



- 1 Addiction Biology Unit, Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, University of Gothenburg, Gothenburg, Sweden
- 2 Beroendekliniken, Sahlgrenska University Hospital, Gothenburg, Sweden
Executive dysfunction is a common symptom among alcohol-dependent individuals. Phencyclidine (PCP) injection induces dysfunction in the prefrontal cortex of animals but little is known about how PCP affects the response to ethanol. Using the in vivo microdialysis technique in male Wistar rats, we investigated how systemic injection of 5 mg/kg PCP would affect the dopamine release induced by local infusion of 300 mM ethanol into the nucleus accumbens. PCP given 60 min before ethanol entirely blocked ethanol-induced dopamine release. However, when ethanol was administered 60 min before PCP, both drugs induced dopamine release and PCP’s effect was potentiated by ethanol (180% increase vs 150%). To test the role of prefrontal cortex dysfunction in ethanol reinforcement, animals were pretreated for 5 days with 2.58 mg/kg PCP according to previously used ‘PFC hypofunction protocols’. This, however, did not change the relative response to PCP or ethanol compared to saline-treated controls. qPCR illustrated that this low PCP dose did not significantly change expression of glucose transporters Glut1 (SLC2A1) or Glut3 (SLC2A3), monocarboxylate transporter MCT2 (SLC16A7), glutamate transporters GLT-1 (SLC1A2) or GLAST (SLC1A3), the immediate early gene Arc (Arg3.1) or GABAergic neuron markers GAT-1 (SLC6A1) and parvalbumin. Therefore, we concluded that PCP at a dose of 2.58 mg/kg for 5 days did not induce hypofunction in Wistar rats. However, PCP and ethanol do have overlapping mechanisms of action and these drugs differentially affect mesolimbic dopaminergic transmission depending on the order of administration.
Introduction
A common symptom among alcohol-dependent individuals is the presence of an executive dysfunction (Kril et al., 1997 ; Dao-Castellana et al., 1998 ). This type of dysfunction is also observed in many other psychiatric disorders such as schizophrenia (Lewis et al., 2005 ), depression (Diamond et al., 2004 ) and obsessive-compulsive disorder (van den Heuvel et al., 2005 ). Chronic use of alcohol decreases prefrontal cortex volume (De Bellis et al., 2005 ; Chanraud et al., 2007 ) and even small doses of ethanol in healthy volunteers decreases prefrontal cortex excitability (Kahkonen et al., 2003 ). At the cellular level, ethanol dose-dependently reduces firing of prefrontal cortex neurons (Tu et al., 2007 ) by inhibition of NMDA receptor-mediated excitation (Weitlauf and Woodward, 2008 ). Previously, we have also found that gene expression of many neurotransmitter receptors in the prefrontal cortex correlates with the amount of alcohol an individual animal consumes in an operant self-administration chamber (Pickering et al., 2007a ,b ).
In order to study whether the prefrontal cortex affects the response to alcohol in dependent individuals, a representative animal model is required. The most ideal solution would be to use ‘alcohol-dependent animals’ but this is controversial and difficult to achieve. Some have observed signs of dependence (e.g. impulsive responding) after self-administration methods (McGregor and Gallate, 2004 ; Belin et al., 2008 ) while others have employed chronic exposure (Wolffgramm and Heyne, 1995 ; Turyabahika-Thyen and Wolffgramm, 2006 ), repeated deprivation periods (Spanagel and Holter, 1999 ), ethanol vapor chamber approaches (Rimondini et al., 2003 ) or severe withdrawal effects (Diana et al., 1993 ) to produce rats which are ‘dependent’ on ethanol. The disadvantage of these procedures is the considerable investment in time and the need for substantial animal facilities to house such large populations of rats for up to a year, something which is beyond the scope of most laboratories. To overcome these difficulties, we approached the problem from a different angle. Rather than using chronic ethanol exposure to induce a prefrontal cortex dysfunction, our goal was to induce dysfunction first in order to determine whether this would affect the response to ethanol.
The schizophrenia field uses phencyclidine (PCP) to mimic the symptoms and cognitive deficits observed clinically in this condition (Allen and Young, 1978 ; Krystal et al., 1994 ). In recent years, studies have used repeated administration of PCP to induce chronic adaptations (Jentsch and Roth, 1999 ). Initially, the paradigm of choice was an injection of 5 mg/kg PCP twice daily for 7 days which selectively reduces dopamine utilization in the prefrontal cortex and disrupts schizophrenia-relevant behaviors and stress response (Jentsch et al., 1997 , 1998a ). Later developmental models used an injection of 10 mg/kg PCP at postnatal day 7, 9 and 11 to produce deficits in cognitive functions (prefrontal cortex-dependent) (Broberg et al., 2008 ) and GABAergic interneurons (Wang et al., 2008 ) in adult animals. Other studies use adolescent animals and repeated injections of PCP for one or more weeks. For example, repeated exposure to 10 mg/kg PCP impairs Y-maze performance, decreases parvalbumin and increases expression of the learning-dependent transcription factor Arc/Arg 3.1 (Thomsen et al., 2009 ). The 10 mg/kg dose is often used when pharmacologically reversing PCP deficits with antipsychotic drugs like quetiapine (Tanibuchi et al., 2009 ), perospirone (Hagiwara et al., 2008 ) or clozapine (Hashimoto et al., 2005 ). High doses of PCP are neurotoxic (Wang and Johnson, 2007 ) and we wanted to avoid this aspect in our model, mainly since we believe that the development of drug dependence is a result of chronic neuroadaptations in brain circuitry rather than direct ethanol toxicity. Based on this, we chose a lower dose of PCP. Repeated injection of 2 mg/kg induces cortical behavioral deficits (Abdul-Monim et al., 2007 ; Amitai et al., 2007 , 2009 ; Amitai and Markou, 2009 ) and reduces parvalbumin expression (Abdul-Monim et al., 2007 ). Of particular interest is the method of Cochran et al. (2003) which produces metabolic hypofunction (i.e. reduced glucose utilization in the prefrontal cortex) in adult Long-Evans rats after injection of 2.58 mg/kg PCP for 5 days. Therefore, to overcome the toxicity aspects, we proceeded with the dose of 2.58 mg/kg PCP for 5 days.
Irrespective of the functional status of the prefrontal cortex, evidence suggests an interaction between ethanol and PCP. Brunet et al. (1985) found a synergistic effect of ethanol/PCP co- administration in terms of the loss of righting reflex while chronic pretreatment with ethanol increases binding of radiolabeled PCP in the brain of rats (Vadlamani et al., 1982 ). Both drugs also share a similar mechanism of action. PCP is a non-competitive NMDA channel antagonist which also has affinity for the sigma1 receptor, serotonin 5-HT2 and dopamine D2 receptors (Takahashi et al., 2001 ; Kapur and Seeman, 2002 ; Seeman et al., 2005 ). Ethanol also acts to antagonize NMDA channel current while potentiating GABAergic transmission (Harris et al., 2008 ). This similar mechanism has been demonstrated in substitution experiments in which animals are trained to press a lever after injection of, for example, ethanol. If another drug is injected and produces the same internal cues as ethanol, the animal will press the ‘ethanol lever’. PCP partially substitutes for ethanol (Hundt et al., 1998 ; Bowen and Grant, 1999 ; Green-Jordan and Grant, 2000 ) however this was not the case in all studies (Grant et al., 1992 ).
Since ethanol and PCP may interact acutely, we began with a study of how the combination of both drugs affects dopamine release in the nucleus accumbens. A systemic injection of 5 mg/kg PCP was chosen for the robust dopamine-elevating effect (150%) which was still within the range of that induced by ethanol (Millan et al., 1999 ; Greenslade and Mitchell, 2004 ). For ethanol, we chose to infuse 300 mM directly into the nucleus accumbens. We have, in a series of microdialysis articles, demonstrated that ethanol infused in this manner acts via glycine receptors and, probably, GABAergic feedback neurons to induce dopamine release in the nucleus accumbens (Ericson et al., 2003 , 2008 ; Larsson et al., 2005 ; Molander and Soderpalm, 2005 ). The first question of this study was to determine whether systemic PCP injection would influence this ethanol-induced accumbal dopamine release. Secondly, we investigated if the acute interactions between ethanol and PCP would change following treatment with 2.58 mg/kg PCP for 5 days. Finally, we performed qPCR to verify that our PCP-treated animals had prefrontal cortex dysfunction.
Materials and Methods
Animals
Male Wistar rats (n = 54, Taconic, Denmark) weighing 270–300 g, were used for the experiments. Upon arrival the rats were housed in groups of four in a humidity (65%) and temperature (22°C) controlled room on a 12/12-h light/dark cycle (on 07:00 off 19:00), with free access to rat food (Lantmännen, Stockholm, Sweden) and tap water. Animals were allowed to adapt for at least 7 days to the animal maintenance facilities of the department prior to the start of the experiments. All experiments were conducted according to the Declaration of Helsinki and were approved by the Ethics Committee for Animals Experiments (Gothenburg, Sweden).
Drugs and Chemicals
Phencyclidine hydrochloride (PCP; Sigma-Aldrich, Stockholm, Sweden) was dissolved in 0.9% NaCl and administered systemically (intraperitoneal, i.p.). Ethanol (Kemetyl AB, Haninge, Sweden) was dissolved in Ringer solution and perfused into the nucleus accumbens. Ringer solution consisted of (in mmol/l): 140 NaCl, 1.2 CaCl2, 3.0 KCl, and 1.0 MgCl2.
Microdialysis Technique
Brain microdialysis experiments were performed in awake and freely-moving animals. Rats were anesthetized by isofluorane (Apoteket AB, Stockholm, Sweden), mounted into a stereotaxic instrument (David Kopf Instruments) and put on a heating pad to prevent hypothermia during the surgery. Holes were superficially drilled for placement of two anchoring screws and one I-shaped dialysis probe, custom made in the laboratory. The dialysis probes were lowered unilaterally into the nAc (A/P: +1.85, M/L: −1.4, D/V: −7.8 relative to bregma; (Paxinos and Watson, 2007 ). The exposed length of the dialysis membrane (active space) was 2 mm. The dialysis probes were placed in the core-shell borderline region (suggesting that sampling was done in both core and shell of the nAc) and probes as well as anchoring screws were fixed to the scull with Harvard cement (DAB Dental AB, Gothenburg, Sweden). After surgery, the rats were allowed to recover for 2 days before the dialysis experiments were initiated. On the experimental day, the sealed inlet and outlet of the probes were cut open and connected to a micro-perfusion pump (U-864 Syringe Pump, AgnTho’s, Lidingö, Sweden) via a swivel allowing the animal to move around freely. The probes were perfused with Ringer solution at a rate of 2 µl/min and dialyzate samples (40 µl) were collected every 20 min. The rats were perfused with Ringer solution for 1 h in order to obtain a balanced fluid exchange before baseline sampling begun. Animals were sacrificed directly after the experiment, brains were removed and probe placement was verified using a vibroslicer (Campden Instruments).
Biochemical assay
To analyze the dopamine content of the samples, a high-pressure liquid chromatography system was used for the separation and detection of dopamine as described in (Lido et al., 2009 ). To identify the dopamine peak, an external standard was used containing 2.64 fmol/µl of dopamine. When at least three consecutive stable values of dopamine were obtained (±5%) the first drug was introduced. All results are presented as a percent of pre-drug treatment baseline.
Experimental procedure
Daily administration of 2.58 mg/kg PCP (i.p.) or 2 ml/kg vehicle (0.9% NaCl) for five consecutive days was followed by 2 days with no injections. On the experimental day, the animals received local perfusion with 300 mM ethanol (continuous administration via the probe from the time point indicated in Figure 1 which either was preceded or followed by an acute 5 mg/kg PCP systemic injection. Based on preliminary studies of the effects of PCP or ethanol alone (Figure 1 C), drugs were administered 60 min apart and sampling continued for a total of 180 min from administration of the first drug.
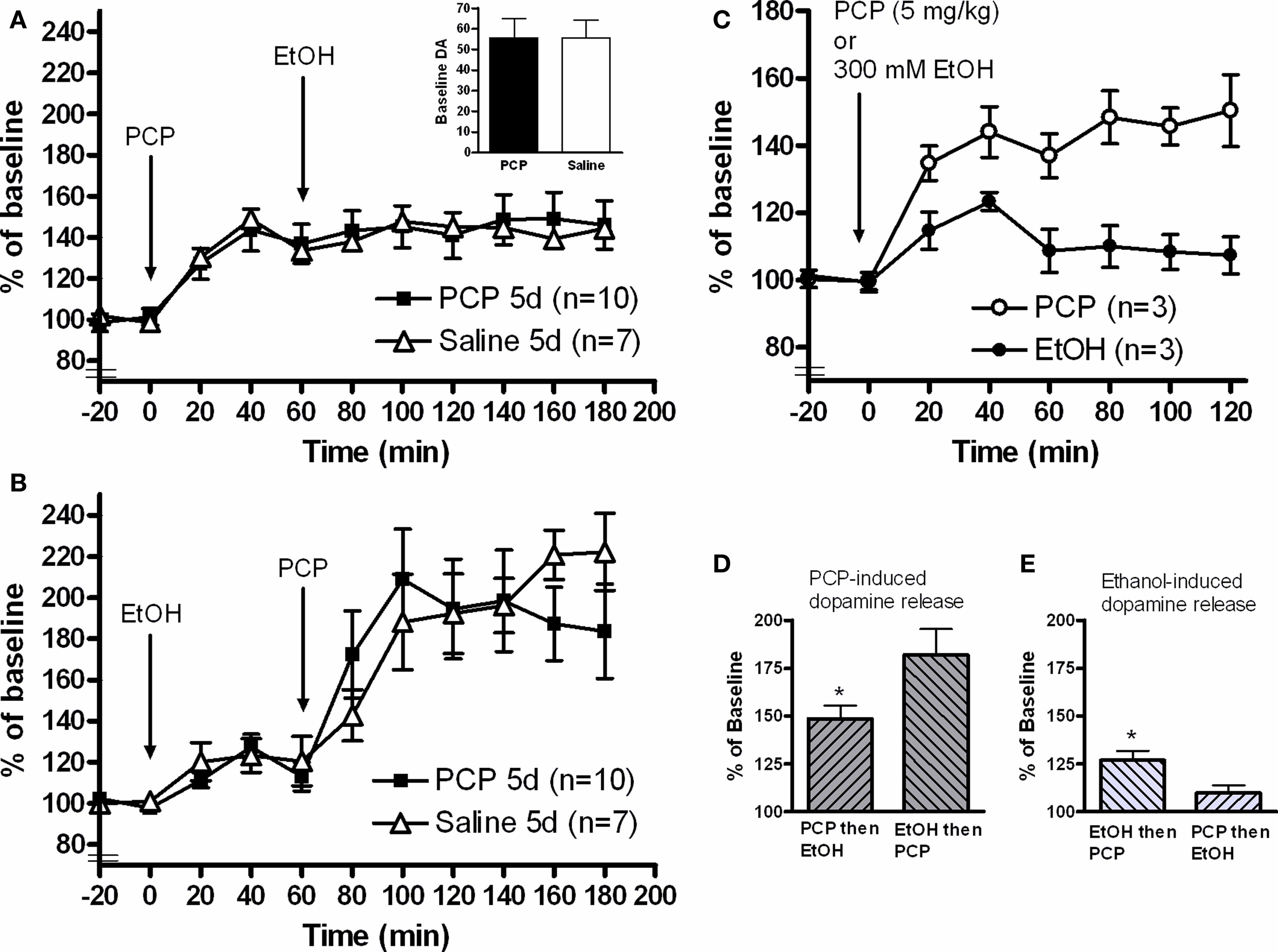
Figure 1. In vivo microdialysis measuring dopamine release in the nucleus accumbens of awake, freely-moving animals. (A) Systemic injection of 5 mg/kg PCP-induced dopamine release but blocked the dopamine-elevating effects of a local 300 mM ethanol infusion. No differences were observed when animals were pretreated for 5 days with 2.58 mg/kg PCP. (B) 300 mM ethanol infusion produced the expected dopamine increase and 5 mg/kg PCP injection led to a potentiated dopamine response compared to the first experiment. Again, no effect of PCP pretreatment was observed. (C) PCP injection (5 mg/kg) alone produced a sustained dopamine release from 40 to 120 min. Three hundred millimolar ethanol infusion into the nucleus accumbens produced a peak dopamine release after 40 min which then fell to levels close to baseline by 60 min. Based on these results, drugs in the combination experiments were administered 60 min after the each other. (D) Comparison of the relative dopamine release stimulated by PCP injection. When ethanol was administered first, the PCP-induced dopamine elevation was greater than PCP alone (t-test, *p < 0.05). (E) Comparison of the relative dopamine release stimulated by ethanol infusion. Ethanol induced significantly less dopamine release when the infusion was proceeded by PCP injection (t-test, *p < 0.05).
qPCR Analysis
Dissection, RNA extraction and cDNA synthesis
In a second group of animals (n = 20), 12 animals were injected with 2.58 mg/kg PCP and 8 animals with 0.9% NaCl. At Day 8 (after 5 days of injection and 2 days of recovery), brains were rapidly removed and the medial prefrontal cortex was dissected according to reference atlases (Heffner et al., 1980 ; Paxinos and Watson, 2007 ). Tissue samples were snap frozen on a dry ice block and stored at −80ºC until further processing. Total RNA extraction was performed with the RNEasy spin column kit (Qiagen) according to manufacturer’s protocols. The TissueRupter (Qiagen) was used to ensure complete homogenization of the individual samples (approximately 1 min for each sample). RNA was quantified by absorbance measurement (260-nm wavelength) (BioRad spectrophotometer) and 1 µg was used for cDNA synthesis (Qiagen kit) which was performed according to standard protocols. This kit also contains a DNase treatment step to remove any genomic DNA contamination which may have occurred during the RNA extraction process. Samples were then diluted to 12.5 ng/µl for direct use in the qPCR method.
qPCR and primers
qPCR analysis was performed on the LightCycler 480 real-time instrument (Roche) in 96-well plates (Roche). All samples were run in duplicate and sometimes this was repeated again if individual values did not concur. The PCR reaction was based on the standard protocol for the Hot Start Taq polymerase enzyme kit (Qiagen) with SYBR green as the fluorescent reporter. The 20 µl sample volume consisted of 10 µl 2 × SYBR Green Master mix (Qiagen), 6 µl RNAse free water, 2 µl primer and 2 µl cDNA. All primers were pre-designed QuantiTect Primer Assays from Qiagen which unfortunately does not disclose the actual sequence of the primer pair but rather gives an approximate gene location on their website. These primers are pre-optimized to give roughly the same PCR efficiency (Table 1 ) for eventual inter-gene comparisons and also allow for the same PCR reaction conditions for the whole experiment. A total of 45 cycles (15-s denaturation at 94°C, 20-s annealing at 55°C, 20-s extension at 72°C) were run for each primer. A melting curve analysis was performed after each PCR program to ensure that a single product was produced in each sample. Samples with primer dimers/multiple products were excluded and re-analyzed.
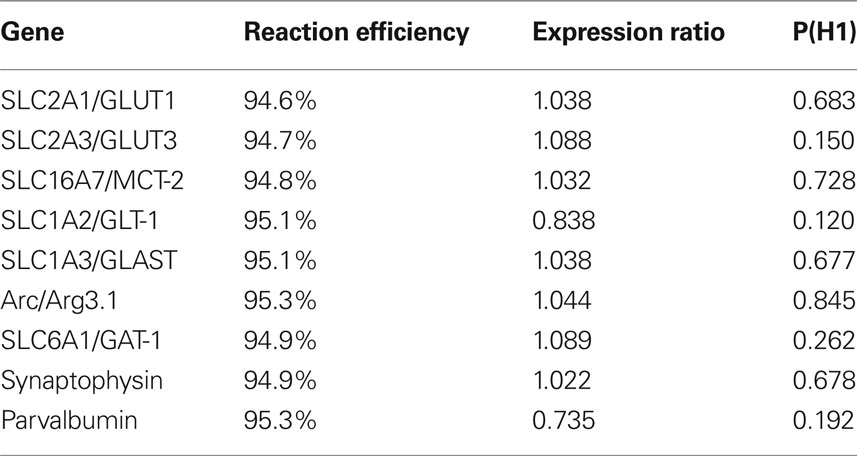
Table 1. Effects of 5 day 2.58 mg/kg PCP injections on the mRNA expression of genes relevant for glucose utilization and interneuron prevalence. Expression ratio of 1 indicates no difference in expression between PCP (n = 12) and Saline (n = 7) groups. Ratios less than 1 indicate downregulation. Since P(H1) was greater than 0.05 for all genes, PCP at this dose, did not significantly affect gene expression. Reaction efficiency is listed as a control for the Qiagen primer assays which are all optimized to 95% efficiency.
Normalization factors, reference genes and analysis
Proper normalization of PCR data and the use of multiple reference genes are critical for the accurate analysis of gene expression (Vandesompele et al., 2002 ). The reference genes beta-actin, GAPDH, RPL19 and SDHA were analyzed in this experiment. Primer efficiencies were calculated using the assumption-free method of Ramakers and colleagues which uses a linear regression of the fluorescence curve (linregPCR v11.1 software) to estimate the efficiency for each run (Ramakers et al., 2003 ; Ruijter et al., 2009 ). Ct values for each sample were calculated with the Light Cycler analysis module provided with the equipment (Roche). To compare Saline and PCP-treated animals, we used the REST2008 gene expression analysis program (Pfaffl et al., 2002 ). Using the calculated primer efficiencies and Ct values, the program performs 5000 iterations to compare whether there is a difference in expression between the groups. This also takes into account the reference genes. Expression values from this program are a ratio such that values above 1 denote an upregulation of gene expression in the treated group while values less than 1 indicate a downregulation.
Statistical analysis
Microdialysis data was compared by two-way repeated measures ANOVA with factor Drug to compare Saline vs PCP effects and factor Time to denote the actual changes in dopamine release induced by the drugs. An unpaired t-test was used to compare relative PCP-induced dopamine release. A significance level of p = 0.05 was used for all analyses in this experiment.
Calculation of the relative dopamine release per drug (Figures 1 D,E) were calculated as follows. Irrespective of drug, the peak dopamine effect was observed 40 min after administration (Figure 1 C). As such, dopamine release for the first drug was defined as the dopamine value at t = 40 divided by the baseline value. Dopamine release of the second drug was the dopamine value at t = 100 divided by the value at t = 60.
Results
Effects of Administration Order on Nucleus Accumbens Dopamine Release
Dopamine release in the nucleus accumbens was measured during injection/infusion of either PCP or ethanol. Both administration orders were tested in order to assess the pharmacological interaction between the two compounds. Probe placement was verified by sectioning (VibroSlice, Camden Instruments) and only correctly-placed probes positioned in the nucleus accumbens were included in this study. Starting with a two-way repeated measures ANOVA of the entire microdialysis experiment, the drugs significantly changed dopamine release over time when either PCP (F = 13.8; p < 0.0001) or ethanol (F = 25.1; p < 0.0001) were administered first.
PCP followed by local accumbal administration of ethanol
A two-way repeated measures ANOVA illustrated that injection of 5 mg/kg PCP significantly induced dopamine release in the nucleus accumbens (F = 22.4; p < 0.0001: Time −20 to 60 min). However, infusion of 300 mM ethanol into the nucleus accumbens after this PCP injection failed to induce dopamine release (F = 0.367; p = 0.869: Time 60 to 180 min) (Figure 1 A). To illustrate this blockade of ethanol- induced dopamine release by PCP, we re-calculated a percent of baseline value relative to the 60 min point (Figure 1 E). When PCP was infused first, ethanol induced significantly less dopamine release (p = 0.0119) compared to normal ethanol induced dopamine release.
Accumbal ethanol infusion followed by PCP injection
Ethanol infusion induced dopamine release in the nucleus accumbens (F = 7.87; p < 0.0001: Time −20 to 60 min) (Figure 1 B). Subsequent injection of 5 mg/kg PCP also induced the release of dopamine (F = 5.71; p = 0002: Time 60 to 180 min) and the Interaction factor was also significantly different (F = 2.63; p = 0.0304). To compare the relative PCP-induced dopamine release, we re-calculated a percent of baseline value relative to the 60 min point (Figure 1 D). When ethanol was infused first, PCP induced a greater relative dopamine release in the nucleus accumbens (p = 0.0334) than PCP injection alone, indicating a synergistic action between ethanol and PCP which was dependent on the order of administration.
Effects of 5-Day PCP Pretreatment
In the second part of this experiment, we investigated whether pretreatment with PCP for 5 days (i.e. prefrontal cortex hypofunction) would change the individual response to ethanol or PCP.
Nucleus accumbens dopamine release
There was no difference in baseline dopamine amount (p = 0.995) between PCP and Saline-treated groups (Figure 1 A, inset) A two-way repeated measures ANOVA was performed (PCP vs Saline) to determine whether pretreatment affected the release of dopamine. When PCP was administered first (Figure 1 A), there was no between-group difference (Factor Drug: F = 0.00606; p = 0.939) and with no significant Interaction (F = 0.256; p = 0.989). When ethanol was infused first (Figure 1 B), there was also no difference between the PCP pretreatment and Saline groups (Factor Drug: F = 0.00838; p = 0.928) with no significant Interaction (F = 1.004; p = 0.442).
Changes in mPFC gene expression following 5-day PCP pretreatment
To verify that injection of 2.58 mg/kg PCP for 5 days would produce a hypofunction, we dissected the medial prefrontal cortex and analyzed mRNA expression of several key energy transporters or components of the GABAergic system. Of the four reference genes analyzed, RPL19 was excluded because it was differentially regulated in the PCP group. One animal in the Saline group was excluded due to poor extraction yield and subsequent difficulties with cDNA synthesis. Pretreatment with PCP did not change the mRNA expression of any gene tested (Table 1 ) although the glutamate transporter SLC1A2/GLT-1 had the strongest trend to significance (p = 0.120). A boxplot of the results produced by the REST2008 program (Figure 2 ) indicated a wider distribution of expression ratios for Arc and parvalbumin which could suggest subgrouping in the population with respect to PCP treatment effect.
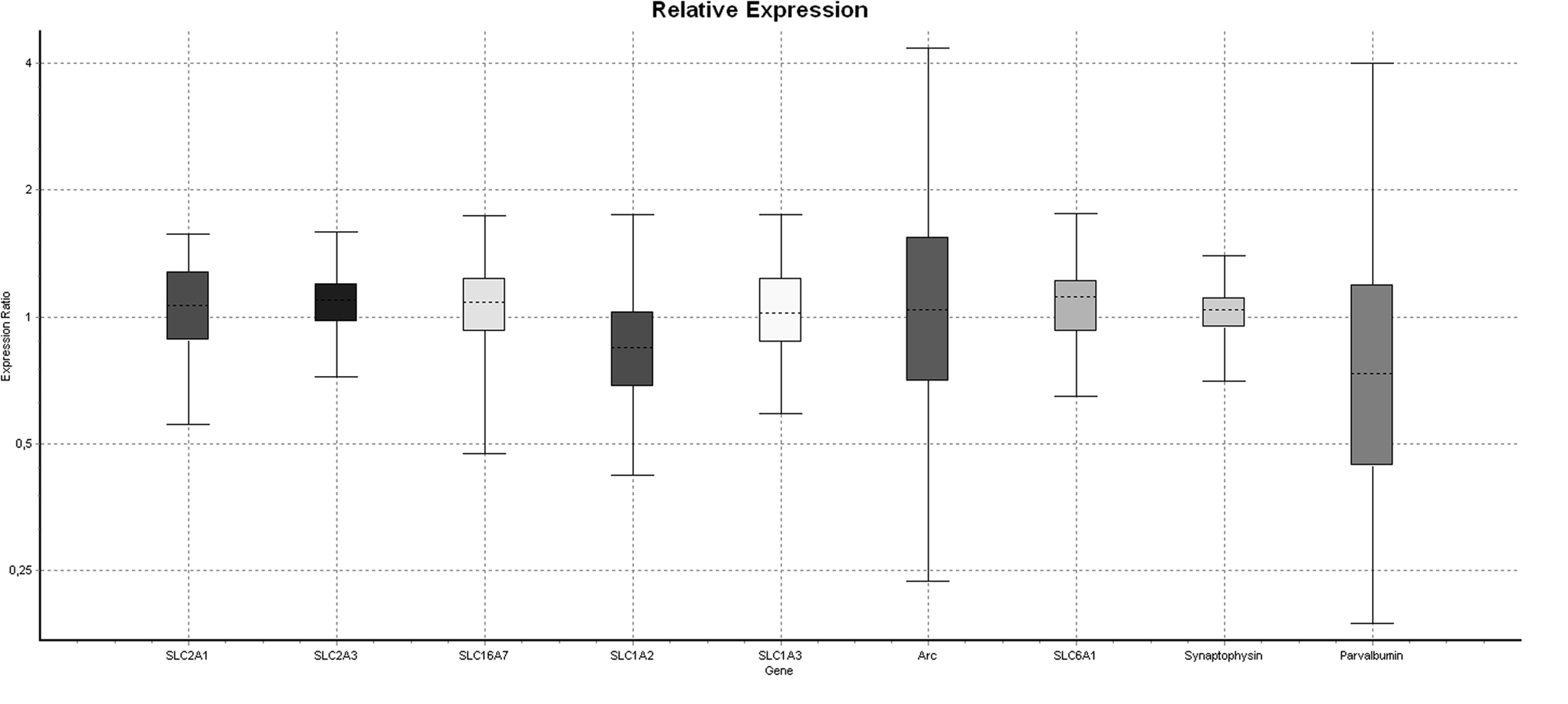
Figure 2. Boxplot of differential gene expression generated by the REST2008 software. The considerable variability in Arc and parvalbumin compared to other genes suggests PCP-induced dysfunction in some but not all individuals.
Discussion
With respect to the induction of dopamine release in the nucleus accumbens, a clear interaction between ethanol and PCP was demonstrated in this study. This interaction, however, was dependent on the order of administration of the two drugs. Intraperitoneal injection of 5 mg/kg PCP increased accumbal dopamine release but blocked any actions of a 300 mM local infusion of ethanol directly into the nucleus accumbens. When ethanol was infused first, the expected dopamine release was produced but this potentiated subsequent PCP-induced dopamine release. Pretreatment for 5 days with 2.58 mg/kg PCP did not affect the response to ethanol or PCP and a further study of the gene expression of markers normally affected by PCP treatment (e.g. GABAergic markers, immediate early genes, etc.) did not provide any evidence of prefrontal cortex hypofunction in these animals after exposure to this low and non-toxic PCP dose.
As mentioned previously, an interaction between ethanol and PCP was expected from previous studies (Brunet et al., 1985 ) but this is the first time an interaction has been demonstrated at the level of dopamine release in the nucleus accumbens. Starting with ethanol’s mechanism of action following local accumbal infusion, our theory proposes that ethanol stimulates inhibitory glycine receptors on GABAergic feedback neurons in the nucleus accumbens and this disinhibition releases an inhibitory ‘brake’ which tonically prevents activation of dopaminergic neurons in the ventral tegmental area (Soderpalm et al., 2009 ). An explanation of how a systemic injection of PCP induces dopamine release is complex given this compound’s actions at the sigma1 receptor (Minabe et al., 1999 ) and other non-NMDA targets (Hundt et al., 1998 ; Svensson, 2000 ; Takahashi et al., 2001 ). A lesion of the prefrontal cortex prevents PCP-induced locomotor stimulation and nucleus accumbens dopamine release, illustrating the necessity of prefrontal cortex projections for PCP’s effects (Jentsch et al., 1998b ). The actual mechanism of PCP may, however, be explained by changes in firing patterns of nucleus accumbens GABAergic neurons. Glutamatergic inputs from the prefrontal cortex, hippocampus and other regions regulate the up and down states of nucleus accumbens neurons since electrical stimulation of these regions will produce an EPSP (O’Donnell and Grace, 1995 ). In particular, these afferents from the hippocampus will, upon stimulation, shift nucleus accumbens neurons to a depolarized ‘up’ state. Since these accumbal neurons are GABAergic, the net result will be an inhibition of target regions of these feedback neurons (e.g. the ventral tegmental area) (O’Donnell and Grace, 1998 ). Acute systemic injection of PCP blocks the transition of nucleus accumbens GABAergic neurons from the inactive ‘down’ state to the depolarized ‘up’ state which reduces the chance of firing, thus removing the strong inhibition of ventral tegmental area dopamine neurons (O’Donnell and Grace, 1998 ). This is how we propose that our order effect can be explained. When PCP is administered first, GABAergic feedback neurons are disinhibited such that the local action of ethanol on these neurons cannot have an increased effect with respect to dopamine release. However, when ethanol is administered first, GABAergic feedback neurons can release dopamine as usual and systemic PCP can still affect the activity of other brain regions. The change in neurotransmission already started by ethanol somehow potentiated the dopamine-releasing ability of PCP. Therefore, a further investigation of this interaction is important in order to determine whether prefrontal cortex dysfunction will affect the response to ethanol.
The acute interaction of PCP and ethanol complicates the question of whether PCP-induced cortical hypofunction affects the response to ethanol. Repeated PCP treatment suggests a decrease in dopamine utilization that is selective to the prefrontal cortex and not the nucleus accumbens (Jentsch et al., 1997 ). It is therefore conceivable that the dopamine response of ethanol following accumbal infusion may not change following PCP treatment. This was, in fact, what we observed in this study. However, we cannot make this conclusion since a clear PCP-induced prefrontal cortex dysfunction could not be demonstrated. In the literature, PCP causes a selective decrease in GABAergic chandelier neurons in the prefrontal cortex (Wang et al., 2008 ). These parvalbumin-positive neurons can be labeled by immunohistochemistry against the GABA transporter GAT-1 (SLC6A1) which visualizes a series of synaptic sites located along the axon of pyramidal neurons (Hardwick et al., 2005 ). These chandelier neurons strongly inhibit pyramidal neuron activity (Lewis et al., 1999 ) so a selective PCP-induced decrease of these neurons should increase the glutamatergic output from the prefrontal cortex to limbic or striatal areas. We pretreated another group of animals and extracted the medial prefrontal cortex for gene expression analysis via qPCR. Using this method, no PCP-induced changes were observed for GAT-1 (SLC6A1), synaptophysin or parvalbumin. Cochran et al. (2003) defined prefrontal cortex hypofunction by a reduction in glucose utilization. Changes in glucose utilization were estimated by analysis of mRNA levels of the glucose transporters Glut1 (SLC2A1) and Glut3 (SLC2A3). In line with the potential role of lactate as an energy source for neurons following activation (Pellerin et al., 2007 ), we also measured expression of the monocarboxylate transporter MCT2 (SLC16A7). PCP did not significantly affect any of these energy transporters. The high-affinity glial-specific glutamate transporters GLT-1 (SLC1A2) and GLAST (SLC1A3) (Shigeri et al., 2004 ) are also integral in the cycling of glucose and lactate between activated neurons and glia (Pellerin et al., 2007 ). Since PCP is an NMDA antagonist, adaptive changes similar to our previous study (Pickering et al., 2006 ) could occur to counteract excessive glutamate release. Again, PCP pretreatment did not significantly change gene expression of these glutamate transporters although GLT-1 did have the strongest trend to significance (p = 0.12) of all the genes analyzed. A similar lack of effect was observed for the transcription factor Arc (Arg 3.1) despite a clear PCP-induced increase of this in other studies (Thomsen et al., 2009 ). We therefore must conclude that treatment for 5 days with 2.58 mg/kg PCP did not induce prefrontal cortex hypofunction.
The absence of a PCP effect in our animals compared to Cochran and colleagues was probably due to the low dose of 2.58 mg/kg PCP but this could also be explained by strain differences or animal age. It is plausible that a subgroup of animals may have had a hypofunction since the distribution of between-group expression for Arc and parvalbumin was much larger than that of other genes (Figure2 ). Nonetheless, a review of the literature indicates several ways to decrease the proportion of non-responder animals. We have chosen adult outbred Wistar rats for this study because we have characterized the effects of ethanol in these animals. Other protocols with demonstrable PCP-induced hypofunction use Long-Evans rats (Cochran et al., 2003 ) or C57BL/6J mice (Thomsen et al., 2009 ). We may also have failed to achieve hypofunction because our animals were too old although some studies suggest increased susceptibility to PCP-induced neurotoxicity with age (Farber et al., 1995 ). PCP may be injected on postnatal day 7, 9 and 11 to produce a subsequent hypofunction in adult animals (Broberg et al., 2008 ; Wang et al., 2008 ). Cochran and colleagues used animals of a weight (180–220 g) corresponding roughly with adolescence in rodents, an age often chosen for the study of developmental effects since several brain areas (e.g. prefrontal cortex) are not yet fully developed and clinical studies often demonstrate the increased use of drugs at this age (Doremus-Fitzwater et al., 2009 ). By fine-tuning the age, strain and dose factors, it will be possible to produce a prefrontal cortex dysfunction in rats. But it was clear from the qPCR results in this study that a higher dose of PCP will be required.
Overall, we have demonstrated that ethanol and PCP interact with respect to the induction of dopamine release in the nucleus accumbens. This acute and order-dependent effect suggests further study of this drug combination with respect to local infusion studies but it also warns for potential difficulties in the interpretation of results in animals with PCP-induced prefrontal cortex dysfunction. In particular, we demonstrated the inability of the 2.58 mg/kg PCP dose to induce a measurable prefrontal cortex dysfunction, again illustrating the importance of a careful selection of animal strain, age, dose and administration pattern when performing studies of this type.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The studies were supported by the Alcohol Research Council of the Swedish Alcohol Retailing Monopoly, the Swedish Research Council – Medicine (2006-4988 and 2006-6385), governmental support under the LUA/ALF agreement, Socialstyrelsen, Lars Hiertas minne, Wilhelm and Martina Lundgrens Vetenskapsfond and Gunnar and Märta Bergendahls Stiftelse. Chris Pickering was supported by the Swedish Brain Research Foundation (Hjärnfonden).
References
Abdul-Monim, Z., Neill, J. C., and Reynolds, G. P. (2007). Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J. Psychopharmacol. (Oxford) 21, 198–205.
Allen, R. M., and Young, S. J. (1978). Phencyclidine-induced psychosis. Am. J. Psychiatry 135, 1081–1084.
Amitai, N., and Markou, A. (2009). Chronic nicotine improves cognitive performance in a test of attention but does not attenuate cognitive disruption induced by repeated phencyclidine administration. Psychopharmacology (Berl.) 202, 275–286.
Amitai, N., Semenova, S., and Markou, A. (2007). Cognitive-disruptive effects of the psychotomimetic phencyclidine and attenuation by atypical antipsychotic medications in rats. Psychopharmacology (Berl.) 193, 521–537.
Amitai, N., Semenova, S., and Markou, A. (2009). Clozapine attenuates disruptions in response inhibition and task efficiency induced by repeated phencyclidine administration in the intracranial self-stimulation procedure. Eur. J. Pharmacol. 602, 78–84.
Belin, D., Mar, A. C., Dalley, J. W., Robbins, T. W., and Everitt, B. J. (2008). High impulsivity predicts the switch to compulsive cocaine-taking. Science 320, 1352–1355.
Bowen, C. A., and Grant, K. A. (1999). Increased specificity of ethanol’s discriminative stimulus effects in an ethanol-pentobarbital-water discrimination in rats. Drug Alcohol Depend. 55, 13–24.
Broberg, B. V., Dias, R., Glenthoj, B. Y., and Olsen, C. K. (2008). Evaluation of a neurodevelopmental model of schizophrenia–early postnatal PCP treatment in attentional set-shifting. Behav. Brain Res. 190, 160–163.
Brunet, B. L., Reiffenstein, R. J., Williams, T., and Wong, L. (1985). Toxicity of phencyclidine and ethanol in combination. Alcohol Drug Res. 6, 341–349.
Chanraud, S., Martelli, C., Delain, F., Kostogianni, N., Douaud, G., Aubin, H. J., Reynaud, M., and Martinot, J. L. (2007). Brain morphometry and cognitive performance in detoxified alcohol-dependents with preserved psychosocial functioning. Neuropsychopharmacology 32, 429–438.
Cochran, S. M., Kennedy, M., McKerchar, C. E., Steward, L. J., Pratt, J. A., and Morris, B. J. (2003). Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology 28, 265–275.
Dao-Castellana, M. H., Samson, Y., Legault, F., Martinot, J. L., Aubin, H. J., Crouzel, C., Feldman, L., Barrucand, D., Rancurel, G., Feline, A., and Syrota, A. (1998). Frontal dysfunction in neurologically normal chronic alcoholic subjects: metabolic and neuropsychological findings. Psychol. Med. 28, 1039–1048.
De Bellis, M. D., Narasimhan, A., Thatcher, D. L., Keshavan, M. S., Soloff, P., and Clark, D. B. (2005). Prefrontal cortex, thalamus, and cerebellar volumes in adolescents and young adults with adolescent-onset alcohol use disorders and comorbid mental disorders. Alcohol. Clin. Exp. Res. 29, 1590–1600.
Diamond, D. M., Campbell, A., Park, C. R., and Vouimba, R. M. (2004). Preclinical research on stress, memory, and the brain in the development of pharmacotherapy for depression. Eur. Neuropsychopharmacol. 14(Suppl. 5), S491–S495.
Diana, M., Pistis, M., Carboni, S., Gessa, G. L., and Rossetti, Z. L. (1993). Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: electrophysiological and biochemical evidence. Proc. Natl. Acad. Sci. U.S.A. 90, 7966–7969.
Doremus-Fitzwater, T. L., Varlinskaya, E. I., and Spear, L. P. (2009). Motivational systems in adolescence: Possible implications for age differences in substance abuse and other risk-taking behaviors. Brain Cogn. 72, 114–123.
Ericson, M., Lof, E., Stomberg, R., Chau, P., and Soderpalm, B. (2008). Nicotinic acetylcholine receptors in the anterior, but not posterior, ventral tegmental area mediate ethanol-induced elevation of accumbal dopamine levels. J. Pharmacol. Exp. Ther. 326, 76–82.
Ericson, M., Molander, A., Lof, E., Engel, J. A., and Soderpalm, B. (2003). Ethanol elevates accumbal dopamine levels via indirect activation of ventral tegmental nicotinic acetylcholine receptors. Eur. J. Pharmacol. 467, 85–93.
Farber, N. B., Wozniak, D. F., Price, M. T., Labruyere, J., Huss, J., St Peter, H., and Olney, J. W. (1995). Age-specific neurotoxicity in the rat associated with NMDA receptor blockade: potential relevance to schizophrenia? Biol. Psychiatry 38, 788–796.
Grant, K. A., Snell, L. D., Rogawski, M. A., Thurkauf, A., and Tabakoff, B. (1992). Comparison of the effects of the uncompetitive N-methyl-D-aspartate antagonist (+-)-5-aminocarbonyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine (ADCI) with its structural analogs dizocilpine (MK-801) and carbamazepine on ethanol withdrawal seizures. J. Pharmacol. Exp. Ther. 260, 1017–1022.
Green-Jordan, K., and Grant, K. A. (2000). Modulation of the ethanol-like discriminative stimulus effects of diazepam and phencyclidine by L-type voltage-gated calcium-channel ligands in rats. Psychopharmacology (Berl.) 149, 84–92.
Greenslade, R. G., and Mitchell, S. N. (2004). Selective action of (-)-2-oxa-4-aminobicyclo[3.1.0] hexane-4,6-dicarboxylate (LY379268), a group II metabotropic glutamate receptor agonist, on basal and phencyclidine- induced dopamine release in the nucleus accumbens shell. Neuropharmacology 47, 1–8.
Hagiwara, H., Fujita, Y., Ishima, T., Kunitachi, S., Shirayama, Y., Iyo, M., and Hashimoto, K. (2008). Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of the antipsychotic drug perospirone: role of serotonin 5-HT1A receptors. Eur. Neuropsychopharmacol. 18, 448–454.
Hardwick, C., French, S. J., Southam, E., and Totterdell, S. (2005). A comparison of possible markers for chandelier cartridges in rat medial prefrontal cortex and hippocampus. Brain Res. 1031, 238–244.
Harris, R. A., Trudell, J. R., and Mihic, S. J. (2008). Ethanol’s molecular targets. Sci. Signal. 1, re7.
Hashimoto, K., Fujita, Y., Shimizu, E., and Iyo, M. (2005). Phencyclidine-induced cognitive deficits in mice are improved by subsequent subchronic administration of clozapine, but not haloperidol. Eur. J. Pharmacol. 519, 114–117.
Heffner, T. G., Hartman, J. A., and Seiden, L. S. (1980). A rapid method for the regional dissection of the rat brain. Pharmacol. Biochem. Behav. 13, 453–456.
Hundt, W., Danysz, W., Holter, S. M., and Spanagel, R. (1998). Ethanol and N-methyl-D-aspartate receptor complex interactions: a detailed drug discrimination study in the rat. Psychopharmacology (Berl.) 135, 44–51.
Jentsch, J. D., and Roth, R. H. (1999). The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 20, 201–225.
Jentsch, J. D., Taylor, J. R., and Roth, R. H. (1998a). Subchronic phencyclidine administration increases mesolimbic dopaminergic system responsivity and augments stress- and psychostimulant-induced hyperlocomotion. Neuropsychopharmacology 19, 105–113.
Jentsch, J. D., Tran, A., Taylor, J. R., and Roth, R. H. (1998b). Prefrontal cortical involvement in phencyclidine-induced activation of the mesolimbic dopamine system: behavioral and neurochemical evidence. Psychopharmacology (Berl.) 138, 89–95.
Jentsch, J. D., Tran, A., Le, D., Youngren, K. D., and Roth, R. H. (1997). Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology 17, 92–99.
Kahkonen, S., Wilenius, J., Nikulin, V. V., Ollikainen, M., and Ilmoniemi, R. J. (2003). Alcohol reduces prefrontal cortical excitability in humans: a combined TMS and EEG study. Neuropsychopharmacology 28, 747–754.
Kapur, S., and Seeman, P. (2002). NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D(2) and serotonin 5-HT(2)receptors-implications for models of schizophrenia. Mol. Psychiatry 7, 837–844.
Kril, J. J., Halliday, G. M., Svoboda, M. D., and Cartwright, H. (1997). The cerebral cortex is damaged in chronic alcoholics. Neuroscience 79, 983–998.
Krystal, J. H., Karper, L. P., Seibyl, J. P., Freeman, G. K., Delaney, R., Bremner, J. D., Heninger, G. R., Bowers, M. B. Jr., and Charney, D. S. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch. Gen. Psychiatry 51, 199–214.
Larsson, A., Edstrom, L., Svensson, L., Soderpalm, B., and Engel, J. A. (2005). Voluntary ethanol intake increases extracellular acetylcholine levels in the ventral tegmental area in the rat. Alcohol Alcohol. 40, 349–358.
Lewis, D. A., Hashimoto, T., and Volk, D. W. (2005). Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 6, 312–324.
Lewis, D. A., Pierri, J. N., Volk, D. W., Melchitzky, D. S., and Woo, T. U. (1999). Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol. Psychiatry 46, 616–626.
Lido, H. H., Stomberg, R., Fagerberg, A., Ericson, M., and Soderpalm, B. (2009). The glycine reuptake inhibitor org 25935 interacts with Basal and ethanol-induced dopamine release in rat nucleus accumbens. Alcohol. Clin. Exp. Res. 33, 1151–1157.
McGregor, I. S., and Gallate, J. E. (2004). Rats on the grog: novel pharmacotherapies for alcohol craving. Addict. Behav. 29, 1341–1357.
Millan, M. J., Brocco, M., Gobert, A., Joly, F., Bervoets, K., Rivet, J., Newman-Tancredi, A., Audinot, V., and Maurel, S. (1999). Contrasting mechanisms of action and sensitivity to antipsychotics of phencyclidine versus amphetamine: importance of nucleus accumbens 5-HT2A sites for PCP-induced locomotion in the rat. Eur. J. Neurosci. 11, 4419–4432.
Minabe, Y., Matsuno, K. and Ashby, C. R. Jr. (1999). Acute and chronic administration of the selective sigma1 receptor agonist SA4503 significantly alters the activity of midbrain dopamine neurons in rats: An in vivo electrophysiological study. Synapse 33, 129–140.
Molander, A., and Soderpalm, B. (2005). Accumbal strychnine-sensitive glycine receptors: an access point for ethanol to the brain reward system. Alcohol. Clin. Exp. Res. 29, 27–37.
O’Donnell, P., and Grace, A. A. (1995). Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J. Neurosci. 15, 3622–3639.
O’Donnell, P., and Grace, A. A. (1998). Phencyclidine interferes with the hippocampal gating of nucleus accumbens neuronal activity in vivo. Neuroscience 87, 823–830.
Paxinos, G., and Watson, C. (2007). The Rat Brain in Stereotaxic Coordinates. Academic Press Inc, USA.
Pellerin, L., Bouzier-Sore, A. K., Aubert, A., Serres, S., Merle, M., Costalat, R., and Magistretti, P. J. (2007). Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia 55, 1251–1262.
Pfaffl, M. W., Horgan, G. W., and Dempfle, L. (2002). Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 30, e36.
Pickering, C., Avesson, L., Lindblom, J., Liljequist, S., and Schioth, H. B. (2007a). Identification of neurotransmitter receptor genes involved in alcohol self-administration in the rat prefrontal cortex, hippocampus and amygdala. Prog. Neuropsychopharmacol. Biol. Psychiatry 31, 53–64.
Pickering, C., Avesson, L., Lindblom, J., Liljequist, S., and Schioth, H. B. (2007b). To press or not to press? Differential receptor expression and response to novelty in rats learning an operant response for reward. Neurobiol. Learn. Mem. 87, 181–191.
Pickering, C., Gustafsson, L., Cebere, A., Nylander, I., and Liljequist, S. (2006). Repeated maternal separation of male Wistar rats alters glutamate receptor expression in the hippocampus but not the prefrontal cortex. Brain Res. 1099, 101–108.
Ramakers, C., Ruijter, J. M., Deprez, R. H., and Moorman, A. F. (2003). Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339, 62–66.
Rimondini, R., Sommer, W., and Heilig, M. (2003). A temporal threshold for induction of persistent alcohol preference: behavioral evidence in a rat model of intermittent intoxication. J. Stud. Alcohol 64, 445–449.
Ruijter, J. M., Ramakers, C., Hoogaars, W. M., Karlen, Y., Bakker, O., van den Hoff, M. J., and Moorman, A. F. (2009). Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37, e45.
Seeman, P., Ko, F., and Tallerico, T. (2005). Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol. Psychiatry 10, 877–883.
Shigeri, Y., Seal, R. P., and Shimamoto, K. (2004). Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res. Brain Res. Rev. 45, 250–265.
Soderpalm, B., Lof, E., and Ericson, M. (2009). Mechanistic studies of ethanol’s interaction with the mesolimbic dopamine reward system. Pharmacopsychiatry 42(Suppl. 1), S87–S94.
Spanagel, R., and Holter, S. M. (1999). Long-term alcohol self- administration with repeated alcohol deprivation phases: an animal model of alcoholism? Alcohol Alcohol. 34, 231–243.
Svensson, T. H. (2000). Dysfunctional brain dopamine systems induced by psychotomimetic NMDA-receptor antagonists and the effects of antipsychotic drugs. Brain Res. Brain Res. Rev. 31, 320–329.
Takahashi, S., Horikomi, K., and Kato, T. (2001). MS-377, a novel selective sigma(1) receptor ligand, reverses phencyclidine-induced release of dopamine and serotonin in rat brain. Eur. J. Pharmacol. 427, 211–219.
Tanibuchi, Y., Fujita, Y., Kohno, M., Ishima, T., Takatsu, Y., Iyo, M., and Hashimoto, K. (2009). Effects of quetiapine on phencyclidine-induced cognitive deficits in mice: a possible role of alpha1-adrenoceptors. Eur. Neuropsychopharmacol. 19, 861–867.
Thomsen, M. S., Christensen, D. Z., Hansen, H. H., Redrobe, J. P., and Mikkelsen, J. D. (2009). alpha(7) Nicotinic acetylcholine receptor activation prevents behavioral and molecular changes induced by repeated phencyclidine treatment. Neuropharmacology 56, 1001–1009.
Tu, Y., Kroener, S., Abernathy, K., Lapish, C., Seamans, J., Chandler, L. J., and Woodward, J. J. (2007). Ethanol inhibits persistent activity in prefrontal cortical neurons. J. Neurosci. 27, 4765–4775.
Turyabahika-Thyen, K., and Wolffgramm, J. (2006). Loss of flexibility in alcohol-taking rats: promoting factors. Eur. Addict. Res. 12, 210–221.
Vadlamani, N. L., Pontani, R. B., and Misra, A. L. (1982). Effect of acute and chronic ethanol pre-treatment on the disposition of phencyclidine (PCP) in the rat. Pharmacol. Biochem. Behav. 16, 847–850.
van den Heuvel, O. A., Veltman, D. J., Groenewegen, H. J., Witter, M. P., Merkelbach, J., Cath, D. C., van Balkom, A. J., van Oppen, P., and van Dyck, R. (2005). Disorder-specific neuroanatomical correlates of attentional bias in obsessive-compulsive disorder, panic disorder, and hypochondriasis. Arch. Gen. Psychiatry 62, 922–933.
Vandesompele, J., De Preter, K., Pattyn, F., Poppe, B., Van Roy, N., De Paepe, A., and Speleman, F. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, RESEARCH0034.
Wang, C. Z., and Johnson, K. M. (2007). The role of caspase-3 activation in phencyclidine-induced neuronal death in postnatal rats. Neuropsychopharmacology 32, 1178–1194.
Wang, C. Z., Yang, S. F., Xia, Y., and Johnson, K. M. (2008). Postnatal phencyclidine administration selectively reduces adult cortical parvalbumin-containing interneurons. Neuropsychopharmacology 33, 2442–2455.
Weitlauf, C., and Woodward, J. J. (2008). Ethanol selectively attenuates NMDAR-mediated synaptic transmission in the prefrontal cortex. Alcohol. Clin. Exp. Res. 32, 690–698.
Keywords: prefrontal cortex hypofunction, microdialysis, alcoholism, schizophrenia, executive dysfunction
Citation: Pickering C, Chau PP, Söderpalm B and Ericson M (2010) Ethanol and phencyclidine interact with respect to nucleus accumbens dopamine release: differential effects of administration order and pretreatment protocol. Front. Behav. Neurosci. 4:32. doi: 10.3389/fnbeh.2010.00032
Received: 26 February 2010;
Paper pending published: 01 April 2010;
Accepted: 17 May 2010;
Published online: 10 June 2010
Edited by:
Amy F. T. Arnsten, Yale University School of Medicine, USAReviewed by:
David Jentsch, University of California, USAA. Del Arco, Universidad Complutense de Madrid, Spain
Copyright: © 2010 Pickering, Chau, Söderpalm and Ericson. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Chris Pickering, Addiction Biology Unit, Department of Psychiatry and Neurochemistry, Institute of Neuroscience and Physiology, University of Gothenburg, Box 410, SE-40530 Gothenburg, Sweden. e-mail:Y2hyaXMucGlja2VyaW5nQG5ldXJvLmd1LnNl