 Daisuke Tashiro1
Daisuke Tashiro1 Shunji Suetaka1
Shunji Suetaka1 Nao Sato1
Nao Sato1 Koji Ooka2
Koji Ooka2 Tomoko Kunihara1
Tomoko Kunihara1 Hisashi Kudo1Junichi Inatomi1
Hisashi Kudo1Junichi Inatomi1 Yuuki Hayashi1
Yuuki Hayashi1 Munehito Arai1,2*
Munehito Arai1,2*- 1Department of Life Sciences, Graduate School of Arts and Sciences, The University of Tokyo, Tokyo, Japan
- 2Department of Physics, Graduate School of Science, The University of Tokyo, Tokyo, Japan
Human epidermal growth factor receptors (HER/ERBB) form dimers that promote cell proliferation, migration, and differentiation, but overexpression of HER proteins results in cancer. Consequently, inhibitors of HER dimerization may function as effective antitumor drugs. An alternatively spliced variant of HER2, called herstatin, is an autoinhibitor of HER proteins, and the intron 8-encoded 79-residue domain of herstatin, called Int8, binds HER family receptors even in isolation. However, the structure of Int8 remains poorly understood. Here, we revealed by circular dichroism, NMR, small-angle X-ray scattering, and structure prediction that isolated Int8 is largely disordered but has a residual helical structure. The radius of gyration of Int8 was almost the same as that of fully unfolded states, although the conformational ensemble of Int8 was less flexible than random coils. These results demonstrate that Int8 is intrinsically disordered. Thus, Int8 is an interesting example of an intrinsically disordered region with tumor-suppressive activity encoded by an intron. Furthermore, we show that the R371I mutant of Int8, which is defective in binding to HER2, is prone to aggregation, providing a rationale for the loss of function.
1 Introduction
Human epidermal growth factor receptors (HER/ERBB) are receptor tyrosine kinases that play crucial roles in the regulation of cell proliferation, migration, and differentiation (Citri and Yarden, 2006). The HER family comprises four members, namely HER1 (EGFR), HER2 (NEU), HER3, and HER4 with differential tissue expression patterns. All four members of the family share a common three-dimensional structure that comprises an extracellular domain (ECD), a transmembrane domain, and an intracellular kinase domain (Figure 1A) (Yarden and Pines, 2012). Homo- or hetero-dimerization of the ECD induces autophosphorylation of the intracellular kinase domain, which in turn results in activation of downstream signaling molecules (Normanno et al., 2003; Mujoo et al., 2014). Genetic variants that disrupt the function of these proteins or lead to their overexpression have been associated with multiple cancers (Howe and Brown, 2011). For instance, HER1 variants are associated with lung, breast, and prostate cancers (Normanno et al., 2003), whereas HER2 variants have been found in approximately 30% of all breast cancers (Tai et al., 2010). Thus, the interruption of HER dimerization by specific inhibitors is an effective strategy to halt the growth of cancer cells. Several antibody therapeutics that function via this mechanism have been developed, including trastuzumab (herceptin) and pertuzumab that target HER2, and cetuximab that targets HER1.
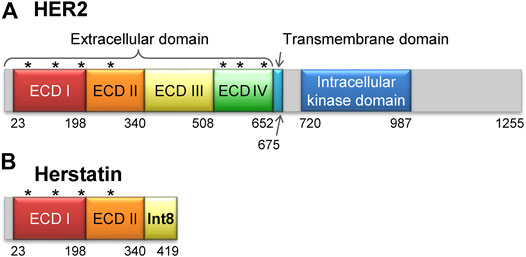
FIGURE 1. Structures of HER2 and herstatin. (A) Structure of HER2. The extracellular domain is composed of four subdomains, ECD I, ECD II, ECD III, and ECD IV. Seven glycosylation sites are shown by asterisks [N68, N124, N187, N259, N530, N571, and N629 according to the UniProt database (Accession number P04626) (UniProt Consortium, 2021)]. (B) Structure of herstatin. ECD I and ECD II are identical to those in HER2. An intron-8 encoded 79-residue domain, named Int8 (or ECD IIIa), is retained at the C-terminal region of herstatin. Four glycosylation sites identical to those in HER2 are shown by asterisks. Note that Int8 does not have a glycosylation site.
Herstatin (dimercept) is an alternatively spliced variant of HER2 that retains intron 8 and is secreted by human cells (Doherty et al., 1999; Stix, 2006; Silipo et al., 2017; Hart et al., 2020). It lacks the intracellular kinase and transmembrane domains, but retains a part of the extracellular domain that binds to the ECDs of intact HER proteins. This results in the suppression of HER autophosphorylation and consequently arrests downstream signal transduction (Doherty et al., 1999; Camenisch et al., 2002; Justman and Clinton, 2002; Jhabvala-Romero et al., 2003; Hu et al., 2006; Gompels et al., 2011). Thus, herstatin can autoinhibit the function of HER proteins by binding them and interfering with their dimer formation (Doherty et al., 1999; Azios et al., 2001; Shamieh et al., 2004). Furthermore, herstatin binds intracellular HER2 and possibly prevents its translocation from the endoplasmic reticulum to the cell surface (Hu et al., 2006). Given that herstatin is expressed in non-cancerous breast tissue (Koletsa et al., 2008), but is absent in about 75% of breast carcinomas, it may function as a tumor-suppressor. Consequently, its role as a growth regulatory factor during normal development can be possibly exploited in the development of antitumor therapies (Staverosky et al., 2005; Stix, 2006; Koletsa et al., 2008; Jackson et al., 2013).
Herstatin is a 68-kDa soluble protein, comprising two of the four subdomains of the ECD of HER2 (ECD I and ECD II, respectively; residues 1–340), and a separate domain encoded by intron 8, referred to as Int8 (or ECD IIIa; residues 341–419) (Figure 1B). The 79-residue Int8 domain binds with high affinity to HER1, HER2, HER4, and the insulin-like growth factor 1 receptor (Shamieh et al., 2004; Lv et al., 2012). Even short peptides derived from the Int8 domain are known to bind HER2 (Cha et al., 2017). Interestingly, the Int8 domain inhibited the growth of HER2-overexpressing cells, suggesting the tumor-suppressing activity of Int8 (Lv et al., 2012).
Previous studies show that the isolated Int8 domain has a high affinity for HER proteins. This is evident from the fact that whereas a dissociation constant, Kd, was 14 nM for the binding between the full-length herstatin and HER2 (Doherty et al., 1999), the Kd for Int8 binding to HER1 and HER2 was 78 ± 10 nM and 50 ± 6 nM, respectively, although Int8 does not have the dimerization arm, which is located in the ECD II (Doherty et al., 1999; Shamieh et al., 2004). In accordance with this, Hu et al. demonstrated that deletion of the Int8 domain from herstatin considerably reduced the affinity with HER2 in vivo, suggesting that the Int8 domain plays a predominant role in the binding of herstatin to HER2 (Hu et al., 2005). Moreover, Shamieh et al. reported loss of interaction between the isolated Int8 domain and HER2 as a consequence of the R371I mutation in Int8 (residue number corresponds to full-length herstatin) (Shamieh et al., 2004). However, while the role of the Int8 domain in HER binding is well established, its structure remains poorly understood.
Here, we characterized the structure of the isolated Int8 domain using circular dichroism (CD), nuclear magnetic resonance (NMR), and small-angle X-ray scattering (SAXS). Our results demonstrate that Int8 is intrinsically disordered with a residual helical structure. The structure prediction and modeling were consistent with experimental results. Furthermore, we show that the R371I mutant, which has a lower affinity for HER2, is prone to aggregation. Therefore, Int8 presents an interesting case, where an intrinsically disordered region (IDR) encoded by an intron possesses tumor-suppressive activity.
2 Materials and Methods
2.1 Protein Expression and Purification
The gene encoding wild-type Int8 (residues 341–419 of herstatin) (Shamieh et al., 2004) was constructed by overlap-extension PCR. To improve solubility, the N-terminus of Int8 was tagged with a 5 × Lys-tag using a Gly-Ser-Ser-Gly linker (MGKKKKKGSSG) (Kato et al., 2007). Additionally, a 6 × His-tag was linked to the C-terminus using a Ser-Ser-Gly linker (SSGHHHHHH) to facilitate protein purification. Codons were optimized for high-level expression in Escherichia coli using the DNAWorks server (Hoover and Lubkowski, 2002). The Int8 DNA fragment was ligated into the NcoI/BamHI sites of the pET-15b vector (MilliporeSigma, Burlington, MA, United States). The R371I mutation was introduced according to the method of the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, United States). The expression levels and solubilities of the wild-type and mutant Int8 proteins were predicted using the ESPRESSO server (Hirose and Noguchi, 2013).
Unlabeled and 15N-labeled Int8, as well as the unlabeled R371I mutant of Int8 were expressed in E. coli BL21(DE3) cells (Nippon Gene, Tokyo, Japan) in 2×YT medium (for unlabeled protein) or M9 minimal medium (for 15N-labeled protein) containing ampicillin (50 μg/ml). The cells were incubated at 37°C and overexpression of Int8 was induced by addition of 1 mM isopropyl β-D-1-thiogalactopyranoside at an optical density of 0.8 at 600 nm. After incubation for an additional 5 h, cells were collected, resuspended in denaturation buffer containing 20 mM Tris-HCl (pH 8.0), 6 M guanidine hydrochloride (GdnHCl), and 20 mM imidazole, and sonicated on ice for 4 min using a Branson Sonifier 250D Advanced (Branson, Danbury, CT, United States). The lysate was subsequently centrifuged at 35,140 × g for 30 min at 4°C. The supernatant was filtered through a 0.45-µm pore size membrane filter and applied to a column containing nickel-nitrilotriacetic acid agarose gel (Qiagen, Hilden, Germany). The column was washed with the denaturation buffer and wash buffer containing 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, and 20 mM imidazole. Int8 was eluted using elution buffer containing 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, and 0.1–1 M imidazole. For CD and light scattering measurements, the eluate was further purified by size-exclusion chromatography using a Superdex 200 pg column (Cytiva, Marlborough, MA, United States) with the buffer containing 10 mM sodium phosphate (pH 6.0) and 50 mM NaCl. For NMR and SAXS measurements, the eluate was desalted using a PD-10 column (Cytiva) in buffer containing 10 mM sodium phosphate (pH 6.0) and 50 mM NaCl. The purity of the Int8 proteins was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Protein concentrations were obtained using the following four methods: 1) absorption measurement at 280 nm in the presence of 6 M GdnHCl (Edelhoch, 1967; Gill and von Hippel, 1989), 2) absorption measurement at 280 nm under native conditions (Pace et al., 1995), 3) absorption measurement at 205 nm (Anthis and Clore, 2013), and 4) the Pierce BCA protein assay kit (Thermo Fisher Scientific, Waltham, MA, United States). Similar protein concentrations were obtained by all methods.
2.2 Circular Dichroism Measurements
Far-UV CD spectra were obtained from a J-805 spectropolarimeter (Jasco, Tokyo, Japan) at 200–250 nm using a quartz cuvette of 1 mm path length at 25°C. The temperature was controlled using a thermostat-circulating water bath. The protein concentrations of the wild type and R371I mutant of Int8 were 30 and 31 μM, respectively. The samples additionally contained 10 mM sodium phosphate (pH 6.0) and 50 mM NaCl, in the presence or absence of 4 M GdnHCl. Mean residue ellipticity (MRE) was calculated as previously described (Arai and Iwakura, 2005). The spectra in the presence of GdnHCl were measured at 211–250 nm. The helix content (fH) was estimated from the MRE at 222 nm according to the following equation (Chen et al., 1972):
2.3 Nuclear Magnetic Resonance Measurements
One-dimensional (1D) pulsed-field gradient (PFG) NMR spectra and two-dimensional (2D) 1H-15N heteronuclear single quantum coherence spectra were recorded using a Bruker Avance500 spectrometer in a buffer containing 10 mM sodium phosphate (pH 6.0), 50 mM NaCl, 10% D2O, and 0.2 mM sodium 3-(trimethylsilyl)-1-propanesulfonate (DSS) at 25°C. All spectra were analyzed using NMRPipe (Delaglio et al., 1995) and NMRViewJ softwares (Johnson and Blevins, 1994). The protein concentrations of the wild-type and R371I mutant of Int8 were 350 and 150 μM, respectively.
PFG NMR measurements were performed using a bipolar longitudinal eddy-current-decay pulse sequence (Wu et al., 1995; Nakamura et al., 2013; Kunihara et al., 2019). The samples contained 0.05% dioxane as a standard for molecular size analysis. The diffusion delay was 100 ms, the gradient pulse width was 6 ms, and the pulse separation was 0.6 ms. The spectral center was 4.701 ppm, and the spectral width was 8012.820 Hz. Five spectra for the peak decay of dioxane and 15 spectra for Int8 were acquired while changing the strength of the diffusion gradient (g) from 5.35 G/cm (5%) to 53.5 G/cm (100%). The peak intensity (s) is related to g as follows:
where s0 is the peak intensity at 0% field gradient and d is the observed decay rate, which is proportional to the diffusion coefficient of the solute. The signal decay was fitted using Eq. 2, and the hydrodynamic radius, Rh of Int8 was calculated using the d value, as follows:
where the Rh of dioxane is 2.12 Å (Wilkins et al., 1999).
2.4 Small-Angle X-Ray Scattering Measurements
SAXS measurements were conducted at beamline (BL)-10C at the Photon Factory of the High Energy Accelerator Research Organization (KEK), Tsukuba, Japan. The camera length (1,996 mm) was calibrated by the diffraction of silver behenate using FIT2D software (Hammersley, 1997). Samples were loaded into a mica-windowed cuvette with a 1 mm path length and were irradiated by a monochromatic X-ray beam (1.488 Å). Int8 concentration was 4.1 mg/ml (390 μM), and sample was centrifuged for 30 min at 4°C before measurement. The temperature in the cuvette was maintained at 25°C using a circulating water bath. Scattering images were acquired by a PILATUS3 300 KW detector (DECTRIS Ltd., Baden, Switzerland) at 10 × 1 s exposures. The scattering intensity, I(Q), was collected from 0.005 to 0.27 Å−1, where Q = 4π sin (θ/λ) (λ, wavelength; 2θ, scattering angle). Scattering of blanks (buffer alone) was measured before and/or after measurement of protein samples. Circular averaging of 2D scattering data was performed using FIT2D. Blanks were subtracted from protein solution scattering data to obtain the scattering profile of protein molecules. The subtracted scattering data were binned per four data points to increase the signal-to-noise ratio. A radius of gyration, Rg, was obtained based on the Guinier approximation within the Guinier region (RgQ < 1.3) (Trewhella et al., 2017):
where I(0) denotes the zero-angle scattering intensity (Glatter and Kratky, 1982; Arai et al., 2007). An Rg was also estimated using the approximation of the Debye function for a random coil within the range of (RgQ)2 < 3:
where a is the y-intercept of the I(Q)−1 versus Q2.206 plot (Calmettes et al., 1994). The pair-distance distribution function, P(r), was calculated using GNOM software (Svergun, 1992).
The SAXS data were further analyzed using the ensemble optimization method (EOM) 2.1 (Bernado et al., 2007; Tria et al., 2015; Vallet et al., 2018). In this analysis, a pool of 10,000 random conformations based on the Int8 amino acid sequence was first generated. Then, a genetic algorithm was run 100 times to select for an ensemble of conformations that best reproduces the experimental scattering curve.
2.5 Size-Exclusion Chromatography and Light Scattering Measurements
Size-exclusion chromatography (SEC) measurements were performed using a high-performance liquid chromatography (HPLC) system (LP-20AP, Shimadzu, Kyoto, Japan) and a Superdex 200 increase 3.2/300 column (Cytiva). The column was pre-equilibrated with 10 mM sodium phosphate (pH 6.0) and 150 mM NaCl, and samples of the wild-type and R371I mutant of Int8 were injected onto the column. The protein concentrations were 129 and 515 μM for the wild type and 138 and 550 μM for the R371I mutant. Molecular weight of the sample was determined on the HPLC system coupled to a Viscotek TDA 305 light scattering detector (Malvern Panalytical, Malvern, United Kingdom) as described previously (Chang et al., 2020). Bovine serum albumin (66.7 kDa) was used as a standard for instrument calibration. Data analysis for estimating molecular weights was performed using OmniSEC software (Malvern Panalytical). The error in molecular weight was calculated from duplicate or triplicate measurements.
3 Results
3.1 Characterization of Int8 Structure
The isolated Int8 domain containing a C-terminal 6 × His-tag and an N-terminal 5 × Lys-tag was overexpressed and purified from E. coli, to characterize its structure (Section 2). The structure of Int8 under native conditions was measured by CD, NMR, and SAXS. The far-UV CD spectrum of Int8 under native conditions had small intensities at ∼222 nm but had a minimum at ∼200 nm, indicating a largely disordered structure (Figure 2A). The CD intensity at ∼222 nm was reduced to almost zero after protein unfolding by the addition of 4 M GdnHCl (Figure 2A). The difference spectrum obtained by subtracting the CD spectrum in the presence of 4 M GdnHCl from that measured under native conditions showed a minimum at 222 nm, indicating the presence of an α-helical structure (Figure 2B). These findings suggest that Int8 is largely disordered under native conditions, but retains a residual helical structure. The helix content as estimated from the CD intensity at 222 nm is ∼4% (Eq. 1 in Section 2).
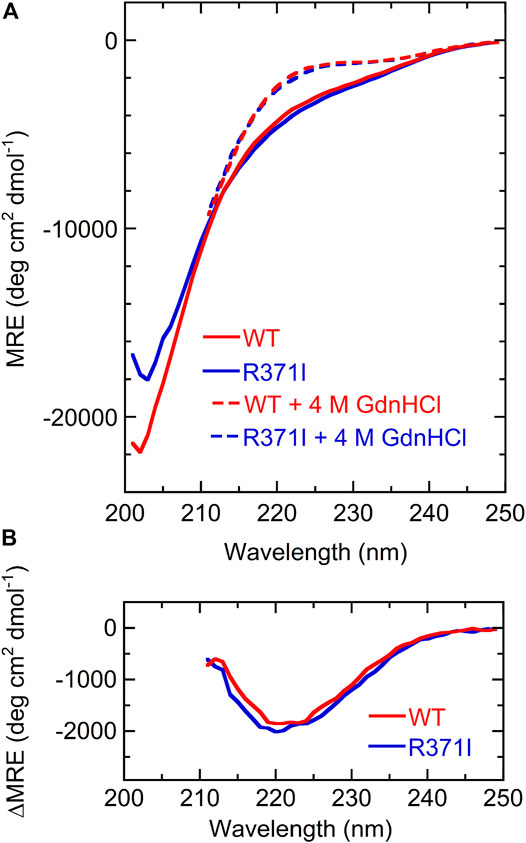
FIGURE 2. CD measurements. (A) Far-UV CD spectra of the wild type (red) and R371I mutant (blue) of Int8 in the absence (continuous lines) and presence (broken lines) of 4 M GdnHCl. The MRE values are shown. The spectra in the presence of 4 M GdnHCl were measured up to 211 nm due to large absorption by GdnHCl at lower wavelengths. (B) Difference CD spectra calculated by subtracting the CD spectrum in the presence of 4 M GdnHCl from that measured in the absence of GdnHCl.
1D and 2D NMR spectra of Int8 showed that chemical shifts of amide protons were confined in a narrow range of 7.7–8.7 ppm (Figures 3A,E). This indicates that the amide protons of Int8 are in similar environments, suggesting that Int8 is predominantly in a disordered state. PFG NMR measurements were performed to characterize the hydrodynamic radius, Rh. From the 1D NMR spectra of Int8, four peaks were selected at ∼1–2 ppm (Figure 3A) and peak intensities were plotted depending on the external magnetic field gradient, g (Figure 3B). By fitting the decay curves (Eq. 2 in Section 2), a parameter d was estimated, which is proportional to the diffusion coefficient. The d values were found to be almost identical for the four selected peaks. Using the Einstein-Stokes equation and the d value of the standard substance dioxane (Rh = 2.12 Å), which was included in the NMR sample, an Rh of 33 ± 2 Å for Int8 was obtained (Eq. 3 in Section 2).
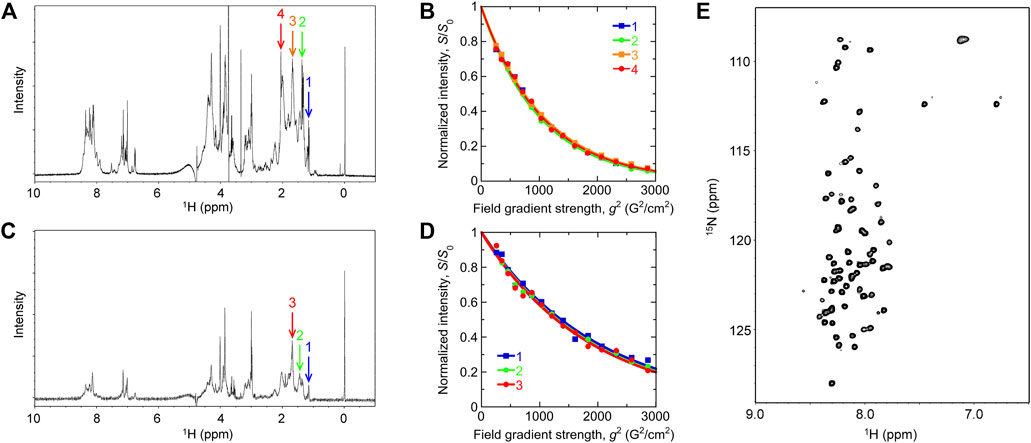
FIGURE 3. NMR measurements. (A,C) One-dimensional NMR spectra of the wild type (A) and R371I mutant (C) of Int8. Arrows show the peaks used for the analysis of pulsed-field gradient (PFG) NMR measurement. The DSS peak is at 0 ppm. (B,D) Peak intensity decay curves obtained by the PFG NMR measurement of the wild type (B) and R371I mutant (D). (E) Two-dimensional 1H−15N heteronuclear single quantum coherence spectrum of the wild-type Int8.
SAXS measurements were performed to characterize the molecular size and shape of Int8 (Figure 4A). Scattering intensities, I(Q), at scattering vectors, Q (Å−1), were analyzed by a Kratky plot [I(Q)Q2 versus Q plot] which provides information on the molecular shape of a protein (Glatter and Kratky, 1982; Arai et al., 2007). The presence of a peak in the plot indicates that the protein has a compact, globular shape, whereas the absence of a peak is indicative of a random coil-like disordered structure (Arai et al., 2007). The Kratky plot of Int8 lacked a peak and instead showed a plateau, indicating that Int8 has a random coil-like disordered structure under native conditions (Figure 4B). The pair-distance distribution function, P(r), calculated by Fourier transformation of I(Q), showed a peak at ∼20 Å, but the maximum distance, Dmax, between two atoms in Int8 was ∼80 Å (Figure 4C). It is known that compact globular particles have a symmetric bell-shaped P(r), whereas unfolded particles have an extended tail in the P(r) function (Kikhney and Svergun, 2015). Thus, the P(r) of Int8 suggests an extended structure.
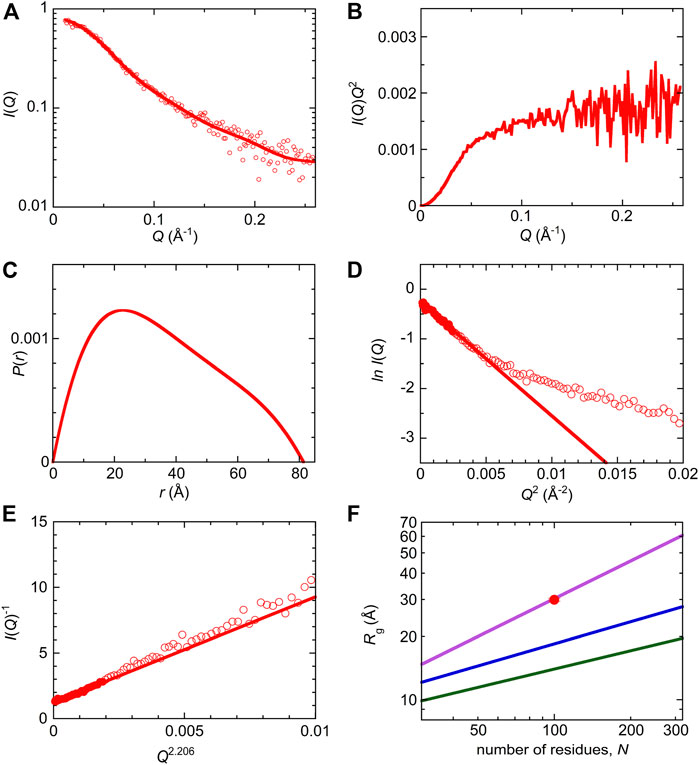
FIGURE 4. SAXS analysis of wild-type Int8. (A) The ln I(Q) versus Q plot. The continuous line was obtained by the EOM fit. The intensity is shown in an arbitrary unit. (B) Kratky plot. (C) A pair-distance distribution function, P(r). (D) Guinier plot. The continuous line was obtained by Guinier approximation. (E) The I(Q)−1 versus Q2.206 plot. The continuous line was obtained by fitting to the Debye function for a random coil. (F) Scaling relationship for the native (green), intermediate (blue), and unfolded state (purple). The red circle shows the Rg of Int8 obtained by fitting to the Debye function.
Next, the SAXS data were analyzed by generating a Guinier plot (i.e., ln I(Q) versus Q2 plot), which provides information on the molecular size of a protein, that is, the radius of gyration Rg, using the Guinier approximation (Eq. 4 in Section 2). The Rg of Int8 was estimated to be 26.2 ± 0.5 Å (Figure 4D). Furthermore, the Rg estimated using the Debye function for a random coil was 29.9 ± 0.5 Å (Figure 4E; Eq. 5 in Section 2). Since the Debye function is applicable to the scattering data at wider angles (RgQ < 1.73) than the Guinier approximation (RgQ < 1.3), the Rg value obtained by the Debye function is less affected by interparticle interference effects (Calmettes et al., 1994). Therefore, we used an Rg of 29.9 Å for Int8. The Rg values of typical native, intermediate (molten globule), and unfolded states of globular proteins are related to the number of residues, N, as Rg = R0Nν, where R0 is the Rg of a single amino acid residue and ν is the scaling exponent (Wilkins et al., 1999; Kohn et al., 2004; Arai et al., 2007). The R0 and ν values are 3.68 ± 0.86 Å and 0.29 ± 0.02 for the native state (Wilkins et al., 1999), 3.68 ± 0.45 Å and 0.35 ± 0.09 for the molten globule-like folding intermediate (Arai et al., 2007), and 1.927 ± 0.271 Å and 0.598 ± 0.028 for the fully unfolded state (Kohn et al., 2004), respectively. A comparison of these values showed that the Rg of Int8 is almost the same as that of the fully unfolded state (Figure 4F).
The Rg/Rh ratio is known to depend on the molecular shape of proteins. For spherical molecules (either folded proteins or disordered proteins in compact conformations) the Rg/Rh ratio is
Then, the ensemble of Int8 conformations that best fitted to the SAXS data was modeled by EOM (Bernado et al., 2007; Tria et al., 2015; Vallet et al., 2018). The ensemble best fitted to the scattering curve (Figure 4A; χ2 = 1.799) contained five representative conformations of Int8 with an averaged Rg of 28.5 Å and Dmax of 79.2 Å, which are consistent with the above analysis (Figure 5). The distributions of Rg and Dmax for Int8 were smaller than those for the completely random pool (Figures 5A,B). Furthermore, Rflex, indicative of the flexibility of the conformational ensemble, was 71.7% for Int8, which was smaller than that for the random pool (86.2%). Thus, the SAXS data suggest that Int8 has a molecular size comparable to the fully unfolded state but is less flexible than random coils probably due to the presence of residual helical structure.
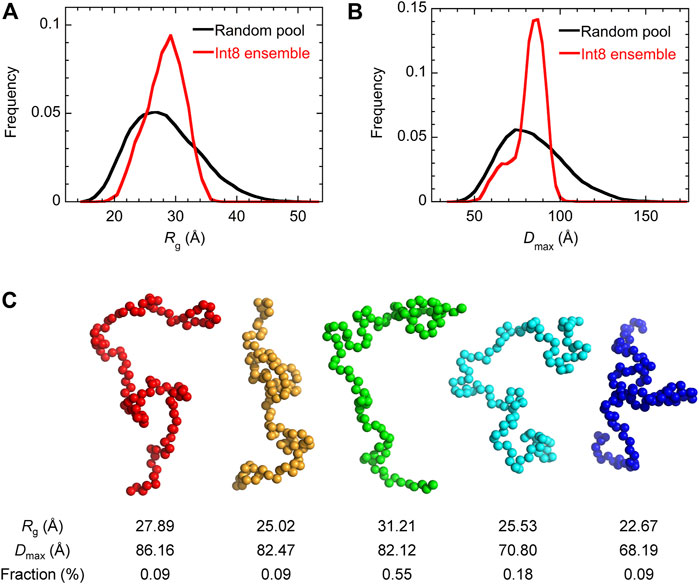
FIGURE 5. EOM analysis of the SAXS data. (A,B) The distribution of Rg (A) and Dmax (B) for the completely random pool (black) and the ensemble of Int8 conformations (red). (C) Five representative conformations of the wild-type Int8 involved in the ensemble that was best fitted to the scattering curve of Int8 (Figure 4A). The Rg, Dmax, and fraction (%) of the conformations are shown at the bottom.
3.2 Structure Prediction of Int8
Secondary structure prediction of Int8 using the PSIPRED server (Buchan and Jones, 2019) showed that most regions of Int8 were in a random coil-like structure (Figure 6A). Only a small number of residues in the middle of Int8 were predicted to be organized into two α-helices [helix 1 (residues 366–370) and helix 2 (residues 374–381)] and one β-strand (residues 410–413). The AGADIR server (Muñoz and Serrano, 1994; Lacroix et al., 1998) predicted that helical propensity was low for both helices, although helix 2 (residues 373–382) had a higher helical propensity than helix 1 (residues 365–369) (Supplementary Figure S1). These predictions are consistent with the CD measurements that showed the presence of only a small amount of helical structure in Int8.
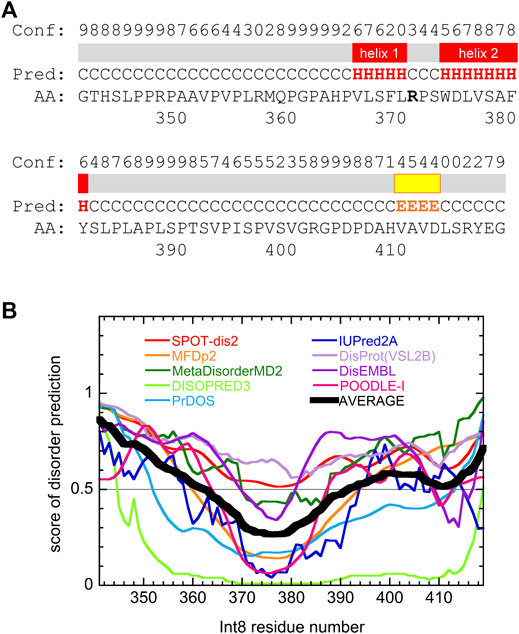
FIGURE 6. Secondary structure and disorder predictions of wild-type Int8. (A) Secondary structure prediction by PSIPRED. Pred indicates the predicted secondary structure (H, α-helix; E, β-sheet; and C, coil). Conf shows the confidence level of the prediction. Regions predicted to form α-helices and β-sheets are shown by red and yellow boxes, respectively. (B) Disorder prediction by nine different prediction servers. Black thick line shows the average of the predictions. Regions with a score larger than 0.5 are predicted to be disordered.
Disorder prediction was performed using nine prediction servers, namely SPOT-Disorder2 (Hanson et al., 2019), MFDp2 (Mizianty et al., 2013), MetaDisorderMD2 (Kozlowski and Bujnicki, 2012), DISOPRED (Jones and Cozzetto, 2015), PrDOS (Ishida and Kinoshita, 2007), IUPred2A (Meszaros et al., 2018), DisProt(VSL2B) (Obradovic et al., 2005), DisEMBL (Linding et al., 2003), and POODLE-I (Shimizu, 2014). Although the results obtained differed between individual prediction servers, many predicted that most regions of Int8, particularly in the N- and C-terminal regions were intrinsically disordered (Figure 6B), which was consistent with the secondary structure predictions. The disordered nature of Int8 is probably due to the presence of many proline and serine residues between residues 346–365 and 384–406. Thus, theoretical predictions suggest that Int8 is intrinsically disordered with a residual helical structure, consistent with our experimental results (Figures 2–5).
3.3 Structure of the R371I Mutant
The structure of the R371I mutant of Int8, which does not bind HER2 (Shamieh et al., 2004), was predicted by PSIPRED and the disorder prediction servers to be similar to that of the wild-type Int8 (Supplementary Figure S2). The AGADIR server also predicted that the helical propensity of the R371I mutant is almost the same as that of the wild-type Int8 (Supplementary Figure S1).
The R371I mutant of Int8 was overexpressed in E. coli and purified. The CD spectrum of the R371I mutant of Int8 was found to be similar to that of wild-type Int8 (Figure 2A). The helix content of the mutant was estimated to be ∼5% (Eq. 1 in Section 2). The difference CD spectrum between those measured in the absence and presence of 4 M GdnHCl had a negative peak at ∼222 nm in the R371I mutant (Figure 2B). These results indicate that the mutation little affected the helix content of Int8, which is consistent with the theoretical predictions (Supplementary Figure S1).
The 1D NMR spectrum of the R371I mutant showed that peaks for amide protons are confined in a narrow range of 7.7–8.7 ppm (Figure 3C), indicating disordered structures. The molecular size of the R371I mutant of Int8 was estimated by PFG NMR measurement. Three peaks at ∼1–2 ppm in the 1D NMR spectra of the mutant were used for the analysis of peak intensity decays (Figures 3C,D). The Rh for the R371I mutant was 72 ± 2 Å. This value is more than two-fold larger than that of wild-type Int8, indicating the formation of soluble aggregates in the mutant. The aggregation was observed despite the use of a lower protein concentration for the R371I mutant (150 μM) than for the wild-type Int8 (350 μM). Therefore, these results indicate that the R371I mutant of Int8 is prone to aggregation.
To further investigate the aggregation propensity of the mutant in more detail, we performed SEC measurements for the wild type and R371I mutant of Int8 (Figure 7). The elution profile of the wild type showed a single peak without aggregates at both low (∼100 μM) and high protein concentrations (∼500 μM). The molecular weights estimated by static right-angle light scattering were 12.6 (±0.1) kDa and 13.2 (±0.1) kDa for the low and high concentration samples of the wild-type Int8, respectively (Supplementary Figure S3), both of which are close to the value expected for an Int8 monomer (10.5 kDa). In contrast, a large amount of aggregates were observed at a high concentration (∼500 μM) of the R371I mutant (Figure 7B) with a molecular weight of 20.3 (±0.5) kDa at the elution peak (Supplementary Figure S3). At a low protein concentration (∼100 μM), the formation of aggregates was suppressed (Figure 7A), and the molecular weight at the elution peak was of 11.3 (±0.6) kDa, which is close to the value for an Int8 monomer (Supplementary Figure S3). However, the mutant started to elute earlier than the wild type, indicating the presence of slight aggregates (Figure 7A). The large Rh of the mutant determined by the PFG NMR measurements at 150 μM may correspond to the molecular size of these aggregates. Taken together, these results clearly demonstrate that the R371I mutant of Int8 is prone to aggregation.
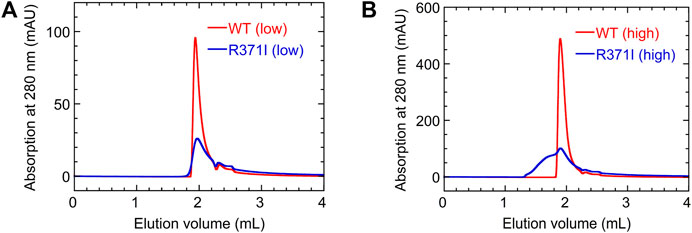
FIGURE 7. SEC measurements. (A,B) Elution profiles of the wild type (red) and R371I mutant (blue) of Int8 measured at low (∼100 μM) (A) and high (∼500 μM) protein concentrations (B).
3.4 Structure Modeling of Int8
Three-dimensional structure prediction of the wild type and R371I mutant of Int8 was performed using the standalone version of AlphaFold2 (Jumper et al., 2021). The predicted structure of wild-type Int8 was largely disordered, but had two α-helices at residues 365–370 and 374–382 corresponding to helices 1 and 2, respectively (Figures 8A,C). The predicted structure of the R371I mutant was similar to that of the wild type (Figure 8B), except that helix 1 (residues 368–370) was shorter (Figure 8D), which may be consistent with the slightly reduced helical propensity predicted by AGADIR for the mutant (Supplementary Figure S1).
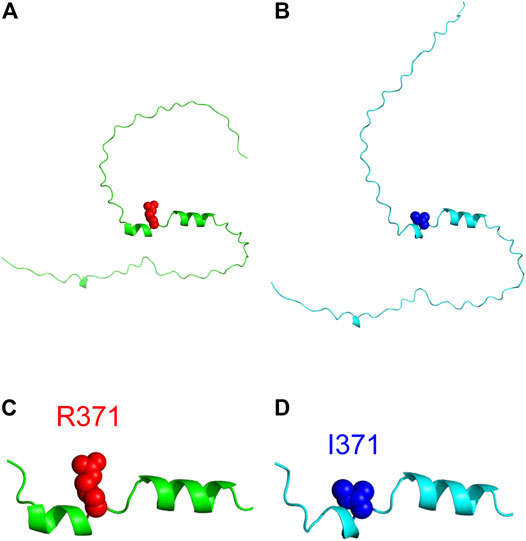
FIGURE 8. Structure modeling of Int8. (A,B) Overall structures of the wild type (A) and R371I mutant (B) of Int8 predicted by AlphaFold2. (C,D) Expanded views of the helical regions of the wild type (C) and R371I mutant (D). R371 and I371 are shown by red and blue balls, respectively. The Figures were drawn using the PyMOL Molecular Graphics System, Version 2.4.0 Schrödinger, LLC.
4 Discussion
In this study, we characterized the structure of Int8, the intron 8-encoded domain of herstatin. Since Int8 binds and interferes with the homo- and hetero-dimerization of HER1, HER2, and HER4, it serves as an intrinsic inhibitor of the HER family proteins. Structure prediction and experimental characterization by CD, NMR, and SAXS indicated that Int8 is largely disordered, but retains a residual helical structure. Moreover, it had a molecular size similar to that of the fully unfolded state, although the conformational ensemble was less flexible than random coils. These results clearly indicate that Int8 is intrinsically disordered. To our knowledge, this is the first report of an IDR encoded by an intron. Further, the structure of the Int8 domain might be classified as a pre-molten globule state, which has an expanded overall structure but with unstable secondary structure (Ptitsyn, 1995; Uversky, 2002a; b; Tcherkasskaya et al., 2003). Pre-molten globule-like structures have also been reported for other intrinsically disordered proteins (IDPs) (Ptitsyn, 1995; Uversky, 2002a; b; Tcherkasskaya et al., 2003; Kunihara et al., 2019). Since Int8 is known to have tumor-suppressive activity (Lv et al., 2012), it is an interesting example of an intron-encoded IDR that may function as an antitumor drug.
Many IDPs exhibit coupled folding and binding behaviors, where binding is accompanied by folding (Arai, 2018). In many cases, the residues involved in coupled folding and binding have a propensity to form a helical structure (Mohan et al., 2006). Therefore, the residual helical structure present in Int8 may serve as the putative HER-binding site of Int8.
Previous reports have shown that intrinsic disorder is involved in protein-protein interactions mediated by HER proteins. The intracellular kinase domain of HER1 contains an IDR at the dimerization interface, which reorganizes into an ordered structure upon dimerization (Shan et al., 2012). Furthermore, HER1 dimerization is facilitated by cancer-related mutations that suppress local disorders at the dimerization interface (Shan et al., 2012). Therefore, intrinsic disorder regulates the dimerization of HER proteins. Thus, the use of intrinsic disorder in the interaction between herstatin and HER proteins is not exceptional. Rather, IDRs may play an important role in regulating protein-protein interactions related to cell proliferation (Uversky et al., 2008).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
DT and MA designed the study and wrote the paper. DT, SS, and NS conducted protein expression and purification and CD measurements. DT, TK, YH, and MA conducted NMR experiment. DT, TK, HK, JI, YH, and MA conducted SAXS experiment. SS, NS, and YH conducted SEC and light scattering measurements. DT, KO, and MA conducted structure prediction. KO and MA conducted structure modeling. DT, SS, NS, KO, YH, and MA analyzed the data. All authors reviewed the results and approved the final version of the manuscript.
Funding
This work was supported by JSPS KAKENHI Grant Numbers JP16H02217, JP19H02521, and JP21K18841 (YH and MA).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
SAXS measurement was performed under the approval of the Photon Factory Program Advisory Committee.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.862910/full#supplementary-material
Abbreviations
1D, one-dimensional; 2D, two-dimensional; CD, circular dichroism; Dmax, maximum distance; DSS, sodium 3-(trimethylsilyl)-1-propanesulfonate; ECD, extracellular domain; EOM, ensemble optimization method; GdnHCl, guanidine hydrochloride; HER, human epidermal growth factor receptor; HPLC, high-performance liquid chromatography; I(0), zero-angle scattering intensity; IDP, intrinsically disordered protein; IDR, intrinsically disordered region; I(Q), scattering intensity; NMR, nuclear magnetic resonance; PFG, pulsed-field gradient; P(r), pair-distance distribution function; Q, scattering vector; Rg, radius of gyration; Rh, hydrodynamic radius; SAXS, small-angle X-ray scattering; SEC, size-exclusion chromatography; UV, ultraviolet.
References
Anthis, N. J., and Clore, G. M. (2013). Sequence-Specific Determination of Protein and Peptide Concentrations by Absorbance at 205 nm. Protein Sci. 22, 851–858. doi:10.1002/pro.2253
Arai, M., and Iwakura, M. (2005). Probing the Interactions between the Folding Elements Early in the Folding of Escherichia coli Dihydrofolate Reductase by Systematic Sequence Perturbation Analysis. J. Mol. Biol. 347, 337–353. doi:10.1016/j.jmb.2005.01.033
Arai, M., Kondrashkina, E., Kayatekin, C., Matthews, C. R., Iwakura, M., and Bilsel, O. (2007). Microsecond Hydrophobic Collapse in the Folding of Escherichia coli Dihydrofolate Reductase, an α/β-Type Protein. J. Mol. Biol. 368, 219–229. doi:10.1016/j.jmb.2007.01.085
Arai, M. (2018). Unified Understanding of Folding and Binding Mechanisms of Globular and Intrinsically Disordered Proteins. Biophys. Rev. 10, 163–181. doi:10.1007/s12551-017-0346-7
Azios, N. G., Romero, F. J., Denton, M. C., Doherty, J. K., and Clinton, G. M. (2001). Expression of Herstatin, an Autoinhibitor of HER-2/Neu, Inhibits Transactivation of HER-3 by HER-2 and Blocks EGF Activation of the EGF Receptor. Oncogene 20, 5199–5209. doi:10.1038/sj.onc.1204555
Bernadó, P., Mylonas, E., Petoukhov, M. V., Blackledge, M., and Svergun, D. I. (2007). Structural Characterization of Flexible Proteins Using Small-Angle X-Ray Scattering. J. Am. Chem. Soc. 129, 5656–5664. doi:10.1021/ja069124n
Buchan, D. W. A., and Jones, D. T. (2019). The PSIPRED Protein Analysis Workbench: 20 Years on. Nucleic Acids Res. 47, W402–W407. doi:10.1093/nar/gkz297
Calmettes, P., Durand, D., Desmadril, M., Minard, P., Receveur, V., and Smith, J. C. (1994). How Random Is a Highly Denatured Protein? Biophysical Chem. 53, 105–113. doi:10.1016/0301-4622(94)00081-6
Camenisch, T. D., Schroeder, J. A., Bradley, J., Klewer, S. E., and McDonald, J. A. (2002). Heart-Valve Mesenchyme Formation Is Dependent on Hyaluronan-Augmented Activation of ErbB2-ErbB3 Receptors. Nat. Med. 8, 850–855. doi:10.1038/nm742
Cha, N., Han, X., Jia, B., Liu, Y., Wang, X., Gao, Y., et al. (2017). Structure-Based Design of Peptides against HER2 with Cytotoxicity on Colon Cancer. Artif. Cell Nanomedicine, Biotechnol. 45, 649–654. doi:10.3109/21691401.2016.1167705
Chang, M., Shimba, K., Hayashi, Y., and Arai, M. (2020). Electrostatic Interactions at the Interface of Two Enzymes Are Essential for Two-step Alkane Biosynthesis in Cyanobacteria. Biosci. Biotechnol. Biochem. 84, 228–237. doi:10.1080/09168451.2019.1677142
Chen, Y.-H., Yang, J. T., and Martinez, H. M. (1972). Determination of the Secondary Structures of Proteins by Circular Dichroism and Optical Rotatory Dispersion. Biochemistry 11, 4120–4131. doi:10.1021/bi00772a015
Citri, A., and Yarden, Y. (2006). EGF-ERBB Signalling: Towards the Systems Level. Nat. Rev. Mol. Cel Biol. 7, 505–516. doi:10.1038/nrm1962
Delaglio, F., Grzesiek, S., Vuister, G., Zhu, G., Pfeifer, J., and Bax, A. (1995). NMRPipe: A Multidimensional Spectral Processing System Based on Unix Pipes. J. Biomol. NMR 6, 277–293. doi:10.1007/BF00197809
Doherty, J. K., Bond, C., Jardim, A., Adelman, J. P., and Clinton, G. M. (1999). The HER-2/Neu Receptor Tyrosine Kinase Gene Encodes a Secreted Autoinhibitor. Proc. Natl. Acad. Sci. U.S.A. 96, 10869–10874. doi:10.1073/pnas.96.19.10869
Edelhoch, H. (1967). Spectroscopic Determination of Tryptophan and Tyrosine in Proteins. Biochemistry 6, 1948–1954. doi:10.1021/bi00859a010
Gill, S. C., and von Hippel, P. H. (1989). Calculation of Protein Extinction Coefficients from Amino Acid Sequence Data. Anal. Biochem. 182, 319–326. doi:10.1016/0003-2697(89)90602-7
Gompels, L. L., Malik, N. M., Madden, L., Jin, P., Feldmann, M., Shepard, H. M., et al. (2011). Human Epidermal Growth Factor Receptor Bispecific Ligand Trap RB200: Abrogation of Collagen-Induced Arthritis in Combination with Tumour Necrosis Factor Blockade. Arthritis Res. Ther. 13, R161. doi:10.1186/ar3480
Hanson, J., Paliwal, K. K., Litfin, T., and Zhou, Y. (2019). SPOT-Disorder2: Improved Protein Intrinsic Disorder Prediction by Ensembled Deep Learning. Genomics, Proteomics & Bioinformatics 17, 645–656. doi:10.1016/j.gpb.2019.01.004
Hart, V., Gautrey, H., Kirby, J., and Tyson-Capper, A. (2020). HER2 Splice Variants in Breast Cancer: Investigating Their Impact on Diagnosis and Treatment Outcomes. Oncotarget 11, 4338–4357. doi:10.18632/oncotarget.27789
Hirose, S., and Noguchi, T. (2013). ESPRESSO: A System for Estimating Protein Expression and Solubility in Protein Expression Systems. Proteomics 13, 1444–1456. doi:10.1002/pmic.201200175
Hoover, D. M., and Lubkowski, J. (2002). DNAWorks: An Automated Method for Designing Oligonucleotides for PCR-Based Gene Synthesis. Nucleic Acids Res. 30, 43e–43. doi:10.1093/nar/30.10.e43
Howe, L. R., and Brown, P. H. (2011). Targeting the HER/EGFR/ErbB Family to Prevent Breast Cancer. Cancer Prev. Res. 4, 1149–1157. doi:10.1158/1940-6207.CAPR-11-0334
Hu, P., Feng, J., Zhou, T., Wang, J., Jing, B., Yu, M., et al. (2005). In Vivo Identification of the Interaction Site of ErbB2 Extracellular Domain with its Autoinhibitor. J. Cel. Physiol. 205, 335–343. doi:10.1002/jcp.20409
Hu, P., Zhou, T., Qian, L., Wang, J., Shi, M., Yu, M., et al. (2006). Sequestering ErbB2 in Endoplasmic Reticulum by its Autoinhibitor from Translocation to Cell Surface: An Autoinhibition Mechanism of ErbB2 Expression. Biochem. Biophysical Res. Commun. 342, 19–27. doi:10.1016/j.bbrc.2006.01.115
Ishida, T., and Kinoshita, K. (2007). PrDOS: Prediction of Disordered Protein Regions from Amino Acid Sequence. Nucleic Acids Res. 35, W460–W464. doi:10.1093/nar/gkm363
Jackson, C., Browell, D., Gautrey, H., and Tyson-Capper, A. (2013). Clinical Significance of HER-2 Splice Variants in Breast Cancer Progression and Drug Resistance. Int. J. Cel Biol. 2013, 1–8. doi:10.1155/2013/973584
Jhabvala-Romero, F., Evans, A., Guo, S., Denton, M., and Clinton, G. M. (2003). Herstatin Inhibits Heregulin-Mediated Breast Cancer Cell Growth and Overcomes Tamoxifen Resistance in Breast Cancer Cells that Overexpress Her-2. Oncogene 22, 8178–8186. doi:10.1038/sj.onc.1206912
Johnson, B. A., and Blevins, R. A. (1994). NMR View: A Computer Program for the Visualization and Analysis of NMR Data. J. Biomol. NMR 4, 603–614. doi:10.1007/bf00404272
Jones, D. T., and Cozzetto, D. (2015). DISOPRED3: Precise Disordered Region Predictions with Annotated Protein-Binding Activity. Bioinformatics 31, 857–863. doi:10.1093/bioinformatics/btu744
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly Accurate Protein Structure Prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Justman, Q. A., and Clinton, G. M. (2002). Herstatin, an Autoinhibitor of the Human Epidermal Growth Factor Receptor 2 Tyrosine Kinase, Modulates Epidermal Growth Factor Signaling Pathways Resulting in Growth Arrest. J. Biol. Chem. 277, 20618–20624. doi:10.1074/jbc.M111359200
Kato, A., Maki, K., Ebina, T., Kuwajima, K., Soda, K., and Kuroda, Y. (2007). Mutational Analysis of Protein Solubility Enhancement Using Short Peptide Tags. Biopolymers 85, 12–18. doi:10.1002/bip.20596
Kikhney, A. G., and Svergun, D. I. (2015). A Practical Guide to Small Angle X-Ray Scattering (SAXS) of Flexible and Intrinsically Disordered Proteins. FEBS Lett. 589, 2570–2577. doi:10.1016/j.febslet.2015.08.027
Kohn, J. E., Millett, I. S., Jacob, J., Zagrovic, B., Dillon, T. M., Cingel, N., et al. (2004). Random-Coil Behavior and the Dimensions of Chemically Unfolded Proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12491–12496. doi:10.1073/pnas.0403643101
Koletsa, T., Kostopoulos, I., Charalambous, E., Christoforidou, B., Nenopoulou, E., and Kotoula, V. (2008). A Splice Variant of HER2 Corresponding to Herstatin Is Expressed in the Noncancerous Breast and in Breast Carcinomas. Neoplasia 10, 687–696. doi:10.1593/neo.08314
Kozlowski, L. P., and Bujnicki, J. M. (2012). MetaDisorder: A Meta-Server for the Prediction of Intrinsic Disorder in Proteins. BMC Bioinformatics 13, 111. doi:10.1186/1471-2105-13-111
Kunihara, T., Hayashi, Y., and Arai, M. (2019). Conformational Diversity in the Intrinsically Disordered HIV-1 Tat Protein Induced by Zinc and pH. Biochem. Biophysical Res. Commun. 509, 564–569. doi:10.1016/j.bbrc.2018.12.126
Lacroix, E., Viguera, A. R., and Serrano, L. (1998). Elucidating the Folding Problem of α-Helices: Local Motifs, Long-Range Electrostatics, Ionic-Strength Dependence and Prediction of NMR Parameters. J. Mol. Biol. 284, 173–191. doi:10.1006/jmbi.1998.2145
Linding, R., Jensen, L. J., Diella, F., Bork, P., Gibson, T. J., and Russell, R. B. (2003). Protein Disorder Prediction. Structure 11, 1453–1459. doi:10.1016/j.str.2003.10.002
Lv, M., Qiao, C., Jiang, N., Li, X., Yu, M., Hou, C., et al. (2012). The Peptide Derived from ErbB2 Auto-Inhibitor Herstatin Shared in the Same Epitope and Function with Functional Antibody 2C4. Mol. Biotechnol. 51, 174–182. doi:10.1007/s12033-011-9454-y
Mészáros, B., Erdős, G., and Dosztányi, Z. (2018). IUPred2A: Context-dependent Prediction of Protein Disorder as a Function of Redox State and Protein Binding. Nucleic Acids Res. 46, W329–W337. doi:10.1093/nar/gky384
Mizianty, M. J., Peng, Z., and Kurgan, L. (2013). MFDp2. Intrinsically Disordered Proteins 1, e24428. doi:10.4161/idp.24428
Mohan, A., Oldfield, C. J., Radivojac, P., Vacic, V., Cortese, M. S., Dunker, A. K., et al. (2006). Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol. 362, 1043–1059. doi:10.1016/j.jmb.2006.07.087
Mujoo, K., Choi, B.-K., Huang, Z., Zhang, N., and An, Z. (2014). Regulation of ERBB3/HER3 Signaling in Cancer. Oncotarget 5, 10222–10236. doi:10.18632/oncotarget.2655
Muñoz, V., and Serrano, L. (1994). Elucidating the Folding Problem of Helical Peptides Using Empirical Parameters. Nat. Struct. Mol. Biol. 1, 399–409. doi:10.1038/nsb0694-399
Nakamura, T., Aizawa, T., Kariya, R., Okada, S., Demura, M., Kawano, K., et al. (2013). Molecular Mechanisms of the Cytotoxicity of Human α-Lactalbumin Made Lethal to Tumor Cells (HAMLET) and Other Protein-Oleic Acid Complexes. J. Biol. Chem. 288, 14408–14416. doi:10.1074/jbc.M112.437889
Normanno, N., Maiello, M. R., and De Luca, A. (2003). Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitors (EGFR-TKIs): Simple Drugs with a Complex Mechanism of Action? J. Cel. Physiol. 194, 13–19. doi:10.1002/jcp.10194
Nygaard, M., Kragelund, B. B., Papaleo, E., and Lindorff-Larsen, K. (2017). An Efficient Method for Estimating the Hydrodynamic Radius of Disordered Protein Conformations. Biophysical J. 113, 550–557. doi:10.1016/j.bpj.2017.06.042
Obradovic, Z., Peng, K., Vucetic, S., Radivojac, P., and Dunker, A. K. (2005). Exploiting Heterogeneous Sequence Properties Improves Prediction of Protein Disorder. Proteins 61 (Suppl. 7), 176–182. doi:10.1002/prot.20735
Pace, C. N., Vajdos, F., Fee, L., Grimsley, G., and Gray, T. (1995). How to Measure and Predict the Molar Absorption Coefficient of a Protein. Protein Sci. 4, 2411–2423. doi:10.1002/pro.5560041120
Ptitsyn, O. B. (1995). Molten Globule and Protein Folding. Adv. Protein Chem. 47, 83–229. doi:10.1016/s0065-3233(08)60546-x
Shamieh, L. S., Evans, A. J., Denton, M. C., and Clinton, G. M. (2004). Receptor Binding Specificities of Herstatin and its Intron 8-Encoded Domain. FEBS Lett. 568, 163–166. doi:10.1016/j.febslet.2004.05.027
Shan, Y., Eastwood, M. P., Zhang, X., Kim, E. T., Arkhipov, A., Dror, R. O., et al. (2012). Oncogenic Mutations Counteract Intrinsic Disorder in the EGFR Kinase and Promote Receptor Dimerization. Cell 149, 860–870. doi:10.1016/j.cell.2012.02.063
Shimizu, K. (2014). “POODLE: Tools Predicting Intrinsically Disordered Regions of Amino Acid Sequence,” in Protein Structure Prediction. Editor D. Kihara (New York, NY: Springer New York), 131–145. doi:10.1007/978-1-4939-0366-5_10
Silipo, M., Gautrey, H., Satam, S., Lennard, T., and Tyson-Capper, A. (2017). How Is Herstatin, a Tumor Suppressor Splice Variant of the Oncogene HER2, Regulated? RNA Biol. 14, 536–543. doi:10.1080/15476286.2016.1267074
Staverosky, J. A., Muldoon, L. L., Guo, S., Evans, A. J., Neuwelt, E. A., and Clinton, G. M. (2005). Herstatin, an Autoinhibitor of the Epidermal Growth Factor Receptor Family, Blocks the Intracranial Growth of Glioblastoma. Clin. Cancer Res. 11, 335–340. doi:10.1158/1078-0432.335.11.1
Svergun, D. I. (1992). Determination of the Regularization Parameter in Indirect-Transform Methods Using Perceptual Criteria. J. Appl. Cryst. 25, 495–503. doi:10.1107/S0021889892001663
Tai, W., Mahato, R., and Cheng, K. (2010). The Role of HER2 in Cancer Therapy and Targeted Drug Delivery. J. Controlled Release 146, 264–275. doi:10.1016/j.jconrel.2010.04.009
Tcherkasskaya, O., Davidson, E. A., and Uversky, V. N. (2003). Biophysical Constraints for Protein Structure Prediction. J. Proteome Res. 2, 37–42. doi:10.1021/pr025552q
Trewhella, J., Duff, A. P., Durand, D., Gabel, F., Guss, J. M., Hendrickson, W. A., et al. (2017). 2017 Publication Guidelines for Structural Modelling of Small-Angle Scattering Data from Biomolecules in Solution: An Update. Acta Cryst. Sect D Struct. Biol. 73, 710–728. doi:10.1107/S2059798317011597
Tria, G., Mertens, H. D. T., Kachala, M., and Svergun, D. I. (2015). Advanced Ensemble Modelling of Flexible Macromolecules Using X-Ray Solution Scattering. Int. Union Crystallogr. J. 2, 207–217. doi:10.1107/S205225251500202X
UniProt Consortium (2021). UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi:10.1093/nar/gkaa1100
Uversky, V. N. (2002a). Natively Unfolded Proteins: A Point where Biology Waits for Physics. Protein Sci. 11, 739–756. doi:10.1110/ps.4210102
Uversky, V. N., Oldfield, C. J., and Dunker, A. K. (2008). Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 37, 215–246. doi:10.1146/annurev.biophys.37.032807.125924
Uversky, V. N. (2002b). What Does it Mean to Be Natively Unfolded? Eur. J. Biochem. 269, 2–12. doi:10.1046/j.0014-2956.2001.02649.x
Vallet, S. D., Miele, A. E., Uciechowska-Kaczmarzyk, U., Liwo, A., Duclos, B., Samsonov, S. A., et al. (2018). Insights into the Structure and Dynamics of Lysyl Oxidase Propeptide, a Flexible Protein with Numerous Partners. Sci. Rep. 8, 11768. doi:10.1038/s41598-018-30190-6
Wilkins, D. K., Grimshaw, S. B., Receveur, V., Dobson, C. M., Jones, J. A., and Smith, L. J. (1999). Hydrodynamic Radii of Native and Denatured Proteins Measured by Pulse Field Gradient NMR Techniques. Biochemistry 38, 16424–16431. doi:10.1021/bi991765q
Wu, D. H., Chen, A. D., and Johnson, C. S. (1995). An Improved Diffusion-Ordered Spectroscopy Experiment Incorporating Bipolar-Gradient Pulses. J. Magn. Reson. Ser. A 115, 260–264. doi:10.1006/jmra.1995.1176
Keywords: human epidermal growth factor receptor, herstatin, intron-encoded protein, intrinsically disordered protein, pre-molten globule state, small-angle X-ray scattering
Citation: Tashiro D, Suetaka S, Sato N, Ooka K, Kunihara T, Kudo H, Inatomi J, Hayashi Y and Arai M (2022) Intron-Encoded Domain of Herstatin, An Autoinhibitor of Human Epidermal Growth Factor Receptors, Is Intrinsically Disordered. Front. Mol. Biosci. 9:862910. doi: 10.3389/fmolb.2022.862910
Received: 26 January 2022; Accepted: 31 March 2022;
Published: 02 May 2022.
Edited by:
Vladimir N. Uversky, University of South Florida, United StatesReviewed by:
Daniel E. Otzen, Aarhus University, DenmarkBogdan Melnik, Institute of Protein Research (RAS), Russia
Arne Raasakka, University of Bergen, Norway
Copyright © 2022 Tashiro, Suetaka, Sato, Ooka, Kunihara, Kudo, Inatomi, Hayashi and Arai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Munehito Arai, YXJhaUBiaW8uYy51LXRva3lvLmFjLmpw