 Arun Chandra Manivannan1
Arun Chandra Manivannan1 Arunkumar Malaisamy2†
Arunkumar Malaisamy2† Murugesh Eswaran3†
Murugesh Eswaran3† Arun Meyyazhagan4
Arun Meyyazhagan4 Vijaya Anand Arumugam5
Vijaya Anand Arumugam5 Kannan R. R. Rengasamy6
Kannan R. R. Rengasamy6 Balamuralikrishnan Balasubramanian7*
Balamuralikrishnan Balasubramanian7* Wen-Chao Liu8*
Wen-Chao Liu8*- 1Department of Microbiology, Alagappa University, Karaikudi, India
- 2Integrative Biology Division, International Centre for Genetic Engineering and Biotechnology, New Delhi, India
- 3Department of Bioinformatics, Bharathiar University, Coimbatore, India
- 4Department of Life Sciences, CHRIST (Deemed to be University), Bengaluru, India
- 5Department of Human Genetics and Molecular Biology, Bharathiar University, Coimbatore, India
- 6Department of Pharmacology, Centre for Transdisciplinary Research, Saveetha Dental College, Saveetha Institute of Medical and Technical Sciences (SIMATS), Chennai, India
- 7Department of Food Science and Biotechnology, College of Life Science, Sejong University, Seoul, South Korea
- 8Department of Animal Science, College of Coastal Agricultural Sciences, Guangdong Ocean University, Zhanjiang, China
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) virus can cause a sudden respiratory disease spreading with a high mortality rate arising with unknown mechanisms. Still, there is no proper treatment available to overcome the disease, which urges the research community and pharmaceutical industries to screen a novel therapeutic intervention to combat the current pandemic. This current study exploits the natural phytochemicals obtained from clove, a traditional natural therapeutic that comprises important bioactive compounds used for targeting the main protease of SARS-CoV-2. As a result, inhibition of viral replication effectively procures by targeting the main protease, which is responsible for the viral replication inside the host. Pharmacokinetic studies were evaluated for the property of drug likeliness. A total of 53 bioactives were subjected to the study, and four among them, namely, eugenie, syzyginin B, eugenol, and casuarictin, showed potential binding properties against the target SARS-CoV-2 main protease. The resultant best bioactive was compared with the commercially available standard drugs. Furthermore, validation of respective compounds with a comprehensive molecular dynamics simulation was performed using Schrödinger software. To further validate the bioactive phytochemicals and delimit the screening process of potential drugs against coronavirus disease 2019, in vitro and in vivo clinical studies are needed to prove their efficacy.
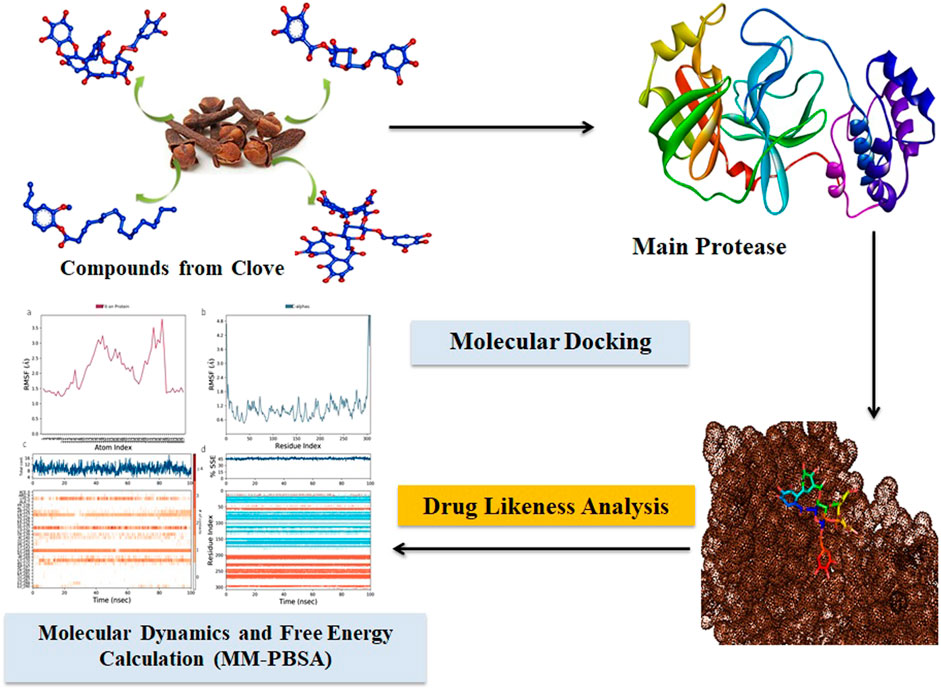
GRAPHICAL ABSTRACT
Introduction
A sudden outbreak of respiratory illness with unknown etiology arose in Wuhan, China, and was later diagnosed as a novel coronavirus (nCoV) with a novel variant called severe acute respiratory syndrome (SARS)-CoV-2. Coronaviruses (CoVs) are a family of enveloped RNA viruses comprising seven human coronaviruses (HCoVs) causing human and animal infection (Biswaranjan, 2022). These are zoonotic obligate intracellular organisms and primarily infect respiratory and associated regions, and this novel virus spreads through air transmission when an infected person coughs or sneezes (Mittal et al., 2020; Majdi et al., 2022). HCoV-229E, HCoV-NL63, HCoV-OC43, and HCoV-HKU1 are usually seasonal, causing mild respiratory diseases that are best known for causing common cold, while other variants like CoV229E and OC43 can provoke pneumonia. Since 3 decades of the virus domination from the first spread of severe strains of middle east respiratory syndrome (MERS)-CoV, SARS-CoV-1, and the current SARS-CoV-2, the lesser known virus is stuck on the headlines for its high pathogenicity and high proliferation rate (Liu et al., 2020; Majdi et al., 2022). The viral infection prevails in one’s body with a highly specific recognition between the virus spike proteins through HCoV-specific receptors. Meanwhile, the main protease (MPro) is the vital enzyme for processing viral polyproteins produced after being translated from RNA. This MPro is structurally present as a homodimer, which is made up of two promoters of three domains, namely I, II, and III, whose amino acid ranges are 8–101, 102–184, and 201–303, respectively, and a long loop (185–200) that connects domains II and III (Kumar et al., 2021; Mengist et al., 2021). Every possible sector has unleashed its potency over research and development to targeting the SARS-CoV-2 virus to wind this raging pandemic (Marcelino et al., 2022). Traditional medicinal practices based on herbs and their extracts are gaining momentum. Their formulations are widely given with supplementary allopathic treatment for the earlier recovery (Ang et al., 2020; Mhya et al., 2021; Vanshylla et al., 2021; Ashande et al., 2022; Nair et al., 2022). Syzygiumaromaticum, a native species of the Maluku Islands in Indonesia, traditionally found its importance as a flavoring additive for food. However, clove finds its use in ayurvedic and Chinese medicine (Cortés-Rojas et al., 2014). These aromatic flower buds are from a tree species of the Myrtaceae family that inhabits tropical climates. The current status of coronavirus disease 2019 (COVID-19) infection on 5 March 2022 crossed 440,807,756 cases with 5,978,096 deaths worldwide, and India’s status was 42,951,556 cases with 514,589 deaths.
This research evaluates the antiviral efficacy of clove-based phytochemicals by computationally using the autodock protocol by targeting the MPro of the SARS-CoV-2 virus (KuchiBhotla et al., 2021; Marcelino et al., 2022). Moreover, the computational world has witnessed a recent trend of large natural compound data retrieval to target various structures of the SARS-CoV-2 virus to define a proper therapeutic. The extensive pathophysiological mechanisms underlying viral infections as well as the related adverse effects of the currently available conventional medicines necessitate the development of a novel drug discovery process. With the conventional methods, finding a new drug is very challenging, and also, it is the costliest method ever. In silico methods were carried out to facilitate the virtual screening of the best drug candidate to overcome the challenging drawbacks. Pharmacological studies were conducted to analyze the bioavailability and dynamics inside the host by profiling the ADMET properties of phytochemicals for the suitable drug candidate through extending absorption, distribution, metabolism, excretion, and toxicity (ADMET) (Kar et al., 2021). The molecular dynamics simulation studies were conducted to dive deep into their extent and efficiency by sustaining the administered system over a defined time of 100 ns of total simulation. Investigation of clove-extracted compounds against a target of the MPro of the SARS-CoV-2 virus leads to effective therapeutic drugs. Further validation of all our compounds was compared with the commercial antiviral drugs to combat COVID-19.
Materials and Methods
Protein Preparation
The MPro acts as a target of treatment for various viral infectious agents, including SARS CoV-2, MERS-CoV, noroviruses, enteroviruses, and rhinoviruses. The MPro is a disparate protein homing in the infected individual, and as it catalyzes most maturation cleavage events, this proteolytic compound is an explicit target for effective lead screening (Luan et al., 2020). The X-ray crystal structure of MPro (PDB ID-6LU7) was retrieved from the RCSB Protein Data Bank Database. The structure was subjected to protein preparation using autodock tools, a graphical user interface program, which was exploited for the preparation, running, and analyzing the docking simulations. Water molecules, ligand groups (inhibitor), and other nonspecific molecules were removed, polar hydrogen was added with the merging nonpolar hydrogens, and partial charges were assigned (Arunkumar et al., 2021). The grids are placed in the region that possesses the nature of an active site since grids direct the ligand toward the binding site (Forli et al., 2016).
Ligand Preparation
Data on active phytochemicals present in clove were acquired from the curated databases of Indian Medicinal Plants, Phytochemistry, and Therapeutics (IMPPAT) (Mohanraj et al., 2018). These subsequent structures were retrieved from the PubChem repository, and the related structures were retried from the Zinc Database in the output format of the structure data file. These were converted to the PDB format using Open Babel software; energy minimization was carried out using pyrxtool applying the molecular mechanics force field and optimized for further exploitation of the ligand. The complete dataset of phytochemical names and their IDs used in this study is provided in Supplementary Table S1.
Molecular Docking
The molecular docking was performed in autodock tools with an extensive suite of python molecular viewers. First, the site-specific docking was carried out with the aid of autodock 4.2; during docking, the protein was placed as a rigid molecule and the ligand was flexible (Trott and Olson., 2010). The studies were carried out using the Lamarckian genetic algorithm with the genetic algorithm parameters comprising 2.5 × 106 energy appraisals and a maximum number of 2.7× 104 generations with a mutation rate of 0.02 with a crossover rate of 0.8. Pseudo Solis and Wets parameters for local search were performed and introduced 300 iterations. Finally, 50 independent runs for each compound were placed, with the grid dimension of 76 × 76 × 76 and with a spacing of 0.375Å (Seeliger and de Groot., 2010).
Pharmacokinetics Evaluation
Target prediction studies compute the probable macromolecular target site of the screened small molecules; this methodology aids in tracing the bioactivity, side effects, and off-targets. In addition, the ADMET analysis divulges the pharmacokinetics that a ligand must boat to establish its function in the administered body (Arunkumar et al., 2022). The top-ranked compound was evaluated for the ADMET analysis using the Qikrop module on Schrödinger’s Maestro platform (Schrödinger Release 2021-2: QikProp, Schrödinger, LLC, New York, NY, 2021).
Molecular Dynamics Simulation
Because molecules are dynamic in nature, studying their motions at the molecular and atomistic levels is critical to comprehending the crucial physicochemical processes. In all other computational applications, molecular dynamics simulation stands alone as the essential computational technique for capturing the dynamic events of scientific interest. Based on the molecular interaction and binding score of the small molecule against the target molecule, the top-ranked complex molecules were selected for the molecular dynamics simulation studies. First, the complex molecule was preprocessed using the protein preparation wizard module; then the structure was refined by optimizing the hydrogen bond and applying the force field OPLS3e for energy minimization. OPLS3e improves the accuracy of small-molecule conformational propensities, solvation, and protein–ligand binding performance benchmarks (Roos et al., 2019). Furthermore, the complex molecule was solvated using a system builder module to a hydration model (TIP3P) in the 3D orthorhombic box with a buffer distance of 10 Å. Finally, the whole system is designated for the simulation time of 100 ns with 1,000 frame trajectory points under a default NPT ensemble of constant pressure, temperature, and atom number. They were performed using the Desmond module on Schrödinger’s Maestro platform (Schrödinger Release 2021-2: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2021. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2021).
Results and Discussion
Despite fast-tracking the research of COVID cure, no potential lead molecules that can effectively break the viral proliferation chain within an individual are identified (Anju et al., 2020). Moreover, many studies are performed on phytochemicals from medicinal plants for their efficacy against the current COVID-19 disease. SARS-CoV-2 viruses are also reported to get disseminated into various body organs and contaminate the environment in more than one route (Xu et al., 2016; Deng-hai Zhang et al., 2020; Garg et al., 2020; Biswaranjan, 2022). The discovery of the SARS-CoV-2 MPro has opened the door for an effective approach to drug discovery that can be enabled via a virtual combinational mode employing computational tools (Guo et al., 2021; Marcelino et al., 2022).
Molecular Docking and Interactions
About 53 phytochemicals extracted from clove were subjected to molecular docking experiments against the main protease; 27 compounds were native to clove, and 23 are chemicals structurally similar to a few native compounds retrieved from the ZINC database. Around 60–90% of clove phytocompositions are eugenol, eugenyl acetate, caryophyllene, and aceteugenol (Xu et al., 2016). The US Food and Drug Administration (FDA) categorized the clove essential oil as generally recognized as safe, and the World Health Organization has drafted the daily intake quantity for cloves as 2.5 mg/kg of an individual (Sink et al., 2007; Kulkarni et al., 2020). The MPro is responsible for proper viral replication in SARS CoV-2. Hence, any potential leads can effectively inhibit viral replication inside the host system (Mothay and Ramesh, 2020; Narkhede et al., 2020). Thus, any compound manifesting the disarming of the MPro can be taken for further clinical studies. Still, various leading laboratories in dry and wet labs worldwide are thriving hard to ace the race to screen for highly efficient drugs for the SARS-CoV-2 virus that could effectively treat any variant of the same.
Recently, large quantities of natural compounds are being exploited to act against the deadly virus. A total of 53 compounds were subjected to robust docking using autodock Vina tools against the main viral protease; four compounds, namely, casuarictin, eugeniin, syzyginin B, and eugenol, are identified to bind with the MPro with the numerically lowest binding energies (most negative) such as −12.2 kcal/mol, −9.8 kcal/mol, −10.4 kcal/mol, and −7.3 kcal/mol, respectively. Since the binding energy score was negative for almost all the compounds, those exhibiting values more than –7 were censored, and the remaining were subjected to further in silico modeling (Figure 1).
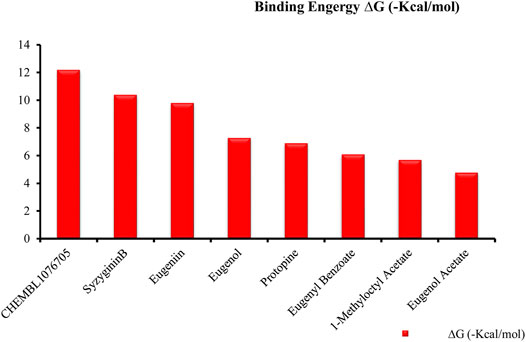
FIGURE 1. Graphical representation of top-ranked phytochemicals docked against the MPro unit of binding energy [∆G (−kcal/mol)].
The complex molecules (protein and ligand) were interrogated for their characters of post docking analysis using the Biovia Discovery studio tool. Hydrogen interaction was the predominant force for bond formation and spatial arrangement of the ligands within interacting pockets. In contrast, hydrophobic and electrostatic interactions are facilitated by a hydrogen bond. Casuarictin demonstrates the highest binding energy with an alliance of five hydrogen bonds with amino acids Thr199 (3.02 Å), Asp197 (3.79 Å), Arg131 (2.76 Å), Lys137 (2.46 Å), and Leu287 (3.53 Å) and three hydrophobic interactions with amino acids Leu287 (5.30Å), Leu272 (5.12 Å), and Tyr239 (5.20 Å).
Next to this, syzyginin B establishes a hydrogen bond with six amino acids, Lys137 (2.70 Å), Arg131 (2.10 Å), Asp289(2.81 Å), Glu228 (3.38 Å), Leu287 (3.33 Å), and Thr199 (2.53 Å), and one amino acid forging the hydrophobic interaction at Leu287 (4.89 Å). Finally, eugeniin with seven hydrogen bonds at locations Ala285 (3.38 Å), Leu287 (3.08 Å), Lys236 (2.67 Å), Asn238(2.57 Å), Lys137 (2.85 Å), Thr199 (3.07 Å), and Arg131 (2.98 Å) fashions a binding energy within the range of −9.8 kcal/mol. Table 1 describes the types of interaction various ligands are experiencing. These bonds are responsible for arresting the ligand efficiently within the active site of the protein. Figures 2, 3, 4 render the delineation of various bonds formed in screened compounds. The remaining 53 complex molecule interactions are listed in Supplementary Table S1, and for the remaining phytochemicals, interactions of their 3D structure with 2D interactions are represented in Supplementary Figures S1–19.
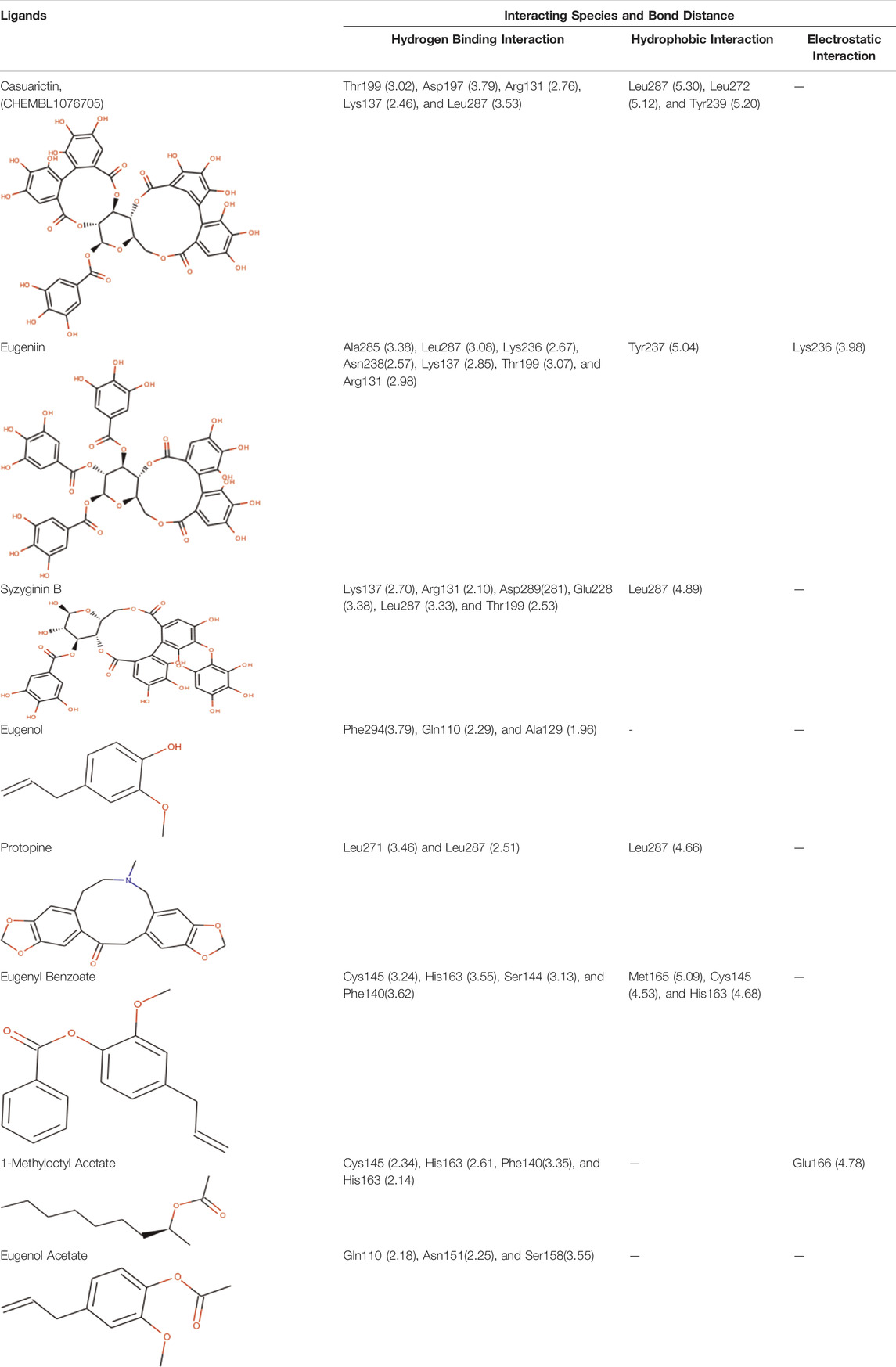
TABLE 1. Interaction between the amino acid residue of COVID -19 main protease and ligands at receptor sites.
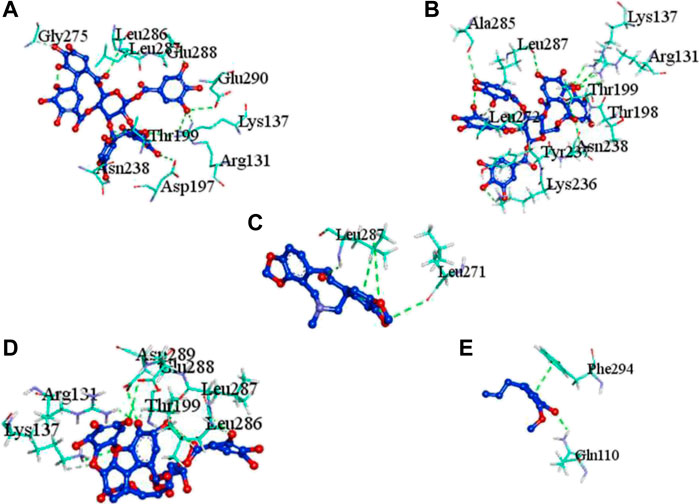
FIGURE 2. Molecular docking interaction of target proteins with clove’s phytochemicals of (A) casuarictin, (B) eugeniin, (C) protopine, (D) syzyginin B, and (E) eugenol.
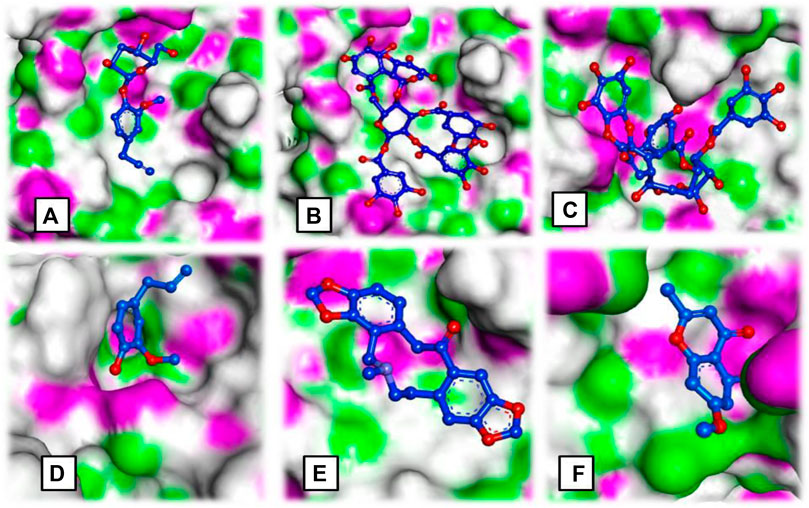
FIGURE 3. Ligand occupancy in an active site of a target protein complex molecule of (A) syringin, (B) eugeniin, (C) syzyginin, (B,D) eugenol acetate, (E) protopine, and (F) 1-methyloctyl acetate.
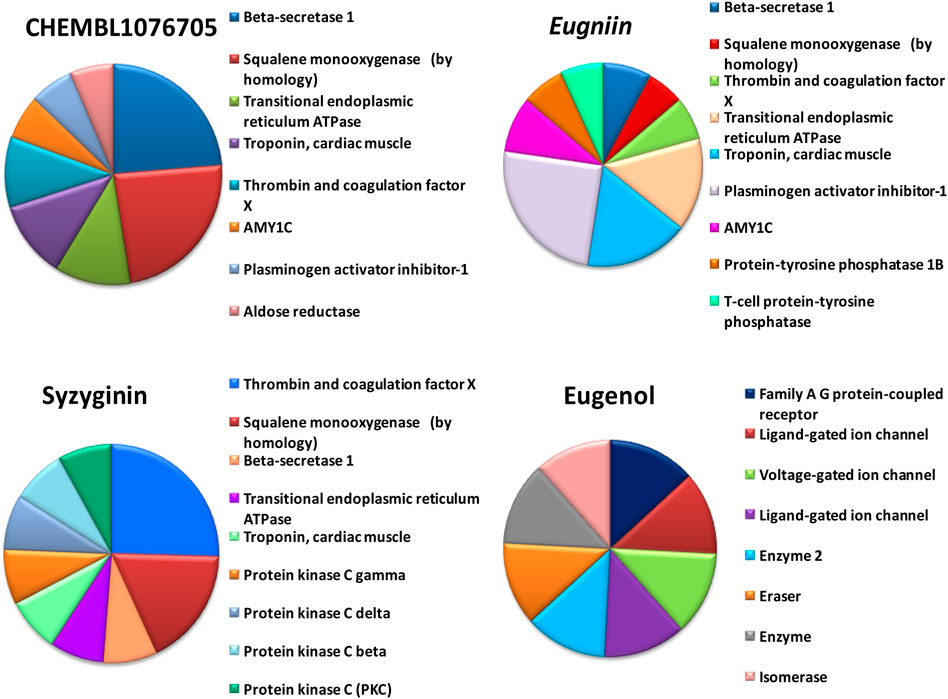
FIGURE 4. Off-target prediction of selected compounds including beta secretase, squalene mono-oxygenase, thrombin, and coagulation factor.
Our top-ranked compound of eugeniin (−9.8 kcal/mol), syzyginin B (−10.4 kcal/mol), eugenol (−7.3 kcal/mol), and casuarictin (−12.2 kcal/mol) has shown higher binding affinity than the commercially available drugs such as remdesivir (−6 kcal/mol), liponavir (−4 kcal/mol), tamiflu (−5.72 kcal/mol), plaquenil (−5.59 kcal/mol), and mycophenolic (−6.02 kcal/mol) (Arunkumar et al., 2021; Khater and Nassar, 2021). Eugenol is extracted from cloves, is abundantly present in the clove oil, is used for antispasmodic treatment, and acts as a carminative to treat gastrointestinal disorders. They are explored in bioactivities of antioxidant, anti-inflammatory, antiviral, insect-repellent, antimicrobial, and antiparasitic properties with various other related infections (Xu et al., 2016). The additional properties of cloves are strong, pungent, spicy odor and pungent combined aromatic taste, and cloves play a role in industrial application in perfumes, soaps, histological cleaning agents, and anesthetic fishes (Taylor and Roberts 1999). Eugenol exploited against spike glycoprotein for the treatment of SARS-CoV-2 has been reported computationally, with the attempted clinical phase in the official Siddha formulation of Kabasura Kudineer (Kiran et al., 2022). Moreover, the crisis on physical health due to continuous steroid supplementation within individuals infected with the SARS-CoV-2 virus stresses the need for alternative medicine.
Prediction of Probable Off-Target Activity
By tracing the probable drug reaction within a host and its assumed interaction, it might undergo intruding the host metabolism, which is provided in hit compound target prediction; this paves the way for the preparedness of any drug that can elicit any adverse reaction for patients. Additionally, it provides the researcher with a putative thought to remodel or restrict the further analysis of screened drug-like molecules (Drwal et al., 2014; Daina et al., 2019). For example, eugenol was predicted to be a class 4 toxic substance. The predicted LD50 is 1.930 mg/kg; it does not elicit carcinogenicity, hepatotoxicity, immunotoxicity, mutagenicity, and cytotoxicity and does not interfere with the signaling and stress response pathways. On the other hand, syzyginin B, casuarictin, and eugeniin are classified as class 5 toxic substances and possess an LD50 value of 2.260 mg/kg. This ternion exhibits mild reactivity and less than the recommended level of reactivity toward phosphoprotein (tumor repressor) p53. Still, the concerning part is that it may result in immunotoxicity under unregulated administration. At the same time, the last couple may interfere with the mitochondrial membrane protein and aryl hydrocarbon receptor and may initiate reactions adding up alarm for carcinogenicity, albeit with negligible probability. These results were obtained by combining two toxicity prediction web tools, Swiss Target prediction tools (Gupta et al., 2013). Casuarictin, syzyginin B, and eugeniin are identified to have common off-target interactions whose extent of integration either overtaking the active antagonist property or least significant can be identified only upon wet-lab studies (Mohamed et al., 2021). The common targets of these three compounds include beta secretase, squalene mono-oxygenase, thrombin and coagulation factor, troponin, and cardiac muscles with varying proportions (Figure 4).
Molecular Dynamics Simulation
The resulting top-ranked docking complex molecule was considered for performing the molecular dynamics simulation for further validation. In these simulation studies, the protein interaction with the ligand molecules was studied throughout the total simulation time of 100 ns with 1,000 projection points (Frames). The macromolecules and ligand causing interactions throughout the simulation time are called contacts, classified based on hydrogen bonds, hydrophobic interactions, ionic bonds, and water bridges. The molecular dynamics simulation output was investigated with a root-mean-square deviation (RMSD) value around 3 Å distance, representing the stability of the complex molecule. Syzyginin B starts the stability at around 60 ns in the first phase with the deviation of around 1 Å distance and remains stable around 2.5 Å. After that, it deviates from the cavity site of 1 Å distance, which holds the stability in 3.5 Å up to a total simulation time of 100 ns. On the other hand, eugeniin deviates in the initial phase up to 3.5 Å and stabilizes with minimal deviation, which remains stable from 20 to 100 ns around 2.8 Å RMSD. Finally, eugenol started the initial phase around 2.9 Å and remains stable across 100 ns of the total simulation time within 2.8 Å. Moreover, all the three complex molecules show better results from the molecular dynamics simulation studies, depicted by the graph in Figure 5.
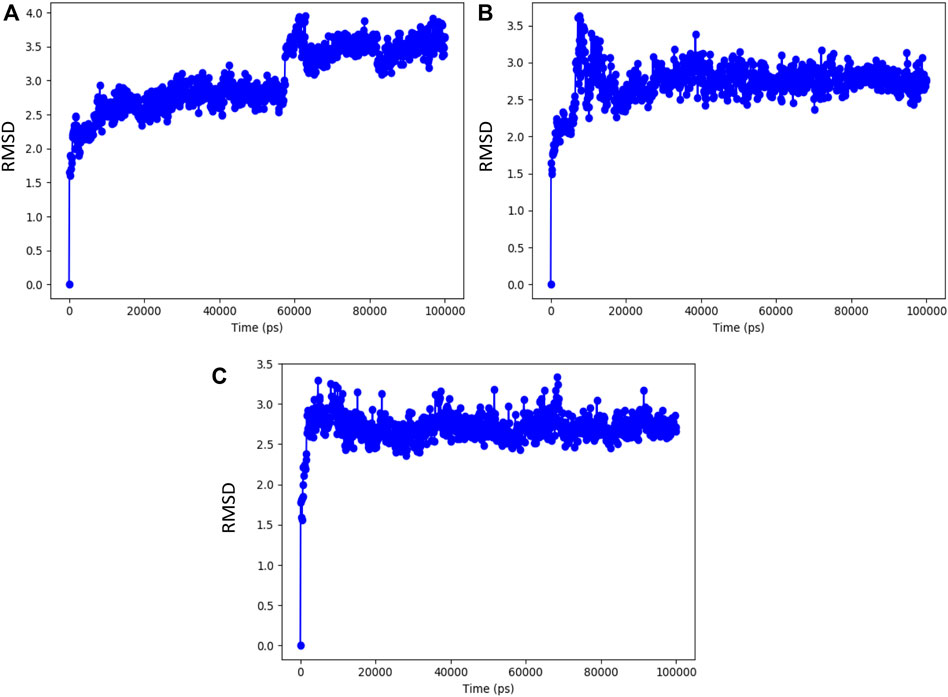
FIGURE 5. RMSD value of the complex molecule of the main protease with (A) syzyginin B, (B) eugeniin, and (C) eugenol.
The protein–ligand contact of clove phytochemical eugeniin showed an interaction of discontinuous contacts in the active site amino acid of LYS5, GLY170, SER139, and PHE140 and continuous contacts with GLU166, LYS137, and GLN27 across the total simulation time. On the other hand, the remaining two complex molecules of syzyginin B and eugenol showed the major discontinuous contacts (Supplementary Figures S20–22).
Pharmacokinetics
Computational ADMET screening can reduce the cost of high capital-consuming wet-lab trials that may end up in failure on many occasions (Hage-Melim et al., 2020; Tongqiang Zhang, et al., 2020). In the current study, not all the selected molecules were following the optimal limit of ADMET properties. With the octanal/water partition coefficient, aqueous solubility, and brain/blood partition coefficient, all compounds align within an acceptable range of −2.0–6.5, −6.5–0.5, and −3.0-2.1, respectively (Gleeson et al., 2009). A major deviation was exhibited by casuarictin (−13.959) and euginiin (−13.959) for skin permeability, where the optimum range is between −8.0 and −1.0. Eugenol exhibited a minimum metabolic reaction limited to 3, followed by protopine with five reactions. At the same time, the next two molecules deviate from an acceptable band of 1–8, where both euginiin and casuarictin exhibit 15 reaction exceptions.
All molecules are efficient enough to bind with human serum albumin within the accepted range of −1.5 to 1.5. Eugenol and protopine are highly capable of oral absorption and hold a good van der Waals surface area of polar nitrogen and oxygen atoms and carbonyl carbon atoms. Euginiin and casuarictin initially exhibited high binding energy (mostly negative). However, they face the most number of violations, albeit their ability to infuse toxicity within an individual is low, which suggests that its efficacy can be taken for the next level of analysis as hit compounds (Liao and Nicklaus, 2009; Zhong et al., 2013) (Table 2).
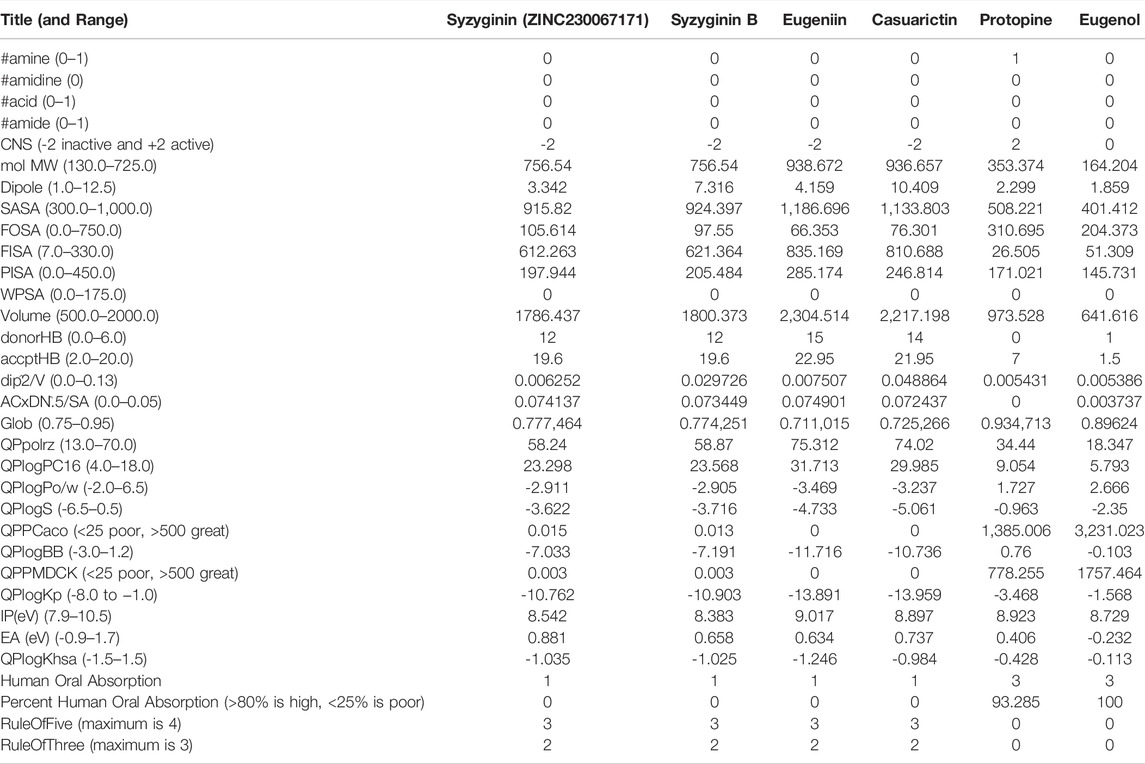
TABLE 2. ADME/T properties of various compounds that exhibited a high binding efficiency.
The ability of candidates to get accommodated within the traditional laws of drug-likeliness and physiochemical property limits are major initial screening processes, aiming to screen out unfit candidates (Mohamed et al., 2021). For example, Lipinski’s rule of five (RO5) is a major factor for drug-likeness that aids in identifying the potential compounds from a pool of drug-like molecules which must have strong gastrointestinal absorption, high oral bioavailability, and descent membrane permeability, with their log p ≤ 5; MW ≤ 500 Da, HBDs ≤5, and HBAs ≤10 (Gleeson et al., 2009). Natural compounds that have already been identified for treatment purposes are reported to violate RO5. On the other hand, due to the significant efficacy of natural compounds, those from marine-based and terrestrial resource-based compounds are accepted, although they have been identified as violating RO5 (Zhong et al., 2013; Mohanraj et al., 2018). The new framework proposed by the FDA over relaxed and diluted norms for FDA approval of drugs supports the idea to migrate the sample for further intensive trials so as to find the therapeutic scope beyond RO5.
Violation of log P is still not a concern for major cancer drugs (DeGoey et al., 2018). Casuarictin violates Veber’s rule for polar surface area (TPSA), resulting in a range ≤ 140 Å by holding a value of 444.18. The existing drugs that are prescribed for being effective against COVID-19 virus-like lopinavir, ritonavir, and remdesivir also violate Lipinski’s RO5, where lopinavir disobeys with MW > 500, many rotatable bonds being >10, ritonavir and remdesivir at features like MW > 500, many rotatable bonds > 10, and TPSA > 140 Å (Kar et al., 2021). Despite few notable fluctuations expressed by these selected compounds, there are updated relaxed norms by FDA over exploiting potential drugs that still disobey drug-likeliness properties and the existing record of supplementing drugs of a high molecular weight, disobeying RO5 (DeGoey et al., 2018).
Conclusion
Using biological sources to find alternative and successful drug candidates could be a long-term strategy for improving the COVID-19 drug discovery process. Cloves are a rich source of bioactive chemicals such as eugenol, which have been shown to have antiviral and immunostimulatory activities. In the current computational approach, clove phytochemicals of a total of 53 compounds were investigated for the molecular docking experiment against the MPro of SARS-CoV-2. Interestingly, among them, eugeniin, syzygininB, eugenol, and casuarictin have shown possible antagonist properties against the MPro with significant binding energies. Furthermore, the top-ranked phytochemicals were validated with the molecular dynamics simulation and revealed three compounds, namely, syzyginin B, eugeniin, and eugenol, as strongly interacting compounds that got stabilized with the least deviation from the site of interaction over the observed total simulation time. Moreover, the phytochemicals were assessed for their pharmacokinetic properties, shown to be druggable with no significant violation of any ADMET profiling parameters. As a result, these cloves’ phytochemicals may be viable candidates against SARS-CoV-2. Eugenol is one of the formulations of official siddha as Kabasura Kudineer for the treatment of COVID-19. However, extensive research is required to determine its efficacy as an antiviral drug, particularly in vitro trials against SARS-CoV-2. Finally, the innovative findings of this study could have a significant impact on the advancement of COVID-19 antiviral drug interventions in the near future (Trott and Olson, 2010; Lin et al., 2020; Khanna et al., 2021).
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
Author Contributions
This research article is written by the collaboration between the authors. Conceptualization, ME, BB, and W-CL; writing the original manuscript, AC, AM; methodology, data curation, and formal analysis, AC, AM, ME, and AM; organizing the working groups, BB, ME; writing review/editing, VA, KR, W-CL; interpretation and review/revision, ME, AM, BB. All Authors revised and approved the final article.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors thank the International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi, for Molecular dynamic simulation and ADMET studies using Schrodinger software.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2022.918101/full#supplementary-material
References
Ang, L., Lee, H. W., Kim, A., and Lee, M. S. (2020). Herbal Medicine for the Management of COVID-19 during the Medical Observation Period: a Review of Guidelines. Integr. Med. Res. 9 (3), 100465. doi:10.1016/j.imr.2020.100465
Anju, M., Sreeush, M. G., Valsala, V., Smitha, B. R., Hamza, F., Bharathi, G., et al. (2020). Understanding the Role of Nutrient Limitation on Plankton Biomass over Arabian Sea via 1‐D Coupled Biogeochemical Model and bio‐Argo Observations. J. Geophys. Res. Oceans 125 (6), e2019JC015502. doi:10.1029/2019jc015502
Arunkumar, M., Gunaseelan, S., Kubendran Aravind, M., Mohankumar, V., Anupam, P., Harikrishnan, M., et al. (2021). Marine Algal Antagonists Targeting 3CL Protease and Spike Glycoprotein of SARS-CoV-2: a Computational Approach for Anti-COVID-19 Drug Discovery. J. Biomol. Struct. Dyn., 1–28. doi:10.1080/07391102.2021.1921032
Arunkumar, M., Mahalakshmi, M., Ashokkumar, V., Aravind, M. K., Gunaseelan, S., Mohankumar, V., et al. (2022). Evaluation of Seaweed Sulfated Polysaccharides as Natural Antagonists Targeting Salmonella typhi OmpF: Molecular Docking and Pharmacokinetic Profiling. Beni-Suef Univ. J. Basic Appl. Sci. 11 (1), 8. doi:10.1186/s43088-021-00192-x
Ashande, C., Masunda, A., Ngbolua, K.-t. -N., Kilembe, J. T., Matondo, A., Liyongo Clément, I., et al. (2022). Glucose Oxidase as a Model Enzyme for Antidiabetic Activity Evaluation of Medicinal Plants: In Vitro and In Silico Evidence. Nat. Resour. Hum. Health 2 (2), 265–273. doi:10.53365/nrfhh/144779
Biswaranjan, P. (2022). Human Health Care against COVID-19 via Environmental Management. Nat. Resour. Hum. Health 2 (2), 142–149. doi:10.53365/nrfhh/143833
Cortés-Rojas, D. F., de Souza, C. R. F., and Oliveira, W. P. (2014). Clove (Syzygium Aromaticum): a Precious Spice. Asian Pac. J. Trop. Biomed. 4 (2), 90–96. doi:10.1016/S2221-1691(14)60215-X
Daina, A., Michielin, O., and Zoete, V. (2019). SwissTargetPrediction: Updated Data and New Features for Efficient Prediction of Protein Targets of Small Molecules. Nucleic Acids Res. 47 (W1), W357–W364. doi:10.1093/nar/gkz382
DeGoey, D. A., Chen, H.-J., Cox, P. B., and Wendt, M. D. (2018). Beyond the Rule of 5: Lessons Learned from AbbVie's Drugs and Compound Collection. J. Med. Chem. 61 (7), 2636–2651. doi:10.1021/acs.jmedchem.7b00717
Deng-hai Zhang, D.-h., Wu, K.-l., Zhang, X., Deng, S.-q., and Peng, B. (2020). In Silico screening of Chinese Herbal Medicines with the Potential to Directly Inhibit 2019 Novel Coronavirus. J. Integr. Med. 18 (2), 152–158. doi:10.1016/j.joim.2020.02.005
Drwal, M. N., Banerjee, P., Dunkel, M., Wettig, M. R., and Preissner, R. (2014). ProTox: a Web Server for the In Silico Prediction of Rodent Oral Toxicity. Nucleic Acids Res. 42 (W1), W53–W58. doi:10.1093/nar/gku401
Forli, S., Huey, R., Pique, M. E., Sanner, M. F., Goodsell, D. S., and Olson, A. J. (2016). Computational Protein-Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 11 (5), 905–919. doi:10.1038/nprot.2016.051
Garg, S., Garg, M., Prabhakar, N., Malhotra, P., and Agarwal, R. (2020). Unraveling the Mystery of Covid‐19 Cytokine Storm: From Skin to Organ Systems. Dermatol. Ther. 33 (6), 13859. doi:10.1111/dth.13859
Gleeson, P., Bravi, G., Modi, S., and Lowe, D. (2009). ADMET Rules of Thumb II: A Comparison of the Effects of Common Substituents on a Range of ADMET Parameters. Bioorg. Med. Chem. 17 (16), 5906–5919. doi:10.1016/j.bmc.2009.07.002
Guo, S., Xie, H., Lei, Y., Liu, B., Zhang, L., Xu, Y., et al. (2021). Discovery of Novel Inhibitors against Main Protease (Mpro) of SARS-CoV-2 via Virtual Screening and Biochemical Evaluation. Bioorg. Chem. 110, 104767. doi:10.1016/j.bioorg.2021.104767
Gupta, S., Kapoor, P., Chaudhary, K., Gautam, A., Kumar, R., and Raghava, G. P. S. (2013). In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 8 (9), e73957. doi:10.1371/journal.pone.0073957
Hage-Melim, L. I. d. S., Federico, L. B., de Oliveira, N. K. S., Francisco, V. C. C., Correia, L. C., de Lima, H. B., et al. (2020). Virtual Screening, ADME/Tox Predictions and the Drug Repurposing Concept for Future Use of Old Drugs against the COVID-19. Life Sci. 256, 117963. doi:10.1016/j.lfs.2020.117963
Kar, P., Sharma, N. R., Singh, B., Sen, A., and Roy, A. (2021). Natural Compounds from Clerodendrum Spp. As Possible Therapeutic Candidates Against SARS-CoV-2: An In Silico Investigation. J. Biomol. Struct. Dyn. 39 (13), 4774–4785. doi:10.1080/07391102.2020.1780947
Khanna, K., Kohli, S. K., Kaur, R., Bhardwaj, A., Bhardwaj, V., Ohri, P., et al. (2021). Herbal Immune-Boosters: Substantial Warriors of Pandemic Covid-19 Battle. Phytomedicine 85, 153361. doi:10.1016/j.phymed.2020.153361
Khater, I., and Nassar, A. (2021). In Silico Molecular Docking Analysis for Repurposing Approved Antiviral Drugs Against SARS-CoV-2 Main Protease. Biochem. Biophys. Rep. 27, 101032.
Kiran, G., Karthik, L., Shree Devi, M. S., Sathiyarajeswaran, P., Kanakavalli, K., Kumar, K. M., et al. (2022). In Silico computational Screening of Kabasura Kudineer - Official Siddha Formulation and JACOM against SARS-CoV-2 Spike Protein. J. Ayurveda Integr. Med. 13 (1), 100324. doi:10.1016/j.jaim.2020.05.009
Kuchi Bhotla, H., Balasubramanian, B., Arumugam, V. A., Pushparaj, K., Easwaran, M., Baskaran, R., et al. (2021). Insinuating Cocktailed Components in Biocompatible-Nanoparticles Could Act as an Impressive Neo-Adjuvant Strategy to Combat COVID-19. Nat. Resour. Hum. Health 1 (1), 3–7. doi:10.53365/nrfhh/140607
Kulkarni, S. A., Nagarajan, S. K., Ramesh, V., Palaniyandi, V., Selvam, S. P., and Madhavan, T. (2020). Computational Evaluation of Major Components from Plant Essential Oils as Potent Inhibitors of SARS-CoV-2 Spike Protein. J. Mol. Struct. 1221, 128823. doi:10.1016/j.molstruc.2020.128823
Kumar, A., Choudhir, G., Shukla, S. K., Sharma, M., Tyagi, P., Bhushan, A., et al. (2021). Identification of Phytochemical Inhibitors against Main Protease of COVID-19 Using Molecular Modeling Approaches. J. Biomol. Struct. Dyn. 39 (10), 3760–3770. doi:10.1080/07391102.2020.1772112
Liao, C., and Nicklaus, M. C. (2009). Comparison of Nine Programs Predicting pKa Values of Pharmaceutical Substances. J. Chem. Inf. Model. 49 (12), 2801–2812. doi:10.1021/ci900289x
Lin, Y., Hu, Z., Alias, H., and Wong, L. P. (2020). Knowledge, Attitudes, Impact, and Anxiety Regarding COVID-19 Infection Among the Public in China. Front. Public Health 8, 236. doi:10.3389/fpubh.2020.00236
Liu, Y.-C., Kuo, R.-L., and Shih, S.-R. (2020). COVID-19: The First Documented Coronavirus Pandemic in History. Biomed. J. 43 (4), 328–333. doi:10.1016/j.bj.2020.04.007
Luan, B., Huynh, T., Cheng, X., Lan, G., and Wang, H.-R. (2020). Targeting Proteases for Treating COVID-19. J. Proteome Res. 19 (11), 4316–4326. doi:10.1021/acs.jproteome.0c00430
Majdi, H., ZarKalai, F., Yeddess, W., and Saidani, M. (2022). Phenolic Compounds as Antiviral Agents: An In-Silico Investigation against Essential Proteins of SARS-CoV-2. Nat. Resour. Hum. Health 2 (1), 62–78. doi:10.53365/nrfhh/143085
Marcelino, R. C., de Araújo, L. P., Bueno de Morais Borba, J., and da Silveira, N. J. F. (2022). Molecular Docking Study Involving Bioactive Natural Compounds against SARS-CoV-2 Proteins. Nat. Resour. Hum. Health. doi:10.53365/nrfhh/147375
Mengist, H. M., Dilnessa, T., and Jin, T. (2021). Structural Basis of Potential Inhibitors Targeting SARS-CoV-2 Main Protease. Front. Chem. 9, 622898. doi:10.3389/fchem.2021.622898
Mhya, D., Nuhu, A., and Mankilik, M. (2021). In-silico Discovery of Antidiabetic Drug Potential of Balanites Aegyptiaca Leaf's Phenolic Compounds. Nat. Resour. Hum. Health 1 (2), 91–97. doi:10.53365/nrfhh/142375
Mittal, A., Manjunath, K., Ranjan, R. K., Kaushik, S., Kumar, S., and Verma, V. (2020). COVID-19 Pandemic: Insights into Structure, Function, and hACE2 Receptor Recognition by SARS-CoV-2. PLoS Pathog. 16 (8), e1008762. doi:10.1371/journal.ppat.1008762
Mohamed, H. S., El-Serwy, W. S., and El-Serwy, W. S. (2021). Synthesis, Molecular Docking, In Silico ADME Predictions, and Toxicity Studies of N-Substituted-5-(4-Chloroquinolin-2-Yl)-1,3,4-Thiadiazol-2-Amine Derivatives as COVID-19 Inhibitors. Russ. J. Bioorg Chem. 47 (1), 158–165. doi:10.1134/S1068162021010155
Mohanraj, K., Karthikeyan, B. S., Vivek-Ananth, R. P., Chand, R. P. B., Aparna, S. R., Mangalapandi, P., et al. (2018). IMPPAT: A Curated Database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci. Rep. 8 (1), 4329. doi:10.1038/s41598-018-22631-z
Mothay, D., and Ramesh, K. v. (2020). Binding Site Analysis of Potential Protease Inhibitors of COVID-19 Using AutoDock. VirusDis. 31 (2), 194–199. doi:10.1007/s13337-020-00585-z
Nair, P. P., Nair, P. G., and Pratap Shankar, K. M. (2022). An Ayurvedic Personalized Prophylactic Protocol in COVID-19. J. Ayurveda Integr. Med. 13 (1), 100351. doi:10.1016/j.jaim.2020.08.004
Narkhede, R. R., Pise, A. v., Cheke, R. S., and Shinde, S. D. (2020). Recognition of Natural Products as Potential Inhibitors of COVID-19 Main Protease (Mpro): In-Silico Evidences. Nat. Prod. Bioprospect. 10 (5), 297–306. doi:10.1007/s13659-020-00253-1
Roos, K., Wu, C., Damm, W., Reboul, M., Stevenson, J. M., Lu, C., et al. (2019). OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 15 (3), 1863–1874. doi:10.1021/acs.jctc.8b01026
Seeliger, D., and de Groot, B. L. (2010). Ligand Docking and Binding Site Analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 24 (5), 417–422. doi:10.1007/s10822-010-9352-6
Sink, T. D., Strange, R. J., and Sawyers, R. E. (2007). Clove Oil Used at Lower Concentrations Is Less Effective Than MS-222 at Reducing Cortisol Stress Responses in Anesthetized Rainbow Trout. North Am. J. Fish. Manag. 27 (1), 156–161. doi:10.1577/m05-103.1
Taylor, P. W., and Roberts, S. D. (1999). Clove Oil: An Alternative Anaesthetic for Aquaculture. North Am. J. Aquac. 61 (2), 150–155. doi:10.1577/1548-8454(1999)061<0150:coaaaf>2.0.co;2
Tongqiang Zhang, T., Cui, X., Zhao, X., Wang, J., Zheng, J., Zheng, G., et al. (2020). Detectable SARS‐CoV‐2 Viral RNA in Feces of Three Children during Recovery Period of COVID‐19 Pneumonia. J. Med. Virol. 92 (7), 909–914. doi:10.1002/jmv.25795
Trott, O., and Olson, A. J. (2010). AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 31 (2), 455–461. doi:10.1002/jcc.21334
Vanshylla, K., Di Cristanziano, V., Kleipass, F., Dewald, F., Schommers, P., Gieselmann, L., et al. (2021). Kinetics and Correlates of the Neutralizing Antibody Response to SARS-CoV-2 Infection in Humans. Cell host microbe 29 (6), 917–929. doi:10.1016/j.chom.2021.04.015
Xu, J.-G., Liu, T., Hu, Q.-P., and Cao, X.-M. (2016). Chemical Composition, Antibacterial Properties and Mechanism of Action of Essential Oil from Clove Buds against Staphylococcus aureus. Molecules 21 (9), 1194. doi:10.3390/molecules21091194
Keywords: clove, casuarictin, phytochemicals, SARS-CoV-2, main protease
Citation: Chandra Manivannan A, Malaisamy A, Eswaran M, Meyyazhagan A, Arumugam VA, Rengasamy KRR, Balasubramanian B and Liu W-C (2022) Evaluation of Clove Phytochemicals as Potential Antiviral Drug Candidates Targeting SARS-CoV-2 Main Protease: Computational Docking, Molecular Dynamics Simulation, and Pharmacokinetic Profiling. Front. Mol. Biosci. 9:918101. doi: 10.3389/fmolb.2022.918101
Received: 12 April 2022; Accepted: 19 May 2022;
Published: 28 June 2022.
Edited by:
Arvind Ramanathan, Argonne National Laboratory (DOE), United StatesReviewed by:
Debsindhu Bhowmik, Oak Ridge National Laboratory (DOE), United StatesKemal Yelekci, Kadir Has University, Turkey
Copyright © 2022 Chandra Manivannan, Malaisamy, Eswaran, Meyyazhagan, Arumugam, Rengasamy, Balasubramanian and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Balamuralikrishnan Balasubramanian, YmFsYS5tLmtAc2Vqb25nLmFjLmty; Wen-Chao Liu, bGl1d2NAZ2RvdS5lZHUuY24=
†These authors have contributed equally to this work