 Karen A. Sap
Karen A. Sap Karlijne W. Geijtenbeek
Karlijne W. Geijtenbeek Sabine Schipper-Krom
Sabine Schipper-Krom Arzu Tugce Guler
Arzu Tugce Guler Eric A. Reits
Eric A. Reits- Department of Medical Biology, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands
Huntington’s disease (HD) is a neurodegenerative disorder caused by a CAG repeat expansion in the N-terminus of the HTT gene. The CAG repeat expansion translates into a polyglutamine expansion in the mutant HTT (mHTT) protein, resulting in intracellular aggregation and neurotoxicity. Lowering the mHTT protein by reducing synthesis or improving degradation would delay or prevent the onset of HD, and the ubiquitin-proteasome system (UPS) could be an important pathway to clear the mHTT proteins prior to aggregation. The UPS is not impaired in HD, and proteasomes can degrade mHTT entirely when HTT is targeted for degradation. However, the mHTT protein is differently ubiquitinated when compared to wild-type HTT (wtHTT), suggesting that the polyQ expansion affects interaction with (de) ubiquitinating enzymes and subsequent targeting for degradation. The soluble mHTT protein is associated with several ubiquitin-modifying enzymes, and various ubiquitin-modifying enzymes have been identified that are linked to Huntington’s disease, either by improving mHTT turnover or affecting overall homeostasis. Here we describe their potential mechanism of action toward improved mHTT targeting towards the proteostasis machinery.
1 Introduction
1.1 Huntington’s disease
Huntington’s disease (HD) is an inherited neurodegenerative disease caused by a CAG repeat expansion in exon1 of the Huntingtin (HTT) gene (MacDonald et al., 1993). The CAG repeat expansion results in a polyglutamine (polyQ) expansion in the HTT protein. N-terminal fragments of the mutant HTT (mHTT) proteins containing the polyQ repeat are aggregation-prone and form intracellular inclusion bodies (IBs) (DiFiglia et al., 1997), as observed in human HD post-mortem brain and in animal or cellular systems. A repeat length of 40 repeats and longer results in HD, whereby the CAG repeat length is inversely correlated with age of onset (Fusilli et al., 2018). The symptoms of HD comprise motor- and cognitive decline and psychological deficits, which aggravate over time, and are lethal approximately 15 years after the onset of the disease. There is no cure for HD. Lowering mHTT by improving its degradation prior to aggregation and toxicity would be a therapeutic strategy to prevent, delay or slow down the onset and progression of disease. The two main intracellular pathways involved in protein degradation are the ubiquitin-proteasome system (UPS) and autophagy. Both pathways play a role in mHTT clearance (Rubinsztein, 2006; Thompson et al., 2009). Although the UPS is active in both the nucleus and the cytoplasm, it is merely capable of degrading unfolded monomeric HTT proteins (Waelter et al., 2001; Li et al., 2010). The autophagic pathway is a cytoplasmic degradation machinery and targets soluble and aggregated HTT proteins for lysosomal destruction (Ravikumar, 2002). The proteasome is not impaired by mHTT fragments, as initially thought, but can degrade mHTT entirely when efficiently targeted towards proteasomes via ubiquitination, and the proteasome can degrade the polyQ sequence itself (Juenemann et al., 2013). In addition, both the proteasome (Schipper-Krom et al., 2014) and ubiquitin (Juenemann et al., 2018) are not sequestered into mHTT aggregates but are dynamically and reversibly recruited. Together, this indicates that the UPS is not impaired in cells expressing mHTT and that efficient targeting of mHTT to proteasomes results in complete degradation. So why is mHTT not efficiently degraded? Recent publications show that the polyQ expansion affects the ubiquitination of mHTT, as shown in different HD mouse and rat models (Sap et al., 2019; Hakim-Eshed et al., 2020), with differences in ubiquitination sites between wtHTT and mHTT. Both HTT degradation and aggregation have been associated with several ubiquitin-modifying enzymes, and we will describe the various ubiquitinating and de-ubiquitinating enzymes that may play a role in mHTT degradation.
1.2 Ubiquitination as a post-translational modification
Ubiquitin is a highly conserved small (8.5 kDa) modular signaling protein that functions as a post-translational modification and can be used to target proteins for degradation. Ubiquitin is attached mainly to internal lysine residues in target proteins (Hershko and Ciechanover, 1998). Additionally, ubiquitin can be attached to target proteins’ N-terminal methionine (M1) (Breitschopf et al., 1998). Ubiquitin can also be a substrate for ubiquitination, where the amino acids M1, K6, K11, K27, K29, K33, K48, and K63 of the ubiquitin protein serve as target residues for polyubiquitin chain formation (Swatek and Komander, 2016). Polyubiquitination with the same linkage type results in polyubiquitin chains, while polyubiquitin branches are formed with mixed linkage types. Polyubiquitin chains and branches formed through K48 and K11 are the “canonical” degradation signals (Jacobson et al., 2009; Min et al., 2015), whereas K63 polyubiquitination plays a role in signaling, protein trafficking, aggregate formation, and autophagy (Tan et al., 2008; Chen and Sun, 2009). Recent insights suggest a “ubiquitination threshold” model for proteasomal degradation where multiple short K48-linked chains or branched structures with K11 and K48 linkages are efficient degradation signals. In this model, the amount of polyubiquitin plays a more important role than the type of polyubiquitin linkage (Swatek and Komander, 2016).
1.3 Ubiquitin-modifying enzymes
Ubiquitin is attached to target proteins through the sequential action of E1 ubiquitin-activating enzymes, E2 ubiquitin-conjugating enzymes, and finally, E3 ubiquitin ligases (Figure 1). Firstly, the E1 enzyme activates the ubiquitin molecule by forming a thiol ester bond between its active site cysteine and the C-terminus of the ubiquitin molecule in an ATP-dependent manner. The E1 enzyme then transfers the activated ubiquitin molecule to the active site cysteine of an E2 enzyme. Subsequently, the E2 enzyme forms a complex with an E3 ubiquitin ligase. The E3 enzyme binds to the target protein and transfers ubiquitin directly or indirectly from the E2 enzyme to the target protein. There are 2 E1, ∼40 E2, and ∼600 E3 enzymes in the ubiquitin pathway (Clague et al., 2015). Deubiquitinating enzymes (DUBs) can remove ubiquitin. There are approximately 100 DUBs (Clague et al., 2019), including DUBs that can remove complete polyubiquitin chains en bloc, DUBs that cleave within chains, and DUBs that remove single ubiquitin molecules from the tip of the polyubiquitin chain (Figure 1).
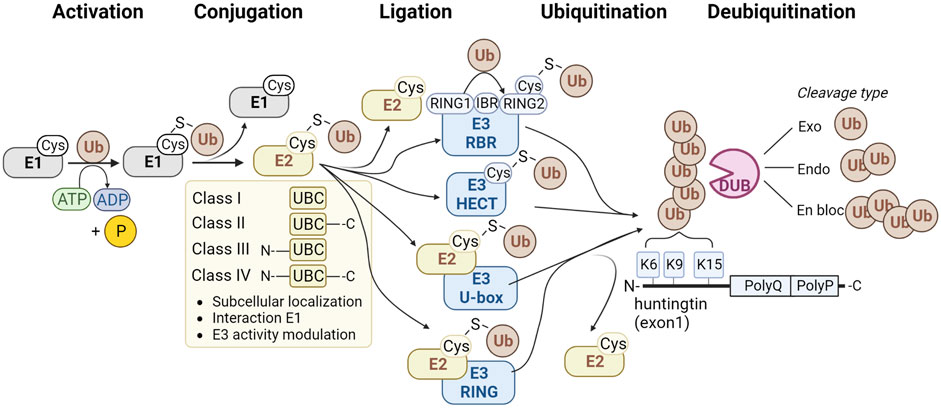
FIGURE 1. Method of action of enzyme classes involved in the ubiquitination pathway. The classes of E1 ubiquitin-activating enzymes are shown (grey), four classes of E2 ubiquitin-conjugating enzymes (yellow), and four families of E3 ubiquitin ligases (blue) and deubiquitinating enzymes (pink). Ubiquitination sites K6, K9 and K15 at the N-terminus of HTT are shown at the right.
1.3.1 E1 ubiquitin-activating enzymes
The E1 family consists of eight members in human, of which two can activate ubiquitin, and the others activate different Ubiquitin-like (UBL) proteins. UBA1 and UBA6 can activate ubiquitin, and contain an N-terminal adenylation domain to bind ATP and ubiquitin, a catalytic cysteine domain, and a C-terminal ubiquitin-fold domain for E2 recruitment. E1 enzymes function on the apex of the ubiquitination pathway to activate ubiquitin with the use of ATP and transfer it to the E2 enzyme (Figure 1). UBA1 seems to play a more general role in activating ubiquitin compared to UBA6. UBA1, for instance, is ∼ ten times more abundant than UBA6 (Yang et al., 2013; Kulak et al., 2014), and this pool is fully charged with ubiquitin, while UBA6 is charged for ∼50% and shows specificity for E2 enzyme UBE2Z/USE1 (Jin et al., 2007) which is involved in targeting proteins with destabilized N-terminal amino acids for degradation (Lee et al., 2011).
1.3.2 E2 ubiquitin-conjugating enzymes
The human genome comprises approximately 40 E2 enzymes (Y. Ye and Rape, 2009). All E2s share a common catalytic core domain, i.e., the Ubiquitin Conjugation (UBC) domain (Wijk and Timmers, 2010), which forms a thioester bond with ubiquitin. The E2 superfamily is divided into four classes (Figure 1). Class I E2 enzymes only contain the UBC domain, while class II E2 enzymes contain the UBC domain plus a C-terminal extension. The class III E2 enzymes contain the UBC domain and an N-terminal extension. Class IV E2 enzymes contain a UBC domain plus an N- and C-terminal extension (Burroughs et al., 2008; Michelle et al., 2009). The different classes show functional diversity and can affect the subcellular localization or modulate the interaction with E1 and E3 enzymes. There is some cross-reactivity among E2 and E3 enzymes, as the same E3 can interact with various E2 enzymes and vice versa, which could be RING and HECT enzymes. Structural characteristics and function of E2 enzymes are reviewed elsewhere in more detail (Wijk and Timmers, 2010).
1.3.3 E3 ubiquitin ligases
E3 ubiquitin ligases are enzymes that recognize substrates that need to be ubiquitinated. Based on differences in structure, E3 ubiquitin ligases can be divided into four types: HECT type, RING-finger type, U-box type, and RBR type (Figure 1).
1.3.3.1 HECT E3 ligases
The human HECT E3 ligase family consists of 28 members, which all vary in their N-terminal domain, used for substrate recognition (Sluimer and Distel, 2018), while they all share a conserved homologous to E6-AP C-terminus domain, or HECT domain, used to bind ubiquitin. Based on the types of N-terminal domains, the HECT E3 ligases can be divided into three major subfamilies: the NEDD4 subfamily with nine members, the HERC subfamily with six members, and the group of ‘other HECTs’ with thirteen members (Rotin and Kumar, 2009). The regulation of the HECT E3 enzymes has been reviewed elsewhere (Sluimer and Distel, 2018). HECT E3 ligases receive a ubiquitin molecule from an E2 enzyme. Next, the ubiquitin-loaded HECT E3 enzyme interacts with the substrate proteins via their N-termini and transfers ubiquitin to the substrates. In contrast, RING-finger E3 ligases do not receive the ubiquitin molecule from the E2 enzyme. RING-finger E3 ligases are described below.
1.3.3.2 RING-finger E3 ubiquitin ligases
The Really Interesting New Gene (RING)-finger E3 ubiquitin ligases comprise, with over 600 members, the largest family of E3 ubiquitin ligases. RING-finger E3s share a RING domain, which is used to interact with E2 enzymes, but this domain does not interact with ubiquitin as it lacks a bona fide catalytic center. Instead, the ubiquitin molecule is transferred directly from the E2 onto the substrate. RING-finger E3 ubiquitin ligases thus function as scaffolds between the E2 and the substrate. Many variations exist between RING-finger E3s as they could function as monomeric proteins but could also be part of large multi-subunit complexes, such as cullin-RING ligases and the anaphase-promoting complex/cyclosome (APC/C) complex. Also, the substrates of RING-finger E3s are highly varied, as they might recognize multiple different substrates, and various E3 ubiquitin ligases could also recognize a specific substrate. The ligase activity and substrate recognition can be modulated via post-translational modifications. RING-type E3 ligases have been reviewed in more detail elsewhere (Metzger et al., 2014).
1.3.3.3 U-box E3 ligases
The number of U-box ubiquitin ligases varies widely per species, with 21 U-box proteins in humans, 101 in Chinese cabbage B. rapa (Wang et al., 2015) and 125 in soybean Glycine max (Wang et al., 2016). The C-terminal U-box domain is conserved from yeast to humans and has a three-dimensional structure similar to the RING-finger domain but lacks the metal chelating residues (Aravind and Koonin, 2000). Similar to RING-finger ubiquitin ligases, the U-box domain is used to interact with E2s, and ubiquitin is directly transferred from the E2 enzyme to the substrate. U-box E3 ligases are often involved in polyubiquitination and some can elongate polyubiquitin chains and are also called E4 ubiquitin ligases. Also, some U-box E3s interact with chaperones to play a role in the protein quality control system. U-box type E3 ubiquitin ligases are reviewed in more detail elsewhere (Hatakeyama and Nakayama, 2003).
1.3.3.4 RBR E3 ligases
Finally, the fourth type of E3 ubiquitin ligases comprises the RING-between-RING (RBR) E3s, and this family has 14 members in humans. RBR ligases contain a RING1 domain, followed by a central in-between-RINGs (IBR) domain and a RING2 domain. The RING1 domain recruits the ubiquitin-loaded E2 and transfers ubiquitin to the catalytic cysteine in the RING2 domain, which transfers it to the substrate (Spratt et al., 2014). Since RBR ligases contain two RING domains but a HECT-type E3 mechanism of work, these enzymes are also called RING-HECT hybrids. Examples of RBR E3 ligases are Parkin (PRKN), involved in Parkinson’s disease, and HOIP, a member of the linear ubiquitin chain assembly complex (LUBAC), responsible for assembling M1-linked linear Ub chains. RBR ligases have been reviewed in more detail elsewhere (Walden and Rittinger, 2018).
1.3.4 Deubiquitinating enzymes
The ubiquitination of protein substrates like HTT becomes reversible through the action of deubiquitinating enzymes (DUBs), which can cleave off ubiquitin in two ways. They can either recognize and target specific protein interaction domains or show specificity for particular polyubiquitin linkage types. The latter type of DUB may hydrolyze polyubiquitin chains but leave the proximal ubiquitin on the substrate. Linkage specificity is determined by the catalytic domain, ubiquitin-binding domains, or by the specificity of interacting proteins. Apart from linkage selectivity, DUBS could also show selectivity in removing off ubiquitin moieties from the end of polyubiquitin chains or cleaving within chains. There are seven different families of DUBs. Six of them are classified as cysteine proteases: USPs, UCHs, OTUs, MJDs (also known as Josephins), MINDYs, and ZUP1. The seventh family is the JAMM family, also known as MPN, and is classified as metalloproteinase. DUB specificity and regulation have been reviewed elsewhere in more detail (Mevissen and Komander, 2017).
1.4 HTT ubiquitination sites
Aberrant splicing and protein cleavage can generate different mHTT protein fragments, including the N-terminal exon1 fragment containing the polyQ expansion. These fragments were shown to be highly pathogenic in HD mouse models and are also observed in fibrillar aggregates in brains of HD patients (DiFiglia et al., 1997; Schilling et al., 2007; Landles et al., 2010). The mHTT fragments can be present as monomers, soluble oligomers, or insoluble aggregates, including the insoluble IBs. Especially soluble oligomeric mHTT species appear to be most toxic to the cell. IBs seem to have a protective role by sequestering toxic oligomeric mHTT species (Arrasate et al., 2004). However, IB formation also results in the sequestration of other proteins from their normal cellular environment, thereby interfering with important processes, including transcriptional regulation (Schaffar et al., 2004) and proteostasis (Park et al., 2013). The N-terminus of the HTT exon1 contains 17 amino acids, including three lysine residues, followed by the polyQ region and a polyproline region (Finkbeiner, 2011). Interestingly, various post-translational modifications (PTMs) of the N17 domain of HTT have been described, including acetylation, SUMOylation, and ubiquitination at lysines 6, 9, and 15 and phosphorylation at threonine 3, serine 13 and serine 16 that affect aggregation, toxicity and degradation (Steffan et al., 2004; Thompson et al., 2009; Maiuri et al., 2013; DeGuire et al., 2018). Within full-length HTT, most PTMs can be found in clusters within predicted unstructured domains but not in the structured HEAT repeats (Arbez et al., 2017). When compared to the many identified phosphorylation and acetylation sites, only nine ubiquitination sites (K6, K9, K15, K132, K337, K631, K804, K837, and K2097) have been identified for HTT (Steffan et al., 2004; Juenemann et al., 2015; Yau et al., 2017; Koyuncu et al., 2018; Sap et al., 2019; Hakim-Eshed et al., 2020). Ubiquitination at K6 and K9 was specific for mHTT in mice and rat brains (Sap et al., 2019; Hakim-Eshed et al., 2020), indicating that polyQ expansion in the HTT protein affects ubiquitination. Differential ubiquitination of wtHTT and mHTT could be the result of different interactions and affinities for (de) ubiquitinating enzymes. In addition, both the proteome and ubiquitinome are changed, including numerous components of the involved proteostasis pathways (Sap et al., 2019). Expression of mHTT exon1 lacking these ubiquitination sites in cells was found to slow down aggregation and reduce IB size. However, it increased both toxicity and the number of much smaller aggregates, suggesting that ubiquitination of the N17 domain of HTT by yet unidentified enzymes affects both aggregation and toxicity (Hakim-Eshed et al., 2020). Studies focusing on soluble and insoluble HTT ubiquitination have been previously reviewed in more detail (Sap and Reits, 2020).
2 Ubiquitin-modifying enzymes linked to HD
Several ubiquitin ligases and DUBs have been linked to HD. These enzymes might be potential therapeutic targets to facilitate the degradation of mHTT by the proteostasis machinery prior to aggregation. Here we give some background information about the enzymes and describe how these enzymes were linked to HD. For instance, the enzymes might affect proteasome-dependent degradation of mHTT, or affect solubilization, polymerization, or aggregation of mHTT. Eventually, the ubiquitin-modifying enzymes could affect cellular pathways and cellular stress that affect HD pathogenicity directly or indirectly. We also describe whether the ubiquitin-modifying enzymes might be a potential target for HD therapy. We start with discussing the involved E1 enzyme, followed by E2 enzymes, and finally E3 enzymes, which are categorized by type, starting with HECT E3s, followed by RING-finger E3s, RBR E3, and finally U-box E3s. The DUBs are described per class and USP type of DUBs in alphabetical order. The effects of these ubiquitin-modifying enzymes on mHTT levels, interactions, aggregation and pathology are summarized in Table 1 and in more detail in Supplementary Table S1. Finally, we discuss difficulties in comparing HD studies, based on different mHTT Q-lengths, fragment lengths, and tags used, but also different methods available to study soluble and insoluble mHTT protein levels and different models of disease.
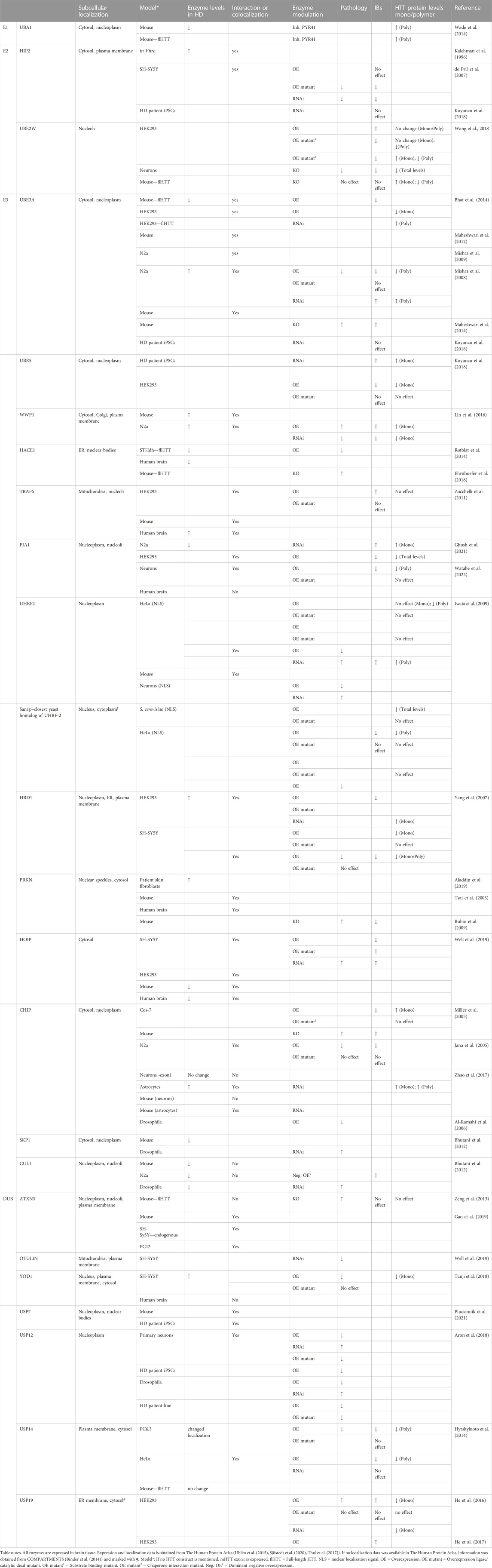
TABLE 1. Effects of ubiquitin-modifying enzymes on cellular mHTT and HD pathology.
2.1 E1, E2 and E3 ubiquitinating enzymes
2.1.1 UBA1
Ubiquitin-activating enzyme E1 (UBA1, also known as UBE1) is an E1 ubiquitin-activating enzyme and catalyzes the first step of the enzymatic ubiquitination cascade; namely activation of the ubiquitin molecules and subsequent transfer to E2 enzymes. Therefore, UBA1 plays a key role in the regulation of ubiquitin homeostasis, hence ubiquitination is involved in all downstream cellular processes, including protein degradation via the UPS. Nollen and colleagues performed a genome-wide RNA interference screen in C. elegans and identified UBA1 as one of the genes that increased polyQ-peptide aggregation upon knockdown (Nollen et al., 2004). Wade and colleagues studied UBA1 concerning the mHTT protein and found that E1 inhibition in brain lysate resulted in increased levels of oligomerized mHTT. Moreover, expression levels of endogenous UBA1 were decreased in striatum and cortex of HD mice, compared to cerebellum and periphery. In addition, UBA1 levels decreased with aging, suggesting that the decrease in enzyme levels contribute to the age-dependent onset of the disease (Wade et al., 2014). Despite the decrease in UBA1 levels during HD, an increase in ubiquitinated proteins was observed when brain lysate was incubated with mHTT. However, it is unclear whether this is a result of increased ubiquitination or decreased clearance (Wade et al., 2014). While UBA1 may play a role in polyQ-peptide and mHTT aggregation, it remains unclear whether UBA1 directly improves the turnover of HTT levels or whether the effect is indirectly mediated by other pathways, given the broad role of UBA1 in cellular pathways. The involvement of an E1 in many processes would limit UBA1 as a suitable target for HD treatment, as modulation of its activity would lead to too many side effects.
2.1.2 HIP2
Ubiquitin-conjugating enzyme HTT Interaction Protein 2 (HIP2, also known as E2-25K/UBE2K) belongs to the class II E2 enzymes, which contain a Ubiquitin-Associated (UBA) domain C-terminal to the UBC domain. A UBA domain binds to ubiquitin, polyubiquitin chains, and ubiquitinated proteins. HIP2 can elongate K48-linked polyubiquitin chains and targets substrates for degradation (Chen and Pickart, 1990), yet the UBA domain of HIP2 preferentially binds to Lys63 polyubiquitin chains and facilitates the selective assembly of branched Lys48/Lys63 ubiquitin structures. In this way, HIP2 can target Lys63 polyubiquitinated substrates for proteasomal degradation via the formation of Lys63/Lys48-linked polyubiquitin signals (Pluska et al., 2021). HIP2 interacts with both wtHTT (Q16) and mHTT (Q44), as was shown with an in vitro binding assay (Kalchman et al., 1996). Colocalization studies suggest an interaction between HTT IBs and HIP2 in SH-SY5Y cells and post-mortem brain tissue, indicating that HIP2 colocalizes with HTT (de Pril et al., 2007). While knockdown of the HIP2 protein or deletion of its catalytic domain reduced mHTT IBs and polyQ-induced cell death in SH-Sy5Y cells, overexpression of HIP2 did not affect mHTT aggregation (de Pril et al., 2007), suggesting that HIP2 is not a limiting factor in controlling mHTT aggregation. Strikingly, knockdown of HIP2 in patient-derived induced pluripotent stem cells (iPSCs) had no effect on the number of mHTT IBs (Koyuncu et al., 2018). Since HIP2 does not colocalize with Q81-YFP IBs (Howard et al., 2007), this suggests that HIP2 requires the HTT protein sequence for interaction, not solely the polyQ tract that lacks lysine residues for ubiquitination. In line with this, silencing of HIP2 resulted in an increase in polyQ-peptide aggregation, yet the number of aggregates remained the same in HEK293 cells, and no change in polyQ solubility or overall levels was observed in a polyQ peptide C. elegans model (Howard et al., 2007). The discrepancy in colocalization between HIP2 and polyQ-peptides or mHTT suggests that HIP2 acts differently on polyQ-peptides and mHTT protein, and it needs to be confirmed whether HIP2 affects aggregate formation indirectly or can directly target mHTT and affect turnover.
2.1.3 UBE2W
UBE2W is a class I E2 enzyme expressed in the nucleus. UBE2W generally does not transfer ubiquitin to free lysine residues, but instead prefers to specifically mono-ubiquitinate the N-terminal α-amino group of various proteins and specifically targets proteins with disordered N-termini independent of the amino acid sequence (Scaglione et al., 2013; Tatham et al., 2013; Vittal et al., 2015). At least 13 target proteins are known, most of which are targeted for proteasomal degradation by N-terminal ubiquitination via K48-linked polyubiquitin. Since the HTT N-terminus is also largely unstructured and disordered (Baias et al., 2017), Wang and colleagues tested the effect of UBE2W deficiency on the HD phenotype in different models. Overexpression of UBE2W in HEK cells increased aggregation, while overexpression of mutant UBE2W decreased aggregation by affecting mHTT solubility, suggesting that its catalytic activity affects mHTT aggregation directly. In line with this, UBE2W KO mouse primary cortical neurons showed reduced aggregation and decreased cell death (Wang et al., 2018), which may be the result of decreased toxic oligomer levels. While UBE2W knockout in a mHTT KI mouse model also led to a shift from oligomers to soluble HTT, it did not alter aggregation levels nor resulted in improved striatal functioning (Wang et al., 2018). Since no increase in ubiquitin levels was found in IBs and no changes in protein quality control pathways were observed in these mice, the mechanism by which UBE2W enhances mHTT solubility is still unclear. The authors suggest that UBE2W could promote aggregation by stabilizing HTT directly by ubiquitination or indirectly by increasing the probability of other post-translational modifications. UBE2W could also exert its effects by regulating other proteins that modify HTT, or the mechanism could be independent of ubiquitination (Wang et al., 2018).
2.1.4 UBE3A
Ubiquitin ligase E3A (UBE3A), also known as E6AP, is a HECT domain E3 ubiquitin ligase with a zinc-binding fold called the AZUL (amino-terminal Zn-finger of UBE3A ligase) domain. UBE3A assembles K48-linked polyubiquitin chains (Wang and Pickart, 2005) and targets proteins, including polyQ proteins, for degradation (Mishra et al., 2008). UBE3A plays a role in synaptic plasticity (Maheshwari et al., 2012) and loss-of-function mutations in the UBE3A gene are associated with Angelman syndrome and Prader-Willi syndrome. In HD cell and mouse models, UBE3A interacts with both soluble and insoluble wtHTT and mHTT, but prefers to interact with longer polyQ-length repeats (Mishra et al., 2008; Bhat et al., 2014). In addition, UBE3A colocalizes with mHTT aggregates (Mishra et al., 2008; Mishra et al., 2009; Maheshwari et al., 2012; Bhat et al., 2014). While the levels of the UBE3A protein decrease in both WT and HD mice brain during aging, this decrease in nuclear UBE3A is more prominent in HD mice than in WT. Consequently, the interaction between UBE3A and the HTT protein is decreased in aged mice (Bhat et al., 2014). In contrast, UBE3A mRNA levels are increased in mHTT-expressing N2A cells (Mishra et al., 2008). Since the decrease in protein levels was observed in aging animals, this discrepancy could result from the lack of aging in immortalized cell lines. Alternatively, mRNA levels might not correlate with protein levels with UBE3A being decreased by post-transcriptional regulation (Mishra et al., 2008). Overexpression of UBE3A reduces mHTT aggregation (Bhat et al., 2014) and promotes mHTT degradation via the proteasome, in accordance with its role in K48-linked ubiquitination (Bhat et al., 2014; Maheshwari et al., 2014), and in parallel K63-linked ubiquitination was reduced (Bhat et al., 2014). Knockdown of UBE3A enhances aggregation (Mishra et al., 2008; Bhat et al., 2014; Maheshwari et al., 2014) and reduces ubiquitination of mHTT aggregates (Maheshwari et al., 2014). In addition, UBE3A knockdown accelerated the HD phenotype and reduced the life span in mice. In line with this, UBE3A overexpression reduced cell death in mHTT-expressing cells (Mishra et al., 2008). Taken together, UBE3A appears to be an important modulator of HD by improving HTT degradation by the proteasome, and maintaining sufficient levels of UBE3A during aging may modify mHTT levels and disease onset in HD patients.
2.1.5 UBR5
UBR5 or EDD1/HHYD/KIAA0896/DD5 is a HECT domain E3 ubiquitin-ligase highly conserved in metazoans and has been implicated in a wide range of cellular processes, including DNA damage response, metabolism, transcription, and apoptosis. UBR5 contains UBA, UBR, and MLLE/PABC domains, with the UBA domain being used to interact with ubiquitin (Kozlov et al., 2007), the zinc finger Ubiquitin Recognin Box (UBR) domain being involved in N-end rule substrate recognition (Tasaki et al., 2005), and the MLLE/PACB domain (homologous to the C-terminal region of Poly-Adenylation Binding Protein) being used for protein-protein interactions and may regulate the transfer of ubiquitin to the substrate (Muńoz-Escobar et al., 2015). UBR5 has been identified as a possible mediator of the age of onset of HD in Genome Wide Association Studies (GWAS) (Lee et al., 2015). Patient-derived iPSCs show no mHTT aggregates or inclusion bodies being formed in time (Noormohammadi et al., 2016), suggesting that iPSCs exhibit an improved proteostasis regulation compared to other cell types. Interestingly, these iPSCs express increased levels of UBR5, which may contribute to the proposed improved proteostasis. Knockdown of UBR5 increased soluble HTT levels and triggered aggregate formation in mHTT-expressing iPSCs. Furthermore, knockdown of UBR5 in an invertebrate polyQ aggregation model also increased aggregation and neurotoxicity, while overexpression of UBR5 reduced aggregate formation in HD-cell models by inducing polyubiquitination and proteasomal degradation (Koyuncu et al., 2018). These results suggest an important role for UBR5 in the regulation of HTT turnover, and the increased ubiquitination of mHTT suggests that direct modifications by UBR5 are responsible for the improved degradation. Indeed, UBR5 is found to be involved in K11/K48-linked ubiquitination of mHTT (Yau et al., 2017). However, UBR5 can also regulate expression levels of UBE3A via polyubiquitination (Tomaić et al., 2011). Since UBE3A is associated with improved HTT degradation, as discussed above, UBR5 overexpression may counteract the beneficial effects of UBE3A.
2.1.6 WWP1 and NEDD4
WWP1 (WW domain-containing E3 ubiquitin protein ligase 1) is a multifunctional HECT domain E3 ubiquitin-ligase and belongs to the NEDD4 subfamily. WWP1 contains an N-terminal C2 domain for membrane and protein binding, four tandem WW binding domains for substrate binding, and a C-terminal catalytic HECT domain for ubiquitin-protein ligase activity for both mono- and polyubiquitination (Courivaud et al., 2015). WWP1 predominantly binds to endosomes (Chen et al., 2008) and regulates various cellular biological processes, including transcription, protein transport, signal transduction, protein degradation, and viral budding. WWP1 has been implicated in aging and several diseases, such as cancers, infectious diseases, and neurological diseases (Zhi and Chen, 2012; Kuang et al., 2021). WWP1 protein levels were elevated in mice and N2a cells expressing mHTT, with WWP1 being recruited to aggregates. Overexpression of WWP1 in N2a cells resulted in increased soluble and insoluble mHTT levels and cell death, while downregulation enhanced cell viability and reduced mHTT levels. Furthermore, overexpression of WWP1 resulted in increased K63-ubiquitination of mHTT in cells, while downregulation decreased ubiquitination and improved degradation by the proteasome (Lin et al., 2016). Together these experiments show that WWP1 is involved in K63-linked ubiquitination and thereby inhibits proteasome-mediated HTT degradation, which depends on K48-linked ubiquitination. NEDD4 is an E3 ubiquitin ligase with the same yeast ortholog as WWP1 (Rsp5). Despite this, NEDD4 does not affect mHTT in the same manner as WWP1. While WWP1 increased soluble mHTT levels, NEDD4 did not alter soluble protein levels. Silencing of NEDD4 in HEK cells led to an increase in small aggregates (Peters et al., 2018). The effects of NEDD4 on total aggregation still need to be resolved.
2.1.7 HACE1
HACE1 (HECT domain and Ankyrin repeat Containing E3 ubiquitin-protein ligase 1) is a HECT-type E3 that contains six Ankyrin repeats at the N-terminus and a C-terminal HECT domain. Ankyrin repeats are relatively common protein domains of ∼33 amino acids in length. These repeats act as scaffolds to facilitate specific protein-protein interactions through variable surface exposure (Mosavi et al., 2004). HACE1 is highly expressed in neuronal and glial cells and is involved in Golgi membrane fusion, the regulation of small GTPases, and protein degradation. Furthermore, HACE1 promotes the stability NRF2 (Da et al., 2021), which is an important regulator of the antioxidative stress response (Ma, 2013). Rotblat and colleagues found that HACE1 is reduced in the striatum of HD patients. However, this reduction was not observed in another highly affected region, the cortex. Expression of mHTT led to reduced HACE1 mRNA in a mouse striatal cell line. HACE1 overexpression made mHTT-expressing striatal cells less sensitive to oxidative stress. This effect was independent of HACE1s E3 ligase activity but rather due to NRF2 induction (Rotblat et al., 2014). NRF2 was previously marked to be involved in mHTT degradation (van Roon-Mom et al., 2008). YAC128 HACE1 knockout mice show an impaired anti-oxidative stress response. Moreover, these mice present with aggravated motor and psychiatric deficits (Ehrnhoefer et al., 2018). However, it must be noted that, although not statistically significant, HACE1 knockout leads to phenotypic impairments. It can, therefore, not be excluded that the aggravated phenotype is a result of the accumulation of the different impairments caused by mHTT expression and HACE1 knockout. Taken together, HACE1 levels decrease in the striatum during HD and due to its role in protecting against HTT-induced oxidative stress, the decrease in HACE1 levels could be responsible for the increased pathology in the striatum.
2.1.8 TRAF6
TNF receptor-Associated Factor 6 (TRAF6) is a RING E3 ligase and member of the TRAF protein family and contains an N-terminal RING domain, four zinc finger (ZF) motifs, and a C-terminal TRAF domain. The N-terminal RING and ZF1 domains are used for K63 polyubiquitination (Yin et al., 2009). The TRAF domain consists of two distinct subdomains: the TRAF N-domain, which is a coiled-coil domain, and a highly conserved TRAF-C domain (Yin et al., 2009). The TRAF-C domain is responsible for protein-protein interactions (H. Ye et al., 2002). TRAF6 promotes polyubiquitination of its substrates (Yin et al., 2009), and is not limited to specific linkage types. It has been shown to promote K63-polyubiquitination (Geetha et al., 2005; Linares et al., 2013), K48/K63 polyubiquitination of ATG9A (Wang et al., 2022), as well as a-typical polyubiquitination via K6-, K27, and K29-linkages of proteins involved in Parkinson’s disease (Zucchelli et al., 2010). TRAF6 interacts with wtHTT and both soluble and aggregated mHTT in HEK cells, mice- and human cortex. In addition, TRAF6 mRNA and protein levels were increased in post-mortem cortex tissue of HD patients (Zucchelli et al., 2011). Overexpression of TRAF6 in HEK cells led to increased aggregation number and size but did not alter total HTT levels. Moreover, overexpression did not affect wtHTT localization. TRAF6 enhanced the ubiquitination of wtHTT and mHTT with atypical ubiquitin chains linked at K6, K27, and K29 (Zucchelli et al., 2011). Despite the role of TRAF6 may not be specific for mHTT, further research needs to be performed on the role of this aberrant ubiquitination in HTT clearance.
2.1.9 PJA1
PJA1 or PRAJA1/RNF70 is a RING ubiquitin ligase with yet unknown function. It is expressed in various tissues with the highest expression levels found in brain and has been associated with neurodevelopmental disorders, such as epilepsy and/or craniofacial abnormalities (Suzuki et al., 2020). The expression of both wtHTT and mHTT leads to decreased mRNA and protein levels of PJA1. While PJA1 interacts and colocalizes with wtHTT and mHTT in cell models (Ghosh et al., 2021; Watabe et al., 2022) aggregates in post-mortem brain samples of HD patients stained negative for PJA1 (Watabe et al., 2022). Overexpression of PJA1 reduced mHTT aggregation and soluble levels of wtHTT (Ghosh et al., 2021; Watabe et al., 2022). Silencing of PJA1, on the other hand, increased both soluble and aggregated mHTT (Ghosh et al., 2018). Although not tested for HD, PJA1 lowers toxicity in a yeast and Drosophila SCA3 model, another polyQ aggregation disorder. To conclude, there seems to be a link between PJA1 and HTT, irrespective of the polyQ repeat length. However, the exact role in HD and the affected mechanism remains to be resolved.
2.1.10 UHRF2
Recently it was shown that cytoplasmic and nuclear aggregates of mHTT have a different biochemical composition, as well as different interactome and structural properties, which suggests that different mechanisms drive aggregate formation, clearance, and toxicity, depending on the subcellular localization (Riguet et al., 2021). When the Protein Quality Control (PQC) system was studied in the nucleus, overexpression of yeast nuclear E3 ligase San1p in yeast and HeLa cells increased cell viability and reduced aggregation of nuclear expressed mHTT by decreasing its half-life. Additionally, total protein levels of wtHTT were also lowered by San1p overexpression (Iwata et al., 2009). Previously it was already found that San1p mediated the ubiquitination of aberrant nuclear proteins leading to their proteasomal degradation. San1p only targeted the mutant aberrant proteins, not the wild-type counterparts, suggesting that San1p is part of a protein quality control system in the nucleus (Gardner et al., 2005). The E3 ubiquitin ligase Ubiquitin Like with PHD and RING Finger Domains 2 (UHRF2) is the closest mammalian homolog of San1p and is a nuclear protein involved in DNA methylation, histone modifications, cell cycle regulation, and DNA repair, and can target proteins for degradation by the proteasome. UHRF2 contains an N-terminal ubiquitin-like domain (UBL) domain, a Tandem-Tudor domain (TTD) and Plant Homeodomain (PHD) for the recognition of histone modifications, a SET and RING associated domain (SRA) for DNA binding, and a RING domain for E3 ubiquitin ligase activity. Similar to San1p, UHRF2 colocalizes with nuclear mHTT aggregates in mammalian cells and R6/2 mice. In addition, overexpression of UHRF2 in cells reduced neurotoxicity and cell death, and reduced nuclear aggregation by decreasing mHTT and wtHTT half-life. Furthermore, increased ubiquitination of mHTT was found after UHRF2 overexpression (Iwata et al., 2009). This data suggests that UHRF2 is involved in HTT turnover, irrespective of its polyQ repeat length, by promoting its ubiquitination. The route of degradation is not investigated.
2.1.11 HRD1
HRD1/Der1 is an ER membrane-spanning E3 ubiquitin ligase that contains five transmembrane domains and a RING finger domain that is facing the cytoplasm, and is induced by the unfolded protein response pathway (UPR) and mediates the degradation of misfolded proteins in ER-associated protein degradation (ERAD). In ERAD, HRD1 functions as a pore and can regulate the transport of misfolded proteins from the ER lumen to the cytoplasm via ubiquitination. Auto-ubiquitination of the HRD1 RING domain generates a high-affinity substrate binding site on the cytoplasmic side of HRD1, where it ubiquitinates these substrates (Bordallo et al., 1998; Vasic et al., 2020). Next, p97/VCP pulls the substrate out of the membrane and hence the substrate can be degraded by the proteasome. Cells overexpressing mHTT exhibit increased levels of HRD1 protein, but not mRNA (Yang et al., 2007), suggesting post-transcriptional regulation of protein levels. By immunoprecipitation, it was shown that HRD1 interacts with both wtHTT and mHTT. Moreover, microscopy assays show that HRD1 recruits HTT to the ER. While HRD1 knockdown increased soluble mHTT levels, overexpression decreased mHTT aggregates and soluble levels, in a polyQ-length dependent manner. The decrease in mHTT half-life depended on the HRD1 RING domain and the ATPase VCP/p97 (Yang et al., 2007). Although HRD1 interacts with both wtHTT and mHTT, improved ubiquitination was only observed in HTT with high polyQ repeat lengths (Yang et al., 2007). HRD1 improved HTT degradation via improved ubiquitination and protected against mHTT-induced cell death, but the exact mechanism needs further study.
2.1.12 Parkin
Parkin (PRKN) is a RING-BETWEEN-RING (RBR) E3 ligase that is located in the cytoplasm. It contains an N-terminal UBL domain and four zinc-coordinating RING-like domains: RING0, RING1, IBR, and RING2. The UBL domain controls cellular PRKN levels and activity in addition to binding to SH3-, ubiquitin interacting motif (IUM) domains and the proteasome complex (Finney et al., 2003; Sakata et al., 2003; Trempe et al., 2009; Chaugule et al., 2011). Polyubiquitin chains formed by PRKN include K6, K11, K48, and K63 linkages (Ordureau et al., 2014). PRKN is upregulated during the UPR and plays a role in the degradation of misfolded ER proteins via the ERAD pathway (Imai et al., 2000). It interacts with HSP70 and ubiquitin ligase CHIP, and in turn, CHIP positively regulates the E3 ligase activity of PRKN (Imai et al., 2002). The E3 activity of PRKN is generally inhibited but is activated by recruitment to the mitochondrial outer membrane (MOM) in response to mitochondrial depolarization (Narendra et al., 2008; Matsuda et al., 2010). Activated PRKN ubiquitinates primarily MOM proteins and promotes their degradation via the UPS (Chan et al., 2011). Increased mRNA and protein levels of PRKN were observed in juvenile HD fibroblasts (Q68 and Q86) (Aladdin et al., 2019), and PRKN colocalizes with mHTT inclusion bodies in the brain of mHTT-Q72 YAC72 mice and with both cytoplasmic and nuclear inclusions in human brain. Co-immunoprecipitation also showed an interaction between PRKN and mHTT, but not with wtHTT, suggesting a preference for PRKN for polyQ-expanded HTT (Tsai et al., 2003). When the HD R6/1 mouse model was crossed with PRKN null mice to study the effect of partial suppression of PRKN on HD, a decreased number of aggregates in the striatum of R6/1/PK+/− mice was observed, while the effect was less severe or absent in other brain regions. Despite the reduction in aggregation, increased striatal cell death and exacerbated motor and behavioral deficits were observed (Rubio et al., 2009). This could indicate that mHTT moves from an aggregation state towards a state with more toxic polymers after PRKN knockdown. Since PRKN knockdown leads to increased LC3II/LC3I ratios, it was suggested that the autophagy pathway becomes more active and may be responsible for the decreased aggregation (Rubio et al., 2009).
2.1.13 HOIP and OTULIN
The E3 ubiquitin ligase HOIP belongs to the RBR family and is the only known E3 ligase that can assemble linear M1-linked polyubiquitin chains and functions as part of the LUBAC complex (Kirisako et al., 2006). HOIP uses its RING2 domain and a newly identified Linear ubiquitin chain Determining Domain (LDD) in its C-terminus to assemble linear ubiquitin chains (Smit et al., 2012). LUBAC assembles linear ubiquitin chains, preferably on pre-existing K63-linked polyubiquitin chains and possibly on other polyubiquitin linkage types (Emmerich et al., 2013). Linear polyubiquitin plays a role in various cellular processes, including immune signaling, defense against intracellular pathogens, protein quality control, and cell death regulation (Iwai et al., 2014; Noad et al., 2017; Well et al., 2019). Aggregates and oligomeric aggregate precursors of mHTT were found to be ubiquitinated with linear ubiquitin and contained several components of the LUBAC complex, including HOIP, HOIL-1L, and SHARPIN (Well et al., 2019). HOIP was recruited to mHTT by p97/VCP, resulting in linear polyubiquitin formation covering the interactive surface on mHTT. Silencing of HOIP reduced mHTT linear ubiquitination and increased the interaction of mHTT with transcription factor Sp1, which resulted in transcriptional dysregulation of LUBAC complex components. HOIP silencing also increased aggregation and proteotoxicity. These effects were counteracted by silencing OTULIN, which is the only described mammalian DUB that specifically hydrolyzes linear polyubiquitin (Keusekotten et al., 2013). Linear ubiquitination also facilitated proteasome-dependent degradation of mHTT (Well et al., 2019). Linear polyubiquitination thus seems to have a protective effect in HD where it decreases the misfolded protein’s toxic potential and stimulates the removal via the proteasome. Since linear ubiquitination is a very specific process, it might be a promising target for drug development for HD and other protein aggregation-based diseases (Well et al., 2019).
2.1.14 CHIP
The C-terminus of HSP70-interacting protein (CHIP) is a U-box ubiquitin ligase (UULs) that contains a U-box domain structurally related to the RING finger domain. CHIP is an E3 ubiquitin ligase that links the protein folding machinery with the UPS, interacts with molecular chaperones HSP70, HSC70, and HSP90 via three tetratrico peptide repeat (TPR) domains, and interacts with the proteasome via its E4/U-box domain. Thus, CHIP possibly acts as a regulator in triage decisions; it sends the substrate for refolding or proteasome-dependent degradation. CHIP uses its TRP domain, not the E4/U-box domain, to reduce aggregation of polyQ-GFP and GFP-tagged N-terminal mHTT in transfected cell lines, suggesting that polyQ proteins are refolded through the action of CHIP in order to keep them in a soluble state and hence reduce aggregation (Miller et al., 2005). CHIP levels increased after mHTT expression in astrocytes but not in neurons, which suggests cell type-specific regulation (Zhao et al., 2017). Interestingly, no interaction or colocalization was found in neurons or a mouse brain region high in neuronal cell content. However, CHIP did interact or colocalize with mHTT in astrocytes and brain regions high in astrocyte abundance (Zhao et al., 2017). Colocalization and interaction of mHTT and CHIP were also observed in N2a cells (Jana et al., 2005). Studies on CHIP overexpression in cellular models of HD showed a reduction of mHTT aggregation and increased soluble levels, which was more prominent when HSC70 was present (Miller et al., 2005). In addition, CHIP overexpression reduced polyQ-induced pathology in cells, zebrafish, and Drosophila (Miller et al., 2005; Al-Ramahi et al., 2006). In line with these results, HD mice that were haploinsufficient for CHIP displayed an accelerated disease phenotype (Miller et al., 2005). While overexpression of a ligase-dead mutant still improved mHTT solubility, overexpression of a chaperone-dead mutant was unable to induce reduced aggregation of a polyQ-expanded protein (Miller et al., 2005). In addition, they found no alterations in mHTT ubiquitination after CHIP knockdown in an HD mouse model or improved mHTT degradation rate after overexpression in cells. This indicates that the chaperone function of CHIP is responsible for the shift in solubility but not mHTT turnover. It is important to note that the role of the ligase-dead mutant was excluded based on only a polyQ-peptide construct and not on the mHTT protein. Al-Ramahi and colleagues found that CHIP could not suppress the toxicity of a bare 127Q stretch but could efficiently suppress the toxicity of an N-terminal mHTT fragment in a Drosophila model for HD (Al-Ramahi et al., 2006). Their conclusion is that the protein context is important for the action of CHIP, maybe due to the presence of lysine residues that can be ubiquitinated. It is worth mentioning that the constructs used for polyQ proteins in the study of Miller and colleagues contain GFP, therefore probably offering a suitable protein context for CHIP. Zhao and colleagues observed decreased K48-linked ubiquitination of mHTT after CHIP downregulation in mouse brain (Zhao et al., 2017). In addition, Jana and colleagues found an increase in ubiquitination of mHTT after CHIP overexpression and found that CHIP ligase mutant could not lower aggregation and accompanied cell death (Jana et al., 2005). The authors conclude that the decreased aggregation is mediated by proteasomal degradation. However, they do not measure mHTT soluble levels or turnover, as this was only tested for ataxin 3, and a proper control condition measuring the sole effect of proteasome inhibition is missing. To conclude, CHIP overexpression decreases aggregation and alleviates pathology, however, further research needs to be performed to draw conclusions on the involved mechanism.
2.1.15 SCF ubiquitin-ligase complex components CUL1, SKP1 and FBXW7
Skp, Cullin, F-box containing complex (or SCF complex) is a multiprotein E3 ubiquitin ligase complex that belongs to the cullin family of RING-finger E3s. The SCF complex consists of three core subunits (the adaptor protein SKP1, a major structural scaffold protein CUL1, an RBX1 RING-finger E3 ligase domain), and a variable F-box protein. Independent of the SCF complex, the F-box protein recognizes and interacts with specific target proteins. Substrate recognition often depends on post-translational modifications, and each F-box protein may recognize several different substrates. Next, the F-box protein binds to the adaptor protein SKP1 that links the F-box protein to CUL1, thereby recruiting the substrate to the E2 enzyme and the RBX1 domain. RBX1 contains a RING-finger domain to which the E2 enzyme binds. Different Cullins and F-Box Proteins combinations can generate a wide variety of E3 ubiquitin ligases targeting different substrates. The SCF complex targets proteins with mono- or polyubiquitin chains and can regulate various molecular processes, including protein localization, protein activity, or protein targeting for degradation, of which the latter one is best studied. In this case, the SCF complex is involved in the polyubiquitination of target proteins. Bhutani and colleagues found that the levels of CUL1 and SKP1 were reduced in HD mice brain. In addition, lower CUL1 levels were observed in a cellular model and a Drosophila model of HD. Expression of a dominant negative mutant of CUL1 resulted in increased aggregation in cell culture. The silencing of CUL1 or SKP1 in Drosophila resulted in increased aggregation and toxicity. Since the combination of mHTT expression and CUL1 or SKP1 silencing was lethal, results were based on the few surviving animals (Bhutani et al., 2012). Gomez-Pastor and colleagues found increased levels of the F-box protein FBXW7 and reduced levels of the stress-protective transcription factor HSF1 in HD models. FBXW7 is an F-box protein of the SCF complex that controls the proteasome-mediated degradation of various oncoproteins. Restoration of HSF1 levels prevented mHTT aggregation. Further research is necessary to understand how mHTT expression drives enhanced expression levels of FBXW7 (Gomez-Pastor et al., 2017). The proposed pathway explaining the effect of FBXW7 on mHTT through HSP1 regulation could also account for the described effects of CUL1/SKP1.
2.2 DUBs
2.2.1 Ataxin 3
Ataxin 3 (ATXN3) belongs to the Josephin family of deubiquitinating enzymes and contains an N-terminal Josephin domain (JD) with a papain-like fold, while the C-terminus is unstructured and contains two or three UIMs and a polyQ stretch (Masino et al., 2003). The UIM motif binds both K48- and K63-linked polyubiquitin chains but prefers to cleave K63-linkages, especially when present in K48- and K63-mixed polyubiquitin chains (Winborn et al., 2008). The JD trims polyubiquitin chains down to four residues (Burnett et al., 2003), while it has weak or no activity against shorter chains. ATXN3 is involved in various biological processes such as transcription, protein homeostasis, cytoskeleton regulation, and degradation of misfolded chaperone substrates. ATXN3 can prevent self-ubiquitination of PRKN, thereby modulating PRKN activity (Durcan et al., 2012). When null mice for ATXN3 were crossed with an HD knock-in mouse model, it was observed that ATXN3 did not contribute significantly to HD progression. Decreased levels of ATXN3 only mildly aggravated age-dependent motor deficits, but no alterations were observed with respect to inclusion body formation, ubiquitination of inclusion bodies, or levels of the mHTT or wtHTT protein (Zeng et al., 2013). ATXN3 was shown to be sequestered in mHTT aggregates in HD brain, and wtHTT was shown to interact with ATXN3 in cell models, together with other subunits of the transcription-coupled DNA repair complex. This complex recognizes DNA damage and mediates DNA repair during transcription elongation. Expression of mHTT decreased the activity of ATXN3, which negatively affected transcription and DNA repair and might trigger neurotoxicity in HD (Gao et al., 2019).
2.2.2 YOD1
YOD1 is a conserved deubiquitinase belonging to the OTU family (Ernst et al., 2009) and prefers cleaving K48- and K63-linked polyubiquitin (Tanji et al., 2018). By associating with p97, it facilitates protein dislocation from the ER (Ernst et al., 2009) and proteasomal degradation of ERAD model substrates (Sasset et al., 2015). YOD1 protein levels were increased in mHTT-expressing cells (Tanji et al., 2018). Overexpression of YOD1 decreased mHTT levels and mHTT-induced cell death. These effects were not observed with catalytically inactive YOD1, indicating it depends on its deubiquitinase activity. YOD1 also altered proteasomal activity. While mHTT induced trypsin and chymotrypsin activity in cell lysate, this increased activity was absent in the presence of YOD1 (Tanji et al., 2018).
2.2.3 USP7
USP7 or HAUSP belongs to the Ubiquitin Specific Protease (USP) family of deubiquitinating enzymes and is involved in various biological pathways, including apoptosis, transcription regulation, DNA replication, and neuronal development. It contains an N-terminal polyQ region, followed by a TRAF domain, a catalytic domain and a C-terminal UBL domain. USP7 has many different interactors, which can function as scaffolds, activity modulators, or substrates for deubiquitination. Well-known substrates for USP7-mediated deubiquitination include the tumor suppressor protein p53 and WASH, a protein that is essential for endosomal protein recycling. USP7 regulation and complex formation is reviewed elsewhere (Kim and Sixma, 2017). USP7 interacts with full-length wtHTT and mHTT in the striatum and frontal cortex of a knock-in mouse model (zQ175) of HD, but USP7 prefers interacting with the mHTT protein. A proximity ligation assay in iPSC cells derived from an HD patient confirmed the interaction between USP7 and HTT in a human model. The data suggest that USP7 preferentially interacts with polyQ-expanded proteins (Pluciennik et al., 2021). It remains to be resolved whether USP7 is involved in the direct de-ubiquitination of HTT. Also, the role of the wtHTT and mHTT interaction with USP7 is still unclear.
2.2.4 USP12
While the role of USP12 is not well established, several substrates for USP12 are identified, including histone H2A/H2B, Notch, MDM2, and androgen receptor (Joo et al., 2011; Moretti et al., 2012; Burska et al., 2013; McClurg et al., 2018). USP12 colocalizes with mHTT IBs in primary neurons. Overexpression of USP12 increases cell survival after mHTT expression in primary neurons and HD patient cells and decreases the mHTT-induced phenotype in Drosophila. Surprisingly, overexpression of a catalytic-dead mutant resulted in the same protective effects. This indicates that the role of USP12 in HD pathology is independent of its deubiquitinase activity. In addition, no change in mHTT half-life was detected, which implies that USP12 is not involved in mHTT turnover. USP12 does lead to autophagy induction, which is needed for the neuroprotective effects of USP12 in HD. The authors suggest that autophagy could be involved by degrading a subpopulation of mHTT that is not measured in their half-life assay. It could also be that autophagy regulates HD pathology by degrading other substrates, and by improving general proteostasis, mHTT-induced toxicity is reduced (Aron et al., 2018).
2.2.5 USP14
The deubiquitinating enzyme USP14 belongs to the USP family and contains N-terminal UBL and C-terminal USP domains. The UBL domain is an important regulator of proteasomal activity, and the USP domain is required for its deubiquitinating activity. USP14 reversibly associates with the 19 S cap of the proteasome. It is involved in the regulation of deubiquitination of proteasome substrates, hence is involved in editing and rejecting substrates for proteasome-dependent degradation. Rapid deubiquitination can reduce the dwell time at the proteasome and prevent substrate degradation. Deubiquitination at the proteasome also enables the recycling of ubiquitin molecules. USP14 can remove single ubiquitin moieties from the tip of polyubiquitin chains (Hu et al., 2005), but it can also remove multiple polyubiquitin chains from a substrate en bloc until a single chain remains (Lee et al., 2016). In summary, the specificity of the proteasome can be regulated by deubiquitination. USP14 interacts with wtHTT and mHTT, as shown by co-immunoprecipitation experiments in cell lysates (Hyrskyluoto et al., 2014). While no change in USP14 total levels was found in BACHD mice and HTT-expressing cells, the localization of USP14 changed from the ER membrane fraction to the cytosolic fraction after mHTT expression. Overexpression of wild-type, but not catalytic inactive, USP14 in cells resulted in reduced aggregation and cell death. The decrease in the aggregation after USP14 expression was blocked by proteasome inhibition, while an autophagy inhibitor did not alter mHTT aggregation. This implies that the proteasome mediates the decrease in aggregates. However, USP14 mainly affected insoluble mHTT levels, not soluble mHTT levels (Hyrskyluoto et al., 2014), suggesting that the effect of USP14 is not mediated by directly improving the degradation of mHTT. USP14 inhibits mHTT-induced caspase-3 activity, which is involved in the regulation of cell death, and mHTT-induced activation of IRE1α, a transmembrane protein in the ER that acts as an ER stress sensor, in cells and BACHD mice (Hyrskyluoto et al., 2014). Although USP14 affects mHTT aggregation, the positive effects on cell viability could also be mediated by caspase-3 or IRE1α.
2.2.6 USP19
USP19 belongs to the USP family of deubiquitinating enzymes. It contains two CHORD-SGT1 domains at the N-terminus, potentially used to interact with the HSP90 chaperone (Zhang et al., 2008). Furthermore, it has a central USP domain with a UBL domain and a MYND zinc finger. Alternative splicing results in the generation of two major isoforms that differ in their C-termini. For instance, one isoform contains a transmembrane domain that anchors USP19 to the endoplasmatic reticulum and is involved in ERAD. The other isoform contains a relatively hydrophilic region and an EEVD motif in the C-terminus and has a cytoplasmic localization. USP19 plays a role in the protein quality control system, protein homeostasis, muscle development, tumorigenesis and controls the half-life of several proteins (Rossi and Rossi, 2022). Overexpression of cytoplasmic localized isoform of USP19 in mHTT-expressing cells increased mHTT levels and aggregation. Overexpression of a catalytic-dead mutant was not able to produce this increase. In addition, USP19 overexpression increased mHTT-induced cell death. Silencing USP19 showed the opposite effects and decreased soluble mHTT levels. USP19 may work together with the chaperone HSP90, which recognizes unfolded mHTT, thereby playing a key role in triage decisions for disease-related polyQ-expanded substrates (He et al., 2016; He et al., 2017).
3 Discussion
The various ubiquitin-modifying enzymes that directly or indirectly affect mHTT aggregation, as described in this review, affect mHTT at various levels, ranging from soluble, monomeric mHTT levels or alter oligomerization and aggregation of mHTT. They can also be involved in targeting mHTT towards the proteasome, or modifying autophagy in HD models, although there might be no direct link with the degradation of mHTT via this pathway. Indirect effects include regulation of ER stress, cell viability and oxidative stress in HD (Figure 2).
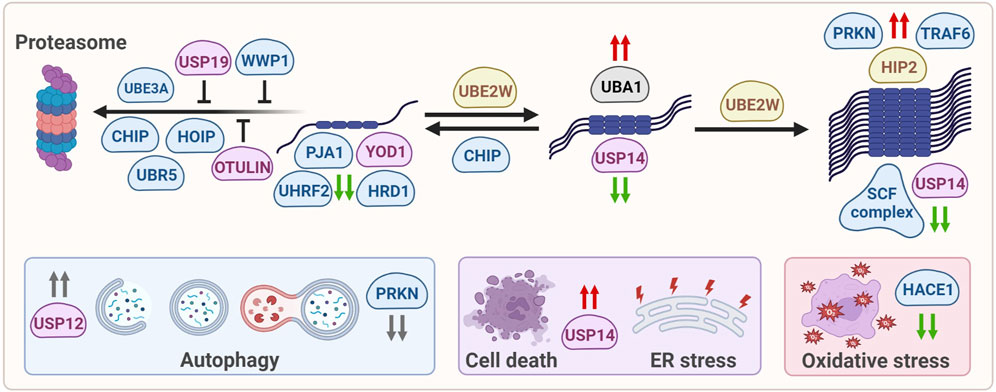
FIGURE 2. Ubiquitin-modifying enzymes that affect mHTT protein levels, solubilization, oligomerization, aggregation, degradation and mHTT-induced cellular stress. E1 enzymes are shown in grey, E2 enzymes are shown in yellow, E3 enzymes are shown in blue, DUBs are shown in pink. Green arrows: enzymes reduce mHTT levels, red arrows: enzymes increase mHTT levels, grey arrows: enzymes affect a process but not mHTT levels.
3.1 Effects of ubiquitin-modifying enzymes on mHTT levels and HD pathology
3.1.1 Ubiquitin-modifying enzymes that affect mHTT aggregation and facilitate degradation by the proteasome
Several ubiquitin ligases improve soluble wtHTT and/or mHTT degradation by the proteasome via poly-ubiquitination, including the E3 ubiquitin ligase UBE3A, UBR5 and HOIP, as well as the U-box ubiquitin ligase and CHIP. Preventing the reported age-related decrease in some of these enzymes would be a therapeutic strategy for HD, for example by enhancing the activity of the remaining pool of UBE3A, or increasing their expression level. Similarly, inhibiting the activity or decreasing the expression levels of particular DUBs including OTULIN and USP19, but also the CHIP inhibiting protein HspBP1 or the ligase WWP1, might be a strategy to reduce or prevent HD progression by improving proteasomal turnover. In addition, there are several ubiquitin-modifying enzymes affecting soluble wtHTT and/or mHTT protein levels, but their mechanism of action remains to be resolved, including the E3 ubiquitin ligase PJA1 and UHRF2. Yet they also affect wtHTT levels, which is not a preferable strategy for treating HD. In contrast, the ligase HRD1 affects soluble and aggregated mHTT levels by interacting with the VCP/p97 complex (Yang et al., 2007) that functions as a disaggregase of misfolded and polyubiquitinated proteins, including mHTT, which may facilitate mHTT accessibility for degradation. Finally, proteasomal degradation of mHTT might be facilitated by enzymes that affect mHTT solubilization. Here, the nuclear E2 enzyme UBE2W and E3 ubiquitin ligase CHIP were found to modify the solubilization of mHTT. However, improving solubilization may also induce mHTT-induced toxicity when not accompanied by improved ubiquitination and subsequent degradation, as shown when UBE2W levels were reduced (Wang et al., 2018). This is different for the ligase CHIP that can improve solubilization of mHTT by chaperone-mediated refolding, as well as proteasome-dependent degradation, thereby reducing pathology (Miller et al., 2005; Al-Ramahi et al., 2006). Therefore, activation of CHIP might be a possible target for drug development, as well as inhibition of HspBP1 as mentioned above. Last but not least is the deubiquitinating enzyme USP14 which targets oligomerized mHTT and subsequently improves proteasome-dependent degradation (Hyrskyluoto et al., 2014), suggesting that even oligomerized mHTT can still become a proteasomal target.
Increased expression of various enzymes described above affect mHTT aggregation and toxicity, including CUL1, SKP1, TRAF6 and PRKN, but since their mechanism-of-action is unknown they are for now no strategic targets for therapy. Other enzymes are known to improve autophagy, as was shown for the deubiquitinating enzyme USP12 that induces neuronal autophagy. Yet while USP12 can rescue mHTT-mediated neurodegeneration in different HD models via induction of autophagy, mHTT IB formation was not affected, suggesting that neuroprotection is obtained by improving clearance of other proteins than mHTT (Aron et al., 2018). In contrast, partial suppression of PRKN improved autophagy flux and reduced mHTT aggregation in HD mice, yet increased neuronal cell death and exacerbated motor and behavioral deficits (Rubio et al., 2009), suggesting that keeping mHTT in a more oligomeric species does not automatically result in improved recognition and clearance by autophagy.
3.1.2 Ubiquitin-modifying enzymes that affect cellular stress and cell death in HD
Reducing oxidative and ER-stress indirectly contributes to HD-related cell stress, and both HACE1 and USP14 play a role in these processes, respectively. Overexpression of HACE1 reduces oxidative stress via NRF2 induction while HACE1 knockout mice show an impaired anti-oxidative stress response and aggravated motor and psychiatric deficits (Ehrnhoefer et al., 2018). The deubiquitinating enzyme USP14 lowers both mHTT aggregation and mHTT-induced cell death, which is mediated by the reduced interaction between USP14 and IRE1α in the ER membrane in mHTT-expressing cells and BACHD mice, thereby reducing ER-stress via IRE1α.
3.2 Comparing HD studies
While numerous enzymes in all stages of the ubiquitination pathway (E1s, E2s, E3s and DUBs) have been identified to influence HTT levels or the HD phenotype (Figure 2) and reviewed in their corresponding sections above, some aspects of the study design require attention when comparing different studies. Studies on the same enzyme can use different versions of the HTT protein, including the full-length protein, but also different truncated N-terminal HTT fragments. Especially shorter N-terminal mHTT fragments are aggregation-prone, while the full-length mHTT protein is mostly soluble. Often HTT fragments are expressed with a tag, which can significantly influence a protein’s localization, interaction with other proteins and stability. It is preferred to use untagged HTT, to exclude the interference of the used tag on the protein’s behavior. In addition, different polyQ-lengths can be used, and differences in length affect the folding, post-translational modifications, protein-protein interaction, aggregation and toxicity. Some studies use polyQ-peptides instead of mHTT peptides and thus lack the HTT sequence. These polyQ-peptides show aggregation properties, but these peptides do not have the mHTT protein context, which can affect protein-protein interactions. All these differences can cause different effects in HD models.
In addition, different methods to analyze soluble and insoluble HTT protein levels are used in different studies. The mHTT protein can be present in different forms ranging from soluble monomers and High Molecular Weight (HMW) polymers to insoluble mHTT aggregates and IBs, and different methods can be used to study these different forms of mHTT. Many studies use fluorescently tagged HTT to quantify the number of IBs by microscopy, including determination of aggregate size, and this is different from biochemical assays using protein lysates. Here, lysate fractions can be divided into Triton-X100 insoluble aggregates, Triton-X100 soluble monomers, and HMW polymers, which can be detected with SDS-PAGE western blot, and it is important to quantify both HTT aggregates and soluble levels to get a complete picture of the effects on total protein levels. Is an intervention responsible for a shift between the soluble and insoluble fraction, or does it affect total protein levels via the proteostasis machinery?
Finally, the role of enzymes acting in the ubiquitination pathway can be investigated in many different models, ranging from in vitro experiments using isolated enzymes to in vivo experiments using small animal models. In cells, the experimental conditions are easily controllable, and it is relatively easy to manipulate enzyme levels. In vivo models, on the other hand, offer the potential to study the effects of enzyme modulation in an entire system and to study the effects on HD pathology. HD pathology only manifests later in life, indicating that aging processes are important in the etiology of the disease. These aging processes are not always present in immortalized cell lines, which could contribute to differences in experimental outcomes. This ‘young’ phenotype is especially present in iPSCs. The fact that most iPSC do not show aggregation already suggests a different proteostasis network, as suggested by Koyuncu et al. (2018). In addition, the brain consists of many cell types, which do not all show the same pathological features. Indeed, Zhao and colleagues showed differences in proteasome-dependent degradation of mHTT via CHIP between astrocytes and neurons, as CHIP activity was inhibited in neurons but not in astrocytes (Zhao et al., 2017). Many enzymes are only described in one or few studies or research groups, while the evidence would be stronger when more independent research groups repeat experiments.
3.3 Conclusion
Ubiquitination is an important process to target proteins for degradation through the UPS or autophagy. Not surprisingly, several enzymes involved in ubiquitination are found to be linked with HD. The levels of many of these enzymes are changed in HD compared to healthy conditions, which suggests a dysregulation of ubiquitin-regulated pathways, including proteostasis. Modulating the ubiquitination system components as therapeutic targets has gained more attention (Cohen and Tcherpakov, 2010). However, clinical agents or small molecule inhibitors have only been developed for a small fraction of these enzymes. A cell-permeable inhibitor for the E1 enzyme UBA1 is developed and used in clinical trials, but given the pleiotropic behavior of E1 enzymes, these inhibitors could only be used in acute settings such as aggressive cancers (Hyer et al., 2018). E3 ubiquitin ligases confer a high substrate specificity and would therefore be attractive targets for drug development. However, the lack of apparent druggable sites in E3 enzymes makes this challenging. On the other hand, due to their well-defined catalytic clefts, DUBs are appealing as prospective therapeutic targets (Cohen and Tcherpakov, 2010). Interestingly, mHTT is ubiquitinated at K6 and K9, while wtHTT is not (Sap et al., 2019; Hakim-Eshed et al., 2020), and the differences in ubiquitination as well as phosphorylation between wtHTT and mHTT might provide new leads for therapeutic strategies to exclusively target mHTT. A selective DUB inhibitor that improves mHTT poly-ubiquitination at its N-terminus might increase the steady-state ubiquitination of mHTT, thereby increasing the degradation of mHTT by the UPS. Indeed, several DUBs have been linked with HD, and two of them, OTULIN and USP19, seem to interfere with proteasomal degradation of mHTT (Figure 2), and might be attractive targets for DUB inhibitor development. Alternatively, compound screens with libraries of DUB inhibitors could lead to selective compounds that modulate mHTT ubiquitination and turnover, or indirectly by modifying involved ubiquitinating enzymes themselves. Inhibiting the DUB ATXN3 would counteract E3 ligase PRKN autoubiquitination (Durcan et al., 2012). Since ATXN3 and the E3 ligase CHIP are both activated by mono-ubiquitination (Todi et al., 2009; Scaglione et al., 2011) preventing their deubiquitination may preserve their activity in HD, which might be especially interesting for CHIP as it can facilitate mHTT degradation via the proteasome (Figure 2). While no DUB inhibitors have been described concerning HD, some DUB inhibitors showed disease-modifying effects in other neurodegenerative diseases. The USP7 inhibitor HBX41108 reduced toxicity in an ALS cell model (Zhang et al., 2020). IU1, a small molecule inhibitor of USP14, increased proteasome activity and proteasomal degradation of TAU, a hallmark protein in AD (Boselli et al., 2017). Despite these promising effects on AD pathology, USP14 inhibition may accelerate HD pathology since this DUB is associated with improved HTT clearance (Figure 2). The discrepancy between these neurodegenerative disorders underscores the importance of research on the effects of DUB inhibitors in HD.
Next to manipulating activity of ubiquitin ligases and DUBs towards mHTT, the degradation of mHTT could be improved using protein-targeting chimeras (PROTACs). PROTACs consist of a target protein-binding site and a binding motif for E3s, connected by a linker. By binding to an E3 and a protein of interest, PROTACs improve the ubiquitination of their target, which results in improved degradation by the UPS (Sakamoto et al., 2001; Békés et al., 2022). Several PROTACs were developed to treat AD and PD (Yao et al., 2022). In addition, Tomoshige and colleagues have shown the effectiveness of PROTACs on improved mHTT clearance in HD cells (Tomoshige et al., 2017; Tomoshige et al., 2018). Unfortunately, PROTACs still face many limitations, such as blood-brain barrier permeability, brain region-specific localization and the need for frequent administration (Farrell and Jarome, 2021). To make their PROTAC better suitable for delivery to the central nervous system, the HTT-targeting PROTAC was converted into a brain-permeable hydrophobic tag, which efficiently lowered HTT levels (Hirai et al., 2022). Another concern of PROTACs is that they need a functional UPS system, which is often impaired in neurodegenerative disorders (Thibaudeau et al., 2018). A possible solution might be to combine PROTAC therapy with proteasome activators (Leestemaker et al., 2017), which would selectively target mHTT, improve its ubiquitination, and activate the proteasome.
Author contributions
KS and KG wrote the article. KG and AG made the figures. SS-K and ER proof-read the article and gave advise for improvement.
Funding
This work was funded by the Campagneteam Huntington, Reggeborgh Medical B. V., and CHDI Foundation (A-16350).
Acknowledgments
Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1107323/full#supplementary-material
SUPPLEMENTARY TABLE S1 | Detailed overview of effects of ubiquitin-modifying enzymes on cellular mHTT and HD pathology.
References
Al-Ramahi, I., Lam, Y. C., Chen, H. K., de Gouyon, B., Zhang, M., Pérez, A. M., et al. (2006). CHIP protects from the neurotoxicity of expanded and wild-type ataxin-1 and promotes their ubiquitination and degradation. J. Biol. Chem. 281 (36), 26714–26724. doi:10.1074/jbc.M601603200
Aladdin, A., Király, R., Boto, P., Regdon, Z., and Tar, K. (2019). Juvenile huntington’s disease skin fibroblasts respond with elevated parkin level and increased proteasome activity as a potential mechanism to counterbalance the pathological consequences of mutant huntingtin protein. Int. J. Mol. Sci. 20 (21), 5338. doi:10.3390/ijms20215338
Aravind, L., and Koonin, E. v. (2000). The U box is a modified RING finger — A common domain in ubiquitination. Curr. Biol. 10 (4), R132–R134. doi:10.1016/S0960-9822(00)00398-5
Arbez, N., Ratovitski, T., Roby, E., Chighladze, E., Stewart, J. C., Ren, M., et al. (2017). Post-translational modifications clustering within proteolytic domains decrease mutant huntingtin toxicity. J. Biol. Chem. 292 (47), 19238–19249. doi:10.1074/jbc.M117.782300
Aron, R., Pellegrini, P., Green, E. W., Maddison, D. C., Opoku-Nsiah, K., Wong, J. S., et al. (2018). Deubiquitinase Usp12 functions noncatalytically to induce autophagy and confer neuroprotection in models of Huntington’s disease. Nat. Commun. 9 (1), 3191. doi:10.1038/s41467-018-05653-z
Arrasate, M., Mitra, S., Schweitzer, E. S., Segal, M. R., and Finkbeiner, S. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431 (7010), 805–810. doi:10.1038/nature02998
Baias, M., Smith, P. E. S., Shen, K., Joachimiak, L. A., Zerko, S., Koźmiński, W., et al. (2017). Structure and dynamics of the huntingtin exon-1 N-terminus: A solution nmr perspective. J. Am. Chem. Soc. 139 (3), 1168–1176. doi:10.1021/jacs.6b10893
Békés, M., Langley, D. R., and Crews, C. M. (2022). PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. Res. 21 (3), 181–200. doi:10.1038/s41573-021-00371-6
Bhat, K. P., Yan, S., Wang, C., Li, S., and Li, X. (2014). Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc. Natl. Acad. Sci. U. S. A. 111 (15), 5706–5711. doi:10.1073/pnas.1402215111
Bhutani, S., Das, A., Maheshwari, M., Lakhotia, S. C., and Jana, N. R. (2012). Dysregulation of core components of SCF complex in poly-glutamine disorders. Cell Death Dis. 3 (11), e428. doi:10.1038/cddis.2012.166
Binder, J. X., Pletscher-Frankild, S., Tsafou, K., Stolte, C., O'Donoghue, S. I., Schneider, R., et al. (2014). Compartments: Unification and visualization of protein subcellular localization evidence. Database J. Biol. databases curation 2014, bau012. doi:10.1093/database/bau012
Bordallo, J., Plemper, R. K., Finger, A., and Wolf, D. H. (1998). Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol. Biol. Cell 9, 209–222. doi:10.1091/mbc.9.1.209
Boselli, M., Lee, B. H., Robert, J., Prado, M. A., Min, S. W., Cheng, C., et al. (2017). An inhibitor of the proteasomal deubiquitinating enzyme USP14 induces tau elimination in cultured neurons. J. Biol. Chem. 292 (47), 19209–19225. doi:10.1074/jbc.M117.815126
Breitschopf, K., Ziv, T., Admon, A., and Ciechanover, A. (1998). BengalA novel site for ubiquitination: The N-terminal residue, and not internal lysines of MyoD, is essential for conjugation and degradation of the protein. EMBO J. 17 (20), 5964–5973. doi:10.1093/emboj/17.20.5964
Burnett, B., Li, F., and Pittman, R. N. (2003). The polyglutamine neurodegenerative protein ataxin-3 binds polyubiquitylated proteins and has ubiquitin protease activity. Hum. Mol. Genet. 12 (23), 3195–3205. doi:10.1093/hmg/ddg344
Burroughs, A. M., Jaffee, M., Iyer, L. M., and Aravind, L. (2008). Anatomy of the E2 ligase fold: Implications for enzymology and evolution of ubiquitin/Ub-like protein conjugation. J. Struct. Biol. 162 (2), 205–218. doi:10.1016/j.jsb.2007.12.006
Burska, U. L., Harle, V. J., Coffey, K., Darby, S., Ramsey, H., O’Neill, D., et al. (2013). Deubiquitinating enzyme Usp12 is a novel co-activator of the androgen receptor. J. Biol. Chem. 288 (45), 32641–32650. doi:10.1074/jbc.M113.485912
Chan, N. C., Salazar, A. M., Pham, A. H., Sweredoski, M. J., Kolawa, N. J., Graham, R. L. J., et al. (2011). Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum. Mol. Genet. 20 (9), 1726–1737. doi:10.1093/hmg/ddr048
Chaugule, V. K., Burchell, L., Barber, K. R., Sidhu, A., Leslie, S. J., Shaw, G. S., et al. (2011). Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J. 30 (14), 2853–2867. doi:10.1038/emboj.2011.204
Chen, Z., and Pickart, C. M. (1990). A 25-kilodalton ubiquitin carrier protein (E2) catalyzes multi-ubiquitin chain synthesis via lysine 48 of ubiquitin. J. Biol. Chem. 265 (35), 21835–21842. doi:10.1016/s0021-9258(18)45815-2
Chen, Z., and Sun, L. J. (2009). Nonproteolytic functions of ubiquitin in cell signaling. Mol. Cell 33 (3), 275–286. doi:10.1016/j.molcel.2009.01.014
Chen, Z., Liu, R., Li, Y., Azmi, P. B., and Seth, A. K. (2008). The WW domain containing E3 ubiquitin protein ligase 1 upregulates ErbB2 and EGFR through RING finger protein 11. Oncogene 27 (54), 6845–6855. doi:10.1038/onc.2008.288
Clague, M. J., Heride, C., and Urbé, S. (2015). The demographics of the ubiquitin system. Trends Cell Biol. 25 (7), 417–426. doi:10.1016/j.tcb.2015.03.002
Clague, M. J., Urbé, S., and Komander, D. (2019). Breaking the chains: Deubiquitylating enzyme specificity begets function. Nat. Rev. Mol. Cell Biol. 20 (6), 338–352. doi:10.1038/s41580-019-0099-1
Cohen, P., and Tcherpakov, M. (2010). Will the ubiquitin system furnish as many drug targets as protein kinases? Cell 143 (5), 686–693. doi:10.1016/j.cell.2010.11.016
Courivaud, T., Ferrand, N., Elkhattouti, A., Kumar, S., Levy, L., Ferrigno, O., et al. (2015). Functional characterization of a WWP1/Tiul1 tumor-derived mutant reveals a paradigm of its constitutive activation in human cancer. J. Biol. Chem. 290 (34), 21007–21018. doi:10.1074/jbc.M115.642314
Da, C., Pu, J., Liu, Z., Wei, J., Qu, Y., Wu, Y., et al. (2021). HACE1-mediated NRF2 activation causes enhanced malignant phenotypes and decreased radiosensitivity of glioma cells. Signal Transduct. Target. Ther. 6 (1), 399. doi:10.1038/s41392-021-00793-z
de Pril, R., Fischer, D. F., Roos, R. A. C., and van Leeuwen, F. W. (2007). Ubiquitin-conjugating enzyme E2-25K increases aggregate formation and cell death in polyglutamine diseases. Mol. Cell. Neurosci. 34 (1), 10–19. doi:10.1016/j.mcn.2006.09.006
DeGuire, S. M., Ruggeri, F. S., Fares, M. B., Chiki, A., Cendrowska, U., Dietler, G., et al. (2018). N-terminal Huntingtin (Htt) phosphorylation is a molecular switch regulating Htt aggregation, helical conformation, internalization, and nuclear targeting. J. Biol. Chem. 293 (48), 18540–18558. doi:10.1074/jbc.RA118.004621
DiFiglia, M., Sapp, E., Chase, K. O., Davies, S. W., Bates, G. P., Vonsattel, J. P., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Sci. (New York, N.Y.) 277 (5334), 1990–1993. doi:10.1126/science.277.5334.1990
Durcan, T. M., Kontogiannea, M., Bedard, N., Wing, S. S., and Fon, E. A. (2012). Ataxin-3 deubiquitination is coupled to parkin ubiquitination via E2 ubiquitin-conjugating enzyme. J. Biol. Chem. 287 (1), 531–541. doi:10.1074/jbc.M111.288449
Ehrnhoefer, D. E., Southwell, A. L., Sivasubramanian, M., Qiu, X., Villanueva, E. B., Xie, Y., et al. (2018). HACE1 is essential for astrocyte mitochondrial function and influences Huntington disease phenotypes in vivo. Hum. Mol. Genet. 27 (2), 239–253. doi:10.1093/hmg/ddx394
Emmerich, C. H., Ordureau, A., Strickson, S., Arthur, J. S. C., Pedrioli, P. G. A., Komander, D., et al. (2013). Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc. Natl. Acad. Sci. U. S. A. 110 (38), 15247–15252. doi:10.1073/pnas.1314715110
Ernst, R., Mueller, B., Ploegh, H. L., and Schlieker, C. (2009). The otubain YOD1 is a deubiquitinating enzyme that associates with p97 to facilitate protein dislocation from the ER. Mol. Cell 36 (1), 28–38. doi:10.1016/j.molcel.2009.09.016
Farrell, K., and Jarome, T. J. (2021). Is PROTAC technology really a game changer for central nervous system drug discovery? Expert Opin. Drug Discov. 16 (8), 833–840. doi:10.1080/17460441.2021.1915979
Finkbeiner, S. (2011). Huntington’s disease. Cold Spring Harb. Perspect. Biol. 3 (6), 0074766–a7524. doi:10.1101/cshperspect.a007476
Finney, N., Walther, F., Mantel, P. Y., Stauffer, D., Rovelli, G., and Dev, K. K. (2003). The cellular protein level of parkin is regulated by its ubiquitin-like domain. J. Biol. Chem. 278 (18), 16054–16058. doi:10.1074/jbc.C300051200
Fusilli, C., Migliore, S., Mazza, T., Consoli, F., de Luca, A., Barbagallo, G., et al. (2018). Biological and clinical manifestations of juvenile huntington’s disease: A retrospective analysis. Lancet Neurology 17 (11), 986–993. doi:10.1016/S1474-4422(18)30294-1
Gao, R., Chakraborty, A., Geater, C., Pradhan, S., Gordon, K. L., Snowden, J., et al. (2019). Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife 8, e42988. doi:10.7554/eLife.42988
Gardner, R. G., Nelson, Z. W., and Gottschling, D. E. (2005). Degradation-mediated protein quality control in the nucleus. Cell 120 (6), 803–815. doi:10.1016/j.cell.2005.01.016
Geetha, T., Kenchappa, R. S., Wooten, M. W., and Carter, B. D. (2005). TRAF6-mediated ubiquitination regulates nuclear translocation of NRIF, the p75 receptor interactor. EMBO J. 24 (22), 3859–3868. doi:10.1038/sj.emboj.7600845
Ghosh, D. K., Roy, A., and Ranjan, A. (2018). The ATPase VCP/p97 functions as a disaggregase against toxic Huntingtin-exon1 aggregates. FEBS Lett. 592 (16), 2680–2692. doi:10.1002/1873-3468.13213
Ghosh, S., Prasad, M., and Mandal, A. K. (2021). Praja1 ubiquitin ligase facilitates degradation of polyglutamine proteins and suppresses polyglutamine-mediated toxicity. Mol. Biol. Cell 32 (17), 1579–1593. doi:10.1091/mbc.E20-11-0747
Gomez-Pastor, R., Burchfiel, E. T., Neef, D. W., Jaeger, A. M., Cabiscol, E., McKinstry, S. U., et al. (2017). Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease. Nat. Commun. 8, 14405. doi:10.1038/ncomms14405
Hakim-Eshed, V., Boulos, A., Cohen-Rosenzweig, C., Yu-Taeger, L., Ziv, T., Kwon, Y. T., et al. (2020). Site-specific ubiquitination of pathogenic huntingtin attenuates its deleterious effects. Proc. Natl. Acad. Sci. U. S. A. 117 (31), 18661–18669. doi:10.1073/pnas.2007667117
Hatakeyama, S., and Nakayama, K. I. I. (2003). U-box proteins as a new family of ubiquitin ligases. Biochem. Biophysical Res. Commun. 302 (4), 635–645. doi:10.1016/S0006-291X(03)00245-6
He, W. T., Xue, W., Gao, Y. G., Hong, J. Y., Yue, H. W., Jiang, L. L., et al. (2017). HSP90 recognizes the N-terminus of huntingtin involved in regulation of huntingtin aggregation by USP19. Sci. Rep. 7 (1), 14797. doi:10.1038/s41598-017-13711-7
He, W. T., Zheng, X. M., Zhang, Y. H., Gao, Y. G., Song, A. X., van der Goot, F. G., et al. (2016). Cytoplasmic ubiquitin-specific protease 19 (USP19) modulates aggregation of polyglutamine-expanded ataxin-3 and huntingtin through the Hsp90 chaperone. PLoS ONE 11 (1), e0147515. doi:10.1371/journal.pone.0147515
Hershko, A., and Ciechanover, A. (1998). The ubiquitin system. Annu. Rev. Biochem. 67, 425–479. doi:10.1146/annurev.biochem.67.1.425
Hirai, K., Yamashita, H., Tomoshige, S., Mishima, Y., Niwa, T., Ohgane, K., et al. (2022). Conversion of a PROTAC mutant huntingtin degrader into small-molecule hydrophobic tags focusing on drug-like properties. ACS Med. Chem. Lett. 13 (3), 396–402. doi:10.1021/acsmedchemlett.1c00500
Howard, R. A., Sharma, P., Hajjar, C., Caldwell, K. A., Caldwell, G. A., du Breuil, R., et al. (2007). Ubiquitin conjugating enzymes participate in polyglutamine protein aggregation. BMC Cell Biol. 8, 32. doi:10.1186/1471-2121-8-32
Hu, M., Li, P., Song, L., Jeffrey, P. D., Chenova, T. A., Wilkinson, K. D., et al. (2005). Structure and mechanisms of the proteasome-associated deubiquitinating enzyme USP14. EMBO J. 24 (21), 3747–3756. doi:10.1038/sj.emboj.7600832
Hyer, M. L., Milhollen, M. A., Ciavarri, J., Fleming, P., Traore, T., Sappal, D., et al. (2018). A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 24 (2), 186–193. doi:10.1038/nm.4474
Hyrskyluoto, A., Bruelle, C., Lundh, S. H., Do, H. T., Kivinen, J., Rappou, E., et al. (2014). Ubiquitin-specific protease-14 reduces cellular aggregates and protects against mutant huntingtin-induced cell degeneration: Involvement of the proteasome and ER stress-activated kinase IRE1α. Hum. Mol. Genet. 23 (22), 5928–5939. doi:10.1093/hmg/ddu317
Imai, Y., Soda, M., Hatakeyama, S., Akagi, T., Hashikawa, T., Nakayama, K.-I., et al. (2002). CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol. Cell 10, 55–67. doi:10.1016/s1097-2765(02)00583-x
Imai, Y., Soda, M., and Takahashi, R. (2000). Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J. Biol. Chem. 275 (46), 35661–35664. doi:10.1074/jbc.C000447200
Iwai, K., Fujita, H., and Sasaki, Y. (2014). Linear ubiquitin chains: NF-κB signalling, cell death and beyond. Nat. Rev. Mol. Cell Biol. 15 (8), 503–508. doi:10.1038/nrm3836
Iwata, A., Nagashima, Y., Matsumoto, L., Suzuki, T., Yamanaka, T., Date, H., et al. (2009). Intranuclear degradation of polyglutamine aggregates by the ubiquitin-proteasome system. J. Biol. Chem. 284 (15), 9796–9803. doi:10.1074/jbc.M809739200
Jacobson, A. D., Zhang, N., Xu, P., Han, K., Noone, S., Peng, J., et al. (2009). The lysine 48 and lysine 63 ubiquitin conjugates are processed differently by the 26 S proteasome * □. J. Biol. Chem. 284 (51), 35485–35494. doi:10.1074/jbc.M109.052928
Jana, N. R., Dikshit, P., Goswami, A., Kotliarova, S., Murata, S., Tanaka, K., et al. (2005). Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280 (12), 11635–11640. doi:10.1074/jbc.M412042200
Jin, J., Li, X., Gygi, S. P., and Harper, J. W. (2007). Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 447 (7148), 1135–1138. doi:10.1038/nature05902
Joo, H. Y., Jones, A., Yang, C., Zhai, L., Smith IV, A. D., Zhang, Z., et al. (2011). Regulation of histone H2A and H2B deubiquitination and xenopus development by USP12 and USP46. J. Biol. Chem. 286 (9), 7190–7201. doi:10.1074/jbc.M110.158311
Juenemann, K., Jansen, A. H. P., van Riel, L., Merkx, R., Mulder, M. P. C., An, H., et al. (2018). Dynamic recruitment of ubiquitin to mutant huntingtin inclusion bodies. Sci. Rep. 8 (1), 1405. doi:10.1038/s41598-018-19538-0
Juenemann, K., Schipper-Krom, S., Wiemhoefer, A., Kloss, A., Sanz, A. S., and Reits, E. A. J. (2013). Expanded polyglutamine-containing N-terminal huntingtin fragments are entirely degraded by mammalian proteasomes. J. Biol. Chem. 288 (38), 27068–27084. doi:10.1074/jbc.M113.486076
Juenemann, K., Wiemhoefer, A., and Reits, E. a. (2015). Detection of ubiquitinated huntingtin species in intracellular aggregates. Front. Mol. Neurosci. 8 (1), 1–8. doi:10.3389/fnmol.2015.00001
Kalchman, M. A., Graham, R. K., Xia, G., Brook Koide, H., Graeme Hodgson, J., Graham, K. C., et al. (1996). Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme. J. Biol. Chem. 271 (32), 19385–19394. doi:10.1074/jbc.271.32.19385
Keusekotten, K., Elliott, P. R., Glockner, L., Fiil, B. K., Damgaard, R. B., Kulathu, Y., et al. (2013). OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1-linked polyubiquitin. Cell 153 (6), 1312–1326. doi:10.1016/j.cell.2013.05.014
Kim, R. Q., and Sixma, T. K. (2017). Regulation of USP7: A high incidence of E3 complexes. J. Mol. Biol. 429 (22), 3395–3408. doi:10.1016/j.jmb.2017.05.028
Kirisako, T., Kamei, K., Murata, S., Kato, M., Fukumoto, H., Kanie, M., et al. (2006). A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 25 (20), 4877–4887. doi:10.1038/sj.emboj.7601360
Koyuncu, S., Saez, I., Lee, H. J., Gutierrez-Garcia, R., Pokrzywa, W., Fatima, A., et al. (2018). The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat. Commun. 9 (1), 2886. doi:10.1038/s41467-018-05320-3
Kozlov, G., Nguyen, L., Lin, T., de Crescenzo, G., Park, M., and Gehring, K. (2007). Structural basis of ubiquitin recognition by the ubiquitin-associated (UBA) domain of the ubiquitin ligase EDD. J. Biol. Chem. 282 (49), 35787–35795. doi:10.1074/jbc.M705655200
Kuang, L., Jiang, Y., Li, C., and Jiang, Y. (2021). “WW domain-containing E3 ubiquitin protein ligase 1: A self-disciplined oncoprotein,” in Frontiers in cell and developmental biology (Lausanne, Switzerland: Frontiers Media S.A), 9. doi:10.3389/fcell.2021.757493
Kulak, N. A., Pichler, G., Paron, I., Nagaraj, N., and Mann, M. (2014). Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 11 (3), 319–324. doi:10.1038/nmeth.2834
Landles, C., Sathasivam, K., Weiss, A., Woodman, B., Moffitt, H., Finkbeiner, S., et al. (2010). Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in huntington disease. J. Biol. Chem. 285 (12), 8808–8823. doi:10.1074/jbc.M109.075028
Lee, B, Lu, Y., Prado, M. A., Shi, Y., Tian, G., Sun, S., et al. (2016). USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 532 (7599), 398–401. doi:10.1038/nature17433
Lee, M. E., Gygi, S. P., and Harper, J. W. (2011). Alternative ubiquitin activation/conjugation cascades interact with N-end rule ubiquitin ligases to control degradation of RGS proteins. Mol. Cell 43 (3), 392–405. doi:10.1016/j.molcel.2011.05.034
Lee, V. C., Chao, M. J., Vonsattel, J. P. G., Pinto, R. M., Lucente, D., Abu-Elneel, K., et al. (2015). Identification of genetic factors that modify clinical onset of huntington’s disease. Cell 162 (3), 516–526. doi:10.1016/j.cell.2015.07.003
Leestemaker, Y., de Jong, A., Witting, K. F., Penning, R., Schuurman, K., Rodenko, B., et al. (2017). Proteasome activation by small molecules. Cell Chem. Biol. 24 (6), 725–736. doi:10.1016/j.chembiol.2017.05.010
Li, X., Wang, C. E., Huang, S., Xu, X., Li, X. J., Li, H., et al. (2010). Inhibiting the ubiquitin-proteasome system leads to preferential accumulation of toxic N-terminal mutant huntingtin fragments. Hum. Mol. Genet. 19 (12), 2445–2455. doi:10.1093/hmg/ddq127
Lin, L., Jin, Z., Tan, H., Xu, Q., Peng, T., and Li, H. (2016). Atypical ubiquitination by E3 ligase WWP1 inhibits the proteasome-mediated degradation of mutant huntingtin. Brain Res. 1643, 103–112. doi:10.1016/j.brainres.2016.03.027
Linares, J. F., Duran, A., Yajima, T., Pasparakis, M., Moscat, J., and Diaz-Meco, M. T. (2013). K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 51 (3), 283–296. doi:10.1016/j.molcel.2013.06.020
Ma, Q. (2013). Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 53, 401–426. doi:10.1146/annurev-pharmtox-011112-140320
MacDonald, M. E., Ambrose, C. M., Duyao, M. P., Myers, R. H., Lin, C., Srinidhi, L., et al. (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72 (6), 971–983. doi:10.1016/0092-8674(93)90585-E
Maheshwari, M., Samanta, A., Godavarthi, S. K., Mukherjee, R., and Jana, N. R. (2012). Dysfunction of the ubiquitin ligase Ube3a may Be associated with synaptic pathophysiology in a mouse model of huntington disease. J. Biol. Chem. 287 (35), 29949–29957. doi:10.1074/jbc.M112.371724
Maheshwari, M., Shekhar, S., Singh, B. K., Jamal, I., Vatsa, N., Kumar, V., et al. (2014). Deficiency of Ube3a in Huntington’s disease mice brain increases aggregate load and accelerates disease pathology. Hum. Mol. Genet. 23 (23), 6235–6245. doi:10.1093/hmg/ddu343
Maiuri, T., Woloshansky, T., Xia, J., and Truant, R. (2013). The huntingtin N17 domain is a multifunctional CRM1 and ran-dependent nuclear and cilial export signal. Hum. Mol. Genet. 22 (7), 1383–1394. doi:10.1093/hmg/dds554
Masino, L., Musi, V., Menon, R. P., Fusi, P., Kelly, G., Frenkiel, T. A., et al. (2003). Domain architecture of the polyglutamine protein ataxin-3: A globular domain followed by a flexible tail. FEBS Lett. 549 (1–3), 21–25. doi:10.1016/S0014-5793(03)00748-8
Matsuda, N., Sato, S., Shiba, K., Okatsu, K., Saisho, K., Gautier, C. A., et al. (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 189 (2), 211–221. doi:10.1083/jcb.200910140
McClurg, U. L., Chit, N. C. T. H., Azizyan, M., Edwards, J., Nabbi, A., Riabowol, K. T., et al. (2018). Molecular mechanism of the TP53-MDM2-AR-AKT signalling network regulation by USP12. Oncogene 37 (34), 4679–4691. doi:10.1038/s41388-018-0283-3
Metzger, M. B., Pruneda, J. N., Klevit, R. E., and Weissman, A. M. (2014). RING-type E3 ligases: Master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochimica Biophysica Acta - Mol. Cell Res. 1843 (1), 47–60. doi:10.1016/j.bbamcr.2013.05.026
Mevissen, T. E. T., and Komander, D. (2017). Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 86 (1), 159–192. doi:10.1146/annurev-biochem-061516-044916
Michelle, C., Vourc’H, P., Mignon, L., and Andres, C. R. (2009). What was the set of ubiquitin and ubiquitin-like conjugating enzymes in the eukaryote common ancestor? J. Mol. Evol. 68 (6), 616–628. doi:10.1007/s00239-009-9225-6
Miller, V. M., Nelson, R. F., Gouvion, C. M., Williams, A., Rodriguez-Lebron, E., Harper, S. Q., et al. (2005). CHIP suppresses polyglutamine aggregation and toxicity in vitro and in vivo. J. Neurosci. 25 (40), 9152–9161. doi:10.1523/JNEUROSCI.3001-05.2005
Min, M., Mevissen, T. E. T., de Luca, M., Komander, D., and Lindon, C. (2015). Efficient APC/C substrate degradation in cells undergoing mitotic exit depends on K11 ubiquitin linkages. Mol. Biol. Cell 26 (24), 4325–4332. doi:10.1091/mbc.E15-02-0102
Mishra, A., Dikshit, P., Purkayastha, S., Sharma, J., Nukina, N., and Jana, N. R. (2008). E6-AP promotes misfolded polyglutamine proteins for proteasomal degradation and suppresses polyglutamine protein aggregation and toxicity. J. Biol. Chem. 283 (12), 7648–7656. doi:10.1074/jbc.M706620200
Mishra, A., Godavarthi, S. K., Maheshwari, M., Goswami, A., and Jana, N. R. (2009). The ubiquitin ligase E6-AP is induced and recruited to aggresomes in response to proteasome inhibition and may be involved in the ubiquitination of Hsp70-bound misfolded proteins. J. Biol. Chem. 284 (16), 10537–10545. doi:10.1074/jbc.M806804200
Moretti, J., Chastagner, P., Liang, C. C., Cohn, M. A., Israël, A., and Brou, C. (2012). The ubiquitin-specific protease 12 (USP12) is a negative regulator of notch signaling acting on notch receptor trafficking toward degradation. J. Biol. Chem. 287 (35), 29429–29441. doi:10.1074/jbc.M112.366807
Mosavi, L. K., Cammett, T. J., Desrosiers, D. C., and Peng, Z. (2004). The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 13 (6), 1435–1448. doi:10.1110/ps.03554604
Muńoz-Escobar, J., Matta-Camacho, E., Kozlov, G., and Gehring, K. (2015). The MLLE domain of the ubiquitin ligase UBR5 binds to its catalytic domain to regulate substrate binding. J. Biol. Chem. 290 (37), 22841–22850. doi:10.1074/jbc.M115.672246
Narendra, D., Tanaka, A., Suen, D. F., and Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 183 (5), 795–803. doi:10.1083/jcb.200809125
Noad, J., von der Malsburg, A., Pathe, C., Michel, M. A., Komander, D., and Randow, F. (2017). LUBAC-synthesized linear ubiquitin chains restrict cytosol-invading bacteria by activating autophagy and NF-κB. Nat. Microbiol. 2, 17063. doi:10.1038/nmicrobiol.2017.63
Nollen, E. A., Garcia, S. M., van Haaften, G., Kim, S., Chavez, A., Morimoto, R. I., et al. (2004). Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U. S. A. 101, 6403–6408. doi:10.1073/pnas.0307697101
Noormohammadi, A., Khodakarami, A., Gutierrez-Garcia, R., Lee, H. J., Koyuncu, S., Konig, T., et al. (2016). Somatic increase of CCT8 mimics proteostasis of human pluripotent stem cells and extends C. elegans lifespan. Nat. Commun. 7, 13649. doi:10.1038/ncomms13649
Ordureau, A., Sarraf, S. A., Duda, D. M., Heo, J. M., Jedrychowski, M. P., Sviderskiy, V. O., et al. (2014). Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 56 (3), 360–375. doi:10.1016/j.molcel.2014.09.007
Park, S. H., Kukushkin, Y., Gupta, R., Chen, T., Konagai, A., Hipp, M. S., et al. (2013). PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154 (1), 134–145. doi:10.1016/j.cell.2013.06.003
Peters, T. W., Nelson, C. S., Gerencser, A. A., Dumas, K. J., Tavshanjian, B., Chang, K. C., et al. (2018). Natural genetic variation in yeast reveals that NEDD4 is a conserved modifier of mutant polyglutamine aggregation. G3 (Bethesda) 8, 3421–3431. doi:10.1534/g3.118.200289
Pluciennik, A., Liu, Y., Molotsky, E., Marsh, G. B., Ranxhi, B., Arnold, F. J., et al. (2021). Deubiquitinase USP7 contributes to the pathogenicity of spinal and bulbar muscular atrophy. J. Clin. Investigation 131 (1), e134565. doi:10.1172/JCI134565
Pluska, L., Jarosch, E., Zauber, H., Kniss, A., Waltho, A., Bagola, K., et al. (2021). The UBA domain of conjugating enzyme Ubc1/Ube2K facilitates assembly of K48/K63-branched ubiquitin chains. EMBO J. 40 (6), e106094. doi:10.15252/embj.2020106094
Ravikumar, B., Duden, R., and Rubinsztein, D. C. (2002). Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11 (9), 1107–1117. doi:10.1093/hmg/11.9.1107
Riguet, N., Mahul-Mellier, A. L., Maharjan, N., Burtscher, J., Croisier, M., Knott, G., et al. (2021). Nuclear and cytoplasmic huntingtin inclusions exhibit distinct biochemical composition, interactome and ultrastructural properties. Nat. Commun. 12 (1), 6579. doi:10.1038/s41467-021-26684-z
Rossi, F. A., and Rossi, M. (2022). “Emerging role of ubiquitin-specific protease 19 in oncogenesis and cancer development,” in Frontiers in cell and developmental biology (Lausanne, Switzerland: Frontiers Media S.A), 10. doi:10.3389/fcell.2022.889166
Rotblat, B., Southwell, A. L., Ehrnhoefer, D. E., Skotte, N. H., Metzler, M., Franciosi, S., et al. (2014). HACE1 reduces oxidative stress and mutant Huntingtin toxicity by promoting the NRF2 response. Proc. Natl. Acad. Sci. U. S. A. 111 (8), 3032–3037. doi:10.1073/pnas.1314421111
Rotin, D., and Kumar, S. (2009). Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10 (6), 398–409. doi:10.1038/nrm2690
Rubinsztein, D. C. (2006). The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443 (7113), 780–786. doi:10.1038/nature05291
Rubio, I., Rodríguez-Navarro, J. A., Tomás-Zapico, C., Ruíz, C., Casarejos, M. J., Perucho, J., et al. (2009). Effects of partial suppression of parkin on huntingtin mutant R6/1 mice. Brain Res. 1281, 91–100. doi:10.1016/j.brainres.2009.05.039
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U. S. A. 98, 8554–8559. doi:10.1073/pnas.141230798
Sakata, E., Yamaguchi, Y., Kurimoto, E., Kikuchi, J., Yokoyama, S., Yamada, S., et al. (2003). Parkin binds the Rpn 10 subunit of 26S proteasomes through its ubiquitin-like domain. EMBO Rep. 4 (3), 301–306. doi:10.1038/sj.embor.embor764
Sap, K. A., Guler, A. T., Bezstarosti, K., Bury, A. E., Juenemann, K., Demmers, J. A. A., et al. (2019). Global proteome and ubiquitinome changes in the soluble and insoluble fractions of Q175 huntington mice brains. Mol. Cell. Proteomics MCP 18 (9), 1705–1720. doi:10.1074/mcp.RA119.001486
Sap, K. A., and Reits, E. A. (2020). Strategies to investigate ubiquitination in huntington’s disease. Sec. Chem. Biol. 8–2020. doi:10.3389/fchem.2020.0048
Sasset, L., Petris, G., Cesaratto, F., and Burrone, O. R. (2015). The VCP/p97 and YOD1 proteins have different substratedependent activities in endoplasmic reticulum-associated degradation (ERAD). J. Biol. Chem. 290 (47), 28175–28188. doi:10.1074/jbc.M115.656660
Scaglione, K. M., Basrur, V., Ashraf, N. S., Konen, J. R., Elenitoba-Johnson, K. S. J., Todi, S. v., et al. (2013). The ubiquitin-conjugating enzyme (E2) ube2w ubiquitinates the N terminus of substrates. J. Biol. Chem. 288 (26), 18784–18788. doi:10.1074/jbc.C113.477596
Scaglione, K. M., Zavodszky, E., Todi, S. v., Patury, S., Xu, P., Rodríguez-Lebrón, E., et al. (2011). Ube2w and ataxin-3 coordinately regulate the ubiquitin ligase CHIP. Mol. Cell 43 (4), 599–612. doi:10.1016/j.molcel.2011.05.036
Schaffar, G., Breuer, P., Boteva, R., Behrends, C., Tzvetkov, N., Strippel, N., et al. (2004). Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol. Cell 15 (1), 95–105. doi:10.1016/j.molcel.2004.06.029
Schilling, G., Klevytska, A., Tebbenkamp, A. T. N., Juenemann, K., Cooper, J., Gonzales, V., et al. (2007). Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J. Neuropathology Exp. Neurology 66 (4), 313–320. doi:10.1097/nen.0b013e318040b2c8
Schipper-Krom, S., Juenemann, K., Jansen, A. H., Wiemhoefer, A., van den Nieuwendijk, R., Smith, D. L., et al. (2014). Dynamic recruitment of active proteasomes into polyglutamine initiated inclusion bodies. FEBS Lett. 588 (1), 151–159. doi:10.1016/j.febslet.2013.11.023
Sjöstedt, E., Zhong, W., Fagerberg, L., Karlsson, M., Mitsios, N., Adori, C., et al. (2020). An atlas of the protein-coding genes in the human, pig, and mouse brain. Sci. (New York, N.Y.) 367 (6482), eaay5947. doi:10.1126/science.aay5947
Sluimer, J., and Distel, B. (2018). Regulating the human HECT E3 ligases. Cell. Mol. Life Sci. 75 (17), 3121–3141. doi:10.1007/s00018-018-2848-2
Smit, J. J., Monteferrario, D., Noordermeer, S. M., van Dijk, W. J., van der Reijden, B. A., and Sixma, T. K. (2012). The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 31 (19), 3833–3844. doi:10.1038/emboj.2012.217
Spratt, D. E., Walden, H., and Shaw, G. S. (2014). RBR E3 ubiquitin ligases: New structures, new insights, new questions. Biochem. J. 458 (3), 421–437. doi:10.1042/BJ20140006
Steffan, J. S., Agrawal, N., Pallos, J., Rockabrand, E., Trotman, L. C., Slepko, N., et al. (2004). SUMO modification of Huntingtin and Huntington’s disease pathology. Sci. (New York, N.Y.) 304 (5667), 100–104. doi:10.1126/science.1092194
Suzuki, T., Suzuki, T., Raveau, M., Miyake, N., Sudo, G., Tsurusaki, Y., et al. (2020). A recurrent PJA1 variant in trigonocephaly and neurodevelopmental disorders. Ann. Clin. Transl. Neurology 7 (7), 1117–1131. doi:10.1002/acn3.51093
Swatek, K. N., and Komander, D. (2016). Ubiquitin modifications. Cell Res. 26 (4), 399–422. doi:10.1038/cr.2016.39
Tan, J. M. M., Wong, E. S. P., Kirkpatrick, D. S., Pletnikova, O., Ko, H. S., Tay, S. P., et al. (2008). Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17 (3), 431–439. doi:10.1093/hmg/ddm320
Tanji, K., Mori, F., Miki, Y., Utsumi, J., Sasaki, H., Kakita, A., et al. (2018). YOD1 attenuates neurogenic proteotoxicity through its deubiquitinating activity. Neurobiol. Dis. 112, 14–23. doi:10.1016/j.nbd.2018.01.006
Tasaki, T., Mulder, L. C. F., Iwamatsu, A., Lee, M. J., Davydov, I. v., Varshavsky, A., et al. (2005). A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol. 25 (16), 7120–7136. doi:10.1128/mcb.25.16.7120-7136.2005
Tatham, M. H., Plechanovová, A., Jaffray, E. G., Salmen, H., and Hay, R. T. (2013). Ube2W conjugates ubiquitin to α-amino groups of protein N-termini. Biochem. J. 453 (1), 137–145. doi:10.1042/BJ20130244
Thibaudeau, T. A., Anderson, R. T., and Smith, D. M. (2018). A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 9 (1), 1097. doi:10.1038/s41467-018-03509-0
Thompson, L. M., Aiken, C. T., Kaltenbach, L. S., Agrawal, N., Illes, K., Khoshnan, A., et al. (2009). IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 187 (7), 1083–1099. doi:10.1083/jcb.200909067
Thul, P. J., Åkesson, L., Wiking, M., Mahdessian, D., Geladaki, A., Ait Blal, H., et al. (2017). A subcellular map of the human proteome. Sci. (New York, N.Y.) 356 (6340), eaal3321. doi:10.1126/science.aal3321
Todi, S. V., Winborn, B. J., Scaglione, K. M., Blount, J. R., Travis, S. M., and Paulson, H. L. (2009). Ubiquitination directly enhances activity of the deubiquitinating enzyme ataxin-3. EMBO J. 28 (4), 372–382. doi:10.1038/emboj.2008.289
Tomaić, V., Pim, D., Thomas, M., Massimi, P., Myers, M. P., and Banks, L. (2011). Regulation of the human papillomavirus type 18 E6/e6ap ubiquitin ligase complex by the HECT domain-containing protein EDD. J. Virology 85 (7), 3120–3127. doi:10.1128/jvi.02004-10
Tomoshige, S., Nomura, S., Ohgane, K., Hashimoto, Y., and Ishikawa, M. (2018). Degradation of huntingtin mediated by a hybrid molecule composed of IAP antagonist linked to phenyldiazenyl benzothiazole derivative. Bioorg. Med. Chem. Lett. 28 (4), 707–710. doi:10.1016/j.bmcl.2018.01.012
Tomoshige, S., Nomura, S., Ohgane, K., Hashimoto, Y., and Ishikawa, M. (2017). Discovery of small molecules that induce the degradation of huntingtin. Angew. Chem. - Int. Ed. 56 (38), 11530–11533. doi:10.1002/anie.201706529
Trempe, J. F., Chen, C. X. Q., Grenier, K., Camacho, E. M., Kozlov, G., McPherson, P. S., et al. (2009). SH3 domains from a subset of BAR proteins define a ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol. Cell 36 (6), 1034–1047. doi:10.1016/j.molcel.2009.11.021
Tsai, Y. C., Fishman, P. S., Thakor, N. v., and Oyler, G. A. (2003). Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J. Biol. Chem. 278, 22044–22055. doi:10.1074/jbc.M212235200
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A., et al. (2015). Proteomics. Tissue-based map of the human proteome. Sci. (New York, N.Y.) 347 (6220), 1260419. doi:10.1126/science.1260419
van Roon-Mom, W. M. C., Pepers, B. A., ’t Hoen, P. A. C., Verwijmeren, C. A. C. M., den Dunnen, J. T., Dorsman, J. C., et al. (2008). Mutant huntingtin activates Nrf2-responsive genes and impairs dopamine synthesis in a PC12 model of Huntington’s disease. BMC Mol. Biol. 9, 84. doi:10.1186/1471-2199-9-84
Vasic, V., Denkert, N., Schmidt, C. C., Riedel, D., Stein, A., and Meinecke, M. (2020). Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol. Res. 22 (3), 274–281. doi:10.1038/s41556-020-0473-4
Vittal, V., Shi, L., Wenzel, D. M., Scaglione, K. M., Duncan, E. D., Basrur, V., et al. (2015). Intrinsic disorder drives N-terminal ubiquitination by Ube2w. Nat. Chem. Biol. 11 (1), 83–89. doi:10.1038/nchembio.1700
Wade, B. E., Wang, C.-E., Yan, S., Bhat, K., Huang, B., Li, S., et al. (2014). Ubiquitin-activating enzyme activity contributes to differential accumulation of mutant huntingtin in brain and peripheral tissues. J. Neurosci. 34 (25), 8411–8422. doi:10.1523/JNEUROSCI.0775-14.2014
Waelter, S., Boeddrich, A., Lurz, R., Scherzinger, E., Lueder, G., Lehrach, H., et al. (2001). Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol. Biol. Cell 12 (5), 1393–1407. doi:10.1091/mbc.12.5.1393
Walden, H., and Rittinger, K. (2018). RBR ligase-mediated ubiquitin transfer: A tale with many twists and turns. Nat. Struct. Mol. Biol. 25 (6), 440–445. doi:10.1038/s41594-018-0063-3
Wang, W., Duan, W., Riquicho, A. R. M., Jing, Z., Liu, T., Hou, X., et al. (2015). Genome-wide survey and expression analysis of the PUB family in Chinese cabbage (Brassica rapa ssp. pekinesis). Mol. Genet. Genomics 290 (6), 2241–2260. doi:10.1007/s00438-015-1075-x
Wang, M, and Pickart, C. M. (2005). Different HECT domain ubiquitin ligases employ distinct mechanisms of polyubiquitin chain synthesis. EMBO J. 24 (24), 4324–4333. doi:10.1038/sj.emboj.7600895
Wang, T. Y., Shen, C. H., Lin, S. Y., Hung, C. C., Hsu, L. C., Chen, G. C., et al. (2022). K48/K63-linked polyubiquitination of ATG9A by TRAF6 E3 ligase regulates oxidative stress-induced autophagy. Cell Rep. 38 (8), 110354. doi:10.1016/j.celrep.2022.110354
Wang, Y., Cong, Y., Wang, T., Zhong, X., Yang, S., Li, Y., et al. (2016). Genome-wide identification of soybean U-box E3 ubiquitin ligases and roles of GmPUB8 in negative regulation of drought stress response in arabidopsis. Plant Cell Physiology 57 (6), 1189–1209. doi:10.1093/pcp/pcw068
Wang, B., Zeng, L., Merillat, S. A., Fischer, S., Ochaba, J., Thompson, L. M., et al. (2018). The ubiquitin conjugating enzyme Ube2W regulates solubility of the Huntington’s disease protein, huntingtin. Neurobiol. Dis. 109, 127–136. doi:10.1016/j.nbd.2017.10.002
Watabe, K., Niida-Kawaguchi, M., Tada, M., Kato, Y., Murata, M., Tanji, K., et al. (2022). Praja1 RING -finger E3 ubiquitin ligase is a common suppressor of neurodegenerative disease-associated protein aggregation. Neuropathology 42, 488–504. doi:10.1111/neup.12840
Well, E. M., Bader, V., Patra, M., Sánchez-Vicente, A., Meschede, J., Furthmann, N., et al. (2019). A protein quality control pathway regulated by linear ubiquitination. EMBO J. 38 (9), e100730. doi:10.15252/embj.2018100730
Wijk, S. J. L., and Timmers, H. T. M. (2010). The family of ubiquitin-conjugating enzymes (E2s): Deciding between life and death of proteins. FASEB J. 24 (4), 981–993. doi:10.1096/fj.09-136259
Winborn, B. J., Travis, S. M., Todi, S. v., Scaglione, K. M., Xu, P., Williams, A. J., et al. (2008). The deubiquitinating enzyme ataxin-3, a polyglutamine disease protein, edits Lys63 linkages in mixed linkage ubiquitin chains. J. Biol. Chem. 283 (39), 26436–26443. doi:10.1074/jbc.M803692200
Yang, J. E., Xu, Q., Zhu, F., Ma, J., Loke, H.-K., Rollins, N., et al. (2013). Absolute quantification of E1, ubiquitin-like proteins and nedd8–mln4924 adduct by mass spectrometry. Cell Biochem. Biophysics 67 (1), 139–147. doi:10.1007/s12013-013-9625-5
Yang, H., Zhong, X., Ballar, P., Luo, S., Shen, Y., Rubinsztein, D. C., et al. (2007). Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin. Exp. Cell Res. 313 (3), 538–550. doi:10.1016/j.yexcr.2006.10.031
Yao, T., Xiao, H., Wang, H., and Xu, X. (2022). Recent advances in PROTACs for drug targeted protein research. Int. J. Mol. Sci. 23, 10328. doi:10.3390/ijms231810328
Yau, R. G., Doerner, K., Castellanos, E. R., Matsumoto, M. L., Dixit, V. M., Rape, M., et al. (2017). Assembly and function of heterotypic ubiquitin chains in cell-cycle and protein quality control. Cell 171, 918–933. doi:10.1016/j.cell.2017.09.040
Ye, H., Arron, J. R., Lamothek{, B., Cirilli, M., Kobayashi, T., Shevdeq, N. K., et al. (2002). Distinct molecular mechanism for initiating TRAF6 signalling. Nature 418, 443–447. doi:10.1038/nature00888
Ye, Y., and Rape, M. (2009). Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10 (11), 755–764. doi:10.1038/nrm2780
Yin, Q., Lin, S. C., Lamothe, B., Lu, M., Lo, Y. C., Hura, G., et al. (2009). E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16 (6), 658–666. doi:10.1038/nsmb.1605
Zeng, L., Tallaksen-Greene, S. J., Wang, B., Albin, R. L., and Paulson, H. L. (2013). The de-ubiquitinating enzyme ataxin-3 does not modulate disease progression in a knock-in mouse model of huntington disease. J. Huntingt. Dis. 2 (2), 201–215. doi:10.3233/JHD-130058
Zhang, G., Lu, Y.-N., and Wang, J. (2020). USP7 regulates ALS-associated proteotoxicity and quality control through the NEDD4L–SMAD pathway. Proc. Natl. Acad. Sci. 117 (45), 28114–28125. doi:10.1073/pnas.2014349117
Zhang, M., Botër, M., Li, K., Kadota, Y., Panaretou, B., Prodromou, C., et al. (2008). Structural and functional coupling of Hsp90-and Sgt1-centred multi-protein complexes. EMBO J. 27 (20), 2789–2798. doi:10.1038/emboj.2008.190
Zhao, T., Hong, Y., Yin, P., Li, S., and Li, X. J. (2017). Differential HspBP1 expression accounts for the greater vulnerability of neurons than astrocytes to misfolded proteins. Proc. Natl. Acad. Sci. U. S. A. 114 (37), E7803–E7811. doi:10.1073/pnas.1710549114
Zhi, X., and Chen, C. (2012). WWP1: A versatile ubiquitin E3 ligase in signaling and diseases. Cell. Mol. Life Sci. 69 (9), 1425–1434. doi:10.1007/s00018-011-0871-7
Zucchelli, S., Codrich, M., Marcuzzi, F., Pinto, M., Vilotti, S., Biagioli, M., et al. (2010). TRAF6 promotes atypical ubiquitination of mutant DJ-1 and alpha-synuclein and is localized to Lewy bodies in sporadic Parkinson’s disease brains. Hum. Mol. Genet. 19 (19), 3759–3770. doi:10.1093/hmg/ddq290
Zucchelli, S., Marcuzzi, F., Codrich, M., Agostoni, E., Vilotti, S., Biagioli, M., et al. (2011). Tumor necrosis factor receptor-associated factor 6 (TRAF6) associates with Huntingtin protein and promotes its atypical ubiquitination to enhance aggregate formation. J. Biol. Chem. 286 (28), 25108–25117. doi:10.1074/jbc.M110.187591
Keywords: Huntington’s disease, huntingtin, ubiquitin, neurodegenerative disease, ligase, deubiquitinating enzyme, proteostasis, proteasome
Citation: Sap KA, Geijtenbeek KW, Schipper-Krom S, Guler AT and Reits EA (2023) Ubiquitin-modifying enzymes in Huntington’s disease. Front. Mol. Biosci. 10:1107323. doi: 10.3389/fmolb.2023.1107323
Received: 24 November 2022; Accepted: 16 January 2023;
Published: 08 February 2023.
Edited by:
Peter Mabbitt, New Zealand Forest Research Institute Limited (Scion), New ZealandReviewed by:
Henry Paulson, Michigan Medicine, University of Michigan, United StatesMinoru Ishikawa, Tohoku University, Japan
Copyright © 2023 Sap, Geijtenbeek, Schipper-Krom, Guler and Reits. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric A. Reits, ZS5hLnJlaXRzQGFtc3RlcmRhbXVtYy5ubA==
†These authors share first authorship