 Davide Silingardi1†
Davide Silingardi1† Andrea Angelucci1† Roberto De Pasquale1† Marco Borsotti2 Giovanni Squitieri2
Andrea Angelucci1† Roberto De Pasquale1† Marco Borsotti2 Giovanni Squitieri2 Riccardo Brambilla3 Elena Putignano1 Tommaso Pizzorusso1,2†
Riccardo Brambilla3 Elena Putignano1 Tommaso Pizzorusso1,2† Nicoletta Berardi1,2*†
Nicoletta Berardi1,2*†- 1 Istituto di Neuroscienze del CNR, Pisa, Italy
- 2 Dipartimento di Psicologia, Università di Firenze, Firenze, Italy
- 3 Università Vita e Salute San Raffaele, Milano, Italy
ERK 1,2 pathway mediates experience-dependent gene transcription in neurons and several studies have identified its pivotal role in experience-dependent synaptic plasticity and in forms of long term memory involving hippocampus, amygdala, or striatum. The perirhinal cortex (PRHC) plays an essential role in familiarity-based object recognition memory. It is still unknown whether ERK activation in PRHC is necessary for recognition memory consolidation. Most important, it is unknown whether by modulating the gain of the ERK pathway it is possible to bidirectionally affect visual recognition memory and PRHC synaptic plasticity. We have first pharmacologically blocked ERK activation in the PRHC of adult mice and found that this was sufficient to impair long term recognition memory in a familiarity-based task, the object recognition task (ORT). We have then tested performance in the ORT in Ras-GRF1 knock-out (KO) mice, which exhibit a reduced activation of ERK by neuronal activity, and in ERK1 KO mice, which have an increased activation of ERK2 and exhibit enhanced striatal plasticity and striatal mediated memory. We found that Ras-GRF1 KO mice have normal short term memory but display a long term memory deficit; memory reconsolidation is also impaired. On the contrary, ERK1 KO mice exhibit a better performance than WT mice at 72 h retention interval, suggesting a longer lasting recognition memory. In parallel with behavioral data, LTD was strongly reduced and LTP was significantly smaller in PRHC slices from Ras-GRF1 KO than in WT mice while enhanced LTP and LTD were found in PRHC slices from ERK1 KO mice.
Introduction
Activation of synapse to nucleus signaling and regulation of gene transcription have been found to be crucial both for long term synaptic plasticity and memory consolidation (Davis and Squire, 1984; Mayford and Kandel, 1999; McGaugh, 2000; Abel and Lattal, 2001).
The ERK 1,2 (also called p42/44 mitogen-activated protein kinase) pathway mediates experience-dependent gene transcription and translational processes in neurons and several studies have identified its pivotal role in experience-dependent synaptic plasticity and in LTM consolidation involving cortex, hippocampus, amygdala, or striatum (Brambilla et al., 1997; Atkins et al., 1998; Blum et al., 1999; Mazzucchelli and Brambilla, 2000; Schafe et al., 2000; Di Cristo et al., 2001; Adams and Sweatt, 2002; Mazzucchelli et al., 2002; Cancedda et al., 2003; Kelleher et al., 2004; Thomas and Huganir, 2004; Doyère et al., 2007; Tsokas et al., 2007; Cohen-Matsliah et al., 2008; Leon et al., 2010; Lin et al., 2010; Dupont et al., 2011).
Many evidences indicate that the perirhinal cortex (PRHC) plays an essential role in familiarity-based object recognition (Suzuki et al., 1993; Zhu et al., 1995; Brown and Xiang, 1998; Murray and Bussey, 1999; Wan et al., 1999; Brown and Aggleton, 2001; Murray et al., 2007) and PRHC long term synaptic plasticity has been implicated in recognition memory (Warburton et al., 2003; Barker et al., 2006; Griffiths et al., 2008; Massey et al., 2008; Seoane et al., 2009).
Involvement of ERK pathway in recognition memory consolidation and reconsolidation has been suggested by two studies employing intraventricular or systemic administration of blockers of ERK1,2 activation (Kelly et al., 2003; Goeldner et al., 2008). It is still unknown whether ERK activation in PRHC is necessary for recognition memory consolidation and whether activation of the ERK pathway affects PRHC synaptic plasticity. Most important, it is unknown whether by modulating the gain of the ERK pathway it is possible to bidirectionally affect recognition memory and PRHC synaptic plasticity, not only causing an impairment by decreasing the gain, but also causing an enhancement by increasing it.
To assess the role of ERK activation in PRHC for recognition memory consolidation we performed a pharmacological block of ERK activation in the PRHC of adult mice and tested long term recognition memory in the object recognition task (ORT).
To assess the possibility that ERK can bidirectionally regulate visual recognition memory and PRHC plasticity we employed two mutant mouse lines. The first, Ras-GRF1 knock-out (KO) mice, has genetic deletion of Ras-GRF1, a guanine exchange factor which catalyzes the exchange of GDP for GTP on Ras, resulting in a reduced activation of ERK by neuronal activity (Brambilla et al., 1997; Fasano et al., 2009). The use of these mice allows to assess both the role of Ras-GRF1 signaling and the effects of reducing ERK activation in visual recognition memory and PRHC synaptic plasticity. The second, ERK1 KO mice (Mazzucchelli et al., 2002), has an increased activation of ERK2 in response to glutamate and exhibit enhanced striatal plasticity and striatal mediated memory. The use of these mice allows to assess both the relative role of ERK 1 and ERK 2 and the effects of increasing ERK 2 activation in visual recognition memory and in PRHC synaptic plasticity.
Materials and Methods
Animals
All mice used were aged between 3 and 6 months of age and were born and housed in our animal house. Lighting conditions were12 h light:12 h dark; animals were housed in standard laboratory cages, groups from five to seven animals per cage, according to the cage size and the animal house regulations. Housing and all experiments were performed in accordance with the Italian Ministry of Health guidelines for care and use of laboratory animals.
We have backcrossed both ERK1 (Mazzucchelli et al., 2002) and Ras-GRF1 KO (Brambilla et al., 1997) mice at least nine times in the C57BL6J mice line present in our animal house. Animals used for this experiments were at least F10 generation. Therefore, differences between the two transgenic lines cannot be ascribed to the genetic background, which is the same.
The breeding scheme for the ERK1 line was to use heterozygous male and female mice for breeding, so that in each litter there were homozygous pups for the deletion of ERK1 (ERK1 KO mice used in this study), heterozygous littermates, and wt littermates (ERK1 WT mice used in this study).
The breeding scheme for Ras-GRF1 KO mice, since this gene shows maternal imprinting (Brambilla et al., 1997) and only the paternal allele is expressed, was the following: we bred heterozygous males with WT females; half of the offsprings are wt (Ras-GRF1 WT used in this study) and half phenotypically KO (Ras-GRF1 KO used in this study).
We have used a total of 23 C57BL6J mice (13 males and 10 females) for the study on the effects of blocking ERK activation in the PRHC on recognition memory (16 mice, 9 males and 7 females) and for the immunohistochemistry of phospho-ERK (pERK; seven mice, four males and three females).
We have used a total of 100 Ras-GRF1 KO mice (54 males and 46 females) for the behavioral tests, 32 (20 males and 12 females) for the ORT test at 1 and 12 h interval and 68 (34 males and 34 females) for the reconsolidation experiments. As controls, 74 Ras-GRF1 WT mice have been used (41 males and 33 females), 29 (15 males and 14 females) for the ORT test at 1 and 12 h interval and 45 (26 males and 19 females) for the reconsolidation experiments. In addition, 20 Ras-GRF1 KO mice (12 males, 8 females) and 18 Ras-GRF1 WT mice (11 males, 7 females) have been used for the electrophysiology.
A total of 52 ERK1 KO mice have been used (22 females, 30 males), 23 for the behavioral experiments (14 males and 9 females), 18 for the electrophysiology (11 males, 7 females), and 11 for biochemistry (6 females and 5 males); as control, 47 ERK1 WT mice have been used (25 males, 22 females), 21 for the behavioral studies (11 males and 10 females), 16 for the electrophysiology (9 males, 7 females), and 10 for biochemistry (5 females and 5 males).
Object Recognition Task
The apparatus consisted of a square arena (60 cm × 60 cm × 30 cm) constructed in PVC with black walls and white floor. The objects were either three-dimensional metal, plastic, glass, or china objects of different shapes, or cubes (15 cm wide) made of transparent Plexiglas that differed for the visual patterns lining the walls. Box and objects were cleaned up between trials to stop the build-up of olfactory cues (De Rosa et al., 2005).
The experimental protocol was modified from Ennaceur and Delacour (1988). Briefly, mice received one session of 5 min duration in the empty arena to help them habituate to the apparatus and test room (habituation phase). Twenty-four hours later, each mouse was placed in the arena and exposed to two identical objects (sample phase) for 5 min and returned to its cage. After a variable delay (1, 12, 48, 72, or 96 h), mice were placed back in the arena and exposed to a familiar object (object identical to those in sample phase) and to a novel object for 5 min (test phase). Objects were placed in the same locations as in sample phase. Time spent exploring each object was recorded for each animal and for each condition and a discrimination index was calculated:

Exploration of an object was defined as directing the nose to the object at a distance of ≤3 cm and/or touching it with the nose or forepaws; time spent turning around the object was not included.
We have used in this study pairs of objects previously validated by us in a large group of wt and mutant animals for not giving differential exploration times. For each retention time interval, the new object for half of the animals, wt, or mutants, was the familiar object for the other half; also the position of the novel and the familiar object (left–right) was balanced. When the same animals performed more than one ORT test, different pairs of objects were used. When different groups of animals performed the same ORT test we used the same pairs of objects.
Reconsolidation protocol
Mice were first habituated to the open arena in the absence of objects for 20 min a day for two consecutive days (“habituation phase”). Twenty-four hours after the second habituation phase, mice were reintroduced in the arena and exposed to two identical objects for either a single familiarization trial of 5 min duration (single sample phase) or eight familiarization trials, duration 5 min each and distributed in four blocks of two sessions each (multiple sample phase); the interval between blocks was 90 min and the interval between sessions within each block was 5 min. During the intersession intervals mice were placed into an holding cage in the experimental room.
Forty-eight hours after the single or the multiple sample phase mice were exposed to a familiar object (identical to those in the sample phase) and to a novel object for 5 min (“test phase”). To test reconsolidation, 24 h after the multiple sample phase mice were exposed again to the same two sample objects explored during the sample phase for a single 5 min period to reactivate the memory trace (reactivation phase). Memory retention after reactivation was tested with novel and familiar objects (test phase) either 10 min, to test post-reactivation short term memory (PR-STM), or 24 h later, to test post-reactivation long term memory (PR-LTM).
Cannulae Implantation and Drug Delivery
Surgery
Mice were anesthetized (avertin 2 ml/100 g), placed in a stereotaxic apparatus and implanted bilaterally with 22 gage indwelling guide cannulae according to the following procedure: two holes were made in the skull through which the guide cannulae were inserted, perpendicular to the horizontal plane, according to the following coordinates: AP −3.05 mm (relative to bregma), L ±4.55 mm (relative to midline), V −1.5 mm (relative to surface of the brain). Cannulae were then fixed to the skull with dental cement. In order to prevent accidental blockage and keep the guide cannulae clear, obdurators cut to extend 1.1 mm beyond the tip of the guide cannulas and with an outer diameter of 0.36 mm were inserted into the guides and remained there except during infusions. Animals were allowed to recover from the surgery for at least a week before the beginning of behavioral testing.
Drug delivery
Immediately after the ORT’s sample phase animals were gently restrained by the experimenter and the infusion process was started. The obdurators were removed, and the 28 gage infusion cannulas, which were cut to extend 1 mm beyond the tip of the guides, were inserted into the guides. ERK inhibitor UO126 (5 mM in 50% DMSO) or vehicle (saline in 50% DMSO) was injected bilaterally (0.5 μl on each side given over 2 min) by means of a 25-μl Hamilton syringe; the infusion cannula was left for an additional minute to allow the full deployment of the drug. The whole process required a total of 10 min for each animal, approximately. Recognition memory was assessed in the test phase 12 h later. Administration of the drug immediately after the sample phase (and not before) should ensure that it affects memory consolidation rather than information encoding (Winters and Bussey, 2005).
Immunohistochemical Procedures
Phospho-ERK immunostaining
Thirty minutes after learning mice were sacrificed and transcardial perfusion was executed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were quickly removed, postfixed for 12 h and cryoprotected in 30% sucrose overnight, and 40 μm coronal sections were cut on cryostat and processed for pERK immunohistochemistry. Free-floating sections were subjected to a 2-h block (PBS containing 10% BSA and 0.4% Triton X-100 at room temperature) followed by overnight incubation with pERK monoclonal antibody (1:1000, Sigma, St. Louis, MO, USA; in PBS containing 1% BSA and 0.1% Triton X-100, at 4°C). pERK was revealed using Alexa 568 labeled secondary antibodies (1:200 Vector Laboratories, Burlingame, CA, USA; diluted in 1% BSA and 0.4% Triton X-100 in PBS). Slices were coded and, for each animal, confocal images (Olympus FV-300) of at least three representative fields (706 μm × 706 μm) for each PRHC were acquired.
After immunohistochemistry for pERK, the staining of inhibitory cells was performed using 1:500 mouse anti GAD67 (Chemicon, USA) monoclonal primary antibody (4°C overnight) followed by 1:200 goat anti-mouse secondary antibody conjugated to Alexa Fluor 488 fluorophore.
Western Blotting
After decapitation, brains were removed rapidly and frozen on dry ice. A cortical area corresponding to PRHC was bilaterally dissected and homogenized in lysis buffer.
Tissue homogenates were centrifuged at 13000 g for 30 min at 4°C. The supernatant (cytoplasmic fraction) was aspirated, and protein concentration was determined in each sample by the Bradford method (Bio-Rad, Milan, Italy). Each sample was boiled, and 25 μg/lane of protein was loaded into 12% acrylamide gels using the Precast Gel System (Bio-Rad). Samples were blotted onto nitrocellulose membrane (Amersham, Bucks, UK). Membranes were blocked in 4% BSA in Tris-buffered saline for 1 h and then incubated overnight at 4°C with antibodies against either Phospho-p42/44 Map Kinase (1:1000, Sigma) or α-Tubulin (1:5000, Sigma). Blots were then rinsed in TTBS and TBS, incubated for 1 h in HRP-conjugated anti-mouse (1:3000 Bio-rad, Italy, in 2% BSA and TTBS), rinsed, incubated in enhanced chemiluminescent substrate (Bio-rad, Italy) and acquired by ChemiDoc (Bio-rad). The immunoblots were analyzed with ImageJ software to measure the optical density of the bands, using α-tubulin as loading control. To minimize variability each sample was loaded in parallel in two lanes and two gels were run simultaneously on the same apparatus.
In vitro Electrophysiology
Slices of the perirhinal and surrounding cortex were prepared from adult (P60–120) mutant mice and their WT littermates. All efforts were made to minimize animal suffering and the number of animals. All experiments were performed in accordance with the Italian Ministry of Health guidelines for care and use of laboratory animals. Animals were anesthetized by isoflurane inhalation and decapitated. The brain was rapidly removed and immersed in ice-cold modified artificial cerebrospinal fluid (composition in mM: NaCl, 132.80; KCl, 3.10; HEPES, 10.00; NaHCO3, 4.00; ascorbic acid, 1.00; myo inositol, 0.50; sodium pyruvate, 2.00; K2HPO4, 1.00; glucose, 5.00; MgCl2, 2.00; and CaCl2, 1.00) bubbled with oxygen (Di Cristo et al., 2001).
A mid-sagittal section was made through the brain, the rostral and caudal parts of the brain were removed by single scalpel cuts made at approximately 45° to the dorsoventral axis (Massey et al., 2001), and each remaining half was glued by its rostral end onto a vibroslice stage Leica VT 1000S microslicer (Leica Microsystems; Nussloch, Germany). Slices (340 μm thick) which included the perirhinal, entorhinal, and temporal cortices were cut and allowed to recover for at least 1 h at 30°C in a submersion chamber containing oxygenated modified artificial cerebrospinal fluid.
A single slice was placed in a submerged recording chamber filled with artificial cerebrospinal fluid (composition in mM 132.80 NaCl, 3.10 KCl, 1.00 K2HPO4, 10.00 HEPES, 1.00 ascorbic acid, 0.50 myo inositol, 2.00 sodium pyruvate, 4.00 NaHCO3, 1 MgCl2, 2 CaCl2, and 5.00 glucose), 30–31°C, flow rate, 1.5 ml/min.
Standard extracellular recording electrodes (1.0 MΩ glass electrodes filled with artificial cerebrospinal fluid) were approximately positioned in layer II, beneath the rhinal fissure, and were used to record field potentials. A stimulating tungsten bipolar electrode was placed in intermediate layers II/III, 0.16–0.36 mm laterally with respect to the recording electrode. Pulses of 100 μs duration were delivered every 30 s (0.033 Hz) to the stimulating electrode. Recorded field potentials were filtered between 0.1 and 3 kHz, amplified 1000 times (Axon CyberAmp), sampled at 10 kHz and fed to a computer using LabVIEW software (National instruments, USA). Stimulation intensity was adjusted to evoke a field potential amplitude 50% of the maximal value. After 15 min of stable baseline obtained with the 0.033-Hz stimulation, either TBS to evoke LTP or charbacol administration to evoke LTD (Massey et al., 2001) were delivered.
Theta burst stimulation protocol consisted in four repetitions, 15 s apart, of 10 trains of 4 pulses at 100 Hz, with an intertrain interval of 200 ms. Field potential amplitudes were recorded every 30 s for 60 min following the cessation of the TBS stimulation protocol. Data from different slices were normalized to baseline amplitudes, averaged, and reported as means ± SEM as a function of time. To assess the final level of potentiation, the mean normalized EPSP amplitude over the last 15′ of recordings was used.
Carbachol (50 μM, Sigma) was applied for 10′ (Massey et al., 2001). Field potential amplitudes were recorded every 30 s for 60 min following the initiation of Carbachol application. Data from different slices were normalized to baseline amplitudes, averaged, and reported as means ± SEM as a function of time. To assess the final level of depression, the mean normalized EPSP amplitude over the last 20′ of recordings was used.
Statistics
All data were analyzed with SigmaStat statistical package. Differences in object exploration times between KO and WT mice were analyzed with the unpaired Student’s t-test or Mann–Whitney U-test, depending to the result of the test for Normal distribution of the data. Differences between the exploration time of the new and that of the familiar object in the test phase was analyzed, for each group of KO or WT mice, with the paired Student’s t-test or the Wilcoxon Signed Rank test, depending to the result of the test for Normal distribution of the data. Differences between the discrimination indexes were analyzed with the Wilcoxon Signed Rank test (for paired groups) or the Mann–Whitney U-test (for unpaired groups).
Field potential amplitudes in LTP and LTD experiments were analyzed by means of repeated measures analysis of variance and appropriate post hoc analysis. p Values less than 0.05 were taken to indicate statistically significant differences.
Results
Activation of ERK in the PRHC is Necessary for Recognition Memory Consolidation
To assess whether activation of the MAP kinase pathway in the PRHC is necessary for visual recognition memory consolidation, we injected into the PRHC an inhibitor of MEK (the ERK activating kinase) UO126, or vehicle for control, immediately after (3 min) the sample (learning) phase of the ORT. UO126 (5 mM in 50% DMSO) or vehicle (saline in 50% DMSO) were injected (0.5 μl) bilaterally through guide cannulae previously implanted into the PRHC (Figures 1A,B).
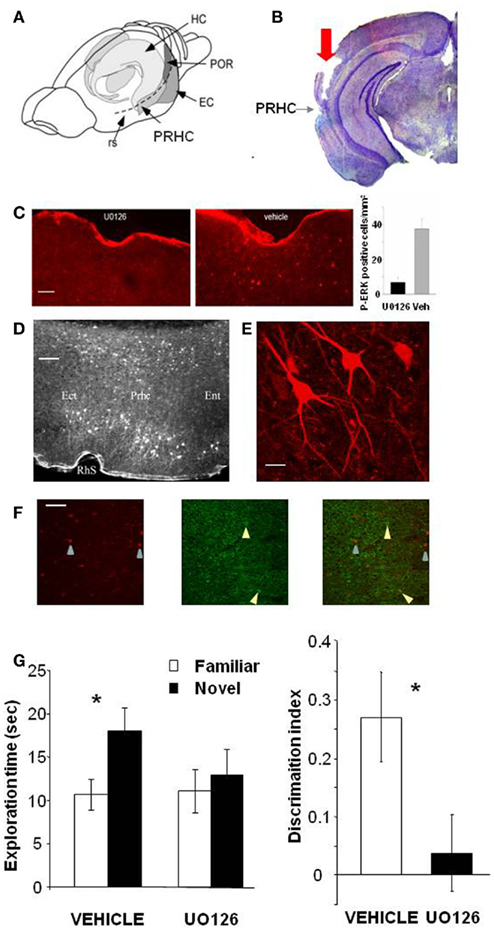
Figure 1. Activation of ERK in the Perirhinal Cortex is necessary for recognition memory consolidation. (A) Location of the perirhinal cortex (PRHC) in a schematic lateral view of the mouse brain. Shading indicates the location of the hippocampal formation (HC) and the perirhinal (PRHC), enthorinal (EC), and postrhinal cortices (POR); rs indicates the rhinal sulcus. (B) Photomicrograph of a coronal brain section showing the track (indicated by red arrow) left by a cannula inserted into the PRHC. The PRHC is also indicated. (C) U0126 infusion is effective in blocking ERK activation in the perirhinal cortex. Example of immunohistochemistry for phospho-ERK 30′ after the sample phase in one animal injected with U0126 in one PRHC and with vehicle in the contralateral PRHC. Phospho-ERK immunopositive cells are clearly present in the PRHC treated with vehicle (right) but are absent from the cortex treated with U0126 (left), showing that U0126 effectively diffused from the injection cannula and blocked ERK activation in the PRHC. In the inset, the mean number of phospho-ERK immunopositive cells per square millimeter counted in the U0126 treated PRHC (7 ± 2.6) and in the vehicle treated PRHC (38 ± 5.7; n = 4 animals) is reported. The difference between U0126 and vehicle treated side is significant (paired t-test, p = 0.035). Calibration bar: 50 μm. (D) Localization of cells immunopositive for phospho-ERK 30′ after exploration of the ORT arena with two new objects. Ect, ectorhinal cortex; Prhc, perirhinal cortex; Ent, entorhinal cortex; RhS, rhinal sulcus. Calibration bar 100 μm. (E) High power image of phospho-ERK immunopositive neurons in the perirhinal cortex. Calibration bar 15 μm. (F) Example of a double staining for phospho-ERK and GAD 67 in the perirhinal cortex. Left, cells immunopositive for phospho-ERK in the PRHC. Arrows point to two stained neurons; center, immunostaining for GAD67. Arrows point to two stained neurons; right, merge of the two images. Calibration bar 80 μm. (G) Perirhinal focal infusion of MAPK blocker UO126 impairs long term (12 h) object recognition memory. UO126 (5 mM in 50% DMSO, n = 16) or vehicle (saline in 50% DMSO, n = 16) were injected (0.5 μl bilaterally) immediately (3 min) after the sample phase of the ORT. Recognition memory was tested (test phase) 12 h after the sample phase. Left: Exploration of novel compared to familiar objects during the test phase. Vehicle injected mice explore the novel object significantly more than the familiar object (asterisk, p < 0.05, paired t-test) while UO126 injected mice do not (paired t-test, p > 0.05). Right: discrimination index in the test phase for vehicle and U0126 injected mice. The latter exhibit a significantly lower discrimination index with respect to vehicle injected mice (Wilcoxon Signed Rank test, p < 0.05, asterisk).
To control that U0126 injections were effective in blocking ERK activation, we performed an immunohistochemistry for pERK 30′ after the sample phase in animals (n = 3) injected with U0126 in one PRHC and with vehicle in the contralateral PRHC. We found that pERK immunopositive cells were clearly present in the PRHC treated with vehicle but were significantly reduced from the cortex treated with U0126, showing that U0126 effectively diffused from the injection cannula and blocked ERK activation (Figure 1C). pERK was localized in the superficial and deep layers of the PRHC (Figure 1D). In the visual cortex, pERK immunostaining after visual stimulation is present mainly in excitatory neurons (Cancedda et al., 2003) and evident in dendrites and cell bodies (Boggio et al., 2007); a similar pattern of dendritic and somatic pERK immunostaining was found in the PRHC (Figures 1E,F).
A total of 16 C57BL/6J mice was employed and each animal performed the ORT twice, with an interval of at least 3 days between the two tests. For both ORT trials, the retention interval between the sample phase and the test phase was 12 h. Half of the animals received vehicle injection during the first ORT and U0126 injections during the second one. The other half of the animals was injected with U0126 during the first ORT and with vehicle during the second one. There was no difference between the total exploration time during sample phase in the second ORT trial between mice treated with U0126 (30.83 ± 7.42 s) at the first ORT trial and those treated with vehicle (25.75 ± 6.06 s; p = 0.222; paired t-test). Also total object exploration time during the test phase did not differ between vehicle (28.73 ± 3.09 s) and UO126 treated mice (24.23 ± 5.25 s), (p = 0.234, paired t-test). This suggests that blockade of ERK activity has no effect on exploratory behavior and locomotor activity.
In the test phase, mice injected with vehicle showed the expected preferential exploration of the novel object (Figure 1G, paired t-test, exploration time of the novel versus the familiar object p = 0.029) while mice injected with U0126 did not (Figure 1G, paired t-test, exploration time of the novel versus the familiar object p = 0.303). As a consequence, the discrimination index in the group treated with UO126 was significantly lower than in the group treated with vehicle (Wilcoxon Signed Rank test, p = 0.011; Figure 1G).
These data suggest that activation of ERK pathway in the PRHC is necessary for consolidation of long term recognition memory.
Ras-GRF1 KO Mice Show Deficits in Consolidation of Visual Recognition Memory
ERK1 and ERK2 activation is the result of the integration of multiple signals deriving from neuronal activity and neurotrophin signaling, such as Ca2+ influx, activation of G-protein coupled receptors, and trk receptor signaling. Central to ERK activation is the increase in the active, GTP bound form of the small G-Protein Ras, which causes MEK phosphorylation. Ras activation, in its turn, is under control of guanine exchange factors, GEF, which catalyze the exchange of GDP for GTP on Ras, such as the Sos and the Ras-GRF families. Ras-GRFs activate Ras in response to Ca2+ signaling and muscarinic receptor activation (Farnsworth et al., 1995; Mattingly and Macara, 1996).To investigate the specific role of Ras-GRF1 signaling and the effects of reducing only the component of ERK activation resulting by Ras-GRF1 on recognition memory, we evaluated the performance in the ORT of Ras-GRF1 KO mice (n = 32) compared to their WT littermates (n = 29).
One group of Ras-GRF1 KO (n = 15) and Ras-GRF1 WT (n = 14) mice performed the ORT with a retention interval of 1 h; a second group of Ras-GRF1 KO (n = 17) and Ras-GRF1 WT mice (n = 15) performed the ORT with a retention interval of 12 h. Total object exploration time during the sample phase was never significantly different between Ras-GRF1 KO and WT mice.
In WT mice, preferential exploration of the novel object during the test phase is present at 1 and 12 h after familiarization (paired t-test, p < 0.001 at 1 h and p < 0.01 at 12 h; Figure 2A); in KO mice preferential exploration of the novel object during the test phase is present only 1 h after familiarization (paired t-test, p < 0.001 at 1 h interval, p = 0.690 at 12 h; Figure 2A), showing a lack of long term retention.
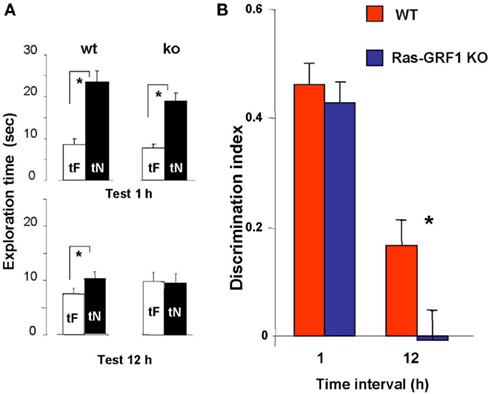
Figure 2. Ras-GRF1 KO mice show long term visual recognition memory deficits. (A) Mean exploration times for the 1- and 12-h interval experiment in Ras-GRF1 KO mice and their WT littermates. The exploration time of the familiar object, tF, and the exploration time of the novel object, tN, in the test phase significantly differ (asterisk) for WT mice both at 1 h (n = 14) and at 12 h interval (n = 15; paired t-test, p < 0.001 at 1 h and p < 0.01 at 12 h) but for KO mice only at 1 h (n = 15) there is a significant difference (paired t-test, p < 0.001). At 12 h KO mice (n = 17) do not show any differential exploration of the new with respect to the familiar object (paired t-test, p = 0.69), suggesting a consolidation deficit. (B) Memory retention curve for ORT in Ras-GRF1 KO mice and their WT littermates. Discrimination index is plotted against time interval between familiarization and test. Asterisk denotes significant difference between WT and KO mice (two-way ANOVA, time × genotype, post hoc Holm–Sidak method).
For both Ras-GRF1 KO and WT mice the discrimination index significantly decreases with increasing retention interval (discrimination index at 12 h significantly lower than at 1 h, p < 0.001 both for WT and Ras-GRF1 KO mice). However, the decrease is faster for KO mice; indeed, the discrimination index of KO mice is significantly lower compared with WT mice after the retention interval of 12 h (p < 0.023; two-way ANOVA, time × genotype, post hoc Holm–Sidak method; Figure 2B).
Thus Ras-GRF1 KO mice have normal memory at 1 h retention interval but exhibit a deficit at 12 h, suggesting that lack of Ras-GRF1 activation of the ERK pathway results in a defect in recognition memory consolidation.
Ras-GRF1 KO Mice Have Deficits in Visual Recognition Memory Reconsolidation
According to the memory reconsolidation hypothesis each time a memory is reactivated/retrieved it becomes labile and must undergo a process of consolidation to be maintained to further long term (see Lee, 2009; Nader and Hardt, 2009). We examined then the possibility that consolidated visual recognition memory traces, after being reactivated by re-exposure to the learning situation, undergo a process of reconsolidation requiring Ras-GRF1.
The consolidation deficit exhibited by Ras-GRF1 KO mice might represent a difficulty, as determining the role of Ras-GRF1 in reconsolidation after retrieval requires that mice form a consolidated long term memory of the objects. However, it has been shown that deficits in visual recognition memory consolidation can be overcome by extended and distributed training in zif 268 KO mice (Bozon et al., 2003). Thus, we first examined whether Ras-GRF1 KO mice could form a long term visual recognition memory if given additional exposures to the objects in a distributed training paradigm. To do so, we compared the performance in the test phase at 48 h after the sample phase in two groups of KO mice, one (n = 20) undergoing the ORT procedure previously employed (one familiarization trial of 5 min during the sample phase) and one (n = 16) receiving extended training, eight familiarization sessions of 5 min each during the sample phase. The eight familiarization sessions were distributed in four blocks of two sessions each; the interval between blocks was 90 min and the interval between sessions within each block was 5 min (Figure 3A). We found that the discrimination index for the group exposed to the extended training was significantly greater than that of the group exposed to a single familiarization session (Mann–Whitney, p = 0.012; Figure 3B). Preferential exploration of the novel object during the test phase was clearly present in Ras-GRF1 KO mice exposed to eight familiarizations (paired t-test, p < 0.001) while it was absent in the group not exposed to extended training (p = 0.252). The discrimination index of Ras-GRF1 KO mice subjected to extended training did not differ from that of Ras-GRF1 WT mice subjected to the same extended training (Figure 3C; p = 0.494 Mann–Whitney). These data indicate that extended training can counteract memory consolidation deficits in Ras-GRF1 KO mice.
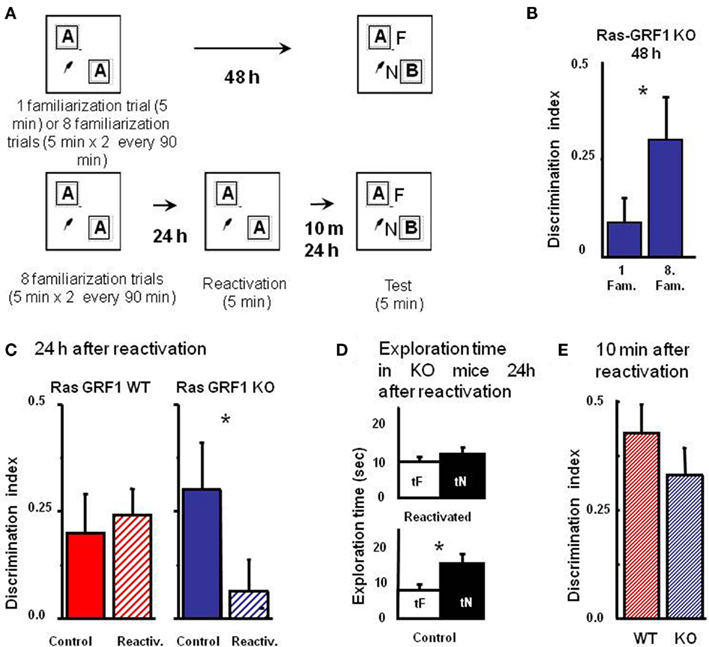
Figure 3. Ras-GRF1 KO mice exhibit a reconsolidation deficit. (A) Protocols for repeated familiarizations (top) and for memory reconsolidation test (bottom). Top: mice are introduced into the arena in the presence of two identical objects for one (single familiarization phase) or eight consecutive sessions of 5 min (multiple familiarization phase). After a delay of 48 h the test phase is performed. Bottom: mice are introduced into the arena in presence of two identical objects for eight consecutive sessions of 5 min each. Twenty-four hours later mice memory trace is reactivated by re-exposing the mice to the same two familiar objects. After a delay of 10 min or of 24 h the test phase is performed. (B) Discrimination index of Ras-GRF1 KO mice 48 h after a single familiarization session or after repeated familiarization sessions. Asterisk denotes significant difference between Ras-GRF1 KO mice (n = 16 for the repeated familiarization, n = 20 for the single familiarization) in the two conditions (Mann–Whitney p = 0.012). Repeated exposure to the stimuli is sufficient to compensate for the deficit of visual recognition memory of Ras-GRF1 KO mice. (C) Discrimination index of Ras-GRF1 KO and WT mice 48 h after repeated familiarization sessions and 24 h after memory reactivation. In wt, re-exposure of the animals to the stimuli did not interfere with recognition of the familiar stimulus 24 h later (control n = 20, reactivated n = 16; p = 0.43 Mann–Whitney). By contrast a single exposure to the familiar stimulus 24 h after its memorization makes the memory trace labile in Ras-GRF1 KO mice: 24 h after reactivation, discrimination index was significantly decreased with respect to that found without reactivation (control n = 15, reactivated n = 17; p = 0.026 Mann–Whitney). (D) Exploration time of novel and familiar object of Ras-GRF1 KO mice 24 h after reactivation: there is no preferential exploration of the novel object (time of exploration of new object, tN versus time exploration familiar object, tF, control p < 0.001; reactivated p = 0.212 paired t-test). Asterisks denote significant differences. (E) Discrimination index 10 min after reactivation; the deficit in Ras-GRF1 KO mice was not present 10 min after reactivation.
Being able to provide both genotypes with long lasting memories, we evaluated the effect of memory reactivation on PR-LTM traces. To do so, mice of both genotypes were provided with the extended training during the sample phase and then, 48 h later, performance in the test phase was assessed with or without intervening memory reactivation, performed 24 h after the sample phase by re-exposing the animals for 5 min to the same objects used in the sample phase (Figure 3A).
As shown in Figure 3C, memory reactivation did not affect subsequent PR-LTM in WT mice (Figure 3C): the discrimination index in the test phase with (0.241 ± 0.064 n = 16) or without (0.214 ± 0.093, n = 20) memory reactivation did not differ, (Mann–Whitney, p = 0.430). In KO mice memory reactivation resulted in a significant reduction of the discrimination index, compared to the condition without reactivation (discrimination index with reactivation 0.062 ± 0.076, n = 17; without reactivation 0.302 ± 0.1, n = 15, Mann–Whitney, p = 0.026; Figure 3C). Accordingly, preferential exploration of the novel object during the test phase for Ras-GRF1 KO mice was only present if memory had not been reactivated (exploration time of new versus familiar object, paired t-test, p < 0.001 in the condition of no reactivation and p = 0.212 in the condition of reactivation; Figure 3D), suggesting that reactivation had canceled the visual recognition memory trace.
In order to ascribe the deficit shown by Ras-GRF1 KO mice 24 h after reactivation to a defect in memory consolidation rather then to an aspecific effect of reactivation we assessed PR-STM. The experimental protocol for the sample and the reactivation phase was as described above but in this case visual recognition memory was assessed 10 min after reactivation. We found that discrimination index in KO and WT mice was not significantly different (Mann–Whitney, p = 0.427; Figure 3E), suggesting that reactivation does not affect PR-STM of KO mice.
These findings suggest the presence of a deficit in memory reconsolidation in Ras-GRF1 KO mice, and imply a critical role for ERK activation by the Ras-GRF1 pathway in this process.
ERK1 KO Mice Have a Longer-Lasting Visual Recognition Memory
To investigate the effects of an enhancement of ERK activation on visual recognition memory, we evaluated the memory performance in a group of 23 ERK1 KO mice compared to their littermates WT (n = 21). ERK1 KO mice present an enhanced activation of ERK2 by glutamate as assessed in vitro (Mazzucchelli et al., 2002). Recently, it has been shown that ERK2 phosphorylation is enhanced in vivo in dorsal hippocampus of ERK1 KO mice with respect to WT mice (Tronson et al., 2008). In line with these results, we found a significantly higher level of ERK2 phosphorylation in the PRHC of ERK1 KO (n = 11) with respect to WT mice (n = 10, t-test, p = 0.039; Figure 4A).
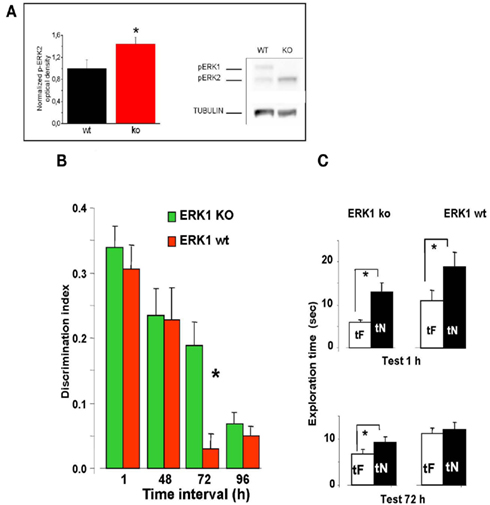
Figure 4. Longer lasting visual recognition memory in ERK1 KO mice. (A) Left, quantification of phospho-ERK2 optical density, normalized to tubulin, in the perirhinal cortex of ERK1 KO (n = 11) and ERK1 WT (n = 10) mice. The difference is statistically significant (t-test, p = 0.039). Right, example of immunoblotting. (B) Memory retention curve for ORT in ERK1 KO mice (n = 23) and their WT littermates (n = 21). Discrimination index is plotted against time interval between familiarization and test. Asterisks denote significant difference between WT and KO mice (two-way ANOVA for repeated measures, time × genotype, genotype, time, and interaction all significant p < 0.001, post hoc Tukey’s test). Performance differs at 72 h. (C) Exploration times in the ORT for ERK1 KO mice (n = 23) and their WT littermates (n = 21). At 1 and 72 h intervals. Asterisks denote significant difference between tF and tN. At 1 h there is a significant difference for both groups but at 72 h only KO mice show a differential exploration of the new with respect to the familiar object (paired t-test, p < 0.001 for KO and p < 0.112 for WT mice).
To assess the time span of retention in the ORT, each animal performed the ORT with a different retention intervals between sample and test phase. For each ORT session, mice went through a new sample phase with two equal objects never used before and then a test phase after the appropriate time interval.
We found that discrimination index in ERK1 KO mice did not differ from that of ERK1 WT mice at 1 or 48 h retention interval but was significantly higher than that of WT for the retention interval of 72 h (two-way ANOVA for repeated measures, time × genotype, genotype, time, and interaction all significant p < 0.001, post hoc Tukey’s test; Figure 4B). In KO mice, but not in WT, preferential exploration of the novel object during the test phase is found up to 72 h from sample phase (paired t-test, exploration time of novel versus familiar object at 72 h p < 0.001 for KO mice and p = 0.112 for WT mice; Figure 4C), indicating that the memory trace persisted for a longer in time. Thus ERK1 KO mice show a longer lasting visual recognition memory.
Genetic Modulation of ERK2 Activation Bidirectionally Affects Synaptic Plasticity in the PRHC
In the previous sections we have shown that the performance in a visual recognition memory task can be enhanced or impaired by genetically interfering with ERK pathway activity. To assess whether the behavioral phenotype of Ras-GRF1 and ERK1 KO was associated with an alteration of synaptic plasticity in the PRHC, we studied LTP and LTD in PRHC of mice belonging to both genetically mutated groups.
LTP was induced in layer II/III horizontal connections using a standard TBS protocol. We found a stable and persistent LTP in all the three mice groups, WT, (littermates of ERK1 KO, and Ras-GRF1 KO, pooled together since there was no statistical difference between the two), ERK1 KO, Ras-GRF1 KO (Figure 5). However, in accordance with the hypothesis of a dependence of synaptic plasticity from genetic modulation of ERK pathway activation, we found that layer II/III PRHC LTP was significantly lower in Ras-GRF1 KO mice [median increase 107.7% of baseline (interquartile ranges 96–115), n = 12] than WT [median increase 136% of baseline (interquartiles 119–149), n = 21], while LTP induced in ERK1 KO [median increase 155.8% ± of baseline (interquartiles 149–169), n = 7] was significantly higher compared with WT (ANOVA, genotype × time, factor genotype: p < 0.001, all three groups differ one from each other, post hoc Student–Newman–Keuls Method).
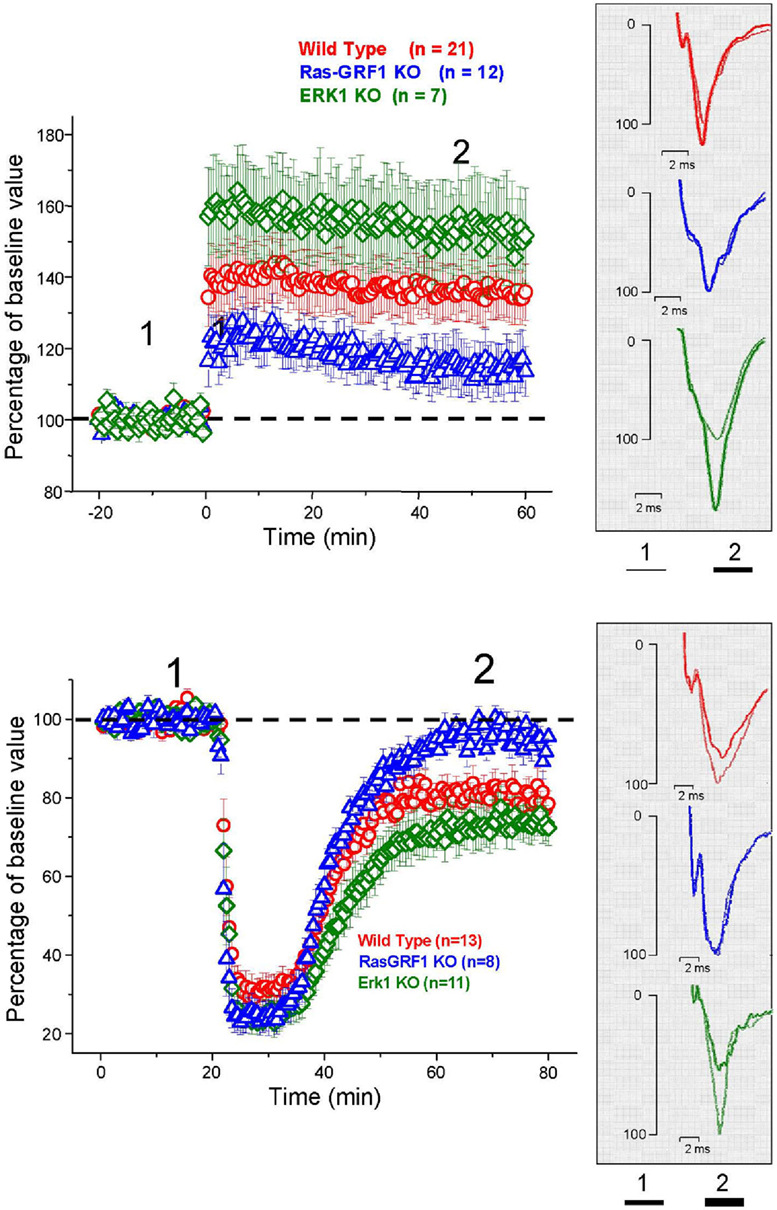
Figure 5. Top: Average time course of field EPSP amplitude recorded from PRHC. The final level attained for LTP is significantly lower in Ras-GRF1 KO mice while it is higher in ERK1 KO mice with respect to WT mice (ANOVA, genotype × time, genotype p < 0.001, post hoc Student–Newman–Keuls Method). Bottom: Average time course of field EPSP amplitude recorded from PRHC. The final level attained for LTD in Ras-GRF1 KO mice is significantly lower than in WT mice; the latter is significantly lower than in ERK1 KO mice (ANOVA, genotype × time, genotype p < 0.001, post hoc Holm–Sidak method).
In another group of animals from the three genotypes LTD was pharmacologically induced by 10 min administration of 50 μM carbachol as in Massey et al. (2001). We found that Ras-GRF1 mutants had a significantly impaired LTD (95.5 ± 3.8% of baseline, n = 8) while ERK1 mutants showed a significantly enhanced LTD (70 ± 3.3% of baseline, n = 11) when compared with WT animals, littermates of ERK1KO and Ras-GRF1 KO, (80.75 ± 3% of baseline, n = 13; ANOVA, genotype × time, factor genotype p < 0.001, all three groups differ one from each other, post hoc Holm–Sidak Method, p < 0.050).
Thus, synaptic plasticity in PRHC can be bidirectionally regulated by genetic manipulating the activation of the ERK pathway. The differences found in PRHC synaptic plasticity of Ras-GRF1 and ERK1 KO mice reflect the differences in behavioral performance observed in the visual recognition memory task: reduced LTP and strongly impaired LTD in Ras-GRF1 KO mice, which have a reduced ERK activation; enhanced LTP and LTD in ERK1 KO mice, which have enhanced ERK2 activation.
Discussion
ERK Activation in the PRHC is Necessary for Long Term Recognition Memory
ERK involvement in recognition memory had been already suggested by Kelly et al. (2003), showing that intraventricular injection before the ORT learning phase of U0126, a blocker of the ERK kinase MEK, impaired consolidation and reconsolidation of recognition memory. This has been subsequently confirmed by Goeldner et al. (2008), employing systemic treatment with SL327, another MEK blocker. These two papers do not allow to assess the role of ERK activation in the PRHC in recognition memory consolidation, since the treatment with blockers of ERK activation was not specifically directed to the PRHC.
Our results show that it is sufficient to block ERK activation in the PRHC after the learning phase to impair long term recognition memory in the ORT. This suggests that ERK activation in the PRHC is necessary for recognition memory consolidation. It also suggests that ERK activation in other medial temporal lobe structures possibly involved in recognition memory, such as the hippocampus, (see Kelly et al., 2003) is not sufficient to mediate the formation of a long term recognition memory. This strengthens the importance of the PRHC and of the plasticity processes triggered within its circuits by learning in the formation of long term, familiarity-based, recognition memory (Warburton et al., 2003; Aggleton and Brown, 2005; Winters and Bussey, 2005; Murray et al., 2007; Griffiths et al., 2008; Massey et al., 2008; Winters et al., 2008).
Ras-GRF1 Signaling is Necessary for Recognition Memory Consolidation and Reconsolidation
ERK1 and ERK2 activation is the result of the integration of multiple signals deriving from neuronal activity and neurotrophin signaling, such as Ca2+ influx, activation of G-protein coupled receptors and trk receptor signaling (Weeber et al., 2002; Sweatt, 2004). Central to ERK activation is the increase in the active, GTP bound form of the small G-Protein Ras, which causes MEK phosphorylation. Ras activation, in its turn, is under control of guanine exchange factors, GEF, which catalyze the exchange of GDP for GTP on Ras, such as the Sos and the Ras-GRF families. Ras-GRFs activate Ras in response to Ca2+ signaling and muscarinic receptor activation (Farnsworth et al., 1995; Mattingly and Macara, 1996). Brambilla et al. (1997) have shown that mice with genetic deletion of Ras-GRF1, a guanine exchange factor which catalyzes the exchange of GDP for GTP on Ras (Ras-GRF1 KO mice) exhibit a reduced activation of ERK by neuronal activity and show deficits in amygdala dependent tasks such as active avoidance, inhibitory avoidance, and cued fear conditioning and also in contextual fear conditioning, which depends both on the amygdala and hippocampus. Giese et al. (2001), in a different mouse line with Ras-GRF1 inactivation (Ras-GRF1 deficient mice), found a clear deficit in spatial memory, which is hippocampus dependent. Nothing was known on the specific role of Ras-GRF1 signaling in visual recognition memory. Our results in Ras-GRF1 KO mice show that these mice have normal short term memory, as tested at 1 h delay from the learning session, but display a long term memory deficit; indeed, their performance differs from that of WT mice at the 12-h interval. This is indicative of a consolidation deficit. Thus the activation of ERK in the PRHC, which our data suggest to be necessary for the consolidation of long term recognition memory, has a crucial component in the Ras-GRF1 signaling.
Since our KO mice are not conditional, we cannot exclude developmental effects, although gross alterations have never been described in these mice (Brambilla et al., 1997; Mazzucchelli et al., 2002) and a recent paper (Tian and Feig, 2005) shows that Ras-GRF signaling is not involved in activating the Ras/ERK pathway in pubescent mice but only in adult mice. However, in absence of an acute interference/rescue of RAS-GRF1 or of ERK1 KO we cannot completely exclude developmental effects.
The consolidation deficit in Ras-GRF1 mice could be rescued by extended training. This suggests that it is possible to compensate for the lack of Ras-GRF1 mediated activation of the Ras-ERK pathway by Ras-GRF2 mediated activation of Ras-ERK (Li et al., 2006), thus preserving the action of neural activity on the Ras-ERK pathway; alternatively, or in addition, in mice subjected to repeated training other ERK activating pathways could be recruited, either impinging on Ras, such as the tyrosine kinase receptors pathways, or independent from Ras, such as the PKA pathway (Weeber et al., 2002; Sweatt, 2004).
It is important to note that also the process of reconsolidation is impaired in Ras-GRF1 KO mice, as shown by the lack of preferential exploration of the novel object 24 h after the reactivation of the memory trace in mice subjected to extended training, which, in absence of the intervening reactivation, would be able to recognize the familiar object up to 48 h after learning. ERK activation was known to be crucial both for consolidation and reconsolidation of recognition memory (Kelly et al., 2003); we now show that ERK activation needs the Ras-GRF1 mediated component to allow learned visual recognition memories to be consolidated and, if recalled, reconsolidated. It has been suggested that consolidation and reconsolidation might involve the activation of both shared pathways and specific pathways: indeed, Lee et al. (2004) found that BDNF and zif 268 were independently required in the hippocampus for consolidation and reconsolidation of contextual fear conditioning. Ras-GRF1 is evidently one of those molecules crucial for both processes, at least as far as recognition memory is concerned.
Long Term Synaptic Plasticity Deficits in the PRHC of Ras-GRF1 KO Mice
In very good correlation with the behavioral deficits, long term synaptic plasticity in the PRHC of Ras-GRF1 KO mice was defective. This lends further support to the role of PRHC plasticity in visual recognition memory (Warburton et al., 2003; Griffiths et al., 2008; Massey et al., 2008) and shows for the first time that ERK activation by Ras-GRF1 is crucial both for the LTD and the LTP type synaptic plasticity in the PRHC.
The LTD induction protocol we employed is dependent on cholinergic activity and it is known that Ras-GRF1 mediates the effects of muscarinic receptor activation (Farnsworth et al., 1995; Mattingly and Macara, 1996). The deleterious effects of Ras-GRF1 deletion on perirhinal LTD and on familiarity-based visual recognition memory are in good agreement with the crucial role of the cholinergic input to the PRHC via muscarinic receptors in this type of memory and in synaptic plasticity of the LTD type in the PRHC (Massey et al., 2001; Warburton et al., 2003).
In addition to the impairment in PRHC LTD type plasticity Ras-GRF1 KO mice also exhibit a deficit in perirhinal LTP. The combined effect on LTP and LTD we have found in Ras-GRF1 KO is reminiscent of the effects of NMDAR block shown by Barker et al. (2006): infusion of the NMDA receptor antagonist AP5 in the PRHC impairs recognition memory and both LTP and LTD. This is consistent with the role of Ras-GRF in mediating the effects of neuronal activity and in particular of Ca++ calcium signaling. Ras-GRF1 function has been indeed linked to Ca++ inflow through NMDA receptors (Li et al., 2006); interestingly, in the Barker et al. (2006) paper, blocking separately NMDA2A or 2B had effects on only one type of plasticity (LTP for the 2A block and LTD for the 2B block) and did not significantly impair recognition memory at the 24-h delay.
Ras-GRF1 could also mediate the effects of Ca++ influx via voltage dependent calcium channels (Farnsworth et al., 1995; Brambilla et al., 1997; Li et al., 2006), which, in a very recent study (Seoane et al., 2009), have been implied in recognition memory: block of a class of voltage dependent Ca++ channels in the PRHC impaired familiarity-based recognition memory and disrupted perirhinal LTD.
The importance of the Ras-GRF1 mediated ERK activation in PRHC plasticity both of the LTP and the LTD type is therefore likely to rest on the integrative action Ras-GRF1 exerts with respect to intracellular signaling in perirhinal neurons. Thus, reduction of ERK activation by neural activity (Fasano et al., 2009) via Ras-GRF1 is sufficient to disrupt both the process of consolidation and reconsolidation of visual recognition memory, and PRHC synaptic plasticity both of the LTP and the LTD type.
Increased ERK2 Activation Enhances Performance in Recognition Memory and Synaptic Plasticity in PRHC
ERK1 KO mice might be considered an in vivo model of an enhanced susceptibility of ERK activation by the many pathways converging on MEK. ERK1 KO mice exhibit a stronger ERK2 activation (Mazzucchelli et al., 2002 and our results); this is explained in terms of the enhanced ERK2 phosphorylation by MEK due to the absence of the competing presence of ERK1. We have shown that this enhanced ERK2 activation is paralleled by an enhanced performance in visual recognition memory and by an enhanced synaptic plasticity in the PRHC. This is in accordance with Mazzucchelli et al. (2002), who found a superior performance of ERK1 KO with respect to wt mice in striatal dependent memory tasks (passive and active avoidance) and an enhanced striatal LTP. Thus, just as reducing ERK activation impairs visual recognition memory and synaptic plasticity in the PRHC, enhancing ERK activation produces the opposite effects.
ERK 1 and ERK 2 have been recently suggested to play different roles in intracellular signaling; in particular, it has been suggested that it is ERK2 which play the major role in ERK signaling (Mazzucchelli et al., 2002; Fremin et al., 2007; Bessard et al., 2008; Samuels et al., 2008). Our data confirm the importance of ERK2 signaling in synaptic plasticity and memory processes. We cannot exclude that in ERK1 KO mice there is a change in other pathways that contributes to recognition memory enhancement. However, intrahippocampal infusion of U0126 prevented contextual fear extinction in ERK1 KOs and their wild-type littermates (Tronson et al., 2008). It appears, therefore, that overactivation of the hippocampal ERK2 isoform by MEK predominantly contributed to the enhanced fear extinction phenotype of ERK1 KOs. In addition, in another recent paper in ERK1 KO mice, it has been shown (Alter et al., 2010) that systemic MEK inhibition with SL327 attenuated pain induced spontaneous behaviors similarly in wild-type and ERK1 KO mice, indicating that unrelated signaling pathways do not functionally compensate for the loss of ERK1.
We are aware that the relation between potentiating a signaling pathway and enhancing a memory process is not straightforward and may depend upon the type of memory or the type and site of intervention on the molecular cascade. For instance, mutation in NF1 (Costa et al., 2001, 2002) causes a constitutive hyperactivation of the Ras pathway which does not produce positive effects on memory. It has to be underlined that the Ras hyperactivation resulting from lack of a GAP protein (NF1) is, in fact, a disregulation of the Ras-ERK pathway and, in terms of ERK activation, it is very much different from the enhanced ERK2 phosphorylation caused by the physiological activation of MEK.
In conclusion, our results contribute to unravel the molecular basis of visual recognition memory, suggesting a pivotal role for Ras-GRF1 activation of ERK in the PRHC in both consolidation and reconsolidation processes and showing that modifying the gain in the ERK activation pathway bidirectionally affects visual recognition memory and synaptic plasticity in the PRHC, producing non-only an impairment, when the gain is reduced, but also an enhancement when the gain is increased.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge the skilled assistance of Mr. Giulio Cappagli and Mr. Carlo Orsini. Funding was provided by FP7 EU projects PLASTICISE and EUROV1SION and by IIT Seed project EXTRAPLAST.
References
Abel, T., and Lattal, K. M. (2001). Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr. Opin. Neurobiol. 11, 180–187.
Adams, J. P., and Sweatt, J. D. (2002). Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu. Rev. Pharmacol. Toxicol. 42, 135–163.
Aggleton, J. P., and Brown, M. W. (2005). Contrasting hippocampal and perirhinal cortex function using immediate early gene imaging. Q. J. Exp. Psychol. B 58, 218–233.
Alter, B. J., Zhao, C., Karim, F., Landreth, G. E., and Gereau, R. W. IV. (2010). Genetic targeting of ERK1 suggests a predominant role for ERK2 in murine pain models. J. Neurosci. 30, 11537–11547.
Atkins, C. M., Selcher, J. C., Petraitis, J. J., Trzaskos, J. M., and Sweatt, J. D. (1998). The MAPK cascade is required for mammalian associative learning. Nat. Neurosci. 1, 602–609.
Barker, G. R., Warburton, E. C., Koder, T., Dolman, N. P., More, J. C., Aggleton, J. P., Bashir, Z. I., Auberson, Y. P., Jane, D. E., and Brown, M. W. (2006). The different effects on recognition memory of perirhinal kainate and NMDA glutamate receptor antagonism: implications for underlying plasticity mechanisms. J. Neurosci. 26, 3561–3566.
Bessard, A., Fremin, C., Ezan, F., Fautrel, A., Gailhouste, L., and Baffet, G. (2008). RNAi-mediated ERK2 knockdown inhibits growth of tumor cells in vitro and in vivo. Oncogene 27, 5315–5325.
Blum, S., Moore, A. N., Adams, F., and Dash, P. K. (1999). A mitogen-activated protein kinase cascade in the CA1/CA2 subfield of the dorsal hippocampus is essential for long-term spatial memory. J. Neurosci. 19, 3535–3544.
Boggio, E. M., Putignano, E., Sassoè-Pognetto, M., Pizzorusso, T., and Giustetto, M. (2007). Visual stimulation activates ERK in synaptic and somatic compartments of rat cortical neurons with parallel kinetics. PLoS One 2, e604.
Bozon, B., Davis, S., and Laroche, S. (2003). A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron 40, 695–701.
Brambilla, R., Gnesutta, N., Minichiello, L., White, G., Roylance, A. J., Herron, C. E., Ramsey, M., Wolfer, D. P., Cestari, V., Rossi-Arnaud, C., Grant, S. G., Chapman, P. F., Lipp, H. P., Sturani, E., and Klein, R. (1997). A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nature 390, 281–286.
Brown, M. W., and Aggleton, J. P. (2001). Recognition memory: what are the roles of the perirhinal cortex and hippocampus? Nat. Rev. Neurosci. 2, 51–61.
Brown, M. W., and Xiang, J. Z. (1998). Recognition memory: neuronal substrates of the judgement of prior occurrence. Prog. Neurobiol. 55, 149–189.
Cancedda, L., Putignano, E., Impey, S., Maffei, L., Ratto, G. M., and Pizzorusso, T. (2003). Patterned vision causes CRE-mediated gene expression in the visual cortex through PKA and ERK. J. Neurosci. 23, 7012–7020.
Cohen-Matsliah, S. I., Seroussi, Y., Rosenblum, K., and Barkai, E. (2008). Persistent ERK activation maintains learning-induced long-lasting modulation of synaptic connectivity. Learn. Mem. 15, 756–761.
Costa, R. M., Federov, N. B., Kogan, J. H., Murphy, G. G., Stern, J., Ohno, M., Kucherlapati, R., Jacks, T., and Silva, A. J. (2002). Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415, 526–530.
Costa, R. M., Yang, T., Huynh, D. P., Pulst, S. M., Viskochil, D. H., Silva, A. J., and Brannan, C. I. (2001). Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat. Genet. 27, 399–405.
Davis, H. P., and Squire, L. R. (1984). Protein synthesis and memory: a review. Psychol. Bull. 96, 518–559.
De Rosa, R., Garcia, A. A., Braschi, C., Capsoni, S., Maffei, L., Berardi, N., and Cattaneo, A. (2005). Intranasal administration of nerve growth factor (NGF) rescues recognition memory deficits in AD11 anti-NGF transgenic mice. Proc. Natl. Acad. Sci. U.S.A. 102, 3811–3816.
Di Cristo, G., Berardi, N., Cancedda, L., Pizzorusso, T., Putignano, E., Ratto, G. M., and Maffei, L. (2001). Requirement of ERK activation for visual cortical plasticity. Science 292, 2337–2340.
Doyère, V., Debiec, J., Monfils, M. H., Schafe, G. E., and LeDoux, J. E. (2007). Synapse-specific reconsolidation of distinct fear memories in the lateral amygdala. Nat. Neurosci. 10, 414–416.
Dupont, E., Stevens, L., Cochon, L., Falempin, M., Bastide, B., and Canu, M. H. (2011). ERK is involved in the reorganization of somatosensory cortical maps in adult rats submitted to hindlimb unloading. PLoS One 6, e17564. doi: 10.1371/journal.pone.0017564
Ennaceur, A., and Delacour, J. (1988). A new one-trial test for neurobiological studies of memory in rats. 1: behavioral data. Behav. Brain Res. 31, 47–59.
Farnsworth, C. L., Freshney, N. W., Rosen, L. B., Ghosh, A., Greenberg, M. E., and Feig, L. A. (1995). Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature 376, 524–527.
Fasano, S., D’Antoni, A., Orban, P. C., Valjent, E., Putignano, E., Vara, H., Pizzorusso, T., Giustetto, M., Yoon, B., Soloway, P., Maldonado, R., Caboche, J., and Brambilla, R. (2009). Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) controls activation of extracellular signal-regulated kinase (ERK) signaling in the striatum and long-term behavioral responses to cocaine. Biol. Psychiatry 66, 758–768.
Fremin, C., Ezan, F., Boisselier, P., Bessard, A., Pages, G., Pouysségur, J., and Baffet, G. (2007). ERK2 but not ERK1 plays a key role in hepatocyte replication: an RNAi-mediated ERK2 knockdown approach in wild-type and ERK1 null hepatocytes. Hepatology 45, 1035–1045.
Giese, K. P., Friedman, E., Telliez, J. B., Fedorov, N. B., Wines, M., Feig, L. A., and Silva, A. J. (2001). Hippocampus-dependent learning and memory is impaired in mice lacking the Ras-guanine-nucleotide releasing factor 1 (Ras-GRF1). Neuropharmacology 41, 791–800.
Goeldner, C., Reiss, D., Wichmann, J., Meziane, H., Kieffer, B. L., and Ouagazzal, A. M. (2008). Nociceptin receptor impairs recognition memory via interaction with NMDA receptor-dependent mitogen-activated protein kinase/extracellular signal-regulated kinase signaling in the hippocampus. J. Neurosci. 28, 2190–2198.
Griffiths, S., Scott, H., Glover, C., Bienemann, A., Ghorbel, M. T., Uney, J., Brown, M. W., Warburton, E. C., and Bashir, Z. I. (2008). Expression of long-term depression underlies visual recognition memory. Neuron 58, 186–194.
Kelleher, R. J. III, Govindarajan, A., Jung, H. Y., Kang, H., and Tonegawa, S. (2004). Translational control by MAPK signaling in long-term synaptic plasticity and memory. Cell 116, 467–479.
Kelly, A., Laroche, S., and Davis, S. (2003). Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J. Neurosci. 23, 5354–5360.
Lee, J. L., Everitt, B. J., and Thomas, K. L. (2004). Independent cellular processes for hippocampal memory consolidation and reconsolidation. Science 304, 839–843.
Leon, W. C., Bruno, M. A., Allard, S., Nader, K., and Cuello, A. C. (2010). Engagement of the PFC in consolidation and recall of recent spatial memory. Learn. Mem. 17, 297–305.
Li, S., Tian, X., Hartley, D. M., and Feig, L. A. (2006). The environment versus genetics in controlling the contribution of MAP kinases to synaptic plasticity. Curr. Biol. 16, 2303–2313.
Lin, P. Y., Wang, S. P., Tai, M. Y., and Tsai, Y. F. (2010). Differential involvement of medial prefrontal cortex and basolateral amygdala extracellular signal-regulated kinase in extinction of conditioned taste aversion is dependent on different intervals of extinction following conditioning. Neuroscience 171, 125–133.
Massey, P. V., Bhabra, G., Cho, K., Brown, M. W., and Bashir, Z. I. (2001). Activation of muscarinic receptors induces protein synthesis-dependent long-lasting depression in the perirhinal cortex. Eur. J. Neurosci. 14, 145–152.
Massey, P. V., Phythian, D., Narduzzo, K., Warburton, E. C., Brown, M. W., and Bashir, Z. I. (2008). Learning-specific changes in long-term depression in adult perirhinal cortex. J. Neurosci. 28, 7548–7554.
Mattingly, R. R., and Macara, I. G. (1996). Phosphorylation-dependent activation of the Ras-GRF/CDC25Mm exchange factor by muscarinic receptors and G-protein beta gamma subunits. Nature 382, 268–272.
Mayford, M., and Kandel, E. R. (1999). Genetic approaches to memory storage. Trends Genet. 15, 463–470.
Mazzucchelli, C., and Brambilla, R. (2000). Ras-related and MAPK signalling in neuronal plasticity and memory formation. Cell. Mol. Life Sci. 57, 604–611.
Mazzucchelli, C., Vantaggiato, C., Ciamei, A., Fasano, S., Pakhotin, P., Krezel, W., Welzl, H., Wolfer, D. P., Pagès, G., Valverde, O., Marowsky, A., Porrazzo, A., Orban, P. C., Maldonado, R., Ehrengruber, M. U., Cestari, V., Lipp, H. P., Chapman, P. F., Pouysségur, J., and Brambilla, R. (2002). Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 34, 807–820.
Murray, E. A., and Bussey, T. J. (1999). Perceptual-mnemonic functions of the perirhinal cortex. Trends Cogn. Sci. (Regul. Ed.) 3, 142–151.
Murray, E. A., Bussey, T. J., and Saksida, L. M. (2007). Visual perception and memory: a new view of medial temporal lobe function in primates and rodents. Annu. Rev. Neurosci. 30, 99–122.
Nader, K., and Hardt, O. (2009). A single standard for memory: the case for reconsolidation. Nat. Rev. Neurosci. 10, 224–234.
Samuels, I. S., Karlo, J. C., Faruzzi, A. N., Pickering, K., Herrup, K., Sweatt, J. D., Saitta, S. C., and Landreth, G. E. (2008). Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. 28, 6983–6995.
Schafe, G. E., Atkins, C. M., Swank, M. W., Bauer, E. P., Sweatt, J. D., and LeDoux, J. E. (2000). Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J. Neurosci. 20, 8177–8187.
Seoane, A., Massey, P. V., Keen, H., Bashir, Z. I., and Brown, M. W. (2009). L-type voltage-dependent calcium channel antagonists impair perirhinal long-term recognition memory and plasticity processes. J. Neurosci. 29, 9534–9544.
Suzuki, W. A., Zola-Morgan, S., Squire, L. R., and Amaral, D. G. (1993). Lesions of the perirhinal and parahippocampal cortices in the monkey produce long-lasting memory impairment in the visual and tactual modalities. J. Neurosci. 13, 2430–2451.
Sweatt, J. D. (2004). Mitogen-activated protein kinases in synaptic plasticity and memory. Curr. Opin. Neurobiol. 14, 311–317.
Thomas, G. M., and Huganir, R. L. (2004). MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci. 5, 173–183.
Tian, X., and Feig, L. A. (2005). Age-dependent participation of Ras-GRF proteins in coupling calcium-permeable AMPA glutamate receptors to Ras/Erk signaling in cortical neuron. J. Biol. Chem. 281, 7578–7582.
Tronson, N. C., Schrick, C., Fischer, A., Sananbenesi, F., Pagès, G., Pouysségur, J., and Radulovic, J. (2008). Regulatory mechanisms of fear extinction and depression-like behavior. Neuropsychopharmacology 33, 1570–1583.
Tsokas, P., Ma, T., Iyengar, R., Landau, E. M., and Blitzer, R. D. (2007). Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J. Neurosci. 27, 5885–5894.
Wan, H., Aggleton, J. P., and Brown, M. W. (1999). Different contributions of the hippocampus and perirhinal cortex to recognition memory. J. Neurosci. 19, 1142–1148.
Warburton, E. C., Koder, T., Cho, K., Massey, P. V., Duguid, G., Barker, G. R., Aggleton, J. P., Bashir, Z. I., and Brown, M. W. (2003). Cholinergic neurotransmission is essential for perirhinal cortical plasticity and recognition memory. Neuron 38, 987–996.
Weeber, E. J., Levenson, J. M., and Sweatt, J. D. (2002). Molecular genetics of human cognition. Mol. Interv. 2, 376–391.
Winters, B. D., and Bussey, T. J. (2005). Transient inactivation of perirhinal cortex disrupts encoding, retrieval, and consolidation of object recognition memory. J. Neurosci. 25, 52–61.
Winters, B. D., Saksida, L. M., and Bussey, T. J. (2008). Object recognition memory: neurobiological mechanisms of encoding, consolidation and retrieval. Neurosci. Biobehav. Rev. 32, 1055–1070.
Keywords: ERK1,2, recognition memory, perirhinal cortex, synaptic plasticity
Citation: Silingardi D, Angelucci A, De Pasquale R, Borsotti M, Squitieri G, Brambilla R, Putignano E, Pizzorusso T and Berardi N (2011) ERK pathway activation bidirectionally affects visual recognition memory and synaptic plasticity in the perirhinal cortex. Front. Behav. Neurosci. 5:84. doi: 10.3389/fnbeh.2011.00084
Received: 09 September 2011; Accepted: 05 December 2011;
Published online: 28 December 2011.
Edited by:
Gilberto Fisone, Karolinska Institutet, SwedenReviewed by:
Kobi Rosenblum, Haifa University, IsraelGilberto Fisone, Karolinska Institutet, Sweden
Copyright: © 2011 Silingardi, Angelucci, De Pasquale, Borsotti, Squitieri, Brambilla, Putignano, Pizzorusso and Berardi. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Nicoletta Berardi, Istituto di Neuroscienze del CNR, Via G. Moruzzi 1, 56124 Pisa, Italy. e-mail:YmVyYXJkaUBpbi5jbnIuaXQ=
†Davide Silingardi and Andrea Angelucci and Roberto De Pasquale and Tommaso Pizzorusso and Nicoletta Berardi have contributed equally to this work.