 Klaudia Dubniewicz
Klaudia Dubniewicz Laura Pardyak
Laura Pardyak Artur Gurgul2
Artur Gurgul2 Zbigniew J. Arent
Zbigniew J. Arent- 1Department of Infectious Diseases and Public Health, Faculty of Veterinary Medicine, University of Agriculture, Krakow, Poland
- 2Department of Basic Sciences, Faculty of Veterinary Medicine, University of Agriculture, Krakow, Poland
Introduction: Leptospirosis, caused by Leptospira spp., is one of the most widespread zoonoses worldwide. It affects both domestic and wild animals, with ruminants serving as a primary reservoir for serovar Hardjo. This serovar causes long-term colonisation of the kidney and genital tract. Hardjo strains are taxonomically assigned to two Leptospira species: Leptospira interrogans and Leptospira borgpetersenii. However, the molecular basis of L. interrogans serovar Hardjo adaptation remains poorly understood. Comparative genomic analysis of L. interrogans strains classified as the Hardjo serovar and other non-Hardjo serovars of the same species may help identify genetic determinants associated with host adaptation and species-specific cellular immune responses. Unfortunately, these pathogens are highly fastidious, and only a limited number of whole genomes have been sequenced to date.
Materials and Methods: Four L. interrogans serovar Hardjo European isolates were sequenced. Using these new sequences alongside publicly available genomes of L. interrogans strains classified as Hardjo and non-Hardjo serovars, we performed comparative genomic analyses.
Results: Hardjo strains formed a distinct phylogenetic clade and harboured unique variants, including an intact cas3 gene and a modified thiM start codon. We identified 88 Hardjo-specific orthologues, some located in putative genomic islands outside rfb locus. Several encoded proteins related to mobile elements, toxin–antitoxin systems or signal transduction. Enhanced biofilm formation in Hardjo strains supports a host-adapted phenotype.
Conclusion: This study expands the genomic dataset for L. interrogans serovar Hardjo and provides novel insights into its genetic distinctiveness, suggesting potential factors that may facilitate colonisation and persistence in ruminant hosts.
1 Introduction
Leptospirosis is a globally distributed zoonotic disease caused by bacteria of the genus Leptospira. Infections occur in a wide range of domestic and wild animals, making it one of the most prevalent zoonoses worldwide (Costa et al., 2015). Pathogenic Leptospira species colonise the kidneys and genital tracts of carrier animals and are shed in urine (Monahan et al., 2009) and reproductive fluids (Dhaliwal et al., 1996; Ellis et al., 1986; Loureiro et al., 2017; Masri et al., 1997), thereby facilitating transmission to new hosts. Although these bacteria can infect many different animal species, each pathogenic serovar is typically maintained within specific animal reservoirs (Ellis, 2015).
Infection in cattle was recognised in 1935 (Mikhin and Azinov, 1935). Bovine infections may result from incidental exposure to serovars (sv.) such as Icterohaemorrhagiae, Pomona, or Grippotyphosa, which are maintained in other animal species (Arent et al., 2017). However, in the 1960s, cattle were recognised as reservoir hosts for the Leptospira serovar Hardjo (Roth and Galton, 1960). This serovar is adapted to cattle and sheep and is capable of long-term colonisation of both the kidneys and reproductive organs. Loureiro and Lilenbaum (Loureiro and Lilenbaum, 2020) proposed that chronic genital leptospirosis in cattle constitutes a distinct syndrome, separate from systemic infection, and thus requires specialised diagnostic and therapeutic strategies.
The introduction of genetic typing methods led to the identification of two genetically distinct but serologically indistinguishable types within serovar Hardjo: Hardjo type Bovis and Hardjo type Prajitno (Thiermann et al., 1986). Based on genomic classification and species definition criteria (Yasuda et al., 1987), these types were assigned to two different species - L. borgpetersenii (Hardjo type Bovis, HB) and L. interrogans (Hardjo type Prajitno, HP). L. interrogans serovar Hardjo was first identified in Scotland and Northern Ireland and has since been isolated from cattle and sheep across Europe and South America (Ellis, 2015; Salgado et al., 2015). Although early isolates primarily belonged to this species, Leptospira borgpetersenii serovar Hardjo is the most prevalent representative of this serovar maintained by cattle and sheep with nearly global distribution (Ellis, 2015).
The ecological adaptability of Leptospira has been linked to the genetic diversity encoded in its relatively large genome - averaging 4.26 Mb, with sizes ranging from 3.89 Mb in L. borgpetersenii to 4.71 Mb in L. interrogans (Fouts et al., 2016; Philip et al., 2021). The reduced genome of L. borgpetersenii is associated with increased host dependence and restricted environmental survival, significantly influencing transmission dynamics (Bulach et al., 2006). In contrast, L. interrogans retains a larger genome, enabling the acquisition of genes involved in immune evasion and host adaptation (Giraud-Gatineau et al., 2024; Xu et al., 2016).
Despite its clinical and epidemiological relevance, the host-pathogen relationship between cattle and Leptospira serovar Hardjo remains poorly understood, limiting progress in vaccine development and effective disease control. Unlike other serovars - such as Icterohaemorrhagiae, Pomona, Canicola, and Grippotyphosa, that primarily induce humoral immunity through antibodies against lipopolysaccharides (LPS), protection against Hardjo appears to rely on strong cell-mediated immune responses (Bolin et al., 1989; Naiman et al., 2001). Although there is compelling evidence for a protective protein antigen (Kunjantarachot et al., 2014; Wunder et al., 2021), no specific antigen has yet been identified (Murray et al., 2013), further underscoring the need to understand serovar-specific immune mechanisms. At the molecular level, the mechanisms of host adaptation by Leptospira sv. Hardjo remain largely uncharacterised. Comparative genomic analyses, particularly between L. interrogans sv. Hardjo and other serovars, could help identify genes involved in host adaptation and immune modulation in cattle and sheep. However, research in this area has been constrained by the fastidious nature of these organisms and the limited availability of fully sequenced genomes, especially for serovar Hardjo. Although previous genomic studies have examined L. interrogans sv. Hardjo (Cosate et al., 2015; Llanes et al., 2016), they lacked European strains and broader representation of Hardjo isolates, leaving important questions unanswered.
This study presents a comparative analysis of L. interrogans serovar Hardjo strains based on whole-genome sequencing. The primary objectives were to sequence and assemble genomes from selected Hardjo isolates, assess their phylogenetic relationships, and identify genes and gene products potentially involved in the specific infection process. Here, we report the genome sequences of four European L. interrogans serovar Hardjo isolates. Our central hypothesis is that comparative genomic analysis of L. interrogans serovar Hardjo with other L. interrogans serovars sharing a similar genomic background could reveal genetic determinants specific to the Hardjo serovar, which may underlie its unique host associations and pathogenic traits. We hope that this research will serve as a foundation for further investigations into the molecular mechanisms underlying Hardjo serovar determination, which may be conserved across both L. interrogans and L. borgpetersenii species.
2 Materials and methods
2.1 Sample characterisation
The study was based on four L. interrogans serovar Hardjo strains: KR40, KR84, KR85, and N116 – each isolated from different host and region in Europe. Strain KR40 was obtained from a horse in Italy in 1997, KR84 from a wallaby in the United Kingdom in 1983, KR85 from a dog in the United Kingdom in 1982, and N116 from a cow in Belgium in 2016. All Leptospira isolates were originally classified on the basis of cross-agglutination tests using serogroup-specific hyperimmune rabbit sera in the microscopic agglutination test (MAT). After genome sequencing, isolate identity was subsequently confirmed using the core-genome multilocus sequence typing (cgMLST) scheme based on 545 highly conserved loci, developed by Guglielmini et al. (2019) and implemented on the Institut Pasteur webstite BIGSdb-Pasteur (https://bigsdb.pasteur.fr/).
2.2 Restriction endonuclease analysis
To enable the preliminary identification of selected isolates as belonging to the L. interrogans serovar Hardjo type Prajitno, we performed restriction endonuclease analysis (REA) following established protocols. Genomic DNA was extracted from 4 L. interrogans isolates (KR40, KR84, KR85, and N116) using the QIAamp DNA Mini Kit (Qiagen) Genomic, according to the manufacturer’s instructions. For comparative purposes, genomic DNA from two Hardjo strains were also included: L. interrogans serovar Hardjo strain Hardjoprajitno and L. borgpetersenii serovar Hardjo strain KR39 (BioProject: PRJNA828006).
DNA samples were digested with the restriction enzyme EcoRI (Thermo Fisher Scientific), using 1 µg of DNA and 10 units of enzyme in a 20 µL reaction volume, incubated at 37 °C for 1 h. The resulting DNA fragments were separated by electrophoresis on a 0.6% agarose gel in 1× TBE buffer at 70 V for 21 h. Gels were stained with ethidium bromide and visualised under UV light using a GelDoc imaging system (G:BOX Chemi system (Syngene) equipped with GeneSys/GeneTools software). Banding patterns were analysed visually and compared with the reference strains following the method described by Ellis et al. (1991). Strains exhibiting REA profiles consistent with the Hardjoprajitno reference were considered to belong to the L. interrogans sv. Hardjo (type Prajitno) lineage.
2.3 Whole genome sequencing and annotation
Genomic DNA was isolated from bacterial cultures using the Genomic Mini AX Bacteria kit (A&A Biotechnology, Poland) and quantified using the NanoDrop 2000 and Qubit systems (Thermo Scientific). Genomic DNA fragmentation was verified by agarose gel electrophoresis. Illumina sequencing libraries for strains under investigation were prepared from 200 ng total DNA according to the manufacturer’s instructions with the Lotus DNA Library Prep Kit (Integrated DNA Technologies, Inc.). Sequencing was performed using single-end 75 bp runs on an Illumina NextSeq 550 platform with a minimum of 40x genome coverage. Oxford Nanopore sequencing libraries, due to objective reasons, were generated only for the KR40 and N116 strains, and were prepared with 3 µg of total DNA using the Ligation Sequencing Kit (SQK-LSK110; Oxford Nanopore Technologies, Oxford, UK) according to the manufacturer’s protocol. The libraries were loaded onto R10.3 flow cells and sequenced on the MinION Mk1C system (Oxford Nanopore Technologies). Basecalling was carried out with Guppy (v5.0.11+2b6dbff) using the super-accuracy model.
For KR84 and KR85 strains, Illumina raw sequencing reads were evaluated for quality using FastQC v0.11.9 (Andrews, 2010) and filtered using TrimGalore software v0.6.10 (Krueger, 2021). Filtering was done through the removal of adapter sequences and trimming of low-quality read ends. Filtered reads were assembled into contigs using Shovill v1.1.0 (https://github.com/tseemann/shovill), an ultra-fast implementation of the SPAdes v3.14.0 algorithm (Nurk et al., 2013). The resulting contigs were polished using Pilon v1.23 (Walker et al., 2014). Contigs statistics were retrieved using QUAST software v5.0.2 (Mikheenko et al., 2018). Scaffolding was carried out by CSAR-web (Chen and Lu, 2018), using the closest available complete genome for the analysis at the time of analysis, determined by 16S rRNA analysis: L. interrogans serovar Hardjo strain Norma (reference assembly: GCA_001293065.1). Genome sequences were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) with the best-placed reference protein set (GeneMarkS-2+) v6.0. The assemblies were deposited in GenBank under BioProject PRJNA809530 (KR84 assembly: GCA_022436545.1; KR85 assembly: GCA_022436605.1).
For KR40 and N116 strains (hybrid assemblies) raw Illumina reads were qualified by FastQC for quality and trimmed using Flexbar v3.5.0 (Dodt et al., 2012), removing adapter sequences and trimming low-quality read ends. Nanopore reads were trimmed using Fastp v0.23.2 (Chen et al., 2018), and adapter sequences were removed using Porechop v0.2.4 (Wick et al., 2017) and filtered with Filtlong v0.2.1 (https://github.com/rrwick/Filtlong), setting a minimum nucleotide length of 500 bp. Low-quality read ends were trimmed using Fastp. Filtered Illumina and Nanopore reads were assembled de novo with a hybrid strategy using Unicycler v0.5.0 (Wick et al., 2017) with default parameters. The obtained assemblies were annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) with best-placed reference protein set (GeneMarkS-2+) v6.1 (strain N116) and v6.2 (KR40). Assembly was deposited in the GenBank under the BioProject number PRJNA828004 (assembly: GCA_023158895.3) and PRJNA828002 (assembly: GCA_023515975.1) for KR40 and N116 respectively.
2.4 Comparative genomics and phylogenetic analyses
Genomic relatedness of analysed strains was compared with 19 publicly available L. interrogans genomes from the NCBI database. All genomes were selected based on complete assembly status, to ensure consistent genome quality and to avoid bias in downstream analyses. They represent a wide range of serovars, geographic regions, host species, and collection years (Supplementary Table S1). The only exception was made for 3 L. interrogans serovar Hardjo strains (str. KR84, KR85, and OV5) which were included despite being high-quality draft assemblies. This was due to the limited availability of complete genomes for serovar Hardjo in public databases at the time of study design. Excluding them would have significantly reduced the representativeness of the Hardjo group.
The relationship between the number of coding sequences (CDS) and total genomic length was assessed using Spearman’s rank correlation, due to non-normal distribution. The average nucleotide identity (ANI) was calculated using JSpecies with ANIm option (Richter et al., 2016). Variant calling analysis (VCA) was performed to identify point mutations and establish population and phylogenetic structure. Filtered sequences were mapped to the reference genome L. interrogans sv. Hardjo str. Norma (Ref Seq: GCF_001293065.1) using BWA v0.7.17 (Li, 2013). Variants were identified using Freebayes v1.3.6 (Garrison and Marth, 2012) and filtered by a quality score cut-off >30 and depth >10. Variants were classified as SNPs (single nucleotide polymorphisms), MNPs (multi-nucleotide polymorphisms), INDELs (insertions/deletions), or mixed variants, which comprised a combination of the above types. The effects of the variants (excluding upstream/downstream, 5′UTR/3′UTR, and intronic changes) were predicted with SnpEff (Cingolani, 2022). Additionally, each of the selected high-impact variants was manually examined to evaluate its phenotypic relevance by determining whether the nucleotide change altered the protein sequence or led to its complete absence. Population structure analysis was conducted with the ADMIXTURE programme (Alexander et al., 2009) for K values from 1 to 10, selecting the optimal K based on cross-validation error. Results were visualised in Clumpak (Kopelman et al., 2015). The phylogenetic tree was constructed using the IQ-TREE web server with the auto-detect best-fit model with 1,000 ultrafast bootstraps (Nguyen et al., 2015) and visualised with iTOL v5 (Letunic and Bork, 2021). Ortholog clusters of the entire set of protein sequences were established using OrthoFinder (v3.0.1) with default parameters (Emms and Kelly, 2019). To assess differences between Hardjo strains and non-Hardjo strains, genomes were divided into two groups: L. interrogans serovar Hardjo group (LiH) and L. interrogans serovar non-Hardjo group (Li-nH). Statistical differences between groups were assessed using Fisher’s Exact Test, which is appropriate for categorical data and small sample sizes. To account for multiple testing, the Bonferroni correction was applied by dividing 0.05 by the number of tests (separately for VCA analysis and ortholog clusters analysis) to establish the initial significance threshold. However, due to the limited sample size and the highly conservative nature of the Bonferroni method, which may lead to a high false-negative rate, we applied a relaxed threshold - filtering based on raw p-values <5 × 10−6 for VCA analysis and 1 × 10−4 for ortholog clusters analysis. All calculations were performed in R (v4.4.2).
2.5 In silico characterisation of genes and proteins
Genes and encoded proteins of interest were further searched in the UniProt database (Bateman et al., 2023) and the STRING database (Szklarczyk et al., 2023). Functional classification was established with the COG database using COGclassifier v1.0.5 (Tatusov et al., 2000). The CELLO programme v.2.5 (Yu et al., 2006), PSORT database v3.0.3 (Venus Lau et al., 2021), and SOSUI GramN programme v1.11 (Imai et al., 2008) were used to predict protein subcellular localisation with a majority voting strategy, as previously described (Techawiwattanaboon et al., 2023). Additionally, proteins that were predicted to be outer membrane, inner membrane or periplasmatic proteins by different programmes, without a majority voting strategy, were assigned to a newly created category labelled as ‘Membrane’. The SignalP 6.0 server (Teufel et al., 2022) was used to search for signal peptides. Proteins not detected by SignalP were further processed by the SecretomeP 2.0 (Bendtsen et al., 2005) to predict non-classical secreted proteins. To improve the prediction of spirochaetal lipoproteins, we additionally applied SpLiP tool (Setubal et al., 2006). All predictions were performed with default settings for Gram-negative bacteria. For genes and proteins lacking predicted information from the described procedures, we employed DeepFRI programme for functional annotation (Gligorijević et al., 2021). Genes and variants of interest were further categorized into those occurring outside the rfb locus and those within the rfb locus. The localisation of the rfb locus was determined based on the region spanning from the MarR regulatory gene to the DASS gene (Fouts et al., 2016; Ferreira et al., 2024). Probable genomic islands associated with horizontal gene transfer were predicted using the IslandViewer 4 platform (Bertelli et al., 2017). Gene localisation and COG categories were visualised using in-house R scripts (v4.4.2).
2.6 Biofilm quantification
To confirm the hypothesis that genetic changes could impact the capacity of Hardjo and non-Hardjo strains of L. interrogans to form biofilm, we evaluated biofilm formation in vitro. Biofilm formation by L. interrogans (serovars Hardjo, Copenhageni, and Bratislava) was evaluated using a static microtiter plate assay according to the protocol of Thibeaux et al. (2020a). Bacteria were grown in EMJH medium at 30 °C under static conditions to the exponential phase, adjusted to a final density of 1 × 106 cells/mL, and seeded into sterile, flat-bottom 96-well polystyrene plates (150 µL per well). Plates were incubated at 30 °C for 21 days without medium replacement to allow for mature biofilm formation. After incubation, planktonic cells were gently removed, and biofilms were fixed with 4% paraformaldehyde for 30 min. Wells were then washed with PBS, stained with 0.1% crystal violet for 10 min, and washed again. Stained biofilm was solubilised using 30% acetic acid, and absorbance was measured at 570 nm using a microplate reader. Biofilm assays were performed in three independent experiments, each with triplicate wells per condition. Statistical comparisons were based on biological replicates (n = 3 per group per time point). Statistical significance between serovars at each time point was assessed using the Kruskal-Wallis test, followed by pairwise Mann-Whitney U tests with Bonferroni correction for multiple comparisons. A p-value <0.05 was considered statistically significant.
3 Results
3.1 Sample characterisation
Serological characterisation indicated that the tested field isolates belonged to the Sejroe serogroup (data not shown). For the preliminary identification of isolates as members of the L. interrogans serovar Hardjo, we applied the restriction endonuclease method (REA). Six strains belonging to serovar Hardjo were included in the analysis. The results revealed that DNA isolated from KR84, KR85, KR40, and N116 strains (Figure 1, lanes 4, 5, 6, 7) showed similar restriction patterns, consistent with that of the reference genome L. interrogans sv. Hardjo str. Hardjoprajitno (Figure 1, lane 3). The pattern for L. interrogans sv. Hardjo differed from that of L. borgpetersenii sv. Hardjo (Figure 1, lane 2). A minor difference was observed in the absence of an extra high molecular weight band of approximately 9,416 bp (Figure 1, white arrow) in the KR84, KR85, and N116 isolates (Figure 1, lanes 4, 5, and 7) compared with strains KR40 and Hardjoprajitno (Figure 1, lanes 3 and 6). Consistent with these REA findings, cgMLST analysis ultimately confirmed the taxonomic assignment - all four isolates grouped in cluster 40 (clonal group 19) of the cgMLST scheme, together with other representatives of L. interrogans serogroup Sejroe serovar Hardjo. The curated records are publicly available in BIGSdb-Pasteur (https://bigsdb.pasteur.fr/leptospira) under IDs: 1234 (KR85), 1271 (N116), 1272 (KR40) and 1336 (KR84).
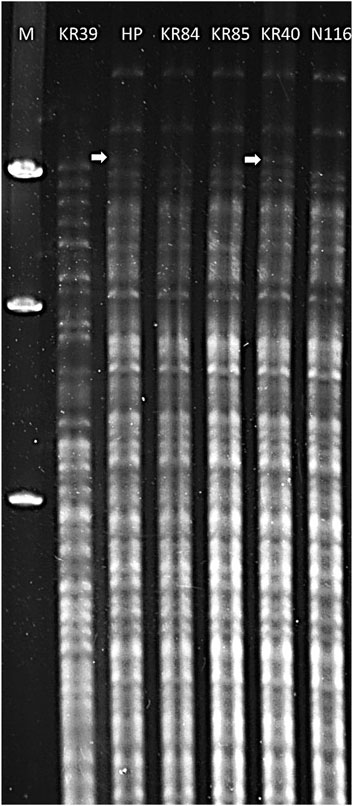
Figure 1. Restriction endonuclease analysis patterns of DNA from Leptospira strains digested with EcoRI. Lanes: 1 – Marker Lambda/Hind III (A&A Biotechnology); 2 – L. borgpetersenii sv. Hardjo str. KR39; 3 – L. interrogans sv. Hardjo str. Hardjoprajitno; 4 – L. interrogans serovar Hardjo str. KR84; 5 – L. interrogans sv. Hardjo str. KR85; 6 – L. interrogans sv. Hardjo str. KR40; 7 - L. interrogans sv. Hardjo str. N116; white arrow - presence of an additional high molecular weight band (∼9,416 bp).
3.2 Assembly and annotation of the sequenced genomes
Sequencing statistics for L. interrogans strains under study are summarised in Supplementary Table S2. The generated sequencing reads provided genome coverage of approximately 42x for assemblies based solely on Illumina reads (strains KR84 and KR85) and over 114x for hybrid assemblies. Using hybrid assembly approaches, complete genomes consisting of five and four circular contigs were obtained for strains N116 and KR40, respectively. For strains KR84 and KR85, draft whole-genome sequences consisted of 177 and 186 contigs, respectively. After scaffolding against the genome of L. interrogans serovar Hardjo str. Norma, nine scaffolds were generated for KR84 and seven scaffolds for KR85. Hybrid assemblies could not be obtained for strains KR84 and KR85 in this study. The genome size and number of predicted genes suggest, however, a high level of the completeness of their genomes, even in non-hybrid assemblies. The high sequencing coverage enabled a nearly complete representation of the genomic content, with only homologous or repetitive regions potentially preventing full assembly into continuous, circularised sequences (str. KR84, KR85).
Table 1 presents the quality control parameters for the four studied strains of L. interrogans, while Table 2 summarises their genome annotation parameters. The annotated genomes displayed a similar number of detected genes, with an average of 3,930 and an interquartile range (IQR) of 111.5; the gene count was slightly higher in the hybrid assemblies. On average, 96% of the genes were protein-coding. Each genome contained between 3 (for whole genome sequences of KR84 and KR85) and 5 complete rRNAs (for complete genome sequences for genomes KR40 and N116), 37 tRNAs, and approximately 156 pseudogenes (IQR: 7.75). The genomic G + C content ranged from 34.96% to 35.05%.
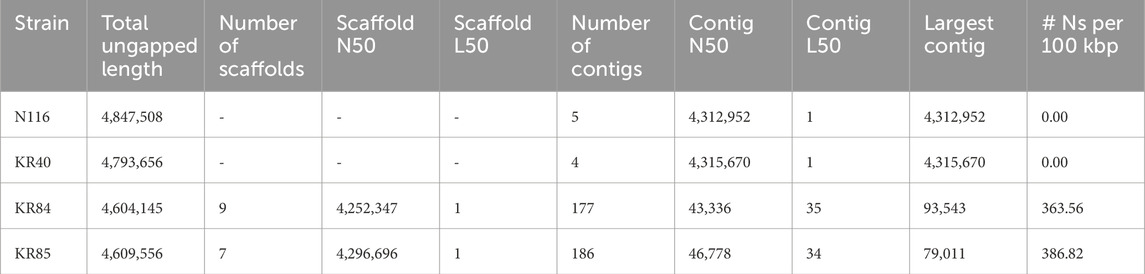
Table 1. Assembly quality control parameters for studied strains of Leptospira interrogans sv. Hardjo.
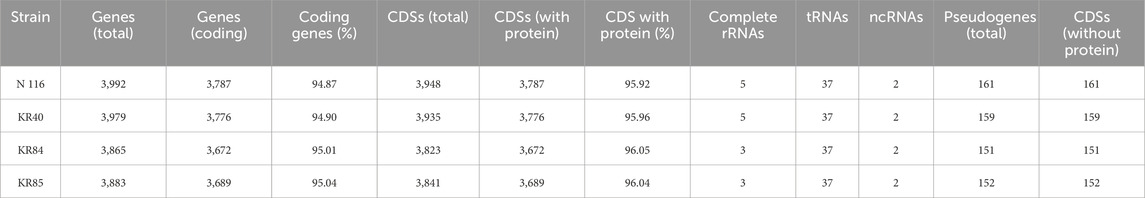
Table 2. Genome annotation parameters for studied strains of Leptospira interrogans sv. Hardjo
3.3 Comparative genomics
Genetic analysis was conducted on the four genomes obtained during this study and on an additional 19 genomes of L. interrogans, selected from the publicly available NCBI database (Supplementary Table S1). The genomic sequence length ranged from 4,630,592 to 5,349,767 bp with an average GC content of 35.01%. The total number of genes per genome ranged from 3,762 to 5,312. The number of coding sequences had a positive Spearman’s correlation of 0.734 (p = 6.73 × 10−5) with the genomic length.
ANI analysis provided a comparative genomic relatedness between various serovars and strains of L. interrogans (Supplementary Figure S1). The results from the JSpecies analysis revealed a high ANI percentage, ranging from 98.15% to 99.98% between different strains in our study. Serovar Hardjo displayed ANI values close to 100% among its strains. Lower ANI values, as shown in the heatmap in blue, suggest increased genetic distance and greater diversity, especially within the L. interrogans sv. Canicola and sv. Manilae, signifying potential genomic variability or divergence from other serovars.
3.4 Population structure and phylogenetic analysis
Population structure analysis and phylogenetic reconstruction were performed based on the output of variant calling analysis (VCA). Population structure analysis (Figure 2A), conducted using the ADMIXTURE programme, indicated an optimal K value of 5 (log-likelihood = −344427.89, CV error: 0.77). The analysis separated all sv. Hardjo strains from other serovars. Since traditional phylogenetic markers such as 16S rRNA lack the resolution to distinguish between closely related serovars within the same species, we employed SNP-based analysis to achieve a higher level of phylogenetic resolution for the analysed L. interrogans strains. A phylogenetic tree was constructed using IqTree with TVM + F substitution model and 1,000 ultrafast bootstrap option (Figure 2B). We obtained strong bootstrap support, particularly for internal nodes connecting major clades. The phylogenetic tree, with some exceptions, revealed serovar-specific branches. The topology of the tree suggest that serovar Hardjo is the most closely related to members of the Icterohaemorrhagiae serogroup. Among all Hardjo representatives, short phylogenetic branches indicate limited genetic diversity.
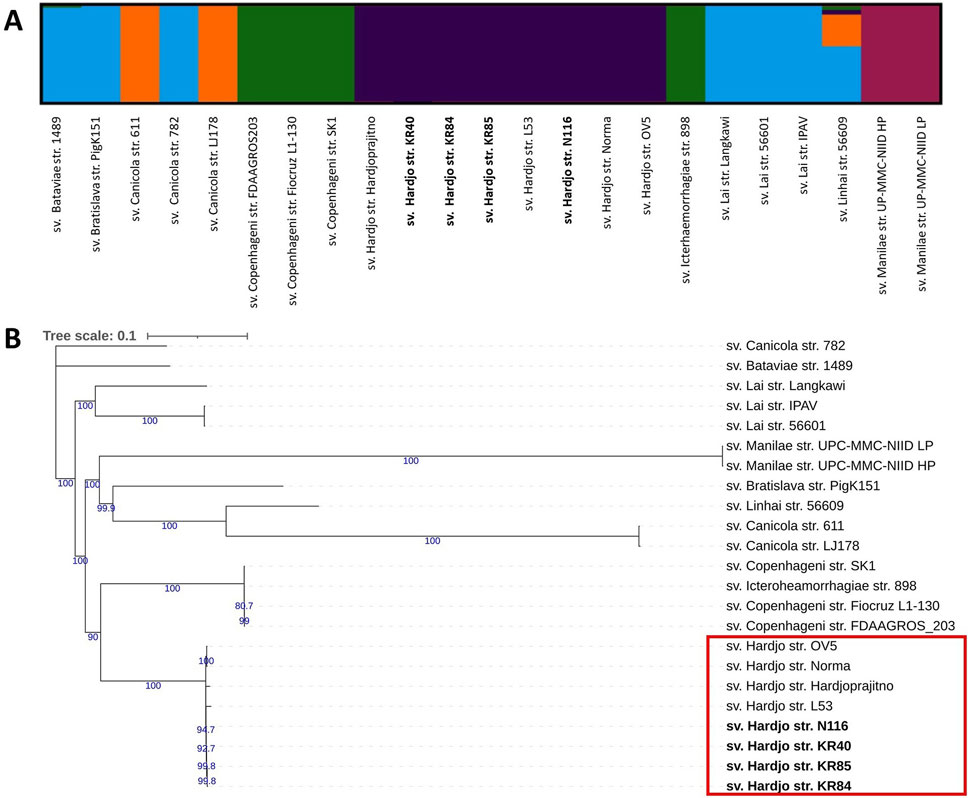
Figure 2. Population structure and phylogenetic relationships of Leptospira interrogans strains show a clear separation of serovar Hardjo. (A) Population structure analysis of L. interrogans strains using ADMIXTURE. Results are shown for K = 5; (B) Maximum likelihood SNP-based phylogenetic tree of L. interrogans with 1,000 ultrafast bootstrap replicates with TVM + F substitution model. Genomes obtained during this study were bolded. Only bootstrap values greater than 60 were shown (blue colour).
3.5 Variant calling analysis and identification of Hardjo-specific effects
For the purposes of this study, VCA using L. interrogans sv. Hardjo str. Norma as the reference genome was chosen. Approximately 99.4%–99.9% of the Hardjo reads were successfully aligned to the reference genome, whereas alignment rates for reads from other serovars varied from 87.9% for sv. Canicola to 96.6% for sv. Copenhageni.
Table 3 provides a summary of all high-quality variants detected across the dataset, as well as the subset of statistically significant differences between LiH and Li-nH groups. In total, the analysis revealed 99,656 genetic variants and 99,771 predicted effects on gene and their encoded proteins. Out of these, 10,266 variants and 10,724 effects were identified as statistically significant as differentiating between the groups, including 6,299 in the coding region and 4,425 in the non-coding region. Statistically significant variants were primarily located outside the rfb locus, although some conserved segments within this region also exhibited polymorphisms. Among genetic variants, SNPs were the most abundant. Overall, the ratio of non-synonymous to synonymous changes (dN/dS) was 0.4336, and was similar for statistically significant differences between the Hardjo and non-Hardjo groups, with a value of 0.4256.
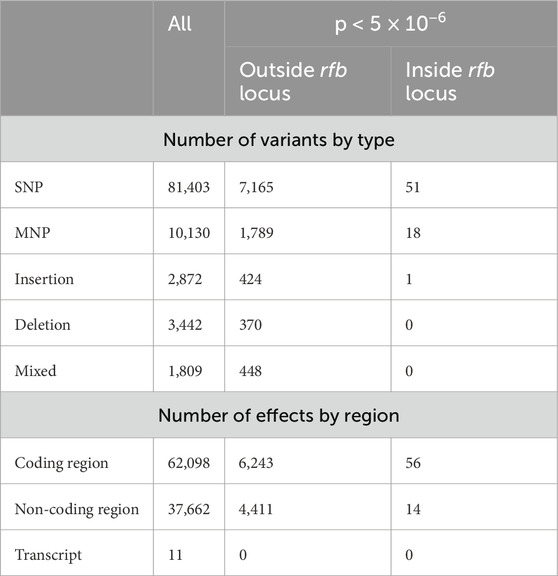
Table 3. The statistics of VCA for results with quality >30 and deep >10. The column “All” presents the total number of all observed variants passing quality thresholds. The two subsequent columns show statistically significant differences between the LiH group and the Li-nH group (p-values <5 × 10−6, calculated using Fisher’s exact test with a relaxed Bonferroni-derived cut-off). Results are further divided by genomic region (coding vs. non-coding) and by their location relative to the rfb locus.
We identified four statistically significant high-impact SNPs with predicted effects on the corresponding protein sequences. One of them, was located in the G436_RS22205 gene (p = 8.7 × 10−12), involving a cytosine (C) to guanine (G) substitution at position 3,936,506 on chromosome I (Figure 3A). This mutation introduces a premature stop codon, resulting in the absence of the encoded protein (WP_002080799.1) in non-Hardjo serovars. According to UniProt, the protein is annotated as a mobile element protein (A0A0M4NZF1), showing 100% identity with a PF07600 domain-containing sequence. Two additional premature stop codons were detected with lower statistical significance (p = 2.8 × 10−6). The first was located at position 3,455,697, involving a cytosine (C) to thymine (T) substitution in gene G436_RS23575 (Figure 3B), affecting a hypothetical protein (WP_002069478.1), suggested by DeepFRI to localise in the cytoplasm and potentially participate in general metabolic processes. The second occurred at position 3,670,902, involving a cytosine (C) to thymine (T) substitution in gene G436_RS15655 (Figure 3C), encoding the CRISPR-associated helicase Cas3 (WP_002189459.1), which appears as a pseudogene or truncated protein in Li-nH group. Additionally, we identified a SNP in G436_RS14945 (Figure 3D), where an adenine (A) to guanine (G) substitution at position 3,497,928 alters a start codon from ATG to GTG in non-Hardjo strains, affecting translation initiation for the thiazole kinase ThiM (WP_002188296.1; p = 1.0 × 10−9).
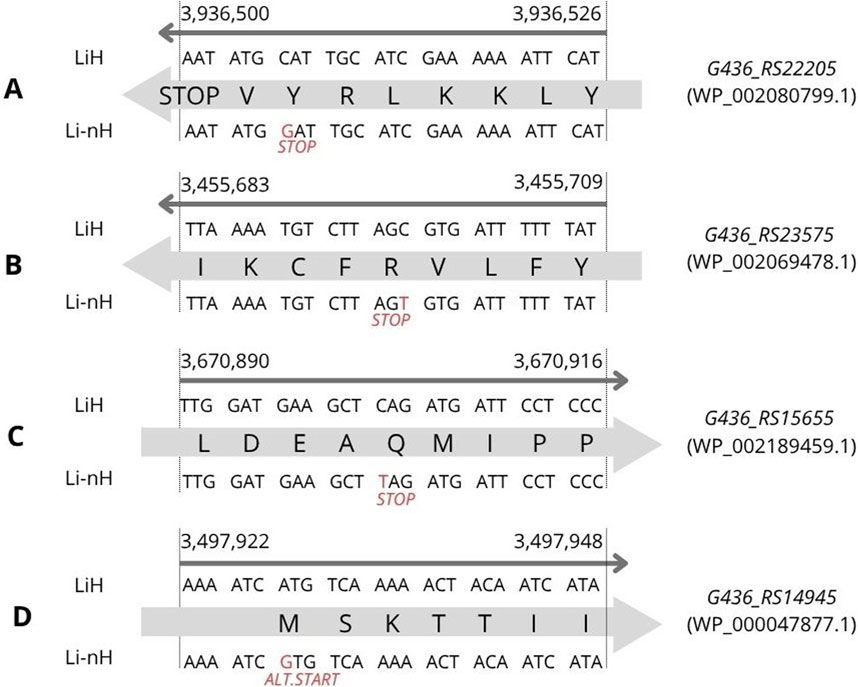
Figure 3. SNP variants identified through variant calling analysis were associated with phenotypic differences observed between the LiH and Li-nH groups (p < 5 × 10−6, calculated using Fisher’s exact test with a relaxed Bonferroni-derived cut-off). Each mutation is annotated based on its location on either the forward or reverse strand. (A) Premature stop codon in G436_RS22205 gene; (B) Premature stop codon in G436_RS23575 gene; (C) Premature stop codon in G436_RS15655; (D) Alternative start codon in G436_RS14945 gene.
In addition, three INDELs were identified with statistically significant frequency differences between LiH and Li-nH groups, each predicted to affect protein sequence. Two of them resulted in the complete loss of protein-coding sequence in the Li-nH group. The first, located in G436_RS23265 (Figure 4A), involves a thymine (T) insertion leading to the absence of protein WP_100224992.1 (p = 8.7 × 10−12). The second, in G436_RS23350 (Figure 4B), results from a guanine (G) insertion and frameshift, leading to disruption of WP_154643182.1 coding sequence in Li-nH (p = 1.0 × 10−9). Both proteins are hypothetical and predicted to localize to the cytoplasm, with putative roles in metabolic processes, as predicted by DeepFRI. The third INDEL, located in G436_RS18190 (Figure 4C), modifies the N-terminal region of the FecR family protein WP_029757821.1, shortening it from 389 to 385 amino acids in non-Hardjo strains (p = 8.7 × 10−12). FecR proteins are implicated in iron uptake via citrate transport, though the functional consequences of this truncation remain unclear.

Figure 4. INDEL variants identified through variant calling analysis were associated with phenotypic differences observed between the LiH and Li-nH groups (p < 5 × 10−6, calculated using Fisher’s exact test with a relaxed Bonferroni-derived cut-off). Each mutation is annotated based on its location on either the forward or reverse strand. (A) Loss of coding sequence due to an insertion in gene G436_RS23265; (B) Loss of coding sequence due to an insertion in gene G436_RS23350; (C) Frameshift caused by an insertion in gene G436_RS18190.
We also identified statistically significant, high-impact mutations, including one start-loss variant, six stop-gain variants, 17 stop-loss variants, 25 frameshift variants, and 13 complex variants occurring in Hardjo pseudogenes (Supplementary Table S3). According to the RefSeq annotation, L. interrogans sv. Hardjo str. Norma has a total of 184 pseudogenes, which constitute 3.7% of all genes. A number of pseudogenes in each newly sequenced genome is shown in Table 2, and varies from 151 to 161.
In total, we identified 1,335 significant missense variants, 159 of which were located in pseudogenes. The remaining 1,176 variants were mapped to 954 distinct genes. Particular attention was given to genes encoding predicted outer membrane proteins (OMPs), due to their surface localisation and possible interaction with the host environment. To identify proteins with N-terminal signal peptides and signal peptidase cleavage sites, SignalP 6.0 was used. Among the 954 genes analysed, 191 were predicted to encode proteins with secretion signal sequences. Of these, 119 had secretory signal peptides transported by the Sec translocon and cleaved by Signal Peptidase I (SP); 66 had lipoprotein signal peptides transported by the Sec translocon and cleaved by Signal Peptidase II (LIPO); four contained Tat signal peptides transported by the Tat translocon and cleaved by Signal Peptidase I (TAT); one had a Tat lipoprotein signal peptide transported by the Tat translocon and cleaved by Signal Peptidase II (TATLIPO); and one had pilin and pilin-like signal peptides transported by the Sec translocon and cleaved by Signal Peptidase III (PilD/PibD) (PILIN). Due to the plasticity of spirochaetal lipobox sequences, SpLiP algorithm, a tool optimised for spirochaetes, was used. 55 proteins were predicted by SpLip as probable lipoproteins and 12 as possible lipoproteins. 80 proteins were predicted by SecretomeP to be exported through the non-classical protein secretion pathway. To further predict protein subcellular localisation, we applied the CELLO programme, the PSORT database, and SOSUI GramN programme applying a majority-voting strategy to enhance prediction accuracy. 29 proteins were predicted as ‘Extracellular’, 49 as ‘Outer membrane’, 130 as ‘Inner membrane’, 12 as ‘Periplasmic’, whereas 56 were assigned to general category ‘Membrane’ (Supplementary Table S4).
3.6 Identification of unique cluster of orthologues
The OrthoFinder programme identified 4,326 orthologous groups across the analysed genomes. The pangenome of L. interrogans comprised 3,168 core orthologous gene clusters, defined as those present in at least 95% of the analysed genomes. 88 orthogroups were classified as unique to the Hardjo serovar (LiH). Conversely, 47 orthogroups were predominantly present in the non-Hardjo group (Li-nH) (Figure 5). A full list of LiH-specific proteins, along with predicted localisation in the genome, potential localisation in the cell compartment, potential prediction of non-classical protein secretion, and prediction of lipoproteins is in Table 4 (for genes located outside the rfb locus) and Supplementary Table S5 (for genes located within the rfb locus).
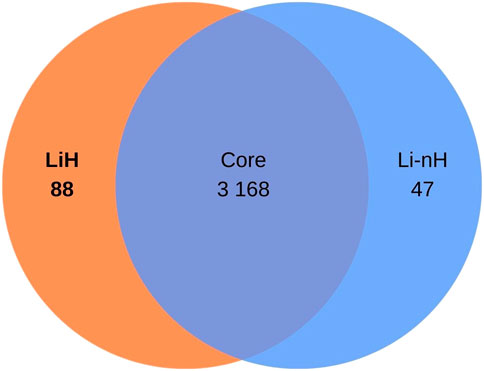
Figure 5. Venn diagram illustrating the distribution of orthogroups identified in Leptospira interrogans. A total of 3 168 orthogroups were classified as core genes. Additionally, 88 orthogroups were identified as specific to the Hardjo serovar (LiH), while 47 orthogroups were specific to other serovars (Li-nH), being absent in the Hardjo serovar (p < 1 × 10−4; calculated using Fisher’s exact test with a relaxed Bonferroni-derived cut-off).
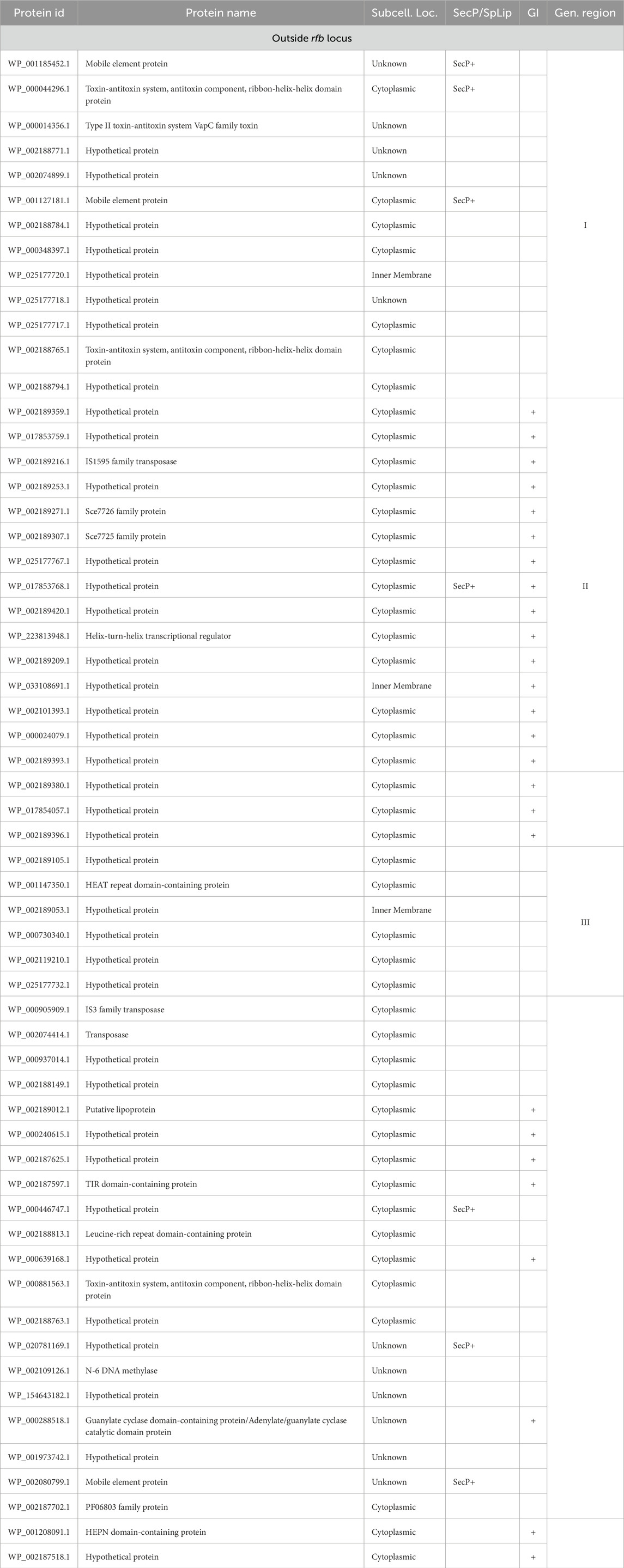
Table 4. A complete list of orthologues proteins specific to LiH group (p < 1 × 10−4; calculated using Fisher’s exact test with a relaxed Bonferroni-derived cut-off) outside rfb locus, along with potential subcellular localisation (Subcell. Loc.) determined using a combination of methods, including CELLO, PSORTdb, and the SOSUI, SpLip for lipoprotein prediction, as well as prediction of non-classical protein secretion type by SecretomeP (SecP). Additionally, the table groups genes based on their location within defined genomic regions (Gen. region) and predicted genomic islands (GI) as identified by IslandViewer 4.
The analysis of orthogroups specific to Hardjo and non-Hardjo serovars, revealed distinct functional profiles, as classified by COG categories. In the LiH group, 36.36% (32/88) of sequences were assigned to COG functional categories, compared to 55.32% (26/47) in the Li-nH group (Figure 6). Among the categories, ‘Cell wall/membrane/envelope biogenesis’ displayed the most significant differences.

Figure 6. COG functional classification of orthgroups unique to LiH and Li-nH groups.
Regarding the predicted subcellular localisation, 70 proteins were classified as ‘Cytoplasmic’, 6 as ‘Inner Membrane’ and 1 as ‘Outer Membrane’. None were predicted by SignalP whereas seven were identified by SecretomeP as candidates for non-classical secretion. However, all were classified as cytoplasmic or of unknown localisation, mostly representing hypothetical or mobile element proteins, often secreted via type IV systems in Gram-negative bacteria (Waksman, 2019). One protein (WP_000808306.1) was predicted as probable lipoprotein by SpLip.
As rfb locus is known for its high variability across Leptospira serogroups and serovars, we examined the localisation of LiH-specific orthogroups with respect to this region and visualised their distribution in the reference genome of strain N116 (Figure 7). Subsequent analyses focused primarily on the LiH-specific genes located outside the rfb locus. Of the 88 LiH-specific genes, 59 were located outside the rfb locus (Table 4). 37 of them were annotated as hypothetical proteins The observed clustering of several genes prompted us to assess their presence within putative genomic islands (GIs) using IslandViewer 4. Three large genomic regions enriched in LiH-specific genes were identified. The first region (positions 2,280,254–2,289,105 bp in str. N116) included genes for eight hypothetical proteins, two mobile element proteins (WP_001185452.1, WP_001127181.1), and three proteins associated with toxin-antitoxin systems (WP_000044296.1, WP_000014356.1, WP_002188765.1). The GC content of this region (39.96%) is noticeably higher than the average genomic GC content of L. interrogans strain N116 (35.05%). A second region (positions 521,196–532,505 bp; GC content 30.29%) comprised fourteen hypothetical proteins, an IS1595 transposase (WP_002189216.1), and two Sce7726 and Sce7725 family proteins (WP_002189271.1 and WP_002189307.1, respectively). Third region (1,463,770–1,468,634 bp) contained mainly hypotethical proteins (GC content 30.98%). In these regions, several interspersed genes were not classified as unique to L. interrogans sv. Hardjo. Additionally, large intergenic distances between some of the genes suggest that these clusters are unlikely to form single operon. While the first region is located entirely on the same strand, the second and third region span both DNA strands. Notably, only the second region was predicted as a genomic island by IslandViewer 4, whereas the first region was not.
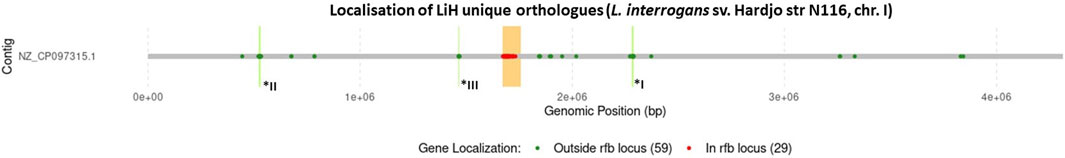
Figure 7. Localisation of genes encoding orhtogroups unique to sv. Hardjo on example of str. N 116. The rfb locus was marked in orange, with genes located within this region shown in red, and those outside it in green. Number of genes in each group is provided in parentheses. Additionally, genomic regions I–III, corresponding to positions I: 2,280,254–2,289,105, II: 521,196–532,002, and III: 1,463,770–1,468,634, are marked in bright green and labelled with asterisks.
Beyond these defined regions, we also detected several LiH-specific genes that either formed small local groupings or were scattered throughout the genome without consistent clustering. Among them were genes encoding a toxin-antitoxin system component (WP_000881563.1), a Toll/Interleukin-1 receptor domain containing protein (WP_002187597.1), a guanylate cyclase domain-containing protein (WP_000288518.1), and a leucine-rich repeat domain-containing protein (WP_002188813.1), positioned near the gene encoding the flagellar motor switch protein FliG. Of note, IslandViewer 4 predicted that some of these individual genes are located within genomic islands (Table 4).
3.7 Biofilm formation
Crystal violet measurements revealed a clear time-dependent increase in biofilm biomass for all 3 L. interrogans serovars (Hardjo, Copenhageni, Bratislava) over the 21-day static incubation (Figure 8A). Serovar Hardjo formed significantly more biofilm than the other two serovars at each time point (p < 0.05), with absorbance values markedly higher by week 3. Serovar Copenhageni exhibited an intermediate biofilm production, whereas Bratislava consistently showed the lowest values. Microscopic examination mirrored these results: phase-contrast images (Figure 8B) demonstrated more extensive and confluent biofilm architecture for Hardjo, especially evident after 3 weeks, while Copenhageni and Bratislava produced progressively less dense microcolonies.
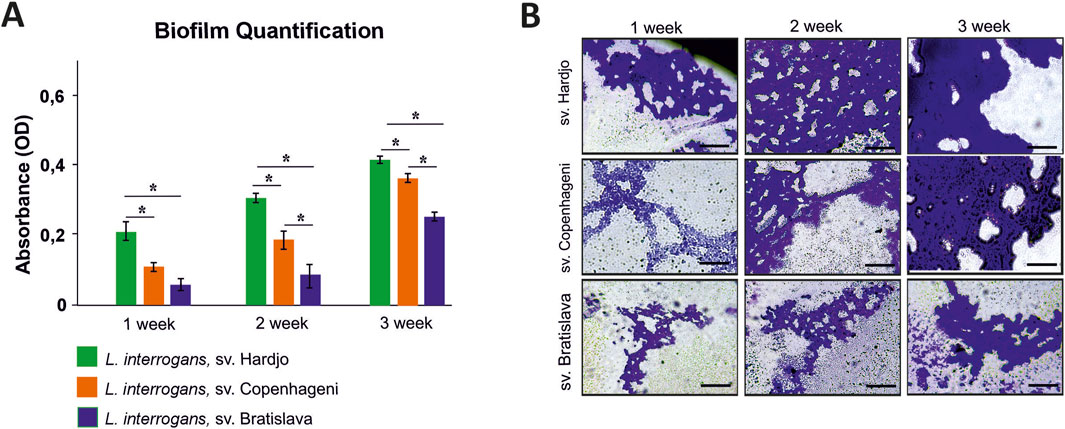
Figure 8. Biofilm formation by Leptospira interrogans serovars Hardjo, Copenhageni, and Bratislava over a 3-week incubation period. (A) Quantification of biofilm biomass by crystal violet staining expressed as optical density (OD) values measured at 570 nm. Bars represent mean ± SD (n = 3). Asterisks indicate statistically significant differences (p < 0.05); (B) Representative phase-contrast micrographs of biofilms stained with crystal violet. Scale bar 200 µm.
4 Discussion
The study examined the genomic features of L. interrogans serovar Hardjo isolates from Europe using a whole-genome sequencing approach, and revealed a high degree of sequence conservation, consistent with limited genetic variation previously described for host-adapted serovars. Although not all of the analysed isolates originated from cattle, it is important to note that Leptospira serovars, including Hardjo, are capable of causing incidental infections in non-maintenance hosts. These incidental infections often present with more pronounced clinical symptoms, increasing the likelihood of detection and subsequent isolation (Ellis, 2015). As such, even isolates obtained from incidental hosts may accurately reflect the genetic characteristics of strains circulating within the animal acting as maintenance host population and the broader environment, providing valuable insights into pathogen diversity.
The use of restriction endonuclease analysis (REA) enabled the preliminary identification of selected isolates as belonging to the L. interrogans serovar Hardjo (Figure 1), while whole-genome sequencing enabled in-depth comparative analysis with other L. interrogans serovars. This approach confirmed a high degree of genomic similarity among Hardjo isolates, with average nucleotide identity (ANI) values close to 100% between Hardjo strains (Supplementary Figure S1), confirming minimal divergence - potentially resulting from long-term infection within a maintenance host.
Using the VCA output, we performed population structure analysis (Figure 2A) and constructed a phylogenetic tree (Figure 2B). Both approaches demonstrated a distinct clustering pattern for serovar Hardjo, clearly separating it from other serovars. This may reflect lineage-specific adaptations that facilitate host association and prolonged colonisation of the reproductive tract in cattle and sheep. The Hardjo clade is most closely related to the serogroup Icterohaemorrhagiae clade (sv. Copenhageni and Icterohaemorrhagiae), which may confirm a previously proposed hypothesis that L. interrogans sv. Hardjo originated through horizontal gene transfer (HGT) from L. borgpetersenii sv. Hardjo to a strain of L. interrogans sv. Copenhageni (de la Peña-Moctezuma et al., 1999; Llanes et al., 2016). Therefore, the observed phylogenetic structure reinforces the value of genome-wide SNP analysis as a high-resolution tool for delineating Leptospira strains and resolving their evolutionary relationships. Such approaches can illuminate the intricate genetic structures within bacterial populations of various genera, thereby supporting phylogenetic studies vital for understanding pathogenicity and epidemiology (Filliol et al., 2006; Girault et al., 2014). For instance, SNP-based genomic analysis of Streptococcus pyogenes populations revealed distinct phylogenetic structures associated with niche-specific infections, such as skin disease and pharyngitis-induced acute rheumatic fever (Bao et al., 2016).
To better understand the genetic basis of this divergence, we next examined specific variants differentiating serovar Hardjo from other L. interrogans serovars. Among statistically significant changes, we identified four high-impact SNPs and three INDELs predicted to affect protein-coding sequences potentially relevant to host adaptation. Particularly noteworthy are the differences found in the gene encoding the CRISPR-associated helicase/endonuclease Cas3, and in the thiM gene.
The variant identified in G436_RS15655 (Figure 3C), encoding the CRISPR-associated helicase/endonuclease Cas3 (WP_002189459.1), affected gene integrity across non-Hardjo serovars. In Hardjo strains, the gene was present in a complete, presumably functional form, whereas in other serovars it appeared truncated, likely leading to protein shortening or pseudogenization. CRISPR-Cas systems, including Cas3, have been found in both pathogenic and intermediate Leptospira species, with pathogenic strains often harbouring multiple types (e.g., subtype I-B and I-E) (Xiao et al., 2019). Cas3, a multi-domain protein with both helicase and nuclease activity, is known to play a role in protecting bacteria from mobile genetic elements. In other bacterial species, studies have implicated Cas3 in functions beyond immune defence, including possible roles in biofilm formation and virulence regulation (Rodrigues et al., 2023). Biofilm formation has been linked to reproductive tract colonisation and abortion, particularly in ruminants (Nesse et al., 2023; Araújo et al., 2024). In other Leptospira serovars, similar associations have been observed (Brihuega et al., 2012). In Leptospira, studies have proposed a link between Cas3 and biofilm formation, suggesting a contribution to bacterial persistence under host-associated stress conditions (Prakash and Kumar, 2022). This raises the hypothesis that Cas3-mediated mechanisms might contribute to the pathogenicity of L. interrogans sv. Hardjo in ruminant hosts, potentially supporting its adaptation and prolonged persistence within reproductive tissues. However, this role remains putative, and experimental validation is required to confirm Cas3’s involvement in host-specific infection dynamics. In line with this hypothesis, our in vitro assay showed that sv. Hardjo formed biofilm more rapidly and in greater amounts than sv. Copenhageni and sv. Bratislava over a 21-day period (Figure 8), suggesting a possible functional consequence of Cas3 variation in the context of tissue colonisation. Quantitative trends were consistent across both techniques, reinforcing the conclusion that serovar Hardjo establishes biofilm more efficiently under these experimental conditions. While this phenotype was observed under controlled laboratory conditions, it may be consistent with enhanced persistence capacity relevant to the reproductive environment; nonetheless, direct causality remains to be investigated.
Another relevant variant was detected in the thiM gene (Figure 3D), which encodes thiazole kinase (WP_000047877.1), and altered the start codon from ATG to GTG. In prokaryotes, the start codon is one of the major translation initiation determinants. The canonical AUG start codon exhibits higher translation efficiency compared to alternative codons, such as GUG (Belinky et al., 2017). In bacteria, thiamine biosynthesis predominantly occurs via the de novo pathway. However, an alternative salvage pathway exists to recycle thiazole, a key intermediate. This salvage pathway ensures continued thiamine production when de novo synthesis is limited (Jurgenson et al., 2009). In our dataset, the canonical ATG start codon was predominantly observed in L. interrogans sv. Hardjo strains, whereas a GTG codon was more common in non-Hardjo strains. This pattern may suggest strain-specific variation in translation efficiency of thiazole kinase. Although the precise phenotypic consequences remain to be confirmed, reduced translation efficiency could theoretically affect thiamine salvage capacity under nutrient-limited conditions. Moreover, ThiM has been considered a potential antibacterial target, as its inhibition could disrupt thiamine biosynthesis and bacterial survival (Chen et al., 2019; Drebes et al., 2016).
The biological relevance of some detected variants remains unclear. An INDEL identified in G436_RS18190 (Figure 4C) alters the N-terminal sequence and length of a FecR family protein (WP_029757821.1), which may be involved in citrate-mediated iron import (Passmore et al., 2020). A premature stop codon in the gene G436_RS22205 (Figure 3A) leads to the absence of protein WP_002080799.1 in non-Hardjo serovars. This protein, annotated as a mobile element protein (PF07600 domain), was predicted to be involved in two-component signal transduction and kinase-related activity, potentially contributing to adaptive responses in L. interrogans sv. Hardjo. Two-component systems are broadly recognised for their role in sensing environmental signals and modulating stress responses in bacteria (Mitrophanov and Groisman, 2008). Additionally, we identified two INDELs (Figure 4A,B) and one SNP (Figure 3B) differing between groups and leading to the loss of protein-coding sequences in Li-nH group. Affected genes encode hypothetical proteins; nevertheless, due to the lack of identifiable domains or homologs, their potential function remains unclear. Although the biological significance of these variants remains uncertain, their presence in Hardjo and absence in other serovars may reflect differences in regulatory pathways that support host or niche adaptation.
In addition to the high-impact variants, we identified several mutations in pseudogenes (Supplementary Table S3) and missense variants in genes encoding outer membrane proteins (OMPs) (Supplementary Table S4). Pseudogenization is often associated with reduced selective pressure during niche adaptation and has been observed across various host-specialised pathogens (Feng et al., 2022; Pink et al., 2011; Yang et al., 2024). In serovar Hardjo, this process may reflect genome streamlining associated with persistence in ruminant hosts. Missense mutations in putative OMPs, in turn - particularly lipoproteins, which are believed to play key roles in infection and immune evasion - may significantly influence host-pathogen interactions. Even single amino acid change may affect protein function, stability, or surface exposure, warranting further investigation into its potential role in the pathogenicity of L. interrogans serovar Hardjo. Therefore, both categories may contribute to further host adaptation, although through different mechanisms.
To complement the SNP and INDEL analyses, we examined orthogroups unique to L. interrogans serovar Hardjo, identifying 88 genes not found in other serovars (Figure 5). Functional classification (Figure 6) highlighted differences in categories linked to membrane biogenesis, which is expected, as all genes assigned to this category were located within the rfb locus, involved in the synthesis of O-antigen of lipopolysaccharides (LPS) (Cosate et al., 2019). Given the known serovar-specific nature of the rfb locus, we focused our analysis on genes located outside this region, where we identified 59 LiH-specific proteins (Table 4). Interestingly, many of the unique LiH-specific genes were found to be clustered in close proximity to the each other, suggesting potential acquisition through horizontal gene transfer (HGT). To explore this hypothesis, we analysed the genomes of serovar Hardjo using IslandViewer 4 to identify putative genomic islands. Despite the presence of mobile element proteins, toxin-antitoxin system components, and a GC content notably higher than the genomic average, the first region could not be classified as a genomic island by IslandViewer 4, and thus no direct evidence of horizontal gene transfer was identified. In contrast, the second region - encompassing multiple hypothetical proteins, an IS1595 transposase, and members of the Sce protein family - was predicted to be a genomic island (GI). This supports the possibility that at least part of the unique genetic content in serovar Hardjo may have been acquired through horizontal gene transfer. Genome plasticity has been shown to contribute to environment and host adaptation in Leptospira (Giraud-Gatineau et al., 2024; Moreno et al., 2017).
Although the majority of the LiH-specific orthogroups (37 out of 59 located outside the rfb locus) encoded hypothetical proteins, partial functional annotation was possible for a subset of the remaining genes. Among these, four proteins were linked to the toxin-antitoxin (TA) system (WP_000044296.1, WP_000014356.1, WP_002188765.1, WP_000881563.1). TA system seems to play a crucial role in cellular survival under stress conditions. These systems have been linked to various infection-related processes, including toxin production, immune system evasion, and metabolic adaptation, underscoring their potential contributions to pathogenicity (Lopes et al., 2014). Although TA modules are associated with the stabilization of GIs (Munshi et al., 2025), none of the identified TA system components in our dataset were located within predicted genomic islands.
Additionally, one of the LiH-specific orthogroups included an adenylate/guanylate cyclase catalytic domain protein (WP_000288518.1), classified within the ‘Signal transduction mechanisms’ category. Adenylate and guanylate cyclases are enzymes responsible for synthesizing cyclic nucleotides, key secondary messengers involved in environmental sensing and gene regulation (Baker and Kelly, 2004). In L. interrogans, cAMP plays a critical function in the regulation of gene expression, particularly during the initial phases of host infection (Matsunaga et al., 2007), whereas c-di-GMP regulates biofilm formation (Thibeaux et al., 2020b). The presence of this gene in the Hardjo-specific set may support its enhanced biofilm-forming capacity, as observed in our in vitro assays.
Furthermore, the gene encoding a LiH-specific leucine-rich repeat (LRR) domain-containing protein (WP_002188813.1) was located adjacent to fliG. Given the role of FliG in motility control, this genomic arrangement raises the possibility that the LRR-domain protein may influence motility of L. interrogans sv. Hardjo, as LRRs have been described as exhibiting a broad spectrum of ligands and having a putative role in bacterial pathogenesis (Foltran et al., 2024).
This study has several limitations that should be acknowledged. First, the analysis was based on a limited number of isolates, which may not fully capture the genomic diversity within the L. interrogans sv. Hardjo. Second, while genomic data provide valuable insights into potential phenotypic traits, such predictions remain inherently uncertain without experimental validation. In particular, functional inferences based on bioinformatic tools should be interpreted with caution, especially for genes annotated as hypothetical. Building on these findings, the next phase of our research will involve transcriptomic and proteomic analyses in an animal infection model. These investigations could help identify key genes and their products essential for infection, which could ultimately contribute to the development of more effective control strategies for these infections.
5 Conclusion
Our study revealed that L. interrogans serovar Hardjo forms a distinct phylogenetic lineage, characterised by specific high-impact mutations and unique gene content. These included a full-length cas3 gene and a start codon substitution in thiM, both of which may contribute to functional differences in Hardjo strains. The Hardjo serovar harbours 88 unique orthologs, most of which are located outside the rfb locus. A subset of these cluster within predicted genomic islands, while others are dispersed throughout the genome. These orthologs include genes associated with mobile genetic elements, toxin-antitoxin systems, and signal transduction pathways. Collectively, these genomic features may contribute to the genomic plasticity and adaptive potential of Hardjo strains. Notably, Hardjo strains exhibit significantly enhanced biofilm formation compared to other L. interrogans serovars, suggesting a possible genetic basis for improved colonisation of host reproductive tissues.
Data availability statement
The genome assemblies were deposited in GenBank (NCBI) under the following BioProject numbers: PRJNA809530 for strains KR84 (assembly: GCA_022436545.1) and KR85 (assembly: GCA_022436605.1); PRJNA828004 for strain KR40 (assembly: GCA_023158895.3); and PRJNA828002 for strain N116 (assembly: GCA_023515975.1). Further inquiries can be directed to the corresponding authors.
Ethics statement
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because this study uses bacterial strains from existing collections. Strains were obtained from Dr Jacek Zmudzki and Dr Artur Jablonski (National Veterinary Research Institute, Pulawy, Poland; strains KR40, KR84, KR85) and Dr Marcella Mori (strain N116). The second Local Institutional Animal Care and Use Committee (IACUC), Krakow, Poland, confirmed that ethical review was not necessary as no live animals were used.
Author contributions
KD: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review and editing. LP: Conceptualization, Formal Analysis, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review and editing. AG: Data curation, Formal Analysis, Methodology, Writing – original draft, Writing – review and editing. IJ: Data curation, Writing – original draft. TS: Data curation, Formal Analysis, Methodology, Software, Writing – review and editing. ZA: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – original draft, Writing – review and editing, Project administration.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grant OPUS 17 2019/33/B/NZ9/02159 from the National Science Centre, Poland.
Acknowledgments
We would like to thank Marcella Mori for kindly providing the bacterial strain N116 used in this study. We also thank Jacek Żmudzki and Artur Jabłoński from the National Veterinary Research Institute (NVRI), Puławy, Poland for the bacterial strains KR40, KR84, and KR85, which were obtained from their collection.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. The authors used ChatGPT (OpenAI, GPT-4, accessed June 2025) to assist with English language editing as a grammar assisant. The authors have verified the accuracy and originality of the final text.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmolb.2025.1648097/full#supplementary-material
References
Alexander, D. H., Novembre, J., and Lange, K. (2009). Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664. doi:10.1101/GR.094052.109
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
Araújo, D., Silva, A. R., Fernandes, R., Serra, P., Barros, M. M., Campos, A. M., et al. (2024). Emerging approaches for mitigating biofilm-formation-associated infections in farm, wild, and companion animals. Pathogens 13, 320. doi:10.3390/PATHOGENS13040320
Arent, Z. J., Gilmore, C., San-Miguel Ayanz, J. M., Neyra, L. Q., and García-Peña, F. J. (2017). Molecular epidemiology of leptospira Serogroup Pomona infections among wild and domestic animals in Spain. Ecohealth 14, 48–57. doi:10.1007/S10393-017-1210-8
Baker, D. A., and Kelly, J. M. (2004). Structure, function and evolution of microbial adenylyl and guanylyl cyclases. Mol. Microbiol. 52, 1229–1242. doi:10.1111/J.1365-2958.2004.04067.X
Bao, Y. J., Liang, Z., Mayfield, J. A., Donahue, D. L., Carothers, K. E., Lee, S. W., et al. (2016). Genomic characterization of a pattern D Streptococcus pyogenes emm53 Isolate reveals a genetic rationale for invasive skin tropicity. J. Bacteriol. 198, 1712–1724. doi:10.1128/JB.01019-15
Bateman, A., Martin, M. J., Orchard, S., Magrane, M., Ahmad, S., Alpi, E., et al. (2023). UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 51, D523–D531. doi:10.1093/NAR/GKAC1052
Belinky, F., Rogozin, I. B., and Koonin, E. V. (2017). Selection on start codons in prokaryotes and potential compensatory nucleotide substitutions. Sci. Rep. 71 (7), 12422–10. doi:10.1038/s41598-017-12619-6
Bendtsen, J. D., Kiemer, L., Fausbøll, A., and Brunak, S. (2005). Non-classical protein secretion in bacteria. BMC Microbiol. 5, 1–13. doi:10.1186/1471-2180-5-58/FIGURES/3
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi:10.1093/NAR/GKX343
Bolin, C. A., Thiermann, A. B., Handsaker, A. L., and Foley, J. W. (1989). Effect of vaccination with a pentavalent leptospiral vaccine containing Leptospira interrogans serovar hardjo type hardjo-bovis on type hardjo-bovis infection of cattle. Am. J. Vet. Res. 50, 2004–2008.
Brihuega, B., Samartino, L., Auteri, C., Venzano, A., and Caimi, K. (2012). In vivo cell aggregations of a recent swine biofilm-forming isolate of Leptospira interrogans strain from Argentina. Rev. Argent. Microbiol. 44, 138–143.
Bulach, D. M., Zuerner, R. L., Wilson, P., Seemann, T., McGrath, A., Cullen, P. A., et al. (2006). Genome reduction in Leptospira borgpetersenii reflects limited transmission potential. Proc. Natl. Acad. Sci. U. S. A. 103, 14560–14565. doi:10.1073/PNAS.0603979103
Chen, K. T., and Lu, C. L. (2018). CSAR-web: a web server of contig scaffolding using algebraic rearrangements. Nucleic Acids Res. 46, W55-W59–W59. doi:10.1093/NAR/GKY337
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi:10.1093/BIOINFORMATICS/BTY560
Chen, Y., Wang, L., Shang, F., Liu, W., Lan, J., Chen, J., et al. (2019). Structural insight of the 5-(Hydroxyethyl)-methylthiazole kinase ThiM involving vitamin B1 biosynthetic pathway from the Klebsiella pneumoniae. Biochem. Biophys. Res. Commun. 518, 513–518. doi:10.1016/J.BBRC.2019.08.086
Cingolani, P. (2022). Variant annotation and functional prediction: Snpeff. Methods Mol. Biol. 2493, 289–314. doi:10.1007/978-1-0716-2293-3_19
Cosate, M. R. V., Soares, S. C., Mendes, T. A., Raittz, R. T., Moreira, E. C., Leite, R., et al. (2015). Whole-Genome sequence of Leptospira interrogans Serovar Hardjo Subtype Hardjoprajitno Strain Norma, isolated from cattle in a leptospirosis outbreak in Brazil. Genome announc. 3, e01302-15. doi:10.1128/GENOMEA.01302-15
Cosate, M. R. V., Sakamoto, T., Moreira, É. C., Ortega, J. M., Leite, R. C., Haddad, J. P., et al. (2019). A potential genomic recombination site upstream of the rfb locus in Leptospira interrogans is associated with serogroup Serjoe and serovar Hardjo classification. bioRxiv, 771931. doi:10.1101/771931
Costa, F., Hagan, J. E., Calcagno, J., Kane, M., Torgerson, P., Martinez-Silveira, M. S., et al. (2015). Global morbidity and mortality of leptospirosis: a systematic review. PLoS Negl. Trop. Dis. 9, e0003898. doi:10.1371/JOURNAL.PNTD.0003898
de la Peña-Moctezuma, A., Bulach, D. M., Kalambaheti, T., and Adler, B. (1999). Comparative analysis of the LPS biosynthetic loci of the genetic subtypes of serovar Hardjo: leptospira interrogans subtype Hardjoprajitno and Leptospira borgpetersenii subtype Hardjobovis. FEMS Microbiol. Lett. 177, 319–326. doi:10.1111/J.1574-6968.1999.TB13749.X
Dhaliwal, G. S., Murray, R. D., Dobson, H., Montgomery, J., and Ellis, W. A. (1996). Presence of antigen and antibodies in serum and genital discharges of cows from dairy herds naturally infected with Leptospira interrogans serovar hardjo. Res. Vet. Sci. 60, 163–167. doi:10.1016/S0034-5288(96)90012-0
Dodt, M., Roehr, J. T., Ahmed, R., and Dieterich, C. (2012). FLEXBAR-Flexible barcode and adapter processing for next-generation sequencing platforms. Biol. (Basel). 1, 895–905. doi:10.3390/BIOLOGY1030895
Drebes, J., Künz, M., Windshügel, B., Kikhney, A. G., Müller, I. B., Eberle, R. J., et al. (2016). Structure of ThiM from Vitamin B1 biosynthetic pathway of Staphylococcus aureus – insights into a novel pro-drug approach addressing MRSA infections. Sci. Rep. 6, 22871. doi:10.1038/SREP22871
Ellis, W. A. (2015). Animal leptospirosis. Curr. Top. Microbiol. Immunol. 387, 99–137. doi:10.1007/978-3-662-45059-8_6
Ellis, W. A., McParland, P. J., Bryson, D. G., and Cassells, J. A. (1986). Boars as carriers of leptospires of the Australis serogroup on farms with an abortion problem. Vet. Rec. 118, 563. doi:10.1136/VR.118.20.563
Ellis, W. A., Montgomery, J. M., and Thiermann, A. B. (1991). Restriction endonuclease analysis as a taxonomic tool in the study of pig isolates belonging to the Australis serogroup of Leptospira interrogans. J. Clin. Microbiol. 29, 957–961. doi:10.1128/JCM.29.5.957-961.1991
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238–14. doi:10.1186/s13059-019-1832-y
Feng, Y., Wang, Z., Chien, K. Y., Chen, H. L., Liang, Y. H., Hua, X., et al. (2022). “Pseudo-pseudogenes” in bacterial genomes: proteogenomics reveals a wide but low protein expression of pseudogenes in Salmonella enterica. Nucleic Acids Res. 50, 5158–5170. doi:10.1093/NAR/GKAC302
Ferreira, L. C. A., Filho, L. de F. F., Cosate, M. R. V., and Sakamoto, T. (2024). Genetic structure and diversity of the rfb locus of pathogenic species of the genus Leptospira. Life Sci. Alliance 7, e202302478. doi:10.26508/LSA.202302478
Filliol, I., Motiwala, A. S., Cavatore, M., Qi, W., Hazbón, M. H., Del Valle, M. B., et al. (2006). Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism (SNP) analysis: insights into tuberculosis evolution, phylogenetic accuracy of other DNA fingerprinting systems, and recommendations for a minimal standard SNP set. J. Bacteriol. 188, 759–772. doi:10.1128/JB.188.2.759-772.2006
Foltran, B. B., Gaspar, J. P., Silva, I. R. M., Pires, H. M., Andrade, F. B., Costa, G. M., et al. (2024). New insights into the putative role of leucine-rich repeat proteins of Leptospira interrogans and their participation in host cell invasion: an in silico analysis. Front. Cell. Infect. Microbiol. 14, 1492352. doi:10.3389/FCIMB.2024.1492352
Fouts, D. E., Matthias, M. A., Adhikarla, H., Adler, B., Amorim-Santos, L., Berg, D. E., et al. (2016). What makes a bacterial species pathogenic?:comparative genomic analysis of the Genus Leptospira. PLoS Negl. Trop. Dis. 10, e0004403. doi:10.1371/JOURNAL.PNTD.0004403
Garrison, E., and Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. doi:10.48550/arXiv.1207.3907
Giraud-Gatineau, A., Nieves, C., Harrison, L. B., Benaroudj, N., Veyrier, F. J., and Picardeau, M. (2024). Evolutionary insights into the emergence of virulent Leptospira spirochetes. PLoS Pathog. 20, e1012161. doi:10.1371/JOURNAL.PPAT.1012161
Girault, G., Blouin, Y., Vergnaud, G., and Derzelle, S. (2014). High-throughput sequencing of Bacillus anthracis in France: investigating genome diversity and population structure using whole-genome SNP discovery. BMC Genomics 15, 288–10. doi:10.1186/1471-2164-15-288
Gligorijević, V., Renfrew, P. D., Kosciolek, T., Leman, J. K., Berenberg, D., Vatanen, T., et al. (2021). Structure-based protein function prediction using graph convolutional networks. Nat. Commun. 12, 3168. doi:10.1038/S41467-021-23303-9
Guglielmini, J., Bourhy, P., Schiettekatte, O., Zinini, F., Brisse, S., and Picardeau, M. (2019). Genus-wide Leptospira core genome multilocus sequence typing for strain taxonomy and global surveillance. PLoS Negl. Trop. Dis. 13, e0007374. doi:10.1371/JOURNAL.PNTD.0007374
Imai, K., Asakawa, N., Tsuji, T., Akazawa, F., Ino, A., Sonoyama, M., et al. (2008). SOSUI-GramN: high performance prediction for sub-cellular localization of proteins in Gram-negative bacteria. Bioinformation 2, 417–421. doi:10.6026/97320630002417
Jurgenson, C. T., Begley, T. P., and Ealick, S. E. (2009). The structural and biochemical foundations of thiamin biosynthesis. Annu. Rev. Biochem. 78, 569–603. doi:10.1146/ANNUREV.BIOCHEM.78.072407.102340
Kopelman, N. M., Mayzel, J., Jakobsson, M., Rosenberg, N. A., and Mayrose, I. (2015). Clumpak: a program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191. doi:10.1111/1755-0998.12387
Krueger, F. (2021). TrimGalore. GitHub repository. Available online at: https://github.com/FelixKrueger/TrimGalore.
Kunjantarachot, A., Yan, W., McDonough, S. P., Prapong, S., Theeragool, G., and Chang, Y. F. (2014). Immunogenicity of Leptospira interrogans outer membrane vesicles in a hamster model. J. Vaccines Vaccin. 5. doi:10.4172/2157-7560.1000239
Letunic, I., and Bork, P. (2021). Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi:10.1093/NAR/GKAB301
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. doi:10.48550/arXiv.1303.3997
Llanes, A., Restrepo, C. M., and Rajeev, S. (2016). Whole genome sequencing allows better understanding of the evolutionary history of leptospira interrogans serovar hardjo. PLoS One 11, e0159387. doi:10.1371/JOURNAL.PONE.0159387
Lopes, A. P. Y., Lopes, L. M., Fraga, T. R., Chura-Chambi, R. M., Sanson, A. L., Cheng, E., et al. (2014). VapC from the leptospiral VapBC toxin-antitoxin module displays ribonuclease activity on the initiator tRNA. PLoS One 9, e101678. doi:10.1371/JOURNAL.PONE.0101678
Loureiro, A. P., and Lilenbaum, W. (2020). Genital bovine leptospirosis: a new look for an old disease. Theriogenology 141, 41–47. doi:10.1016/J.THERIOGENOLOGY.2019.09.011
Loureiro, A. P., Pestana, C., Medeiros, M. A., and Lilenbaum, W. (2017). High frequency of leptospiral vaginal carriers among slaughtered cows. Anim. Reprod. Sci. 178, 50–54. doi:10.1016/J.ANIREPROSCI.2017.01.008
Masri, S. A., Nguyen, P. T., Gale, S. P., Howard, C. J., and Jung, S. C. (1997). A polymerase chain reaction assay for the detection of Leptospira spp. in bovine semen. J. Vet. Res. 61, 15–20.
Matsunaga, J., Lo, M., Bulach, D. M., Zuerner, R. L., Adler, B., and Haake, D. A. (2007). Response of Leptospira interrogans to physiologic osmolarity: relevance in signaling the environment-to-host transition. Infect. Immun. 75, 2864–2874. doi:10.1128/IAI.01619-06
Mikheenko, A., Prjibelski, A., Saveliev, V., Antipov, D., and Gurevich, A. (2018). Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 34, i142–i150. doi:10.1093/BIOINFORMATICS/BTY266
Mikhin, N. A., and Azinov, S. A. (1935). Spirochaetal jaundice in cattle in North Caucasus. Sov. Vet. 10, 23–37.
Mitrophanov, A. Y., and Groisman, E. A. (2008). Signal integration in bacterial two-component regulatory systems. Genes Dev. 22. 2601–2611. doi:10.1101/GAD.1700308
Monahan, A. M., Callanan, J. J., and Nally, J. E. (2009). Review paper: Host-pathogen interactions in the kidney during chronic leptospirosis. Vet. Pathol. 46, 792–799. doi:10.1354/VP.08-VP-0265-N-REV
Moreno, L. Z., Kremer, F. S., Jaeger, L. H., Loureiro, A. P., Miraglia, F., Eslabao, M. R., et al. (2017). Genomic characterization and comparative analysis of Leptospira interrogans serogroup Australis isolated from swine, Pathog. Dis. Genomic Charact. Comp. analysis Leptospira interrogans Serogr. Aust. Isol. swine 75, 119. doi:10.1093/femspd/ftx119
Munshi, I. D., Mathuria, A., Sharma, H., Acharya, M., Chaudhary, A., Jain, K., et al. (2025). Emerging concept of genomic islands in bacterial adaptation and pathogenicity. Res. Microbiol. 104303. doi:10.1016/J.RESMIC.2025.104303
Murray, G. L., Lo, M., Bulach, D. M., Srikram, A., Seemann, T., Quinsey, N. S., et al. (2013). Evaluation of 238 antigens of Leptospira borgpetersenii serovar Hardjo for protection against kidney colonisation. Vaccine 31, 495–499. doi:10.1016/J.VACCINE.2012.11.028
Naiman, B. M., Alt, D., Bolin, C. A., Zuerner, R., and Baldwin, C. L. (2001). Protective killed Leptospira borgpetersenii vaccine induces potent Th1 immunity comprising responses by CD4 and gammadelta T lymphocytes. Infect. Immun. 69, 7550–7558. doi:10.1128/IAI.69.12.7550-7558.2001
Nesse, L. L., Osland, A. M., and Vestby, L. K. (2023). The role of biofilms in the pathogenesis of animal bacterial infections. Microorganisms 11, 608. doi:10.3390/MICROORGANISMS11030608
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi:10.1093/MOLBEV/MSU300
Nurk, S., Bankevich, A., Antipov, D., Gurevich, A., Korobeynikov, A., Lapidus, A., et al. (2013). Assembling genomes and mini-metagenomes from highly chimeric reads. Lect. Notes Comput. Sci. Incl. Subser. Lect. Notes Artif. Intell. Lect. Notes Bioinforma. 7821 LNBI, 158–170. doi:10.1007/978-3-642-37195-0_13
Passmore, I. J., Dow, J. M., Coll, F., Cuccui, J., Palmer, T., and Wren, B. W. (2020). Ferric citrate regulator FecR is translocated across the bacterial inner membrane via a unique twin-arginine transport-dependent mechanism. J. Bacteriol. 202, e00541-19. PDF. doi:10.1128/JB.00541-19
Philip, N., Jani, J., Azhari, N. N., Sekawi, Z., and Neela, V. K. (2021). In vivo and in silico virulence analysis of Leptospira species isolated from environments and rodents in leptospirosis outbreak areas in Malaysia. Front. Microbiol. 12, 753328. doi:10.3389/fmicb.2021.753328
Pink, R. C., Wicks, K., Caley, D. P., Punch, E. K., Jacobs, L., and Carter, D. R. F. (2011). Pseudogenes: Pseudo-functional or key regulators in health and disease? RNA 17, 792–798. doi:10.1261/RNA.2658311
Prakash, A., and Kumar, M. (2022). Transcriptional analysis of CRISPR I-B arrays of Leptospira interrogans serovar Lai and its processing by Cas6. Front. Microbiol. 13, 960559. doi:10.3389/fmicb.2022.960559
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi:10.1093/BIOINFORMATICS/BTV681
Rodrigues, R. C., Tagliaferri, T. L., and Mendes, T.A. de O. (2023). Potential of the endogenous and artificially inserted CRISPR-Cas system for controlling virulence and antimicrobial resistance of food pathogens. Food Chem. Adv. 2, 100229. doi:10.1016/J.FOCHA.2023.100229
Roth, E. E., and Galton, M. M. (1960). Isolation and identification of Leptospira hardjo from cattle in Louisiana. Am. J. Vet. Res. 21, 422–427.
Salgado, M., Otto, B., Moroni, M., Sandoval, E., Reinhardt, G., Boqvist, S., et al. (2015). Isolation of Leptospira interrogans serovar Hardjoprajitno from a calf with clinical leptospirosis in Chile. BMC Vet. Res. 11, 66–4. doi:10.1186/s12917-015-0369-x
Setubal, J. C., Reis, M., Matsunaga, J., and Haake, D. A. (2006). Lipoprotein computational prediction in spirochaetal genomes. Microbiology 152, 113–121. doi:10.1099/MIC.0.28317-0
Szklarczyk, D., Kirsch, R., Koutrouli, M., Nastou, K., Mehryary, F., Hachilif, R., et al. (2023). The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 51, D638–D646. doi:10.1093/NAR/GKAC1000
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi:10.1093/NAR/28.1.33
Techawiwattanaboon, T., Phanchamnan, E., Iadsee, N., Makjaroen, J., Pisitkun, T., and Patarakul, K. (2023). Proteomic profile of naturally released extracellular vesicles secreted from Leptospira interrogans serovar Pomona in response to temperature and osmotic stresses. Sci. Rep. 13, 18601. doi:10.1038/S41598-023-45863-0
Teufel, F., Almagro Armenteros, J. J., Johansen, A. R., Gíslason, M. H., Pihl, S. I., Tsirigos, K. D., et al. (2022). SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 407 40, 1023–1025. doi:10.1038/s41587-021-01156-3
Thibeaux, R., Kainiu, M., and Goarant, C. (2020a). “Biofilm Formation and quantification using the 96-Microtiter plate,” in Leptospira Spp.: methods and protocols. Editors N. Koizumi, and M. Picardeau (New York, NY: Springer US), 207–214. doi:10.1007/978-1-0716-0459-5_19
Thibeaux, R., Soupé-Gilbert, M. E., Kainiu, M., Girault, D., Bierque, E., Fernandes, J., et al. (2020b). The zoonotic pathogen Leptospira interrogans mitigates environmental stress through cyclic-di-GMP-controlled biofilm production. npj Biofilms Microbiomes 61 (6), 1–11. doi:10.1038/s41522-020-0134-1
Thiermann, A. B., Handsaker, A. L., Foley, J. W., White, F. H., and Kingscote, B. F. (1986). Reclassification of North American leptospiral isolates belonging to serogroups Mini and Sejroe by restriction endonuclease analysis. Am. J. Vet. Res. 47, 61–66. doi:10.2460/ajvr.1986.47.01.61
Venus Lau, W. Y., Hoad, G. R., Jin, V., Winsor, G. L., Madyan, A., Gray, K. L., et al. (2021). PSORTdb 4.0: expanded and redesigned bacterial and archaeal protein subcellular localization database incorporating new secondary localizations. Nucleic Acids Res. 49, D803–D808. doi:10.1093/NAR/GKAA1095
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9, e112963. doi:10.1371/JOURNAL.PONE.0112963
Waksman, G. (2019). From conjugation to T4S systems in Gram-negative bacteria: a mechanistic biology perspective. EMBO Rep. 20. doi:10.15252/EMBR.201847012
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Completing bacterial genome assemblies with multiplex MinION sequencing. Microb. genomics 3, e000132. doi:10.1099/MGEN.0.000132
Wunder, E. A., Adhikarla, H., Hamond, C., Bonner, K. A. O., Liang, L., Rodrigues, C. B., et al. (2021). A live attenuated-vaccine model confers cross-protective immunity against different species of the Leptospira genus. Elife 10, e64166–20. doi:10.7554/ELIFE.64166
Xiao, G., Yi, Y., Che, R., Zhang, Q., Imran, M., Khan, A., et al. (2019). Characterization of CRISPR-Cas systems in Leptospira reveals potential application of CRISPR in genotyping of Leptospira interrogans. APMIS 127, 202–216. doi:10.1111/APM.12935
Xu, Y., Zhu, Y., Wang, Y., Chang, Y. F., Zhang, Y., Jiang, X., et al. (2016). Whole genome sequencing revealed host adaptation-focused genomic plasticity of pathogenic Leptospira. Sci. Rep. 61 (6), 20020–11. doi:10.1038/srep20020
Yang, Y., Wang, P., Qaidi, S.El, Hardwidge, P. R., Huang, J., and Zhu, G. (2024). Loss to gain: pseudogenes in microorganisms, focusing on eubacteria, and their biological significance. Appl. Microbiol. Biotechnol. 108, 328. doi:10.1007/S00253-023-12971-W
Yasuda, P. H., Steigerwalt, A. G., Sulzer, K. R., Kaufmann, A. F., Rogers, F., and Brenner, D. J. (1987). Deoxyribonucleic acid relatedness between serogroups and serovars in the family Leptospiraceae with proposals for seven new Leptospira species. Int. J. Syst. Bacteriol. 37, 407–415. doi:10.1099/00207713-37-4-407
Keywords: Leptospira interrogans, Hardjo, Comparative genomics, whole genome sequencing, Europe
Citation: Dubniewicz K, Pardyak L, Gurgul A, Jasielczuk I, Szmatoła T and Arent ZJ (2025) Serovar-specific genomic features of Leptospira interrogans Hardjo: implications for host adaptation. Front. Mol. Biosci. 12:1648097. doi: 10.3389/fmolb.2025.1648097
Received: 16 June 2025; Accepted: 06 August 2025;
Published: 10 September 2025.
Edited by:
Shisheng Li, Louisiana State University, United StatesReviewed by:
Jasmin Wenderlein, German Federal Institute for Risk Assessment, GermanyMohd Abdullah, Cornell University Cornell Population Center, United States
Anissa Desmoulin, Centre Hospitalier Universitaire Sud Réunion, Saint-Pierre, Réunion
Copyright © 2025 Dubniewicz, Pardyak, Gurgul, Jasielczuk, Szmatoła and Arent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Klaudia Dubniewicz, a2xhdWRpYS5kdWJuaWV3aWN6QHVyay5lZHUucGw=; Zbigniew J. Arent, emJpZ25pZXcuYXJlbnRAdXJrLmVkdS5wbA==