 Miriam B. Virgolini
Miriam B. Virgolini Mara S. Mattalloni
Mara S. Mattalloni Paula A. Albrecht
Paula A. Albrecht Romina Deza-Ponzio
Romina Deza-Ponzio Liliana M. Cancela
Liliana M. Cancela- IFEC-CONICET, Departamento de Farmacología, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Córdoba, Argentina
This review article provides evidence of the impact of the environmental contaminant lead (Pb) on the pattern of the motivational effects of ethanol (EtOH). To find a mechanism that explains this interaction, the focus of this review article is on central EtOH metabolism and the participating enzymes, as key factors in the modulation of brain acetaldehyde (ACD) accumulation and resulting effect on EtOH intake. Catalase (CAT) seems a good candidate for the shared mechanism between Pb and EtOH due to both its antioxidant and its brain EtOH-metabolizing properties. CAT overactivation was reported to increase EtOH consumption, while CAT blockade reduced it, and both scenarios were modified by Pb exposure, probably as the result of elevated brain and blood CAT activity. Likewise, the motivational effects of EtOH were enhanced when brain ACD metabolism was prevented by ALDH2 inhibition, even in the Pb animals that evidenced reduced brain ALDH2 activity after chronic EtOH intake. Overall, these results suggest that brain EtOH metabolizing enzymes are modulated by Pb exposure with resultant central ACD accumulation and a prevalence of the reinforcing effects of the metabolite in brain against the aversive peripheral ACD accumulation. They also support the idea that early exposure to an environmental contaminant, even at low doses, predisposes at a later age to differential reactivity to challenging events, increasing, in this case, vulnerability to acquiring addictive behaviors, including excessive EtOH intake.
Introduction
“The Barker hypothesis” (Osmond and Barker, 2000) first popularized the concept that parameters related to fetal, infant and childhood growth may be predictors of disease in later life. The original hypothesis has been extended to a range of components of the developmental environment such as the mother’s nutrition, stress levels, lifestyle and exposure to chemicals, all factors that may play a powerful role in influencing later susceptibility to challenging events. Based on these considerations, this review article provides behavioral and biochemical evidence that aims to support the idea that early-life exposure to lead (Pb), an environmental neurotoxicant, produces an “imprint” in CNS functionality. We propose that this experience has health consequences over the life span, increasing vulnerability to addictive behaviors, in this case to the motivational responses to ethanol (EtOH), with brain acetaldehyde (ACD) and EtOH metabolizing enzymes playing a crucial role.
Lead, Ethanol and the Two Faces of Reinforcement
Although a non-essential metal and with widely restricted industrial uses, Pb is present in the environment and in living organisms. Alarmingly, early-life Pb exposure even in trace amounts induces neurobehavioral manifestations that may not be evident until later in life (Vorvolakos et al., 2016). They include hyperactivity, cognitive deficits and altered responses to drugs of abuse including EtOH.
From the clinical perspective, a relationship between Pb and EtOH has been described (Cezard et al., 1992). Animals chronically exposed to high Pb levels during adulthood, showed higher, although less efficient, lever pressing for EtOH in a self-administration test, associated with increased EtOH intake (Nation et al., 1987). More recently, it was shown EtOH-induced hyperlocomotion after acute Pb administration (Correa et al., 2001). Similarly, perinatal low-level Pb exposure enhanced EtOH intake in a daily 2-h EtOH/water free-choice sessions. Moreover, Pb-exposed animals also showed elevated response rates in a FR-2 schedule of behavior associated with a higher breaking point compared to controls, evidencing their motivation to self-administer EtOH (Mattalloni et al., 2013).
Thus, to find a mechanism that explains these differential effects, the concepts of positive and negative reinforcing must be introduced. It is known that drug addiction is a process that progresses from an early condition of positive reinforcement, evidenced by the euphorizing and stimulant effects of the drug (compulsive desire for pleasure), to a later state of negative reinforcement, evidenced as dysphoria and anxiety as a result of drug removal (compulsive desire for relief). Thus, the two main sources of reinforcement play key roles in the allostatic processes that lead to drug abuse (Wise and Koob, 2014). Both aspects will be mentioned in this review article, with a focus on the positive reinforcement perspective, particularly related to EtOH and its bioproducts, with developmental Pb exposure as a determinant factor in the vulnerability of these animals to the motivational effects of EtOH.
A putative explanation for the Pb/EtOH interaction supported by the negative reinforcement perspective is based on the tension-reduction hypothesis (Pohorecky, 1990). This proposes that the anxiolytic properties of EtOH are the main factors that lead some individuals to consume excessive amounts of the drug to relieve negative emotionality. This mechanism involves both the hypothalamic-pituitary adrenal axis with corticosterone secretion as the final output (Fahlke et al., 1994), as well as the extrahypothalamic systems including the extended amygdala (Koob, 2008). The increased susceptibility to EtOH-anxiolytic effects and the enhanced EtOH intake reported in perinatally Pb-exposed animals was associated with elevated basal corticosterone levels (Virgolini et al., 1999). Thus, it is proposed that Pb-treated animals would ingest EtOH to diminish their basal anxiety in an attempt to cope with stressful situations. This line of research was not further investigated and deserves future endeavors.
The positive reinforcement view, on the other hand, is related to the motivational and stimulant effects of EtOH, mediated through the reported ability of EtOH, ACD and salsolinol (a tetrahydroisoquinoline product of dopamine (DA) and ACD condensation) to facilitate DA neurotransmission in the mesolimbic circuit. Moreover, central administration of EtOH (or its bioproducts) induces hyperlocomotion, conditioned place preference and promotes EtOH intake (reviewed in: Quertemont et al., 2005; Correa et al., 2012; Hipólito et al., 2012; Deehan et al., 2013; Israel et al., 2015; Peana et al., 2016). Therefore, the present review will particularly emphasize the modulation that the environmental neurotoxicant Pb exerts on the enzymes involved in central EtOH metabolism, given the positive reinforcing properties of EtOH, ACD and salsolinol.
With the reported low ADH activity in the brain, the CAT-H2O2 system in addition to being a peroxisomal redox regulator is the key enzyme involved in H2O2-dependent brain EtOH oxidation to ACD (Vetrano et al., 2005). It should be mentioned that blood catalase (CAT) activity is positively correlated with EtOH consumption in both rats (Amit and Aragon, 1988) and humans (Koechling and Amit, 1992). Several reports indicate that CAT activity is decreased after chronic adult Pb exposure in the brain (Jindal and Gill, 1999), liver (Flora et al., 2012a) and blood (Sajitha et al., 2010). Interestingly, developmental exposure to high Pb doses is able to increase CAT activity (brain: Valenzuela et al., 1989; brain, liver and heart: Somashekaraiah et al., 1992). Moreover, cumulative evidence demonstrated that acute (but not chronic) Pb administration raises brain CAT levels and increases the locomotor response to EtOH in mice (Correa et al., 1999, 2001). Similarly, we have reported that developmental Pb exposure increased basal blood CAT activity in periadolescent rats, an effect that persisted throughout their lifetime and was potentiated by EtOH intake. Although no differences between groups were observed in whole brain CAT activity, there was a region-specific increase in the Pb-exposed hippocampus and cerebellum, indicating that CAT-mediated EtOH oxidation is not homogeneous throughout the brain (Mattalloni et al., 2013, 2017).
On the other hand, ACD removal is mediated by ALDH2, a mitochondrial enzyme that belongs to the ALDH superfamily and catalyzes both brain and liver ACD oxidation to acetic acid (Crabb et al., 2004). The two evidences of an interaction between Pb and ALDH2 have shown that adult Pb exposure reduced liver ALDH2 (Flora and Tandon, 1987) whereas developmental Pb-exposure reduced brain ALDH2 activity (Mattalloni et al., 2017) after chronic EtOH consumption.
Thus, based on the premise that early-life Pb exposure will interfere with EtOH metabolism, brain ACD may be noted as the common site of action of the two neurotoxicants. Pharmacological manipulations of EtOH-metabolizing enzymes attempting to modulate brain ACD accumulation will therefore be described below, with the resultant changes evidenced at both behavioral and biochemical levels.
Pharmacological Interference of Ethanol Metabolism
The next section follows the two-dimensional model of alcohol consumption hypothesized over 30 years ago, supporting the idea that “both brain CAT and ALDH may represent a biological marker system underlying the affinity of the animals to consume ethanol” (Aragon and Amit, 1985). Evidence is provided that Pb induces dynamic changes in the two main enzymes involved in brain EtOH metabolism, which may account for differential EtOH intake in response to pharmacological manipulations in these animals (Figure 1).
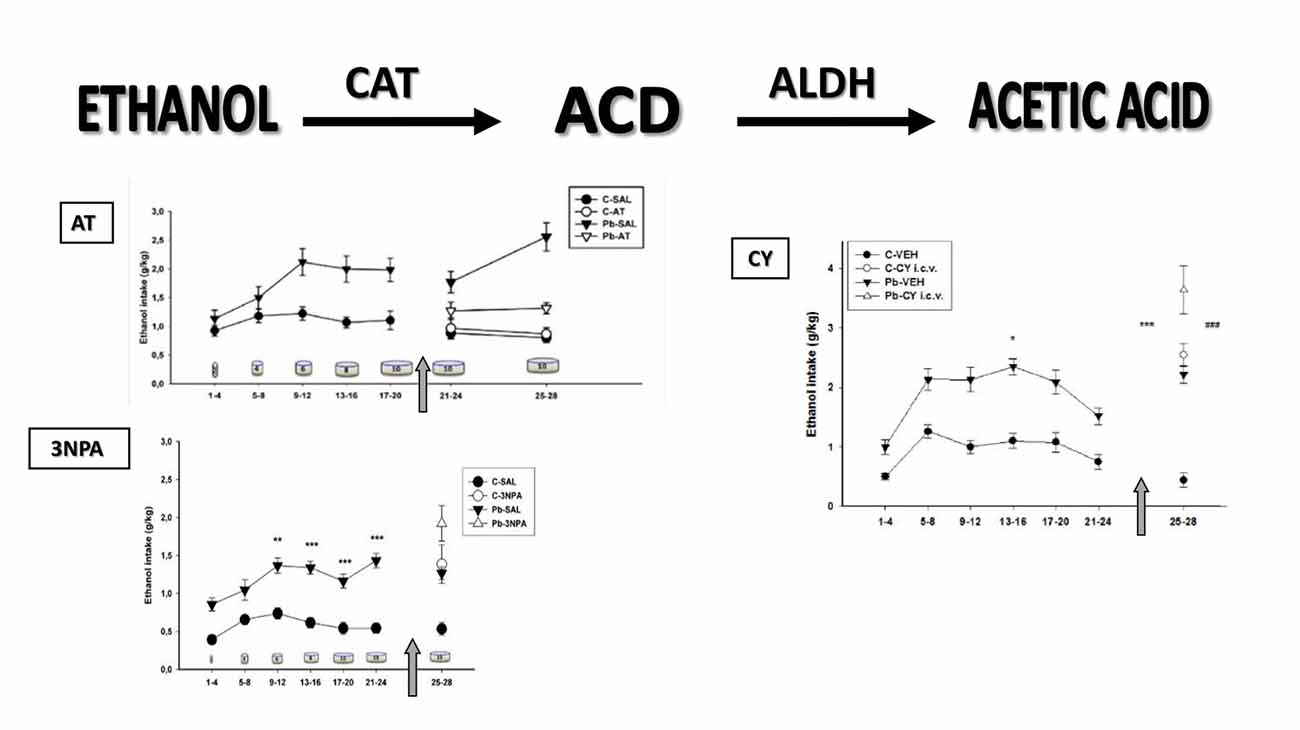
Figure 1. Voluntary ethanol (EtOH) consumption measured in Wistar rats. Data (mean expressed as grams of EtOH per kilogram of body weight ± SE) grouped in 4-day blocks along the horizontal axis (in days) that correspond to EtOH intake in response to increasing EtOH concentrations symbolized as cylinders (days 1–4: 2%; days 5–8: 4%; days 9–12: 6%; days 13–16: 8%; and days 17–28: 10%). C, control; Pb, lead; CAT, catalase; SAL, saline; VEH, vehicle. Top, left: EtOH intake in response to 3-amino 1,2,4-triazole (AT) administration. The arrow signifies the start of SAL or AT administration (days 21–24 and 25–28; 250 mg/kg i.p.). C-SAL = 10; C-AT = 11; Pb-SAL = 11; Pb-AT = 9 animals per group (Mattalloni et al., 2013). Bottom, left: EtOH intake in response to 3-nitropropionic acid (3NPA) administration. The arrow signifies the start of SAL or 3NPA administration (days 25–28; 20 mg/kg s.c.). Baseline: *denotes statistical difference compared to controls at **p < 0.01 and ***p < 0.001. C-SAL = 8; C-3NPA = 11; Pb-SAL = 9; Pb-3NPA = 9 animals per group (Mattalloni et al., 2013). Right: EtOH intake in response to intracerebroventricular cyanamide (CY) administration. The arrow signifies the start of VEH or CY administration (days 25–28; 0.3 mg i.c.v.). Baseline: *denotes differences compared to controls at *p < 0.05. CY administration: *denotes differences between the C and Pb-exposed animals injected with VEH at ***p < 0.001; #denotes differences between the VEH and corresponding CY groups for both C and Pb-exposed animals at ###p < 0.001. C-VEH = 11; C-CY i.c.v.= 14; Pb-VEH = 8; Pb-CY i.c.v.= 8 animals per group (Mattalloni et al., 2017).
Brain Acetaldehyde Formation
One of the most commonly used CAT blockers employed to modulate stimulant responses to EtOH is 3-amino 1,2,4-triazole (AT), a fungicide that produces irreversible inhibition of the CAT-H2O2 site, thereby preventing in vivo brain EtOH oxidation to ACD (Aragon et al., 1989). Interestingly, the only report of an interaction among Pb, CAT, AT and EtOH showed that AT was able to reverse the increase in EtOH-induced hyperlocomotion and brain CAT activity observed after acute Pb administration (Correa et al., 2001). Similarly, we have reported that AT pretreatment prevented both elevated EtOH intake and blood and brain (hippocampus and cerebellum) CAT activity in developmentally Pb-exposed animals (Mattalloni et al., 2013). The absence of these effects in the control group suggests that the enzyme inhibition requires either high H2O2 (and ROS) levels that are increased as a result of Pb exposure (Flora et al., 2012b) or the excessive EtOH intake evidenced in Pb-exposed animals.
On the other hand, CAT overactivation can be achieved by the administration of 3-nitropropionic acid (3NPA), a mycotoxin that produces an irreversible inhibition of the succinate dehydrogenase (SDH) enzyme, along with ROS elevation and increased CAT activity, with resultant EtOH-induced hyperlocomotion (Manrique et al., 2006). Thus, 3NPA induced-CAT elevation was able to increase EtOH consumption in both, the Pb-exposed and the control animals, accompanied by higher blood and brain (striatal) CAT activity in the Pb group (Mattalloni et al., 2013).
Brain Acetaldehyde Removal
Cyanamide (CY) is a drug prescribed in some countries as a deterrent for alcoholics due to its ability to increase peripheral (aversive) ACD as a result of ALDH inhibition (Koppaka et al., 2012). Central CY administration enhanced EtOH intake in rats that had never consumed EtOH, an effect highly dependent on the CY dose (Critcher and Myers, 1987). We have demonstrated that i.c.v. CY administration inhibited brain ALDH2 and increased EtOH intake in control animals, whereas the Pb-exposed group also showed elevated EtOH intake although in the absence of brain ALDH2 inhibition (Mattalloni et al., 2017). This finding may be related to the reduced basal brain ALDH2 activity present in the Pb-exposed group, or to the fact that CY is a prodrug that, to convert itself to the active metabolite requires CAT and H2O2, a system that is modified by Pb-exposure.
Conclusion
This review article provides evidence of Pb modulation on the enzymes involved in either the production or the removal of brain ACD, i.e., CAT and ALDH2, the activities of which have been proposed as trait biomarkers of excessive EtOH intake (Aragon and Amit, 1985). The data demonstrate differential CAT and ALDH2 functionality in the developmentally Pb-exposed animals, with high blood and brain CAT activity and low brain ALDH2 activity, thereby promoting central ACD accumulation (Figure 2). This effect would directly influence EtOH self-administration, with Pb exposure representing a crucial variable in the behavioral and biochemical outputs described here. It can thus be postulated that one of the shared mechanisms between Pb and EtOH could be the result of differential EtOH metabolism in brain areas related to reward. Possibly, an imbalance towards a prevalence of the reinforcing effects of brain ACD vs. aversive peripheral ACD accumulation may play a key role in the differential motivational response to EtOH evidenced in Pb-exposed animals. Moreover, immunohistochemical studies have demonstrated that ALDH2 is widely expressed in the brain, with low activity in the aminergic neurons, which coincidentally are the richest in CAT expression (Zimatkin, 1991; Zimatkin and Lindros, 1996), a fact that promotes brain ACD accumulation in the mesolimbic circuit, site of the reinforcing properties of addictive drugs. Immunostaining experiments are desirable for brain CAT and ALDH2 expression in the Pb-exposed animals.
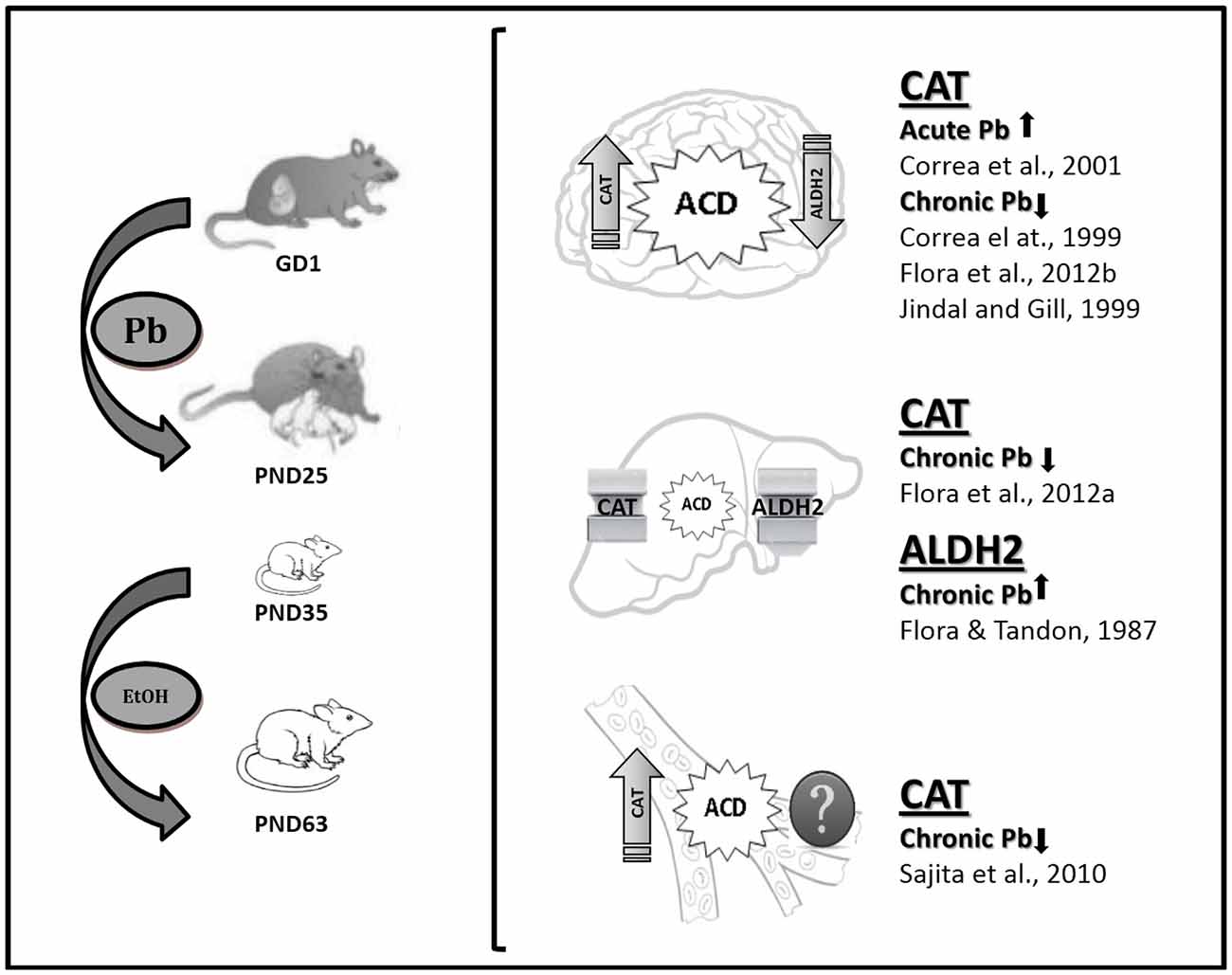
Figure 2. Lead (Pb) exposure and EtOH intake with emphasis in EtOH metabolizing enzymes status. The square bracket comprises pictures for brain, liver and blood CAT and ALDH2 status and putative acetaldehyde (ACD) accumulation in the experimental model described in Mattalloni et al. (2013, 2017; as shown on the left). The references point-out CAT and ALDH2 data reported elsewhere as result of adult acute or chronic Pb exposure in animals with chronic EtOH intake. GD, gestational day; PND, postnatal day.
Interestingly, there are differences in EtOH metabolism over the lifetime. CAT-H2O2 system activity is higher in pups than in adults (Hamby-Mason et al., 1997), thus promoting central ACD accumulation. Brain ALDH2 activity also increases gradually, reaching the activity specific for mature animals by periadolescence (Zamatkin and Lis, 1990). Hence, Pb exposure during development may have affected the functionality of these enzymes at a time of high ACD accumulation. This assumption has important clinical implications provided that the neurobehavioral outcomes showed no evidence of a safe threshold for Pb exposure in immature organisms and the ubiquity of this environmental neurotoxicant. Thus, these results indicate the existence of prenatal programming as a consequence of early Pb exposure, an experience that would leave an imprint that later in life may be responsible for differential responsiveness to events that generate a conflict in the individual, such as the initiation in addictive behaviors.
Author Contributions
LMC and MBV conceived and designed the experiments. MSM, RD-P and PAA performed the experiments and analyzed the data. MBV wrote and LMC contributed to the writing of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants to MBV by the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET); Secretaría de Ciencia y Tecnología (SeCyT-UNC); Ministerio de Ciencia y Tecnología de la Provincia de Córdoba (MinCyT), and Fondo para la Investigación Científica y Tecnológica (FONCyT) from Argentina.
References
Amit, Z., and Aragon, C. M. G. (1988). Catalase activity measured in rats naive to ethanol correlates with later voluntary ethanol consumption: possible evidence for a biological marker system of ethanol intake. Psychopharmacology 95, 512–515. doi: 10.1007/bf00172965
Aragon, C. M. G., and Amit, Z. (1985). A two-dimensional model of alcohol consumption: possible interaction of brain catalase and aldehyde dehydrogenase. Alcohol 2, 357–360. doi: 10.1016/0741-8329(85)90075-8
Aragon, C. M. G., Spivak, K., and Amit, Z. (1989). Effects of 3-amino-1,2,4-triazole on ethanol-induced open field activity: evidence for brain catalase mediation of ethanol’s effects. Alcohol. Clin. Exp. Res. 13, 104–108. doi: 10.1111/j.1530-0277.1989.tb00293.x
Cezard, C., Demarquilly, C., Boniface, M., and Haguenoer, J. M. (1992). Influence of the degree of exposure to lead on relations between alcohol consumption and the biological indices of lead exposure: epidemiological study in a lead acid battery factory. Br. J. Ind. Med. 49, 645–647. doi: 10.1136/oem.49.9.645
Correa, M., Miquel, M., Sanchis-Segura, C., and Aragon, C. M. G. (1999). Effects of chronic lead administration on ethanol-induced locomotor and brain catalase activity. Alcohol 19, 43–49. doi: 10.1016/s0741-8329(99)00023-3
Correa, M., Salamone, J. D., Segovia, K. N., Pardo, M., Longoni, R., Spina, L., et al. (2012). Piecing together the puzzle of acetaldehyde as a neuroactive agent. Neurosci. Biobehav. Rev. 36, 404–430. doi: 10.1016/j.neubiorev.2011.07.009
Correa, M., Sanchis-Segura, C., and Aragon, C. M. G. (2001). Brain catalase activity is highly correlated with ethanol-induced locomotor activity in mice. Physiol. Behav. 73, 641–647. doi: 10.1016/s0031-9384(01)00511-x
Crabb, D. W., Matsumoto, M., Chang, D., and You, M. (2004). Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc. Nutr. Soc. 63, 49–63. doi: 10.1079/pns2003327
Critcher, E. C., and Myers, R. D. (1987). Cyanamide given ICV or systemically to the rat alters subsequent alcohol drinking. Alcohol 4, 347–353. doi: 10.1016/0741-8329(87)90066-8
Deehan, G. A., Brodie, M. S., and Rodd, Z. A. (2013). What is in that drink: the biological actions of ethanol, acetaldehyde, and salsolinol. Curr. Top. Behav. Neurosci. 13, 163–184. doi: 10.1007/7854_2011_198
Fahlke, C., Engel, J. A., Eriksson, C. J. P., Hrd, E., and Söderpalm, B. (1994). Involvement of corticosterone in the modulation of ethanol consumption in the rat. Alcohol 11, 195–202. doi: 10.1016/0741-8329(94)90031-0
Flora, S. J., Gautam, P., and Dwivedi, N. (2012a). Dose-dependent effects of ethanol on lead-induced oxidative stress in rats. J. Environ. Pathol. Toxicol. Oncol. 31, 61–73. doi: 10.1615/jenvironpatholtoxicoloncol.v31.i1.70
Flora, S. J., Gautam, P., and Kushwaha, P. (2012b). Lead and ethanol co-exposure lead to blood oxidative stress and subsequent neuronal apoptosis in rats. Alcohol Alcohol. 47, 92–101. doi: 10.1093/alcalc/agr152
Flora, S. J. S., and Tandon, S. K. (1987). Effect of combined exposure to lead and ethanol on some biochemical indices in the rat. Biochem. Pharmacol. 36, 537–541. doi: 10.1016/0006-2952(87)90363-7
Hamby-Mason, R., Chen, J. J., Schenker, S., Perez, A., and Henderson, G. I. (1997). Catalase mediates acetaldehyde formation from ethanol in fetal and neonatal rat brain. Alcohol. Clin. Exp. Res. 21, 1063–1072. doi: 10.1097/00000374-199709000-00020
Hipólito, L., Sánchez-Catalán, M. J., Martí-Prats, L., Granero, L., and Polache, A. (2012). Revisiting the controversial role of salsolinol in the neurobiological effects of ethanol: old and new vistas. Neurosci. Biobehav. Rev. 36, 362–378. doi: 10.1016/j.neubiorev.2011.07.007
Israel, Y., Quintanilla, M. E., Karahanian, E., Rivera-Meza, M., and Herrera-Marschitz, M. (2015). The “first hit” toward alcohol reinforcement: role of ethanol metabolites. Alcohol. Clin. Exp. Res. 39, 776–786. doi: 10.1111/acer.12709
Jindal, V., and Gill, K. D. (1999). Ethanol potentiates lead-induced inhibition of rat brain antioxidant defense systems. Pharmacol. Toxicol. 85, 16–21. doi: 10.1111/j.1600-0773.1999.tb01057.x
Koechling, U. M., and Amit, Z. (1992). Relationship between blood catalase activity and drinking history in a human population, a possible biological marker of the affinity to consume alcohol. Alcohol Alcohol. 27, 181–188. doi: 10.1093/oxfordjournals.alcalc.a045218
Koob, G. F. (2008). A role for brain stress systems in addiction. Neuron 59, 11–34. doi: 10.1016/j.neuron.2008.06.012
Koppaka, V., Thompson, D. C., Chen, Y., Ellermann, M., Nicolaou, K. C., Juvonen, R. O., et al. (2012). Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 64, 520–539. doi: 10.1124/pr.111.005538
Manrique, H. M., Miquel, M., and Aragon, C. M. G. (2006). Acute administration of 3-nitropropionic acid, a reactive oxygen species generator, boosts ethanol-induced locomotor stimulation. New support for the role of brain catalase in the behavioural effects of ethanol. Neuropharmacology 51, 1137–1145. doi: 10.1016/j.neuropharm.2006.07.022
Mattalloni, M. S., De Giovanni, L. N., Molina, J. C., Cancela, L. M., and Virgolini, M. B. (2013). Participation of catalase in voluntary ethanol consumption in perinatally low-level lead-exposed rats. Alcohol. Clin. Exp. Res. 37, 1632–1642. doi: 10.1111/acer.12150
Mattalloni, M. S., Deza-Ponzio, R., Albrecht, P. A., Cancela, L. M., and Virgolini, M. B. (2017). Developmental lead exposure induces opposite effects on ethanol intake and locomotion in response to central vs. systemic cyanamide administration. Alcohol 58, 1–11. doi: 10.1016/j.alcohol.2016.11.002
Nation, J. R., Baker, D. M., Fantasia, M. A., Ruscher, A. E., and Clark, D. E. (1987). Ethanol consumption and free operant avoidance performance following exposure to dietary lead. Neurotoxicology 8, 561–568.
Osmond, C., and Barker, D. J. P. (2000). Fetal, infant, and childhood growth are predictors of coronary heart disease, diabetes and hypertension in adult men and women. Environ. Health Perspect. 108, 545–553. doi: 10.2307/3454545
Peana, A. T., Rosas, M., Porru, S., and Acquas, E. (2016). From ethanol to salsolinol: role of ethanol metabolites in the effects of ethanol. J. Exp. Neurosci. 10, 137–146. doi: 10.4137/jen.s25099
Pohorecky, L. A. (1990). Interaction of ethanol and stress: research with experimental animals—an update. Alcohol Alcohol. 25, 263–276. doi: 10.1093/oxfordjournals.alcalc.a045000
Quertemont, E., Tambour, S., and Tirelli, E. (2005). The role of acetaldehyde in the neurobehavioral effects of ethanol: a comprehensive review of animal studies. Prog. Neurobiol. 75, 247–274. doi: 10.1016/j.pneurobio.2005.03.003
Sajitha, G. R., Jose, R., Andrews, A., Ajantha, K. G., Augustine, P., and Augusti, K. T. (2010). Garlic oil and vitamin E prevent the adverse effects of lead acetate and ethanol separately as well as in combination in the drinking water of rats. Indian J. Clin. Biochem. 25, 280–288. doi: 10.1007/s12291-010-0042-x
Somashekaraiah, B. V., Padmaja, K., and Prasad, A. R. (1992). Lead-induced lipid peroxidation and antioxidant defense components of developing chick embryos. Free Radic. Biol. Med. 13, 107–114. doi: 10.1016/0891-5849(92)90071-n
Valenzuela, A., Lefauconnier, J. M., Chaudiere, J., and Bourre, J. M. (1989). Effects of lead acetate on cerebral glutathione peroxidase and catalase in the suckling rat. Neurotoxicology 10, 63–69.
Vetrano, A. M., Heck, D. E., Mariano, T. M., Mishin, V., Laskin, D. L., and Laskin, J. D. (2005). Characterization of the oxidase activity in mammalian catalase. J. Biol. Chem. 280, 35372–35381. doi: 10.1074/jbc.m503991200
Virgolini, M. B., Cancela, L. M., and Fulginiti, S. (1999). Behavioral responses to ethanol in rats perinatally exposed to low lead levels. Neurotoxicol. Teratol. 21, 551–557. doi: 10.1016/s0892-0362(99)00020-3
Vorvolakos, T., Arseniou, S., and Samakouri, M. (2016). There is no safe threshold for lead exposure: a literature review. Psychiatriki 27, 204–214.
Wise, R. A., and Koob, G. F. (2014). The development and maintenance of drug addiction. Neuropsychopharmacology 39, 254–262. doi: 10.1038/npp.2013.261
Zamatkin, S. M., and Lis, R. E. (1990). Aldehyde dehydrogenase activity in the rat brain during ontogenesis. Arkh. Anat. Gistol. Embriol. 98, 27–33.
Zimatkin, S. M. (1991). Histochemical study of aldehyde dehydrogenase in the rat CNS. J. Neurochem. 56, 1–11. doi: 10.1111/j.1471-4159.1991.tb02555.x
Keywords: ethanol, acetaldehyde, lead-exposure, catalase, ALDH2
Citation: Virgolini MB, Mattalloni MS, Albrecht PA, Deza-Ponzio R and Cancela LM (2017) Modulation of Ethanol-Metabolizing Enzymes by Developmental Lead Exposure: Effects in Voluntary Ethanol Consumption. Front. Behav. Neurosci. 11:95. doi: 10.3389/fnbeh.2017.00095
Received: 25 January 2017; Accepted: 04 May 2017;
Published: 23 May 2017.
Edited by:
Merce Correa, Jaume I University, SpainReviewed by:
Marcelo Tadeu Marin, Universidade Estadual Paulista Júlio Mesquita Filho, BrazilEduardo Karahanian, Universidad Autonoma de Chile, Chile
Copyright © 2017 Virgolini, Mattalloni, Albrecht, Deza-Ponzio and Cancela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miriam B. Virgolini, bXZpcmdvbGlAZmNxLnVuYy5lZHUuYXI=