 Dean Nizetic
Dean Nizetic Christopher L. Chen4,5
Christopher L. Chen4,5 Edward H. Koo
Edward H. Koo- 1Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore, Singapore
- 2The LonDownS Consortium, Wellcome Trust, London, UK
- 3The Blizard Institute, Barts and The London School of Medicine, Queen Mary University of London, London, UK
- 4Department of Psychological Medicine and Memory Aging and Cognition Centre, National University Health System, Singapore, Singapore
- 5Department of Pharmacology, National University of Singapore, Singapore, Singapore
- 6Agency for Science, Technology and Research (A*Star), Institute of Molecular Cell Biology, Singapore, Singapore
- 7Departments of Medicine and Physiology, Yong Loo Lin School of Medicine, National University of Singapore, Singapore, Singapore
- 8Department of Neurosciences, University of California, San Diego, San Diego, CA, USA
People with Down syndrome (DS) virtually all develop intellectual disability (ID) of varying degree of severity, and also have a high risk of early Alzheimer's disease (AD). ID prior to the onset of dementia, and its relationship to the onset of dementia in DS is a complex phenomenon influenced by many factors, and scarcely understood. Unraveling the causative factors and modulators of these processes remains a challenge, with potential to be informative for both ID and AD, for the development of early biomarkers and/or therapeutic approaches. We review the potential relative and inter-connected roles of the chromosome 21 gene for amyloid precursor protein (APP), in both pathological conditions. Rare non-DS people with duplication of APP (dupAPP) get familial early onset AD (FEOAD) with virtually 100% penetrance and prominent cerebrovascular pathology, but don't suffer from ID before dementia onset. All of these features appear to be radically different in DS. On the other hand, rare individuals with partial trisomy 21 (T21) (with APP, but not DS-critical region in trisomy) have been described having ID. Likewise, partial T21 DS (without APP trisomy) show a range of ID, but no AD pathology. We review the multi-faceted roles of APP that might affect cognitive functioning. Given the fact that both Aβ secretion and synaptic maturation/plasticity are dependent on neuronal activity, we explore how this conflicting inter-dependency might affect cognitive pathogenesis in a dynamic way in DS, throughout the lifespan of an individual.
Introduction
Virtually all people with Down syndrome (DS) show some degree of intellectual disability (ID), due to many factors, including a certain degree of cognitive dysfunction caused by the pathobiology of trisomy 21 (T21) (Epstein, 2002; Head et al., 2012). On the other hand, T21 is the most common known genetic cause of obligatory development of pathological hallmarks of AD in the brain tissue (Mann, 1988a; Lott and Head, 2005). This happens extremely early in virtually all people with DS (in the 30-s) (Mann, 1988a,b). The cause of this is apparently an extra copy of APP (amyloid precursor protein gene located on chromosome 21), as one individual with DS at age 74 (with no dementia, and no amyloid pathology in the brain) has been described who was born with partial chromosome 21 trisomy that did not include APP (Prasher et al., 1998). In rare families of non-DS (euploid) people, APP micro-duplications (dupAPP) are responsible for Familial Early Onset Alzheimer's Disease (FEOAD), with virtually 100% penetrance by age 60 and a median onset age of clinical dementia of 41–51, and dupAPP has so far never been seen in any human unlinked to FEOAD (Rovelet-Lecrux et al., 2006, 2007; Sleegers et al., 2006; Kasuga et al., 2009; Thonberg et al., 2011; Cohn-Hokke et al., 2012; McNaughton et al., 2012; Wallon et al., 2012). In adults with DS, the average age at onset of dementia varies greatly, with ~25% starting extremely early (in early thirties), but a good 25–50% having a much delayed onset (compared to dupAPP), or not developing dementia at all by age >60 (Holland et al., 1998; Sekijima et al., 1998; Tyrrell et al., 2001; Coppus et al., 2006; McCarron et al., 2014). Also, cerebrovascular pathology, mainly intra-cerebral hemorrhage is a prominent symptom of dupAPP (McCarron et al., 1998; Rovelet-Lecrux et al., 2006; Sleegers et al., 2006; Kasuga et al., 2009; McNaughton et al., 2012; Wallon et al., 2012), whereas it is rarely seen in DS (Belza and Urich, 1986). This is in spite of abundant deposits of amyloid in blood vessels (congophilic angiopathy) in both conditions (see below). On the other hand, people with dupAPP largely have a normal development, intellectual and social functioning (families) prior to dementia, in contrast to people with DS.
This indicates that mechanisms are in action in DS individuals (that dupAPP individuals don't have), that cause ID, can accelerate clinical onset of dementia, as well as factors that can delay onset or protect from overt dementia and associated intra-cerebral hemorrhage.
While dupAPP patients, by definition, are seen in families exhibiting FEOAD, this implies that this is a self-selected group of people that largely lack ID prior to dementia. On the other hand, rare individuals with partial T21 (with APP, but not DS-critical chr21 region in trisomy) have been described having I.Q. ranging from ~30 to <80, prior to dementia-prone age (Korbel et al., 2009; summarized in Table 1). This could be caused by an overdosed action of APP, but equally other genes included in the partial trisomy, mostly located in the “gene poor” proximal half of chromosome 21 (Groet et al., 1998). We review potential roles for APP, that go beyond the AD paradigm, and could contribute to the modulation of ID.
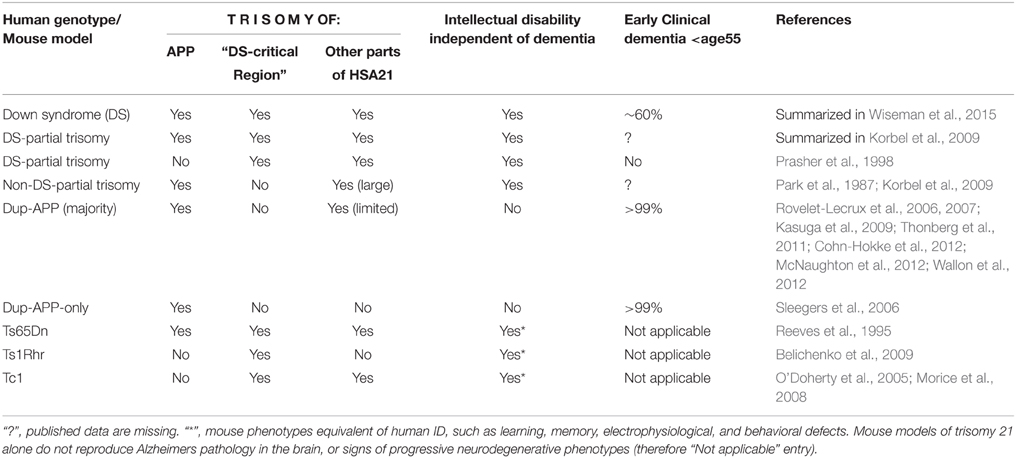
Table 1. Effects of the trisomy of APP and other segments of human chromosome 21 on development of intellectual disability (independently of dementia), and on presence of early clinical dementia.
Beyond Alzheimer's: Multiple Roles of APP Potentially Affecting Cognitive Dysfunction in DS (Summarized in Table 2)
T21 causes synaptic plasticity defects and dendritic spine abnormalities in human brains (Marin-Padilla, 1976; Ferrer and Gullotta, 1990) as early as 19 weeks in utero (Weitzdoerfer et al., 2001). This pathology can be modeled using mouse models of DS (Haas et al., 2013). Dendritic spines and their plasticity are also the site of important pathology in neurodevelopmental conditions such as Rett and FraX syndromes (Troca-Marín et al., 2012; Chang et al., 2013). Their loss contributes to pathogenesis of AD and PD (McGowan et al., 2006; Schulz-Schaeffer, 2010), though specific morphological differences and causes are likely unique to each condition. Generation and deposition of beta-amyloid peptides (Aβ40 and Aβ42) is linked with destruction of dendritic spines and synaptic loss in AD (McGowan et al., 2006; Shrestha et al., 2006). APP protein (Kamenetz et al., 2003), as well as other chromosome-21 encoded gene products, may have important functions in synaptic biology (Wang et al., 2013).
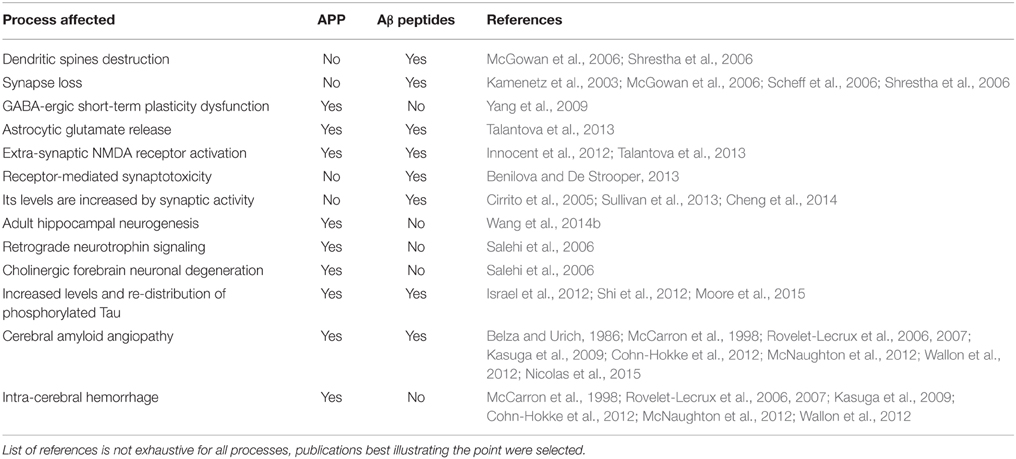
Table 2. An overview of a variety of processes affected by an increased dose of APP protein and/or its derivative Aβ peptides, that may contribute in DS to pathogenesis of Alzheimer's dementia or cognitive impairment (I.D.), or both.
Down's syndrome causes overexpression of miR-155, a chromosome 21–encoded microRNA that negatively regulates C/EBPb, thereby reducing sorting nexin 27 (SNX27) expression and resulting in synaptic dysfunction (Wang et al., 2013). SNX27 is a novel activity-dependent signaling molecule that has the ability to decode the Ras signal and transduce the plasticity stimuli to the delivery of postsynaptic AMPA receptors (Loo et al., 2014). So, SNX27 signaling is also activity-dependent: the more neuronal activity, the bigger chances of seeing pathology due to inability to raise sufficient SNX27 levels. On the other hand, SNX27 acts as a γ-secretase interaction partner to promote dissociation of the γ-secretase complex, thus decreasing its proteolytic activity, thereby reducing the generation of all Aβ peptides (Wang et al., 2014a). The effects of the apparent reduction of SNX27 in DS would therefore be expected to further increase Aβ levels, (by lessening the dissociation of the γ-secretase complex), and therefore worsen AD-pathology. However, the end results of this on AD pathogenesis are far from simple, and need to be carefully further investigated. While inhibition of γ-secretase activity might reduce the levels of neurotoxic Aβ42, intriguingly, very recent results on hiPSC-derived neurons show that chemical inhibition of γ-secretase activity in T21 neurons dramatically increased levels of Tau, the principal constituent of neurofibrillary tangles. This could actually have pro-dementia effects (Moore et al., 2015). The picture is even more intriguing, as the same study found that addition of a γ-secretase modulator (GSM), E2012, decreased Tau levels in T21 neurons (that showed an otherwise increased Tau levels; Moore et al., 2015).
Synaptic dysfunction is an early feature of AD, likely much before significant Aβ deposition (Arendt, 2009). There is a line of thought that AD neurodegenerative processes begin by alterations in synapse function/structure leading to synapse loss, prior to significant neuronal loss. Consistent with this, disturbance in synaptic integrity is detected in patients with mild cognitive impairment (MCI), which is sometimes perceived as an early stage of AD (Scheff et al., 2006). Loss of synaptic markers is a predictor of disease progression in AD (Selkoe, 2002). Aβ, generated from proteolytic processing of APP, has been shown to disrupt synapses (Kamenetz et al., 2003; Scheff et al., 2006; Shrestha et al., 2006) and, conversely, synaptic activity is an important factor regulating Aβ levels (Cirrito et al., 2005; Sullivan et al., 2013; Cheng et al., 2014). Aβ was also found to induce astrocytic glutamate release, extra-synaptic NMDA receptor activation, and synaptic loss (Talantova et al., 2013).
APP and Aβ have other, direct and indirect roles in functioning of synapses. Overexpression of APP affects Cav1.2 L-type calcium channel levels and through this influence GABAergic short-term plasticity (Yang et al., 2009). APP may contribute to postsynaptic mechanisms via the regulation of the surface trafficking of excitatory N-methyl-D-aspartate (NMDA) receptors (Innocent et al., 2012). Aβ was also shown binding to a number of receptors embedded in neuronal plasma membrane, (such as PrP, EphB2, FcγRIIb, and PirB), potentially contributing to receptor-mediated synaptotoxic pathways (Benilova and De Strooper, 2013).
There are also compelling evidence for alterations in synaptic function/plasticity in DS mouse models. For instance, altered excitatory/inhibitory balance is known to modify synaptic plasticity in DS mouse models (Kleschevnikov et al., 2004; Souchet et al., 2014). Importantly, increased APP expression in mouse models of DS has other deleterious effects that could be linked with neurodevelopmental milestone delays (and resulting ID): APP controls adult hippocampal neurogenesis, maintaining the tone of action of inhibitory GABAergic interneurons (Wang et al., 2014b). Interestingly, in the Ts65Dn partial trisomy 16 mice, defects in retrograde neurotrophin signaling and cholinergic forebrain neuronal degeneration are specifically related to extra APP gene dosage (Salehi et al., 2006). Similarly, endosomal abnormalities in the form of enlarged organelles which are characteristic changes in brains of both AD and DS individuals and also a consequence of increased APP expression (Cataldo et al., 2003). Whether these alterations are due to elevated APP or Aβ, or both, is not clear although there are suggestions from cultured neurons that endosomal dysfunction may be Aβ-independent (Jiang et al., 2010). Also, in lymphoblastoid cell lines carrying amyloid precursor protein (APP) microduplications causing autosomal dominant EOAD, enlarged endosomes were absent, suggesting that APP overexpression alone is not involved in the modification of early endosomes, but overexpression of other chromosome 21 genes plays an important role (Cossec et al., 2012). Finally, APP whole protein was also shown binding to Aβ, adding to the complexity of the potentially pathological interactions (Lorenzo et al., 2000).
Why are Vascular and Mixed Dementia not Prevalent in DS?
Mixed dementia (MD)—defined as the coexistence of Alzheimer's disease (AD) and cerebrovascular disease (CVD) (Rockwood, 2003) has been identified as one of the most common subtypes of dementia by autopsy-based epidemiological studies (Skoog et al., 1993; Snowdon et al., 1997). The presence and degree of CVD modulates the cognitive picture of AD (Dong et al., 2013). Neuropathological studies have shown that infarcts increase the odds of dementia in patients with equivalent AD burden by adding to the deleterious effects of AD pathology (Schneider et al., 2007). While most common in hypertensive individuals, intracerebral hemorrhage has been reported in 20–50% of APP-Dup cases (Rovelet-Lecrux et al., 2006, 2007; Kasuga et al., 2009; Cohn-Hokke et al., 2012; McNaughton et al., 2012; Wallon et al., 2012), whereas individuals with DS are generally protected from this pathology. Reasons for this protection in DS individuals are unknown, but are potentially very important. Both dupAPP and DS show severe cerebral amyloid angiopathy (CAA) (Belza and Urich, 1986; McCarron et al., 1998; Rovelet-Lecrux et al., 2006, 2007; Kasuga et al., 2009; Cohn-Hokke et al., 2012; McNaughton et al., 2012; Wallon et al., 2012) which renders blood vessels more susceptible to vessel wall breakdown and subsequent hemorrhage. It remains to be answered whether more general disturbances of vascular physiology seen in DS (but not in dupAPP) have anything to do with the apparent protection from vascular dementia and higher frequency of cerebral hemorrhages. Several effects caused by T21 have been described that could affect vascular biology (Vis et al., 2009; Draheim et al., 2010). T21 has a powerful effect on inhibition of angiogenesis, and a reduced response to pro-angiogenic cytokines, such as VEGF-A (Arron et al., 2006). This biological feature of T21 is one of the explanations for the reduced incidence of solid tissue tumors in DS (Baek et al., 2009; Yang and Reeves, 2011; Nizetic and Groet, 2012). This is attributed to at least 7 genes on HSA21, and full mechanisms are not completely understood (Zorick et al., 2001; Ryeom et al., 2003; Arron et al., 2006; Baek et al., 2009; Reynolds et al., 2010; Yang and Reeves, 2011; Nizetic and Groet, 2012).
As regards atherosclerotic cardiovascular disease (Vis et al., 2009), although abnormalities in lipid metabolism, which are associated with high risk of premature atherosclerosis in the general population (increased triglycerides, decreased HDL), are frequently seen in patients with DS (Dörner et al., 1984; Bocconi et al., 1997; Corsi et al., 2005), and despite reduced physical activity and high rates of obesity (de Winter et al., 2012), atherosclerosis and coronary artery disease related mortality is surprisingly low (Baird and Sadovnick, 1988; Corsi et al., 2005; Lott and Head, 2005), a finding that led some authors to conclude that DS may represent an atheroma-free model of disease (Murdoch et al., 1977). Interestingly, hyper-triglyceridaemia, increased obesity and low exercise rates are common in adults with DS (Haas et al., 2013), and high cholesterol levels have been associated with risk of developing dementia (Chang et al., 2013). However, some cardiovascular risk factors, including hypertension, atherosclerosis, and smoking (McGowan et al., 2006; Troca-Marín et al., 2012) that are thought to contribute to the development of dementia in the general population (Schulz-Schaeffer, 2010), are lower among adults with DS.
Other DS-specific biological features may also play a role. While DS neurons show a severe down-regulation of mitochondrial function (Busciglio and Yankner, 1995; Roat et al., 2007; Murray et al., 2015), multiple studies indicate that reducing mitochondrial function can protect against aging and age-associated diseases (Trifunovic and Ventura, 2014). DS cells demonstrate adaptive down-regulation of mitochondrial function for survival under increased ROS conditions (Helguera et al., 2013).
Is Intensified Neuronal Activity Good or Bad for Life-long DS Cognitive Functioning?
Recent work on mouse models has shown that hyperactivity of GABAergic interneurons in mouse models of DS over-inhibits hippocampal cortical excitatory neurons (Fernandez et al., 2007; Kleschevnikov et al., 2011). This has resulted in the first clinical trials in adults with DS for the improvement of cognitive functions, for the cognitive enhancement with GABA-α5 inverse agonists (Martínez-Cué et al., 2014). So, increased activity of one type of neurons is causing a decreased activity of another type of neurons in DS.
However, by increasing the neuronal activity of hippocampal cortical excitatory neurons, in theory, we also increase Aβ production and release. In fact, it has been demonstrated that both Aβ secretion, and synaptic plasticity are dependent on neuronal activity (Kamenetz et al., 2003; Cirrito et al., 2005; Scheff et al., 2006; Shrestha et al., 2006; Cheng et al., 2014; Lundgren et al., 2014). It remains unclear though whether inhibitory neurons have as much APP as excitatory neurons, or, whether over activity of inhibitory vs. excitatory neurons drive more Aβ generation. A proteolytic fragment (p25) of the cdk5-activator is generated as a function of neuronal activity, and it regulates synaptic plasticity and Aβ-induced cognitive impairment (Seo et al., 2014). Aβ-generation, endosomal trafficking and secretion (Wu et al., 2011; Sullivan et al., 2013; Lundgren et al., 2014) and Aβ-dependent Tau translocation to excitatory synapses (Frandemiche et al., 2014) are all neuronal activity-dependent. On the other hand, synaptic plasticity and synaptic maturation have also been shown to be neuronal activity-dependent processes (Fukazawa et al., 2003; Segal, 2005, 2010; Bosch and Hayashi, 2012; Heimer-McGinn et al., 2013; Ramiro-Cortés and Israely, 2013).
This seemingly controversial roles of neuronal activity level in DS pathology have also repercussions when it comes to therapeutic management approaches: sleep deprivation contributes to increased Aβ generation/secretion (Kang et al., 2009) as well as reduced clearance (Xie et al., 2013), and sleep deprivation in DS was shown to affect cognitive function (Brooks et al., 2015). On the other hand, deep brain stimulation was proposed as one of the intervention approaches to ameliorate cognitive dysfunction in AD (Boggio et al., 2011). More research is needed in this direction, as clearly opposing consequences could be reached, if the approaches are not better understood, and accordingly fine-tuned.
In conclusion, there are clearly many open questions on the inter-relation between pathogenic processes that affect neuronal development, synaptic plasticity, neuronal aging and longevity, and AD in DS. We have not had the space or scope in this mini-review to mention the potential modulating action of all other chromosome 21 genes that might influence this process, besides APP. Much more research is needed using high-resolution dissection of individual chr21 gene contributions using mouse models and human iPSC modeling. Such efforts should be coordinated and inter-disciplinary, including clinical dementia and cognitive assessments, and imaging studies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors research is supported by: Lee Kong Chian School of Medicine, Nanyang Technological University Start-up Grant, Singapore Ministry of Education Academic Research Fund Tier 1(2014-T1-001-173) and The Wellcome Trust “LonDownS Consortium” Strategic Funding Award (098330/Z/12/Z) (DN), Singapore National Medical Research Council Senior Clinician Scientist Award (NMRC/CSA/032/2011), (CC), and NIH NS 84324 and Singapore NMRC/StaR/009/2012 (EK).
References
Arendt, T. (2009). Synaptic degeneration in Alzheimer's disease. Acta Neuropathol. 118, 167–179. doi: 10.1007/s00401-009-0536-x
Arron, J. R., Winslow, M. M., Polleri, A., Chang, C. P., Wu, H., Gao, X., et al. (2006). NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441, 595–600. doi: 10.1038/nature04678
Baek, K. H., Zaslavsky, A., Lynch, R. C., Britt, C., Okada, Y., Siarey, R. J., et al. (2009). Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 459, 1126–1130. doi: 10.1038/nature08062
Baird, P. A., and Sadovnick, A. D. (1988). Causes of death to age 30 in Down syndrome. Am. J. Hum. Genet. 43, 239–248.
Belichenko, N. P., Belichenko, P. V., Kleschevnikov, A. M., Salehi, A., Reeves, R. H., and Mobley, W. C. (2009). The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J. Neurosci. 29, 5938–5948. doi: 10.1523/JNEUROSCI.1547-09.2009
Belza, M. G., and Urich, H. (1986). Cerebral amyloid angiopathy in Down's syndrome. Clin. Neuropathol. 5, 257–260.
Benilova, I., and De Strooper, B. (2013). Neuroscience. Promiscuous Alzheimer's amyloid: yet another partner. Science 341, 1354–1355. doi: 10.1126/science.1244166
Bocconi, L., Nava, S., Fogliani, R., and Nicolini, U. (1997). Trisomy 21 is associated with hypercholesterolemia during intrauterine life. Am. J. Obstet. Gynecol. 176, 540–543. doi: 10.1016/S0002-9378(97)70544-1
Boggio, P. S., Valasek, C. A., Campanhã, C., Giglio, A. C., Baptista, N. I., and Lapenta, O. M. (2011). Non-invasive brain stimulation to assess and modulate neuroplasticity in Alzheimer's disease. Neuropsychol. Rehabil. 21, 703–716. doi: 10.1080/09602011.2011.617943
Bosch, M., and Hayashi, Y. (2012). Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 22, 383–388. doi: 10.1016/j.conb.2011.09.002
Brooks, L. J., Olsen, M. N., Bacevice, A. M., Beebe, A., Konstantinopoulou, S., and Taylor, H. G. (2015). Relationship between sleep, sleep apnea, and neuropsychological function in children with Down syndrome. Sleep Breath. 19, 197–204. doi: 10.1007/s11325-014-0992-y
Busciglio, J., and Yankner, B. A. (1995). Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature 378, 776–779. doi: 10.1038/378776a0
Cataldo, A. M., Petanceska, S., Peterhoff, C. M., Terio, N. B., Epstein, C. J., Villar, A., et al. (2003). App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of down syndrome. J. Neurosci. 23, 6788–6792.
Chang, K. T., Ro, H., Wang, W., and Min, K. T. (2013). Meeting at the crossroads: common mechanisms in Fragile X and Down syndrome. Trends Neurosci. 36, 685–694. doi: 10.1016/j.tins.2013.08.007
Cheng, X., Wu, J., Geng, M., and Xiong, J. (2014). The role of synaptic activity in the regulation of amyloid beta levels in Alzheimer's disease. Neurobiol. Aging 35, 1217–1232. doi: 10.1016/j.neurobiolaging.2013.11.021
Cirrito, J. R., Yamada, K. A., Finn, M. B., Sloviter, R. S., Bales, K. R., May, P. C., et al. (2005). Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron 48, 913–922. doi: 10.1016/j.neuron.2005.10.028
Cohn-Hokke, P. E., Elting, M. W., Pijnenburg, Y. A., and van Swieten, J. C. (2012). Genetics of dementia: update and guidelines for the clinician. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 628–643. doi: 10.1002/ajmg.b.32080
Coppus, A., Evenhuis, H., Verberne, G. J., Visser, F., van Gool, P., and Eikelenboom, P. (2006). Dementia and mortality in persons with Down's syndrome. J. Intellect. Disabil. Res. 50(Pt 10), 768–777. doi: 10.1111/j.1365-2788.2006.00842.x
Corsi, M. M., Malavazos, A. E., Passoni, D., and Licastro, F. (2005). LDL receptor expression on T lymphocytes in old patients with Down syndrome. Immun. Ageing 2:3. doi: 10.1186/1742-4933-2-3
Cossec, J. C., Lavaur, J., Berman, D. E., Rivals, I., Hoischen, A., Stora, S., et al. (2012). Trisomy for synaptojanin1 in Down syndrome is functionally linked to the enlargement of early endosomes. Hum. Mol. Genet. 21, 3156–3172. doi: 10.1093/hmg/dds142
de Winter, C. F., Bastiaanse, L. P., Hilgenkamp, T. I., Evenhuis, H. M., and Echteld, M. A. (2012). Overweight and obesity in older people with intellectual disability. Res. Dev. Disabil. 33, 398–405. doi: 10.1016/j.ridd.2011.09.022
Dong, Y., Gan, D. Z., Tay, S. Z., Koay, W. I., Collinson, S. L., Hilal, S., et al. (2013). Patterns of neuropsychological impairment in Alzheimer's disease and mixed dementia. J. Neurol. Sci. 333, 5–8. doi: 10.1016/j.jns.2013.05.011
Dörner, K., Gaethke, A. S., Tolksdorf, M., Schumann, K. P., and Gustmann, H. (1984). Cholesterol fractions and triglycerides in children and adults with Down's syndrome. Clin. Chim. Acta 142, 307–311.
Draheim, C. C., Geijer, J. R., and Dengel, D. R. (2010). Comparison of intima-media thickness of the carotid artery and cardiovascular disease risk factors in adults with versus without the Down syndrome. Am. J. Cardiol. 106, 1512–1516. doi: 10.1016/j.amjcard.2010.06.079
Epstein, C. J. (2002). William Allan Award Address. From Down syndrome to the “human” in “human genetics.” Am. J. Hum. Genet. 70, 300–313. doi: 10.1086/338915
Fernandez, F., Morishita, W., Zuniga, E., Nguyen, J., Blank, M., Malenka, R. C., et al. (2007). Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 10, 411–413. doi: 10.1038/nn1860
Ferrer, I., and Gullotta, F. (1990). Down's syndrome and Alzheimer's disease: dendritic spine counts in the hippocampus. Acta Neuropathol. 79, 680–685. doi: 10.1007/BF00294247
Frandemiche, M. L., De Seranno, S., Rush, T., Borel, E., Elie, A., Arnal, I., et al. (2014). Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-Beta oligomers. J. Neurosci. 34, 6084–6097. doi: 10.1523/JNEUROSCI.4261-13.2014
Fukazawa, Y., Saitoh, Y., Ozawa, F., Ohta, Y., Mizuno, K., and Inokuchi, K. (2003). Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 38, 447–460. doi: 10.1016/S0896-6273(03)00206-X
Groet, J., Ives, J. H., South, A. P., Baptista, P. R., Jones, T. A., Yaspo, M. L., et al. (1998). Bacterial contig map of the 21q11 region associated with Alzheimer's disease and abnormal myelopoiesis in Down syndrome. Genome Res. 8, 385–398.
Haas, M. A., Bell, D., Slender, A., Lana-Elola, E., Watson-Scales, S., Fisher, E. M., et al. (2013). Alterations to dendritic spine morphology, but not dendrite patterning, of cortical projection neurons in Tc1 and Ts1Rhr mouse models of Down syndrome. PLoS ONE 8:e78561. doi: 10.1371/journal.pone.0078561
Head, E., Silverman, W., Patterson, D., and Lott, I. T. (2012). Aging and down syndrome. Curr. Gerontol. Geriatr. Res. 2012:412536. doi: 10.1155/2012/412536
Heimer-McGinn, V., Murphy, A. C., Kim, J. C., Dymecki, S. M., and Young, P. W. (2013). Decreased dendritic spine density as a consequence of tetanus toxin light chain expression in single neurons in vivo. Neurosci. Lett. 555, 36–41. doi: 10.1016/j.neulet.2013.09.007
Helguera, P., Seiglie, J., Rodriguez, J., Hanna, M., Helguera, G., and Busciglio, J. (2013). Adaptive downregulation of mitochondrial function in down syndrome. Cell Metab. 17, 132–140. doi: 10.1016/j.cmet.2012.12.005
Holland, A. J., Hon, J., Huppert, F. A., Stevens, F., and Watson, P. (1998). Population-based study of the prevalence and presentation of dementia in adults with Down's syndrome. Br. J. Psychiatry 172, 493–498. doi: 10.1192/bjp.172.6.493
Innocent, N., Cousins, S. L., and Stephenson, F. A. (2012). NMDA receptor/amyloid precursor protein interactions: a comparison between wild-type and amyloid precursor protein mutations associated with familial Alzheimer's disease. Neurosci. Lett. 515, 131–136. doi: 10.1016/j.neulet.2012.03.029
Israel, M. A., Yuan, S. H., Bardy, C., Reyna, S. M., Mu, Y., Herrera, C., et al. (2012). Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 482, 216–220. doi: 10.1038/nature10821
Jiang, Y., Mullaney, K. A., Peterhoff, C. M., Che, S., Schmidt, S. D., and Boyer-Boiteau, A. (2010). Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. U.S.A. 107, 1630–1635. doi: 10.1073/pnas.0908953107
Kamenetz, F., Tomita, T., Hsieh, H., Seabrook, G., Borchelt, D., and Iwatsubo, T. (2003). APP processing and synaptic function. Neuron 37, 925–937. doi: 10.1016/S0896-6273(03)00124-7
Kang, J. E., Lim, M. M., Bateman, R. J., Lee, J. J., Smyth, L. P., Cirrito, J. R., et al. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007. doi: 10.1126/science.1180962
Kasuga, K., Shimohata, T., Nishimura, A., Shiga, A., Mizuguchi, T., Tokunaga, J., et al. (2009). Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J. Neurol. Neurosurg. Psychiatry 80, 1050–1052. doi: 10.1136/jnnp.2008.161703
Kleschevnikov, A. M., Belichenko, P. V., Gall, J., George, L., Nosheny, R., Maloney, M. T., et al. (2011). Increased efficiency of the GABAA and GABAB receptor-mediated neurotransmission in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 45, 683–691. doi: 10.1016/j.nbd.2011.10.009
Kleschevnikov, A. M., Belichenko, P. V., Villar, A. J., Epstein, C. J., Malenka, R. C., and Mobley, W. C. (2004). Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 24, 8153–8160. doi: 10.1523/JNEUROSCI.1766-04.2004
Korbel, J. O., Tirosh-Wagner, T., Urban, A. E., Chen, X. N., Kasowski, M., Dai, L., et al. (2009). The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc. Natl. Acad. Sci. U.S.A. 106, 12031–12036. doi: 10.1073/pnas.0813248106
Loo, L. S., Tang, N., Al-Haddawi, M., Dawe, G. S., and Hong, W. (2014). A role for sorting nexin 27 in AMPA receptor trafficking. Nat. Commun. 5, 3176. doi: 10.1038/ncomms4176
Lorenzo, A., Yuan, M., Zhang, Z., Paganetti, P. A., Sturchler-Pierrat, C., Staufenbiel, M., et al. (2000). Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat. Neurosci. 3, 460–464. doi: 10.1038/74833
Lott, I. T., and Head, E. (2005). Alzheimer disease and Down syndrome: factors in pathogenesis. Neurobiol. Aging 26, 383–389. doi: 10.1016/j.neurobiolaging.2004.08.005
Lundgren, J. L., Ahmed, S., Winblad, B., Gouras, G. K., Tjernberg, L. O., and Frykman, S. (2014). Activity-independent release of the amyloid beta-peptide from rat brain nerve terminals. Neurosci. Lett. 566, 125–130. doi: 10.1016/j.neulet.2014.02.050
Mann, D. M. (1988a). Alzheimer's disease and Down's syndrome. Histopathology 13, 125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x
Mann, D. M. (1988b). Calcification of the basal ganglia in Down's syndrome and Alzheimer's disease. Acta Neuropathol. 76, 595–598. doi: 10.1007/BF00689598
Marin-Padilla, M. (1976). Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome. A Golgi study. J. Comp. Neurol. 167, 63–81. doi: 10.1002/cne.901670105
Martínez-Cué, C., Delatour, B., and Potier, M. C. (2014). Treating enhanced GABAergic inhibition in Down syndrome: use of GABA alpha5-selective inverse agonists. Neurosci. Biobehav. Rev. 46(Pt 2), 218–227. doi: 10.1016/j.neubiorev.2013.12.008
McCarron, M., McCallion, P., Reilly, E., and Mulryan, N. (2014). A prospective 14-year longitudinal follow-up of dementia in persons with Down syndrome. J. Intellect. Disabil. Res. 58, 61–70. doi: 10.1111/jir.12074
McCarron, M. O., Nicoll, J. A., and Graham, D. I. (1998). A quartet of Down's syndrome, Alzheimer's disease, cerebral amyloid angiopathy, and cerebral haemorrhage: interacting genetic risk factors. J. Neurol. Neurosurg. Psychiatry 65, 405–406. doi: 10.1136/jnnp.65.3.405
McGowan, E., Eriksen, J., and Hutton, M. (2006). A decade of modeling Alzheimer's disease in transgenic mice. Trends Genet. 22, 281–289. doi: 10.1016/j.tig.2006.03.007
McNaughton, D., Knight, W., Guerreiro, R., Ryan, N., Lowe, J., Poulter, M., et al. (2012). Duplication of amyloid precursor protein (APP), but not prion protein (PRNP) gene is a significant cause of early onset dementia in a large UK series. Neurobiol. Aging 426, e13–e21. doi: 10.1016/j.neurobiolaging.2010.10.010
Moore, S., Evans, L. D., Andersson, T., Portelius, E., Smith, J., Dias, T. B., et al. (2015). APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep. 11, 689–696. doi: 10.1016/j.celrep.2015.03.068
Morice, E., Andreae, L. C., Cooke, S. F., Vanes, L., Fisher, E. M. C., and Tybulewicz, V. L. J. (2008). Preservation of long-term memory and synaptic plasticity despite short-term impairments in the Tc1 mouse model of Down syndrome. Learn. Mem. 15, 492–500. doi: 10.1101/lm.969608
Murdoch, J. C., Rodger, J. C., Rao, S. S., Fletcher, C. D., and Dunnigan, M. G. (1977). Down's syndrome: an atheroma-free model? Br. Med. J. 2, 226–228. doi: 10.1136/bmj.2.6081.226
Murray, A., Letourneau, A., Canzonetta, C., Stathaki, E., Gimelli, S., and Sloan-Bena, F. (2015). Isogenic induced pluripotent stem cell lines from an adult with mosaic down syndrome model accelerated neuronal ageing and neurodegeneration. Stem Cells 33, 2077–2084. doi: 10.1002/stem.1968
Nicolas, G., Wallon, D., Goupil, C., Richard, A. C., Pottier, C., Dorval, V., et al. (2015). Mutation in the 3'untranslated region of APP as a genetic determinant of cerebral amyloid angiopathy. Eur. J. Hum. Genet. doi: 10.1038/ejhg.2015.61. [Epub ahead of print].
Nižetic, D., and Groet, J. (2012). Tumorigenesis in Down's syndrome: big lessons from a small chromosome. Nat. Rev. Cancer 12, 721–732. doi: 10.1038/nrc3355
O'Doherty, A., Ruf, S., Mulligan, C., Hildreth, V., Errington, M. L., Cooke, S., et al. (2005). An aneuploid mouse strain carrying human chromosome 21 with down syndrome phenotypes. Science 309, 2033–2037. doi: 10.1126/science.1114535
Park, J. P., Wurster-Hill, D. H., Andrews, P. A., Cooley, W. C., and Graham, J. M. Jr. (1987). Free proximal trisomy 21 without the Down syndrome. Clin. Genet. 32, 342–348.
Prasher, V. P., Farrer, M. J., Kessling, A. M., Fisher, E. M., West, R. J., Barber, P. C., et al. (1998). Molecular mapping of Alzheimer-type dementia in Down's syndrome. Ann. Neurol. 43, 380–383. doi: 10.1002/ana.410430316
Ramiro-Cortés, Y., and Israely, I. (2013). Long lasting protein synthesis- and activity-dependent spine shrinkage and elimination after synaptic depression. PLoS ONE 8:e71155. doi: 10.1371/journal.pone.0071155
Reeves, R. H., Irving, N. G., Moran, T. H., Wohn, A., Kitt, C., Sisodia, S. S., et al. (1995). A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 11, 177–184. doi: 10.1038/ng1095-177
Reynolds, L. E., Watson, A. R., Baker, M., Jones, T. A., D'Amico, G., Robinson, S. D., et al. (2010). Tumour angiogenesis is reduced in the Tc1 mouse model of Down's syndrome. Nature 465, 813–817. doi: 10.1038/nature09106
Roat, E., Prada, N., Ferraresi, R., Giovenzana, C., Nasi, M., Troiano, L., et al. (2007). Mitochondrial alterations and tendency to apoptosis in peripheral blood cells from children with Down syndrome. FEBS Lett. 581, 521–525. doi: 10.1016/j.febslet.2006.12.058
Rockwood, K. (2003). Mixed dementia: Alzheimer's and cerebrovascular disease. Int. Psychogeriatr. 15(Suppl. 1), 39–46. doi: 10.1017/S1041610203008949
Rovelet-Lecrux, A., Frebourg, T., Tuominen, H., Majamaa, K., Campion, D., and Remes, A. M. (2007). APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J. Neurol. Neurosurg. Psychiatry 78, 1158–1159. doi: 10.1136/jnnp.2006.113514
Rovelet-Lecrux, A., Hannequin, D., Raux, G., Le Meur, N., Laquerrière, A., Vital, A., et al. (2006). APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 38, 24–26. doi: 10.1038/ng1718
Ryeom, S., Greenwald, R. J., Sharpe, A. H., and McKeon, F. (2003). The threshold pattern of calcineurin-dependent gene expression is altered by loss of the endogenous inhibitor calcipressin. Nat. Immunol. 4, 874–881. doi: 10.1038/ni966
Salehi, A., Delcroix, J. D., Belichenko, P. V., Zhan, K., Wu, C., Valletta, J. S., et al. (2006). Increased App expression in a mouse model of Down's syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron 51, 29–42. doi: 10.1016/j.neuron.2006.05.022
Scheff, S. W., Price, D. A., Schmitt, F. A., and Mufson, E. J. (2006). Hippocampal synaptic loss in early Alzheimer's disease and mild cognitive impairment. Neurobiol. Aging 27, 1372–1384. doi: 10.1016/j.neurobiolaging.2005.09.012
Schneider, J. A., Boyle, P. A., Arvanitakis, Z., Bienias, J. L., and Bennett, D. A. (2007). Subcortical infarcts, Alzheimer's disease pathology, and memory function in older persons. Annals Neurol. 62, 59–66. doi: 10.1002/ana.21142
Schulz-Schaeffer, W. J. (2010). The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 120, 131–143. doi: 10.1007/s00401-010-0711-0
Segal, M. (2005). Dendritic spines and long-term plasticity. Nat. Rev. Neurosci. 6, 277–284. doi: 10.1038/nrn1649
Segal, M. (2010). Dendritic spines, synaptic plasticity and neuronal survival: activity shapes dendritic spines to enhance neuronal viability. Eur. J. Neurosci. 31, 2178–2184. doi: 10.1111/j.1460-9568.2010.07270.x
Sekijima, Y., Ikeda, S., Tokuda, T., Satoh, S., Hidaka, H., Hidaka, E., et al. (1998). Prevalence of dementia of Alzheimer type and apolipoprotein E phenotypes in aged patients with Down's syndrome. Eur. Neurol. 39, 234–237.
Selkoe, D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298, 789–791. doi: 10.1126/science.1074069
Seo, J., Giusti-Rodríguez, P., Zhou, Y., Rudenko, A., Cho, S., Ota, K. T., et al. (2014). Activity-dependent p25 generation regulates synaptic plasticity and abeta-induced cognitive impairment. Cell 157, 486–498. doi: 10.1016/j.cell.2014.01.065
Shi, Y., Kirwan, P., Smith, J., MacLean, G., Orkin, S. H., and Livesey, F. J. (2012). A human stem cell model of early Alzheimer's disease pathology in Down syndrome. Sci. Transl. Med. 4, 124ra29. doi: 10.1126/scitranslmed.3003771
Shrestha, B. R., Vitolo, O. V., Joshi, P., Lordkipanidze, T., Shelanski, M., and Dunaevsky, A. (2006). Amyloid beta peptide adversely affects spine number and motility in hippocampal neurons. Mol. Cell. Neurosci. 33, 274–282. doi: 10.1016/j.mcn.2006.07.011
Skoog, I., Nilsson, L., Palmertz, B., Andreasson, L. A., and Svanborg, A. (1993). A population-based study of dementia in 85-year-olds. N. Engl. J. Med. 328, 153–158. doi: 10.1056/NEJM199301213280301
Sleegers, K., Brouwers, N., Gijselinck, I., Theuns, J., Goossens, D., Wauters, J., et al. (2006). APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain 129(Pt 11), 2977–2983. doi: 10.1093/brain/awl203
Snowdon, D. A., Greiner, L. H., Mortimer, J. A., Riley, K. P., Greiner, P. A., and Markesbery, W. R. (1997). Brain infarction and the clinical expression of Alzheimer disease. JAMA 277, 813–817. doi: 10.1001/jama.1997.03540340047031
Souchet, B., Guedj, F., Sahún, I., Duchon, A., Daubigney, F., Badel, A., et al. (2014). Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol. Dis. 69, 65–75. doi: 10.1016/j.nbd.2014.04.016
Sullivan, S. E., Dillon, G. M., Sullivan, J. M., and Ho, A. (2013). Mint proteins are required for synaptic activity-dependent APP trafficking and Aβ generation. J. Biol. Chem. 289, 15374–15383. doi: 10.1074/jbc.M113.541003
Talantova, M., Sanz-Blasco, S., Zhang, X., Xia, P., Akhtar, M. W., Okamoto, S., et al. (2013). Abeta induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. U.S.A. 110, E2518–E2527. doi: 10.1073/pnas.1306832110
Thonberg, H., Fallström, M., Björkström, J., Schoumans, J., Nennesmo, I., and Graff, C. (2011). Mutation screening of patients with Alzheimer disease identifies APP locus duplication in a Swedish patient. BMC Res. Notes 4:476. doi: 10.1186/1756-0500-4-476
Trifunovic, A., and Ventura, N. (2014). Mitochondria and metabolic control of the aging process. Exp. Gerontol. 56, 1–2. doi: 10.1016/j.exger.2014.05.009
Troca-Marín, J. A., Alves-Sampaio, A., and Montesinos, M. L. (2012). Deregulated mTOR-mediated translation in intellectual disability. Prog. Neurobiol. 96, 268–282. doi: 10.1016/j.pneurobio.2012.01.005
Tyrrell, J., Cosgrave, M., McCarron, M., McPherson, J., Calvert, J., Kelly, A., et al. (2001). Dementia in people with Down's syndrome. Int. J. Geriatr. Psychiatry 16, 1168–1174. doi: 10.1002/gps.502
Vis, J. C., Duffels, M. G., Winter, M. M., Weijerman, M. E., Cobben, J. M., and Huisman, S. A. (2009). Down syndrome: a cardiovascular perspective. J. Intellect. Disabil. Res. 53, 419–425. doi: 10.1111/j.1365-2788.2009.01158.x
Wallon, D., Rousseau, S., Rovelet-Lecrux, A., Quillard-Muraine, M., Guyant-Maréchal, L., and Martinaud, O. (2012). The French series of autosomal dominant early onset Alzheimer's disease cases: mutation spectrum and cerebrospinal fluid biomarkers. J. Alzheimers Dis. 30, 847–856. doi: 10.3233/JAD-2012-120172
Wang, B., Wang, Z., Sun, L., Yang, L., Li, H., Cole, A. L., et al. (2014b). The amyloid precursor protein controls adult hippocampal neurogenesis through GABAergic interneurons. J. Neurosci. 34, 13314–13325. doi: 10.1523/JNEUROSCI.2848-14.2014
Wang, X., Huang, T., Zhao, Y., Zheng, Q., Thompson, R. C., Bu, G., et al. (2014a). Sorting nexin 27 regulates Abeta production through modulating gamma-secretase activity. Cell Rep. 9, 1023–1033. doi: 10.1016/j.celrep.2014.09.037
Wang, X., Zhao, Y., Zhang, X., Badie, H., Zhou, Y., Mu, Y., et al. (2013). Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat. Med. 19, 473–480. doi: 10.1038/nm.3117
Weitzdoerfer, R., Dierssen, M., Fountoulakis, M., and Lubec, G. (2001). Fetal life in Down syndrome starts with normal neuronal density but impaired dendritic spines and synaptosomal structure. J. Neural Transm. Suppl. 61, 59–70. doi: 10.1007/978-3-7091-6262-0_5
Wiseman, F. K., Al-Janabi, T., Hardy, J., Karmiloff-Smith, A., Nizetic, D., and Tybulewicz, V. L. J. (2015). A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 16, 564–574. doi: 10.1038/nrn3983
Wu, J., Petralia, R. S., Kurushima, H., Patel, H., Jung, M. Y., Volk, L., et al. (2011). Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell 147, 615–628. doi: 10.1016/j.cell.2011.09.036
Xie, L., Kang, H., Xu, Q., Chen, M. J., Liao, Y., Thiyagarajan, M., et al. (2013). Sleep drives metabolite clearance from the adult brain. Science 342, 373–377. doi: 10.1126/science.1241224
Yang, A., and Reeves, R. H. (2011). Increased survival following tumorigenesis in Ts65Dn mice that model Down syndrome. Cancer Res. 71, 3573–3581. doi: 10.1158/0008-5472.CAN-10-4489
Yang, L., Wang, Z., Wang, B., Justice, N. J., and Zheng, H. (2009). Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J. Neurosci. 29, 15660–15668. doi: 10.1523/JNEUROSCI.4104-09.2009
Keywords: Down syndrome, vascular dementia, neuron activity-dependent, cognitive dysfunction, amyloid beta-peptides, amyloid beta-protein precursor
Citation: Nizetic D, Chen CL, Hong W and Koo EH (2015) Inter-Dependent Mechanisms Behind Cognitive Dysfunction, Vascular Biology and Alzheimer's Dementia in Down Syndrome: Multi-Faceted Roles of APP. Front. Behav. Neurosci. 9:299. doi: 10.3389/fnbeh.2015.00299
Received: 08 August 2015; Accepted: 27 October 2015;
Published: 01 December 2015.
Edited by:
Marie-Claude Potier, Centre National de la Recherche Scientifique, FranceReviewed by:
Jorge Busciglio, University of California, Irvine, USADominique Campion, Institut National de la Santé et de la Recherche Médicale, France
Copyright © 2015 Nizetic, Chen, Hong and Koo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dean Nizetic, ZC5uaXpldGljQG50dS5lZHUuc2c=