 Dirson J. Stein1,2
Dirson J. Stein1,2 Mailton F. Vasconcelos3
Mailton F. Vasconcelos3 Lucas Albrechet-Souza3
Lucas Albrechet-Souza3 Keila M. M. Ceresér1,2
Keila M. M. Ceresér1,2 Rosa M. M. de Almeida3*
Rosa M. M. de Almeida3*- 1Laboratory of Molecular Psychiatry, Hospital de Clínicas de Porto Alegre, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 2Post-Graduate Program in Psychiatry and Behavioral Sciences, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
- 3Psychology Institute, Universidade Federal do Rio Grande do Sul, Porto Alegre, Brazil
Hyper activation of the neuroimmune system is strongly related to the development of neuropsychiatric disorders. Psychosocial stress has been postulated to play an important role in triggering anxiety and major depression. In preclinical models, there is mounting evidence that social defeat stress activates microglial cells in the central nervous system. This type of stress could be one of the major factors in the development of these psychopathologies. Here, we reviewed the most recent literature on social defeat and the associated immunological reactions. We focused our attention on microglial cells and kept the effect of social defeat over microglia separate from the effect of this stressor on other immune cells and the influence of peripheral immune components in priming central immune reactions. Furthermore, we considered how social defeat stress affects microglial cells and the consequent development of anxiety- and depressive-like states in preclinical studies. We highlighted evidence for the negative impact of the over-activation of the neuroimmune system, especially by the overproduction of pro-inflammatory mediators and cytotoxins. Overproduction of these molecules may cause cellular damage and loss or decreased function of neuronal activity by excessively pruning synaptic connections that ultimately contribute to the development of anxiety- and depressive-like states.
Introduction
Neuropsychiatric disorders, such as anxiety and major depression (MD), are highly prevalent and contribute significantly to the worldwide burden of diseases (Ferrari et al., 2013; Whiteford et al., 2013). As a major contributor to the development of affective and neuropsychiatric disorders in humans, psychosocial stress has been reported to induce central and peripheral immune pathway signaling by repeated activation of the neuroendocrine and neurovegetative systems (Glaser and Kiecolt-Glaser, 2005; Lehmann et al., 2016). When the individual is repeatedly exposed to stress, the brain homeostatic environment alters and may give rise to various cognitive and mood disorders that impair everyday functioning and overall quality of life (McKim et al., 2016a). Within the central nervous system (CNS) immunological defense, microglia are the key immune players and acquire a reactive profile to cope with altered homeostasis (Hanisch and Kettenmann, 2007). When activated, these cells are supposed to trigger anxiety- and depressive-like behaviors (Lehmann et al., 2016), mainly by increasing the expression of pro-inflammatory mediators and neurotoxins in stress-sensitive brain regions (Reader et al., 2015; Ramirez and Sheridan, 2016), and can ultimately influence the overall cellular functions and survival, from neurons to glial cells.
Brief and prolonged episodes of social defeat (SD) have been correlated with anxiety- and depressive-like behaviors, respectively. While brief episodes can increase self-grooming, locomotion in novel environments, risk assessment and binge-like cocaine self-administration, prolonged episodes induce anhedonic behaviors such as reduced sweet solution preference, reduced mounting in copulatory behavior, reduced climbing in the forced swimming test (FST), lower general activity and sociability and suppressed cocaine intake (Razzoli et al., 2009; Miczek et al., 2011; Hollis and Kabbaj, 2014; Vasconcelos et al., 2015). Despite the clear evidence of the role of social stress triggering mood disorder-related behaviors, to the best of our knowledge, the exclusive contribution of SD to microglial over-activation has never been reviewed. Here, we discuss the emerging field of social stress-induced microglial over-activation, providing an overview of how microglial reactions can lead to these mood disorders, and briefly discuss some relevant translational significance of the findings. We hypothesized that acute/repeated and chronic social defeat (CSD) stress can induce microglial activation and over-activation that can engender anxiety and depressive-like states, respectively. The repeated social defeat (RSD) paradigm reported in this review is characterized by the introduction of an aggressive intruder male into the cages of established male cohorts of mice for three or six consecutive nights, leading to the establishment of dominance over the original colony (Wohleb et al., 2014b). CSD varied from 14 to 20 days of a 24 h/day dyadic social housing, exposing the defeated animal to continuous psychological stress via sensory interaction with the aggressor, accompanied by a 5 min/day agonistic encounter between the aggressor and the defeated animal (Brachman et al., 2015; Lehmann et al., 2016; Tong et al., 2017).
Articles used in this mini-review were selected from the PubMed, Embase and ScienceDirect databases between March and April 2017. Search terms were “microglia” and “SD”, without any time limitation. Of the 23 selected articles, 11 were excluded for the following reasons: not an original article, no clear effect of stress over microglia and the use of mixed stress protocols.
Microglia: The First Defence of The CNS
Microglia comprise about 10%–15% of all brain cells and are crucial players in normal development through the regulation of functional and structural processes, contributing to plasticity from individual synapses to neural circuits and behavior (Wake et al., 2013; Salter and Beggs, 2014; Verkhratsky et al., 2015). Microglial cells originate from extra-embryonic yolk sac progenitors, establish unique CNS cell populations and are maintained throughout life by local proliferation (Ginhoux et al., 2010, 2013). As tissue-resident macrophages in the CNS, along with other mononuclear phagocytes, microglia are critical effectors and regulators of changes in CNS homeostasis during development, in health and disease (Hanisch and Kettenmann, 2007; Prinz and Priller, 2014).
Some evidence points to new and fundamental roles for microglia in the control of neuronal proliferation and differentiation, as well as in the formation of synaptic connections (Kettenmann et al., 2011; Ginhoux et al., 2013). These cells are distributed in the brain parenchyma, have small delineated processes and actively screen the inter-neuronal space for incoming threats, exhibiting immune regulatory functions, from local surveillance to the removal of debris (Prinz and Priller, 2014). Microglial activation is the main neuroinflammatory element in the CNS, providing the front line defense whenever injury, disease or infection occurs (Lehnardt, 2010; Tang and Le, 2016).
Inflammatory processes are usually self-limited, culminating with tissue repair; damage to the CNS occurs when the system is over-activated for a long time, extending the release of pro-inflammatory mediators and neurotoxins. This process can worsen tissue damage and negatively impact disease outcome, leading to anxiety- and depressive-like states (Reader et al., 2015; Ramirez and Sheridan, 2016). Increasing evidence points to a heterogeneous status of microglial activation in the CNS. Although it is not a consensus, some authors categorize microglia into two opposite activation states, M1 and M2 phenotypes, which can produce either cytotoxic or neuroprotective effects (Tang and Le, 2016). M1-polarized microglia are associated with the production of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), superoxide, nitric oxide, reactive oxygen species and proteases (Ajmone-Cat et al., 2013), whereas M2-polarized microglia express cytokines and receptors that are implicated in the inhibition of inflammation and restoration of homeostasis by tissue repair and extracellular matrix reconstruction (Nakagawa and Chiba, 2014; Tang and Le, 2016). Nevertheless, as this nomenclature is not fully accepted and some authors consider microglia polarization to have derived from studying peripheral macrophages rather than microglia (Ransohoff, 2016), it is important to carefully use and interpret these terms to avoid misunderstandings.
The SD Paradigm as a Valid Stressor
Most stressors in human life arise from interactions within the social environment. In fact, social stress encompasses various types of significant life events, ranging from maternal separation (Meaney, 2001; Nishi et al., 2014), brief episodes of social confrontations in adolescence and adulthood, to continuous subordination stress (Miczek et al., 2008). In preclinical studies, some models of stress are often criticized as being artificial and not representative of human stress (Björkqvist, 2001; Almeida et al., 2002).
The SD paradigm is recognized as an ethological valid method to engender social stress in rodents (Vasconcelos et al., 2015; Henriques-Alves and Queiroz, 2016; Koolhaas et al., 2017). RSD is a stressor that recapitulates key physiological, immunological and behavioral alterations observed in humans exposed to chronic psychosocial stress (McKim et al., 2016a). Models of psychosocial stress rely on innate social behavior among pairs or groups of male rodents allowing the formation of stable dominant-subordinate relationships (Krishnan and Nestler, 2011). Another strong point of these models is the lack of habituation; despite repeated exposures, animals continue to generate emotional stress responses (Tidey and Miczek, 1997).
SD stress activates the hypothalamic-pituitary-adrenal axis and sympathetic nervous system, increasing systemic glucocorticoids that trigger the release of catecholamines and pro-inflammatory cytokines (Avitsur et al., 2001; Herman et al., 2016). Although there are distinct models of social stress, this review will focus on the role of SD in the development of anxiety and MD, tracking the contribution of the over-activation of the main CNS immune component, microglia, in triggering these psychiatric diseases.
Effects of SD Stress on Microglial Cells
One of the major advances in the field of the study of psychiatric disorders came from the notion that the immune system and inflammatory processes can be activated by psychosocial stressors (Miller and Raison, 2015). Despite the well-established evidence that the peripheral and central immune systems act in concert to promote the stress reaction, greater attention has been given to immune cells of the CNS, in particular, microglia. Social stress may activate microglial cells in a way different from other stressors (Glaser and Kiecolt-Glaser, 2005; Calcia et al., 2016) and seems to exert a direct effect over microglia activity through the activation of glucocorticoid and mineralocorticoid (Sierra et al., 2008) and β-adrenergic receptors (Walker et al., 2013; Calcia et al., 2016). Considering these factors, we directed our attention to microglial reactions induced by SD; the evidence is presented in Table 1.
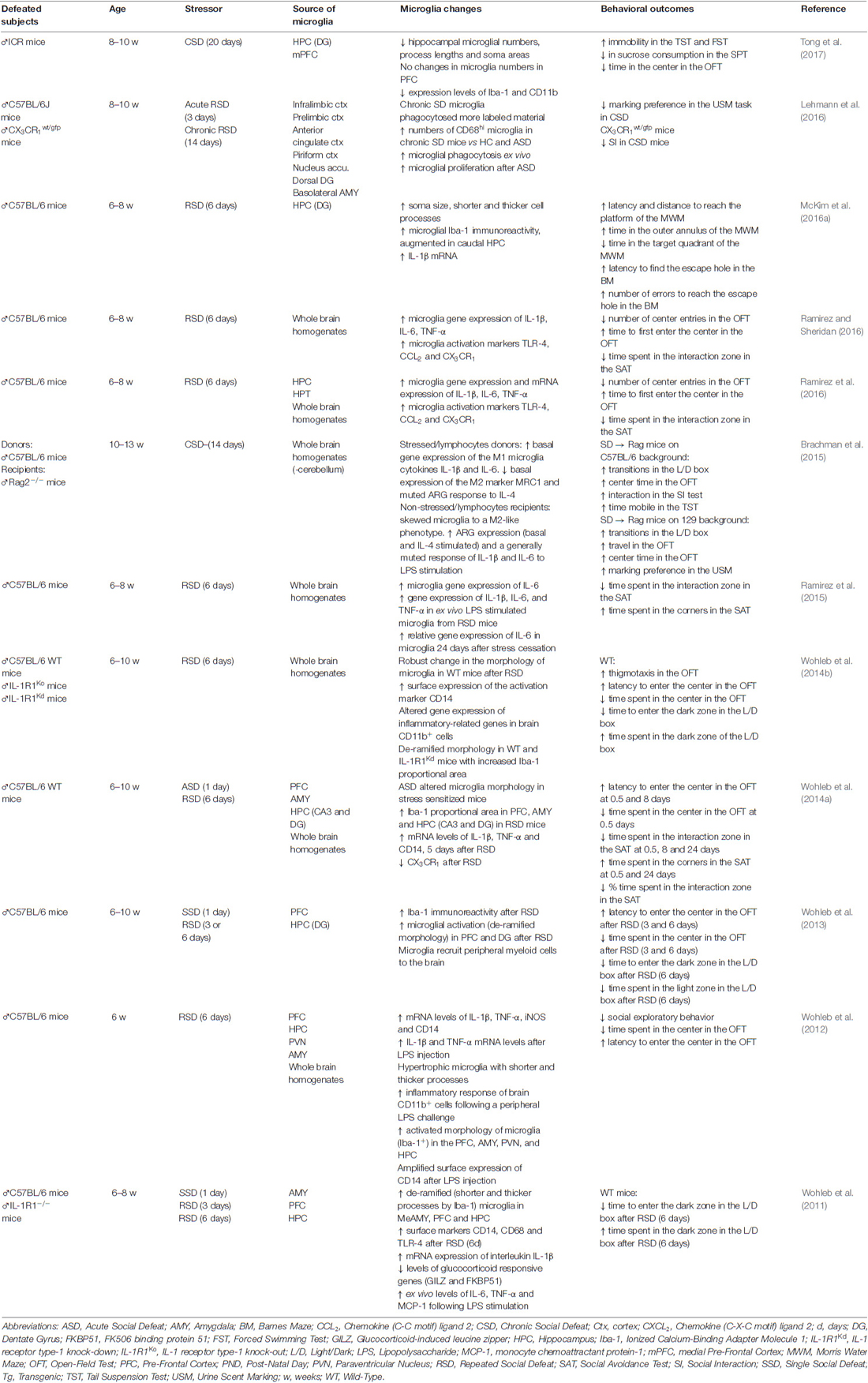
Table 1. Microglial activation profile induced by SD stress and related behavioral outcomes.
Microglia present increased activation status after SD (Wohleb et al., 2014b; Ramirez and Sheridan, 2016) and the effects are mainly observed within brain regions associated with fear, anxiety and threat appraisal (Wohleb et al., 2015). From a ramified aspect found in the immunosurveillant state, microglia change robustly to a de-ramified state with shorter and thicker processes (Wohleb et al., 2011, 2012, 2013, 2014b), leading to increased soma size after acute, RSD and CSD (McKim et al., 2016a; Figure 1). Changes in soma and processes are usually analyzed by increases in ionised calcium-binding adapter molecule 1 (Iba-1) or cluster of differentiation 11b (CD11b) immunoreactivity. However, although the vast majority of studies report results similar to those described above, decreases in microglial Iba-1, CD11b and consequently soma areas were found by others in the dentate gyrus (DG), but not in the medial prefrontal cortex, in a stress protocol that consisted of 20 days of exposure to SD (Tong et al., 2017). These controversial data could be attributed to differences in stress chronicity.
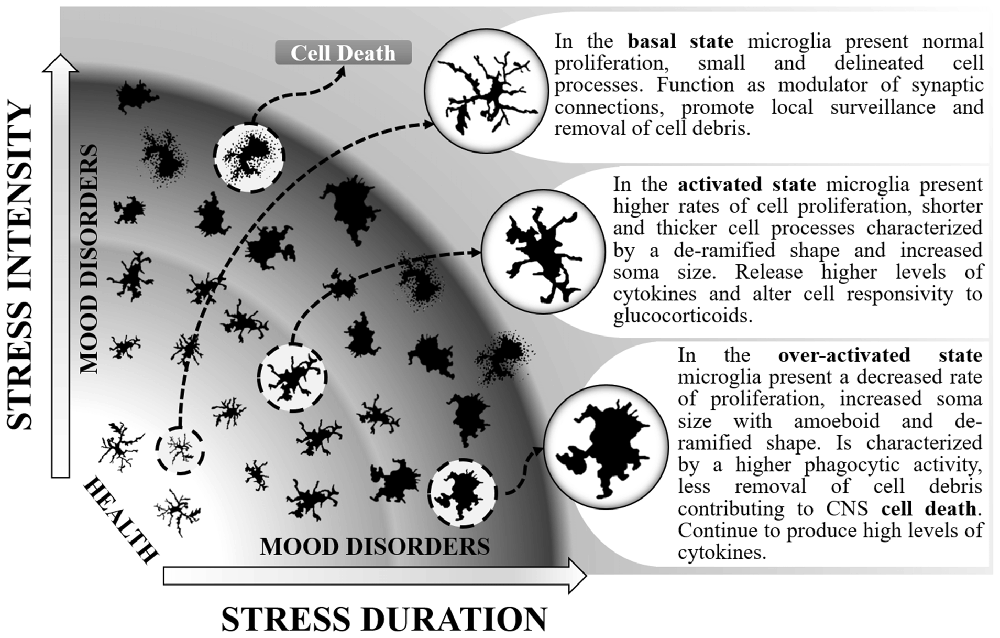
Figure 1. Different stages of microglial activation caused by changes in the intensity and duration of social stressors that can maintain individuals in a healthy state or contribute to both anxiety and depressive-like behaviors. In the basal state, microglia can be distinguished by normal levels of immunoreactivity to ionized calcium-binding adapter molecule 1 (Iba-1) and CD11b. In this state, enable proper coping to stress situations. When activated, microglia proliferate, release higher levels of interleukin-1β (IL-1β), interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α), present higher expression of toll-like receptor 4 (TLR-4), chemokine (C-C motif) ligand 2 (CCL2), CX3CR1) and decreased levels of glucocorticoid-induced leucine zipper (GILZ) and FK506 binding protein 51 (FKBP51). They can be distinguished by higher immunoreactivity to Iba-1, CD11b, CD14 and CD68. During the activated state, the release of pro-inflammatory mediators and the altered response to glucocorticoids may lead to anxiety. The activated state can also be protective by resuming stress effects. As a consequence, microglia return to their basal state. Otherwise, persistent stress shifts microglia to an over-activated state. Overactive microglia continue to release pro-inflammatory mediators (IL-1β and IL-6). They can be distinguished by higher immunoreactivity to CD68hi and possibly lower levels of CX3CR1 and iNOS antibodies. Along with the microglial phagocytic activity occurs a higher rate of cell death, including microglia as well as neuronal and other glial cells. This effect reduces their capacity to remove cell debris and surveillance of the inter-neuronal space, altogether leading to depression.
One additional way to identify changes in microglia activity is through the analysis of activation markers such as the chemokine (C-C motif) ligand 2 (CCL2), toll-like receptor 4 (TLR-4) or the CX3 chemokine receptor 1 (CX3CR1) which are expressed by microglial cells. SD induces an increase in the gene expression of TLR-4, CCL2 and CX3CR1 (Ramirez et al., 2015, 2016; Ramirez and Sheridan, 2016). However, decreases in CX3CR1 gene expression were also observed after SD, although in enriched brain CD11b+ cells (Wohleb et al., 2014a). One of the most evident reactions to SD observed in microglial cells is the rise in gene expression and mRNA levels of the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α and expression of the surface activation marker CD14. Increases of these inflammatory mediators were observed after acute, RSD and CSD (Wohleb et al., 2011, 2012, 2014a; Brachman et al., 2015; Ramirez et al., 2015, 2016; McKim et al., 2016a; Ramirez and Sheridan, 2016), even 24 days after stress cessation (Ramirez et al., 2015). The importance of these findings is reinforced by the results obtained from either microglial cells analyzed in fresh CNS tissue, isolated from socially defeated animals (Wohleb et al., 2012) or in ex vivo SD-sensitized microglia stimulated with lipopolysaccharide (LPS; Wohleb et al., 2011). Additionally, reduced levels of glucocorticoid responsive genes (GILZ and FKBP51) are evident after exposure to SD (Wohleb et al., 2011). Chronically SD stress-activated microglial cells increase their phagocytic activity. This effect is achieved by increasing the expression of CD68hi (a marker for phagocytic activity; Lehmann et al., 2016). The increasing phagocytic activity of microglia from CSD animals suggests that cellular debris or cell damage or death may be a hallmark of chronic stress effects on the brain. SD can also change microglial cell numbers; while acute SD enhances the number of microglia (Lehmann et al., 2016), CSD diminishes these cells (Tong et al., 2017), mainly in the hippocampus. It seems that a crucial factor is the intensity of activation of microglia by stress, which can lead to different psychiatric disorder outcomes (Figure 1). Taken together, these data highlight the broad spectrum of effects that can be observed in microglial cells when activated by SD.
The Link Between Microglial Activation, Anxiety- and Depressive-Like Behaviors
It is now well known that disturbances in microglial functioning has an etiological role in mood disorders (Frick et al., 2013; Kreisel et al., 2014). However, if the effect of social stress on these deregulated behaviors can be mainly attributed to microglial over-activation or if the participation of other CNS immune cells and/or the peripheral immune system plays a major role remains controversial. While researchers have shown in some studies that SD stress-induced anxiety- and depressive-like states are mediated by the activation of microglia with the involvement of peripheral macrophages and trafficking of monocytes to the brain (Wohleb et al., 2013, 2014b, 2015), other studies excluded the direct involvement of peripheral monocytes triggering these behaviors (Lehmann et al., 2016). Stress chronicity and/or peripheral wounds (triggers of peripheral immune reactions), which can usually be observed in defeated animals after confrontation with an aggressor, could be major determinants. This is one of the main reasons that led researchers to choose alternative stress protocols, such as variable unpredictable stress and foot shocks to study microglial activation in neuropsychiatric disorders, even though these procedures present lower ethological relevance.
Studies in humans have shown that microglial activation is positively correlated with psychiatric disorders. For example, individuals experiencing a major depressive episode present enhanced positron emission topography labeling of the translocator protein (TSPO), a putative marker of neuroinflammation and microglia activation (Setiawan et al., 2015). It has also been speculated that there is a causal link between microglial activation and suicidal behavior (Schnieder et al., 2014); neuroendocrine factors, cytokines and nitric oxide, which are released from microglial cells and are known to modulate noradrenergic or serotonergic neurotransmission, may trigger suicidal behavior (Steiner et al., 2008). Pro-inflammatory cytokines including IL-1β and TNF-α, can reduce the availability of serotonin, dopamine and noradrenaline by increasing the expression and function of reuptake transporters, reducing synthesis or decreasing monoamine precursors (Miller and Raison, 2015). Activated microglia can also act on the glutamate pathway and together with astrocytes stimulate the increased release of this neurotransmitter and decreased brain-derived neurotrophic factor, which ultimately leads to excitotoxicity (Steiner et al., 2012; Miller and Raison, 2015). Additionally, it has been shown that elevated pro-inflammatory cytokine levels caused by microglia activation associated with the recruitment of monocytes to the brain contribute to the development and persistent anxiety-like behavior (Wohleb et al., 2014b, 2015). Moreover, chronic microglial activation in particular can result in neuronal apoptosis, neurogenesis inhibition, hippocampal volume reduction, lower neurotransmitters synthesis and cytotoxicity (Ascoli et al., 2016), which is ultimately related to depressive behavior.
Although microglia are not the only effectors of the immune system, it has been suggested that the anti-inflammatory effect of antidepressants may have protective effects by silencing RSD-induced priming and activation of microglia, thus down-regulating the biosynthesis of high levels of pro-inflammatory cytokines (Ramirez et al., 2015). Recently, microglia have been recognized as important targets for pharmaceutical research. Brain diseases, including depression and anxiety, could potentially be treated with drugs that are capable of inhibiting or restoring specific microglial functions (Biber et al., 2016). Anti-inflammatory drugs such as COX2 inhibitors or minocycline, aimed at inhibiting the pro-inflammatory status of microglia, have been suggested as therapeutics for inflammatory brain diseases (Biber et al., 2016). The CX3CR1, as an exclusive microglial marker, could also be a potential target. Since the activation of microglia is not consistent for all patients, it has been recently proposed that anti-inflammatory treatment targeting microglial activation could specifically be more effective in patients with increased microglial activation, leading to the idea that microglial activation may be a marker for severe and untreatable psychiatric disorders (Mondelli et al., 2017).
Social stress can alter the number of microglial cells (Lehmann et al., 2016; Tong et al., 2017), mainly dependent on the duration of stress exposure. While acute, but not CSD is supposed to increase microglial proliferation selectively in telencephalic stress-related brain areas (Lehmann et al., 2016), a loss of hippocampal microglia was observed and is supposed to promote the development of MD, indicating that the restoration of microglial functions and/or numbers may be beneficial for the therapy of MD (Tong et al., 2017). Since pro-inflammatory cytokines can also modify neurogenesis in the hippocampus (Koo and Duman, 2009), RSD has been shown to induce anxiety-like behavior by impairing the neuronal differentiation of neural progenitor cells in the hippocampus that proliferated during stress exposure. These data were positively correlated to an impairment in performance on working and spatial memory in the Morris water maze (MWM) and transiently disrupted short-term memory recall in the Barnes maze (BM; McKim et al., 2016a). Overall, these data highlight the magnitude of the microglial over-activation-induced deficits in monoamine neurotransmission, cytotoxicity, cellular loss and reduced neurogenesis, ultimately leading to memory impairment and behaviors that are observed in both, anxiety and depression.
Conclusion Remarks
Exposure to SD induces microglial cells to assume an activated state, which initially may be considered beneficial. RSD and CSD can induce microglia to assume over-activated states that, by persistently releasing pro-inflammatory mediators, cytotoxins and reactive oxygen species, may cause cellular dystrophy and a loss or decreased function of neuronal activity through excessively pruned synaptic connections. All of these stress effects over microglia worsen memory and behaviors that are important factors in psychiatric disorders. The SD paradigm is an important tool to induce anxiety- and depressive-like states in laboratory animals for investigating stress-induced immunological and behavioral alterations.
It seems that the development of anxiety and MD is, besides microglial activation, dependent on peripheral monocyte recruitment to the brain (McKim et al., 2016b), attaching importance to the bidirectional communication between the brain and peripheral immune system. However, since the activation of microglia by psychosocial stress might be different from that of physical injury (Glaser and Kiecolt-Glaser, 2005), more attention must be given to peripheral wounds when studying SD stress effects over central immune reactions. SD protocols that allow physical injuries to the defeated animal during confrontations with an opponent may contribute to the participation of peripheral immune cells in the final outcome. Alternatively, stress protocols that do not involve physical injuries, such as chronic unpredictable stress, can be used to overcome this issue. Contradictory findings have shown that microglial over-activation, as well as microglial dystrophy and loss, can mediate the development of MD. Depression is considered to be a disorder that is associated with microglial over-activation. That leads to an interpretation that suppressed microglial hyperactivity should be the focus to treat depressive symptoms (Tong et al., 2017). However, since microglia in its basal state is also critical for brain normal function, microglial dystrophy and loss would also mediate the development of this disorder (Kreisel et al., 2014; Tong et al., 2017). Therefore, over-inhibition or over-down-regulation of microglial function will inevitably produce detrimental effects as well. Focusing on microglial cells as therapeutic targets for pharmacological interventions, especially by restoring functions and/or basal levels, may be a promising strategy for anxiety and depression therapy.
Author Contributions
All authors contributed equally to this study. All of them contributed to the conception and design of the work, literature analyses and interpretation, drafting the article, critical revision and final approval of the version to be published.
Funding
This research was supported by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—CAPES-Brazil (PAJT 88887.096822/2015-00 LA-S), by Conselho Nacional de Desenvolvimento Científico e Tecnológico—CNPq-Brazil (475176/2012-0 RMMA) and by Fundo de Incentivo à Pesquisa e Eventos (FIPE-HCPA/UFRGS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Ajmone-Cat, M. A., Mancini, M., De Simone, R., Cilli, P., and Minghetti, L. (2013). Microglial polarization and plasticity: evidence from organotypic hippocampal slice cultures: microglial polarization and tolerization. Glia 61, 1698–1711. doi: 10.1002/glia.22550
Almeida, D. M., Wethington, E., and Kessler, R. C. (2002). The daily inventory of stressful events: an interview-based approach for measuring daily stressors. Assessment 9, 41–55. doi: 10.1177/1073191102009001006
Ascoli, B. M., Géa, L. P., Colombo, R., Barbé-Tuana, F. M., Kapczinski, F., and Rosa, A. R. (2016). The role of macrophage polarization on bipolar disorder: identifying new therapeutic targets. Aust. N Z J. Psychiatry 50, 618–630. doi: 10.1177/0004867416642846
Avitsur, R., Stark, J. L., and Sheridan, J. F. (2001). Social stress induces glucocorticoid resistance in subordinate animals. Horm. Behav. 39, 247–257. doi: 10.1006/hbeh.2001.1653
Biber, K., Möller, T., Bodekke, E., and Prinz, M. (2016). Central nervous system myeloid cells as drug targets: current status and translational challenges. Nat. Rev. Drug Discov. 15, 110–124. doi: 10.1038/nrd.2015.14
Björkqvist, K. (2001). Social defeat as a stressor in humans. Physiol. Behav. 73, 435–442. doi: 10.1016/s0031-9384(01)00490-5
Brachman, R. A., Lehmann, M. L., Maric, D., and Herkenham, M. (2015). Lymphocytes from chronically stressed mice confer antidepressant-like effects to naive mice. J. Neurosci. 35, 1530–1538. doi: 10.1523/JNEUROSCI.2278-14.2015
Calcia, M. A., Bonsall, D. R., Bloomfield, P. S., Selvaraj, S., Barichello, T., and Howes, O. D. (2016). Stress and neuroinflammation: a systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology 233, 1637–1650. doi: 10.1007/s00213-016-4218-9
Ferrari, A. J., Charlson, F. J., Norman, R. E., Patten, S. B., Freedman, G., Murray, C. J. L., et al. (2013). Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 10:e1001547. doi: 10.1371/journal.pmed.1001547
Frick, L. R., Williams, K., and Pittenger, C. (2013). Microglial dysregulation in psychiatric disease. Clin. Dev. Immunol. 2013:608654. doi: 10.1155/2013/608654
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330, 841–845. doi: 10.1126/science.1194637
Ginhoux, F., Lim, S., Hoeffel, G., Low, D., and Huber, T. (2013). Origin and differentiation of microglia. Front. Cell. Neurosci. 7:45. doi: 10.3389/fncel.2013.00045
Glaser, R., and Kiecolt-Glaser, J. K. (2005). Stress-induced immune dysfunction: implications for health. Nat. Rev. Immunol. 5, 243–251. doi: 10.1038/nri1571
Hanisch, U.-K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Henriques-Alves, A. M., and Queiroz, C. M. (2016). Ethological evaluation of the effects of social defeat stress in mice: beyond the social interaction ratio. Front. Behav. Neurosci. 9:364. doi: 10.3389/fnbeh.2015.00364
Herman, J. P., McKlveen, J. M., Ghosal, S., Kopp, B., Wulsin, A., Makinson, R., et al. (2016). “Regulation of the hypothalamic-pituitary-adrenocortical stress response,” in Comprehensive Physiology, ed. R. Terjung (Hoboken, NJ: John Wiley and Sons, Inc.), 603–621.
Hollis, F., and Kabbaj, M. (2014). Social defeat as an animal model for depression. ILAR J. 55, 221–232. doi: 10.1093/ilar/ilu002
Kettenmann, H., Hanisch, U.-K., Noda, M., and Verkhratsky, A. (2011). Physiology of microglia. Physiol. Rev. 91, 461–553. doi: 10.1152/physrev.00011.2010
Koo, J. W., and Duman, R. S. (2009). Interleukin-1 receptor null mutant mice show decreased anxiety-like behavior and enhanced fear memory. Neurosci. Lett. 456, 39–43. doi: 10.1016/j.neulet.2009.03.068
Koolhaas, J. M., de Boer, S. F., Buwalda, B., and Meerlo, P. (2017). Social stress models in rodents: towards enhanced validity. Neurobiol. Stress 6, 104–112. doi: 10.1016/j.ynstr.2016.09.003
Kreisel, T., Frank, M. G., Licht, T., Reshef, R., Ben-Menachem-Zidon, O., Baratta, M. V., et al. (2014). Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 19, 699–709. doi: 10.1038/mp.2013.155
Krishnan, V., and Nestler, E. J. (2011). “Animal models of depression: molecular perspectives,” in Molecular and Functional Models in Neuropsychiatry, (Vol. 7) ed. J. J. Hagan (Berlin, Heidelberg: Springer Berlin Heidelberg), 121–147.
Lehmann, M. L., Cooper, H. A., Maric, D., and Herkenham, M. (2016). Social defeat induces depressive-like states and microglial activation without involvement of peripheral macrophages. J. Neuroinflammation 13:224. doi: 10.1186/s12974-016-0672-x
Lehnardt, S. (2010). Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia 58, 253–263. doi: 10.1002/glia.20928
McKim, D. B., Niraula, A., Tarr, A. J., Wohleb, E. S., Sheridan, J. F., and Godbout, J. P. (2016a). Neuroinflammatory dynamics underlie memory impairments after repeated social defeat. J. Neurosci. 36, 2590–2604. doi: 10.1523/JNEUROSCI.2394-15.2016
McKim, D. B., Patterson, J. M., Wohleb, E. S., Jarrett, B. L., Reader, B. F., Godbout, J. P., et al. (2016b). Sympathetic release of splenic monocytes promotes recurring anxiety following repeated social defeat. Biol. Psychiatry 79, 803–813. doi: 10.1016/j.biopsych.2015.07.010
Meaney, M. J. (2001). Maternal care, gene expression, and the transmission of individual differences in stress reactivity across generations. Annu. Rev. Neurosci. 24, 1161–1192. doi: 10.1146/annurev.neuro.24.1.1161
Miczek, K. A., Nikulina, E. M., Shimamoto, A., and Covington, H. E. III. (2011). Escalated or suppressed cocaine reward, tegmental BDNF, and accumbal dopamine caused by episodic versus continuous social stress in rats. J. Neurosci. 31, 9848–9857. doi: 10.1523/JNEUROSCI.0637-11.2011
Miczek, K., Yap, J., and Covington, H. III. (2008). Social stress, therapeutics and drug abuse: preclinical models of escalated and depressed intake. Pharmacol. Ther. 120, 102–128. doi: 10.1016/j.pharmthera.2008.07.006
Miller, A. H., and Raison, C. L. (2015). The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 16, 22–34. doi: 10.1038/nri.2015.5
Mondelli, V., Vernon, A. C., Turkheimer, F., Dazzan, P., and Pariante, C. M. (2017). Brain microglia in psychiatric disorders. Lancet Psychiatry 4, 563–572. doi: 10.1016/S2215-0366(17)30101-3
Nakagawa, Y., and Chiba, K. (2014). Role of microglial M1/M2 polarization in relapse and remission of psychiatric disorders and diseases. Pharmaceuticals 7, 1028–1048. doi: 10.3390/ph7121028
Nishi, M., Horii-Hayashi, N., and Sasagawa, T. (2014). Effects of early life adverse experiences on the brain: implications from maternal separation models in rodents. Front. Neurosci. 8:166. doi: 10.3389/fnins.2014.00166
Prinz, M., and Priller, J. (2014). Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat. Rev. Neurosci. 15, 300–312. doi: 10.1038/nrn3722
Ramirez, K., Niraula, A., and Sheridan, J. F. (2016). GABAergic modulation with classical benzodiazepines prevent stress-induced neuro-immune dysregulation and behavioral alterations. Brain Behav. Immun. 51, 154–168. doi: 10.1016/j.bbi.2015.08.011
Ramirez, K., Shea, D. T., McKim, D. B., Reader, B. F., and Sheridan, J. F. (2015). Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain Behav. Immun. 46, 212–220. doi: 10.1016/j.bbi.2015.01.016
Ramirez, K., and Sheridan, J. F. (2016). Antidepressant imipramine diminishes stress-induced inflammation in the periphery and central nervous system and related anxiety- and depressive- like behaviors. Brain Behav. Immun. 57, 293–303. doi: 10.1016/j.bbi.2016.05.008
Ransohoff, R. M. (2016). A polarizing question: do M1 and M2 microglia exist? Nat. Neurosci. 19, 987–991. doi: 10.1038/nn.4338
Razzoli, M., Carboni, L., and Arban, R. (2009). Alterations of behavioral and endocrinological reactivity induced by 3 brief social defeats in rats: relevance to human psychopathology. Psychoneuroendocrinology 34, 1405–1416. doi: 10.1016/j.psyneuen.2009.04.018
Reader, B. F., Jarrett, B. L., McKim, D. B., Wohleb, E. S., Godbout, J. P., and Sheridan, J. F. (2015). Peripheral and central effects of repeated social defeat stress: monocyte trafficking, microglial activation and anxiety. Neuroscience 289, 429–442. doi: 10.1016/j.neuroscience.2015.01.001
Salter, M. W., and Beggs, S. (2014). Sublime microglia: expanding roles for the guardians of the CNS. Cell 158, 15–24. doi: 10.1016/j.cell.2014.06.008
Schnieder, T. P., Trencevska, I., Rosoklija, G., Stankov, A., Mann, J. J., Smiley, J., et al. (2014). Microglia of prefrontal white matter in suicide. J. Neuropathol. Exp. Neurol. 73, 880–890. doi: 10.1097/NEN.0000000000000107
Setiawan, E., Wilson, A. A., Mizrahi, R., Rusjan, P. M., Miler, L., Rajkowska, G., et al. (2015). Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72, 268–275. doi: 10.1001/jamapsychiatry.2014.2427
Sierra, A., Gottfried-Blackmore, A., Milner, T. A., McEwen, B. S., and Bulloch, K. (2008). Steroid hormone receptor expression and function in microglia. Glia 56, 659–674. doi: 10.1002/glia.20644
Steiner, J., Bielau, H., Brisch, R., Danos, P., Ullrich, O., Mawrin, C., et al. (2008). Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J. Psychiatr. Res. 42, 151–157. doi: 10.1016/j.jpsychires.2006.10.013
Steiner, J., Bogerts, B., Sarnyai, Z., Walter, M., Gos, T., Bernstein, H.-G., et al. (2012). Bridging the gap between the immune and glutamate hypotheses of schizophrenia and major depression: potential role of glial NMDA receptor modulators and impaired blood-brain barrier integrity. World J. Biol. Psychiatry 13, 482–492. doi: 10.3109/15622975.2011.583941
Tang, Y., and Le, W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53, 1181–1194. doi: 10.1007/s12035-014-9070-5
Tidey, J. W., and Miczek, K. A. (1997). Acquisition of cocaine self-administration after social stress: role of accumbens dopamine. Psychopharmacology 130, 203–212. doi: 10.1007/s002130050230
Tong, L., Gong, Y., Wang, P., Hu, W., Wang, J., Chen, Z., et al. (2017). Microglia loss contributes to the development of major depression induced by different types of chronic stresses. Neurochem. Res. doi: 10.1007/s11064-017-2270-4 [Epub ahead of print].
Vasconcelos, M., Stein, D. J., and de Almeida, R. M. M. (2015). Social defeat protocol and relevant biomarkers, implications for stress response physiology, drug abuse, mood disorders and individual stress vulnerability: a systematic review of the last decade. Trends Psychiatry Psychother. 37, 51–66. doi: 10.1590/2237-6089-2014-0034
Verkhratsky, A., Noda, M., and Parpura, V. (2015). “Microglia: structure and function,” in Brain Mapping, ed. A. W. Toga (New York, NY: Elsevier), 109–113.
Wake, H., Moorhouse, A. J., Miyamoto, A., and Nabekura, J. (2013). Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 36, 209–217. doi: 10.1016/j.tins.2012.11.007
Walker, F., Nilsson, M., and Jones, K. (2013). Acute and chronic stress-induced disturbances of microglial plasticity, phenotype and function. Curr. Drug Targets 14, 1262–1276. doi: 10.2174/13894501113149990208
Whiteford, H. A., Degenhardt, L., Rehm, J., Baxter, A. J., Ferrari, A. J., Erskine, H. E., et al. (2013). Global burden of disease attributable to mental and substance use disorders: findings from the Global Burden of Disease study 2010. Lancet 382, 1575–1586. doi: 10.1016/S0140-6736(13)61611-6
Wohleb, E. S., Fenn, A. M., Pacenta, A. M., Powell, N. D., Sheridan, J. F., and Godbout, J. P. (2012). Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 37, 1491–1505. doi: 10.1016/j.psyneuen.2012.02.003
Wohleb, E. S., Hanke, M. L., Corona, A. W., Powell, N. D., Stiner, L. M., Bailey, M. T., et al. (2011). β-adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 31, 6277–6288. doi: 10.1523/JNEUROSCI.0450-11.2011
Wohleb, E. S., McKim, D. B., Shea, D. T., Powell, N. D., Tarr, A. J., Sheridan, J. F., et al. (2014a). Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol. Psychiatry 75, 970–981. doi: 10.1016/j.biopsych.2013.11.029
Wohleb, E. S., Patterson, J. M., Sharma, V., Quan, N., Godbout, J. P., and Sheridan, J. F. (2014b). Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J. Neurosci. 34, 2583–2591. doi: 10.1523/JNEUROSCI.3723-13.2014
Wohleb, E. S., McKim, D. B., Sheridan, J. F., and Godbout, J. P. (2015). Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front. Neurosci. 8:447. doi: 10.3389/fnins.2014.00447
Keywords: microglia, neuroimmunity, immune cells, psychosocial stress, neuropsychiatric disorders, inflammatory processes
Citation: Stein DJ, Vasconcelos MF, Albrechet-Souza L, Ceresér KMM and de Almeida RMM (2017) Microglial Over-Activation by Social Defeat Stress Contributes to Anxiety- and Depressive-Like Behaviors. Front. Behav. Neurosci. 11:207. doi: 10.3389/fnbeh.2017.00207
Received: 18 July 2017; Accepted: 10 October 2017;
Published: 24 October 2017.
Edited by:
Mathias V. Schmidt, Max Planck Institute of Psychiatry (MPG), GermanyReviewed by:
Barbara Di Benedetto, University of Regensburg, GermanyJaime Fornaguera, University of Costa Rica, Costa Rica
Copyright © 2017 Stein, Vasconcelos, Albrechet-Souza, Ceresér and de Almeida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa M. M. de Almeida, cm9zYS5hbG1laWRhQHVmcmdzLmJy