 Stephanie Seneff
Stephanie Seneff Anthony M. Kyriakopoulos
Anthony M. Kyriakopoulos- 1Computer Science and Artificial Intelligence Laboratory, Massachusetts Institute of Technology, Cambridge, MA, United States
- 2Laboratory of Molecular Biology and Immunology, Department of Pharmacy, University of Patras, Rio-Patras, Greece
- 3Department of Research and Development, Nasco AD Biotechnology Laboratory, Piraeus, Greece
Deuterium is a natural heavy isotope of hydrogen, containing an extra neutron. Eukaryotic organisms have devised complex metabolic policies that restrict the amount of deuterium reaching the mitochondria, because it damages the ATPase pumps, leading to release of excessive reactive oxygen species and inefficiencies in ATP production. Human metabolism relies heavily on the gut microbiome to assure an abundant supply of deuterium depleted (deupleted) nutrients to the host. Mitochondrial dysfunction is a hallmark of many chronic diseases, and deuterium overload, often due to gut dysbiosis, may be a major factor contributing to this issue. In this paper, we explore the potential role of certain amyloidogenic proteins, including amylin, amyloid beta, the prion protein, huntingtin, and α-synuclein, in disease processes that result in the accumulation of deposits of protein fibrils, along with lipid membrane components of damaged mitochondria, which we argue may be a mechanism to sequester deuterium in order to reduce the deuterium burden in the tissues. We show how cardiolipin, an anionic lipid synthesized in mitochondria and localized to the mitochondrial membrane, may play a central role both in trapping deuterium in the mitochondrial membrane and in inducing protein misfolding to facilitate the formation of deuterium-rich deposits. We focus on the potential role of the amino acid histidine and its interaction with the mineral copper, both to catalyze certain essential reactions and to facilitate the misfolding of amyloidogenic proteins triggered by contact with anionic phospholipids, particularly cardiolipin, and especially in the outer mitochondrial membrane of deuterium-damaged mitochondria.
1 Introduction
Deuterium is a natural nonradioactive isotope of hydrogen. It contains a neutron as well as a proton, making it twice as heavy, and this change alters its biochemical and biophysical properties, compared to hydrogen. Deuterium is present in seawater at 155 parts per million, which seems small, but, because hydrogen is so common, deuterium is actually present in the blood at 5 or 6 times the concentration of calcium, and its level is much higher than the level of trace minerals such as zinc and copper. Eukaryotic cells have developed complex metabolic strategies to severely restrict deuterium levels in the mitochondria, due to the fact that it damages the ATPase pumps, which produce adenosine triphosphate (ATP), the energy currency of the cell (Boros et al., 2024; Olgun, 2007; Seneff and Kyriakopoulos, 2025a; Seneff and Kyriakopoulos, 2025b).
Nicotinamide adenine dinucleotide (NADH) plays an essential role in supplying deuterium depleted (deupleted) protons to the mitochondrial intermembrane space, through the action of NADH dehydrogenase, a flavoprotein enzyme complex also known as Complex I. The proton that it delivers has been previously scrubbed of deuterium through the action of isomerases to exchange deuterium with hydrogen in the cytoplasmic water and techniques exploited by flavoproteins such as proton tunneling to support a high deuterium kinetic isotope effect (KIE) (Seneff and Kyriakopoulos, 2025a). The mitochondria are constantly producing deupleted metabolic water from oxygen via oxidative phosphorylation, capturing the deupleted protons that pass through the ATPase pumps from the intermembrane space into the matrix (Seneff and Kyriakopoulos, 2025a). Maintaining low deuterium levels in mitochondrial water is an essential aspect of metabolism, and a failure in this regard leads to severe mitochondrial dysfunction and oxidative stress, associated with many chronic diseases (Bhatti et al., 2017).
Cytochrome P450 enzymes (CYPs), primarily localized to the endoplasmic reticulum or the mitochondria, oxidize a variety of different substrates, replacing a proton with a hydroxyl group in the substrate, splitting molecular oxygen, and producing water as the other product. The first step is usually the abstraction of hydrogen from a C-H bond. Many studies have shown that CYPs typically have a high deuterium KIE for hydrogen abstraction, which means that the water molecule will be deupleted. The KIE can be greater than 10, suggesting the involvement of proton-coupled electron transfer and/or proton tunneling (Atkinson et al., 1994; Guengerich, 2017).
There is considerable recent interest in the idea of treating cancer with DDW (Lu and Chen, 2024). Long-term consumption of DDW can protect mice from cancer, and DDW has been shown to prolong survival of human cancer patients and to protect from relapse. Deuterium depletion improved the median survival time of glioblastoma multiforme patients (Somlyai et al., 2023). It has been hypothesized that a reduction in deuterium levels in the sugar-phosphates in the DNA backbone preserves the stability of hydrogen bond networks, protecting against aneuploidy and strand breaks (Boros et al., 2016). A study comparing 56 patients with pancreatic cancer who were treated with deuterium depleted water with 86 untreated control patients showed a strong, statistically significant Pearson correlation between survival time and length and frequency of DDW treatment (Boros et al., 2021).
Exposure of cells to deuterium-hydrogen ratios that differ from the natural ratio of 1/6600 alter the expression of hundreds of cancer-related and kinase genes, reflecting the fact that deuterium levels play a significant role in metabolic policy. One hundred percent of 124 cancer-related genes were upregulated in cultured cells exposed to 300 ppm deuterium (nearly twice the normal level) (Kovács et al., 2022). In fact, a case has been made that cancer cells paradoxically play a protective role in the organism by sequestering deuterium and releasing deupleted nutrients such as lactate to support mitochondrial recovery of resident immune cells (Seneff and Kyriakopoulos, 2025a). It has been hypothesized that eukaryotic cells must have a sensor for the D/H ratio in their environment that can act as a signaling mechanism to control metabolic policy (Yaglova et al., 2020).
Short chain fatty acids (SCFAs) are essential metabolites normally produced in abundance by the gut microbes, and they play a pivotal role in gut-brain communication and in maintaining human health (Silva et al., 2020). A recent study on the importance of metabolites produced by the gut microbiome in regulating neurological disorders defined the “gut-brain axis” as follows: “The microbiota gut-brain axis (MGBA) is a term used to describe the link between the gut and the brain involving the interplay of several systems, mainly the autonomic, central, and enteric nervous systems and the hypothalamic-pituitary axis” (Swer et al., 2023).
The interplay between hydrogen-producing fermentative bacteria and hydrogen consuming reductive acetogens, methanogenic archaea and sulfate-reducing bacteria becomes imbalanced in association with many gut disorders, such as inflammatory bowel disease (IBD), small intestinal bacterial overgrowth (SIBO), irritable bowel syndrome (IBS), and colon cancer (Carbonero et al., 2012). Interestingly, it has been shown experimentally that the molecular hydrogen gas H2, produced by hydrogenogenic microbes, is severely depleted in deuterium, relative to the levels of deuterium in the molecules from which it was extracted. It was found that microbially produced H2 contained only 20% as much deuterium as is normally found in water (Krichevsky et al., 1961). The acetogenic bacteria, such as Faecalibacterium prausnitzii, use H2 to reduce carbon dioxide to acetate, and to produce the derived SCFAs propionate and butyrate (Parsaei et al., 2021). These SCFAs, especially butyrate, are normally the primary nutrients for the colonocytes, and they can be expected to be extremely deupleted due to the deupleted protons they sourced from microbially-produced H2. An imbalance in the gut microbiome can lead to a deficiency in butyrate, which can cause metabolic stress for the colonocytes, and can even impact neurons in the brain via the gut-brain axis (Hodgkinson et al., 2023). Butyrate, functioning both as a histone deacetylase inhibitor and as a ligand for G-protein coupled receptors, can alter gene expression in the brain, preventing neurodegeneration and promoting regeneration (Bourassa et al., 2016).
In our recent publication, we developed a hypothetical but novel argument that there are a small number of organic molecules that may be uniquely configured to support deuterium trapping on carbon atoms (Seneff et al., 2025a). One class of molecules that we considered are those that contain an imidazole ring, most notably, the amino acid histidine. C2 in the imidazole ring is positioned between two nitrogen atoms, and it has a unique ability to exchange its proton with a deuteron in an irreversible spontaneous reaction. A study on peptide dendrimers containing histidine residues demonstrated experimentally that C2 of the imidazole ring of histidine was the only carbon atom in the dendrimer that was able to displace its proton with a deuteron when the dendrimer was immersed in heavy water. By contrast, all of the nitrogen and oxygen atoms were rapidly fully deuterated. However, once the molecule was placed in regular water under acidic conditions, all of the oxygen and nitrogen atoms immediately lost their bound deuterons, whereas the histidine residues retained theirs (Sheveleva et al., 2019). This property of the imidazole ring suggests to us the possibility that it might be useful in biological organisms as a deuterium trap. We have proposed that certain molecules that contain an imidazole ring, particularly one with a high pKa, may be able to permanently sequester deuterium, helping to reduce the deuterium burden in mitochondria (Seneff et al., 2025a).
Notably, the hydrogen/deuterium exchange (HDX) rate of a given imidazole ring depends on the concentration of OD− (O(2H)-; the deuteroxyl anion that is the equivalent of a hydroxyl anion) in the medium, but it is also dramatically reduced when histidine residues are phosphorylated. The rate is also substantially slower in metal-bound histidine residues. Furthermore, the maximum rate of exchange occurs when the pH is high, and its value exponentially increases with the increase in the pKa of the imidazole ring itself (Miyagi and Nakazawa, 2024). Histidine residues that are buried within hydrophobic regions of the protein have poor access to water and therefore exchange very slowly. It is also conceivable that enzymatic action might greatly accelerate HDX rates. All these factors could play a role in controlling whether a histidine residue in a protein might be able to capture and secure a deuteron, given sufficient time, even under physiological conditions.
Another interesting class of carbon atoms that may serve as deuterium traps are the bis-allylic carbon atoms, notably present in both microbial breakdown products of haem and in polyunsaturated fatty acids (PUFAs). Bis-allylic carbon atoms are carbon atoms in a carbon chain that are single-bonded on both sides to carbon atoms that are double-bonded to their other neighbor. PUFAs, particularly those found in the mitochondrially-localized lipid, cardiolipin, are particularly attractive candidates for deuterium trapping. Cardiolipin is tightly integrated with the ATPase pumps in the inner membrane of the intermembrane space. It is plausible that it may be able to trap deuterons that pass through the membrane before they reach the pumps, serving as a shield or a filter (Seneff et al., 2025b).
In this paper, we develop some more new hypotheses concerning a potential role for PUFAs in protecting mitochondria from deuterium overload. In particular, cardiolipin, a novel phospholipid synthesized in the mitochondria, may serve to trap deuterium that enters the intermembrane space before it reaches the ATPase pumps. When cardiolipin becomes oxidized, it migrates to the outer membrane, where it can interact with cytoplasmic amyloidogenic proteins to facilitate the formation of pores in the mitochondrial membrane, and, ultimately, to trap mitochondrial debris from aged mitochondria within fibrillar extracellular deposits such as Lewy bodies (Seneff et al., 2025b). There is a compelling story around amylin, a protein whose misfolding is a core feature of diabetes. α-Synuclein, amyloid-β, and huntingtin are other proteins that may follow the same principles for trapping deuterium. We review the role of histidine residues, with particular emphasis on their interaction with copper, in facilitating the cascade of events that arise, we argue, due to severe deuterium overload in the mitochondria. Whether the imidazole rings in these amyloidogenic proteins can capture and sequester deuterium is an open question, but further experimental research will be necessary to resolve it. We will not address this question further in this paper, as peer reviewed papers on that topic are lacking.
2 Hydrogen peroxide in the peroxisome and the mitochondria: a source of deupleted water?
In this section, we will argue that hydrogen peroxide plays an important role in delivering deupleted water to the mitochondria, through a coordinated action between the peroxisome and the mitochondria. Furthermore, the supply of hydrogen peroxide from the peroxisome to the mitochondria increases as cells age (Titorenko and Terlecky, 2010).
Hydrogen peroxide (H2O2) is a non-radical reactive oxygen species that plays a powerful role as a signaling molecule to mediate redox reactions. The peroxisome is a hub for the H2O2 signaling network (Lismont et al., 2019). The peroxisome is responsible for the β-oxidation of long chain fatty acids and the synthesis of ether-phospholipids such as plasmalogens, bile acids and the PUFA docosahexaenoic acid (DHA) (Van Veldhoven, 2010). Many of the reactions that take place in the peroxisome ultimately reduce molecular oxygen to H2O2, and subsequently to H2O.
Catalase (CAT) is an essential enzyme in the peroxisome that converts two molecules of H2O2 into two molecules of water and one molecule of molecular oxygen. H2O2 is commonly produced as a consequence of the action of superoxide dismutase (SOD), which converts two molecules of superoxide and two protons from the water to H2O2 and molecular oxygen. Hence the net result of a cascade reaction of SOD and CAT is to convert 4 molecules of superoxide and four protons to 2 molecules of water and 3 molecules of molecular oxygen. This is an important process for protection from superoxide damage to tissues, but it also may serve to supply deupleted water to the organelles.
It can therefore be predicted that the water that is produced will be deupleted, for two reasons. First, when HDO ionizes, it is more inclined to split into OD− and H+, rather than OH− and D+, because deuterium binds more strongly to oxygen than does hydrogen. Therefore, protons in water will carry less deuterium than hydroxyls. More importantly, SOD has a very high solvent deuterium KIE, due to a proton-coupled electron transport mechanism. A study on a nickel-catalyzed SOD enzyme showed that it had a solvent KIE of 20 – favoring protons over deuterons by a factor of 20. The authors argued that a similar effect would be expected for CuZn-SOD, the SOD enzyme that operates in the peroxisome (Shearer, 2013).
Mitochondria and lysosomes interact through direct membrane contact sites, enabling bidirectional communication and regulation of both organelles. The contacts are dynamic, constantly forming and then breaking. Impairments in the formation of these contacts are associated with age-related diseases, specifically neurodegeneration and lysosomal storage diseases (Rizzollo and Agostinis, 2025). Membranes of organelles represent a diffusion barrier for H2O2, but do not block its transport (Laporte et al., 2020). Since H2O2 freely diffuses across membranes, it is easy for H2O2 produced in the peroxisome to reach a mitochondrion in close contact. In mitochondria, treatment with H2O2 rapidly, but in an indirect way, inactivates mitochondrial aconitase, an essential enzyme in the citric acid cycle (Bulteau et al., 2003). Evidence that the peroxisomal H2O2 reaches the mitochondria comes from experiments that showed that the suppression of CAT in the peroxisome decreased the mitochondrial aconitase activity by 85% within 24 h (Walton and Pizzitelli, 2012).
The mitochondria normally have a good defense mechanism against H2O2, which involves the oxidation of glutathione by glutathione peroxidase and its subsequent reduction by glutathione reductase. Glutathione reductase delivers one proton from the solution and the other from NADPH to the two sulfur atoms of glutathione disulfide (GSSG). Mitochondrial NADH kinase converts NADH to NADPH, consuming one ATP molecule in the process. Since mitochondrial NADH carries a deupleted proton (which is delivered to the intermembrane space by NADH dehydrogenase), the proton in NADPH will also be deupleted (Seneff and Kyriakopoulos, 2025a). Furthermore, glutathione reductase is a flavoprotein, and flavoproteins generally have a high deuterium KIE, as has been confirmed experimentally for glutathione reductase (Vanoni et al., 1990). What this all means is that the water molecules that are produced by glutathione reductase will be deupleted.
In general, oxidative stress mediated by H2O2 is considered as a key pathogenesis factor in neurodegenerative diseases. Relevant studies on the use of deuterium depleted water (DDW) against H2O2-mediated oxidative stress show that cells pretreated with DDW resist apoptosis, have a reduced production of ROS, and show increased activities of SOD, Cu/ZnSOD and CAT when exposed to ROS (Wu et al., 2020). This indicates that cells, and specifically mitochondria, that suffer from deuterium overload would be more susceptible to oxidative stress mediated by H2O2. It would therefore be essential for the cell to use the mechanism of deupletion proposed in this section to provide neuroprotection and counterweigh neurodegeneration mediated by deuterium overload.
Thus, we can argue that one important role that the peroxisome plays is to extract deupleted protons from solution and deliver them to the mitochondria, and, ultimately, to the ATPase pumps, in the form of H2O2. The mitochondria provide two more deupleted protons to synthesize metabolic water.
Figure 1 schematizes the process by which the peroxisome generates abundant deuterium depleted water through β-oxidation of fatty acids, followed by its conversion to deupleted water and oxygen via the catalytic activity of SOD and CAT in the peroxisome, as well as glutathione peroxidase in the mitochondria.
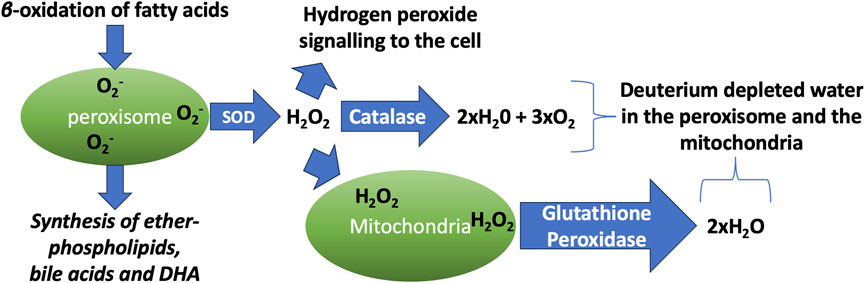
Figure 1. Schematic illustration of the processes that take place in the peroxisome and the mitochondria, which produce vital metabolites while generating abundant deuterium depleted water. DHA: docosahexaenoic acid. SOD: superoxide dismutase.
Age-associated mitochondrial dysfunction is regarded as a pivotal factor in the aging process. As cells age, CAT forms a tetrameric molecule in the cytosol and remains inactive. This results in increased levels of H2O2 in the peroxisome, which can then damage mitochondria, particularly if they have insufficient antioxidant defenses, such as depleted glutathione levels (Walton and Pizzitelli, 2012). However, this increase provides a mechanism to deliver deupleted water to the mitochondria, which may be a beneficial aspect to compensate for insufficient synthesis of metabolic water by the ATPase pumps, due to deuterium overload.
3 Mitochondria, cardiolipin, pyroptosis, and deuterium
Mitochondria serve broad functions in the cell, including cellular metabolism, calcium buffering, signaling pathways and regulating apoptotic cell death. Cardiolipin is a unique glycerophospholipid that is synthesized in the mitochondrial matrix and then inserted into the inner membrane of the intermembrane space. There, it stabilizes protein complexes and facilitates oxidative phosphorylation, and it also regulates the release of cytochrome c from mitochondria to launch an apoptotic cascade. Upon oxidation, cardiolipin migrates to the outer membrane, where it can come into close contact with amyloidogenic proteins in the cytoplasm. The interaction of cardiolipin with amyloid oligomers favors pore formation in the membrane, which causes osmotic swelling, loss of the voltage potential, and release of pro-apoptogenic factors. Besides the amyloidogenic proteins, several other proteins have also been shown to be able to punch holes in mitochondria through interaction with cardiolipin, including members of the Bcl-2 family, cobra venom cardiotoxins and bacterial toxins (Vassallo, 2025).
Pyroptosis is a cell death program that involves disruption of the plasma membrane and the release of pro-inflammatory cytokines into the external milieu. Gasdermin D (GSDMD) is a central protein controlling pyroptosis. It is activated by caspase-1, and it forms pores in the plasma membrane, inducing cell lysis (Liu et al., 2016). In response to endotoxemia, cardiolipin becomes oxidized by reactive oxygen species produced by complex II in cardiomyocytes. Oxidation and externalization of cardiolipin then induces GSDMD oligomerization and pore formation in mitochondria, a very early stage in the pyroptosis cascade (Tang et al., 2024).
Cardiolipin is tightly associated with ATPase pumps (Eble et al., 1990). It has been proposed that it buffers protons as they enter the membrane (Haines and Dencher, 2002). However, it may also be the case that it is able to trap and sequester deuterons before they reach the ATPase motor, where they can cause a stutter and induce reactive oxygen release. In neurons, cardiolipin is especially enriched in long-chain PUFAs, particularly docosahexaenoic acid (DHA), which has five bis-allylic carbon atoms. Bis-allylic carbon atoms are defined as carbon atoms in a chain that are single-bonded to the left and to the right to carbons that are double-bonded to their other neighbor: -C=C-C*-C=C-. These carbon atoms are unusual in that they are far more willing to give up their bound protons than carbon atoms in other configurations. They are the carbons in PUFAs that initiate and sustain the peroxidation chain reaction that produces a number of reactive products with a powerful role in the inflammatory response. In each step in the cascade reaction, a bis-allylic hydrogen is removed by a hydroxyl radical, and molecular oxygen is then added to form a highly reactive lipid peroxyl radical that propagates the chain reaction (Ng et al., 2022).
Remarkably, PUFAs that are deuterated at their bis-allylic carbon atoms have been found to be highly effective at quenching the chain reaction and suppressing the inflammatory response. Deuterium doping of PUFAs can have a profound effect in protecting from inflammation, as has been shown experimentally (Li et al., 2022). Cardiolipin may be able to situate itself in the inner membrane in such a way as to be able to eventually populate its bis-allylic carbon atoms with deuterium, and in this fashion protect the mitochondrion from oxidative damage (Seneff et al., 2025a). This also implies that, once it migrates to the outer membrane, it may have succeeded in permanently trapping one or more deuterium atoms, because deuterons bound to bis-allylic carbon atoms very rarely exchange back with hydrogen (Navratil et al., 2018).
Persistent accumulation of damaged mitochondria due to impaired mitophagy is a likely antecedent to neurodegenerative disease. Cardiolipin externalization activates mitophagy, and this is essential for rapidly sequestering injured mitochondria for clearance in the autophagosomes (Kagan et al., 2016). A team of researchers in Beijing, China conducted elegant experiments involving PrP106-126 exposure to mouse neuroblastoma cells grown in culture. They found that inhibition of the externalization of cardiolipin during treatment with Prp106-126 peptide aggravates mitochondrial oxidative stress by significantly intensifying the dysfunction of complex I and complex III. This demonstrates that externalization of cardiolipin affords protection from oxidative stress (Yang et al., 2023).
4 The two faces of lipid peroxidation: Good or bad for human health?
Peroxidation of lipids is a physiological process continuously ongoing in the organism. When it extends beyond limits, however, it leads to a wide spectrum of disease pathogenesis. The products of oxidized lipids, especially those derived from the PUFAs in lipid membranes that are attacked by reactive oxygen species (ROS) and reactive nitrogen species (RNS) are the ones that produce harm (Ramana et al., 2019). Unambiguously, the main diseases produced by excessive lipid peroxidation range from neurodegenerative diseases (manifesting as Alzheimer’s disease (AD), Parkinson’s disease (PD) and others), cardiovascular diseases (manifesting as atherosclerosis, ischemia and heart failure), autoimmune disorders, cancer, kidney and respiratory disease, and other inflammatory and immunological disorders (Ramana et al., 2019).
However, while excessive peroxidation of lipids is implicated in the pathogenesis of myriads of diseases, the same peroxidation of lipids provides the necessary signalling molecules for the regulation of very important cellular functions that incorporate the balance and regulation of autophagy (Gęgotek and Skrzydlewska, 2024) and the decision on survival or cell death (Zheng et al., 2024). Autophagy, i.e., the internal mechanism for recycling, biosynthesis and degradation of constituents and organelles via lysosomes, is probably the most essential universal process for the survival of all kinds of cells (He and Klionsky, 2009). Central to an organism’s homeostasis is regulated cell death. It is the same kinds of ROS and NOS modalities that oxidize and alter the structures of lipids in cellular membranes that signal for various kinds of cell death, apoptosis, ferroptosis, pyroptosis and many other death pathways (Zheng et al., 2024). Deviations in this important signalling disbalances the homeostasis that protects from tumorigenesis, infection and excessive forms of inflammation (Lee et al., 2023).
The study of A. Ayala et al. excellently describes the signalling molecules generated by lipoxygenase, cyclooxygenase and cytochrome-P40 peroxidation activities on lipids that regulate the whole of an organism’s homeostasis, from blood pressure to promotion of cell survival and proliferation, to reproduction and metabolism (Ayala et al., 2014). Under physiologic lipid peroxidation activities, the cells promote their antioxidant activities to remain alive and functional. It is only when the process of lipid peroxidation accelerates beyond the normal limits that the cells do not have the necessary repair mechanisms to survive the oxidative damage and therefore die (Volinsky and Kinnunen, 2013).
A good example of a PUFA lipoxygenase-peroxidation secondary end product that regulates the survival or the death of cells is 4-hydroxynonenal (4-HNE). 4-HNE in high concentrations is one of the most toxic and mutagenic metabolites of lipid peroxidation for the cell (Esterbauer et al., 1990). It is accused of causing mitochondrial dysfunction that mediates premature ageing, diabetes, and neurodegeneration, leading to AD (Bartman et al., 2024). However, at normal concentrations, 4-HNE is a major cell survivor-transcription factor regulator. It induces nuclear erythroid 2-related factor (Nrf-2), activating protein-1 (AP-1), the family of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and the peroxisome proliferator-activated receptors (PPAR). Moreover, it activates the mitogen-activated protein kinases (MAPK) and Epidermal Growth Factor Receptor (EGFR)/Akt pathways, and protein kinase C. In this way the formation and release of 4-HNE through lipid peroxidation orchestrates the anti-oxidant-cell stress response, which is an ancient cellular defense mechanism, from microbes to humans, for survival and to counterweight disease (Nicolaou et al., 2010; Rath and Haller, 2011). Apart from cellular antioxidant defense, 4-HNE is a major regulator of normal proliferation and the cell cycle. Importantly, there are numerous studies showing, through the regulation of the aforementioned transcription factors and more, that 4-HNE causes cell cycle arrest and inhibits the proliferation of malignant cells through the p53 and p21 dependent pathways (for review see (Ayala et al., 2014)). Furthermore, in the same review, several studies describe the importance of normal 4-HNE concentrations in the regulation of autophagy and senescence.
Another important signalling metabolite that is generated through the cyclooxygenase-mediated peroxidation of PUFAs is malondialdehyde (MDA). MDA is a relatively stable oxidizing molecule that shows similar cytotoxicity and mutagenic potential with 4-HNE, reacting with various proteins and biomolecules. MDA is often used as an oxidative stress biomarker for the prognosis of various pathologic conditions (including cancer) (Gaweł et al., 2004; Fraile-Martínez et al., 2023). However, low levels of MDA are significant for the regulation of gene expression. These studies indicate that moderate levels of MDA are important for the regulation of insulin secretion (Wang et al., 2014), and the production of collagen (García-Ruiz et al., 2002). Significantly the transcription factors SP1 and SP3 that are influenced by MDA regulate the maturation of a cell and are involved in apoptosis and cell cycle arrest (chromatin remodelling). Their loss leads to severe and lethal forms of thrombocytopenia (Meinders et al., 2015).
Summarizing, although the peroxidation of lipids is responsible for the development of many diseases, it is a natural process and moderate to low levels of products have beneficial effects for the organism’s health, holding the wheel for a regulated autophagy, cell proliferation, haematologic homeostasis and a balanced programmed cell death. One way of reducing or even diminishing the capability of PUFAs to become oxidized is to deuterate them, i.e., to replace and exchange their hydrogen atoms with deuterium. In vitro and in vivo studies in animals have shown that the use of deuterated PUFAs can lower the peroxidation effects on producing atherosclerosis, control hyperlipidaemia, and reduce cholesterol levels in blood (García-Ruiz et al., 2002).
Although these are beneficial signals for human health, many questions are raised for the whole of the organism’s health since the deuterated lipids will accumulate in a vast number of cells and in their membranes, as in the case of myelin, and will cause a significant alteration in the metabolism of the deuterated host organism (Baumann et al., 2022). Most worrisome is the possibility that supplementation with pre-deuterated lipids will interfere with the ability of cardiolipin to filter out deuterons before they reach the ATPase pumps. This will have significant effects on essentials for life activities such as gestation. Moreover, the excessive accumulation of deuterated lipids in cell membranes will alter their physiologic characteristics of thickness and lamellar spacing (structure of membranes), and this is tightly linked to the development of neurodegeneration, infectious disease pathology, and protein lipid tumorgenicity amongst many others (Torres et al., 2021). These are similar pathological factors that are tightly regulated by the forementioned secondary peroxidation PUFA signalling metabolites 4-HNE and MDA, when their concentrations are moderate to low.
It would therefore be crucial to re-think the use of deuterated lipids to counterweight lipid, especially PUFA, peroxidation, as their application may seem promising but can hide severe and unpredictable pathologies raised in the recipient host. This is especially concerning for humans, as the molecular signalling in human disease is far more complex than in many animal models (Akhtar, 2015).
Figure 2 schematizes the two faces of lipid peroxidation.
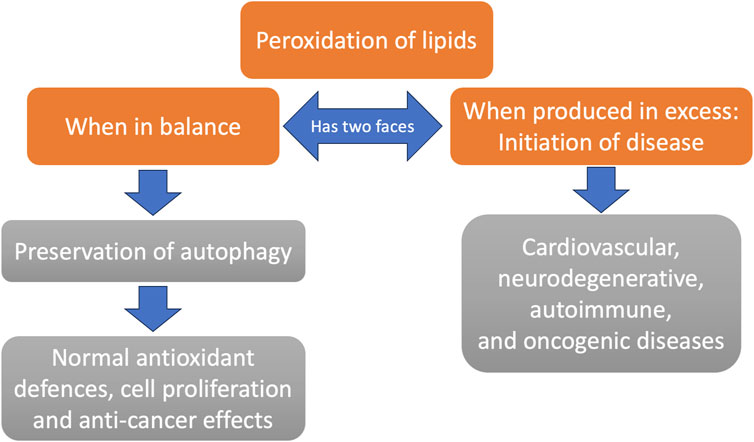
Figure 2. The two faces of lipid peroxidation. While membrane lipid peroxidation is generally viewed as a disease process, there are powerful signaling mechanisms in place that lead to a healthy outcome (e.g., preservation of autophagy) unless it occurs in excessive amounts. Excessive membrane peroxidation is associated with many chronic diseases.
5 Copper’s roles in the Mitochondria and in protein misfolding
Copper is a highly reactive metal, but it is an essential cofactor for several vital enzymes, notably two that are active in the mitochondria. Copper, zinc superoxide dismutase (CuZn-SOD) detoxifies superoxide by converting it to the less reactive hydrogen peroxide and oxygen. It depends on both copper and zinc as cofactors, but only copper regulates its activity (Harris, 1992). The copper ion is bound to either three or four histidine side chains in the enzyme (Hart et al., 1999). When exposed to high levels of its own reaction product, H2O2, CuZn-SOD’s activity is suppressed, via oxidation of His-118 to form 2-oxo-histidine (Uchida and Kawakishi, 1994).
Cytochrome c oxidase, also known as complex IV, at a final step in the respiratory chain, catalyzes the reduction of oxygen to water. In cytochrome c oxidase, the catalytic core is formed by three subunits containing three copper atoms. Two copper atoms are held in place in a bimetallic center in subunit 2 by six histidine residues that are highly conserved among multiple family members. A third copper atom is associated with the haem group of subunit 1 (Horn and Barrientos, 2008). Histidine residue 503 (His-503) is crucial for the enzyme’s catalytic action. A water molecule is securely fixed between His-503 and Asp-91 and is linked to two water arrays that reach the surface of the molecule. These two residues together trap the proton that is transferred from the surface through the water arrays. Once oxidized, His-503 releases the proton to Asp-91, and Asp-91 then donates the proton to the dioxygen reduction site (Muramoto et al., 2007).
Biological organisms have developed sophisticated strategies for copper transport and copper delivery to the enzymes that require it, but these mechanisms can become defective, leading to both copper toxicity and copper deficiency, sometimes even co-occurring. Min et al. wrote in the abstract of a paper published in 2024: “The pathophysiology of ALS [Amyotrophic Lateral Sclerosis] involves many signs of a disruption in copper homeostasis, with both excess free levels and functional deficiency likely occurring simultaneously” (Min et al., 2024). Copper interacts with the key aggregation-prone proteins associated with ALS, including TAR DNA binding protein 43 (TDP-43), CuZn-SOD, and fused in sarcoma (FUS). Mutant SOD1 aggregates more readily when copper is present (Tokuda and Y Furukawa, 2016). Unbound copper and excessive ROS can accelerate protein aggregation. This can lead to copper sequestration in misfolded fibrils along with copper deficiency in the mitochondria, causing impaired energy production (Min et al., 2024).
At least one histidine residue is present in the majority of amyloidogenic proteins, and these histidine residues interact with copper in important ways, influencing their conformational shape. Histidine is a unique amino acid with respect to its interactions with copper. A study on cultured HaCaT keratinocytes exposed to CuSO4 assessed the potential for each of the 20 coding amino acids to ameliorate copper toxicity (Ha et al., 2023). Although cysteine binds more strongly to copper than does histidine, histidine was the only amino acid that protected the cells from death due to copper overload. Despite its potent ability to chelate copper, cysteine had no cytoprotective effects. Cysteine is one of the three amino acids in glutathione, and it has been shown that glutathione greatly facilitates copper uptake by cells via its ability to bind copper (Maryon et al., 2013). But, unlike histidine, it is not able to protect from copper toxicity.
Amyloid beta (Abeta) is the peptide that is associated with Alzheimer’s disease. High levels of copper are accumulated near Abeta peptide-containing plaque in the brain. Several histidine residues are involved in Cu2+ coordination in Abeta. The N-terminus and His-13 are crucial for Cu2+ binding, and His-6 and His-14 are also implicated. Copper accumulates in the senile plaque where it is tightly bound to Abeta (Syme et al., 2004). This suggests that the plaque may serve a role in sequestering free copper to protect from its toxic effects. In vitro, the Abeta-Cu2+ complex, in the presence of a reducing agent, reduces oxygen to produce H2O2, and this involves recycling copper from Cu2+ to Cu1+ and back to Cu2+ along with the oxidation of the reducing agent (Jiang et al., 2010). Abeta has a very high affinity for Cu2+. It has been well established that Abeta, when incubated with copper and oxygen, produces H2O2, while reducing Cu2+ to Cu1+, during the period when it slowly transforms into soluble oligomers (Huang et al., 1999). However, in later stages of the incubation period, the H2O2, levels decrease, suggesting that the fibrils that are ultimately formed may clear the H2O2. Abeta also binds to zinc, but at a lower affinity, and it does not bind at all to manganese or iron (Mayes et al., 2014).
In 2014, Mayes et al. decided to specifically investigate the ability of Abeta(1–42) peptide fibrils to remove H2O2 and to determine the exact reaction mechanisms (Mayes et al., 2014). Incubation of Abeta(1–42) fibrils with Cu2+ at a one-to-one ratio with H2O2 revealed that these fibrils were able to clear the H2O2, while causing the formation of carbonyl groups in the peptide. Carbonyl formation is routinely exploited to detect the production of reactive oxygen species such as the hydroxyl radical. Thus, these authors concluded that Abeta fibrils may not be completely innocuous, as is widely believed (Mayes et al., 2014).
In early work, excess copper has been linked to neurotoxicity in association with PD (Cruces-Sande et al., 2019) and Alzheimer’s disease (X Li et al., 2023), and in a Parkinsonian model in studies on C. elegans (Weishaupt et al., 2024). A comprehensive review of copper’s role in neurodegenerative disease, published in 2024, found substantial evidence that disrupted copper homeostasis can be linked to AD, PD, Huntington’s disease, ALS, Wilson’s disease, Menkes disease, prion diseases, and multiple sclerosis (Zhong et al., 2024). There is a complex network of copper chelators and transporters that regulate cellular copper uptake, distribution, and metabolism (Zhong et al., 2024).
Free copper is extremely reactive through Fenton chemistry, and it stimulates the secretion of pro-inflammatory cytokines such as interleukin-1 (IL-1), IL-4, and tumor necrosis factor α (TNFα) in both the brain and the blood (Tokuda et al., 2009). Figure 3 is a schematic of the processes by which impaired copper homeostasis can lead to neurodegenerative disease, and the hypothesis that copper trapping in plaque regions may be protective.
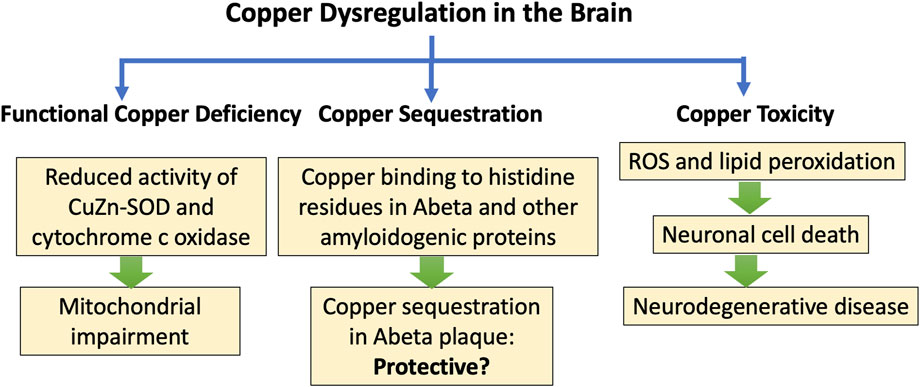
Figure 3. Schematic of processes leading to neurodegenerative disease and protein misfolding in association with impairments in copper transport and delivery. A proposed hypothesis is that copper sequestration in plaque regions may be protective.
6 The role of copper-histidine binding in amyloidogenic prion proteins
Histidine residues of proteins are unique in their function. Due to their imidazole ring, they possess unique acid-base abilities and under pH 6 are usually in a state of protonation, i.e., being positively charged (Dokainish and Kitao, 2016). However, the acid-base dissociation constant (Ka) of imidazole rings of histidine residues often reflects the overall protein catalysis functions. The pKa, of imidazole rings varies according to the microenvironment that protein folds in, and this huge pKa fluctuation enables the protein’s histidine binding with metals (Miyagi and Nakazawa, 2024). One of the most biologically significant protein-histidine residue/imidazole binding is with copper (Cu2+). In fact, the Cu2+-histidine interaction (binding) in nature can match the importance of iron (Fe2+) binding with proteins, and the consequent haem interactions that support the very existence of life (Walton et al., 2023).
Intriguingly, the interactions of N-terminal histidine residues of the prion protein (PrP) mainly with Cu2+ sequestration and accumulation in diseased brain (T Frączyk, 2021) has long been identified to be responsible for the neurotoxic effects of PrP in neurons producing prion disease. Each of the eight amino acid sequences that are repeated four times in the octarepeat domain of the N terminal of PrP contains one histidine. The four histidine residues, through their imidazole rings, coordinate the occupancy and binding of one atom of Cu2+ at neutral pH (7.4) (Chattopadhyay et al., 2005). However, by all means, the binding of Cu2+ is not necessarily pathogenic since it has also been found to produce neuroprotection due to the cis interaction with the C terminal of PrP.
The histidine residues provide a tertiary structural interaction with the two domains of PrP and copper, that is also protective in function due to involvement of the conserved central region of the protein (Roseman et al., 2020). Moreover, recent experimental results show that two more histidine residues located in the C terminal region of PrP provide protection against neurotoxicity, when this is exerted from the N terminal region (Schilling et al., 2020). Also, histidine 96 and histidine 111 (human PrP numbering) that are located outside the octarepeat region, are also actively involved in PrP-Cu2+ binding (Sánchez-López et al., 2018). Therefore, Cu2+ coordination by the histidine residues in PrP is central to either neuroprotection or neurodegeneration. Conformational changes that correspond to the binding of PrP with Cu2+ are associated with prion disease, and these are usually induced by alterations to Cu2+-to-histidine occupancy within the PrP molecule. These structural deformations occur during native PrP (PrPc) misfolding to give the pathogenic PrPsc variant that produces amyloidogenesis (Sánchez-López et al., 2018).
Native human PrP can bind up to six Cu2+ ions, in a manner involving all the imidazole groups of the histidine residues located in the N-terminal repeats as well as His96 and His111. The prerequisite for this interaction is that, in these locations of the PrPc backbone, two of the amide groups are deprotonated. This makes the histidine-Cu2+ interactions and subsequent conformational changes of PrPc upon Cu2+ binding pH dependent (Staś et al., 2023). Moreover, the histidine imidazole Cu2+ binding (anchoring) is followed when Cu2+ is in a coordinated state, and this is achieved by the presence of two deprotonated backbone amides (Rivillas-Acevedo et al., 2013). The same counts when the occupancy of Cu2+ involves less than 6 ions. In this situation, multiple histidine residues of the octarepeats are involved in the binding reaction. However, the involvement of a deprotonated amide chelation reaction preceding the Cu2+ anchoring is still necessary. Moreover, other histidine residues of the neighboring PrPs can be involved in Cu2+ anchoring, with an end result of PrP formation of oligomers (Yang et al., 2008). When the α-helices of PrPc refold into β-sheets, this gives the pathogenic PrPsc misfolded variants, and then the prion proteins become pathogenic, resulting in neurodegeneration (Derreumaux, 2001).
7 Akkermansia, butyrate, GLP-1, diabetes and obesity
In this section, we will describe the role that the gut microbes, particularly Akkermansia muciniphila, play in protecting the host from diabetes and obesity, via the release of a protein that induces the secretion of glucagon-like peptide 1 (GLP-1) from intestinal L-cells (Rodrigues et al., 2022). This is important not only because GLP-1 receptor agonists have become very popular as pharmaceutical drugs to treat diabetes and obesity (JY Wang et al.), but also because, as we will see later, GLP-1 prevents amyloidosis of the amyloidogenic small peptide hormone amylin, which is normally released along with insulin from the pancreatic β-cells when blood sugar levels are elevated. Amylin misfolding is a strong feature of type II diabetes (Asthana et al., 2018).
A muciniphila is a commensal bacterium in the human gut that feeds only on the sulfomucins produced by the colonic goblet cells. It breaks down the sulfomucins into small organic metabolites that can then be used by hydrogenogenic bacteria, mainly Bacteroidetes and Firmicutes strains, as a source for hydrogen gas production through fermentation (Wolf et al., 2016). The hydrogen gas that is produced is then used as a reducing agent to convert carbon dioxide into organic matter, primarily the three major SCFAs, acetate, propionate and butyrate. This process of hydrogen gas recycling is a crucial component of gut microbial metabolism that is essential for the wellbeing of the host. A. muciniphila colonizes the infant gut shortly after birth, and, in the healthy gut, their colonization is maintained throughout the lifespan. Butyrate is a favored fuel of the colonocytes (Salvi and Cowles, 2021), likely in part because it is expected to be an excellent source of low-deuterium protons. Butyrate is also passed into the circulation from the gut, so it or its derivatives can also fuel mitochondria in distant tissues, including the brain (Bourassa et al., 2016).
There is an inverse correlation between the abundance of A. muciniphila in the gut and metabolic disease, and there is recent excitement among practitioners about the potential of A. muciniphila probiotics to treat several different metabolic diseases, including obesity, type II diabetes, cardiovascular disease, and nonalcoholic fatty liver disease (Yan et al., 2021; Rodrigues et al., 2022). Despite the fact that it consumes sulfomucins, its presence in the gut is usually associated with a greater abundance of sulfomucins, perhaps because it facilitates the supply of butyrate to the colonocytes, supplying them with the energy they need to produce sulfomucins, and also, importantly, maintaining low deuterium levels in the mitochondria of the colonocytes (Seneff and Kyriakopoulos, 2025b).
A. muciniphila secretes a protein called P9 that has been shown to upregulate the expression of glucagon-like peptide-1 (GLP-1), an incretin hormone secreted by the intestinal L-cells (Buteau, 2008). GLP-1 has many positive effects on gut homeostasis, including stimulation of insulin secretion, slowing of gastric emptying, appetite suppression, and increasing β-cell proliferation (Buteau, 2008; Müller et al., 2019). GLP-1 receptor agonists are a promising therapy for diabetes and have also recently enjoyed extraordinary success as a therapy to support weight loss, by reducing the appetite (Wang et al., 2023).
GLP-1 is known as an insulinotropic hormone, because it induces the release of insulin from pancreatic β-cells in response to rising serum glucose levels (Müller et al., 2019). In a study on a rat insulinoma cell line, a commonly used model for pancreatic β-cells, treatment with GLP-1 induced mitochondrial biogenesis, through a mechanism that involved increased cAMP activation. The expression of a key regulator of mitochondrial biogenesis was increased dramatically after 1 h of treatment of the cultured cells with GLP-1, and the expression level remained elevated for 48 h after treatment (Kang et al., 2015). Methylglyoxal is a highly reactive glycating agent produced as a by-product of glycolysis, and excess exposure can induce apoptosis in pancreatic β-cells. GLP-1 was shown in an in vitro experiment to protect β-cells from methylglyoxal-induced apoptosis, by improving mitochondrial function and suppressing prolonged activation of AMP-dependent protein kinase (AMPK) (Chang et al., 2016).
Upon glucose stimulation, the pancreatic β-cells release insulin into the circulation from secretory granules, where it is temporarily stored, along with another short peptide called amylin (islet amyloid polypeptide; IAPP), a 37-residue peptide hormone that slows gastric emptying and promotes satiety. In type II diabetes, the number of docked granules is about 1/3 lower than in normal cells, and the rate of glucose-stimulated exocytosis is only 20% of that in normal cells (Gandasi et al., 2018). Remarkably, GLP-1 mobilizes insulin secretory granules to dock at the plasma membrane, which then increases the rate of release of insulin from the granules (Kwan and Gaisano, 2005). IAPP is of great interest here, because, like amyloid β, it is an amyloidogenic protein (Press et al., 2019). In the next section, we review what is currently known about IAPP and its critical role in pancreatic β-cell destruction and type II diabetes.
8 IAPP and diabetes
Type II diabetes can be characterized as a protein-misfolding disease, and the protein involved in its pathology is hIAPP/amylin. More than 90% of patients with type II diabetes have amyloid deposits of IAPP in their pancreas, as determined by post-mortem studies. As discussed previously, monomeric hIAPP binds to specific receptors in the membranes of cells to regulate glucose homeostasis and gastric emptying (Engel, 2009).
hIAPP has several positively charged residues on its N-terminal side, which are involved in the interaction of hIAPP with negatively charged lipids (Engel, 2009). His-18 is central to the misfolding potential of hIAPP. In fact, the isoform found in both rats and mice is missing His-18, and their versions of the protein are immune to the formation of toxic oligomers or fibrils (Asthana et al., 2018). Mice with transgenic overexpression of human IAPP develop toxic IAPP oligomers intracellularly within the secretory pathway, whereas overexpression in mice of rat IAPP does not (Gurlo et al., 2010). His-18 is important for both fibril formation and membrane interaction. Protonation of His-18 is crucial for its interaction with and entry into lipid membranes, and this leads to membrane disruption and toxicity (Cao et al., 2013).
Cu2+ binds to amylin, promoting the formation of toxic oligomers. Cu2+ induces a conformational change into a more compact structure containing α-helices that resists HDX and is protected from enzymatic hydrolysis by insulin degrading enzyme (IDE). Preincubation with copper sulfate increases its toxic effects on neurons (Sinopoli et al., 2014). An imidazole nitrogen atom in His-18 is the main binding site for Cu2+ (Magrì et al., 2016). Membranes, especially those with an abundance of negatively charged lipids, are able to catalyze hiAPP transformation into an α-helical structure, especially in the presence of free copper, which then leads to membrane penetration and pore formation (Engel, 2009). Anionic lipids drastically accelerate aggregation, an effect that is substantially weaker for sphingophospholipid and zwitterionic phospholipids (Sitton et al., 2025). Even low levels of anionic lipids promote membrane permeabilization due to pore formation. Cholesterol at physiological levels significantly reduces the rate of amyloid formation and protects membranes from leakage (Zhang et al., 2017).
A study on human kidney embryonic cells grown in vitro revealed that culturing the cells with copper and amylin resulted in greater stress to the cells than culturing with amylin alone. Multilayer lamellar compact structures were observed on the cell membranes and were associated with amylin aggregates. Copper ions were localized only in the vicinity of the amylin aggregates. Fibrillar structures were also observed, but they were not associated with copper ions, suggesting that copper promotes the formation of toxic oligomers that disrupt membranes (Congiu et al., 2021).
Impaired autophagy plays an important role in IAPP toxicity, due to the accumulation of excessive amounts of IAPP oligomers intracellularly. Autophagosomes can efficiently dispose of IAPP small oligomers in p62-containing vacuoles to keep them from becoming toxic (Gupta and Leahy, 2014).
A seminal paper by Gurlo et al. published in 2010 provides tremendous insight into the early steps in the misfolding of IAPP that eventually leads to β-cell destruction. These authors found that toxic IAPP oligomers initially appear within the granules in the secretory pathway, where they disrupt the membranes of the granules, leading to their release into the cytoplasm. They then come into close proximity with mitochondrial membranes, which they also penetrate and disrupt. The destruction of the mitochondria may indeed be an early step in the pathology, eventually leading to β-cell death and a reduction in the number of functioning β-cells. These authors used antibody labelling of islets isolated from hIAPP transgenic mice to confirm the hypothesis that toxic IAPP oligomers form at all steps of the secretory pathway. They also confirmed that toxic oligomers were identified intracellularly in β-cells from humans with type II diabetes. Mitochondria adjacent to the oligomers exhibited disrupted membranes, whereas mitochondria that were remote from the aggregates were relatively normal. The authors concluded in the abstract that “IAPP toxic oligomers are formed intracellularly within the secretory pathway in T2DM. Most striking, IAPP toxic oligomers appear to disrupt membranes of the secretory pathway, and then when adjacent to mitochondria, disrupt mitochondrial membranes” (Gurlo et al., 2010).
Figure 4 illustrates schematically the process by which Akkermansia spp protect the host from type II diabetes. This is one of the best examples available of a direct pathway between the gut microbiome and disease caused by protein misfolding.
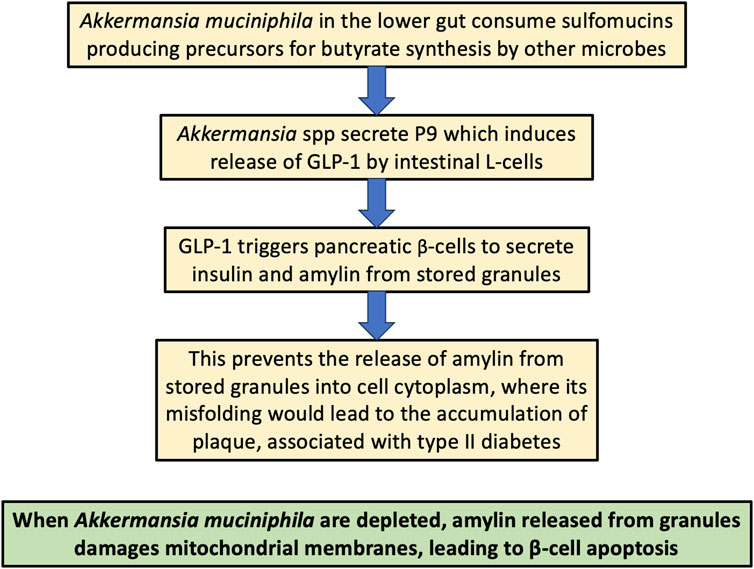
Figure 4. Schematic of the process by which Akkermansia muciniphila in the gut protect the host from type II diabetes, both through supplying deupleted butyrate to the colonocytes and by stimulating L-cells to release the hormone GLP-1, which in turn facilitates the release of amylin from stored granules into the extracellular space, protecting from its build-up in the cytoplasm, which can lead to destruction of mitochondria and β-cells through its amyloidosis.
Mitochondria are central to the process that couples glucose metabolism to insulin exocytosis, and their dysfunction is an early feature of diabetes (Supale et al., 2012). Mitochondrial autophagy (mitophagy) is critical for the clearance of damaged mitochondria, and it is primarily regulated by two proteins, PTEN-induced putative kinase 1 (PINK1) and the E3 ubiquitin ligase Parkin. Genetic mutations in these proteins are linked to PD, but they have also been found to play a role in diabetes pathogenesis and associated β-cell dysfunction (Qu et al., 2011; Jin and Youle, 2012).
The antioxidant defenses of β-cells are weak compared to other cell types, which leaves them more susceptible to oxidative stress (Kaufman et al., 2015). 12-Lipoxygenase overexpression in pancreatic islets induces oxidative stress, which can damage membrane lipids through a lipid peroxidation chain reaction (Tersey et al., 2014). We will have more to say later in this paper about mitochondrial membranes and how they are altered in response to oxidative stress, specifically focusing on a potential role for the anionic lipid cardiolipin both in protecting the ATPase pumps from deuterium toxicity and in facilitating the removal of dysfunctional mitochondria through the action of amyloidogenic proteins.
9 Amyloid-beta and Alzheimer’s disease
Amyloid beta (Abeta) is the amyloidogenic peptide that is associated with Alzheimer’s disease. It is derived by cleavage of the transmembrane amyloid precursor protein (APP), and it is found as both a 40-peptide and a 42-peptide sequence. Abeta-42 is considered to be much more toxic than Abeta-40 (O'Brien and Wong, 2011). Amyloid plaque is primarily formed of Abeta fibrils, and much research on potential therapies has centered on creating anti-amyloid monoclonal antibodies, with the aim of clearing the plaque by targeting the peptide for degradation (Cummings et al., 2024).
A fascinating paper by Serra-Vidal et al. examined the dynamics of Abeta aggregation over time using HDX. They found that, at the beginning of the aggregation process, nearly all of Abeta-40’s amides had access to free exchange, whereas only about 33 of the 41 amides available in Abeta-42 underwent exchange. Over time, both forms experienced further reduction in the availability of amides for exchange, with Abeta-42 showing a faster timeline for reduction. They stated that this observation provided evidence that Abeta-42 is more prone to aggregation than Abeta-40. As time elapsed, they identified three phases where different populations became dominant. They associated the early phase with monomers, the middle phase with soluble oligomers, and the final phase with insoluble fibrils. In parallel, they tracked the neurotoxicity of the peptides, and neuronal viability showed a clear dip during the period when the oligomers were most prominent. This experiment was an elegant approach to demonstrating that it is likely the oligomers that are the toxic species (Serra-Vidal et al., 2014).
As early as 1993, it was proposed that Abeta has the ability to form channels within lipid membranes, and this may be its primary toxic effect (Arispe et al., 1993). Lipids are able to uniquely modify the secondary structure and toxicity of Abeta oligomers and fibrils. Zhaliazka et al. (2023) examined the effects of phosphatidylcholine, cardiolipin, and cholesterol on Abeta structure. They found that all three of these molecules strongly accelerated the rate of fibril formation compared to a lipid-free environment. They wrote: “Remarkably, under the optimized micelle conditions, Aβ42 assembles into oligomers that insert into lipid bilayers as well-defined pores and adopt a specific structure with characteristics of a β-barrel arrangement” (Zhaliazka et al., 2023). Both cardiolipin and cholesterol drastically increased β-sheet formation in Abeta-42 oligomers, which greatly increased the toxicity compared to lipid-free environments (Zhaliazka et al., 2023). Anionic cardiolipin had the strongest effect. By handling Abeta oligomers as if they were membrane proteins, Serra-Batiste et al. were able to demonstrate that Abeta-42 assembles into stable oligomers that get incorporated into membranes as pores, causing neurotoxicity (Serra-Batiste et al., 2016).
A remarkable study published in 2015 used extensive and sophisticated imaging techniques to assess whether there is a localized relationship between regions of the brain showing hypometabolism and regions with plaque buildup. The last sentence in their abstract read “Thus we conclude that regional fibrillar amyloid deposition has little to no association with regional hypometabolism.” (Altmann et al., 2015). Increasingly, researchers in the field are concluding that the fibrillar plaque is not toxic, and, in fact, may serve a useful role by clearing the toxic form of Abeta. This also explains why drugs that have been shown to reduce plaque burden do not actually improve disease symptoms, and they come with a significant side-effect burden (Karran and Hardy, 2014; Waite, 2015).
The transition metals copper, zinc and iron are all enriched in Abeta plaques. Abeta interactions with copper and zinc can control the aggregation state of Abeta. Abeta-Cu2+ complexes are redox-active, and therefore they can release reactive oxygen (Smith et al., 2006). The coordination sphere around a bound Cu2+ ion in Abeta resembles the active site of superoxide dismutase. Methylation of either of the nitrogen atoms in the imidazole rings of the histidine residues 6, 13 and 14 prevents formation of bridging histidine moieties. Remarkably, the modified peptides were four times more effective at generating H2O2 (behaving like a SOD), but they were not neurotoxic. This was probably due to the fact that the modification inhibited the interaction between the peptides and cell surface membranes. This suggests that the toxicity of Abeta may be due to its effects at the membrane (Tickler et al., 2005).
Abeta plaque accumulates in the extracellular space, but Alzheimer’s is also strongly associated with another misfolded protein, phosphorylated tau. Neurofibrillary tangles, containing abundant tau, accumulate inside the neurons. Tau pathology depends upon the presence of cardiolipin in the outer mitochondrial membrane. Tau oligomers bind to cardiolipin, and this binding leads to the creation of membrane pores, causing mitochondrial swelling, cytochrome c release, and a reduction in membrane potential (Camilleri et al., 2020). We therefore propose that it is plausible that the migration of oxidized cardiolipin to the outer membrane, due to excessive release of reactive oxygen species by the ATPase pumps, is the initiating factor in tau tangle formation and an early event in AD progression.
Figure 5 illustrates schematically the processes that eventually lead to Alzheimer’s disease, where the major players are Abeta, phosphorylated tau, and cardiolipin.
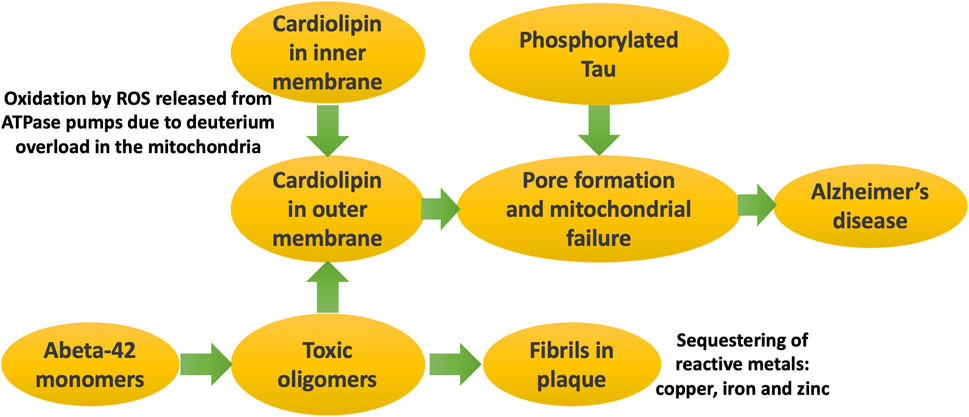
Figure 5. Schematic of processes that eventually lead to Alzheimer’s disease. It is the soluble oligomers that are the toxic form of Abeta. They disrupt the mitochondrial outer membrane, especially when cardiolipin has migrated there due to its oxidation by ROS generated during ATP synthesis. Phosphorylated tau forms pores in the mitochondrial membrane, triggered by cardiolipin accumulation there. Mitochondrial dysfunction in critical brain regions leads to symptoms of Alzheimer’s disease.
10 Huntingtin and Huntington’s disease
Mitochondrial dysfunction plays a major role in neurodegenerative diseases, including Huntington’s disease, AD, and PD. Disruption of synaptic connectivity and impaired neuronal signaling are early signs of these diseases, and this could be due to defective processing by synaptic mitochondria, which supply the ATP needed as a source of energy for these functions (Petersen et al., 2019).
Huntington’s disease is a rare progressive hereditary neurodegenerative disorder whose symptoms include uncontrolled movements, cognitive decline, and psychiatric issues. It is caused by a mutation in the huntingtin gene (HTT), which causes an expansion of a glutamine-containing repeat sequence (polyQ) beyond 36 repeats. Htt inclusion bodies are composed of amyloid fibrils. One way in which Htt aggregates could cause toxicity is through depleting ubiquitin, a protein that binds to misfolded proteins to promote their clearance through the ubiquitin-proteasome system (Ciechanover and Schwartz, 1998). Folger and Wang propose a model that “misfolded protein aggregation soaks up free ubiquitin like a sponge, leading to cytotoxicity by slowing ubiquitin-dependent proteasomal degradation of other proteins” (Folger and Wang, 2021).
Notably, the N-terminal 17-residue domain stretch of huntingtin folds into an amphipathic α-helix in the presence of membranes (Pandey et al., 2018). N-terminal fragments form aggregates and induce neuritic degeneration in cultured striatal neurons (Li et al., 2000). There appear to be three distinct aggregation pathways for Huntingtin: (1) unseeded in solution, (2) seeded in solution, and (3) membrane-mediated. Initiation of aggregation into an oligomer requires an expanded polyQ region and an α-helix formation at the N-terminus. β-sheets form in the second phase. The slowest step is the structural maturation of the proline-rich domain. Seeding accelerates aggregation and is a prerequisite for prion-like spreading. Remarkably, membranes can catalyze aggregation even more potently than seeding, and this also causes extensive membrane damage. The N-terminal α-helix governs membrane-mediated aggregation by anchoring the protein into the membrane (Pandey et al., 2018). Similar phenomena of membranes promoting α-helix formation followed by membrane damage have been observed for at least two other amyloidogenic proteins: α-synuclein, linked to Parkinson’s disease (Dikiy and D Eliezer, 2012) and IAPP, associated with diabetes (Jayasinghe and Langen, 2005; Jayasinghe and Langen, 2007).
Mitochondrial dysfunction in the striatum is a primary feature of Huntington’s disease (Oliveira, 2010). In Huntington’s disease, mutant Htt accumulates in the synapses in the striatum, where it is often associated with mitochondria (Petersen et al., 2019). Mitochondria in striatal synapses in mice with mutated Htt show an increased rate of proton leakage compared to wild type mice. This is associated with a reduced capacity to handle large calcium loads and increased depolarization in response to calcium (Petersen et al., 2019). Researchers have also noted significantly decreased ATP levels in synaptosomes isolated from the forebrains in a mouse model of Huntington’s disease (Orr et al., 2008). It is tempting to speculate that mutant Htt entering mitochondrial membranes via the N-terminal α-helix in the synapses within the striatum is an important factor facilitating mitochondrial dysfunction.
While Huntington’s disease is widely considered to be due to a gain-of-function toxic effect, it has been demonstrated through selective deletion of Htt in striatal projection neurons of mice that these neurons require Htt for motor regulation, synaptic development, cell health, and survival during aging (Burrus et al., 2020). This suggests that a loss-of-function mechanism cannot be ruled out.
Free copper enhances the aggregational toxicity of Htt (Lobato et al., 2024). Elevation of copper and iron in the brain striata has been found at early stages in Huntington’s disease (Rosas et al., 2012; Pfalzer et al., 2022). Copper plays an important role in the depletion and oligomerization/aggregation of wild type Htt. The addition of equimolar or higher concentrations of Cu2+ to Htt induces oligomerization, along with the rapid reduction of Cu2+ to Cu1+ (Neupane et al., 2025). Histidine residues 82 and 98 are both essential for copper interaction, and they appear to coordinate a single copper ion. Thus, Htt reduces Cu2+ to Cu1+, and copper promotes its aggregation, mediated by histidine residues (Fox et al., 2007).
Figure 6 schematizes the pathways that lead to symptoms of Huntington’s disease as a consequence of misfolded mutant Htt.
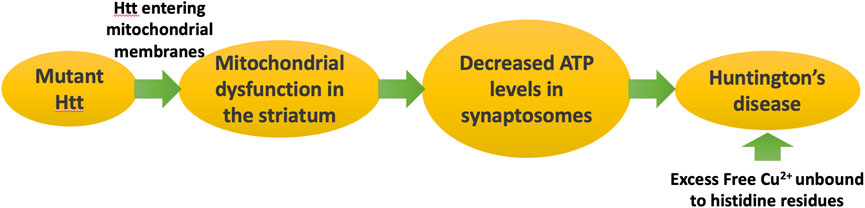
Figure 6. Huntington’s disease is caused by a genetic defect in Huntingtin (Htt), which leads to formation of mitochondrial membrane pores, disrupting ATP synthesis. Deficiencies in ATP in the synaptosome lead to symptoms of Huntington’s disease. Huntingtin traps copper and reduces it from Cu2+ to Cu1+, potentially protecting from copper toxicity.
10.1 Multiple proteins interact with cardiolipin to induce pore formation
While several amyloidogenic proteins have been known to disrupt the plasma membrane through pore formation, recently there has been growing interest in the concept that pore formation in the mitochondrial membrane may be the primary factor that initiates neurodegenerative disease, as well as type II diabetes. An emerging concept is that cardiolipin migration to the outer membrane of the mitochondrion is the trigger that induces misfolding into soluble oligomers of various proteins present in the cytoplasm. Subsequent pore formation following binding to cardiolipin may then facilitate entry of the oligomers into the intermembrane space, where they then gain access to much more cardiolipin present on the inner membrane. This can launch an apoptotic cascade through the release of cytochrome c, resulting in neuronal cell death. A recent review paper provides a great deal of detail about various pathways where cardiolipin plays a central role in driving pore formation in a variety of different pathogenic mechanisms (Vassallo, 2025).
Figure 7 illustrates schematically the steps that we propose take place over a long period of time, with cardiolipin initially protecting the ATPase pumps from deuterium, but ultimately, as the diseased state progresses, sequestering deuterium trapped in cardiolipin along with amyloid deposits.
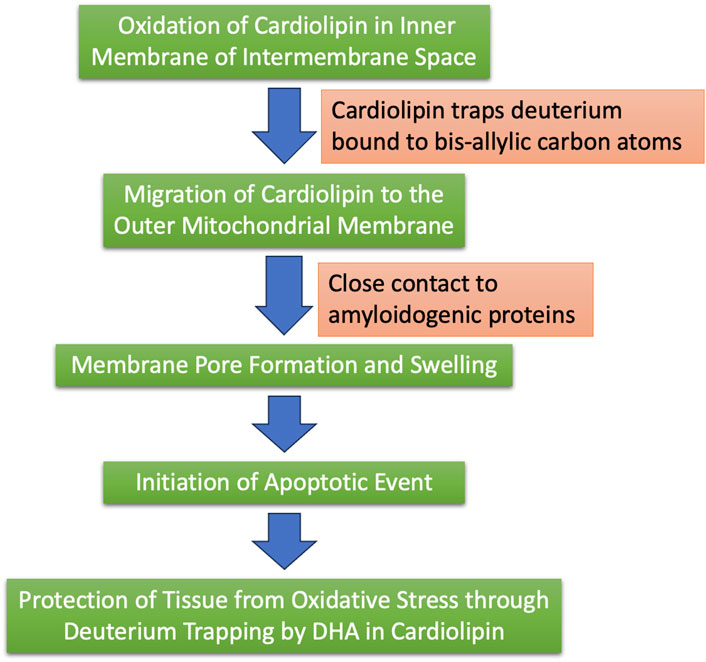
Figure 7. Schematic of the process by which we hypothesize that cardiolipin, synthesized in the mitochondria, may trap deuterium and ultimately sequester it in plaque containing amyloid proteins.
Bax is a Bcl-2 family protein that plays a crucial role in apoptosis. Research has shown that Bax by itself is capable of inducing pore formation in the mitochondrial membrane, following interaction with cardiolipin (Lai et al., 2019). Cytochrome c also binds to cardiolipin, and the Cytochrome c-cardiolipin conjugate opens up lipid pores in the membrane, resulting in the release of cytochrome c into the cytoplasm, thus facilitating its own escape and initiating an apoptosis cascade (Bergstrom et al., 2013).
Table 1 provides a list of proteins that can form oligomers and interact with cardiolipin to induce pore formation and disease processes, with associated references.
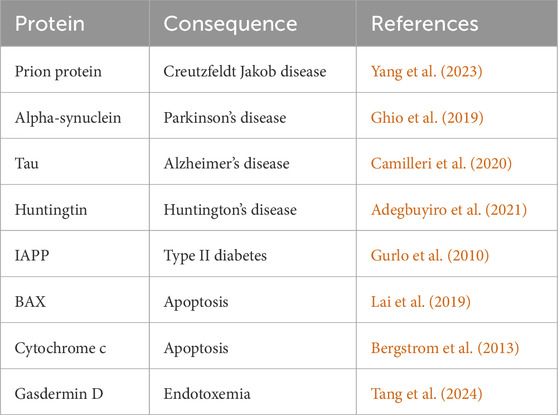
Table 1. Examples of proteins that have been shown to be induced to form pores in the mitochondrial outer membrane when exposed to cardiolipin.
11 Conclusion
In this paper, we have investigated the role of deuterium in mitochondrial dysfunction, and the potential role of amyloidogenic proteins in sequestering deuterium within plaque deposits, both in the brain and in the pancreas. We have also considered the important role of copper, both as a catalyst and as an inducer of oxidative stress. We propose that amyloidogenic proteins may protect from copper overload and facilitate copper delivery to the enzymes that depend on it. Copper may also be sequestered, along with deuterium, in the plaque deposits, under conditions of copper overload, which is toxic for the cell.
Gut microflora, by supplying deuterium depleted nutrients to the host, safeguard normal mitochondrial function in the organism. Furthermore, bis-allylic carbon-containing PUFAs, such as the ones found in cardiolipin, may function as a filter in the inner membrane of the mitochondrial intermembrane space to trap deuterium before it reaches the ATPase pumps, protecting the cells from mitochondrial-respiratory chain dysfunction.
Figure 8 summarizes the main points of this paper, where we claim that excessive levels of deuterium in the mitochondria due to gut dysbiosis may be the driving force that ultimately results in cardiolipin oxidation and migration to the outer membrane, following its accumulation of deuterium bound to the bis-allylic carbon atoms. Amyloidogenic proteins then interact with cardiolipin in the outer membrane to induce an apoptotic cascade, resulting in neuronal cell death and, ultimately, neurodegenerative disease.
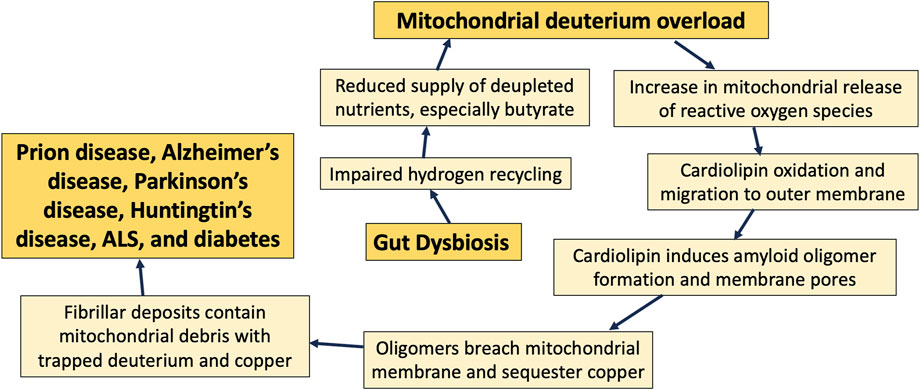
Figure 8. Schematic of the sequence of events that transpire over decades, beginning with an imbalance in gut microbes and eventually leading to neurodegenerative disease.
Cardiolipin migrates to the outer membrane when it becomes oxidized, and it may ameliorate oxidative stress there through prior deuterium doping. Cardiolipin in the outer membrane also facilitates the transformation of multiple amyloidogenic proteins into pore-forming oligomers that can disrupt the membrane potential, leading to destruction of the mitochondrion, and, in the extreme case, initiation of an apoptotic cascade. Mitochondrial membrane debris is commonly found in plaque deposits in the brain, and we hypothesize that the fibrils sequester deuterium-doped lipids, resulting in reduced deuterium burden in the cell and the tissue.
Copper acts as a catalyst in critical proteins involved in oxidative phosphorylation and antioxidant defenses in the mitochondria. The process of safely delivering copper to these enzymes is complex, and disruption of this process can lead to disease, particularly neu-rodegenerative disease.
Histidine residues in amyloid proteins play an essential role in copper homeostasis. The copper interaction anomalies in these proteins contribute to misfolding and aggregation that is linked to neurodegeneration. Impaired copper and deuterium homeostasis has neurotoxic effects via amyloidogenic proteins in prion disease, Parkinson’s disease, Alzheimer’s disease, ALS, and Huntington’s disease. Similar mechanisms of amyloidogenesis in the pancreas contribute to the development of diabetes.
Pure speculation based on experiments with heavy water suggests that histidine may be able to trap deuterium in its imidazole ring, and copper may serve as a catalyst for this by inducing proton currents. However, we were unable to find any literature on the notion that histidine traps deuterium under physiological conditions. This remains a topic for future investigation.
Author contributions
SS: Conceptualization, Funding acquisition, Investigation, Writing – original draft, Writing – review and editing. AK: Conceptualization, Investigation, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. SS received funding from Quanta Computer, Inc. in Taoyuan, Taiwan, as part of the AIR project, under grant #6950759. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article, or the decision to submit it for publication.
Conflict of interest
Author AK was employed by Nasco AD Biotechnology Laboratory.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2025.1639327/full#supplementary-material
References
Adegbuyiro, A., Sedighi, F., Jain, P., Pinti, M. V., Siriwardhana, C., Hollander, J. M., et al. (2021). Mitochondrial membranes modify mutant huntingtin aggregation. Biochim. Biophys. Acta Biomembr. 1863 (10), 183663. doi:10.1016/j.bbamem.2021.183663
Akhtar, A. (2015). The flaws and human harms of animal experimentation. Camb. Q. Healthc. Ethics. 24 (4), 407–419. doi:10.1017/S0963180115000079
Altmann, A., Ng, B., Landau, S. M., Jagust, W. J., and Greicius, M. D.Alzheimer’s disease-neuroimaging initiative (2015). Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 138 (Pt 12), 3734–3746. doi:10.1093/brain/awv278
Arispe, N., Rojas, E., and Pollard, H. B. (1993). Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. U. S. A. 90 (2), 567–571. doi:10.1073/pnas.90.2.567
Asthana, S., Mallick, B., Alexandrescu, A. T., and Jha, S. (2018). IAPP in type II diabetes: basic research on structure, molecular interactions, and disease mechanisms suggests potential intervention strategies. Biochim. Biophys. Acta Biomembr. 1860 (9), 1765–1782. doi:10.1016/j.bbamem.2018.02.020
Atkinson, J. K., Hollenberg, P. F., Ingold, K. U., Johnson, C. C., Le Tadic, M. H., Newcomb, M., et al. (1994). Cytochrome P450-catalyzed hydroxylation of hydrocarbons: Kinetic deuterium isotope effects for the hydroxylation of an ultrafast radical clock. Biochemistry 33, 10630–10637. doi:10.1021/bi00201a009
Ayala, A., Muñoz, M. F., and Argüelles, S. (2014). Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 360438. doi:10.1155/2014/360438
Bartman, S., Coppotelli, G., and Ross, J. M. (2024). Mitochondrial dysfunction: a key player in brain aging and diseases. Curr. Issues Mol. Biol. 46 (3), 1987–2026. doi:10.3390/cimb46030130
Baumann, A., Denninger, A. R., Domin, M., Demé, B., and Kirschner, D. A. (2022). Metabolically-incorporated deuterium in myelin localized by neutron diffraction and identified by mass spectrometry. Curr. Res. Struct. Biol. 4, 231–245. doi:10.1016/j.crstbi.2022.06.003
Bergstrom, C. L., Beales, P. A., Lv, Y., Vanderlick, T. K., and Groves, J. T. (2013). Cytochrome c causes pore formation in cardiolipin-containing membranes. Proc. Natl. Acad. Sci. U. S. A. 110 (16), 6269–6274. doi:10.1073/pnas.1303819110
Bhatti, J. S., Bhatti, G. K., and Reddy, P. H. (2017). Mitochondrial dysfunction and oxidative stress in metabolic disorders -- A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 1863 (5), 1066–1077. doi:10.1016/j.bbadis.2016.11.010
Boros, L. G., D'Agostino, D. P., Katz, H. E., Roth, J. P., Meuillet, E. J., and Somlyai, G. (2016). Submolecular regulation of cell transformation by deuterium depleting water exchange reactions in the tricarboxylic acid substrate cycle. Med. Hypotheses. 87, 69–74. doi:10.1016/j.mehy.2015.11.016
Boros, L. G., Seneff, S., Túri, M., Palcsu, L., and Zubarev, R. A. (2024). Active involvement of compartmental, inter-and intramolecular deuterium disequilibrium in adaptive biology. PNAS 121 (37), e2412390121. doi:10.1073/pnas.2412390121
Boros, L. G., Somlyai, I., Kovács, B. Z., Puskás, L. G., Nagy, L. I., Dux, L., et al. (2021). Deuterium depletion inhibits cell proliferation, RNA and nuclear membrane turnover to enhance survival in pancreatic cancer. Cancer Control. 28, 1073274821999655. doi:10.1177/1073274821999655
Bourassa, M. W., Alim, I., Bultman, S. J., and Ratan, R. R. (2016). Butyrate, neuroepigenetics and the gut microbiome: can a high fiber diet improve brain health? Neurosci. Lett. 625, 56–63. doi:10.1016/j.neulet.2016.02.009
Bulteau, A. L., Ikeda-Saito, M., and Szweda, L. I. (2003). Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry 42 (50), 14846–14855. doi:10.1021/bi0353979
Burrus, C. J., McKinstry, S. U., Kim, N., Ozlu, M. I., Santoki, A. V., Fang, F. Y., et al. (2020). Striatal projection neurons require huntingtin for synaptic connectivity and survival. Cell. Rep. 30 (3), 642–657. doi:10.1016/j.celrep.2019.12.069
Buteau, J. (2008). GLP-1 receptor signaling: effects on pancreatic beta-cell proliferation and survival. Diabetes Metab. 34 (Suppl. 2), S73–S77. doi:10.1016/S1262-3636(08)73398-6
Camilleri, A., Ghio, S., Caruana, M., Weckbecker, D., Schmidt, F., Kamp, F., et al. (2020). Tau-induced mitochondrial membrane perturbation is dependent upon cardiolipin. Biochim. Biophys. Acta Biomembr. 1862 (2), 183064. doi:10.1016/j.bbamem.2019.183064
Cao, P., Abedini, A., Wang, H., Tu, L. H., Zhang, X., Schmidt, A. M., et al. (2013). Islet amyloid polypeptide toxicity and membrane interactions. Proc. Natl. Acad. Sci. U. S. A. 110 (48), 19279–19284. doi:10.1073/pnas.1305517110
Carbonero, F., Benefiel, A. C., and Gaskins, H. R. (2012). Contributions of the microbial hydrogen economy to colonic homeostasis. Nat. Rev. Gastroenterol. Hepatol. 9 (9), 504–518. doi:10.1038/nrgastro.2012.85
Chang, T. J., Tseng, H. C., Liu, M. W., Chang, Y. C., Hsieh, M. L., and Chuang, L. M. (2016). Glucagon-like peptide-1 prevents methylglyoxal-induced apoptosis of beta cells through improving mitochondrial function and suppressing prolonged AMPK activation. Sci. Rep. 6, 23403. doi:10.1038/srep23403
Chattopadhyay, M., Walter, E. D., Newell, D. J., Jackson, P. J., Aronoff-Spencer, E., Peisach, J., et al. (2005). The octarepeat domain of the prion protein binds Cu(II) with three distinct coordination modes at pH 7.4. J. Am. Chem. Soc. 127 (36), 12647–12656. doi:10.1021/ja053254z
Ciechanover, A., and Schwartz, A. L. (1998). The ubiquitin-proteasome pathway: the complexity and myriad functions of proteins death. Proc. Natl. Acad. Sci. U. S. A. 95 (6), 2727–2730. doi:10.1073/pnas.95.6.2727
Congiu, T., Alghrably, M., Emwas, A. H., Jaremko, L., Lachowicz, J. I., Piludu, M., et al. (2021). Undercover toxic Ménage à Trois of amylin, copper(II) and metformin in human embryonic kidney cells. Pharmaceutics 13 (6), 830. doi:10.3390/pharmaceutics13060830
Cruces-Sande, A., Rodrguez-Prez, A. I., Herbello-Hermelo, P., Bermejo-Barrera, P., Mndez-lvarez, E., Labandeira-Garca, J. L., et al. (2019). Copper increases brain oxidative stress and enhances the ability of 6-hydroxydopamine to cause dopaminergic degeneration in a rat model of Parkinson’s disease. Mol. Neurobiol. 56 (4), 28452854. doi:10.1007/s12035-018-1274-7
Cummings, J., Osse, A. M. L., Cammann, D., Powell, J., and Chen, J. (2024). Anti-amyloid monoclonal antibodies for the treatment of Alzheimer’s disease. BioDrugs 38 (1), 5–22. doi:10.1007/s40259-023-00633-2
Derreumaux, P. (2001). Evidence that the 127-164 region of prion proteins has two equi-energetic conformations with beta or alpha features. Biophys. J. 81 (3), 1657–1665. doi:10.1016/S0006-3495(01)75819-5
Dikiy, I., and Eliezer, D. (2012). Folding and misfolding of alpha-synuclein on membranes. Biochim. Biophys. Acta. 1818 (4), 1013–1018. doi:10.1016/j.bbamem.2011.09.008
Dokainish, H. M., and Kitao, A. (2016). Computational assignment of the histidine protonation state in (6-4) photolyase enzyme and its effect on the protonation step. ACS Catal. 6, 5500–5507. doi:10.1021/acscatal.6b01094
Eble, K. S., Coleman, W. B., Hantgan, R. R., and Cunningham, C. C. (1990). Tightly associated cardiolipin in the bovine heart mitochondrial ATP synthase as analyzed by 31P nuclear magnetic resonance spectroscopy. J. Biol. Chem. 265 (32), 19434–19440. doi:10.1016/s0021-9258(17)45391-9
Engel, M. F. (2009). Membrane permeabilization by islet amyloid polypeptide. Chem. Phys. Lipids. 160 (1), 1–10. doi:10.1016/j.chemphyslip.2009.03.008
Esterbauer, H., Eckl, P., and Ortner, A. (1990). Possible mutagens derived from lipids and lipid precursors. Mutat. Res. 238 (3), 223–233. doi:10.1016/0165-1110(90)90014-3
Folger, A., and Wang, Y. (2021). The cytotoxicity and clearance of mutant huntingtin and other misfolded proteins. Cells 10 (11), 2835. doi:10.3390/cells10112835
Fox, J. H., Kama, J. A., Lieberman, G., Chopra, R., Dorsey, K., Chopra, V., et al. (2007). Mechanisms of copper ion mediated Huntington’s disease progression. PLoS One 2 (3), e334. doi:10.1371/journal.pone.0000334
Frączyk, T. (2021). Cu(II)-binding N-terminal sequences of human proteins. Chem. Biodivers. 18 (4), e2100043. doi:10.1002/cbdv.202100043
Fraile-Martínez, O., Gómez-Lahoz, A. M., Boaru, D. L., de Leon-Oliva, D., Guijarro, L. G., Atienza-Perez, M., et al. (2023). Prognostic value of malondialdehyde (MDA) in the temporal progression of chronic spinal cord injury. J. Pers. Med. 13 (4), 626. doi:10.3390/jpm13040626
Gandasi, N. R., Yin, P., Omar-Hmeadi, M., Ottosson Laakso, E., Vikman, P., and Barg, S. (2018). Glucose-dependent granule docking limits insulin secretion and is decreased in human type 2 diabetes. Cell. Metab. 27 (2), 470–478. doi:10.1016/j.cmet.2017.12.017
García-Ruiz, I., de la Torre, P., Díaz, T., Esteban, E., Fernández, I., Muñoz-Yagüe, T., et al. (2002). Sp1 and Sp3 transcription factors mediate malondialdehyde-induced collagen alpha 1(I) gene expression in cultured hepatic stellate cells. J. Biol. Chem. 277 (34), 30551–30558. doi:10.1074/jbc.M203368200
Gaweł, S., Wardas, M., Niedworok, E., and Wardas, P. (2004). Malondialdehyde (MDA) as a lipid peroxidation marker. Wiad. Lek. 57 (9-10), 453–455. Available online at: https://pubmed.ncbi.nlm.nih.gov/15765761/.
Gęgotek, A., and Skrzydlewska, E. (2024). Lipid peroxidation products’ role in autophagy regulation. Free Rad. Biol. Med. 212, 375–383. doi:10.1016/j.freeradbiomed.2024.01.001
Ghio, S., Camilleri, A., Caruana, M., Ruf, V. C., Schmidt, F., Leonov, A., et al. (2019). Cardiolipin promotes pore-forming activity of alpha-synuclein oligomers in mitochondrial membranes. ACS Chem. Neurosci. 10 (8), 3815–3829. doi:10.1021/acschemneuro.9b00320
Guengerich, F. P. (2017). Kinetic deuterium isotope effects in cytochrome P450 reactions. Methods Enzymol. 596, 217–238. doi:10.1016/bs.mie.2017.06.036
Gupta, D., and Leahy, J. L. (2014). Islet amyloid and type 2 diabetes: overproduction or inadequate clearance and detoxification? J. Clin. Invest. 124 (8), 3292–3294. doi:10.1172/JCI77506
Gurlo, T., Ryazantsev, S., Huang, C.-J., Yeh, M. W., Reber, H. A., Hines, O. J., et al. (2010). Evidence for proteotoxicity in beta-cells in type 2 diabetes, toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol. 176, 861–869. doi:10.2353/ajpath.2010.090532
Ha, J. W., Choi, J. Y., and Boo, Y. C. (2023). Differential effects of histidine and histidinamide versus cysteine and cysteinamide on copper ion-induced oxidative stress and cytotoxicity in HaCaT keratinocytes. Antioxidants (Basel) 12 (4), 801. doi:10.3390/antiox12040801
Haines, T. H., and Dencher, N. A. (2002). Cardiolipin: a proton trap for oxidative phosphorylation. FEBS Lett. 528 (1-3), 35–39. doi:10.1016/s0014-5793(02)03292-1
Harris, E. D. (1992). Copper as a cofactor and regulator of copper,zinc superoxide dismutase. J. Nutr. 122 (3 Suppl. l), 636–640. doi:10.1093/jn/122.suppl_3.636
Hart, P. J., Balbirnie, M. M., Ogihara, N. L., Nersissian, A. M., Weiss, M. S., Valentine, J. S., et al. (1999). A structure-based mechanism for copper-zinc superoxide dismutase. Biochemistry 38 (7), 2167–2178. doi:10.1021/bi982284u
He, C., and Klionsky, D. J. (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 43, 67–93. doi:10.1146/annurev-genet-102808-114910
Hodgkinson, K., El Abbar, F., Dobranowski, P., Manoogian, J., Butcher, J., Figeys, D., et al. (2023). Butyrate’s role in human health and the current progress towards its clinical application to treat gastrointestinal disease. Clin. Nutr. 42 (2), 61–75. doi:10.1016/j.clnu.2022.10.024
Horn, D., and Barrientos, A. (2008). Mitochondrial copper metabolism and delivery to cytochrome c oxidase. IUBMB Life 60 (7), 421–429. doi:10.1002/iub.50
Huang, X., Cuajungco, M. P., Atwood, C. S., Hartshorn, M. A., Tyndall, J. D., Hanson, G. R., et al. (1999). Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 274 (52), 37111–37116. doi:10.1074/jbc.274.52.37111
Jayasinghe, S. A., and Langen, R. (2005). Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 44 (36), 12113–12119. doi:10.1021/bi050840w
Jayasinghe, S. A., and Langen, R. (2007). Membrane interaction of islet amyloid polypeptide. Biochim. Biophys. Acta. 1768 (8), 2002–2009. doi:10.1016/j.bbamem.2007.01.022
Jiang, D., Li, X., Liu, L., Yagnik, G. B., and Zhou, F. (2010). Reaction rates and mechanism of the ascorbic acid oxidation by molecular oxygen facilitated by Cu(II)-containing amyloid-beta complexes and aggregates. J. Phys. Chem. B 114 (14), 4896–4903. doi:10.1021/jp9095375
Jin, S. M., and Youle, R. J. (2012). PINK1- and Parkin-mediated mitophagy at a glance. J. Cell. Sci. 125 (Pt 4), 795–799. doi:10.1242/jcs.093849
Kagan, V., Jiang, J., Huang, Z., Tyurina, Y., Desbourdes, C., Cottet-Rousselle, C., et al. (2016). NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell. Death Differ. 23, 1140–1151. doi:10.1038/cdd.2015.160
Kang, M. Y., Oh, T. J., and Cho, Y. M. (2015). Glucagon-like Peptide-1 increases mitochondrial biogenesis and function in INS-1 rat insulinoma cells. Endocrinol. Metab. Seoul. 30 (2), 216–220. doi:10.3803/EnM.2015.30.2.216
Karran, E., and Hardy, J. (2014). Antiamyloid therapy for Alzheimer’s disease – are we on the right road? N. Engl. J. Med. 370 (4), 377–378. doi:10.1056/NEJMe1313943
Kaufman, B. A., Li, C., and Soleimanpour, S. A. (2015). Mitochondrial regulation of β-cell function: maintaining the momentum for insulin release. Mol. Asp. Med. 42, 91–104. doi:10.1016/j.mam.2015.01.004
Kovács, B. Z., Puskás, L. G., Nagy, L. I., Papp, A., Gyöngyi, Z., Fórizs, I., et al. (2022). Blocking the increase of intracellular deuterium concentration prevents the expression of cancer-related genes, tumor development, and tumor recurrence in cancer patients. Cancer Control. 29, 10732748211068963. doi:10.1177/10732748211068963
Krichevsky, M. I., Friedman, I., Newell, M. F., and Sisler, F. D. (1961). Deuterium fractionation during molecular hydrogen formation in a marine pseudomonad. J. Biol. Chem. 236, 2520–2525. doi:10.1016/s0021-9258(18)64032-3
Kwan, E. P., and Gaisano, H. Y. (2005). Glucagon-like peptide 1 regulates sequential and compound exocytosis in pancreatic islet beta-cells. Diabetes 54 (9), 2734–2743. doi:10.2337/diabetes.54.9.2734
Lai, Y. C., Li, C. C., Sung, T. C., Chang, C. W., Lan, Y. J., and Chiang, Y. W. (2019). The role of cardiolipin in promoting the membrane pore-forming activity of BAX oligomers. Biochim. Biophys. Acta Biomembr. 1861 (1), 268–280. doi:10.1016/j.bbamem.2018.06.014
Laporte, A., Lortz, S., Schaal, C., Lenzen, S., and Elsner, M. (2020). Hydrogen peroxide permeability of cellular membranes in insulin-producing cells. Biochim. Biophys. Acta Biomembr. 1862 (2), 183096. doi:10.1016/j.bbamem.2019.183096
Lee, E., Song, C. H., Bae, S. J., Ha, K. T., and Karki, R. (2023). Regulated cell death pathways and their roles in homeostasis, infection, inflammation, and tumorigenesis. Exp. Mol. Med. 55 (8), 1632–1643. doi:10.1038/s12276-023-01069-y
Li, H., Li, S., Johnston, H., Shelbourne, P. F., and Li, X. (2000). Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat. Genet. 25 (4), 385–389. doi:10.1038/78054
Li, H., Zhang, O., Hui, C., Huang, Y., Shao, H., Song, M., et al. (2022). Deuterium-reinforced polyunsaturated fatty acids prevent diet-induced nonalcoholic steatohepatitis by reducing oxidative stress. Med. Kaunas. 58 (6), 790. doi:10.3390/medicina58060790
Li, X., Chen, X., and Gao, X. (2023). Copper and cuproptosis: new therapeutic approaches for alzheimers disease. Front. Aging Neurosci. 15, 1300405. doi:10.3389/fnagi.2023.1300405
Lismont, C., Revenco, I., and Fransen, M. (2019). Peroxisomal hydrogen peroxide metabolism and signaling in health and disease. Int. J. Mol. Sci. 20 (15), 3673. doi:10.3390/ijms20153673
Liu, X., Zhang, Z., Ruan, J., Pan, Y., Magupalli, V. G., Wu, H., et al. (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. doi:10.1038/nature18629
Lobato, A. G., Ortiz-Vega, N., Zhu, Y., Neupane, D., Meier, K. K., and Zhai, R. G. (2024). Copper enhances aggregational toxicity of mutant huntingtin in a drosophila model of huntington's disease. Biochim. Biophys. Acta Mol. Basis Dis. 1870 (1), 166928. doi:10.1016/j.bbadis.2023.166928
Lu, Y., and Chen, H. (2024). Deuterium-depleted water in cancer therapy: a systematic review of clinical and experimental trials. Nutrients 16 (9), 1397. doi:10.3390/nu16091397
Magrì, A., La Mendola, D., Nicoletti, V. G., Pappalardo, G., and Rizzarelli, E. (2016). New insight in copper-ion binding to human islet amyloid: the contribution of metal-complex speciation to reveal the polypeptide toxicity. Chemistry 22 (37), 13287–13300. doi:10.1002/chem.201602816
Maryon, E. B., Molloy, S. A., and Kaplan, J. H. (2013). Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. Cell. Physiol. 304 (8), C768–C779. doi:10.1152/ajpcell.00417.2012
Mayes, J., Tinker-Mill, C., Kolosov, O., Zhang, H., Tabner, B. J., and Allsop, D. (2014). β-amyloid fibrils in alzheimer disease are not inert when bound to copper ions but can degrade hydrogen peroxide and generate reactive oxygen species. J. Biol. Chem. 289 (17), 12052–12062. doi:10.1074/jbc.M113.525212
Meinders, M., Kulu, D. I., van de Werken, H. J., Hoogenboezem, M., Janssen, H., Brouwer, R. W., et al. (2015). Sp1/Sp3 transcription factors regulate hallmarks of megakaryocyte maturation and platelet formation and function. Blood 125 (12), 1957–1967. doi:10.1182/blood-2014-08-593343
Min, J. H., Sarlus, H., and Harris, R. A. (2024). Copper toxicity and deficiency: the vicious cycle at the core of protein aggregation in ALS. Front. Mol. Neurosci. 17, 1408159. doi:10.3389/fnmol.2024.1408159
Miyagi, M., and Nakazawa, T. (2024). Significance of histidine hydrogen-deuterium exchange mass spectrometry in protein structural biology. Biol. (Basel) 13 (1), 37. doi:10.3390/biology13010037
Müller, T. D., Finan, B., Bloom, S. R., D’Alessio, D., Drucker, D. J., Flatt, P. R., et al. (2019). Glucagon-like peptide 1 (GLP-1). Mol. Metab. 30, 72–130. doi:10.1016/j.molmet.2019.09.010
Muramoto, K., Hirata, K., Shinzawa-Itoh, K., Yoko-o, S., Yamashita, E., Aoyama, H., et al. (2007). A histidine residue acting as a controlling site for dioxygen reduction and proton pumping by cytochrome c oxidase. Proc. Natl. Acad. Sci. U. S. A. 104 (19), 7881–7886. doi:10.1073/pnas.0610031104
Navratil, A. R., Shchepinov, M. S., and Dennis, E. A. (2018). Lipidomics reveals dramatic physiological kinetic isotope effects during the enzymatic oxygenation of polyunsaturated fatty acids ex vivo. J. Am. Chem. Soc. 140 (1), 235–243. doi:10.1021/jacs.7b09493
Neupane, D., Santos-Fernandez, M., Fernandez-Lima, F., and Meier, K. K. (2025). Multiple copper ions bind to and promote the oligomerization of huntingtin protein with nonpathological repeat expansions. Biochemistry 64 (5), 1121–1135. doi:10.1021/acs.biochem.5c00012
Ng, S. C. W., Furman, R., Axelsen, P. H., and Shchepinov, M. S. (2022). Free radical chain reactions and polyunsaturated fatty acids in brain lipids. ACS Omega 7 (29), 25337–25345. doi:10.1021/acsomega.2c02285
Nicolaou, S. A., Gaida, S. M., and Papoutsakis, E. T. (2010). A comparative view of metabolite and substrate stress and tolerance in microbial bioprocessing: from biofuels and chemicals, to biocatalysis and bioremediation. Metab. Eng. 12 (4), 307–331. doi:10.1016/j.ymben.2010.03.004
O’Brien, R. J., and Wong, P. C. (2011). Amyloid precursor protein processing and Alzheimer’s disease. Annu. Rev. Neurosci. 34, 185–204. doi:10.1146/annurev-neuro-061010-113613
Olgun, A. (2007). Biological effects of deuteronation: ATP synthase as an example. Theor. Biol. Med. Model 4, 9. doi:10.1186/1742-4682-4-9
Oliveira, J. M. (2010). Nature and cause of mitochondrial dysfunction in Huntington’s disease: focusing on huntingtin and the striatum. J. Neurochem. 114 (1), 1–12. doi:10.1111/j.1471-4159.2010.06741.x
Orr, A. L., Li, S., Wang, C. E., Li, H., Wang, J., Rong, J., et al. (2008). N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 28 (11), 2783–2792. doi:10.1523/JNEUROSCI.0106-08.2008
Pandey, N. K., Isas, J. M., Rawat, A., Lee, R. V., Langen, J., Pandey, P., et al. (2018). The 17-residue-long N terminus in huntingtin controls stepwise aggregation in solution and on membranes via different mechanisms. J. Biol. Chem. 293 (7), 2597–2605. doi:10.1074/jbc.M117.813667
Parsaei, M., Sarafraz, N., Moaddab, S. Y., and Ebrahimzadeh Leylabadlo, H. (2021). The importance of Faecalibacterium prausnitzii in human health and diseases. New Microbes New Infect. 43, 100928. doi:10.1016/j.nmni.2021.100928
Petersen, M. H., Willert, C. W., Andersen, J. V., Waagepetersen, H. S., Skotte, N. H., and Nrremlle, A. (2019). Functional differences between synaptic mitochondria from the striatum and the cerebral cortex. Neuroscience 406, 432–443. doi:10.1016/j.neuroscience.2019.02.033
Pfalzer, A. C., Yan, Y., Kang, H., Totten, M., Silverman, J., Bowman, A. B., et al. (2022). Alterations in metal homeostasis occur prior to canonical markers in huntington disease. Sci. Rep. 12 (1), 10373. doi:10.1038/s41598-022-14169-y
Press, M., Jung, T., Knig, J., Grune, T., and Höhn, A. (2019). Protein aggregates and proteostasis in aging: Amylin and β-cell function. Mech. Ageing Dev. 177, 46–54. doi:10.1016/j.mad.2018.03.010
Qu, Y., Sun, L., Yang, Z., and Han, R. (2011). Variation in the PTEN-Induced putative kinase 1 gene associated with the increase risk of type 2 diabetes in northern Chinese. J. Genet. 90 (1), 125–128. doi:10.1007/s12041-011-0020-y
Ramana, K. V., Srivastava, S., and Singhal, S. S. (2019). Lipid peroxidation products in human health and disease. Oxid. Med. Cell. Longev. 2019, 7147235. doi:10.1155/2019/7147235
Rath, E., and Haller, D. (2011). Inflammation and cellular stress: a mechanistic link between immune-mediated and metabolically driven pathologies. Eur. J. Nutr. 50 (4), 219–233. doi:10.1007/s00394-011-0197-0
Rivillas-Acevedo, L., Maciel-Barón, L., García, J. E., Juaristi, E., and Quintanar, L. (2013). Insertion of beta-alanine in model peptides for copper binding to His96 and His111 of the human prion protein. J. Inorg. Biochem. 126, 104–110. doi:10.1016/j.jinorgbio.2013.05.016
Rizzollo, F., and Agostinis, P. (2025). Mitochondria-lysosome contact sites: emerging players in cellular homeostasis and disease. Contact (Thousand Oaks) 8, 25152564251329250. doi:10.1177/25152564251329250
Rodrigues, V. F., Elias-Oliveira, J., Pereira, S., Pereira, J. A., Barbosa, S. C., Machado, M. S. G., et al. (2022). Akkermansia muciniphila and gut immune system: a good friendship that attenuates inflammatory bowel disease, obesity, and diabetes. Front. Immunol. 13, 934695. doi:10.3389/fimmu.2022.934695
Rosas, H. D., Chen, Y. I., Doros, G., Salat, D. H., Chen, N. K., Kwong, K. K., et al. (2012). Alterations in brain transition metals in huntington disease: an evolving and intricate story. Arch. Neurol. 69 (7), 887–893. doi:10.1001/archneurol.2011.2945
Roseman, G. P., Wu, B., Wadolkowski, M. A., Harris, D. A., and Millhauser, G. L. (2020). Intrinsic toxicity of the cellular prion protein is regulated by its conserved central region. FASEB J. 34 (6), 8734–8748. doi:10.1096/fj.201902749RR
Salvi, P. S., and Cowles, R. A. (2021). Butyrate and the intestinal epithelium: modulation of proliferation and inflammation in homeostasis and disease. Cells 10 (7), 1775. doi:10.3390/cells10071775
Sánchez-López, C., Rossetti, G., Quintanar, L., and Carloni, P. (2018). Structural determinants of the prion protein N-Terminus and its adducts with copper ions. Int. J. Mol. Sci. 20 (1), 18. doi:10.3390/ijms20010018
Schilling, K. M., Tao, L., Wu, B., Kiblen, J. T. M., Ubilla-Rodriguez, N. C., Pushie, M. J., et al. (2020). Both N-Terminal and C-Terminal histidine residues of the prion protein are essential for copper coordination and neuroprotective self-regulation. J. Mol. Biol. 432 (16), 4408–4425. doi:10.1016/j.jmb.2020.05.020
Seneff, S., and Kyriakopoulos, A. M. (2025a). Cancer, deuterium, and gut microbes: a novel perspective. Endocr. Metab. Sci. 17, 100215. doi:10.1016/j.endmts.2025.100215
Seneff, S., and Kyriakopoulos, A. M. (2025b). Taurine prevents mitochondrial dysfunction and protects mitochondria from reactive oxygen species and deuterium toxicity. Amino Acids 57, 6. doi:10.1007/s00726-024-03440-3
Seneff, S., Nigh, G., and Kyriakopoulos, A. M. (2025a). Is deuterium sequestering by reactive carbon atoms an important biological mechanism to reduce deuterium content in biological water? FASEB Bioadv 7, E70019. doi:10.1096/fba.2025-00032
Seneff, S., Nigh, G., and Kyriakopoulos, A. M. (2025b). Mitochondrial dysfunction in Parkinson’s disease: is impaired deuterium-depleted nutrient supply by gut microbes a primary factor? Biocell 2025, 1–10. doi:10.32604/biocell.2025.066687
Serra-Vidal, B., Pujadas, L., Rossi, D., Soriano, E., Madurga, S., and Carulla, N. (2014). Hydrogen/Deuterium exchange-protected oligomers populated during A fibril formation correlate with neuronal cell death. ACS Chem. Biol. 9 (11), 2678–2685. doi:10.1021/cb500621x
Serra-Batiste, M., Ninot-Pedrosa, M., Bayoumi, M., Gair, M., Maglia, G., and Carulla, N. (2016). A42 assembles into specific -barrel pore-forming oligomers in membrane-mimicking environments. Proc. Natl. Acad. Sci. U. S. A. 113 (39), 10866–10871. doi:10.1073/pnas.1605104113
Shearer, J. (2013). Use of a metallopeptide-based mimic provides evidence for a proton-coupled electron-transfer mechanism for superoxide reduction by nickel-containing superoxide dismutase. Angew. Chem. Int. Ed. Engl. 52 (9), 2569–2572. doi:10.1002/anie.201209746
Sheveleva, N. N., Markelov, D. A., Vovk, M. A., Tarasenko, I. I., Mikhailova, M. E., Ilyash, M. Y., et al. (2019). Stable deuterium labeling of histidine-rich lysine-based dendrimers. Molecules 24 (13), 2481. doi:10.3390/molecules24132481
Silva, Y. P., Bernardi, A., and Frozza, R. L. (2020). The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front. Endocrinol. (Lausanne). 11, 25. doi:10.3389/fendo.2020.00025
Sinopoli, A., Magrì, A., Milardi, D., Pappalardo, M., Pucci, P., Flagiello, A., et al. (2014). The role of copper (II) in the aggregation of human amylin. Metallomics 6 (10), 1841–1852. doi:10.1039/c4mt00130c
Sitton, J., Pickett, D., Rodriguez, A., and Kurouski, D. (2025). Lipids determine the toxicity of human islet polypeptide aggregates in vivo. J. Biol. Chem. 301 (1), 108029. doi:10.1016/j.jbc.2024.108029
Smith, D. P., Smith, D. G., Curtain, C. C., Boas, J. F., Pilbrow, J. R., Ciccotosto, G. D., et al. (2006). Copper-mediated amyloid-beta toxicity is associated with an intermolecular histidine bridge. J. Biol. Chem. 281 (22), 15145–15154. doi:10.1074/jbc.M600417200
Somlyai, G., Kovács, B. Z., Papp, A., and Somlyai, I. (2023). A preliminary study indicating improvement in the median survival time of glioblastoma multiforme patients by the application of deuterium depletion in combination with conventional therapy. Biomedicines 11 (7), 1989. doi:10.3390/biomedicines11071989
Staś, M., Najgebauer, P., and Siodak, D. (2023). Imidazole-amino acids. Conformational switch under tautomer and pH change. Amino Acids 55 (1), 33–49. doi:10.1007/s00726-022-03201-0
Supale, S., Li, N., Brun, T., and Maechler, P. (2012). Mitochondrial dysfunction in pancreatic cells. Trends Endocrinol. Metab. 23 (9), 477–487. doi:10.1016/j.tem.2012.06.002
Swer, N. M., Venkidesh, B. S., Murali, T. S., and Mumbrekar, K. D. (2023). Gut microbiota-derived metabolites and their importance in neurological disorders. Mol. Biol. Rep. 50 (2), 1663–1675. doi:10.1007/s11033-022-08038-0
Syme, C. D., Nadal, R. C., Rigby, S. E., and Viles, J. H. (2004). Copper binding to the amyloid-beta (abeta) peptide associated with Alzheimer’s disease: folding, coordination geometry, pH de-pendence, stoichiometry, and affinity of Abeta-(1-28): insights from a range of complementary spectroscopic techniques. J. Biol. Chem. 279 (18), 18169–18177. doi:10.1074/jbc.M313572200
Tang, Y., Wu, J., Sun, X., Tan, S., Li, W., Yin, S., et al. (2024). Cardiolipin oxidized by ROS from complex II acts as a target of gasdermin D to drive mitochondrial pore and heart dysfunction in endotoxemia. Cell. Rep. 43 (5), 114237. doi:10.1016/j.celrep.2024.114237
Tersey, S. A., Maier, B., Nishiki, Y., Maganti, A. V., Nadler, J. L., and Mirmira, R. G. (2014). 12-lipoxygenase promotes obesity-induced oxidative stress in pancreatic islets. Mol. Cell. Biol. 34 (19), 3735–3745. doi:10.1128/MCB.00157-14
Tickler, A. K., Masters, C. L., Bush, A. I., Cherny, R. A., Cappai, R., Wade, J. D., et al. (2005). Methylation of the imidazole side chains of the alzheimer disease amyloid-beta peptide results in abolition of superoxide dismutase-like structures and inhibition of neurotoxicity. J. Biol. Chem. 280 (14), 13355–13363. doi:10.1074/jbc.M414178200
Titorenko, V. I., and Terlecky, S. R. (2010). Peroxisome metabolism and cellular aging. Traffic 12 (3), 252–259. doi:10.1111/j.1600-0854.2010.01144.x
Tokuda, E., and Furukawa, Y. (2016). Copper homeostasis as a therapeutic target in amyotrophic lateral sclerosis with SOD1 mutations. Int. J. Mol. Sci. 17 (5), 636. doi:10.3390/ijms17050636
Tokuda, E., Okawa, E., and Ono, S. (2009). Dysregulation of intracellular copper trafficking pathway in a mouse model of mutant copper/zinc superoxide dismutase-linked familial amyotrophic lateral sclerosis. J. Neurochem. 111 (1), 181–191. doi:10.1111/j.1471-4159.2009.06310.x
Torres, M., Parets, S., Fernández-Díaz, J., Beteta-Göbel, R., Rodríguez-Lorca, R., Román, R., et al. (2021). Lipids in pathophysiology and development of the membrane lipid therapy: new bioactive lipids. Membr. (Basel) 11 (12), 919. doi:10.3390/membranes11120919
Uchida, K., and Kawakishi, S. (1994). Identification of oxidized histidine generated at the active site of Cu,Zn-superoxide dismutase exposed to H2O2. Selective generation of 2-oxo-histidine at the histidine 118. J. Biol. Chem. 269 (4), 2405–2410. doi:10.1016/s0021-9258(17)41960-0
Vanoni, M. A., Wong, K. K., Ballou, D. P., and Blanchard, J. S. (1990). Glutathione reductase: comparison of steady-state and rapid reaction primary kinetic isotope effects exhibited by the yeast, spinach, and Escherichia coli enzymes. Biochemistry 29 (24), 5790–5796. doi:10.1021/bi00476a021
Van Veldhoven, P. P. (2010). Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J. Lipid Res. 51 (10), 2863–2895. doi:10.1194/jlr.R005959
Vassallo, N. (2025). Poration of mitochondrial membranes by amyloidogenic peptides and other biological toxins. J. Neurochem. 169 (1), e16213. doi:10.1111/jnc.16213
Volinsky, R., and Kinnunen, P. K. (2013). Oxidized phosphatidylcholines in membrane-level cellular signaling: from biophysics to physiology and molecular pathology. FEBS J. 280 (12), 2806–2816. doi:10.1111/febs.12247
Waite, L. M. (2015). Treatment for Alzheimer’s disease: has anything changed? Aust. Prescr. 38 (2), 60–63. doi:10.18773/austprescr.2015.018
Walton, P. A., and Pizzitelli, M. (2012). Effects of peroxisomal catalase inhibition on mitochondrial function. Front. Physiol. 3, 108. doi:10.3389/fphys.2012.00108
Walton, P. H., Davies, G. J., Diaz, D. E., and Franco-Cairo, J. P. (2023). The histidine brace: nature’s copper alternative to haem? FEBS Lett. 597 (4), 485–494. doi:10.1002/1873-3468.14579
Wang, J. Y., Wang, Q. W., Yang, X. Y., Yang, W., Li, D. R., Jin, J. Y., et al. (2023). GLP-1 receptor agonists for the treatment of obesity: role as a promising approach. Front. Endocrinol. (Lausanne). 14, 1085799. doi:10.3389/fendo.2023.1085799
Wang, X., Lei, X. G., and Wang, J. (2014). Malondialdehyde regulates glucose-stimulated insulin secretion in murine islets via TCF7L2-dependent wnt signaling pathway. Mol. Cell. Endocrinol. 382 (1), 8–16. doi:10.1016/j.mce.2013.09.003
Weishaupt, A.-K., Ruecker, L., Meiners, T., Schwerdtle, T., Avila, D. S., Aschner, M., et al. (2024). Copper-mediated neurotoxicity and genetic vulnerability in the background of neurodegenerative diseases in C. elegans. Toxicol. Sci. 201 (2), 254–262. doi:10.1093/toxsci/kfae092
Wolf, P. G., Biswas, A., Morales, S. E., Greening, C., and Gaskins, H. R. (2016). H2 metabolism is widespread and diverse among human colonic microbes. Gut Microbes 7 (3), 235–245. doi:10.1080/19490976.2016.1182288
Wu, Y., Qin, D., Yang, H., Wang, W., Xiao, J., Zhou, L., et al. (2020). Neuroprotective effects of deuterium-depleted water (DDW) against H2O2-induced oxidative stress in differentiated PC12 cells through the PI3K/Akt signaling pathway. Neurochem. Res. 45 (5), 1034–1044. doi:10.1007/s11064-020-02978-4
Yaglova, N. V., Timokhina, E. P., Obernikhin, S. S., and Yaglov, V. V. (2023). Emerging role of deuterium/protium disbalance in cell cycle and apoptosis. Int. J. Mol. Sci. 24 (4), 3107. doi:10.3390/ijms24043107
Yan, J., Sheng, L., and Li, H. (2021). Akkermansia Muciniphila: is it the holy grail for ameliorating metabolic diseases? Gut Microbes 13 (1), 1984104. doi:10.1080/19490976.2021.1984104
Yang, D., Li, J., Li, Z., Zhao, M., Wang, D., Sun, Z., et al. (2023). Cardiolipin externalization mediates prion protein (PrP) peptide 106-126-associated mitophagy and mitochondrial dysfunction. Front. Mol. Neurosci. 16, 1163981. doi:10.3389/fnmol.2023.1163981
Yang, S., Thackray, A. M., Fitzmaurice, T. J., and Bujdoso, R. (2008). Copper-induced structural changes in the ovine prion protein are influenced by a polymorphism at codon 112. Biochim. Biophys. Acta. 1784 (4), 683–692. doi:10.1016/j.bbapap.2008.01.011
Zhaliazka, K., Matveyenka, M., and Kurouski, D. (2023). Lipids uniquely alter the secondary structure and toxicity of amyloid beta 1-42 aggregates. FEBS J. 290 (12), 3203–3220. doi:10.1111/febs.16738
Zhang, X., St Clair, J. R., London, E., and Raleigh, D. P. (2017). Islet amyloid polypeptide membrane interactions: effects of membrane composition. Biochemistry 56 (2), 376–390. doi:10.1021/acs.biochem.6b01016
Zheng, Y., Sun, J., Luo, Z., Li, Y., and Huang, Y. (2024). Emerging mechanisms of lipid peroxidation in regulated cell death and its physiological implications. Cell. Death Dis. 15 (11), 859. doi:10.1038/s41419-024-07244-x
Keywords: deuterium, copper, histidine, amyloidogenic proteins, cardiolipin, mitochondrial dysfunction, neurodegeneration
Citation: Seneff S and Kyriakopoulos AM (2025) Deuterium trafficking, mitochondrial dysfunction, copper homeostasis, and neurodegenerative disease. Front. Mol. Biosci. 12:1639327. doi: 10.3389/fmolb.2025.1639327
Received: 07 June 2025; Accepted: 04 July 2025;
Published: 22 July 2025.
Edited by:
Venkaiah Betapudi, United States Department of Health and Human Services, United StatesReviewed by:
Anderson Oliveira Souza, Federal University of Mato Grosso, BrazilRaffaella Scanga, University of Calabria, Italy
Danish Idrees, University of Iowa Healthcare, United States
Rahul Patil, Augusta State University, United States
Copyright © 2025 Seneff and Kyriakopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie Seneff, c2VuZWZmQGNzYWlsLm1pdC5lZHU=
†ORCID: Stephanie Seneff, orcid.org/0000-0001-8191-1049; Anthony M. Kyriakopoulos, orcid.org/0000-0002-0749-0060