 Miriam A. Vogt
Miriam A. Vogt Dragos Inta
Dragos Inta Alessia Luoni2
Alessia Luoni2 Marco A. Riva
Marco A. Riva Peter Gass
Peter Gass- 1Department of Psychiatry and Psychotherapy, RG Animal Models in Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim / Heidelberg University, Mannheim, Germany
- 2Department of Pharmacological and Biomolecular Sciences, University of Milan, Milan, Italy
The cyclic AMP (cAMP)-response element binding protein (CREB) is an activity-dependent transcription factor playing a role in synaptic plasticity, learning and memory, and emotional behavior. However, the impact of Creb ablation on rodent behavior is vague as e.g., memory performance of different Creb mutant mice depends on the specific type of mutation per se but additionally on the background and learning protocol differences. Here we present the first targeted ablation of CREB induced during adulthood selectively in principal forebrain neurons in a pure background strain of C57BL/6 mice. All hippocampal principal neurons exhibited lack of CREB expression. Mutant mice showed a severe anxiety phenotype in the openfield and novel object exploration test as well as in the Dark-Light Box Test, but unaltered hippocampus-dependent long-term memory in the Morris water maze and in context dependent fear conditioning. On the molecular level, CREB ablation led to CREM up regulation in the hippocampus and frontal cortex which may at least in part compensate for the loss of CREB. BDNF, a postulated CREB target gene, was down regulated in the frontal lobe but not in the hippocampus; neurogenesis remained unaltered. Our data indicate that in the adult mouse forebrain the late onset of CREB ablation can, in case of memory functionality, be compensated for and is not essential for memory consolidation and retrieval during adulthood. In contrast, the presence of CREB protein during adulthood seems to be pivotal for the regulation of emotional behavior.
Introduction
Inhibition of transcription factors or protein synthesis is hypothesized to block consolidation of short-term memory into long-term memory and affect emotional and depression-related circuits. The role of cAMP signaling in simple forms of learning and memory was described for the first time in the sea snail Aplysia (Brunelli et al., 1976), a result confirmed by comparable results in Drosophila some years later (Byers et al., 1981; Yin et al., 1994). To date, a multitude of studies revealed CREB, the cAMP-response element binding protein as the main element in converting short- to long-term memory (Barco et al., 2003; Kim et al., 2013). Apart from its role in learning and memory, CREB is known to modify the sensitivity to rewarding and aversive drugs within the Nucleus accumbens (Dinieri et al., 2009; Bilbao et al., 2014), resets the circadian clock by its phosphorylation at SER142 (Gau et al., 2002) and is upregulated in the hippocampus by chronic antidepressant treatment, linking Creb activity to the pathogenesis and therapy of depression and regulation of emotion (Gass and Riva, 2007).
CREB is part of the family of activating transcription factors including besides CREB the cAMP responsive element modulator (CREM) and the activating transcription factors 1–7 (ATF; Brindle and Montminy, 1992). Due to the complex structure of the Creb gene with multiple exons and introns, the encoded proteins vary with splicing variants and different properties (Blendy et al., 1996; Mayr and Montminy, 2001). This creates a wide range of possibilities of genetic approaches to modify expression, formation or function of Creb (for an overview see Kida and Serita, 2014). The homozygous null mutation of Creb is perinatally lethal, therefore no behavioral analysis is available, the mice suffer from apoptosis and degeneration of sensory neurons combined with reduced axonal growth and projections (Rudolph et al., 1998; Lonze et al., 2002). The development of temporal and spatially restricted mutants using the Cre/loxP system allowed generating mutant mice with CREB ablation in forebrain neurons only (Mantamadiotis et al., 2002), however, this line was developed on a Crem negative background to avoid compensatory effects of Crem regulation, although it is known that Crem ablation leads to altered emotional behavior and hyperactivity (Maldonado et al., 1999). Behavioral phenotyping aiming on emotional regulation was mostly focusing on depression-related changes, although CrebαΔ mice, which carry a constitutive deletion of the α and Δ isoforms of CREB, show baseline alterations in anxiety-like behavior (Conti et al., 2002). In humans, a rare genomic variant could link bipolar disorder including co-morbid anxiety to intracellular pathways under the regulation of Creb (Kerner et al., 2013). Mice with virus-mediated hippocampal Creb ablation demonstrate no baseline anxiety alterations but enhanced neurogenesis, which could lead in longer time frames to emotional changes (Gundersen et al., 2013), an aspect which we wanted to evaluate in our mice by assessing adult neurogenesis. Learning and memory tasks were conducted for the first time 1992 in mice generated by using the tetracycline-controlled transactivator/operator system expressing a dominant negative inhibitor of Creb (Walton et al., 1992) active in CamKIIα-positive cells of the forebrain only. As predicted from studies with invertebrates, long-term memory but not short-term memory was impaired, although the mutant mice exhibited no contextual fear conditioning deficit (Pittenger et al., 2002). The blockade of Creb using a tamoxifen-inducible expression of a dominant negative Creb repressor was used by Kida et al. (2002) to dissect the crucial role of CREB in consolidation of contextual fear conditioning.
Even though a plethora of mice with alterations of Creb expression or CREB function is available (Kida et al., 2002), the behavioral phenotype has not been consistent across lines. The direct comparison of the multiple studies was aggravated by the fact that the mouse lines differ in their genetic system, e.g., if the line was generated with partial gene deletions as the CrebαΔ mice (Bourtchuladze et al., 1994) tissue specific deletions as deletions only in the forebrain (Pittenger et al., 2002), time-dependent induction (Kida et al., 2002), or if additional genes were knocked down to avoid compensatory effects e.g., of Crem (Mantamadiotis et al., 2002). While studies about emotional behavior regarding anxiety were almost missing, learning performance was, already within single lines, dependent on protocol (with or without spaces between trials in the Morris water maze) or background strains. We have chosen here the inducible Cre/loxP recombination system with tamoxifen-controlled gene manipulation in mice on a pure C57BL/6 background suitable for behavioral phenotyping. Creb ablation was induced during adulthood to analyze emotional behavior, learning and memory and additionally, mRNA levels of CREM, ATF-1 and BDNF in hippocampus and frontal cortex.
Materials and Methods
Generation of Crebflox/floxCamKCreERT2 Mice and Induction of Cre-Mediated Recombination with Tamoxifen
Transgenic mice expressing the tamoxifen-inducible fusion protein composed of the Cre recombinase and the mutated ligand binding domain of the human estrogen receptor under the control of the αCamKII promoter (CamKCreERT2; Erdmann et al., 2007), were crossed with homozygous Creb1flox/flox mice (Mantamadiotis et al., 2002) to generate Crebflox/floxCamKCreERT2 mice. Systemic tamoxifen injection to all mice led to the excision of the exon 10 of the Creb 1 allele in excitatory neurons of the forebrain region including hippocampus, amygdala, cortex and striatum only in mutant mice. Crebflox/floxCamKCreERT2 mice were treated with tamoxifen at the age of 9–12 weeks. Tamoxifen (Sigma, Deisenhofen, Germany) was dissolved in ethanol absolute (100 mg tamoxifen per 1 ml ethanol) and then diluted 1:10 with sunflower seed oil (Sigma). Crebflox/floxCamKCreERT2 mice (“mutants”) and Crebflox/flox mice (“controls”) were injected intraperitoneally twice a day with 100 μl (i.e., 1 mg) tamoxifen, for 5 days. All mouse lines were bred for at least 10 generations on a C57BL6/N background.
Behavioral Experiments
The behavioral testing started when the animals were approximately 5 months old. Only male mice were tested. Two weeks prior to and during experiments, animals were single-housed in a 12 h reversed dark-light cycle with lights on at 7 pm and supplied with food and water ad libitum. All experiments were performed during the dark phase. All genotypes were verified with immunohistochemistry controlling for CREB ablation. All experimental procedures were approved by the Animal Welfare Committee (Regierungspräsidium Karlsruhe, 35-9185-81-G/193/11) and carried out according to the European Communities Council Directive of 24 November 1986 (86/609/EEC). One month after termination of the behavioral experiments, n = 12 mice per genotype, respectively, received a intraperitoneal injection of BrdU (50 mg/kg) and were sacrificed 1 day later to evaluate neurogenesis.
Openfield and novel object exploration test
Activity monitoring was conducted in a square shaped, white open field, measuring 50 × 50 cm2 and illuminated from above by 25 lx. Mice were placed individually into the arena and monitored for 20 min by a Video camera (Sony CCD IRIS). The resulting data were analyzed using the image processing system EthoVision 3.1 (Noldus Information Technology, Wageningen, the Netherlands). Parameters assessed were total distance moved, velocity, and time in center, which was defined as the area 10 cm distant from the walls. After 10 min Open field Test, a novel object was introduced into the middle of the arena. Object exploration was assessed for the subsequent 10 min, assessing latency of first approach, and the total number of approaches (N = 24 per genotype) as previously described (Berkel et al., 2012).
Dark-Light Box
The Dark-Light-Box consisted of two plastic chambers, connected by a small tunnel. The dark chamber measured 20 × 15 cm2 and was covered by a lid. The adjacent chamber, measuring 30 × 15 cm2, was white and illuminated from above by 600 Lux. Mice were placed into the dark compartment and latency to first exit, number of exits and total time in the light compartment were recorded for 5 min (N = 24 per genotype) as described earlier (Fuss et al., 2010).
Contextual fear conditioning
For the contextual conditioning, mice were individually placed into the conditioning chamber (58 × 30 × 27 cm3, TSE, Bad Homburg, Germany) and allowed to habituate for 2 min before subjecting them to the unconditioned stimulus (2 s of continuous footshock of 0.8 mA). 24 h after training, context conditioning was assessed by measuring freezing, defined as a complete lack of movements apart from respiration. Context learning was tested in the same conditioning chamber which was used during the training. Freezing behavior was scored manually at intervals of 10 s for 5 min (N = 14 Crebflox/floxCamKCreERT2 mutant mice, n = 10 controls) as described earlier (Fleischmann et al., 2003).
Morris water maze
Animals (n = 14 controls, n = 10 mutants) were trained for 4 days with a total of 24 trials (6 trials per day) to swim in a water-filled circular pool (diameter: 150 cm) and find a platform (14 × 14 cm2, plexiglas) (acquisition phase). In each swim trial during the acquisition phase, in which the position of the platform was kept unchanged, mice were left in the pool for a maximum of 120 s or they found the platform. On the fifth day the platform was removed and the mouse was recorded for 60 s (probe trial). With a video camera suspended above the center of the pool, the swim tracks of the mice were analyzed using the image processing system EthoVision X8 (Noldus Information Technology, Wageningen, the Netherlands). The following variables from the recorded paths were analyzed: time to find platform (s), length of swim path (m), velocity (cm/s), percent of time spent moving, percent of time spent within a rim of 20 cm from the wall, in a ring in which all 4 possible platform positions are included and additionally in the center. For the probe trial, additionally percent of time in target and other quadrants, as well as number of crossings of the former platform area in comparison to 3 other possible platform positions were analyzed as formerly described (Vogt et al., 2008). Animals showing movement less than 80% of the total time (mean over all acquisition trials and the probe trial) were excluded from the analysis of the experiment, which resulted in N = 8 controls and N = 7 mutants.
Hotplate Test
The mice were tested on the hotplate test (ATLab, Vendargues, France). Temperature was set at 53°C (±0.3°C) and a 45 s cut-off was determined to prevent injury of mice. Latency to first reaction, i.e., licking hind paws or jumping, was assessed (N = 24 per genotype) as described elsewhere (Chourbaji et al., 2008).
Immunohistochemistry
One day after the BrdU injection all animals were perfused transcardially with 4% paraformaldehyde (PFA), brains were postfixed overnight and 40 μm thick coronal sections were cut on a vibratome as described elsewhere (Böttiger et al., 1999). Every sixth section of each animal was processed free-floating.
Dividing cells were visualized using primary rat monoclonal anti-BrdU-antibody (1:1000, MAK 2060, Linaris, Wertheim-Bettingen, Germany). To determine the absolute number of BrdU-labeled cells, we used the peroxidase method (ABC system, Vectastain, Vector Laboratories, Burlingame, CA, USA) with biotinylated anti-rat antibodies (1:500; Dianova, Hamburg, Germany). Nickel-intensified diaminobenzidine (DAB, Sigma) was used as chromogen substrate (Römer et al., 2010).
CREB expression was analyzed by incubating the sections overnight with polyclonal rabbit anti-CREB antibody (1:50000, Cell Signaling). After washes with PBST, sections were incubated with secondary antibody (CREB: biotinylated anti-rabbit IgG, Vector laboratories), 1:400 for 2 h at room temperature. After washes, sections were processed with avidin-biotinylated horseradish peroxidase complex (Vectastain ABC kit; Vector Laboratories) in PBST for 1 h at RT, and the reaction was visualized using Nickel-3,30-diaminobenzidine (DAB; Strekalova et al., 2003).
Cell Counts
To evaluate proliferation, the total number of BrdU-positive cells was assessed in 1-in-6 series of sections (240 μm apart) from all animals. BrdU-positive cells were counted throughout the rostro-caudal extent of the granule cell layer (GCL) using a 40 × objective. The optical dissector method was modified as described previously, in that cells appearing in the uppermost focal plane, when focusing into the section, were not counted. The resulting numbers were then multiplied by six to obtain the estimated total cell number (N = 12 per genotype).
RNA Preparation for qRT-PCR and Analysis of mRNA Levels
Naïve male mice not used for behavioral analyses (N = 12 per genotype) were killed by cervical dislocation and the brain was quickly removed. Hippocampus and frontal cortex were dissected on ice, immediately frozen on dry ice and stored by −80°C until further analyses. Total RNA was isolated by single step guanidinium isothiocyanate/phenol extraction using PureZol RNA isolation reagent (Bio-Rad Laboratories, Italy), according with the manufacturer’s instructions and quantified by spectrophotometric analysis. Following total RNA extraction, the samples were processed for real-time polymerase chain reaction (qPCR) to assess mRNA levels. An aliquot of each sample was treated with DNase to avoid DNA contamination.
RNA was analyzed by TaqMan qRT-PCR instrument (CFX384 real time system, Bio-Rad Laboratories) using the iScriptTM one-step RT-PCR kit for probes (Bio-Rad Laboratories). Samples were run in 384 well formats in triplicate as multiplexed reactions with a normalizing internal control (36B4) as described elsewhere (Chourbaji et al., 2012).
TaqMan gene expression assays were purchased from Eurofins MWG-Operon (Germany) and have the following sequences:

Thermal cycling was initiated with an incubation at 50°C for 10 min (RNA retrotranscription) and then at 95°C for 5 min (TaqMan polymerase activation). After this initial step, 39 cycles of PCR were performed. Each PCR cycle consisted of heating the samples at 95°C for 10 s to enable the melting process and then for 30 s at 60°C for the annealing and extension reaction. Relative target gene expression was calculated according to the 2(-Delta Delta C(T)) method.
Statistical Analysis
Statistical analyses for behavioral tests, the mRNA levels and the cell counts were performed using SPSS Statistics 20 for Windows. Inter-group comparisons were calculated by Student’s t-tests. Where appropriate, the model was complemented by within subject factors to explore the dependence of genotype effects on time (data from acquisition phase of the Morris Water Maze). Significance for all tests was assumed for p < 0.05. Data are presented as means ± standard error (S.E.M.).
Results
Crebflox/floxCamKCreERT2 mice were treated with tamoxifen at the age of 9–12 weeks (adulthood), which resulted in the ablation of CREB in excitatory forebrain neurons including all principal neurons of the hippocampus (Figures 1A,B). We detected a reduction of CREB expression in additional regions of the limbic system important e.g., for reward mechanisms (nucleus accumbens, Figures 1C,D) and for fear memory (basolateral amygdala, Figures 1E,F).
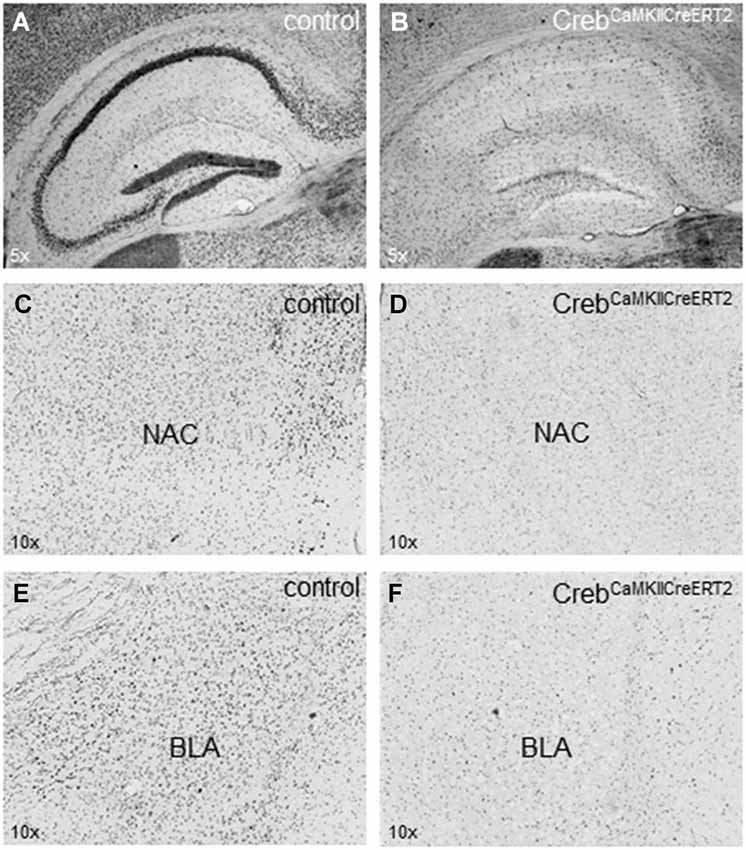
Figure 1. Expression of CREB in hippocampus, nucleus accumbens and amygdala. (A) Hippocampal expression of CREB in (tamoxifen-treated) control animals with regular CREB expression and (B) in CrebCamKCreERT2 mutant mice after induced ablation with tamoxifen in the adulthood with a definite loss of CREB expression in the pyramidal cell layer but not in putative GABAergic interneurons sparsely distributed in all hippocampal layers. (C) Expression of CREB in the nucleus accumbens (NAC) important for rewarding mechanisms in (tamoxifen-treated) control animals with regular CREB expression and (D) in CrebCamKCreERT2 mutant mice after induced ablation with tamoxifen in the adulthood. (E) Expression of CREB in the basolateral amygdala (BLA) important for fear-related memory in control and (F) in CrebCamKCreERT2 mutant mice after induced ablation with tamoxifen in the adulthood. N = 12 per genotype.
Mice with a CREB ablation during adulthood thrived well and did not show defects concerning body growth or bodyweight after induction and especially during testing, and gained weight normally (repeated measurement ANOVA after onset of treatment: Ftime(3,66) = 39,197 p < 0.001).
In the openfield and novel object exploration test, Crebflox/floxCamKCreERT2 mice, when compared to controls, did not show locomotor disturbances with regard to the distance traveled and velocity (Figures 2A,B). Concerning the spatial pattern of movement, Crebflox/floxCamKCreERT2 mutant mice displayed reduced time in center and distance from the nearest wall (Figures 2C,D; repeated measurement ANOVA center time: Fgenotype(1,42) = 6.814 p = 0.012; distance to walls: Fgenotype(1,42) = 5.429 p = 0.025), reflecting an anxiety-like behavior, accompanied by an increased latency to explore the novel object (Figure 2E; student t-test: p = 0.059) and reduced number of approaches (Figure 2F; p = 0.017).
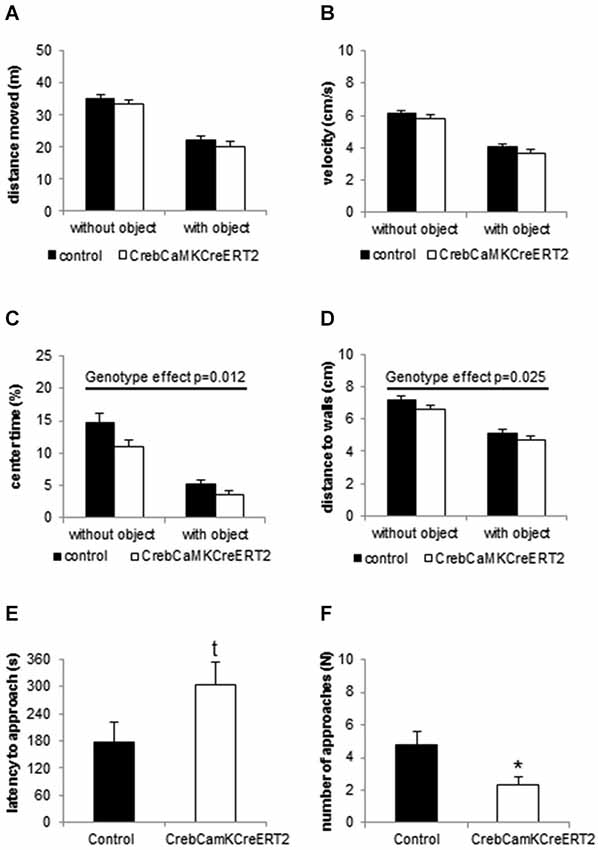
Figure 2. Locomotion and Novel object exploration in the openfield. (A) Distance moved and (B) velocity is unaltered in CrebCamKCreERT2 mutant mice compared to controls in the openfield without object (first 10 min) and after introduction of the novel object. (C) Center time and (D) mean distance to walls is reduced in CrebCamKCreERT2 mutant mice, reflecting anxiety-like behavior. (E) Latency to approach the novel object is enhanced (p = 0.059) and (F) the number of approaches is decreased in CrebCamKCreERT2 mutant mice compared to controls. Black bars: controls, white bars: CrebCamKCreERT2 mutant mice, * p < 0.05, N = 24 per genotype.
In the Dark-Light Box, Crebflox/floxCamKCreERT2 mutant mice demonstrated anxiety-like behavior by entering the bright compartment with a significantly increased latency (Figure 3A; p = 0.019), displayed significantly less exits to the bright compartment (Figure 3B; p < 0.001) and stayed less (but non-significantly) in the light (Figure 3C; p = 0.193).
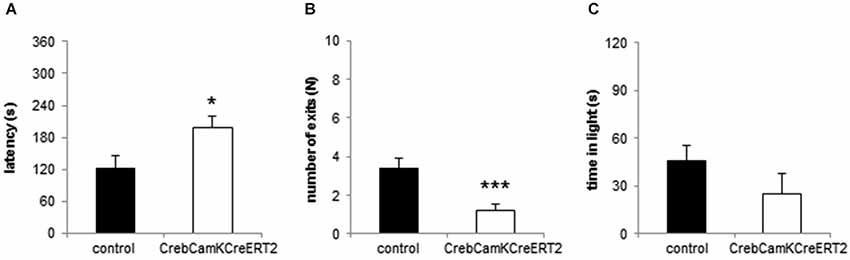
Figure 3. Anxiety-like behavior in the Dark-Light Box. (A) The latency to enter the light compartment is enhanced and the (B) number of exits is reduced in CrebCamKCreERT2 mutant mice compared to controls. (C) Time in the bright compartment is unaltered in CrebCamKCreERT2 mutant mice compared to controls. Black bars: controls, white bars: CrebCamKCreERT2 mutant mice, * p < 0.05, *** p < 0.001, N = 24 per genotype.
In the Morris water maze, irrespective of the genotype, all mice demonstrated an improved path length (Figure 4A) and a diminished latency to find the platform over time (repeated measurement ANOVA: path length Ftime(11,143) = 21.441 p < 0.001, latency to find platform: Ftime(11,143) = 9.674 p < 0.001, velocity: Ftime(11,143) = 6.732 p < 0.001). Concerning the spatial swimming patterns, both groups focused their swimming to the relevant areas of the pool and decreased wall time and the time in the center over trials (repeated measurement ANOVA: Time in center: Ftime(11,143) = 2.232 p = 0.016, Time in wall zone: Ftime(11,143) = 6.203 p < 0.001, Time in platform ring zone: Ftime(11,143) = 5.050 p < 0.001). No significant differences between Creb mutant mice and the control group were found for all measured parameters including velocity and spatial swimming pattern in different zones of the pool, including wall zone and center. During the probe trial, both groups exhibited a significant preference for the trained platform position, measured as time spent in the target quadrant (Figure 4B) and crossings above the previous platform position (two-way ANOVA: time in quadrant: Fplace(1,13) = 7.093 p = 0.020, crossings Fplace(1,13) = 11.122 p = 0.005). Both Creb mutant mice and controls preferred the target quadrant to the same extent and showed comparable number of crossings (two-way ANOVA: time in quadrant: Fgenotype(1,13) = 0.238 p = 0.634, crossings Fgenotype(1,13) = 1.445 p = 0.251).
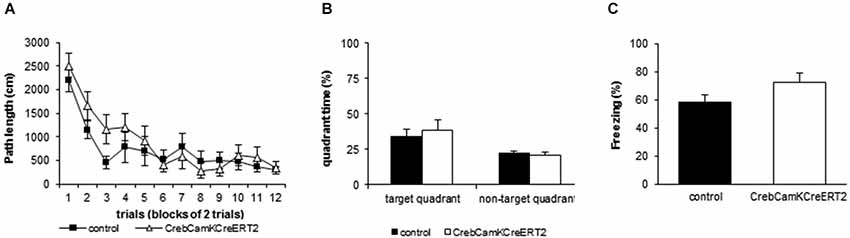
Figure 4. Learning and Memory performance in Morris water maze and contextual fear conditioning. (A) Mutant mice with a loss of Creb in the adulthood do not show disturbed learning during acquisition in the Morris water maze as represented by the distance moved to reach the platform (repeated measurement ANOVA factor time: F(11,132) = 18.975, p < 0.001). All data points represent the mean (±SEM), N = 8 controls and N = 7 mutants. (B) Induced loss of Creb in the adulthood does not affect spatial reference memory in the Morris water maze as represented by the time spent in the target zone vs. the means of the 3 other quadrants during the probe trial (two-way ANOVA factor place: F(1,13) = 19.122, p < 0.001, factor genotype: F(1,13) = 1.752, p = 0.208). All data points represent the mean (+SEM), N = 8 controls and N = 7 mutants. (C) Induced loss of Creb in the adulthood does not affect hippocampus-dependent associative learning in the fear conditioning paradigm. All data points represent the mean (+SEM) percent time spent freezing during context replacement 24 h after training. N = 14 controls, N = 10 mutants.
Pain perception, measured as latency to the first reaction on the hotplate (licking hind paws or jumping), was unaltered in Creb mutant mice (controls: 23.30 ±2.02 s; mutants: 23.67 ±2.61 s).
In the Fear Conditioning paradigm, mice were exposed to a single foot shock (0.8 mA) in a certain context. When replaced in this context 24 h later, all mice showed context memory to the same extent as measured by the time spent freezing (Figure 4C; controls: 58.10 ± 5.3%; mutants: 72.08 ± 6.9%).
Crem, ATF-1 and BDNF mRNA level were analyzed in the hippocampus and the frontal cortex of naïve male mice. Crem mRNA level were significantly up regulated in Crebflox/floxCamKCreERT2 mutant mice when compared to control animals (Figure 5A; student t-tests: hippocampus: p < 0.001, frontal cortex p < 0.001). Conversely, ATF-1 level remained unaltered in both hippocampus and frontal cortex (Figure 5B). In contrast, BDNF mRNA level were slightly, although significantly, decreased in the frontal cortex of mutant mice (Figure 5C; p = 0.0017), whereas its expression was unchanged in the hippocampus.
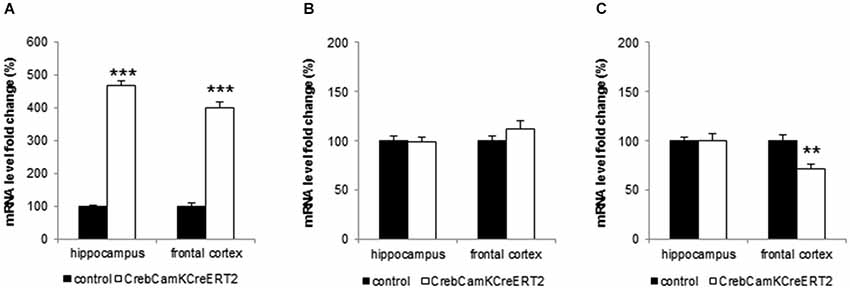
Figure 5. Analysis of CREM, ATF-1 and BDNF- mRNA level in hippocampus and frontal cortex. (A) Mutant mice with a loss of Creb at adulthood exhibit significant higher mRNA levels of CREM in the hippocampus and frontal cortex. (B) Induced loss of Creb did not affect ATF-1 mRNA level in hippocampus and frontal cortex. (C) Mutant mice express significantly less BDNF mRNA in the frontal cortex but its levels remain unchanged in the hippocampus. All data points represent mean (+SEM), stated as fold change of the control level. Black bars: controls, white bars: CrebCamKCreERT2 mutant mice, ** p < 0.01, *** p < 0.001, N = 12 per genotype.
Proliferation, measured as number of BrdU-positive cells, was unaltered in Crebflox/floxCamKCreERT2 mice (data not shown).
Discussion
We used an improved Cre/loxP recombination system with tamoxifen-controlled gene manipulation, for time- and region-restricted deletion of Creb (Erdmann et al., 2007). Gene ablation was induced at early adulthood (9–12 weeks) to avoid vulnerable phases as puberty and especially to exclude adaptations through compensatory mechanisms affecting neurodevelopment (Aguado et al., 2009; Nonaka, 2009). All animals were treated with tamoxifen to exclude treatment effects by tamoxifen, which had been confirmed in wild-type mice in particular in depression-related tasks (Vogt et al., 2008). The tamoxifen treatment led to an ablation of CREB protein in excitatory forebrain neurons (including hippocampus, nucleus accumbens and basolateral amygdala) expressing regulatory elements of the CamKIIα gene. The induction and the subsequent ablation of the CREB protein in tamoxifen-injected Crebflox/floxCamKCreERT2 mice occurred within a few days, as described in detail for the glucocorticoid receptor (GR) by Erdmann et al. (2007). The tamoxifen injections led to an expression pattern comparable to those described by Gundersen et al. (2013), where Crebflox/flox mice were injected with AAV-Cre virus in the hippocampus to induce the deletion.
To dissect anxiety-like behavior the animals underwent an openfield and Novel object exploration task as well as the Dark-Light Box test, classical approach- avoidance conflict tasks. Whereas locomotion was not altered, Creb mutant mice avoided the central part of the arena, and approached a novel object placed in the middle later and less often. The Dark-Light Box test revealed correspondingly higher anxiety levels in our Crebflox/floxCamKCreERT2 mice.
Several studies have shown that altered levels of CREB or pCREB, the phosphorylated active form of CREB, affect anxiety levels directly or indirectly. Mice with early onset of Creb ablation throughout the nervous system (CrebNesCre mice) and CrebαΔ knock-out mice with a hypomorphic allele and highly reduced CREB levels show in accordance with the present data higher anxiety levels in elevated zero maze, elevated plus maze Dark-Light Box and openfield (Valverde et al., 2004; Gur et al., 2007). Virus-mediated expression of a dominant negative form of CREB increased anxiety-like behavior, however in this case the target of gene ablation was the Nucleus accumbens shell only (Barrot et al., 2002), an effect which could be reversed by anxiolytic drug treatment or overexpression of Creb in the Nucleus accumbens (Barrot et al., 2005). In the mice used in this study, the nucleus accumbens was affected by the reduction of CREB levels, but it is rather unlikely that the complex anxiety phenotype found here is based on one affected brain structure as the shell of the Nucleus accumbens, although the Nucleus accumbens might be a crucial brain structure. Chronic social defeat reduced CREB and BDNF levels in the raphe, and increased in parallel anxiety levels (Boyarskikh et al., 2013). The knockdown of the corticotropin-releasing hormone receptor 2 (CRH2), known to phosphorylate CREB, resulted, as expected, in reduced pCREB and, additionally, higher anxiety levels (Kishimoto et al., 2000). Reduced pCREB levels via indirect effects combining several studies from different fields of neuroscience all resulted in increased anxiety levels analyzed in standard test of anxiety as openfield, elevated plus maze and/or Dark-Light Box test (for review of altered anxiety in CREB-, BDNF- and CRH1-mutant mice see also Urani et al., 2005).
Animal studies aiming on Alzheimers’ disease either focusing on the enzyme 12/15–lipoxygenase catalyzing the deoxygenation of polyunsaturated fatty acids, or focusing on the carboxy terminal fragments of the human amyloid precursor protein (betaCTF99) revealed CREB alterations and corresponding anxiety changes (Lee et al., 2006; Joshi et al., 2014). Correspondingly, the upregulation of Creb expression or pCREB levels was followed by reduced anxiety in studies using pharmacological approaches or knockout mice. Thus, the knockdown of the regulator of calcineurin 1 (RCAN1)—a protein regulating the calcium/calmodulin-dependent phosphatase calcineurin implicated in human anxiety disorders—increased in mice pCREB and BDNF levels. Moreover, these mice displayed innate reduced anxiety levels, an effect which could not be reversed by treatment with the selective serotonin reuptake inhibitor (SSRI) fluoxetine (Hoeffer et al., 2013). Interestingly, Mombereau et al. (2010) showed in CrebαΔ knock-out mice that Citalopram, another SSRI, could only be effective (and consequently decrease anxiety) when the animals expressed normal CREB levels. Serotonergic pathways driving not only SSRI response but furthermore CREB signaling were known to be affected as shown by Stewart et al. (2014). In this study, the endogenous regulator of G-protein signaling 6 (RGS6) implicated as inhibitor of 5-HT1a receptors, was knocked out which resulted in anxiety and pCREB level alterations. Interrelations of 5HT1a and Creb are furthermore demonstrated by Mombereau et al. (2010) who could show that CrebαΔ mice react with a blunted response in the 5-HT1a agonist-induced hypothermia test following a single administration of 8-OHDPAT.
BDNF levels appear to play an important role in Creb-dependent regulation of emotional behavior, since peripherally administered BDNF was observed to increase pCREB and, in parallel, to reduce anxiety (Schmidt and Duman, 2010). BDNF is widely discussed as factor altering emotional states including anxiety and depression (for a detailed review see Chourbaji et al., 2011). We analyzed BDNF mRNA levels and could indeed find a reduction in the frontal cortex, but unaltered levels in the hippocampus. In previous studies, we could show that a heterozygous deletion of BDNF was not per se responsible for changed emotional states (Chourbaji et al., 2004) but rather dependent on gene-environment interactions like different housing conditions in combination with the heterozygous deletion (Chourbaji et al., 2008). In general, the findings of BNDF levels affecting the emotional state is diverse (for a detailed review see Chourbaji et al., 2011) with positive or negative findings concerning the role of BDNF on anxiety dependent on the generated mouse lines (Monteggia et al., 2007; Autry et al., 2009). Keeping this in mind, the slightly reduced BDNF levels in the frontal cortex only could be a consequence of the Creb ablation but are an insufficient explanation for the altered anxiety levels seen in the Creb mutants analyzed here.
Another assumption could be that CREB changes (possibly via altered BDNF levels) neurogenesis or neurodegeneration (Mantamadiotis et al., 2002). Our data did not show altered proliferation in the mice; however our mice were over 5 months old, so proliferation rate after one BrdU injection was already quite low. In a study of (Li et al., 2009), the phosphodiesterase 4 inhibitor rolipram increased neurogenesis, pCREB levels and surprisingly decreased anxiety levels, an effect which could be completely stopped by Methylazoxymethanol (MAM) treatment, which blocks proliferation. This, additionally to the study of Gundersen et al. (2013), where virus-induced ablation in the hippocampus led to severely increased neurogenesis, could be a hint that neurogenesis is causative for the alterations although we could not show a correlation in these mice at a single time point.
CREB deficiency was often accompanied by adaptive up regulation of the Cre modulator CREM (Hummler et al., 1994; Gundersen et al., 2013), being in line, Crem knockout mice demonstrated reduced anxiety (Maldonado et al., 1999). To overcome adaptive effects of CREB/CREM up or down regulation, some studies used knockout strategies by deleting both Creb and Crem (Mantamadiotis et al., 2002); if effects of Creb knockout were therefore driven by adaptive Crem regulations could not be excluded, because both factors were absent. In our mice, we could detect a strong up regulation of CREM in both hippocampus and frontal cortex. Mice with a knockout of Crem show low anxiety levels in several anxiety tests like the elevated Plus Maze, Open Field or elevated Zero Maze (Maldonado et al., 1999). This altered emotional phenotype was accompanied by a severe hyperactivity (Maldonado et al., 1999), a behavioral feature which we did not discover in the mice used in this study. Due to the high expression of Crem mRNA, it could be assumed that these levels may contribute to behavioral consequences as the altered emotional state found here. The missing activity/locomotor changes may be explained by a lack of developmental effects due to the late onset of the Creb ablation. ATF-1, the third player besides CREB and CREM in the transcriptional unit stated by Pittenger et al. (2002) was up or down regulated in the hippocampus or frontal cortex. This may not completely exclude the possibility of a role of ATF-1 but the possibility of playing a leading role is rather small.
In conclusion, there are several plausible mechanisms which can explain the anxious phenotype of Crebflox/floxCamKCreERT2 mice. Future studies will have to clarify the molecular downstream mechanisms responsible for the phenotype seen here.
As Creb is known to play a role in synaptic plasticity, learning and memory, we analyzed hippocampus-dependent spatial reference memory by conducting a Morris water maze (Vogt et al., 2008). Both acquisition of the task and memory retrieval was not impaired in Crebflox/floxCamKCreERT2 mice.
In previous studies, various Creb mutant mice displayed deficits in both acquisition and spatial reference memory of the water maze task (Bourtchuladze et al., 1994; Gass et al., 1998; Pittenger et al., 2002; Balschun et al., 2003). CrebαΔ knock-outs exhibited profound memory deficits (Bourtchuladze et al., 1994), which depended strongly, however, on the genetic background (Gass et al., 1998; Graves et al., 2002). CrebNesCre mice also showed strong impairment in memory acquisition and retrieval, while mice with ablation later in development and restricted to the forebrain (CrebCamKCre7 mice) did not exhibit alterations during acquisition or probe trial (Balschun et al., 2003). This suggests that early induction of Creb ablation leads to developmental deficits that cannot be compensated in adulthood.
To further determine the role of CREB during adulthood in hippocampus-dependent associative learning, we exposed Crebflox/floxCamKCreERT2 mice to a foot shock-context conditioning procedure and tested the consolidation into long-term memory. To exclude alterations in pain perception as confounding factors, a hotplate test was performed, revealing an unaltered nociception of Creb mutants. In previous studies fear conditioning paradigms have demonstrated robust deficits in several Creb mutants (Bourtchuladze et al., 1994; Gass et al., 1998; Graves et al., 2002). When placed into the same context 24 h after conditioning, Crebflox/floxCamKCreERT2 mice revealed the same level of memory performance as controls.
Similar to the water maze task, the extent of memory deficits in CrebαΔ knock-out mice in contextual fear conditioning was dependent on the genetic background (Bourtchuladze et al., 1994; Gass et al., 1998; Graves et al., 2002). In contrast, neither CrebNesCre nor CrebCamKCre7 mice were impaired in contextual fear conditioning (Balschun et al., 2003). However, the tamoxifen-inducible expression of a dominant negative CREB repressor 6–12 h before conditioning impaired memory consolidation during adulthood (Kida et al., 2002). The present study demonstrates that the loss of Creb during adulthood can be fully compensated when the ablation is not immediately induced before the conditioning procedure. A re-balanced network could be the reason that we could not find alterations as recent studies showed that the network state during learning, e.g., the level of CREB in single neurons of the lateral amygdala, enhances the possibility of the cell to be part of a neuronal ensemble encoding fear memory (Kim et al., 2013).
Up to date, it had not been possible to breed at least one of the different Creb mutant mouse lines on an isogenic background. Therefore, all CrebαΔ knock-out mice investigated had been on different, sometimes randomly mixed backgrounds (Bourtchuladze et al., 1994; Gass et al., 1998; Graves et al., 2002; Briand and Blendy, 2013). Similarly, CrebNesCre and CrebCamKCre7 mice were on a randomly mixed C57BL/6 and 129SvEv background (Balschun et al., 2003), while the tamoxifen-inducible repressor mice were bred as F1 generation of C57BL/6 and C3H (Kida et al., 2002) or of C57BL/6 and129 mice (Graves et al., 2002). Notably, the mice used in our study allow for the first time an induced Creb ablation in a pure inbred strain regularly used for behavioral analyses.
The behavioral data of the anxiety tests and two gold standard tests assessing hippocampal learning and memory have demonstrated that the induction of Creb ablation during adulthood does impair neither spatial reference memory nor context dependent conditioning, but highly impairs emotional behavior. It can be stated that the induced disturbances of CREB pathways can lead to behavioral and molecular consequences. In humans, this relation towards emotional regulation seems also to be relevant as stated by Kerner et al. (2013) linking rare genomic variants of patients with bipolar disorder and co-morbid anxiety as well as panic disorders (Domschke et al., 2003) to CREB signaling pathways.
Author Contributions
Miriam A. Vogt, Dragos Inta, Hasan Elkin and Peter Gass conceived this work, Miriam A. Vogt, Dragos Inta, Hasan Elkin, Natascha Pfeiffer and Peter Gass designed it, Miriam A. Vogt, Dragos Inta, Natascha Pfeiffer and Alessia Luoni acquired data, Miriam A. Vogt, Dragos Inta, Natascha Pfeiffer and Alessia Luoni analyzed data and interpreted data together with Hasan Elkin, Peter Gass and Marco A. Riva. Miriam A. Vogt and Dragos Inta drafted the work; all authors critically revised it and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was funded in part by grants to Peter Gass from the Deutsche Forschungsgemeinschaft (SFB636-TP3) and from the German Ministry for Education and Research (BMBF, 01GQ1003B) National Bernstein Network for Computational Neuroscience.
References
Aguado, F., Díaz-Ruiz, C., Parlato, R., Martínez, A., Carmona, M. A., Bleckmann, S., et al. (2009). The CREB/CREM transcription factors negatively regulate early synaptogenesis and spontaneous network activity. J. Neurosci. 29, 328–333. doi: 10.1523/JNEUROSCI.5252-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Autry, A. E., Adachi, M., Cheng, P., and Monteggia, L. M. (2009). Gender-specific impact of brain-derived neurotrophic factor signaling on stress-induced depression-like behavior. Biol. Psychiatry 66, 84–90. doi: 10.1016/j.biopsych.2009.02.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Balschun, D., Wolfer, D. P., Gass, P., Mantamadiotis, T., Welzl, H., Schütz, G., et al. (2003). Does cAMP response element-binding protein have a pivotal role in hippocampal synaptic plasticity and hippocampus-dependent memory? J. Neurosci. 23, 6304–6314.
Barco, A., Pittenger, C., and Kandel, E. R. (2003). CREB, memory enhancement and the treatment of memory disorders: promises, pitfalls and prospects. Expert Opin. Ther. Targets 7, 101–114. doi: 10.1517/14728222.7.1.101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrot, M., Olivier, J. D., Perrotti, L. I., Dileone, R. J., Berton, O., Eisch, A. J., et al. (2002). CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc. Natl. Acad. Sci. U S A 99, 11435–11440. doi: 10.1073/pnas.172091899
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Barrot, M., Wallace, D. L., Bolaños, C. A., Graham, D. L., Perrotti, L. I., Neve, R. L., et al. (2005). Regulation of anxiety and initiation of sexual behavior by CREB in the nucleus accumbens. Proc. Natl. Acad. Sci. U S A 102, 8357–8362. doi: 10.1073/pnas.0500587102
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Berkel, S., Tang, W., Treviño, M., Vogt, M., Obenhaus, H. A., Gass, P., et al. (2012). Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum. Mol. Genet. 21, 344–357. doi: 10.1093/hmg/ddr470
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bilbao, A., Rieker, C., Cannella, N., Parlato, R., Golda, S., Piechota, M., et al. (2014). CREB activity in dopamine D1 receptor expressing neurons regulates cocaine-induced behavioral effects. Front. Behav. Neurosci. 8:212. doi: 10.3389/fnbeh.2014.00212
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blendy, J. A., Kaestner, K. H., Schmid, W., Gass, P., and Schutz, G. (1996). Targeting of the CREB gene leads to up-regulation of a novel CREB mRNA isoform. EMBO J. 15, 1098–1106.
Böttiger, B. W., Teschendorf, P., Krumnikl, J. J., Vogel, P., Galmbacher, R., Schmitz, B., et al. (1999). Global cerebral ischemia due to cardiocirculatory arrest in mice causes neuronal degeneration and early induction of transcription factor genes in the hippocampus. Brain Res. Mol. Brain Res. 65, 135–142. doi: 10.1016/s0169-328x(98)00298-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bourtchuladze, R., Frenguelli, B., Blendy, J., Cioffi, D., Schutz, G., and Silva, A. J. (1994). Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79, 59–68. doi: 10.1016/0092-8674(94)90400-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Boyarskikh, U. A., Bondar, N. P., Filipenko, M. L., and Kudryavtseva, N. N. (2013). Downregulation of serotonergic gene expression in the Raphe nuclei of the midbrain under chronic social defeat stress in male mice. Mol. Neurobiol. 48, 13–21. doi: 10.1007/s12035-013-8413-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Briand, L. A., and Blendy, J. A. (2013). Not all stress is equal: CREB is not necessary for restraint stress reinstatement of cocaine-conditioned reward. Behav. Brain Res. 246, 63–68. doi: 10.1016/j.bbr.2013.02.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brindle, P. K., and Montminy, M. R. (1992). The CREB family of transcription activators. Curr. Opin. Genet. Dev. 2, 199–204. doi: 10.1016/S0959-437X(05)80274-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brunelli, M., Castellucci, V., and Kandel, E. R. (1976). Synaptic facilitation and behavioral sensitization in Aplysia: possible role of serotonin and cyclic AMP. Science 194, 1178–1181. doi: 10.1126/science.186870
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Byers, D., Davis, R. L., and Kiger, J. A. Jr. (1981). Defect in cyclic AMP phosphodiesterase due to the dunce mutation of learning in Drosophila melanogaster. Nature 289, 79–81. doi: 10.1038/289079a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chourbaji, S., Brandwein, C., and Gass, P. (2011). Altering BDNF expression by genetics and/or environment: impact for emotional and depression-like behaviour in laboratory mice. Neurosci. Biobehav. Rev. 35, 599–611. doi: 10.1016/j.neubiorev.2010.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chourbaji, S., Brandwein, C., Vogt, M. A., Dormann, C., Hellweg, R., and Gass, P. (2008). Nature vs. nurture: can enrichment rescue the behavioural phenotype of BDNF heterozygous mice? Behav. Brain Res. 192, 254–258. doi: 10.1016/j.bbr.2008.04.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chourbaji, S., Hellweg, R., Brandis, D., Zorner, B., Zacher, C., Lang, U. E., et al. (2004). Mice with reduced brain-derived neurotrophic factor expression show decreased choline acetyltransferase activity, but regular brain monoamine levels and unaltered emotional behavior. Brain Res. Mol. Brain Res. 121, 28–36. doi: 10.1016/j.molbrainres.2003.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chourbaji, S., Hörtnagl, H., Molteni, R., Riva, M. A., Gass, P., and Hellweg, R. (2012). The impact of environmental enrichment on sex-specific neurochemical circuitries—effects on brain-derived neurotrophic factor and the serotonergic system. Neuroscience 220, 267–276. doi: 10.1016/j.neuroscience.2012.06.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Conti, A. C., Cryan, J. F., Dalvi, A., Lucki, I., and Blendy, J. A. (2002). cAMP response element-binding protein is essential for the upregulation of brain-derived neurotrophic factor transcription, but not the behavioral or endocrine responses to antidepressant drugs. J. Neurosci. 22, 3262–3268.
Dinieri, J. A., Nemeth, C. L., Parsegian, A., Carle, T., Gurevich, V. V., Gurevich, E., et al. (2009). Altered sensitivity to rewarding and aversive drugs in mice with inducible disruption of cAMP response element-binding protein function within the nucleus accumbens. J. Neurosci. 29, 1855–1859. doi: 10.1523/JNEUROSCI.5104-08.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Domschke, K., Kuhlenbäumer, G., Schirmacher, A., Lorenzi, C., Armengol, L., Dibella, D., et al. (2003). Human nuclear transcription factor gene CREM: genomic organization, mutation screening and association analysis in panic disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 117B, 70–78. doi: 10.1002/ajmg.b.10018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Erdmann, G., Schütz, G., and Berger, S. (2007). Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 8:63. doi: 10.1186/1471-2202-8-63
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fleischmann, A., Hvalby, O., Jensen, V., Strekalova, T., Zacher, C., Layer, L. E., et al. (2003). Impaired long-term memory and NR2A-type NMDA receptor-dependent synaptic plasticity in mice lacking c-Fos in the CNS. J. Neurosci. 23, 9116–9122.
Fuss, J., Ben Abdallah, N. M., Hensley, F. W., Weber, K. J., Hellweg, R., and Gass, P. (2010). Deletion of running-induced hippocampal neurogenesis by irradiation prevents development of an anxious phenotype in mice. PLoS One 5:e12769. doi: 10.1371/journal.pone.0012769
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gass, P., and Riva, M. A. (2007). CREB, neurogenesis and depression. Bioessays 29, 957–961. doi: 10.1002/bies.20658
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gass, P., Wolfer, D. P., Balschun, D., Rudolph, D., Frey, U., Lipp, H. P., et al. (1998). Deficits in memory tasks of mice with CREB mutations depend on gene dosage. Learn. Mem. 5, 274–288.
Gau, D., Lemberger, T., von Gall, C., Kretz, O., Le Minh, N., Gass, P., et al. (2002). Phosphorylation of CREB Ser142 regulates light-induced phase shifts of the circadian clock. Neuron 34, 245–253. doi: 10.1016/s0896-6273(02)00656-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Graves, L., Dalvi, A., Lucki, I., Blendy, J. A., and Abel, T. (2002). Behavioral analysis of CREB αΔ mutation on a B6/129 F1 hybrid background. Hippocampus 12, 18–26. doi: 10.1002/hipo.10003.abs
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gundersen, B. B., Briand, L. A., Onksen, J. L., Lelay, J., Kaestner, K. H., and Blendy, J. A. (2013). Increased hippocampal neurogenesis and accelerated response to antidepressants in mice with specific deletion of CREB in the hippocampus: role of cAMP response-element modulator tau. J. Neurosci. 33, 13673–13685. doi: 10.1523/JNEUROSCI.1669-13.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gur, T. L., Conti, A. C., Holden, J., Bechtholt, A. J., Hill, T. E., Lucki, I., et al. (2007). cAMP response element-binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J. Neurosci. 27, 7860–7868. doi: 10.1523/jneurosci.2051-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoeffer, C. A., Wong, H., Cain, P., Levenga, J., Cowansage, K. K., Choi, Y., et al. (2013). Regulator of calcineurin 1 modulates expression of innate anxiety and anxiogenic responses to selective serotonin reuptake inhibitor treatment. J. Neurosci. 33, 16930–16944. doi: 10.1523/JNEUROSCI.3513-12.2013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hummler, E., Cole, T. J., Blendy, J. A., Ganss, R., Aguzzi, A., Schmid, W., et al. (1994). Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc. Natl. Acad. Sci. U S A 91, 5647–5651. doi: 10.1073/pnas.91.12.5647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Joshi, Y. B., Di Meco, A., and Praticò, D. (2014). Overexpression of 12/15-lipoxygenase increases anxiety behavior in female mice. Neurobiol. Aging 35, 1032–1036. doi: 10.1016/j.neurobiolaging.2013.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kerner, B., Rao, A. R., Christensen, B., Dandekar, S., Yourshaw, M., and Nelson, S. F. (2013). Rare genomic variants link bipolar disorder with anxiety disorders to CREB-regulated intracellular signaling pathways. Front. Psychiatry 4:154. doi: 10.3389/fpsyt.2013.00154
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kida, S., Josselyn, S. A., Peña de Ortiz, S., Kogan, J. H., Chevere, I., Masushige, S., et al. (2002). CREB required for the stability of new and reactivated fear memories. Nat. Neurosci. 5, 348–355. doi: 10.1038/nn819
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kida, S., and Serita, T. (2014). Functional roles of CREB as a positive regulator in the formation and enhancement of memory. Brain Res. Bull. 105, 17–24. doi: 10.1016/j.brainresbull.2014.04.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, J., Kwon, J. T., Kim, H. S., and Han, J. H. (2013). CREB and neuronal selection for memory trace. Front. Neural Circuits 7:44. doi: 10.3389/fncir.2013.00044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kishimoto, T., Radulovic, J., Radulovic, M., Lin, C. R., Schrick, C., Hooshmand, F., et al. (2000). Deletion of crhr2 reveals an anxiolytic role for corticotropin-releasing hormone receptor-2. Nat. Genet. 24, 415–419. doi: 10.1038/74271
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, K. W., Im, J. Y., Song, J. S., Lee, S. H., Lee, H. J., Ha, H. Y., et al. (2006). Progressive neuronal loss and behavioral impairments of transgenic C57BL/6 inbred mice expressing the carboxy terminus of amyloid precursor protein. Neurobiol. Dis. 22, 10–24. doi: 10.1016/j.nbd.2005.09.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, Y. F., Huang, Y., Amsdell, S. L., Xiao, L., O’Donnell, J. M., and Zhang, H. T. (2009). Antidepressant- and anxiolytic-like effects of the phosphodiesterase-4 inhibitor rolipram on behavior depend on cyclic AMP response element binding protein-mediated neurogenesis in the hippocampus. Neuropsychopharmacology 34, 2404–2419. doi: 10.1038/npp.2009.66
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lonze, B. E., Riccio, A., Cohen, S., and Ginty, D. D. (2002). Apoptosis, axonal growth defects and degeneration of peripheral neurons in mice lacking CREB. Neuron 34, 371–385. doi: 10.1016/s0896-6273(02)00686-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maldonado, R., Smadja, C., Mazzucchelli, C., and Sassone-Corsi, P. (1999). Altered emotional and locomotor responses in mice deficient in the transcription factor CREM. Proc. Natl. Acad. Sci. U S A 96, 14094–14099. doi: 10.1073/pnas.96.24.14094
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mantamadiotis, T., Lemberger, T., Bleckmann, S. C., Kern, H., Kretz, O., Martin Villalba, A., et al. (2002). Disruption of CREB function in brain leads to neurodegeneration. Nat. Genet. 31, 47–54. doi: 10.1038/ng882
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mayr, B., and Montminy, M. (2001). Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2, 599–609. doi: 10.1038/35085068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mombereau, C., Gur, T. L., Onksen, J., and Blendy, J. A. (2010). Differential effects of acute and repeated citalopram in mouse models of anxiety and depression. Int. J. Neuropsychopharmacol. 13, 321–334. doi: 10.1017/s1461145709990630
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monteggia, L. M., Luikart, B., Barrot, M., Theobold, D., Malkovska, I., Nef, S., et al. (2007). Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol. Psychiatry 61, 187–197. doi: 10.1016/j.biopsych.2006.03.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nonaka, M. (2009). A Janus-like role of CREB protein: enhancement of synaptic property in mature neurons and suppression of synaptogenesis and reduced network synchrony in early development. J. Neurosci. 29, 6389–6391. doi: 10.1523/JNEUROSCI.1309-09.2009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pittenger, C., Huang, Y. Y., Paletzki, R. F., Bourtchouladze, R., Scanlin, H., Vronskaya, S., et al. (2002). Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus-dependent spatial memory. Neuron 34, 447–462. doi: 10.1016/s0896-6273(02)00684-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Römer, B., Pfeiffer, N., Lewicka, S., Ben-Abdallah, N., Vogt, M. A., Deuschle, M., et al. (2010). Finasteride treatment inhibits adult hippocampal neurogenesis in male mice. Pharmacopsychiatry 43, 174–178. doi: 10.1055/s-0030-1249095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rudolph, D., Tafuri, A., Gass, P., Hämmerling, G. J., Arnold, B., and Schütz, G. (1998). Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc. Natl. Acad. Sci. U S A 95, 4481–4486. doi: 10.1073/pnas.95.8.4481
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schmidt, H. D., and Duman, R. S. (2010). Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology 35, 2378–2391. doi: 10.1038/npp.2010.114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stewart, A., Maity, B., Wunsch, A. M., Meng, F., Wu, Q., Wemmie, J. A., et al. (2014). Regulator of G-protein signaling 6 (RGS6) promotes anxiety and depression by attenuating serotonin-mediated activation of the 5-HT1A receptor-adenylyl cyclase axis. FASEB J. 28, 1735–1744. doi: 10.1096/fj.13-235648
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strekalova, T., Zörner, B., Zacher, C., Sadovska, G., Herdegen, T., and Gass, P. (2003). Memory retrieval after contextual fear conditioning induces c-Fos and JunB expression in CA1 hippocampus. Genes Brain Behav. 2, 3–10. doi: 10.1034/j.1601-183x.2003.00001.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Urani, A., Chourbaji, S., and Gass, P. (2005). Mutant mouse models of depression: candidate genes and current mouse lines. Neurosci. Biobehav. Rev. 29, 805–828. doi: 10.1016/j.neubiorev.2005.03.020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Valverde, O., Mantamadiotis, T., Torrecilla, M., Ugedo, L., Pineda, J., Bleckmann, S., et al. (2004). Modulation of anxiety-like behavior and morphine dependence in CREB-deficient mice. Neuropsychopharmacology 29, 1122–1133. doi: 10.1038/sj.npp.1300416
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vogt, M. A., Chourbaji, S., Brandwein, C., Dormann, C., Sprengel, R., and Gass, P. (2008). Suitability of tamoxifen-induced mutagenesis for behavioral phenotyping. Exp. Neurol. 211, 25–33. doi: 10.1016/j.expneurol.2007.12.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Walton, K. M., Rehfuss, R. P., Chrivia, J. C., Lochner, J. E., and Goodman, R. H. (1992). A dominant repressor of cyclic adenosine 3′,5′-monophosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol. Endocrinol. 6, 647–655. doi: 10.1210/me.6.4.647
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yin, J. C., Wallach, J. S., Del Vecchio, M., Wilder, E. L., Zhou, H., Quinn, W. G., et al. (1994). Induction of a dominant negative CREB transgene specifically blocks long-term memory in Drosophila. Cell 79, 49–58. doi: 10.1016/0092-8674(94)90399-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: mouse, CREB, anxiety, tamoxifen induction, learning, memory, CREM, BDNF
Citation: Vogt MA, Inta D, Luoni A, Elkin H, Pfeiffer N, Riva MA and Gass P (2014) Inducible forebrain-specific ablation of the transcription factor Creb during adulthood induces anxiety but no spatial/contextual learning deficits. Front. Behav. Neurosci. 8:407. doi: 10.3389/fnbeh.2014.00407
Received: 18 July 2014; Accepted: 10 November 2014;
Published online: 27 November 2014.
Edited by:
Riccardo Brambilla, San Raffaele Scientific Institute and University, ItalyReviewed by:
Angel Barco, Instituto de Neurociencias de Alicante, SpainDimitri De Bundel, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2014 Vogt, Inta, Luoni, Elkin, Pfeiffer, Riva and Gass. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam A. Vogt, Department of Psychiatry and Psychotherapy, RG Animal Models in Psychiatry, Central Institute of Mental Health, Medical Faculty Mannheim / Heidelberg University, J5, 68159 Mannheim, Germany e-mail:bWlyaWFtLnZvZ3RAemktbWFubmhlaW0uZGU=