortalized rat liver stellate cell line as an in vitro cell model of hepatic fibrosis (Greenwel et al., 1995). We observed that Antrodin C inhibited TGF-β1 or PDGF-BB stimulated CFSC-8B cell activation, migration and ECM production.
Current anti-fibrotic therapeutic strategies include inhibition of HSCs migration, TGF-β expression and activation, blocking TGF-β canonical or non-canonical signaling pathways (Rosenbloom et al., 2013). TGF-β signaling pathways are critical for the fibrotic response. The canonical TGF-β1 signaling is mediated through receptor binding, then through receptor activating Smads, Smad2, or Smad3 (Kastanis et al., 2011). In addition to the Smad dependent pathway, TGF-β1 also activate PI3K/AKT, ERK, JNK, and P38 MAPK signaling pathways, which play an important role in regulating HSC activation and ECM synthesis (Voloshenyuk et al., 2011). Our data showed that TGF-β1 stimulated the canonical or non-canonical signaling indicated by the up regulation of phosphorylation of Smad2, Akt, ERK, and p38 MAPK, which could be inhibited by Antrodin C.
PDGF signaling plays a critical role in various cellular responses including proliferation, chemotaxis and actin reorganization. PDGFs bind and activate tyrosine kinase receptors, PDGFRα and PDGFRβ, which leads to overlapping signaling, including Src family kinases, PI3K/AKT and Ras-MAPK. PDGF can affect the fibrotic progression of several organs, including liver, lung and kidney (Steiner and Higgins, 2018). Several reports have implicated that there was a crosstalk between TGF-β1 and PDGF signaling (Novosyadlyy et al., 2006). Our results demonstrated that Antrodin C might suppress the PDGF induced activation of HSCs through blocking phosphorylation of Akt and ERK.
CCl4-induced hepatic fibrosis in mice is a well-known in vivo model for screening anti-fibrotic drugs (Kuwahata et al., 2012). The metabolites of CCl4 cause the apoptosis of hepatocyte and liver injury. Repeated injection of CCl4 causes chronic injury to the liver, eventually results in liver fibrosis. Hepatocyte integrity indicators ALT and AST were both up regulated after CCl4 treatment. We found that the treatment with Antrodin C significantly diminished the liver fibrosis accompanied by the decreasing AST and ALT levels in blood circulation. Antrodin C also inhibited the activation of HSC in vivo revealed by the decreased expression level of α-SMA. In terms of mechanism, it was found that repeated CCl4 injections could induce the phosphorylation of Smad2, Akt and MAPK in the mouse liver tissues (Yoshida et al., 2005), whereas the oral administration with Antrodin C suppressed the phosphorylation of Smad2, Akt, ERK and P38 in the fibrotic livers. Further studies are needed to identify the underlying receptor-dependent. Moreover, Antrodin C may inhibits liver fibrosis by other Signaling pathways like TNF-α-NF-κB, AMPK, and β-Catenin Signaling pathway (Yang et al., 2019).
In conclusion, Antrodin C suppresses HSC activation, migration and ECM production, partially through inhibition of PDGF and TGF-β1 signaling pathways, that are the two most potent stimuli of liver fibrosis. Our study provides new insights for the development of therapeutic drugs against liver fibrosis. However, further studies are needed to fully understand the cellular and molecular mechanisms of Antrodin C in liver fibrosis.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Ethics Statement
The animal study was reviewed and approved by the Experimental Animal ethics committee of jiangnan unversity.
Author Contributions
X-YX and YG conceived and designed this research. X-YX and YR performed most of the experiments and analyzed data. YM and H-XX contributed to reagents and materials. X-YX and YG contributed to the manuscript editing. All authors contributed and reviewed the results and approved the final version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (Grant No. 31970746, 31771514, 31201020) and the Qing Lan Project in Jiangsu Province.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Xiao-Qian Xie and Qing Sun for their technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.835508/full#supplementary-material
References
Aydin, M. M., and Akcali, K. C. (2018). Liver Fibrosis. Turk J. Gastroenterol. 29 (1), 14–21. doi:10.5152/tjg.2018.17330
Brinda, B. J., Zhu, H.-J., and Markowitz, J. S. (2012). A Sensitive LC-MS/MS Assay for the Simultaneous Analysis of the Major Active Components of Silymarin in Human Plasma. J. Chromatogr. B 902, 1–9. doi:10.1016/j.jchromb.2012.06.003
Caligiuri, A., Gentilini, A., Pastore, M., Gitto, S., and Marra, F. (2021). Cellular and Molecular Mechanisms Underlying Liver Fibrosis Regression. Cells 10 (10). doi:10.3390/cells10102759
David, V., and George, J. (2008). Disease-Specific Mechanisms of Fibrosis: Hepatitis C Virus and Nonalcoholic Steatohepatitis. Clin. Liver Dis. 12 (4), 805–824.
Deng, X. X., Li, H., Zhang, Z. F., and Gu, Q. L. (2020). Research Progress of Liver Sinusoidal Endothelial Cells in the Regulation of Liver Microenvironment to Affect Liver Fibrosis. Zhonghua Gan Zang Bing Za Zhi 28 (4), 357–360. doi:10.3760/cma.j.cn501113-20190715-00247
Dhar, D., Baglieri, J., Kisseleva, T., and Brenner, D. A. (2020). Mechanisms of Liver Fibrosis and its Role in Liver Cancer. Exp. Biol. Med. (Maywood) 245 (2), 96–108. doi:10.1177/1535370219898141
Geethangili, M., and Tzeng, Y. M. (2010). Review of Pharmacological Effects of Antrodia Camphorata and its Bioactive Compounds. Evid. Based Complement. Alternat Med. 2011 (1741-427X), 212641. doi:10.1093/ecam/nep108
Geng, Y., Wang, J., Sun, Q., Xie, M., Lu, Z. M., Xu, H. Y., et al. (2016). Identification of Antrodin B from Antrodia Camphorata as a New Anti-hepatofibrotic Compound Using a Rapid Cell Screening Method and Biological Evaluation. Hepatol. Res. 46, E15–E25. doi:10.1111/hepr.12516
George, J., Tsuchishima, M., and Tsutsumi, M. (2019). Molecular Mechanisms in the Pathogenesis of N-Nitrosodimethylamine Induced Hepatic Fibrosis. Cel Death Dis. 10. doi:10.1038/s41419-018-1272-8
Goldstein, N. S., Hastah, F., Galan, M. V., and Gordon, S. C. (2005). Fibrosis Heterogeneity in Nonalcoholic Steatohepatitis and Hepatitis C Virus Needle Core Biopsy Specimens. Am. J. Clin. Pathol. 123 (3), 382–387. doi:10.1309/ey72f1en9xcb1kxx
Guerra, S., Mamede, A. C., Carvalho, M. J., Laranjo, M., Tralhão, J. G., Abrantes, A. M., et al. (2016). Liver Diseases: what Is Known So Far about the Therapy with Human Amniotic Membrane? Cell Tissue Bank 17 (4), 653–663. doi:10.1007/s10561-016-9579-0
Greenwel, P., Schwartz, M., Rosas, M., Peyrol, S., Grimaud, J. A., and Rojkind, M. (1991). . Characterization of Fat-Storing Cell Lines Derived From Normal and CCl4-Cirrhotic Livers. Differences in the Production of Interleukin-6. Laboratory Investigation, 1991. 65(6): p. 644-653. 65 (6), 644–653.
Huang, T. T., Lan, Y. W., Chen, C. M., Ko, Y. F., Ojcius, D. M., Martel, J., et al. (2019). Antrodia Cinnamomea Induces Anti-tumor Activity by Inhibiting the STAT3 Signaling Pathway in Lung Cancer Cells. Sci. Rep. 9, 5145. doi:10.1038/s41598-019-41653-9
Ikeda, K., Wakahara, T., Wang, Y. Q., Kadoya, H., Kawada, N., and Kaneda, K. (2010). In Vitro migratory Potential of Rat Quiescent Hepatic Stellate Cells and its Augmentation by Cell Activation. Hepatology 29 (6), 1760–1767. doi:10.1002/hep.510290640
Jin, C. H., Jang, J., and Min, K. T. (2020). The Role of Hepatic Stellate Cells in Fibrotic Liver Diseases. J. Anim. Reproduciton Biotechnol. 35 (2), 113–118.
Kang, K.-H., Qian, Z.-J., Ryu, B., Karadeniz, F., Kim, D., and Kim, S.-K. (2013). Hepatic Fibrosis Inhibitory Effect of Peptides Isolated from Navicula Incerta on TGF-β Induced Activation of LX-2 Human Hepatic Stellate Cells. Jfn 18 (2), 124–132. doi:10.3746/pnf.2013.18.2.124
Kastanis, G. J., Hernandez-Nazara, Z., Nieto, N., Rincón-Sanchez, A. R., Popratiloff, A., Dominguez-Rosales, J. A., et al. (2011). The Role of Dystroglycan in PDGF-BB-dependent Migration of Activated Hepatic Stellate Cells/myofibroblasts. Am. J. Physiology-Gastrointestinal Liver Physiol. 301 (3), G464–G474. doi:10.1152/ajpgi.00078.2011
Kumar, K. J. S., Vani, M. G., Chueh, P. J., Mau, J. L., and Wang, S. Y. (2015). Antrodin C Inhibits Epithelial-To-Mesenchymal Transition and Metastasis of Breast Cancer Cells via Suppression of Smad2/3 and Beta-Catenin Signaling Pathways. Plos One 10 (2). doi:10.1371/journal.pone.0117111
Kuwahata, M., Kubota, H., Kanouchi, H., Ito, S., Ogawa, A., Kobayashi, Y., et al. (2012). Supplementation with Branched-Chain Amino Acids Attenuates Hepatic Apoptosis in Rats with Chronic Liver Disease. Nutr. Res. 32 (7), 522–529. doi:10.1016/j.nutres.2012.06.007
Lee, A. S., Jung, Y. J., Kim, D., Nguyen-Thanh, T., Kang, K. P., Lee, S., et al. (2014). SIRT2 Ameliorates Lipopolysaccharide-Induced Inflammation in Macrophages. Biochem. Biophysical Res. Commun. 450 (4), 1363–1369. doi:10.1016/j.bbrc.2014.06.135
Nakamura, N., Hirakawa, A., Gao, J.-J., Kakuda, H., Shiro, M., Komatsu, Y., et al. (2004). Five New Maleic and Succinic Acid Derivatives from the Mycelium of Antrodia Camphorata and Their Cytotoxic Effects on LLC Tumor Cell Line. J. Nat. Prod. 67 (1), 46–48. doi:10.1021/np030293k
Novosyadlyy, R., Dudas, J., and Pannem, R. (2006). Crosstalk Between PDGF and IGF-I Receptors in Rat Liver Myofibroblasts: Implication for Liver Fibrogenesis. Lab. Invest. 86, 710–723.
Odiba, A. S., Ukegbu, C., Okechukwu, I., and Iruoghene, O. (2014). Transaminase [Alanine aminotransferase (ALT) and Aspartate aminotransferase (AST)] Activity of HIVFemale Patients on Drugs and Female Patients Not on Drugs. IOSR J. Pharm. Biol. Sci. 9 (7), 60–65.
Rosenbloom, J., Mendoza, F. A., and Jimenez, S. A. (2013). Strategies for Anti-fibrotic Therapies. Biochim. Biophys. Acta 1832 (7), 1088–1103. doi:10.1016/j.bbadis.2012.12.007
Schyman, P., Printz, R. L., Estes, S. K., O'Brien, T. P., Shiota, M., and Wallqvist, A. (2019). Assessing Chemical-Induced Liver Injury In Vivo from In Vitro Gene Expression Data in the Rat: The Case of Thioacetamide Toxicity. Front. Genet. 10, 1233. doi:10.3389/fgene.2019.01233
Selenina, A., Tomilin, A., and Tsimokha, A. (2019). Evidences against Vesicle-dependent Trafficking and Involvement of Extracellular Proteasomes into Cell-To-Cell Communications. Biochem. Biophysical Res. Commun. 508 (2), 368–373.
Steiner, C. A., and Higgins, P. (2018). Anti-Fibrotic Therapies from Other Organs: What the Gut Can Learn from the Liver, Skin, Lung and HeartFibrostenotic Inflammatory Bowel Disease.
Suwanabol, P. A., Seedial, S. M., Zhang, F., Shi, X., Si, Y., Liu, B., et al. (2012). TGF-β and Smad3 Modulate PI3K/Akt Signaling Pathway in Vascular Smooth Muscle Cells. Am. J. Physiology-Heart Circulatory Physiol. 302 (11), H2211–H2219. doi:10.1152/ajpheart.00966.2011
Tan, Z., Sun, H., Xue, T., Gan, C., Liu, H., Xie, Y., et al. (2021). Liver Fibrosis: Therapeutic Targets and Advances in Drug Therapy. Front. Cel Developmental Biol. 9, 730176. doi:10.3389/fcell.2021.730176
Tricarico, C., Pinzani, P., Bianchi, S., Paglierani, M., Distante, V., Pazzagli, M., et al. (2002). Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction: Normalization to rRNA or Single Housekeeping Genes Is Inappropriate for Human Tissue Biopsies. Anal. Biochem. 309 (2), 293–300. doi:10.1016/s0003-2697(02)00311-1
Trivedi, P., Wang, S., and Friedman, S. L. (2021). The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cel Metab. 33 (2), 242–257. doi:10.1016/j.cmet.2020.10.026
Tsuchida, T., and Friedman, S. L. (2017). Mechanisms of Hepatic Stellate Cell Activation. Nat. Rev. Gastroenterol. Hepatol. 14 (7), 397–411. doi:10.1038/nrgastro.2017.38
Voloshenyuk, T. G., Landesman, E. S., Khoutorova, E., Hart, A. D., and Gardner, J. D. (2011). Induction of Cardiac Fibroblast Lysyl Oxidase by TGF-Β1 Requires PI3K/Akt, Smad3, and MAPK Signaling. Cytokine 55 (1), 90–97. doi:10.1016/j.cyto.2011.03.024
Yang, H., Bai, X., Zhang, H., Zhang, J., Wu, Y., Tang, C., et al. (2019). Antrodin C, an NADPH Dependent Metabolism, Encourages Crosstalk between Autophagy and Apoptosis in Lung Carcinoma Cells by Use of an AMPK Inhibition-independent Blockade of the Akt/mTOR Pathway. Molecules 24 (5). doi:10.3390/molecules24050993
Yoon, Y.-C., Fang, Z., Lee, J. E., Park, J. H., Ryu, J.-K., Jung, K. H., et al. (2020). Selonsertib Inhibits Liver Fibrosis via Downregulation of ASK1/MAPK Pathway of Hepatic Stellate Cells. Biomolecules Ther. 28 (6), 527–536. doi:10.4062/biomolther.2020.016
Yoshida, K., Matsuzaki, K., Mori, S., Tahashi, Y., Yamagata, H., Furukawa, F., et al. (2005). Transforming Growth Factor-β and Platelet-Derived Growth Factor Signal via C-Jun N-Terminal Kinase-dependent Smad2/3 Phosphorylation in Rat Hepatic Stellate Cells after Acute Liver Injury. Am. J. Pathol. 166 (4), 1029–1039. doi:10.1016/s0002-9440(10)62324-3
Zhao, Z., He, D., Ling, F., Chu, T., Huang, D., Wu, H., et al. (2020). CD4+ T Cells and TGFβ1/MAPK Signal Pathway Involved in the Valvular Hyperblastosis and Fibrosis in Patients with Rheumatic Heart Disease. Exp. Mol. Pathol. 114, 104402. doi:10.1016/j.yexmp.2020.104402
Keywords: liver disease, cell migration, hepatic stellate cells, extracellular matrix, cell signaling
Citation: Xu X-Y, Geng Y, Xu H-X, Ren Y, Liu D-Y and Mao Y (2022) Antrodia camphorata-Derived Antrodin C Inhibits Liver Fibrosis by Blocking TGF-Beta and PDGF Signaling Pathways. Front. Mol. Biosci. 9:835508. doi: 10.3389/fmolb.2022.835508
Received: 14 December 2021; Accepted: 18 January 2022;
Published: 15 February 2022.
Edited by:
Leming Sun, Northwestern Polytechnical University, ChinaCopyright © 2022 Xu, Geng, Xu, Ren, Liu and Mao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Geng, Z2VuZ3lhbkBqaWFuZ25hbi5lZHUuY24=; Yong Mao, OTgxMjAxNTI1MkBqaWFuZ25hbi5lZHUuY24=