


- Department of Psychology, University of Wisconsin-Milwaukee, Milwaukee, WI, USA
Recent work has led to a better understanding of the neural mechanisms underlying the extinction of Pavlovian fear conditioning. Long-term synaptic changes in the medial prefrontal cortex (mPFC) are critical for extinction learning, but very little is currently known about how the mPFC and other brain areas interact during extinction. The current study examined the effect of drugs that impair the extinction of fear conditioning on the activation of the extracellular-related kinase/mitogen-activated protein kinase (ERK/MAPK) in brain regions that likely participate in the consolidation of extinction learning. Inhibitors of opioid and N-methyl-D-aspartic acid (NMDA) receptors were applied to the ventrolateral periaqueductal gray matter (vlPAG) and amygdala shortly before extinction training. Results from these experiments show that blocking opioid receptors in the vlPAG prevented the formation of extinction memory, whereas NMDA receptor blockade had no effect. Conversely, blocking NMDA receptors in the amygdala disrupted the formation of fear extinction memory, but opioid receptor blockade in the same brain area did not. Subsequent experiments tested the effect of these drug treatments on the activation of the ERK/MAPK signaling pathway in various brain regions following extinction training. Only opioid receptor blockade in the vlPAG disrupted ERK phosphorylation in the mPFC and amygdala. These data support the idea that opiodergic signaling derived from the vlPAG affects plasticity across the brain circuit responsible for the formation of extinction memory.
Introduction
Pavlovian fear conditioning involves the presentation of a neutral conditional stimulus (CS) that predicts the occurrence of an aversive unconditional stimulus (UCS). Repeated exposure to the CS alone after initial training results in a gradual decrease in fear responses, which is referred to as extinction (Pavlov, 1927). Extinction both refers to the initial decrement in performance that occurs as the CS is being presented alone and also to the long-term retention of this learning across days. Considerable evidence indicates that extinction involves new learning rather than unlearning of the original training (Pavlov, 1927; Rescorla and Heth, 1975; Bouton and King, 1983). Work on the neural mechanisms of fear extinction has demonstrated that this form of learning likely involves plasticity in both the amygdala and mPFC (Falls et al., 1992; Milad and Quirk, 2002).
One question that has received little attention involves how the mPFC and amygdala are signaled during extinction learning. Prior work suggests that opioid receptors may regulate the formation of Pavlovian associations during fear learning (Fanselow and Bolles, 1979; McNally and Westbrook, 2003). These studies have been understood in terms of the notion that learning is a function of the discrepancy between the expected and actual outcome of a conditioning trial (for review see Fanselow, 1998; McNally and Cole, 2006). For example, during fear extinction learning animals expect to receive shock when presented with the CS because during initial training shock always followed the CS. When the CS is repeatedly presented without being followed by shock there is a violation of expectancy, which drives new learning. The vlPAG, which is rich in opioid receptors and critical for aversive learning, is crucial to mediating these effects (Hammer and Kapp, 1986; McNally et al., 2004, 2005; Cole and McNally, 2007a). However, these studies have not looked at whether blocking opioid receptors in the vlPAG changes activity in structures important for fear learning in a manner consistent with an effect on expectancy. We propose that if opioid receptors in the vlPAG are critical for reacting to changes in expectancy during fear learning, cellular activity in the mPFC and amygdala consequential to the extinction learning process should be prevented by opioid antagonists applied to the vlPAG.
We tested this idea by infusing opioid and NMDA receptor antagonists into the vlPAG and amygdala and examining the effect they had on behavior and the phosphorylation of ERK in several brain structures following extinction training. We looked at ERK activity because there is considerable evidence that this signaling pathway is critical for the consolidation of extinction memory in both the amygdala and medial prefrontal cortex (Lu et al., 2001; Hugues et al., 2004, 2006; Herry et al., 2006; Kim, et al., 2009). The decision to examine opioid receptors was based on evidence that they may mediate prediction errors during fear learning (Fanselow, 1998; McNally and Cole, 2006). Dozens of studies have demonstrated that NMDA receptors are critically important for fear learning (e.g., Falls et al., 1992; Walker et al., 2002), perhaps by controlling the rate of learning (Cole and McNally, 2007b) and thus the two receptor systems may play complementary roles in fear learning. Our results indicate that whereas opioid receptor blockade in the vlPAG resulted in a disruption of ERK phosphorylation in the mPFC and the amygdala following extinction training, blocking the activity of NMDA receptors in the amygdala had no effect.
Materials and Methods
Subjects
All experiments were approved by the University of Wisconsin-Milwaukee Institutional Animal Care and Use Committee and were in accordance with the National Institutes of Health Guidelines for the Care and Use of Experimental Animals. The subjects were male Long-Evans rats weighing 300–375 g and obtained from Harlan (Madison, WI, USA). The rats were housed individually in standard shoebox cages and had free access to water and food throughout the experiments. The colony room was maintained on a 14:10 h light–dark cycle.
Surgery and Histology
All rats were handled daily for several minutes prior to the start of any procedures. Rats that underwent surgery were prepared with bilateral stainless steel 26-gauge guide cannulae (Plastics One, Roanoke, VA, USA) aimed at the basolateral (AP = −3.0/L = 5.0/DV = −7.2) nucleus of the amygdala or unilateral cannulae aimed at the ventrolateral periaqueductal gray (AP = −7.6/L = ±0.8/DV = −5.6). Cannulae were secured to the skull with the aid of stainless steel screws, superglue, and dental cement. Stainless steel dummy cannulae remained in place when the rats were not being infused to prevent the guide from becoming occluded. The rats were allowed to recover for at least 7 days before behavioral testing began. After behavioral testing, rats not processed for western blot were deeply anesthetized using isoflurane and the brains were processed for histology (see Parsons et al., 2006). In some cases, the vlPAG from rats processed for western blot was taken fresh, sectioned using a cryostat, and images were captured using Nomarski optics. Injection sites were marked measuring approximately 0.5–0.7 mm (i.e., the distance the internal cannulae protruded from the guides) beyond the cannulae track and were determined with the aid of a rat brain atlas (Paxinos and Watson, 1998). Placements for the amygdala were considered acceptable if the injection sites on both sides were determined to be within the amygdala. Over 90% of the placements met these criteria and of those nearly all were centered in the lateral or basolateral nuclei. For the vlPAG placements were considered good if the injection sites terminated in the vlPAG without damaging the central aqueduct. Again, over 90% of the placements met these criteria.
Apparatus
Fear conditioning took place in a set of four identical observation chambers (28 cm × 20.5 cm × 21 cm). The floor of each chamber (Context A) was composed of stainless steel rods spaced 1.5 cm apart through which the footshock was delivered. Each chamber was connected to its own shock generator-scrambler (Grason-Stadler, W. Concord, MA, USA), and was illuminated by a 7.5-W white light bulb. Ventilation fans provided a constant background of approximately 60–62 dB, and these chambers were wiped down with a 5% ammonium hydroxide solution before each set of animals. A separate set of chambers (Context B) housed in a different room were used as the context where extinction training and retention testing occurred. These chambers differed from the conditioning context in lighting, texture, and odor. The floors of each chamber were made of Plexiglas and each chamber was illuminated with infrared light. Fans provided a background noise of approximately 58 dB. The chambers in Context B were wiped down with a 5% acetic acid solution before each set of rats was tested.
Infusion Procedure and Drugs
All infusions were 0.5 μl/site and delivered at a rate of 0.5 μl/min. The injection cannulae were cut to extend approximately 0.5–0.7 mm beyond the guide cannulae and remained in place for 90 s following infusion to ensure adequate diffusion. All infusions took place approximately 5 min prior to extinction training, and the rats remained in transport cages between infusions and testing. The NMDA antagonist 3-(2-Carboxypiperazin-4-yl) propyl-1-phosphonic acid (Sigma Chemical, St Louis, MO, USA) was diluted in sterile saline to a concentration of 120 ng/μl. This dose of CPP has been shown to disrupt the consolidation of fear extinction when delivered into the mPFC (Burgos-Robles et al., 2007). Naloxone (Sigma Chemical) was diluted in sterile saline to a concentration of 5 μg/μl, a dose that been demonstrated to attenuate the retention of extinction when delivered into the vlPAG (McNally et al., 2004).
Behavioral Procedures
For acquisition, all rats were placed in the training chamber and after 6 min received two parings of white noise (10 s, 72 dB) and foot shock (1 s, 0.6 mA) spaced 90 s apart. The rats were in the chambers for a total of 11 min during acquisition training. Extinction training occurred the day following acquisition. Depending on the experiment, rats were exposed to one of three different procedures. Rats in the 40EXT groups were given full extinction training which consisted of a 1-min baseline followed by 40 presentations of the white noise (30 s) separated by 60 s. Other rats (4 EXT) received only four extinction trials which were presented with the same timing as the first four trials of rats in the 40 EXT group. Finally, some of the rats (NO-EXT) were trained as described above, but were only exposed to the chamber for an equivalent period of time without being exposed to the CS. In those rats not sacrificed following extinction training, a test for extinction memory took place approximately 24 h after extinction training. The testing procedure was identical to extinction training except that there were only eight presentations of the CS. The extinction training and memory tests all occurred in context B.
In experiments testing the effects of extinction and various drug treatments on the phosphorylation of the extracellular-related kinase/mitogen-activated protein kinase a slightly different set of procedures were employed. Rats were put through acquisition and extinction training as described previously, but were sacrificed 45 min from the end of the extinction training session. Some of the experiments had a group which was given acquisition, but sacrificed from their home cage the next day. Other experiments were preceded by an infusion of drugs into the vlPAG or BLA as described above.
Analysis of Behavioral Data
The behavior of each rat was recorded on digital video, and the amount of movement was determined in real-time by calculating the frame-by-frame changes in pixels using the FreezeScan 1.0 software (Clever Sys Inc., Reston, VA, USA). The computer scoring parameters have been chosen such that the scored activity closely matches hand-scoring methods previously used in our lab to measure freezing behavior. Freezing detection parameters were as follows (noise filtering radius = 1; Inter-frame motion = 400 pixels; automata parameters–freeze = N-25, M-22; move = N-10, M-8). The freezing analysis used a static threshold of 12. Tests in our laboratory typically show high inter-rater reliability between human and computer scored data (r values of 0.95 or greater). Data during acquisition were analyzed by separating out the session into a baseline period (i.e., the 6 min before shock), a 2-min period during which the CS and UCS occurred, and a 3-min block of time after CS-UCS pairings. The mean percent time freezing was determined for each group of rats during these three time periods. The mean percent freezing was also calculated for each four trial block of CS presentations during the extinction training and extinction memory test sessions. The data were analyzed by performing separate one-way ANOVAs on the first four trials of the extinction training and extinction memory test sessions. Data during the extinction training session had to be hand scored by a trained observer. The behavior of each rat was scored as freezing or not freezing once every 5 s during the 30-s CS trials. It was necessary to hand score the data for these sessions because during the relatively long extinction training sessions it was not uncommon for rats to lie down, which the computer would score as freezing behavior. Data from the extinction training sessions were divided into 10 blocks of 4 trials and analyzed using a repeated measure ANOVA with Block as a within-subjects factor and Drug as a between-subjects factor. With some of the data we also computed the mean change in percent freezing between the first four trials of extinction training and the extinction memory test and statistically compared these values using t-tests or an ANOVA. It was possible to use this as a measure of long-term extinction memory because the drug infusions did not affect the expression of freezing behavior during the first block of extinction training trials in any experiment.
Western Blot Procedure
To sacrifice the animals, rats were removed from their home cages and placed in an inhalation narcosis chamber (Harvard Apparatus, Holliston, MA, USA) that had isoflurane vapors in it. Approximately 30 s later, the rats were decapitated and their brains were rapidly removed and frozen at −80°C until they were dissected. Tissue from the mPFC and amygdala was dissected out by blocking the brain in a rat brain matrix (Harvard Apparatus, Holliston, MA, USA). For dissection of the mPFC an initial coronal cut was made to reach the anterior section of the medial prefrontal cortex approximately 3.5 mm anterior to bregma. Another coronal cut was made about 2 mm posterior of the first cut. This section of tissue was taken and the entire medial prefrontal cortex was dissected out making a horizontal cut between the ventral tips of the forceps minor of the corpus callosum and vertical cuts running along medial border of this same structure to the dorsal surface of the brain. This tissue sample included infralimbic, prelimbic, and anterior cingulate cortex. Both sides of the whole amygdala were dissected out from the blocked tissue by making a cut along the external capsule and a diagonal cut along the optic tract. Our pilot data using immunocytochemistry showed that subregions of the mPFC (infralimbic, prelimbic, and cingulate gyrus) all showed increased pERK upon extinction training. Thus, it is unlikely that the whole dissections of this area we used for western blot analysis affected our results.
All of the samples were homogenized in buffer (all in 100 ml DDH20; Tris–HCL 0.605 g; sodium deoxycholate 0.25 g; NaCl 0.876 g; EDTA 0.038 g; 0.0042 g NaF; PMSF 1 μg/ml; leupeptin 1 μg/ml; aprotinin 1 μg/ml; 10 ml 10% SDS, 1 mM sodium orthovanadate) and immediately placed on dry ice. Samples were stored at −80°C until they were thawed and then centrifuged at 4000 rpm for 20 min. The supernatant was removed and protein levels were measured using a Bradford protein assay kit (Bio-Rad laboratories, Hercules, CA, USA). Protein samples were normalized and loaded on a 7.5% SDS/PAGE gel. Proteins were then transferred from the gel to a membrane using a semidry transfer apparatus (Bio-Rad). Membranes were incubated in blocking buffer for 1 h and then overnight at 4°C in primary antibody (1:500) against phospho-ERK (Cell Signaling Technology, Danvers, MA, USA). Following primary antibody exposure, all membranes were incubated in goat anti-rabbit secondary antibody (1:2000) (Millipore, Billerica, MA, USA) for 90 min at room temperature. Membranes were washed three times, placed face down in an enhanced chemiluminescence (ECL) solution for 3 min (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and exposed to ECL sensitive film (GE Biosciences, Piscataway, NJ, USA). Following development of the film, the membranes were washed several times and exposed to an antibody (1:500, Cell Signaling) for total ERK overnight at 4°C.
Analysis of Western Blot Data
Western blot data were analyzed by taking images from the film and densitometry was performed using Image J (NIH). A single mean optical density value was determined for each sample by positioning a properly sized sample box over each band on the film and subtracting out the background of an equivalently sized area just below the target band. These values were averaged across the various treatment groups. ANOVA, Student’s t-tests, and appropriate post hoc comparisons were used to test for differences between groups.
Results
Fear Extinction Depends on Opioid but not NMDA Receptors in the Ventrolateral Periaqueductal Gray Matter
The first series of experiments were designed to determine the effectiveness of opioid and NMDA receptor antagonists on memory for extinction when delivered to the vlPAG and BLA prior to extinction training. Data were analyzed by comparing the mean percent freezing during the first four CS trials of extinction training across the different drug treatment groups. This analysis allowed us to determine if the drug treatments disrupted the expression of freezing behavior during extinction training, which is important considering that both of the targeted structures have been implicated in the expression of fear CRs (Helmstetter, 1992; Kim et al., 1993). We also performed an identical analysis on the first four trials of the extinction memory test in order to determine the effectiveness of the drug treatments on long-term extinction memory. Data during the extinction training session were analyzed with a mixed-factor ANOVA using Drug as a between-subject factor and Block as a within-subject factor.
Figure 1 shows the effect of the opioid receptor antagonist naloxone (NAL) on long-term extinction memory when infused into the vlPAG. No differences were seen before drug infusion during acquisition (Figure 1B), or during the first four trials of extinction training. Data from the extinction training session revealed no significant effect of Drug, but a significant effect of Block (F(1,10) = 19.84, p < 0.01), and no Drug × Block interaction. These results argue that both groups showed normal within-session extinction. During testing, rats infused with NAL in the vlPAG showed a disruption in the retention of extinction (Figure 1D, right side, Figure 1E). This is supported by a Student’s t-tests indicating that whereas rats treated with NAL froze similarly to controls during the first four CS presentations of extinction training (t < 1.0), they showed significantly more freezing to the CS during the extinction memory test [t(10) = 2.843, p < 0.05]. Change scores were computed for rats in each group by subtracting freezing during extinction training from freezing during the extinction memory test, resulting in a value reflecting the difference in freezing behavior between sessions (Figure 1E). A t-test on these values show that compared to saline infused rats, NAL treated animals showed little decrease between sessions [t(10) = 3.287, p < 0.01] indicating of a disruption in long-term extinction memory.
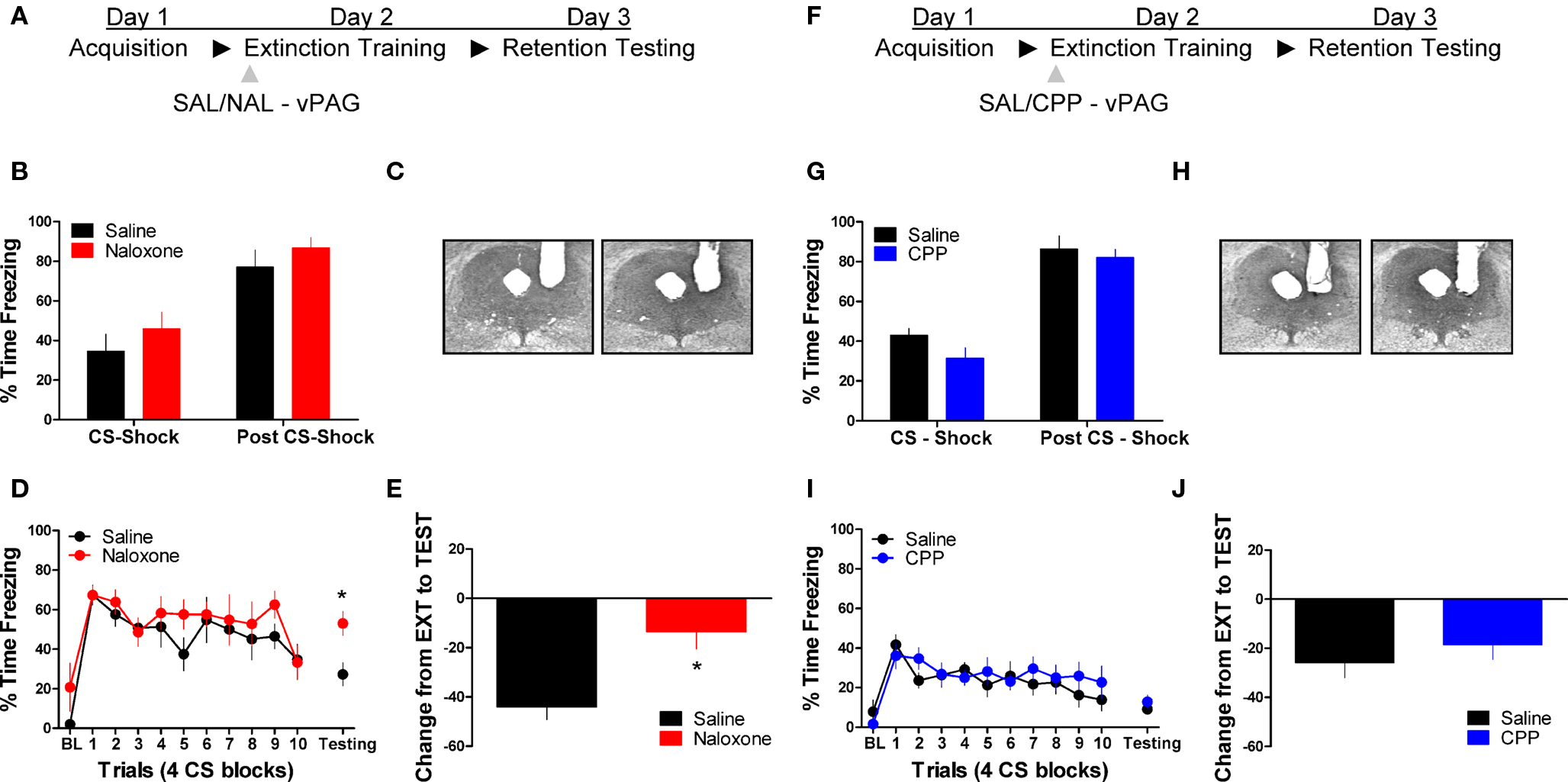
Figure 1. Opioid but not NMDA receptor blockade in the vlPAG disrupts memory for fear extinction. (A,F) A schematic depicting the behavioral procedure. (C,H) Photomicrographs showing gray scale Nissl stained images of representative cannulae placements into vlPAG for both experiments. (B,G) Freezing levels during CS-US (CS-shock) presentations and after CS-US (Post CS-shock) for the fear acquisition session in both experiments. (D) Freezing behavior during extinction training and the first four trials of the test for extinction retention in rats infused with saline (N = 6, black) or naloxone (N = 6, red). (E) Extinction retention reflected as a function of the decrease between extinction training and testing in these same rats. (I) Freezing during extinction training and retention testing in rats infused with CPP (N = 9, blue) or saline (N = 9, black), and (J) the change scores in the same animals. Graphs show the average time spent freezing (±SEM) to the CS extinction training and the first 4 trials of testing. Data were also analyzed by (E,J) computing the average change in freezing levels from extinction to test (*p < .0.05).
We also tested the effects of the NMDA receptor antagonist CPP on extinction memory when applied to the vlPAG. There were no differences during acquisition of fear conditioning before application of the drug (Figure 1G). During the extinction training session tests revealed no main effect of Drug or a Drug × Block interaction, but a significant effect of Block [F(1,16) = 9.20, p < 0.01]. Analysis of data from the extinction memory test session showed that CPP had no effect (t < 1.0) on freezing to the CS during the extinction memory test (Figure 1I, left side) or when change score values (Figure 1J) were assessed (t < 1.0). These data demonstrate that opioid receptors in the vlPAG are critical for the formation of extinction memory, whereas NMDA receptors are not involved.
NMDA but not Opioid Receptors in the Amygdala are Necessary for the Extinction of Fear Conditioning
Next we tested the effect of these same drug treatments when applied to the BLA prior to extinction training. No differences were seen between the different groups prior to drug treatment during acquisition (Figure 2B). An ANOVA on data from the extinction training session yielded a significant effect of Drug (2,17) = 3.75, p < 0.05) and Block [F(2,17) = 6.45, p < 0.05], but no Drug × Block interaction (Figure 2D, left side). Post hoc tests showed that the group treated with CPP showed more freezing than rats treated with saline (p < 0.05). As the graph shows, this difference seems to be driven by a disruption in within-session extinction in the CPP treated rats, as the difference emerges after the first block of trials. An ANOVA performed on data from the extinction memory test yielded no differences between groups during the first four trials of this session (F < 1.9), although the CPP treated rats showed a strong trend towards more freezing behavior at test. To explore this further, we performed an ANOVA on the change score data (Figure 2E) which yielded a significant group effect [F(2,17) = 3.742, p < 0.05]. Post hoc tests showed that the CPP treated group showed significantly less change compared to the SAL (p < 0.05) and NAL (p < 0.05) treated groups indicating that CPP treated rats had a disruption in memory for extinction.
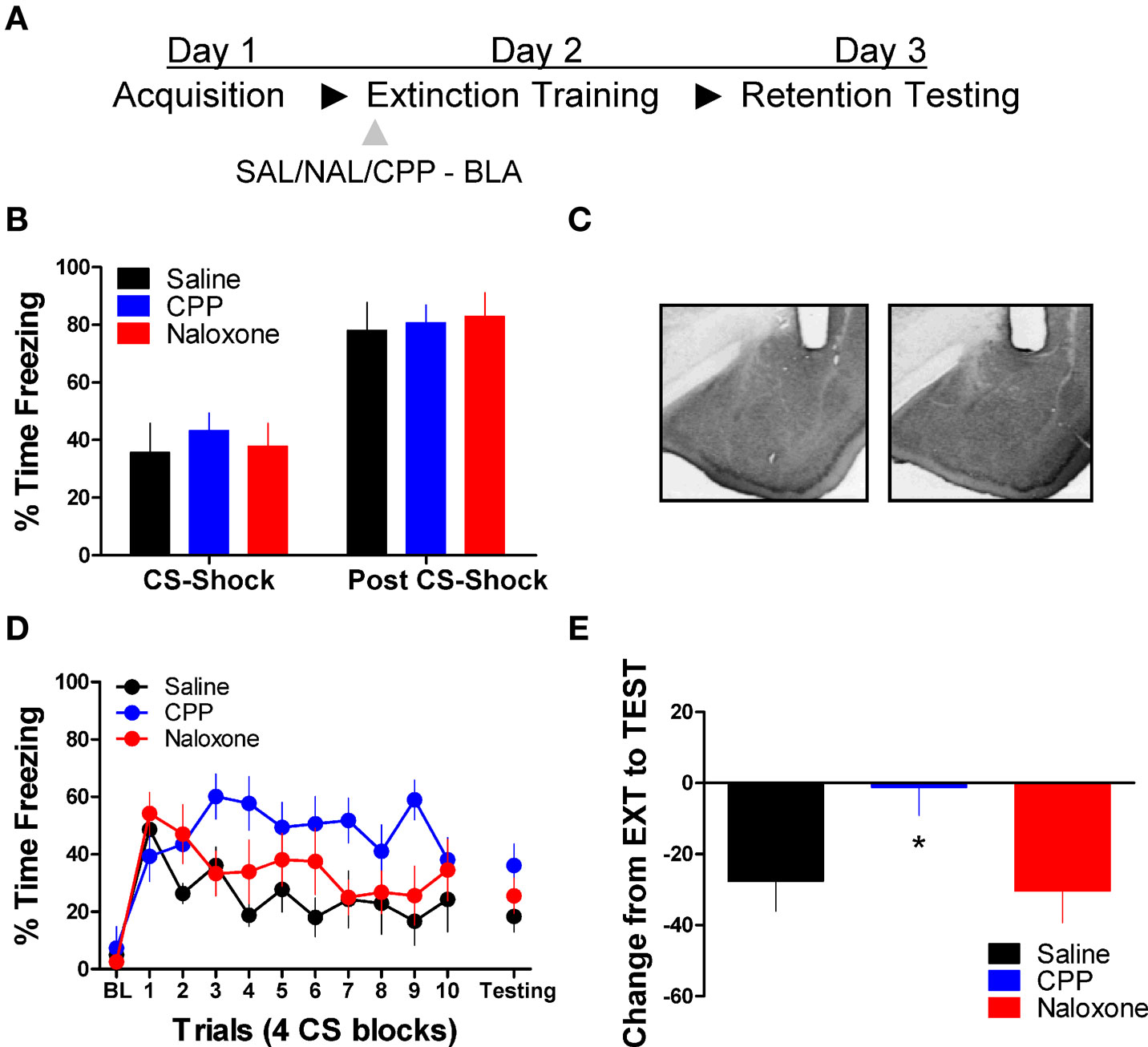
Figure 2. Blockade of NMDA but not opioid receptors in the amygdala disrupts memory for fear extinction. (A) A schematic showing the behavior procedures in this experiment. (B) Freezing behavior during the fear acquisition session in all three groups of rats. (C) Representative cannulae placements in the amygdala for two rats in this experiment. (D) Freezing behavior during the extinction training session and during a test for the retention of extinction in rats infused with saline (N = 6, black), CPP (N = 7, blue), or naloxone (N = 7, red). (E) Decrease in freezing from extinction to testing in the same set of rats (*p < 0.05).
Activation of ERK Signaling Following Extinction Training
The next experiment characterized the pattern of ERK/MAPK phosphorylation in the amygdala and mPFC following extinction training. The day after acquisition, rats were either exposed to 40 trials of extinction (40 EXT), 4 trials of extinction (4 EXT), or no trials of extinction (NO EXT). Most of the rats were killed 45 min after this session, although some rats were given an extinction memory test the following day (Figure 3A). Freezing during the first 4 extinction trials was compared between the 4 EXT and 40 EXT groups in both sets of rats. No differences were seen between groups on this measure (Figure 3B,C). Data from rats given the full extinction training were also subjected to a repeated measure ANOVA. In both the set of rats tested and those sacrificed after extinction training there was a significant effect of Block [F(1,5) = 11.68, p < 0.05, tested rats; F(1,9) = 10.60, p < 0.05, sacrificed rats] seemingly driven by the decrease in freezing seen over the course of extinction training. A one-way ANOVA on data from the behavioral test session (Figure 3B, right side) yielded a significant group effect [F(2,13) = 7.819, p < 0.01]. Post hoc tests confirmed that rats in the 40 EXT group showed less freezing compared to rats in both the 4 EXT (p < 0.01) and NO EXT (p < 0.01) groups, indicating that only the 40 EXT group showed evidence of extinction learning. These data provide the basis to understand the changes in ERK phosphorylation that occurred following extinction training.
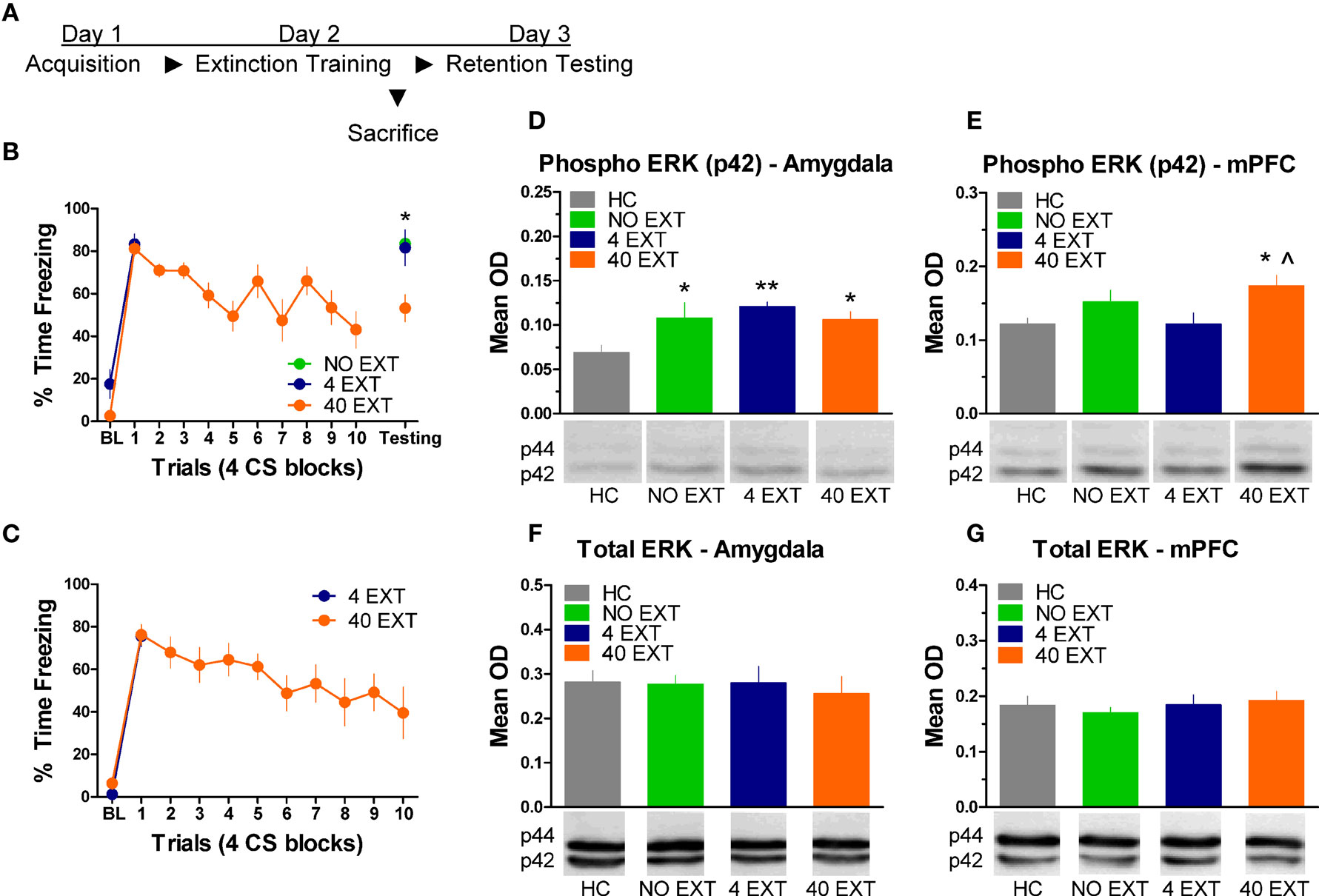
Figure 3. Extracellular-related kinase phosphorylation in the amygdala and mPFC following extinction training. (A) Schematic showing the behavioral procedures in this experiment. (B) Freezing during the CS for animals (N = 16) given extinction training and testing. (C) Freezing during the extinction training session for rats included in the western blot analysis. Expression of phosphorylated ERK in the amygdala (D) and mPFC (E) in rats given no extinction (N = 8, green), 4 trials of extinction (N = 7, blue) or 40 trials of extinction (N = 9, orange) compared to home cage rats (N = 8, gray). Levels of total ERK in the amygdala (F) and mPFC (G) in the same set of rats. The graphs show mean optical density (OD) values (±SEM) for each treatment group (*p < 0.05 relative to HC; **p < 0.01 relative to HC; ^p < 0.05 relative to 4 EXT).
Tissue samples from the amygdala and medial prefrontal cortex were processed for western blot. A one-way ANOVA on the mean optical density (OD) values from amygdala tissue (Figure 3D) yielded a significant group effect [F(3,29) = 3.535, p < 0.05]. Follow up post hoc tests showed that compared to home cage (HC) rats, animals exposed to 4 extinction trials (p < 0.01), 40 extinction trials (p < 0.05), or those rats given no extinction (p < 0.05) all showed increased p-ERK activity. The same membranes were exposed to an antibody that recognizes total ERK protein. There were no differences (F < 1.0) observed in the relative level of total ERK between groups (Figure 3F), indicating that the differences seen in p-ERK were not due to differences in loading the samples or in overall levels or ERK.
Samples from the mPFC (Figure 3E) in the same set of rats were also processed for western blot analysis. A one-way ANOVA on OD values from these data uncovered a significant effect between groups [F(3,29) = 3.347, p < 0.05]. Post hoc tests on the same data showed that rats given 40 EXT trials showed increased ERK phosphorylation compared to rats in the 4 EXT group (p < 0.01) and HC rats (p < 0.05). An ANOVA on the total ERK values yielded no significant differences (F < 1.0) between groups (Figure 3G). Collectively, our results argue that whereas ERK phosphorylation increased in the amygdala non-specifically, the increases observed in the mPFC were selective to rats exposed to a procedure that results in long-term memory for extinction.
Naloxone Delivered to the vlPAG Blocks Extinction-Related Increases in ERK Phosphorylation In The mPFC and Amygdala
The next experiment tested the effect of NAL delivered to the vlPAG (Figure 4C) prior to extinction training on the phosphorylation of ERK in the amygdala and mPFC (Figure 4A). An ANOVA revealed no significant difference in freezing behavior between groups during extinction training (Figure 4B). However, within-subject contrasts showed a significant effect of Block [F(1,14) = 15.75, p < 0.01) with no Block × Drug interaction indicating both groups showed a decrease in freezing behavior over the session. All rats were sacrificed 45 min following extinction training, except for the group of rats sacrificed from their home cage (HC). Levels of phosphorylated ERK were compared across the three groups using a one-way ANOVA. In the amygdala, there was a significant difference between groups [F(2,20) = 4.296, p < 0.05]. Post hoc tests showed that rats given SAL into the vlPAG showed increased ERK phosphorylation in the amygdala (Figure 4D) compared to HC rats (p < 0.05). Saline treated rats also had elevated pERK levels compared to those rats treated with NAL (p < 0.05), indicating that NAL prevented the extinction-induced activation of ERK. There was no difference between rats treated with NAL and HC rats, nor were there any differences (F < 1.0) in levels of total ERK in the amygdala (Figure 4F). A similar pattern of data emerged in the mPFC (Figure 4E). An ANOVA uncovered a significant difference in ERK phosphorylation between the three different groups [F(2,20) = 7.571, p < 0.01]. Post hoc t-tests confirmed that rats given SAL into the vlPAG and exposed to 40 extinction trials showed increased ERK phosphorylation compared to HC rats (p < 0.01) and rats given NAL prior to extinction training (p < 0.01) (Figure 4E). An ANOVA confirmed (F < 1.1) that levels of total ERK were unchanged between the three groups (Figure 4G). These data argue that opioid receptors in the vlPAG are involved in regulating plasticity in the mPFC and BLA related to the extinction of fear conditioning.
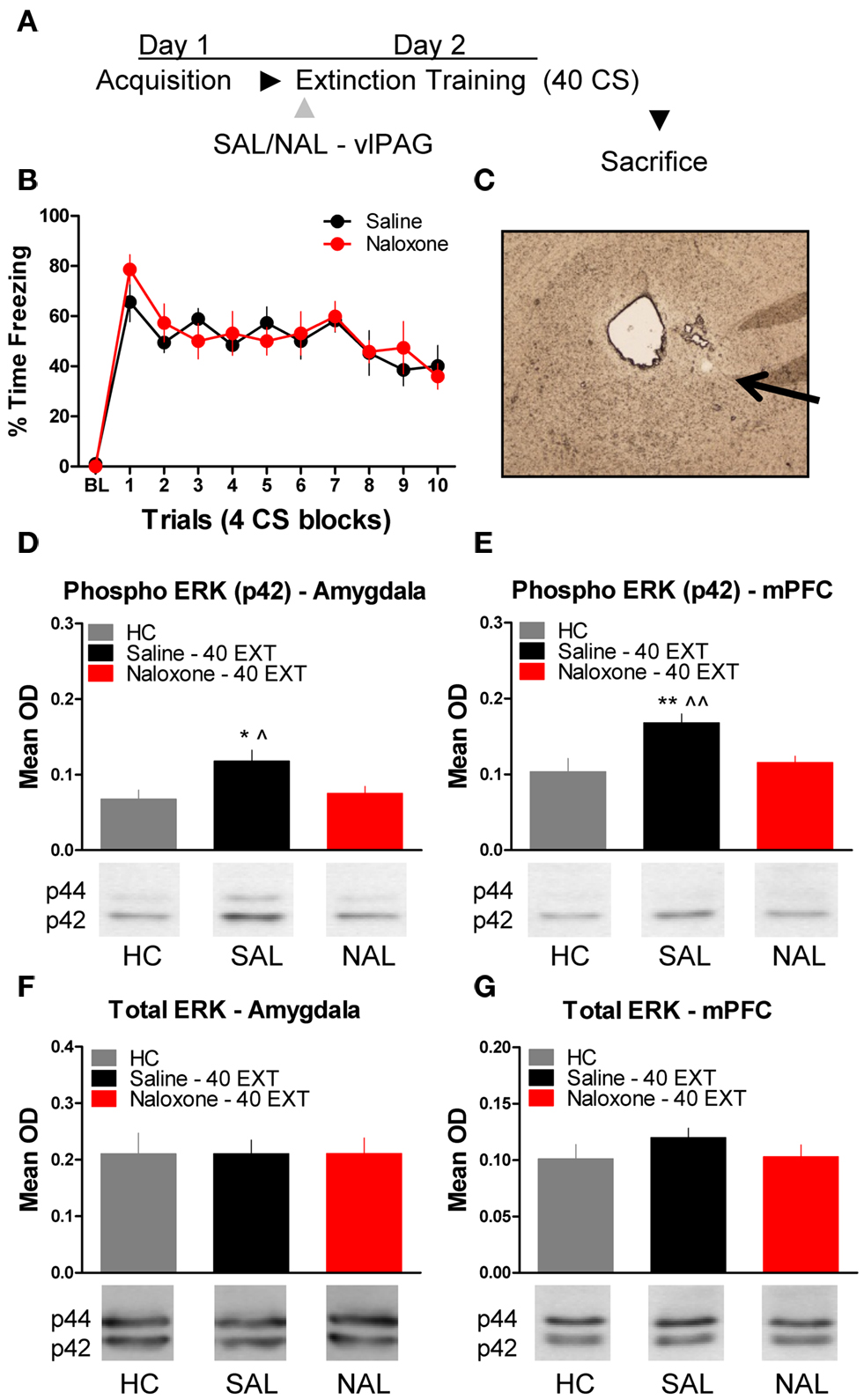
Figure 4. Extracellular-related kinase phosphorylation following extinction training is blocked by opioid receptor antagonism in the vlPAG. (A) Schematic showing the behavioral procedures used in this experiment. (B) Freezing to the CS during the extinction training session. (C) Representative image taken using Nomarski optics at 10×. Arrow indicates the approximate infusion site in the vlPAG. Expression of phosphorylated ERK in the amygdala (D) and mPFC (E) of rats infused with saline (N = 9, black) or naloxone (N = 9) and given extinction training. HC rats (N = 6, gray) were trained, but sacrificed from their home cage. Total ERK levels in the amygdala (F) and mPFC (G) in the same rats. (*p < 0.05 relative to HC; **p < 0.01 relative to HC; ^p < 0.05 relative to Naloxone 40 EXT; ^^p < 0.01 relative to Naloxone 40 EXT).
To determine whether or not the disruption in ERK phosphorylation by opioid receptor blockade in the vlPAG was specific to extinction learning, we infused rats with SAL or NAL into the vlPAG and exposed them to only four extinction training trials. This experiment allowed us to determine if the decrease in ERK phosphorylation in the amygdala and mPFC seen in the previous experiment following NAL infusion is specific to a procedure that results in the formation of extinction memory or if NAL decreases ERK phosphorylation related to retrieval or reconsolidation of memory. Rats received infusions of SAL or NAL into the vlPAG (Figure 5C) prior to four trials of extinction training (Figure 5A). Both groups of rats in this experiment showed normal levels of freezing to the CS during the extinction training session (Figure 5B). ERK phosphorylation was assessed in the amygdala and mPFC 45 min after the extinction training session. t-Tests comparing the two groups indicate that ERK phosphorylation did not change in either the amygdala (t < 1.0) or mPFC (t < 1.0) following NAL infusion and extinction training with four trials (Figure 5D,E). These data indicate that NAL does not affect ERK phosphorylation using a procedure that results in negligible extinction learning.
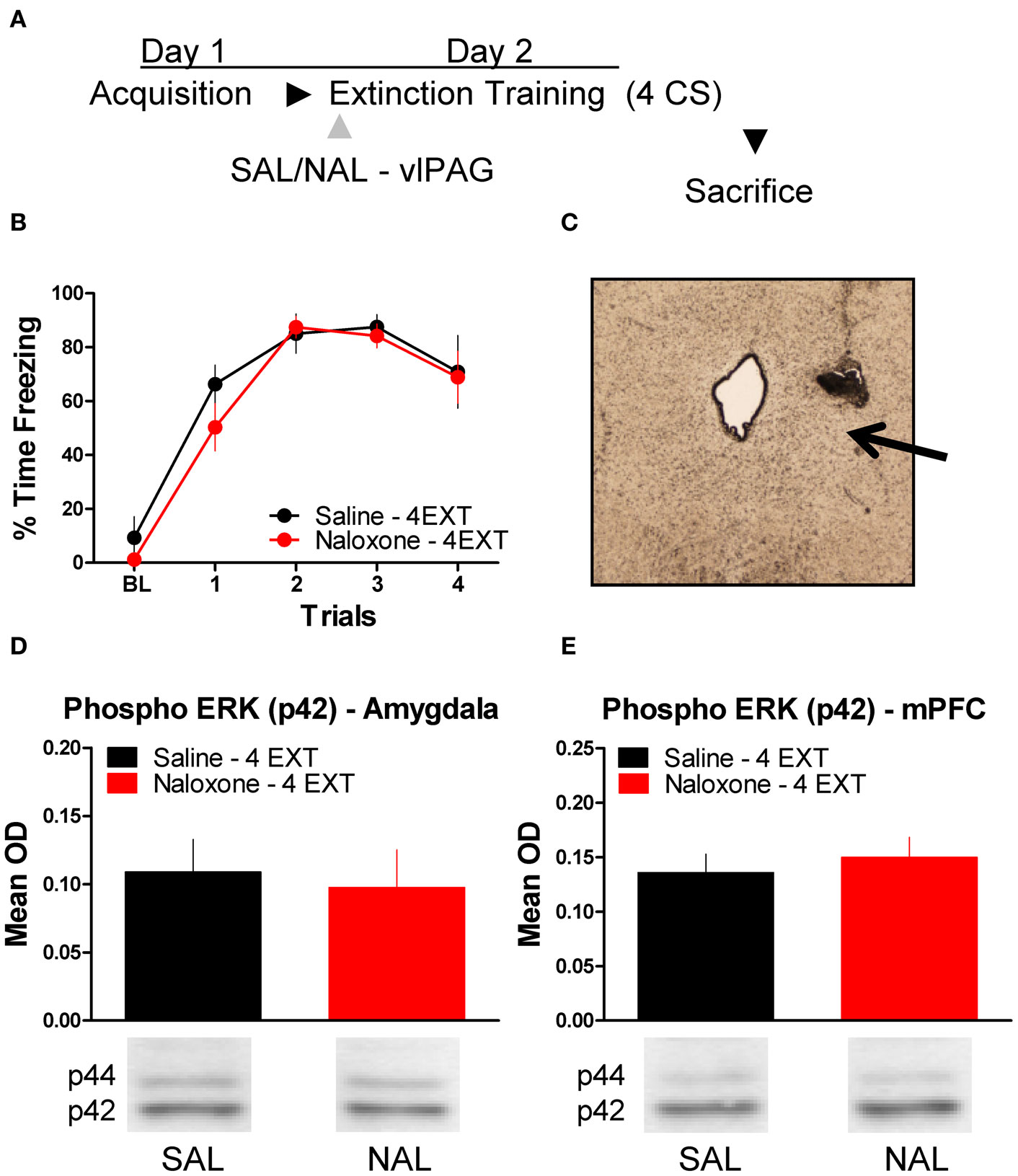
Figure 5. Opioid receptor blockade does not affect ERK phosphorylation following four trials of extinction. (A) Schematic showing the behavioral procedures employed in this experiment. (B) Freezing during the four trials of extinction training. (C) Representative image taken using Nomarski optics at 10×. Arrow indicates the approximate infusion site in the vlPAG. Expression of phosphorylated ERK in the amygdala (D) and mPFC (E) for rats infused with saline (N = 6, black) or naloxone (N = 6, red) and given four trials of extinction training.
CPP Delivered to the Amygdala does not Affect ERK Phosphorylation in mPFC Following Extinction Training
Finally, we tested whether or not ERK phosphorylation in the mPFC is disrupted by treatment with drugs which prevent the formation of extinction memory when given into the amygdala (Figure 2). A mixed-factor ANOVA was used to test for differences during the extinction training session, with Drug as a between-subject factor and Block as a within-subject comparison. This analysis found no significant effect of Drug, but there was as significant effect of Block [F(1,14) = 7.13, p < 0.05], and a significant Drug × Block interaction [F(1,14) = 4.84, p < 0.05] (Figure 6B). The significant interaction is indicative of a disruption in within-session extinction in the rats treated with CPP. ERK phosphorylation in these animals was examined in the mPFC 45 min later. t-Tests showed that ERK phosphorylation did not change (t < 1.0) in the mPFC following NMDA receptor blockade in the amygdala (Figure 6C). Although CPP delivered to the BLA disrupted extinction learning both within (Figures 2 and 6) and across session (Figure 2), blocking NMDA receptors had no affect on ERK phosphorylation in the mPFC.
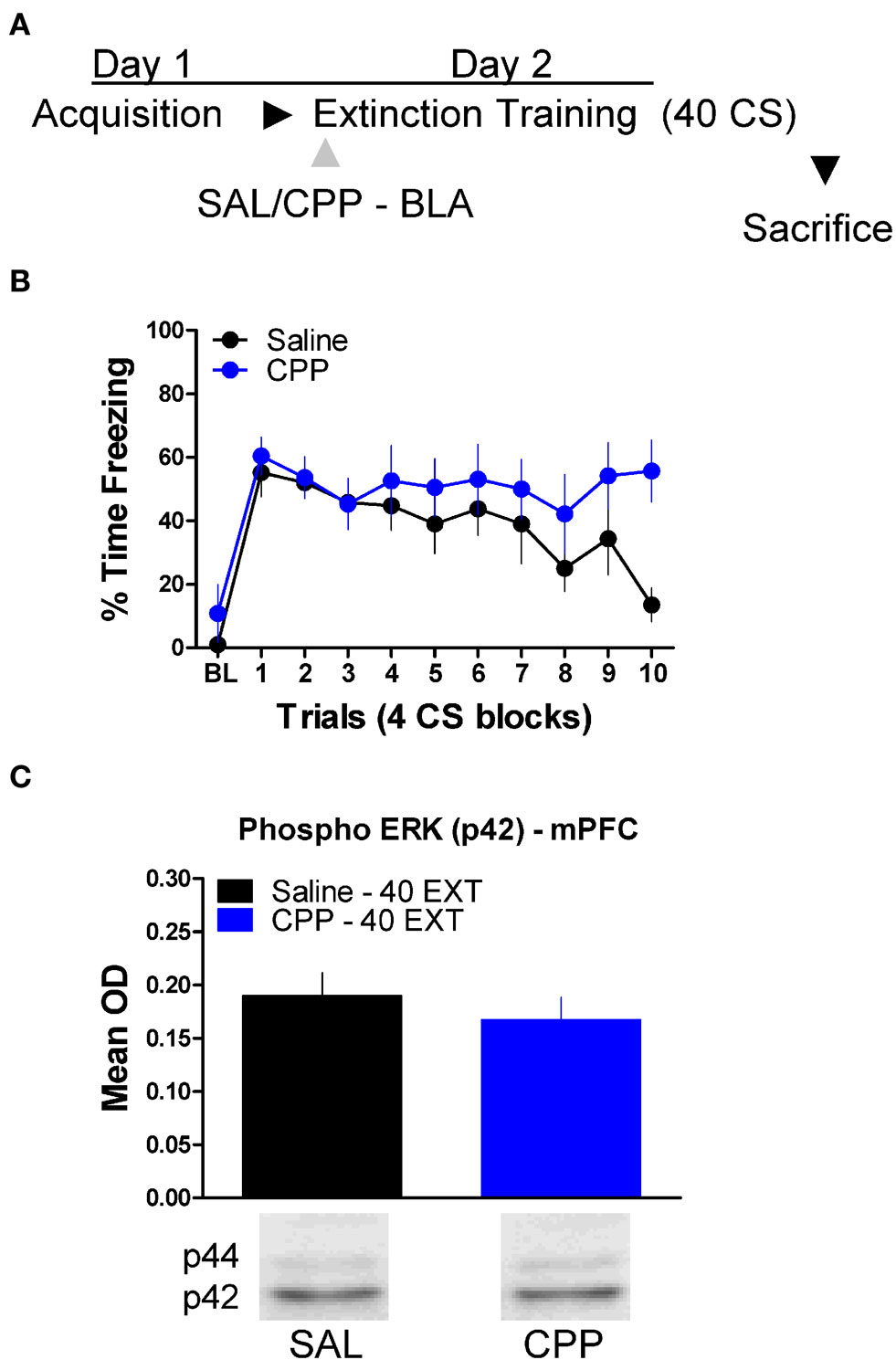
Figure 6. N-methyl-D-aspartic acid receptor blockade in the amygdala has no effect on ERK phosphorylation in the mPFC related to extinction training. (A) Schematic showing the behavioral procedures employed in this experiment. (B) Freezing to the CS during the 40 trials of extinction training. Expression of phosphorylated ERK in the amygdala mPFC (C) of rats infused with saline (N = 8, black) or CPP (N = 8, blue) into the amygdala and given 40 trials of extinction training.
Discussion
This study provides a critical step towards understanding how the brain signals extinction learning. Insight into how the neural system underlying extinction interacts is critical not only for a basic understanding of the mechanisms supporting this form of learning, but knowledge from studies of fear extinction are potentially useful towards understanding and treating various anxiety disorders and phobias. Previous work has indicated that the amygdala, mPFC, and vlPAG are all involved in fear extinction, but our results indicate that the vlPAG can regulate the function of the mPFC and amygdala as it relates to fear extinction and that it specifically involves the action of the opioid system in the vlPAG.
Consistent with prior published work (McNally et al., 2004) we showed that opioid receptor blockade in the vlPAG prevents the retention of extinction, although blocking the activity of NMDA receptors had no effect. Importantly, our work showed that opioid receptor blockade in the vlPAG also prevented increases in ERK phosphorylation in the mPFC and amygdala after extinction training. We also tested the effect of NMDA and opioid receptor blockade in the amygdala on the retention of extinction memory. Supporting prior work we showed that NMDA receptor activity in the amygdala disrupted the formation of extinction memory (see also, Falls et al., 1992; Walker et al., 2002; Sotres-Bayon et al., 2007), however opioid receptor blockade had no effect. Again, we tested whether or not CPP applied to the amygdala would affect activity in the mPFC related to the consolidation of extinction memory. Despite the fact that CPP disrupted extinction both within and between sessions, it did not affect ERK phosphorylation in the mPFC.
The ERK signaling pathway is critical for neuronal changes supporting long-term memory (e.g., Atkins et al., 1998), including memory for the extinction of fear conditioning (Lu et al., 2001; Hugues et al., 2004). Therefore, it might initially be surprising in the current study that phosphorylation of ERK in the amygdala was not specific to behavioral manipulations that result in the formation of extinction memory. We observed that exposure to four extinction trials significantly increased ERK phosphorylation, even though this manipulation did not result in any extinction. It is likely that this increased expression of ERK is related to the retrieval and subsequent reconsolidation of the original memory for training. Supporting this idea, ERK inhibitors delivered to the amygdala around the time of retrieval disrupt memory reconsolidation (Duvarci et al., 2005). We also observed an increase in ERK phosphorylation in rats simply exposed to the extinction context, which might be related to generalized fear to the chamber. Because ERK activity is not simply related to one aspect of learning or memory, we tested the effect of NAL in the vlPAG on ERK phosphorylation driven by four presentations of the CS. This experiment was critical, because it allowed us to test whether the influence of the vlPAG on the amygdala and mPFC is evident only when an extinction memory is formed, or if opioid receptor blockade in the vlPAG affects ERK phosphorylation related to retrieval and/or reconsolidation of memory. The data clearly show that NAL has no effect on ERK phosphorylation driven by four exposures to the CS, strongly suggesting the amygdala and mPFC only come under the influence of the vlPAG when significant extinction learning occurs.
Significantly elevated pERK levels in the mPFC were specific to the group that received 40 extinction trials. These results are very similar to previous data showing that FOS protein expression increases in the mPFC following extinction training (Santini et al., 2004), and recent results examining CREB activity in the mPFC following extinction training of different lengths (Mamiya et al., 2009). Our results are also generally in agreement with the recent study (Kim et al., 2009) showing that activation of ERK is seen in both the prelimbic and infralimbic regions of the mPFC following 30 extinction trials, but not after 6 trials. Thus, even though recording studies (e.g., Milad and Quirk, 2002) have shown functional heterogeneity in the mPFC as it relates to extinction learning, these expression studies have not. Therefore, it is unlikely that our decision to analyze mPFC significantly changed our findings. The observation that increased pERK activity was not seen in rats exposed to four extinction trials rules out the possibility that activity in this area is simply driven by the performance of freezing behavior or recall of the acquisition memory (see also, Kim et al., 2009). Consistent with the idea that ERK activity in mPFC is necessary to the consolidation of extinction memory, previous work has shown that ERK inhibitors in the mPFC disrupt the consolidation of extinction memory (Hugues et al., 2004, 2006).
The effects of NAL on fear extinction and the phosphorylation of ERK might reflect a disruption in reacting to changes in expectancy during learning (McNally and Cole, 2006). Other studies have demonstrated that NAL injected systemically immediately following fear extinction has no effect on subsequent retention, ruling out a direct effect of the drug on consolidation processes (McNally and Westbrook, 2003; Kim and Richardson, 2009). The fact that NAL must be on board during extinction training to be effective supports the idea that it is affecting the learning process, but it also raises issues with interpreting the data because drugs delivered into the vlPAG have the potential to affect freezing behavior. However, other studies have shown that opioid antagonists delivered to the vlPAG before testing do not simply enhance freezing behavior (Helmstetter and Landeira-Fernandez, 1990) and there was no evidence in any of the current experiments that NAL affected freezing behavior. These observations rule out the possibility that the effects of NAL on ERK activity and extinction were the result of an acute effect on freezing.
A challenge for our findings is to describe the anatomical and physiological substrates that would allow the vlPAG to regulate the mPFC and the amygdala in such a manner. The study of the vlPAG has traditionally been framed by its identification as a structure that mediates select fear responses and regulates nociception via descending connections to the rostral ventral medulla (Basbaum and Fields, 1984; Fendt and Fanselow, 1999). However, there is evidence demonstrating that the vlPAG sends projections into forebrain areas including central nucleus of the amygdala (Rizvi et al., 1991). It is possible that the regulation of ERK activity in the amygdala by the vlPAG is achieved via this vlPAG-central nucleus connection. However, we feel it is more likely that vlPAG is influencing mPFC and amygdala through other neural pathways. There is considerable evidence that the vlPAG sends especially dense projections to several medial thalamic nuclei including the central lateral and central medial nuclei, mediodorsal, and reuniens nucleus (Hoover and Vertes, 2007). These thalamic areas in turn project to mPFC regions important for fear extinction learning (Krout and Loewy, 2000). In addition to these indirect connections, at least one paper has shown that vlPAG projection neurons may directly reach areas in the mPFC (Lu et al., 2006). Therefore, the changes we observed in ERK expression in the mPFC as a result of NAL administration could be mediated by the direct or indirect connections from the vlPAG to mPFC.
Another challenge is to explain how opioid receptor blockade in the vlPAG might regulate neural activity in such a way that it prevents ERK activity in the amygdala and mPFC. Several studies have shown that one of the predominant effects of opioids applied to the vlPAG is the inhibition of tonically active GABAergic interneurons (Basbaum and Fields, 1984; Vaughan et al., 1997). When opioids are released, these interneurons are inhibited and this results in disinhibition of output neurons in the vlPAG. Blocking these actions with NAL or similar compounds antagonizes the effects opioids have in the vlPAG. Most of the data which supports this model centers on how the vlPAG mediates nociception through its effects on the rostral ventral medulla. However, if the vlPAG is influencing the amygdala or mPFC through a similar mechanism during fear extinction learning it readily explains how opioid receptors in the vlPAG regulate activity in these structures.
Finally, we propose a model (Figure 7) meant to account for the findings of the current study and synthesize them with prior published work. According to our model, opioids are released in the vlPAG when the animals are exhibiting fear to the CS during the early phases of extinction. As the animals keep responding during extinction training the cumulative effect of opioid release is a disinhibition of vlPAG output neurons, including those connected directly or indirectly to the mPFC. The net result of this is in an increase in activity in the mPFC. Other groups have described in detail the effects mPFC can have on the amygdala, in particular the physiology of this circuit as it relates to extinction learning, and our model incorporates these (Royer and Pare, 2002; Quirk et al., 2003; Likthtik et al., 2008). Briefly, excitation of mPFC increases activity of excitatory projection neurons from the mPFC to GABAergic intercalated cells in the amygdala. Accordingly, an increase in these inhibitory neurons dampens the output of the central amygdala, which is responsible for driving the various fear responses. NMDA-dependent processes occur in the amygdala allowing for these changes. When opioid receptors are blocked in the vlPAG, the mPFC does not receive the appropriate signals that allow it to decrease the activity of the amygdala. In addition to accounting for the results of the current and many published studies, a number of predictions are derived from this model. Any number of the other changes in the mPFC and amygdala related to extinction learning or the consolidation of extinction should be blocked by naloxone infusions into the vlPAG. For example, bursting of mPFC neurons associated with consolidation of extinction learning (Burgos-Robles et al., 2007) should be disrupted by NAL in vlPAG. Furthermore, any change in the excitability of the intercalated cells of the amygdala should also be interrupted by vlPAG opioid receptor blockade. Finally, given the effects of opioid receptor antagonists in this study and others, agonists of this receptor applied to the vlPAG should augment extinction learning by enhancing the activity of mPFC inputs into the amygdala. Future experiments should provide answers to these and other important questions.
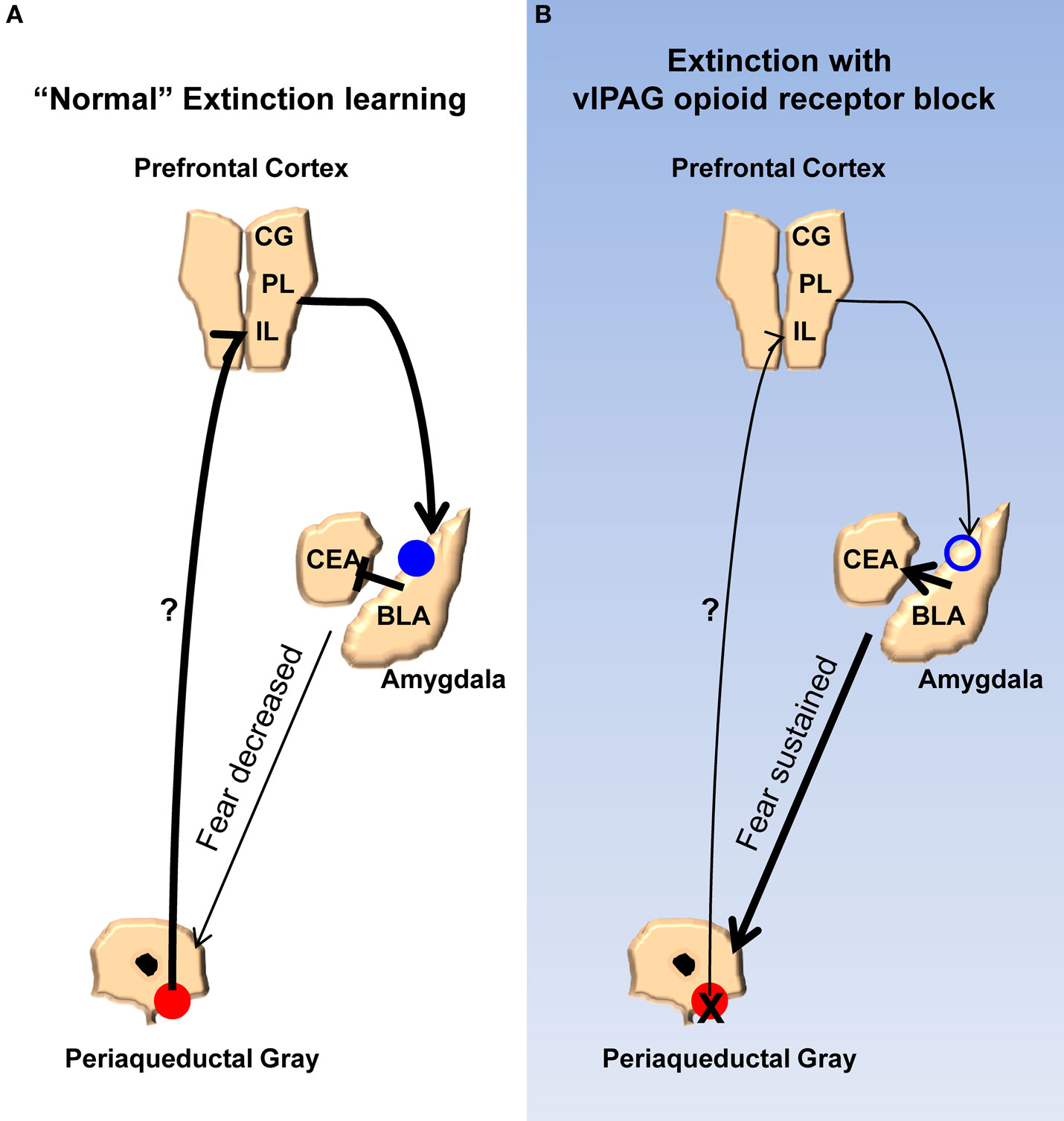
Figure 7. Model for the extinction of fear memories. (A) Diagram showing the activity of the ventrolateral PAG, medial prefrontal cortex, and amygdala during the extinction of fear memory. Activity of opioid receptors in the vlPAG (red circles) causes an increase in activity in the mPFC. Augmented excitability of the mPFC drives NMDA-mediated plasticity (blue circles) in the basolateral nucleus and/or GABAergic intercalated cells (ITC) of the amygdala. Enhancement of BLA/ITC activity results in an inhibition of the central nucleus of the amygdala and a decreased ability to produce fear responses. (B) When opioid receptors are blocked during extinction learning there is no change in excitability of the mPFC or the BLA/ITC, the output of the central nucleus is not affected, and fear responses are sustained.
Conflict of Interest Statement
The authors declare that research in this manuscript was conducted without any commercial or financial interests that could be interpreted as a conflict of interest.
Acknowledgments
This work was supported by grants from the National Institutes of Health (MH069558 to Fred J. Helmstetter) and a fellowship from the University of Wisconsin-Milwaukee Graduate School (to Ryan G. Parsons). The authors would also like to thank Tim Jarome and Janine Kwapis for technical assistance.
References
Atkins, C. M., Selcher, J. C., Petraitis, J. J., Trzaskos, J. M., and Sweatt, J. D. (1998). The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1, 602–609.
Basbaum, A. I., and Fields, H. L. (1984). Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu. Rev. Neurosci. 7, 309–338.
Bouton, M. E., and King, D. A. (1983). Contextual control of the extinction of conditioned fear: tests for the associative value of the context. J. Exp. Psychol. Anim. Behav. Process 9, 248–265.
Burgos-Robles, A., Vidal-Gonzalez, I., Santini, E., and Quirk, G. J. (2007). Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron 53, 871–880.
Cole, S., and McNally, G. P. (2007a). Opioid receptors mediate direct predictive fear learning: evidence from one-trial blocking. Learn. Mem. 14, 229–235.
Cole, S., and McNally, G. P. (2007b). Temporal difference prediction errors and pavlovian fear conditioning: role of NMDA and opioid receptors. Behav. Neurosci. 121, 1043–1052.
Duvarci, S., Nader, K., and Ledoux, J. E. (2005). Activation of extracellular signal-regulated kinase-mitogen-activated protein kinase cascade in the amygdala is required for memory reconsolidation of auditory fear conditioning. Eur. J. Neurosci. 21, 283–289.
Falls, W. A., Miserendino, M. J., and Davis, M. (1992). Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J. Neurosci. 12, 854–863.
Fanselow, M. S. (1998). Pavlovian conditioning, negative feedback, and blocking: mechanisms that regulate association formation. Neuron 20, 625–627.
Fanselow, M. S., and Bolles, R. C. (1979). Naloxone and shock-elicited freezing in the rat. J. Comp. Physiol. Psychol. 93, 736–744.
Fendt, M., and Fanselow, M. S. (1999). The neuroanatomical and neurochemical basis of conditioned fear. Neurosci. Biobehav. Rev. 23, 743–760.
Hammer, G. D., and Kapp, B. S. (1986). The effects of naloxone administered into the periaqueductal gray on shock-elicited freezing behavior in the rat. Behav. Neural Biol. 46, 189–195.
Helmstetter, F. J. (1992). Contribution of the amygdala to learning and performance of conditional fear. Physiol. Behav. 51, 1271–1276.
Helmstetter, F. J., and Landeira-Fernandez, J. (1990). Conditional hypoalgesia is attenuated by naltrexone applied to the periaqueductal gray. Brain Res. 537, 88–92.
Herry, C., Trifilieff, P., Micheau, J., Luthi, A., and Mons, N. (2006). Extinction of auditory fear conditioning requires MAPK/ERK activation in the basolateral amygdala. Eur. J. Neurosci. 24, 261–269.
Hoover, W. B., and Vertes, R. P. (2007). Anatomical analysis of afferent projections to the medial prefrontal cortex in the rat. Brain Struct. Funct. 212, 149–179.
Hugues, S., Chessel, A., Lena, I., Marsault, R., and Garcia, R. (2006). Prefrontal infusion of PD098059 immediately after fear extinction training blocks extinction-associated prefrontal synaptic plasticity and decreases prefrontal ERK2 phosphorylation. Synapse 60, 280–287.
Hugues, S., Deschaux, O., and Garcia, R. (2004). Postextinction infusion of a mitogen-activated protein kinase inhibitor into the medial prefrontal cortex impairs memory of the extinction of conditioned fear. Learn. Mem. 11, 540–543.
Kim, J. H., Hamlin, A. S., and Richardson, R. (2009). Fear extinction across development: the involvement of the medial prefrontal cortex as assessed by temporary inactivation and immunohistochemistry. J. Neurosci. 29, 10802–10808.
Kim, J. H., and Richardson, R. (2009). The effect of the mu-opioid receptor antagonist naloxone on extinction of conditioned fear in the developing rat. Learn. Mem. 16, 161–166.
Kim, J. J., Rison, R. A., and Fanselow, M. S. (1993). Effects of amygdala, hippocampus, and periaqueductal gray lesions on short- and long-term contextual fear. Behav. Neurosci. 107, 1093–1098.
Krout, K. E., and Loewy, A. D. (2000). Periaqueductal gray matter projections to midline and intralaminar thalamic nuclei of the rat. J. Comp. Neurol. 424, 111–141.
Lu, J., Jhou, T. C., and Saper, C. B. (2006). Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J. Neurosci. 26, 193–202.
Lu, K. T., Walker, D. L., and Davis, M. (2001). Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J. Neurosci. 21, RC162.
Likthtik, E., Popa, D., Apergis-Schoute, J., Fidacaro, G. A., and Paré, D. (2008). Amygdala intercalated neurons are required for expression of fear extinction. Nature 454, 642–646.
Mamiya, N., Fukushima, H., Suzuki, A., Matsuyama, Z., Homma, S., Frankland, P. W., and Kida, S. (2009). Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J. Neurosci. 29, 402–413.
McNally, G. P., and Cole, S. (2006). Opioid receptors in the midbrain periaqueductal gray regulate prediction errors during pavlovian fear conditioning. Behav. Neurosci. 120, 313–323.
McNally, G. P., Lee, B. W., Chiem, J. Y., and Choi, E. A. (2005). The midbrain periaqueductal gray and fear extinction: opioid receptor subtype and roles of cyclic AMP, protein kinase A, and mitogen-activated protein kinase. Behav. Neurosci. 119, 1023–1033.
McNally, G. P., Pigg, M., and Weidemann, G. (2004). Opioid receptors in the midbrain periaqueductal gray regulate extinction of pavlovian fear conditioning. J. Neurosci. 24, 6912–6919.
McNally, G. P., and Westbrook, R. F. (2003). Opioid receptors regulate the extinction of Pavlovian fear conditioning. Behav. Neurosci. 117, 1292–1301.
Milad, M. R., and Quirk, G. J. (2002). Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420, 70–74.
Parsons, R. G., Riedner, B. A., Gafford, G. M., and Helmstetter, F. J. (2006). The formation of auditory fear memory requires the synthesis of protein and mRNA in the auditory thalamus. Neuroscience 141, 1163–1170.
Paxinos, G., and Watson, C. (1998). The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press.
Quirk, G. J., Likhtik, E., Pelletier, J. G., and Pare, D. (2003). Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J. Neurosci. 23, 8800–8807.
Rescorla, R. A., and Heth, C. D. (1975). Reinstatement of fear to an extinguished conditioned stimulus. J. Exp. Psychol. Anim. Behav. Process 1, 88–96.
Rizvi, T. A., Ennis, M., Behbehani, M. M., and Shipley, M. T. (1991). Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: topography and reciprocity. J. Comp. Neurol. 303, 121–131.
Royer, S., and Pare, D. (2002). Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 115, 455–462.
Santini, E., Ge, H., Ren, K., Pena d. O., and Quirk, G. J. (2004). Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J. Neurosci. 24, 5704–5710.
Sotres-Bayon, F., Bush, D. E., and Ledoux, J. E. (2007). Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology 32, 1929–1940.
Vaughan, C. W., Ingram, S. L., Connor, M. A., and Christie, M. J. (1997). How opioids inhibit GABA-mediated neurotransmission. Nature 390, 611–614.
Keywords: fear extinction, opioid receptors, amygdala, medial prefrontal cortex, ventrolateral periaqueductal gray matter, extracellular signal-related kinase
Citation: Parsons RG, Gafford GM and Helmstetter FJ (2010) Regulation of extinction-related plasticity by opioid receptors in the ventrolateral periaqueductal gray matter. Front. Behav. Neurosci. 4:44. doi: 10.3389/fnbeh.2010.00044
Received: 30 March 2010;
Paper pending published: 24 April 2010;
Accepted: 26 June 2010;
Published online: 03 August 2010
Edited by:
Jeansok J. Kim, University of Washington, USAReviewed by:
Gregory Quirk, University of Puerto Rico, USAHongjoo J. Lee, University of Texas, USA
Copyright: © 2010 Parsons, Gafford, Helmstetter. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Fred J. Helmstetter, Department of Psychology, University of Wisconsin-Milwaukee, P.O. Box 413, Milwaukee, WI 53201, USA. e-mail:ZmpoQHV3bS5lZHU=