 Juyi Li
Juyi Li Chengzhi He2†
Chengzhi He2† Chao Liu
Chao Liu Aiping Deng
Aiping Deng Lin Zhu
Lin Zhu- 1Department of Pharmacy, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China
- 2Department of Gastrointestinal Surgery, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
- 3Department of Gastroenterology, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China
- 4Department of Pain, The Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China
- 5Hubei Key Laboratory of Diabetes and Angiopathy, Hubei University of Science and Technology, Xianning, Hubei, China
- 6Department of Pediatrics, Tongji Hospital, Huazhong University of Science and Technology, Wuhan, Hubei, China
Introduction: Familial adenomatous polyposis (FAP) is the second most commonly inherited colorectal cancer (CRC) predisposition caused by germline mutations within the adenomatous polyposis coli (APC) gene. The molecular defects and clinical manifestations of two FAP families were analyzed, and individual prevention strategies suitable for mutation carriers in different families were proposed.
Methods and results: The pathogenic gene mutations were identified among the two families using whole-exome sequencing and verified with Sanger sequencing or quantitative polymerase chain reaction (qPCR). One novel (GRCh37:Chr5: 112145676–112174368, del, 28,692 bp) and a known (c.C847T:p.R283X) mutation in the APC gene were pathogenic mutations for FAP, according to the sequencing data and tumorigenesis pattern among the family members. The two mutations led to a premature translational stop signal, synthesizing an absent or disrupted protein product.
Conclusion: Our findings expand the known germline mutation spectrum of the APC gene among the Chinese population. This reaffirms the importance of genetic testing in FAP. Genetic consultation and regular follow-ups are necessary for the individualized treatment of cancer-afflicted families with APC expression deficiency. Additional work is required to develop safe and effective chemotherapy and immunotherapy for FAP based on the mutation type.
1 Introduction
Familial adenomatous polyposis (FAP) is a hereditary colorectal disease subtype with a poor prognosis. Colorectal cancer (CRC) predisposition syndrome is rare, characterized by 100 s–1,000 s of adenomas developing in the colon and rectum with their onset in childhood and adolescence. Moreover, CRC possesses associated extracolonic manifestations, such as desmoid tumors, dental and skin abnormalities, retinal spots, and malignant tumors from other organs (Carr and Kasi, 2022). Surgery is an effective therapy for FAP patients with colonic disorders, and regular chemotherapy has been shown to benefit FAP patients with the postoperative pathological diagnosis of adenocarcinoma (Tougeron et al., 2020). Recent colorectal cancer guidelines indicate that the type of pathogenic gene mutation is related to the patient’s prognosis and responses to chemotherapy and immunotherapy (Stjepanovic et al., 2019).
FAP follows an autosomal dominant inheritance pattern caused by the monoallelic mutation in the adenomatous polyposis coli (APC) gene (Recio-Boiles and Cagir, 2022). APC is a tumor suppressor gene located on chromosome 5q21, encoding a large scaffolding protein with functions in cell cycle regulation, apoptosis, transcription, and cell migration (Kim and Bodmer, 2021). APC comprises 16 exons; the last exon encodes nearly 70% of the APC protein. FAP frequency is approximately 1:8,000 in the general population with almost complete penetrance, affecting multiple family generations (Bisgaard et al., 1994).
Currently, more than 2,000 pathogenic variations in the APC gene are found, the majority of which are observed in the 5′-ends of exon 15, also called the mutation cluster region (MCR) (Schirosi et al., 2013; Jung et al., 2016), and are situated between 1286 and 1513 codons (Cetta and Dhamo, 2007). Codons 1309 and 1061 are hotspots for mutation, accounting for nearly 17% and 11% of all germline APC mutations, respectively (Leoz et al., 2015). Compared to mutation patients outside the MCR, those with MCR mutations typically have a worse prognosis, manifesting their condition early (Ghadamyari et al., 2021). In a family, identifying the specific APC mutation can help targeted sequencing testing for presymptomatic at-risk family members (Cruz-Correa et al., 2017).
Identifying patients and families at an extremely high risk of developing cancer can help reduce cancer occurrence and mortality. Several studies described an association between APC mutation localization and the phenotype among FAP patients (Heinen, 2010). The diagnosis and patient follow-up could be improved by connecting the genotypes to the phenotypes. However, there is a paucity of information on the genotypic spectrum and clinical characterization of FAP in China. One novel and one known APC mutation were reported in two Chinese families with FAP. Our study aimed to analyze the molecular defects and clinical manifestations in the two families, for appropriate personalized prevention strategies against all mutation carriers.
2 Results
2.1 Clinical characteristics
The detailed pedigree of family I is given in Figure 1A. The proband was a 34-year-old female, whose first symptom of altered bowel movement appeared at 31 years (July 2019). She underwent her first gastrointestinal endoscopy and treatment that year. This involved the endoscopic dissection of colonic polyps and gastric polypectomy. Biopsy indicated multiple tubular adenomas inside the ascending colon and rectum. The patient underwent three more gastrointestinal endoscopy examinations and treatment in the following 2 years. The last endoscopy examination (6 December 2021, Figure 1) showed multiple glandular polyps inside the gastric body and fundus, with tubular adenoma and adenomatous polyp inside the ascending colon and tubular adenoma in the transverse colon with mild-to-moderate heteroplasia of the focal glandular epithelium. However, no extracolonic manifestations appeared, such as desmoid tumors or dental and skin abnormalities. The possibility of classic FAP was highly suspected with her clinical manifestations, so whole-exome sequencing was conducted.

FIGURE 1. Clinical data of family I. (A) Pedigree structure of family I: the red ring depicts the proband, squares indicate male members, and circles represent female members. Black shading represents FAP individuals. The small diamonds denote the relatives whose DNA was available for testing. (B–D) Gastroscopy of the proband. (E–G) Colonoscopy of the proband. (H,I) Sanger sequencing analysis of the proband’s APC gene (c.C847T:p.R283X): the C base at position 847 of the APC gene is replaced by the T base. (J) Location of the p.R283X mutation in the secondary structures of the APC protein.
Family II is a typical and interesting large family. The detailed pedigree is given in Figure 2A, and the clinical characteristics of the family members are listed in Supplementary Table S2. The proband of family II sought medical attention for bloody stools at 41 years (July 2019). Gastrointestinal endoscopy showed multiple new organisms in the stomach and colon (Figure 2), and biopsy suggested adenocarcinoma of the transverse colon. The glandular epithelium of the mucosa showed moderate-to-severe dysplasia of approximately 40 cm from the anal margin. A fragmentary villous or serrated glandular epithelium was observed with low-grade intraepithelial neoplasia of approximately 10 cm from the anal margin. In August 2019, laparoscopic total colectomy and ileostomy were conducted. The pathological examination suggested the following: 1. multiple neoplasm and ulcerated areas were carcinomatous, with medium well-differentiated adenocarcinoma. The deepest cancer infiltration reached the outer membrane (the largest mass was 4 × 3.5 cm), with tumor plugs in the vascular area. 2. There were no cancer cells in the colon tissues of the two broken ends, and the local glandular epithelium of the broken ends indicated low-grade intraepithelial neoplasia. 3. Metastatic cancer was found in 3/14 of the peri-intestinal lymph nodes. Oxaliplatin chemotherapy was performed several times after the operation. Over the next year or so, multiple gastroscopies and colonoscopies were performed.
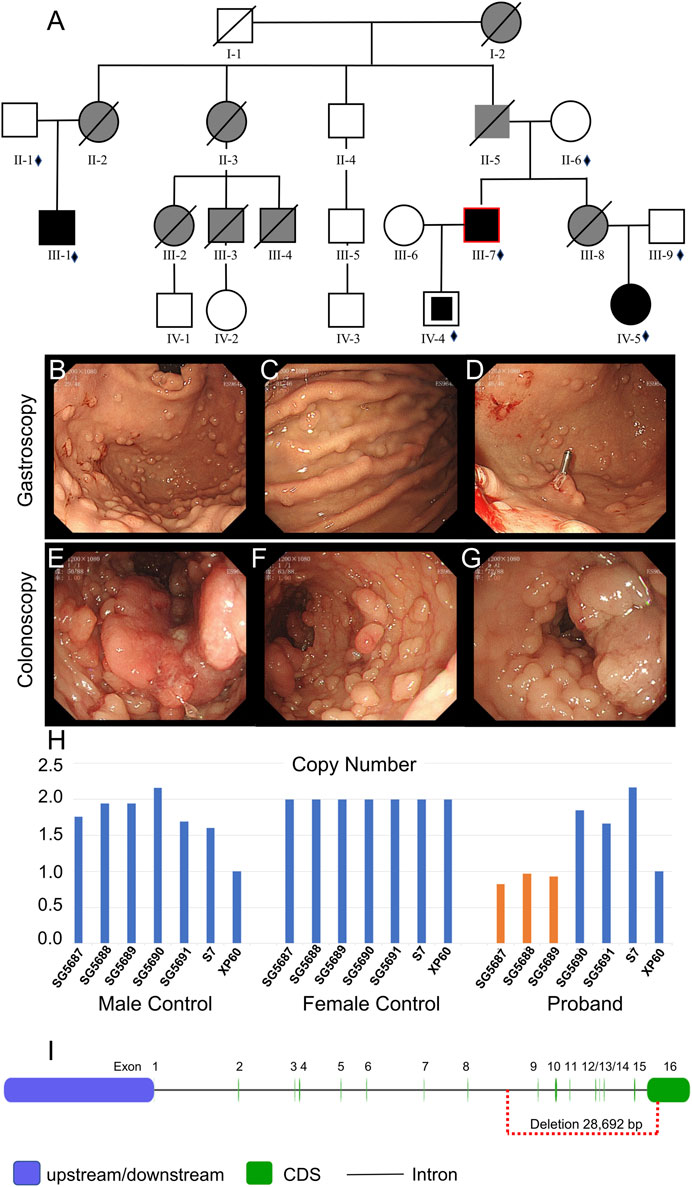
FIGURE 2. Clinical data of family II. (A) Pedigree structure of family II: the red ring indicates the proband, squares indicate male members, circles indicate female members, and crosses indicate deceased individuals. Dark shading represents individuals with FAP, partly dark shading represents mutation carriers, and gray shading represents CRC without the gene test. The small diamonds denote the relatives whose DNA was available for testing. (B–D) Gastroscopy of the proband. (E–G) Colonoscopy of the proband. (H) qPCR of the proband’s peripheral blood DNA to verify the CNV in APC (GRCh37:Chr5: 112145676–112174368, del, 28,692 bp). (I) Location of the GRCh37:Chr5: 112145676–112174368 delete mutation in the APC gene.
The father (II-5), aunts (II-2 and II-3), sister (III-8), and cousins (III-2 and III-3) of the proband were all diagnosed with colon or rectal cancer and were deceased. The brother of the proband (III-1) was hospitalized at 32 years (December 2020) due to irregular stools, diarrhea, and stool blood. Five gastrointestinal endoscopy examinations and treatments were performed, among which the gastrointestinal endoscopy examination performed in December 2020 is depicted in Figure 3. Gastroscopy (Figures 3A–C) indicated duodenal bulb ulcer (stage A2) and erosive gastritis (grade 2). Biopsy suggested severe chronic inflammation of the gastric antrum mucosa accompanied by erosive gastritis with mild activity. Colonoscopy (Figure 3D–F) revealed multiple neoplasms of the large intestine. EMR and argon coagulation were performed. Biopsy of the ascending colon revealed adenomatous polyps with multiple adenomatous polyps at the rectum and sigmoid colon junction. Rectal eminence was observed under a rectoscope (Figures 3G–I), and a biopsy revealed low-grade intraepithelial neoplasia of superficial mucosal glands with polyps inside the anal canal.
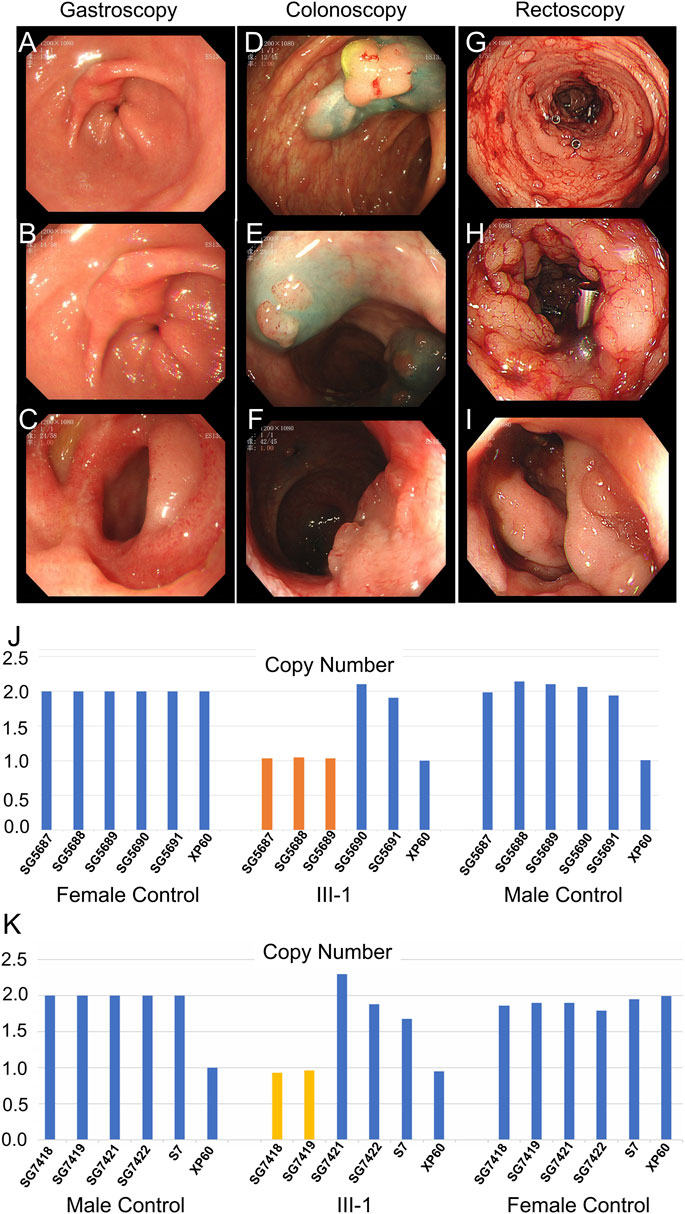
FIGURE 3. Clinical data of III-1 (the proband’s brother) of family II. (A–C) Gastroscopy of III-1. (D–F) Colonoscopy of III-1. (G–I) Rectoscopy of III-1. (J) qPCR of III-1’s peripheral blood DNA to verify the CNV in APC (GRCh37:Chr5: 112145676–112174368, del, 28,692 bp). (K) qPCR of III-1’s DNA extracted from newly developed polyps to verify the CNV in APC (GRCh37:Chr5: 112145676–112174368, del, 28,692 bp).
The 22-year-old niece (IV-5) of the proband underwent gastrointestinal polyp extraction at 17 years. After surgery, gastrointestinal endoscopy was performed several times. Gastroscopy indicated multiple gastric polyps treated with argon coagulation and erosive gastritis (grade 1) (Figures 4A–C). Biopsy depicted mild-to-moderate chronic inflammation of the gastric antrum mucosa showing mild activity. A colonoscopy (March 2021) showed multiple large intestine polyps, and EMR and argon coagulation were conducted (Figures 4D–F). Biopsy revealed adenomatous polyps inside the transverse colon. The 6-year-old son (IV-4) of the proband is currently asymptomatic; he is too young to develop clinical symptoms. Whole-exome sequencing was performed for proband II and quantitative polymerase chain reaction (qPCR) for the other family members associated with the disease characteristics of family II members.
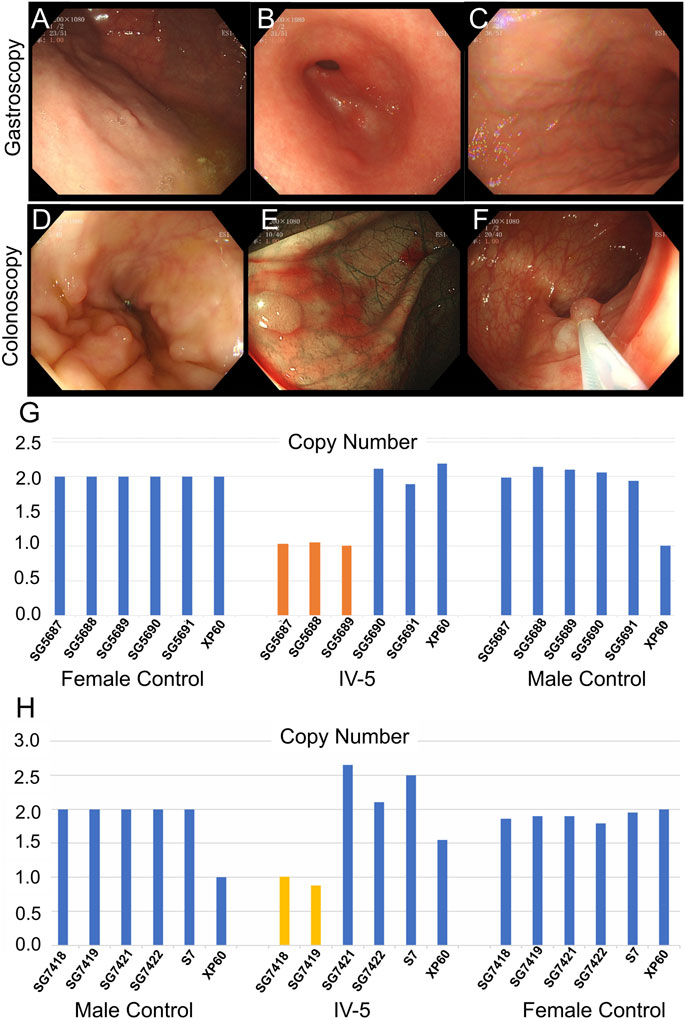
FIGURE 4. Clinical data of IV-5 (the proband’s niece) in family II. (A–C) Gastroscopy of IV-5. (D–F) Colonoscopy of IV-5. (G) qPCR of IV-5’s peripheral blood DNA to verify the CNV in APC (GRCh37:Chr5: 112145676–112174368, del, 28,692 bp). (H) qPCR of IV-5’s DNA extracted from newly developed polyps to verify the CNV in APC (GRCh37:Chr5: 112145676–112174368, del, 28,692 bp).
2.2 Genetic test results
The whole-exome sequencing data of the proband in family I are described in Supplementary Table S3A. Approximately 117,621 mutations were identified with 106,580 SNPs and 11,041 indels. The number of synonymous and missense mutations, new SNPs, and indels was 11,015, 9,886, 628, and 676, respectively. We identified a known mutation NM_000038:exon9:c.C847T:p.R283X of APC (rs786201856; accession: VCV000836957.3) (Hong et al., 2021a; Hong et al., 2021b). In this mutation, the C base at position 847 of the APC gene was replaced by the T base (Figure 1H/I). This led to the premature emergence of a stop codon, premature APC protein translation termination, and protein function loss (Figure 1J), showing the specific clinical significance of this mutation.
Sanger sequencing helped detect the APC mutation (NM_000038:exon9:c.C847T:p.R283X) in the proband and family members of the patient to provide genetic counseling. The germline mutation in APC was not observed in other family members (I-1, I-2, II-1, and III-1). Thus, the APC mutation of the proband is spontaneous and not passed on to her son.
Whole-exome sequencing was also performed on the proband in family II (Supplementary Table S3B). Approximately 114,508 mutations with 104,007 SNPs, 10,485 indels, and 16 copy number variations (CNVs) were identified. The number of new SNPs and indels was 656 and 656, respectively. We verified the proband harboring a new CNV loss within the APC tumor suppressor gene (GRCh37:Chr5: 112145676–112174368, del, 28,692 bp) (Figure 2I) using qPCR (Figure 2H). The brother (III-1), niece (IV-5), and son (IV-4) of the proband possessed the same mutation (Figure 3J; Figure 4G; Supplementary Figure S1A). The mutation was detected in the first two polypous tissues (Figures 3K, 4H). In contrast, this mutation was not observed in the other family members (Supplementary Figures S1B–D). Therefore, the substantial deletion of the APC gene in this family is the causative mutation for polyposis. FAP is a dominant syndrome caused by truncating mutations or large deletions.
3 Discussion
We identified one novel mutation (GRCh37:Chr5: 112145676–112174368, del) and a known mutation (NM_000038:exon9:c.C847T:p.R283X, rs786201856) within the APC gene in two typical FAP families, which were pathogenic for FAP.
APC is a tumor suppressor gene, promoting the rapid degradation of CTNNB1 (cadherin-associated protein, beta 1). APC participates in Wnt signaling as a negative regulator. Currently, more than 10,000 APC mutations have been identified. There are several important observations regarding APC mutations: 1) the vast majority of mutations discovered would lead to APC product truncation; 2) most mutations occurred in the first half of the coding sequence, and somatic mutations in colorectal tumors were concentrated in the MCR; 3) transition of cytosine to other nucleotides was the most common point mutations in the APC gene; and 4) there was a correlation between the germline mutations of FAP1 patients and colorectal polyps. For most adenomas and carcinomas in the colon and rectum and some in the stomach, inactivating both alleles of the APC gene is necessary as an early event.
During the diagnosis of colorectal cancer in the 34-year-old female patient, our patient from family I had more than 100 adenomatous colorectal polyps with gastric polyps. However, there were no additional extracolonic signs linked to FAP (desmoids, osteomas, cutaneous soft-tissue tumors, dental abnormalities, and CHRPE). The APC mutation is denoted by APC c.847C>T in the proband at the cDNA level and p.Arg283Ter (R283X) at the protein level. It has been reported in sporadic and syndromic adenocarcinoma patients within the COSMIC database (Tate et al., 2019) and is considered pathogenic by ClinVar (Landrum et al., 2018). Additionally, this mutation has been clarified in FAP patients as having adamantinomatous craniopharyngiomas.
The c.847C>T pathogenic mutation, also called p.R283X, affects the coding exon 8 of the APC gene and is caused by a C-to-T substitution at the nucleotide position 847. This mutation converts the amino acid within coding exon 8 from an arginine to a stop codon. This harmful variation exists in many families across several ethnic groups with familial adenomatous polyposis (Owen et al., 2018; de Oliveira et al., 2019). Other than the clinical information in the literature, this change is anticipated to cause function loss by nonsense-mediated mRNA decay (NMD) or premature protein truncation. This variant has been reported in 15 probands, meeting eight phenotype points, and in 50 probands with FAP not otherwise specified, meeting more than 16 phenotype points in total. It has been reported to segregate with FAP in five meioses from one family and in 31 members from one large FAP family (Mohamed et al., 2003). In summary, this variant meets the criteria to be classified as pathogenic for FAP based on the ACMG/AMP criteria applied. The mutation of the proband was not passed on to her son, and she is expecting her second child. However, the second child probably carried the disease-causing mutation. Genetic counseling was recommended according to her genetic mutation.
Family II is a typical FAP family. The genomic region of the APC gene is deleted out-of-frame in family II (GRCh37:Chr5: 112145676–112174368, del, 28,692 bp). The consequence could be a missing or disrupted protein product with a premature translational stop signal. This variation has not been documented among people with APC-related diseases. Pathogenic loss-of-function mutations in APC exist (Lagarde et al., 2010), which are labeled pathogenic based on these factors. The elder brother (III-1) and niece (IV-5) of the proband were mutant gene carriers; both were of marriageable age, but neither was married. They were concerned that their future children would carry the same gene mutation. Therefore, according to the principle of eugenics, genetic counseling is necessary, for the third generation of test-tube babies before pregnancy.
Nearly all FAP patients develop CRC if not identified and treated early (Half et al., 2009). Adjuvant chemotherapy and immunotherapy are required in most instances of FAP after total colectomy, similar to sporadic colorectal cancer and Lynch syndrome (Zhang et al., 2019; Koskenvuo et al., 2020; Kemp Bohan et al., 2021; Rohani et al., 2022). Cancer research significantly emphasizes the therapeutic targeting of abnormal beta-catenin activity. Creating medicines that target cancer cells, while having acceptable safety profiles, has remained a pharmacological challenge since Wnt signaling is a highly conserved system in normal cellular physiology. Early clinical trials of several medicines that inhibit upstream effectors in the Wnt signaling pathway have been undertaken, albeit with worries about off-target effects. In the phase 1 clinical trial of paclitaxel and vantictumab, a monoclonal antibody against Fzd receptors, was used to suppress Wnt signaling by preventing binding with all Wnt ligands. The trial indicated moderate efficacy among Wnt-upregulated metastatic breast (Diamond et al., 2020) and pancreatic (Davis et al., 2020) cancer. However, concerns have been raised about the safety of the drug concerning the bones. Furthermore, porcupine inhibitors, an enzyme that processes Wnt signaling proteins, have been created to combat Wnt-driven cancers. Most notably, WNT974 has demonstrated safety but limited efficacy in advanced solid tumors (Rodon et al., 2021). However, the effectiveness of focusing on upstream targets is still debatable due to tumor resistance with more downstream alterations, including CTNNB1- or APC-mutated malignancies. There are serious concerns about off-target consequences, such as damage to normal intestinal tissues and directly inhibiting beta-catenin or APC (Kahn, 2014; Zhang et al., 2016; Lai and Kahn, 2021; Wang et al., 2021). Hence, much research on cancer is required to create safe and efficient Wnt signaling inhibitors.
Our study contributes to identifying FAP genotypes in China, with significant implications for genetic counseling, diagnosis, cancer prevention, and treatment. Whole-exome sequencing is a rapid, accurate, and reliable technique to identify genetic variants in suspected FAP patients. It has various potential applications in the genetic testing of FAP-related tumors. Thus, abnormal Wnt signaling, including APC mutations, can be a promising target for the development of chemotherapy and immunotherapy against FAP.
4 Materials and methods
4.1 Patients
The Ethics Committee of the Central Hospital of Wuhan approved the study, with the ethical approval code of 2020-192. All subjects included in this analysis were informed in person, and their written informed consent was obtained. The two probands, diagnosed with FAP, were recruited from the Department of Gastrointestinal Surgery at the Central Hospital of Wuhan. The clinical diagnosis of FAP was confirmed by a gastroenterologist using multiple gastroscopic and colonoscopic biopsy reports, clinical investigations, and a detailed family pedigree.
4.2 Mutation analysis
We extracted the genomic DNA from the peripheral blood or newly developed polyps of each proband and part of the family members. DNA fragments were sequenced using a high-throughput sequencer (Illumina HiSeq 2500 Analyzer; Illumina, CA, United States). As previously demonstrated, single-nucleotide variant, insertion, and deletion queries were performed (Li et al., 2017). The reference genome for whole-exome sequencing was UCSC hg19, NCBI build 37. PCR amplification and Sanger sequencing confirmed the detected variants, which helped in segregation analysis.
Samples were detected using next-generation sequencing for suspected CNVs. The quantitative polymerase chain reaction system using Roche LightCycler® II 480 Probes Master mix, as per the manufacturer’s instructions, was applied to verify the result accuracy and determine the breakpoint positions. Primer sequences used are listed in Supplementary Table S1.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
The studies involving human participants were reviewed and approved by the Wuhan Central Hospital Ethics Committee. Written informed consent to participate in this study was provided by the participants’ legal guardian/next of kin.
Author contributions
AD and LZ conceived and designed the experiments. JL, CH, JG, and XW performed the experiments. JL, CH, JG, XW, and CL analyzed the data. JL and LZ wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Wuhan Municipal Health Commission (No. WX18M02), Central Guiding Local Science and Technology Development Special Project (No. 2022BGE272), and Guangdong Yiyang Healthcare Charity Foundation (JZ2022011).
Acknowledgments
The authors thank the patients, their families, referral physicians, and investigators for their cooperation and contribution.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2023.1234296/full#supplementary-material
References
Bisgaard, M. L., Fenger, K., Bülow, S., Niebuhr, E., and Mohr, J. (1994). Familial adenomatous polyposis (FAP): Frequency, penetrance, and mutation rate. Hum. Mutat. 3 (2), 121–125. doi:10.1002/humu.1380030206
Carr, S., and Kasi, A. (2022). “Familial adenomatous polyposis,” in StatPearls (Treasure Island (FL: StatPearls Publishing LLC).StatPearls publishing copyright © 2022.
Cetta, F., and Dhamo, A. (2007). Inherited multitumoral syndromes including colorectal carcinoma. Surg. Oncol. 16 (Suppl. 1), S17–S23. doi:10.1016/j.suronc.2007.10.013
Cruz-Correa, M., Pérez-Mayoral, J., Dutil, J., Echenique, M., Mosquera, R., Rivera-Román, K., et al. (2017). Hereditary cancer syndromes in latino populations: Genetic characterization and surveillance guidelines. Hered. Cancer Clin. Pract. 15, 3. doi:10.1186/s13053-017-0063-z
Davis, S. L., Cardin, D. B., Shahda, S., Lenz, H. J., Dotan, E., O'Neil, B. H., et al. (2020). A phase 1b dose escalation study of Wnt pathway inhibitor vantictumab in combination with nab-paclitaxel and gemcitabine in patients with previously untreated metastatic pancreatic cancer. Invest. New Drugs 38 (3), 821–830. doi:10.1007/s10637-019-00824-1
de Oliveira, J. C., Viana, D. V., Zanardo, C., Santos, E. M. M., de Paula, A. E., Palmero, E. I., et al. (2019). Genotype-phenotype correlation in 99 familial adenomatous polyposis patients: A prospective prevention protocol. Cancer Med. 8 (5), 2114–2122. doi:10.1002/cam4.2098
Diamond, J. R., Becerra, C., Richards, D., Mita, A., Osborne, C., O'Shaughnessy, J., et al. (2020). Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 184 (1), 53–62. doi:10.1007/s10549-020-05817-w
Ghadamyari, F., Heidari, M. M., Zeinali, S., Khatami, M., Merat, S., Bagherian, H., et al. (2021). Mutational screening through comprehensive bioinformatics analysis to detect novel germline mutations in the APC gene in patients with familial adenomatous polyposis (FAP). J. Clin. Lab. Anal. 35 (5), e23768. doi:10.1002/jcla.23768
Half, E., Bercovich, D., and Rozen, P. (2009). Familial adenomatous polyposis. Orphanet J. Rare Dis. 4, 22. doi:10.1186/1750-1172-4-22
Heinen, C. D. (2010). Genotype to phenotype: Analyzing the effects of inherited mutations in colorectal cancer families. Mutat. Res. 693 (1-2), 32–45. doi:10.1016/j.mrfmmm.2009.09.004
Hong, C. S., Elsamadicy, A. A., Fisayo, A., Inzucchi, S. E., Gopal, P. P., Vining, E. M., et al. (2021b). Comprehensive genomic characterization of A case of granular cell tumor of the posterior pituitary gland: A case report. Front. Endocrinol. (Lausanne) 12, 762095. doi:10.3389/fendo.2021.762095
Hong, C. S., Omuro, A., An, Y., Inzucchi, S. E., Kohli, A. A., McGuone, D., et al. (2021a). Sporadic adamantinomatous craniopharyngioma with double-hit somatic APC mutations. Neurooncol Adv. 3 (1), vdab124. doi:10.1093/noajnl/vdab124
Jung, S. M., Yoon, Y. S., Lim, S. B., Yu, C. S., and Kim, J. C. (2016). Clinicopathological features of familial adenomatous polyposis in Korean patients. World J. Gastroenterol. 22 (17), 4380–4388. doi:10.3748/wjg.v22.i17.4380
Kahn, M. (2014). Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13 (7), 513–532. doi:10.1038/nrd4233
Kemp Bohan, P. M., Mankaney, G., Vreeland, T. J., Chick, R. C., Hale, D. F., Cindass, J. L., et al. (2021). Chemoprevention in familial adenomatous polyposis: Past, present and future. Fam. Cancer 20 (1), 23–33. doi:10.1007/s10689-020-00189-y
Kim, J. C., and Bodmer, W. F. (2021). Genotypic and phenotypic characteristics of hereditary colorectal cancer. Ann. Coloproctol. 37 (6), 368–381. doi:10.3393/ac.2021.00878.0125
Koskenvuo, L., Ryynänen, H., and Lepistö, A. (2020). Timing of prophylactic colectomy in familial adenomatous polyposis. Colorectal Dis. 22 (11), 1553–1559. doi:10.1111/codi.15151
Lagarde, A., Rouleau, E., Ferrari, A., Noguchi, T., Qiu, J., Briaux, A., et al. (2010). Germline APC mutation spectrum derived from 863 genomic variations identified through a 15-year medical genetics service to French patients with FAP. J. Med. Genet. 47 (10), 721–722. doi:10.1136/jmg.2010.078964
Lai, K. K. Y., and Kahn, M. (2021). Pharmacologically targeting the WNT/β-Catenin signaling cascade: Avoiding the sword of damocles. Handb. Exp. Pharmacol. 269, 383–422. doi:10.1007/164_2021_523
Landrum, M. J., Lee, J. M., Benson, M., Brown, G. R., Chao, C., Chitipiralla, S., et al. (2018). ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062–d1067. doi:10.1093/nar/gkx1153
Leoz, M. L., Carballal, S., Moreira, L., Ocaña, T., and Balaguer, F. (2015). The genetic basis of familial adenomatous polyposis and its implications for clinical practice and risk management. Appl. Clin. Genet. 8, 95–107. doi:10.2147/TACG.S51484
Li, C. G., Jin, P., Yang, L., Zang, W. C., Kang, Q., et al. (2017). Germline mutations in patients with multiple colorectal polyps in China. J. Gastroenterol. Hepatol. 32 (10), 1723–1729. doi:10.1111/jgh.13776
Mohamed, Z., Ahmad, R., Yoke, N. S., Zakaria, Z., Ahmad, H., and Yew, T. H. (2003). A nonsense mutation in exon 8 of the APC gene (Arg283Ter) causes clinically variable FAP in a Malaysian Chinese family. Cancer Sci. 94 (8), 725–728. doi:10.1111/j.1349-7006.2003.tb01509.x
Owen, D. R., Wong, H. L., Bonakdar, M., Jones, M., Hughes, C. S., Morin, G. B., et al. (2018). Molecular characterization of ERBB2-amplified colorectal cancer identifies potential mechanisms of resistance to targeted therapies: A report of two instructive cases. Cold Spring Harb. Mol. Case Stud. 4 (2), a002535. doi:10.1101/mcs.a002535
Recio-Boiles, A., and Cagir, B. (2022). “Colon cancer,” in StatPearls (Treasure Island (FL: StatPearls Publishing LLC). StatPearls publishing copyright © 2022.
Rodon, J., Argilés, G., Connolly, R. M., Vaishampayan, U., de Jonge, M., Garralda, E., et al. (2021). Phase 1 study of single-agent WNT974, a first-in-class Porcupine inhibitor, in patients with advanced solid tumours. Br. J. Cancer 125 (1), 28–37. doi:10.1038/s41416-021-01389-8
Rohani, S., Suhada, N., Mohammad, Z., and Azhar, S. (2022). Traditional herbal medicine as adjunctive therapy for colorectal cancer: A scoping review. Traditional Med. Res. 7, 15–18. doi:10.53388/tmr20220127260
Schirosi, L., Pellegrino, M., Tarantino, P., Mauro, S., Tinelli, A., and Greco, M. (2013). A new germline stop codon mutation in exon 15 of the APC gene predisposing to familial adenomatous polyposis. Int. J. Biol. Markers 28 (4), e405–e408. doi:10.5301/jbm.5000042
Stjepanovic, N., Moreira, L., Carneiro, F., Balaguer, F., Cervantes, A., Balmaña, J., et al. (2019). Hereditary gastrointestinal cancers: ESMO clinical practice guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 30 (10), 1558–1571. doi:10.1093/annonc/mdz233
Tate, J. G., Bamford, S., Jubb, H. C., Sondka, Z., Beare, D. M., Bindal, N., et al. (2019). Cosmic: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 47 (D1), D941–d947. doi:10.1093/nar/gky1015
Tougeron, D., Sueur, B., Zaanan, A., de la Fouchardiére, C., Sefrioui, D., Lecomte, T., et al. (2020). Prognosis and chemosensitivity of deficient MMR phenotype in patients with metastatic colorectal cancer: An AGEO retrospective multicenter study. Int. J. Cancer 147 (1), 285–296. doi:10.1002/ijc.32879
Wang, Z., Li, Z., and Ji, H. (2021). Direct targeting of β-catenin in the Wnt signaling pathway: Current progress and perspectives. Med. Res. Rev. 41 (4), 2109–2129. doi:10.3390/foods10092109
Zhang, L., Jiang, B., Zhu, N., Tao, M., Jun, Y., Chen, X., et al. (2019). Mitotic checkpoint kinase Mps1/TTK predicts prognosis of colon cancer patients and regulates tumor proliferation and differentiation via PKCα/ERK1/2 and PI3K/Akt pathway. Med. Oncol. 37 (1), 5. doi:10.1007/s12032-019-1320-y
Keywords: familial adenomatous polyposis, adenomatous polyposis coli gene, whole-exome sequencing, copy number variations, genetic counseling
Citation: Li J, He C, Gong J, Wang X, Liu C, Deng A and Zhu L (2023) Identification of a novel CNV at the APC gene in a Chinese family with familial adenomatous polyposis. Front. Mol. Biosci. 10:1234296. doi: 10.3389/fmolb.2023.1234296
Received: 04 June 2023; Accepted: 10 July 2023;
Published: 27 July 2023.
Edited by:
Lin Zhang, Hefei Comprehensive National Science Center, ChinaReviewed by:
Farah Ballout, University of Miami Health System, United StatesMarco Bono, University of Palermo, Italy
Copyright © 2023 Li, He, Gong, Wang, Liu, Deng and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lin Zhu, emh1bGluaHVzdEBodXN0LmVkdS5jbg==; Aiping Deng, ZGFweXhiQDE2My5jb20=
†These authors have contributed equally to this work