 Ketan K. Marballi
Ketan K. Marballi Amelia L. Gallitano
Amelia L. Gallitano- Department of Basic Medical Sciences and Psychiatry, University of Arizona College of Medicine—Phoenix, Phoenix, AZ, United States
While the causes of myriad medical and infectious illnesses have been identified, the etiologies of neuropsychiatric illnesses remain elusive. This is due to two major obstacles. First, the risk for neuropsychiatric disorders, such as schizophrenia, is determined by both genetic and environmental factors. Second, numerous genes influence susceptibility for these illnesses. Genome-wide association studies have identified at least 108 genomic loci for schizophrenia, and more are expected to be published shortly. In addition, numerous biological processes contribute to the neuropathology underlying schizophrenia. These include immune dysfunction, synaptic and myelination deficits, vascular abnormalities, growth factor disruption, and N-methyl-D-aspartate receptor (NMDAR) hypofunction. However, the field of psychiatric genetics lacks a unifying model to explain how environment may interact with numerous genes to influence these various biological processes and cause schizophrenia. Here we describe a biological cascade of proteins that are activated in response to environmental stimuli such as stress, a schizophrenia risk factor. The central proteins in this pathway are critical mediators of memory formation and a particular form of hippocampal synaptic plasticity, long-term depression (LTD). Each of these proteins is also implicated in schizophrenia risk. In fact, the pathway includes four genes that map to the 108 loci associated with schizophrenia: GRIN2A, nuclear factor of activated T-cells (NFATc3), early growth response 1 (EGR1) and NGFI-A Binding Protein 2 (NAB2); each of which contains the “Index single nucleotide polymorphism (SNP)” (most SNP) at its respective locus. Environmental stimuli activate this biological pathway in neurons, resulting in induction of EGR immediate early genes: EGR1, EGR3 and NAB2. We hypothesize that dysfunction in any of the genes in this pathway disrupts the normal activation of Egrs in response to stress. This may result in insufficient electrophysiologic, immunologic, and neuroprotective, processes that these genes normally mediate. Continued adverse environmental experiences, over time, may thereby result in neuropathology that gives rise to the symptoms of schizophrenia. By combining multiple genes associated with schizophrenia susceptibility, in a functional cascade triggered by neuronal activity, the proposed biological pathway provides an explanation for both the polygenic and environmental influences that determine the complex etiology of this mental illness.
Introduction
Since the start of the 21st century, tremendous advances have been made in the identification of genes associated with risk for major neuropsychiatric illnesses such as schizophrenia, bipolar disorder and depression. However, the field of psychiatry still lags behind other areas of medicine in identifying even a single gene that has been definitively demonstrated to cause any of these mental illnesses. This is due in large part to the complex genetics that underlie these disorders. Now, 15 years after the landmark publication that identified Neuregulin 1 (NRG1) as a candidate gene for schizophrenia (Stefansson et al., 2002), there remain two unanswered questions at the forefront of the field of psychiatric genetics: (1) how can the polygenic nature of susceptibility to schizophrenia be explained? and (2) how do genes implicated in risk for schizophrenia interact with environmental factors to give rise to the disorder?
To address both of these questions, we have proposed the hypothesis that numerous schizophrenia susceptibility genes form a critical signaling pathway that is responsive to environmental stress. Dysfunction of any of the proteins in this pathway would reduce the normal activation of a key transcription factor, early growth response 3 (EGR3), an immediate early gene that is both associated with schizophrenia in humans (Yamada et al., 2007; Kim et al., 2010; Ning et al., 2012; Zhang et al., 2012; Huentelman et al., 2015), and expressed at reduced levels in patients’ brains (Mexal et al., 2005; Yamada et al., 2007). As a critical mediator of numerous biological processes disrupted in schizophrenia, such as growth factor signaling, myelination, vascularization, immune function, memory formation and synaptic plasticity (Gallitano-Mendel et al., 2007; Jones et al., 2007; Suehiro et al., 2010; Li et al., 2012; Kurosaka et al., 2017), insufficient activation of EGR3 may result in neuropathology that gives rise to schizophrenia.
Schizophrenia risk is influenced by many genes in addition to environmental factors. The illness has a prevalence rate of roughly 1% worldwide, and its cause remains unknown. Studies show concordance rates of approximately 50% in monozygotic twins, roughly twice that of dizygotic twins, indicating that there are both genetic and non-genetic determinants of schizophrenia (McGue and Gottesman, 1991). Stressful events are associated with schizophrenia risk. These include prenatal stress such as nutritional deficiency, or exposure to famine, infection (e.g., rubella, influenza, Toxoplasma gondii and herpes simplex virus), or maternal stress. Stress during the perinatal period and early life also increase risk for the illness. Examples include obstetric complications and perinatal trauma, and stressful life events such as childhood trauma (Corcoran et al., 2001, 2003; Mittal et al., 2008; van Winkel et al., 2008; Brown and Derkits, 2010; Brown, 2011). Adding to the complex etiology of this illness, the most recent genome-wide association study (GWAS) of single nucleotide polymorphisms (SNPs) identified 108 genomic loci that influence schizophrenia susceptibility (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). To date, there is no consensus on a mechanism to explain how so many genetic variations interact with environmental factors to cause schizophrenia.
Identifying A Pathway
Immediate early genes are a class of genes that are rapidly induced in response to a stimulus, in a manner that is independent of protein synthesis. In the brain, they are expressed within minutes of neuronal activity triggered by environmental stimuli. A large number of immediate early genes encode proteins that function as transcription factors (termed immediate early gene transcription factors (Curran and Morgan, 1995)). These genes are thus poised to translate changes in the environment into long-term changes in the brain through the regulation of their target genes. This presumably underlies the critical role of many immediate early gene transcription factors in memory formation, a process that requires long-term encoding of environmental experiences.
We have hypothesized that this function of immediate early gene transcription factors, as key regulators of the brain’s gene-expression response to experience, uniquely positions them to mediate the dual genetic and environmental influences on schizophrenia susceptibility (Gallitano-Mendel et al., 2008). We focus on the Egr family of immediate early genes since they are activated in response to changes in the environment (Senba and Ueyama, 1997; Martinez et al., 2002), and regulate fundamental processes in the nervous system that are known to be dysfunctional in schizophrenia. These include myelination, vascularization, learning and memory, and synaptic plasticity (Paulsen et al., 1995; Guzowski et al., 2001; Nagarajan et al., 2001; Bozon et al., 2002, 2003; Flynn et al., 2003; Crabtree and Gogos, 2014). In addition, Egrs are activated downstream of N-methyl-D-aspartate receptors (NMDARs; Cole et al., 1989) and growth factors (Schulze et al., 2008; Shin et al., 2010), dysfunction of which have each been hypothesized to contribute to schizophrenia susceptibility (Olney et al., 1999; Moises et al., 2002; Calabrese et al., 2016).
We hypothesize that variations that reduce the normal amount of Egr gene expression in response to environmental stimuli would result in lower than normal levels function of these processes. Specifically, this would result in insufficient activation of Egr target genes, such as brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeleton associated protein (Arc/Arg3.1: hereafter referred to as “Arc”; Li et al., 2005; Maple et al., 2017; Meyers et al., 2017a,b). Lower than normal activation of these genes could account for the reduced levels of white matter, reduced synaptic spine density, and deficiencies in memory and cognitive processing, that are seen in patients with schizophrenia, or hypothesized to underlie its pathogenesis (Ingvar and Franzen, 1974; Saykin et al., 1991; Glantz and Lewis, 2000; Moises et al., 2002; Davis et al., 2003).
Several findings led us to focus specifically on Egr family member Egr3 as we investigated this hypothesis. First, NRG1 was identified as a schizophrenia candidate gene in a large-scale genetic association study (Stefansson et al., 2002). In mice, Nrg1 was found to be essential to maintain Egr3 expression in the peripheral muscle spindle (Hippenmeyer et al., 2002). Subsequently the protein phosphatase calcineurin (CN) was identified as a schizophrenia candidate protein based on the phenotype of CN−/− mice, and the association of the gene encoding one of its subunits (PPP3CC) with schizophrenia in a Japanese population (Gerber et al., 2003). CN had also been shown to regulate expression of Egr3 in the immune system (Mittelstadt and Ashwell, 1998). Together, these findings indicated that Egr3 was regulated downstream of three proteins independently implicated in schizophrenia risk: NMDARs, NRG1 and CN.
To answer whether Egr3 may play a role in schizophrenia, we investigated the behavior and physiology of Egr3-deficient (−/−) mice. Our studies revealed that Egr3−/− mice display behavioral abnormalities consistent with animal models of schizophrenia. These include locomotor hyperactivity, a behavior produced in rodents treated with drugs that cause psychosis in humans, such as NMDAR antagonists—phencyclidine and ketamine, and dopaminergic agents including amphetamines (Lipska and Weinberger, 2000; Tamminga et al., 2003). The fact that this activity is reversible with antipsychotic medications further validates this as a phenotype relevant to schizophrenia (Freed et al., 1984; O’Neill and Shaw, 1999). Egr3−/− mice show locomotor hyperactivity in response to a novel environment that is reversed with antipsychotic treatment (Gallitano-Mendel et al., 2008). Schizophrenia is also characterized by cognitive deficits, which a feature that led to Emil Kraepelin to define the syndrome “Dementia Praecox” (Kraepelin and Robertson, 1919). Egr3−/− mice have deficits in spatial memory as well as defects in hippocampal long-term depression (LTD), a form of synaptic plasticity (Gallitano-Mendel et al., 2007, 2008) normally activated in response to stress and novelty (Manahan-Vaughan and Braunewell, 1999; Kemp and Manahan-Vaughan, 2007). Notably, both NMDARs and CN, proteins that regulate induction of Egr3, are also required for memory formation and LTD. This suggested that EGR3 was functioning in a signal transduction cascade of proteins that were activated in the brain in response to novelty and stress, and required for both memory formation and LTD (Gallitano-Mendel et al., 2007).
We subsequently described that numerous of the key proteins in this pathway were associated with risk for schizophrenia (Gallitano-Mendel et al., 2008). This led us to hypothesize that other genes in this pathway that shared the characteristics of regulating memory and hippocampal LTD, such as the EGR3 target gene ARC, should also be candidates for a role in schizophrenia susceptibility (Gallitano, 2008). Indeed, numerous genome wide association studies of copy number variations (CNVs), de novo mutations and SNPs (Kirov et al., 2012; Fromer et al., 2014; Purcell et al., 2014), as well as our own resequencing study that identified a schizophrenia associated ARC SNP (Huentelman et al., 2015), have subsequently supported this hypothesis.
Here we present the key proteins that comprise our proposed biological pathway, shown in Figure 1. We begin with a brief review of hippocampal LTD, highlighting its response to stress. We follow with a section devoted to each protein in the pathway. For each protein we: (1) summarize the evidence demonstrating the regulatory relationships that support its position in the pathway; (2) outline the findings indicating its roles in processes that are disrupted in schizophrenia; (3) mention supporting studies that demonstrate its genetic association with risk for the illness (summarized in Supplementary Table S1); and (4) if known, altered levels in the brains of patients with schizophrenia. When available, we will also review the role for each in synaptic plasticity, focusing on LTD, and define the relationship of the protein being discussed with EGR3. We will include a brief section on additional proteins for which there is new evidence indicating a potential contribution to this pathway. We briefly review the role of LTD in the neurobiological response to stress and novelty. In the Discussion we propose our hypothesis that this biological pathway mediates protective neurobiological responses to stress. We then present a model to explain how genetic variations in genes of this pathway may produce a predisposition to schizophrenia that results in illness in a manner dependent upon the stress history of an individual.
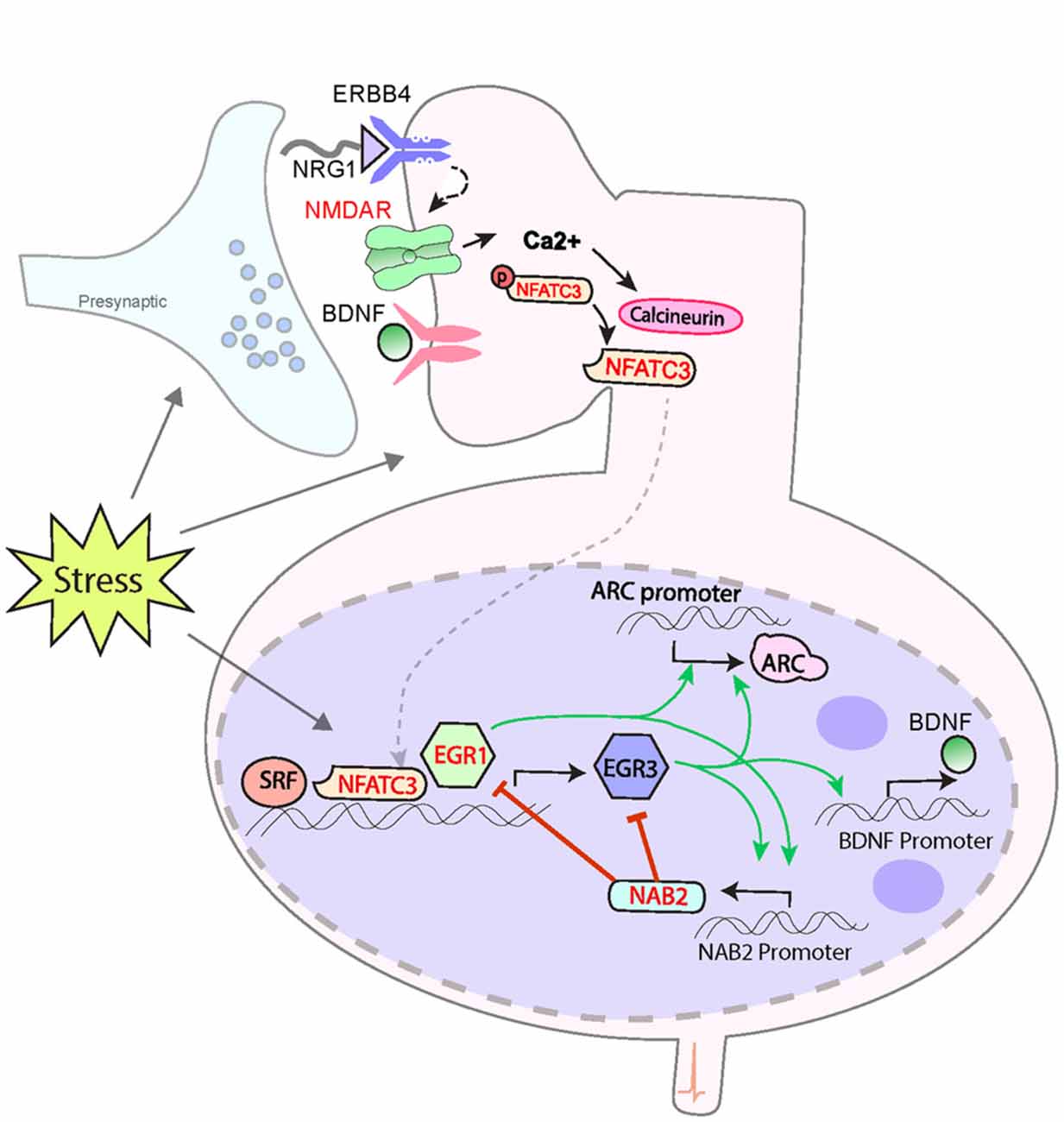
Figure 1. Model for a biological pathway regulating memory, synaptic plasticity and schizophrenia risk. Numerous schizophrenia-risk associated proteins act either upstream or downstream of immediate early genes EGR3 and ARC. Red labels indicate proteins encoded by genes that map to the 108 loci for schizophrenia risk. Both EGR3 and ARC are activated by environmental events, such as stress, via activity-dependent activation of NMDARs. The resulting influx of Ca++ triggers Calcineurin/(CN) nuclear factors of activated T-cell (NFAT) signaling which can induce EGR3 expression. EGR3 positively regulates, and maintains expression of, ARC. NGFI-A Binding Protein 2 (NAB2), which is a transcriptional target of EGR3 and EGR1, binds to EGR proteins (EGR1, 2 and 3) to co-regulate expression of target genes. This action includes feedback inhibition of EGR1 and EGR3, and NAB2 itself, a critical element of the temporal regulation of activity-dependent genes. Additional proteins that interact in the pathway include NRG1 which, via binding to ERBB receptors, maintains expression of EGR3 (in muscle spindles), and has been shown to activate NMDARs. BDNF, via binding to Trk-B receptors, regulates EGR3-mediated expression of NMDAR subunit NR1. EGR3, in turn, is required for activity-dependent expression of BDNF. SRF is a transcription factor required for memory formation and long-term depression (LTD), which regulates expression of EGR3 (and EGR1). Perturbations in any components of this biological pathway are expected to result in deficient induction of EGR3, and its target genes, in response to neuronal activity, including that triggered by stress. Insufficient activation of this pathway in response to stimuli may result in poor memory formation and contribute to cognitive deficits. Deficient activation of the pathway in response to stress may also result in insufficient neuroprotective processes and thereby contribute, over time, to the neuropathology that gives rise to schizophrenia (see Figure 2). Note that many interactions represented are taken from literature on studies in immune system and have yet to be validated in the brain. The shared roles of these proteins in the immune system may contribute to association between schizophrenia and immune dysfunction. Abbreviations: NMDAR, N-methyl d-aspartate receptor; NFATC3, nuclear factor of activated T-cells; EGR1, 3, Early growth response 1, 3; ARC, Activity-regulated cytoskeleton-associated protein. Additional potential contributing proteins are indicated in gray include: SRF, serum response factor; BDNF, brain-derived neurotrophic factor; NRG1, neuregulin 1; ErbB4, ErbB2 receptor tyrosine kinase 4, Trk-B neurotrophic receptor tyrosine kinase 2. References: Yamagata et al. (1994); Senba and Ueyama (1997); Mittelstadt and Ashwell (1998); Rengarajan et al. (2000); Hao et al. (2003); Jacobson et al. (2004); Kumbrink et al. (2005, 2010); Li et al. (2005); Ramanan et al. (2005); Etkin et al. (2006); Lindecke et al. (2006); Bjarnadottir et al. (2007); Gallitano-Mendel et al. (2007, 2008); Gallitano (2008); Marrone et al. (2012); Herndon et al. (2013); Ramirez-Amaya et al. (2013); Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014); Maple et al. (2015); Pouget et al. (2016); Meyers et al. (2017a,b); Ramanan (personal communication).
Long-Term Depression, Stress and Memory
LTD
LTD is a form of synaptic plasticity in which stimulation results in a prolonged decrease in strength of the synaptic connection (reviewed in Malenka and Bear, 2004; Kemp and Manahan-Vaughan, 2007; Collingridge et al., 2010). One of the most well-investigated forms of LTD is that produced by low frequency stimulation of the hippocampal Schaffer Collateral pathway, the axonal projections from CA3 neurons that synapse onto CA1 pyramidal neurons. This process is essential for spatial memory consolidation, and induces expression of immediate early genes. Moreover, the stimulation that induces the highest levels of immediate early genes also results in the longest duration of LTD (Abraham et al., 1994) suggesting an important functional role for immediate early genes in this form of synaptic plasticity. The low frequency stimulation that induces LTD also requires the function of NMDARs, as well as numerous proteins in our proposed biological pathway for schizophrenia susceptibility.
Compared to its counterpart long-term potentiation (LTP), LTD has received much less investigative attention, and is thus less well understood. NMDAR activation is an essential step in establishment of low frequency stimulation-induced hippocampal LTD (Dudek and Bear, 1992; Mulkey and Malenka, 1992). Both GluN1 and GluN2B subunits have also been implicated in mediating this form of LTD (Kutsuwada et al., 1996; Manahan-Vaughan and Braunewell, 1999; Liu et al., 2004). Early studies demonstrated that mice lacking the specific GluN2B-containing NMDAR isoform had deficits in LTD (Kutsuwada et al., 1996; Brigman et al., 2010). However, recent work investigating the role of GLUN2B in this form of synaptic plasticity have produced differing results, some of which may be explained by technical differences in study methodologies (Chen and Bear, 2007; Bartlett et al., 2011; Shipton and Paulsen, 2014).
As described in the sections below, numerous downstream effectors of NMDAR activation have also been shown to be critical for activity dependent LTD. These include CN, EGR3, ARC, serum response factor (SRF) and BDNF (Zeng et al., 2001; Etkin et al., 2006; Plath et al., 2006; Gallitano-Mendel et al., 2007; Beazely et al., 2009; Mizui et al., 2015; Novkovic et al., 2015; Bukalo et al., 2016; Kojima and Mizui, 2017). The roles of several of the proteins that comprise our proposed pathway for schizophrenia risk are not limited to this form of synaptic plasticity. For example, NMDARs, BDNF and ARC are also required for LTP. Interestingly, dysfunction in LTP has also been hypothesized to play a role in schizophrenia (Rison and Stanton, 1995). However, the shared role of numerous core pathway genes in the process of LTD stood out to us early on in the recognition of this biological cascade (Gallitano-Mendel et al., 2007, 2008). Since LTD had received so much less emphasis, at that time, this seemed a coincidence worthy of further investigation.
Several features of LTD make this form of synaptic plasticity of particular interest for a potential contribution to the development of schizophrenia. Stress is one of the major environmental risk factors for this schizophrenia (Holtzman et al., 2013). Numerous studies have demonstrated a relationship between stress and LTD (Rowan et al., 1998). For example, while stress inhibits establishment of LTP in the hippocampal CA1 region, it augments LTD (Kim et al., 1996; Xu et al., 1997; Artola et al., 2006). The association of stress with schizophrenia susceptibility suggests the possibility that disruption of the normal LTD response to stress could result in pathology that may increase risk for this, and potentially other, psychiatric illness.
A second intriguing role of LTD is that of encoding long-term plasticity in response to novelty. Studies involving numerous genes in the proposed biological pathway have demonstrated an association between acquisition and retention of novel information and establishment of LTD (Manahan-Vaughan and Braunewell, 1999; Artola et al., 2006; Etkin et al., 2006; Plath et al., 2006; Gallitano-Mendel et al., 2007; Kemp and Manahan-Vaughan, 2007). Exposure to novelty enhances LTD (Manahan-Vaughan and Braunewell, 1999) and also induces expression of pathway genes such as Egr3 and Arc, the latter of which persists in the hippocampal dentate gyrus for up to 8 h (Marrone et al., 2012; Ramirez-Amaya et al., 2013).
We have recently reported that this persistent expression of Arc induced by a brief novel exposure requires Egr3 (Maple et al., 2017). We have also reported that Egr3 deficient mice, which have deficits in hippocampal LTD, also demonstrate hyper-reactivity to novel environments. Not only does exploration of a novel location induce LTD, but the reverse is also true; that low frequency stimulation of the hippocampus in vivo facilitates the exploratory behavior of rodents (Manahan-Vaughan and Braunewell, 1999). It is intriguing to consider whether dysfunction in these processes in humans may contribute to the cognitive and memory deficits that are a key feature of schizophrenia. These findings suggest it will be of great interest to investigate whether other genes recently identified as candidates for influencing schizophrenia risk may also play a role in this specific form of hippocampal synaptic plasticity.
Response to Stress and Novelty
Stress and exposure to novelty are two of the major stimuli that activate expression of immediate early genes, including EGR3 and ARC, key output proteins in our proposed biological pathway (Senba and Ueyama, 1997; Marrone et al., 2012; Ramirez-Amaya et al., 2013; Maple et al., 2015). These stimuli also facilitate induction of hippocampal LTD, a form of synaptic plasticity associated with the formation and retention of immediate memories of novel experiences (Rowan et al., 1998; Manahan-Vaughan and Braunewell, 1999; Xiong et al., 2003; Artola et al., 2006; Kemp and Manahan-Vaughan, 2007; Collingridge et al., 2010).
Our prior work characterizing the phenotypes of Egr3−/− mice revealed that Egr3 is required for the normal response to stress and novelty (Gallitano-Mendel et al., 2007). Similar behavioral abnormalities in the stress and novelty responses have been reported in preclinical studies of other genes in the proposed pathway including CN, SRF and ARC (Miyakawa et al., 2003; Etkin et al., 2006; Kozlovsky et al., 2008; Bramham et al., 2010; Weinstock, 2017).
Schizophrenia is also characterized by a heightened sensitivity to stress, abnormal sensorimotor gating in response to stressful stimuli, and abnormalities in habituation to novel and stressful stimuli (Braff et al., 1992; Corcoran et al., 2001; van Os et al., 2010; Holtzman et al., 2013; Kahn et al., 2015). The fact that LTD is facilitated by novelty and stress, and is disrupted when genes of our proposed pathway, numerous of which are associated with schizophrenia risk, are dysfunctional, suggests an intriguing link between this form of synaptic plasticity and the processes that are awry in schizophrenia.
Proteins That Form the Pathway
NMDA Receptor
NMDARs are activated in response to neuronal depolarization, which occurs when the brain responds to stimuli in the environment. NMDARs are one of three major classes of ligand-gated ionotropic receptors for glutamate in the brain. They are multi-subunit Ca++ channels, formed by hetero-tetramers of two glycine-binding (GLUN1), and two glutamate-binding (GLUN2), subunits (Papadia and Hardingham, 2007). The GLUN1 subunit, which has eight isoforms, is encoded by the GRIN1 gene. In contrast, there are four subtypes of GLUN2 subunits (GLUN2A, GLUN2B, GLUN2C and GLUN2D), which are encoded by four different genes (GRIN2A, GRIN2B, GRIN2C and GRIN2D, respectively). Each subunit has three distinct domains, an external N-terminal domain, a transmembrane domain that forms the channel pore, and an internal C terminal domain (Carvajal et al., 2016). The NMDAR is activated when glutamate binds to the GLUN2 subunit, coincident with binding of co-agonists magnesium and glycine/ D-serine to GLUN1 (Balu, 2016). Depolarization of the neuron is essential for NMDAR function, as this change in charge is required to remove the Mg++ ion that blocks the channel pore region, thereby allowing calcium to enter the neuron.
Evidence for a Role in Schizophrenia
The NMDAR hypofunction model of schizophrenia was proposed in the 1990’s (Javitt and Zukin, 1991; Olney et al., 1999) based on studies highlighting the ability of NMDAR antagonists, phencyclidine and ketamine, to cause psychosis and memory impairments in humans. The effect of these drugs in rodents were the basis for subsequently defining “schizophrenia-like” behavioral abnormalities in animal models (Malhotra et al., 1997; Lahti et al., 2001; Amitai and Markou, 2010; van den Buuse, 2010).
Mice deficient for the GluN1 subunit of the NMDAR show schizophrenia-like behavioral abnormalities that are reversed with antipsychotic treatment (Mohn et al., 1999). Studies employing a conditional knockout of the GluN1subunit have been used to identify specific cell types in which disruption of the receptor results in cognitive and behavioral defects. Selective disruption of the GluN1 gene in excitatory neurons of layer II/III of the prefrontal and sensory cortices of mice causes impairments in short-term memory and prepulse inhibition (Rompala et al., 2013), and GluN1 gene inactivation in parvalbumin interneurons caused deficits in habituation, working memory and associative learning (Carlén et al., 2012).
Genetic studies also support the importance of NMDAR genes in schizophrenia. Genome wide significant associations with schizophrenia have been reported for at least two of the NMDAR genes- GRIN2A and GRIN2B (Taylor et al., 2016). GRIN2A was recently shown to be associated with schizophrenia in the largest GWAS to date (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). Moreover, it is the sole gene at the locus (108 loci-region 82), and thereby also contains the “Index SNP”, defined as the SNP with the greatest significance at the disease-associated genomic locus (Please refer to Supplementary Table S1 for details of all index SNPs found in genes from our biological pathway). Genetic variation in the GRIN2B gene has also been reported to be associated with schizophrenia (Awadalla et al., 2010; Demontis et al., 2011; Kenny et al., 2014).
A large exome sequencing study of de novo mutations in schizophrenia conducted by Fromer et al. (2014) used DNA from 623 family “trios” (affected individual and both parents) to identify novel mutations in schizophrenia patients. They found that de novo mutations are increased in schizophrenia patients compared to the general population and, that these mutations were enriched in genes encoding proteins forming the NMDAR complex (Fromer et al., 2014), further supporting the hypothesis that dysfunction of NMDARs, or their downstream processes, increases risk for schizophrenia.
Postmortem brain tissue studies have shown region-specific alterations in NMDAR subunits at both the gene and protein levels. Weickert et al. (2013) showed decreased GRIN1 gene and protein expression, and decreased GRIN2C gene expression, in the dorsolateral prefrontal cortex regions of schizophrenia patients compared to controls. In a separate study, Gao et al. (2000) demonstrated high levels of GRIN1 and low levels of GRIN2C mRNA in postmortem hippocampal samples from schizophrenia patients compared with healthy controls.
Role in LTD
NMDARs have long been recognized to play critical roles in hippocampal long-term potentiation (LTP) and LTD, forms of synaptic plasticity that correlate with learning and memory (Bear and Malenka, 1994; Malenka and Bear, 2004). LTP is defined as the activity-dependent strengthening of synapses (Scharf et al., 2002), while LTD refers to an activity-dependent decrease in synaptic strength (Mulkey et al., 1994). Although NMDARs influence both forms of synaptic plasticity, a role in LTD appears to be a feature of numerous genes functioning both upstream and downstream of EGR3, and is therefore the focus of this review.
LTD mediated by NMDARs is crucial for consolidation of hippocampal dependent memory (Wong et al., 2007; Brigman et al., 2010; Ge et al., 2010), and alterations in the ratio of GLUN2A:GLUN2B subunits affects levels of LTD induced by a low frequency (3–5 Hz) electrical stimulation (Cui et al., 2013). Mice that lack GluN2B in pyramidal neurons in the cortex and CA1 regions of the hippocampus show impaired NMDAR-mediated LTD, and decreased dendritic spine density in CA1, compared to wildtype mice (Brigman et al., 2010).
Relationship to EGR3
The connection between NMDAR activation and Egr3 gene expression was first shown by Yamagata et al. (1994), who demonstrated that high frequency stimulation of rat hippocampal neurons that induces LTP also activates expression of Egr3. Both Egr3 expression and establishment of LTP are blocked by pretreatment with the NMDAR antagonist MK-801. Thus, this form of activity-dependent expression of Egr3 in the hippocampus requires NMDAR function.
Interestingly, Egr3 also influences the function of NMDARs. Egr3−/− mice display deficits in the electrophysiologic response of hippocampal neurons to drugs selective for GLUN2B-containing NMDARs (Gallitano-Mendel et al., 2007). However, the fact that quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analysis did not reveal differences in the levels of Grin2b mRNA in Egr3−/− mice compared with controls, suggests that EGR3 may not be directly regulating expression of this NMDAR subunit (Gallitano-Mendel et al., 2007). Others have reported that overexpression of Egr3 (but not Egr1) in vitro regulates expression of an NMDAR subunit NR1-promoter-driven reporter construct, a response that is BDNF dependent (Kim et al., 2012). Thus, Egr3 expression is induced by neuronal activity in an NMDAR-dependent fashion. In addition, Egr3 is also required for function of GLUN2B-containing NMDARs, though the mechanisms underlying this regulation have not yet been established.
Calcineurin
A key process triggered by calcium influx through NMDARs is the activation of calcium-dependent phosphatases, including CN (Horne and Dell’Acqua, 2007). CN is a multi-subunit protein consisting of two main subunits: the catalytic subunit calcineurin A, and the calcium binding regulatory subunit calcineurin B (Rusnak and Mertz, 2000). CN plays important roles in both the immune system and the brain. In T-cells, mitogenic stimuli increase levels of intracellular calcium, causing calmodulin-mediated activation of CN. CN then dephosphorylates nuclear factors of activated T-cell (NFAT) in the cytoplasm, causing its translocation to the nucleus where it regulates expression of target genes (Rao et al., 1997). In the nervous system, CN acts in conjunction with NFAT proteins to regulate numerous critical processes—reviewed elegantly by Kipanyula et al. (2016). These range from developmental roles in corticogenesis (Artegiani et al., 2015) and myelination (Kao et al., 2009), to activity dependent synaptogenesis (Ulrich et al., 2012), to glial cell activation, particularly in the context of neuroinflammation (Neria et al., 2013).
Evidence for a Role in Schizophrenia
An increasing body of in vivo and genetic evidence suggests a role for CN in the pathogenesis of schizophrenia. Immunosuppressive drugs that block CN, such as cyclosporin A and FK-506 produce side effects reminiscent of the symptoms of schizophrenia. These include memory impairment, auditory and visual hallucinations, paranoia, depression, and flattened affect (Bechstein, 2000; Porteous, 2008; American Psychiatric Association, 2013). Forebrain specific CN knockout mice show cognitive and behavioral abnormalities consistent with animal models of schizophrenia, such as working memory deficits (Zeng et al., 2001), increased locomotor activity and abnormalities in attention, social interaction and nesting behavior (Miyakawa et al., 2003).
Human genetic studies have identified associations between variations in PPP3CC, the gene that encodes the α-1 subunit of calcineurin A, and schizophrenia, in both Caucasian (Gerber et al., 2003) and Asian populations (Horiuchi et al., 2007; Liu et al., 2007; Yamada et al., 2007). Yamada et al. (2007) noted that EGR3 was located near PPP3CC (within a 252 kb interval) on the short arm of chromosome 8, and showed through linkage disequilibrium studies that they form distinct regions of schizophrenia susceptibility. Although PPP3CC is not listed as a candidate gene at one of the 108 loci associated with schizophrenia, numerous other non-calcineurin protein phosphatase genes do map to the loci identified in that GWAS. These include: PPP1R13B, PPP1R16B, PPP2R3A and PPP4C. This suggests the importance of protein-phosphatases in the etiology of schizophrenia. Notably serine/threonine phosphatase PP1/PP2A is a crucial player in synaptic plasticity and LTD (Mulkey et al., 1994).
Role in LTD
The forebrain specific CN knockout mice that display schizophrenia-like behavioral abnormalities also have disrupted hippocampal LTD, as well as deficits in hippocampal working and episodic memory (Zeng et al., 2001). Notably, this hippocampal LTD deficit phenotype is also seen in Egr3−/− mice (Gallitano-Mendel et al., 2007). Several additional lines of evidence suggest CN to be a crucial regulator of LTD. CN regulates dephosphorylation, and subsequent removal, of a membrane bound AMPAR subunit during LTD (Mulkey et al., 1994; Sanderson et al., 2016), which is induced by transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor activation (Chávez et al., 2010). These observations collectively highlight CN as an important regulator of behavior, memory, and LTD and as a schizophrenia candidate gene that regulates EGR3, making this protein an integral component of our proposed biological pathway.
Relationship to EGR3
Evidence that CN regulates expression of Egr3 comes from the immune system. T-cell activation induces expression of Egr3, which, in turn, directly regulates expression of Fas-ligand (Fasl). Expression of both Egr3 and its target gene is blocked by the CN inhibitor cyclosporin A (Mittelstadt and Ashwell, 1998). Although a similar interaction has not been investigated in the brain, the remarkable similarity in the behavioral and electrophysiologic phenotypes of CN knockout, and Egr3−/−, mice strongly suggests this molecular pathway is conserved in the brain (Zeng et al., 2001; Miyakawa et al., 2003; Gallitano-Mendel et al., 2007). Both CN knockout and Egr3−/− mice show deficits in LTD, spatial learning (Zeng et al., 2001; Gallitano-Mendel et al., 2007), and heightened responsiveness to handling (Gallitano-Mendel et al., 2007; supplemental data, Miyakawa et al., 2003). These intriguing initial findings support the need to determine whether this regulatory relationship between CN and Egr3 found in the immune system is also present in the brain.
NFATc3 (NFAT4)
The NFATs comprise a family of transcription factors that regulate activity-dependent gene expression in the immune system and the brain (Macian, 2005; Vihma et al., 2016). They reside in the cytoplasm in an inactive, phosphorylated state. Activity-triggered calcium entry into the cell stimulates CN to dephosphorylate NFAT proteins, allowing them to translocate to the nucleus where they activate expression of immediate early genes (Abdul et al., 2010). NFATs can be shuttled back into the cytoplasm via nuclear export sequences (nuclear localization signal) that, once exposed, lead to rephosphorylation of NFATs by kinases (Okamura et al., 2000) and subsequent inhibition of NFAT-mediated gene transcription (Hogan et al., 2003).
The NFAT family comprises five genes, four of which are calcium regulated and include NFAT1-4, and one that is osmotic tension-regulated, namely NFAT5, in both humans and mice (Vihma et al., 2008). NFAT proteins contain a C-terminal Rel homology domain that enables them to interact with other transcription factors (including EGRs) to co-regulate expression of downstream genes. Additional protein regions include an N-terminal domain that contains two CN binding sites (Aramburu et al., 1998; Park et al., 2000), the nuclear localization sequence, and serine residues that are sites of phosphorylation (Kiani et al., 2000).
As transcription factors, NFATs regulate expression of a wide array of genes ranging from cytokines (Klein et al., 2006), to growth factors (Hernández et al., 2001), to cell-cycle regulators (Caetano et al., 2002). In conjunction with CN, NFATs mediate a variety of physiological processes such as angiogenesis (Courtwright et al., 2009), osteogenesis (Winslow et al., 2006), and cardiovascular system development (Schulz and Yutzey, 2004). In the nervous system the NFATs have numerous roles, which include regulation of synaptic plasticity (Schwartz et al., 2009) and neurotrophin signaling (Groth et al., 2007; reviewed in Kipanyula et al., 2016).
We focus our discussion on NFATC3 (a.k.a. NFAT4), as it regulates expression of Egr3, and it is the only family member to show genome wide association with schizophrenia (Rengarajan et al., 2000; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Pouget et al., 2016). Nfatc3 is expressed in the adult mouse cerebellum, hippocampus, choroid plexus and ependymal cells as shown by in situ hybridization studies, and in the midbrain, pons, striatum, and thalamus using reverse transcription polymerase chain reaction (RT-PCR; Vihma et al., 2008). The level of expression varies across these regions with the highest expression in the mouse cerebellum and granular cell layer of the dentate gyrus in human brain tissue (Vihma et al., 2008). In vitro studies demonstrate that NFATC3 is one of the most highly activated NFAT members following neuronal depolarization (Vihma et al., 2016). To date, NFATC3 remains the only NFAT family member to show genome wide association with schizophrenia and regulate gene expression of immediate early genes, both of which are discussed below.
Evidence for a Role in Schizophrenia
NFATC3 maps to locus 85 of the 108 loci identified by the psychiatric genetics consortium and is, in fact, the index SNP for that locus (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). This initial finding has subsequently been supported by a follow-up GWAS that examined immune system-related genes from the original consortium study in an extended cohort (Pouget et al., 2016). Results of this validation study identified both NFATC3 and EGR1 among the six immune genes that showed genome-wide significance. Pouget et al. (2016) propose that NFATC3, which, like each of the 5 candidate genes they followed up, is known to have a predominantly “immune” role in peripheral organs, may be playing a non-immune role in the brain.
The vast majority of work investigating the activity and function of NFATC3 has focused on its roles in the immune system. Studies of Nfatc3 knockout mice show reduced numbers of CD4 and CD8 cells in the thymus and spleen, and increased apoptosis of T-cells, suggesting the importance of Nfatc3 in T-cell development and survival. No brain or nervous system phenotypes were reported in this study (Oukka et al., 1998). However emerging data suggest that NFATC3 has neuroprotective functions. It mediates astrocyte activation in response to brain damage (Serrano-Pérez et al., 2011; Yan et al., 2014), protection against apoptosis in neuronal cells in vitro (Butterick et al., 2010), and growth and differentiation of neuronal precursors (Serrano-Pérez et al., 2015). NFATC3 also mediates neurodegenerative effects including methamphetamine-induced apoptosis (Jayanthi et al., 2005), and apoptosis via Fas activation in lithium-induced neurotoxicity in vivo (Gómez-Sintes and Lucas, 2010). The latter of these is reminiscent of Egr3-mediated regulation of Fasl in T-cells (Mittelstadt and Ashwell, 1998, 1999; Rengarajan et al., 2000). Although there are no published studies investigating the role of NFATC3 in LTD, its position as an activity-dependent regulator of Egr3 in this pathway leads us to hypothesize that NFATC3 will be necessary for this form of synaptic plasticity.
Relationship to EGR3
In the immune system, CN regulates Egr3 expression in response to T-cell activation, an interaction that is mediated via NFATs. NFATC3 regulates expression of Egr3 in T-cells in mice. Egr3 expression is reduced in amount and duration in T-cells of mice lacking either NFATC2 or NFATC3, and is nearly absent in NFATC2/C3 double knockout mice. This regulation is direct, as both NFATC2 and NFATC3 are able to transactivate the Egr3 promoter in vitro (Rengarajan et al., 2000). We hypothesize that, in neurons, activity-dependent activation of CN dephosphorylates NFATC3, allowing it to enter the nucleus where it binds to the Egr3 promoter and activates its expression. While NFATC2 may also regulate neuronal expression of Egr3, as well as of Egr2, and potentially Egr1, the strong evidence for NFATC3 interacting with EGR3 is the basis for including this family-member as a critical component of our proposed biological pathway.
EGR3
EGR3 is a member of the Egr family of immediate early gene zinc finger transcription factors, and is activated downstream of numerous schizophrenia candidate proteins, including NRG1, NMDAR and CN (Yamagata et al., 1994; Mittelstadt and Ashwell, 1998; Hippenmeyer et al., 2002). This family consists of four genes, Egr1-4, that are activated in response to a wide range of environmental stimuli, including stress (Senba and Ueyama, 1997).
Egr3 was identified through a screen for genes homologous to Egr1. Like the founding family member, Egr3 is also highly expressed throughout the brain, including in the cortex, hippocampus, basal ganglia (Yamagata et al., 1994) and suprachiasmatic nucleus (Morris et al., 1998), as well as in the immune system and other organs (Tourtellotte and Milbrandt, 1998; Mittelstadt and Ashwell, 1999; Tourtellotte et al., 2001) Egr3 mRNA is highly induced in response to electroconvulsive seizure in hippocampal and cortical neurons, and in hippocampal dentate gyrus granule cells, by NMDAR activation (Yamagata et al., 1994), prompting further research into its role in synaptic plasticity and behavior (Gallitano-Mendel et al., 2007, 2008).
Evidence for a Role in Schizophrenia and LTD
Investigations published by our group revealed that Egr3−/− mice display schizophrenia-like behavioral abnormalities, including locomotor hyperactivity that is reversed by antipsychotic treatment. In addition, these mice show immediate memory deficits, heightened reactivity to novelty, and disrupted hippocampal LTD (Gallitano-Mendel et al., 2007). Induction of LTD has recently been shown to inactivate fear-related memory in vivo; however, we do not know whether Egr3 plays a role in this process (Nabavi et al., 2014). Egr3−/− mice also display a marked resistance to the sedating effect of clozapine, and other second-generation antipsychotics, which parallels the resistance that schizophrenia patients show to the side-effects of these medications (Cutler, 2001). This phenotype may be explained, at least in part, but the reduced level of serotonin 2A receptors found in the frontal cortex of Egr3−/− mice, a feature also seen in the brains of patients with schizophrenia (Williams et al., 2012; Selvaraj et al., 2014).
Our research also showed that Egr3−/− mice exhibit increased aggression in response to a foreign intruder (Gallitano-Mendel et al., 2008). This response was abrogated by chronic clozapine administration, despite the fact that the Egr3−/− mice are resistant to the sedative effects of this antipsychotic (Gallitano-Mendel et al., 2008). A similar phenomenon is seen in schizophrenia patients treated with clozapine (Hector, 1998; Chalasani et al., 2001; Chengappa et al., 2002), in whom the anti-aggressive effect of the drug can be distinguished from its sedating effect (Krakowski et al., 2006). These roles that Egr3 plays in memory, synaptic plasticity, and behavior, and the response to antipsychotics that mimics that of patients, suggest that abnormal function of EGR3 in humans may contribute to schizophrenia pathogenesis or symptomatology.
Genetic association of EGR3 with schizophrenia has been shown in Chinese (Ning et al., 2012; Zhang et al., 2012), Japanese (Yamada et al., 2007) and Korean (Kim et al., 2010) populations, and more recently in a population of European descent (Huentelman et al., 2015). It was also reported that decreased EGR3 mRNA levels were observed in postmortem dorsolateral prefrontal cortex samples of schizophrenia patients compared with controls (Yamada et al., 2007). In addition, EGR3 was identified in a screen for genes that are expressed at reduced levels in the brains of schizophrenia patients that do not smoke tobacco, and are normalized to control levels in patients that smoke. Notably, EGR3 levels followed a pattern identical to that of CN in the study, consistent with its regulation by CN in the proposed biological pathway (Mexal et al., 2005).
The 8p chromosomal region, where EGR3 resides, is a long-recognized hub for schizophrenia associations (Suarez et al., 2006; Lohoff et al., 2008). A recent review highlighted that EGR3 was among 20 genes of interest for schizophrenia, map to the 8p locus (Tabarés-Seisdedos and Rubenstein, 2009). Association of an EGR3 SNP with decreased prefrontal hemodynamic response was observed during a verbal fluency task in both healthy controls and schizophrenia patients (Nishimura et al., 2014).
Both human and in vivo animal studies suggest that overexpression of EGR3 may also negatively impact non-neural physiological processes, including immune system function. High levels of EGR3 positively correlate with levels of proinflammatory cytokines in peripheral monocytes of schizophrenia patients (Drexhage et al., 2010). This may be mediated through the Triggering receptor expressed on myeloid cells 1 (TREM-1), a key regulator of inflammation in both brain microglia and peripheral monocytes, as EGR3 directly binds the TREM-1 promoter in monocytes (Weigelt et al., 2011).
These data support the hypothesis that EGR3 may function as a master regulator of multiple processes that are relevant to the pathophysiology of schizophrenia. In line with this theory, bioinformatic analyses have predicted EGR3 to be a central modulator of a regulatory network of microRNAs and transcription factors associated with schizophrenia (Guo et al., 2010). Despite these findings, EGR3 was not identified as a gene residing at one of the 108 loci associated with schizophrenia. Genome-wide association studies from the PGC schizophrenia studies suggest that none of the SNPs in the region including EGR3 showed a significant association. The SNP that showed the lowest p value was an intergenic SNP, rs12541654 (p = 0.23, OR = 0.99)1. However other EGR family members, such as EGR1, and EGR coregulatory binding factor NGFI-A Binding Protein 2 (NAB2), are each index SNPs at one of the 108 loci (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). EGR3 binds to NAB2 to co-regulate expression of target genes, and is involved in co-regulatory feedback relationships with EGR1, as well EGR1, EGR2 and EGR3 can regulate NAB2 expression, demonstrating a functional interaction of both of these schizophrenia associated genes in the proposed pathway (Svaren et al., 1996; Mechta-Grigoriou et al., 2000; Kumbrink et al., 2005, 2010; Srinivasan et al., 2007).
ARC
One of the first EGR3-target genes to be elucidated was Arc (Li et al., 2005). Arc was originally identified as a novel transcript in the adult rat brain that was rapidly and strongly induced in response to electroconvulsive seizures (Link et al., 1995; Lyford et al., 1995). Induction of Arc was observed in vitro in PC12 cells following exposure to growth factors such as nerve growth factor (NGF) and epidermal growth factor (EGF; Lyford et al., 1995). Arc mRNA and protein are enriched in neuronal dendrites, and ARC protein colocalizes with the actin cytoskeletal matrix (F-actin; Lyford et al., 1995). These observations led to its name “ARC”. Further characterization of the role of ARC protein in cytoskeletal function revealed that it also associates with microtubules and microtubule associated protein (MAP2; Fujimoto et al., 2004). ARC was also shown to maintain phosphorylation of cofilin, the actin depolymerization factor, and to promote F-actin formation (Messaoudi et al., 2007).
ARC is capable of localizing to NMDAR and postsynaptic density (PSD) 95 protein complexes (Husi et al., 2000; Donai et al., 2003) suggesting that it may be involved in synaptic neurotransmission. ARC also binds to multiple components of the clathrin-dependent endocytic machinery including endophilin and dynamin, and decreases total and surface AMPAR (GluR1) protein levels via endocytosis in hippocampal neurons in vitro (Chowdhury et al., 2006). Numerous proteins bind to ARC to mediate its many important roles in the nervous system. Notably 4–8 of these binding partners, termed “ARC complex proteins”, were identified in schizophrenia GWAS (Kirov et al., 2012; Fromer et al., 2014; Purcell et al., 2014) and are discussed in depth in the next section.
Evidence for a Role in Schizophrenia
The behavioral phenotype of Arc−/− mice was recently characterized and revealed schizophrenia-like abnormalities. These include deficits in prepulse inhibition and recency discrimination (indicative of cognitive dysfunction), impairments in response to social novelty, hyperactivity in response to amphetamine administration, and region-specific alterations in dopamine (Managò et al., 2016). Though they retain the ability to form intact short-term memory, Arc−/− mice fail to form long lasting memories.
The necessity of Arc for normal memory and behavior in mice suggest that dysfunction of ARC in humans could result in abnormalities that increase risk to develop psychiatric illness. Indeed, ARC mRNA expression is decreased in the frontal cortex of schizophrenia patients (Guillozet-Bongaarts et al., 2014). In addition, genes encoding proteins that bind to ARC, “ARC-complex proteins”, were identified in several large-scale, genome-wide schizophrenia genetic association studies. Kirov et al. (2012) reported that de novo CNVs in the genes DLG1, DLG2, DLGAP1, CYFIP1, each of which encode ARC-complex proteins, show significant enrichment in individuals with schizophrenia. A separate case-control study showed an enrichment of disruptive mutations in genes encoding ARC-complex proteins in schizophrenia cases vs. controls (Purcell et al., 2014). The nine mutations identified in this study included nonsense, essential splice site, and framseshift mutations, that occurred in nine genes, and were found only in schizophrenia cases, with none occurring in controls (Purcell et al., 2014). In addition, the study by Fromer et al. (2014) that identified increased de novo mutations in NMDAR-complex gene in schizophrenia patients also revealed that these mutations are enriched in genes encoding proteins associated with the ARC-complex. Fromer et al. (2014) showed that loss of function mutations are 17-fold enriched in genes encoding ARC-complex proteins in their cohort and 19-fold enriched in the data set from the Purcell et al. (2014) study. They concluded that “ARC disruption has particularly strong effects on disease risk” (Fromer et al., 2014).
These and other findings implicated ARC as a critical protein in a synaptic pathway involving voltage gated calcium channels, NMDARs, PSD95, FMRP, mGLuR and AMPARs, in schizophrenia risk (Hall et al., 2015). Although these studies did not report genetic association of variations in the ARC gene, itself, with schizophrenia, our group subsequently reported the first association of a SNP in the ARC gene with schizophrenia in two separate populations (discussed below; Huentelman et al., 2015). More recently, hypermethylation of eight CpG sites, and presence of several rare variants that reduce reporter gene activity, were reported in the putative ARC promoter region of schizophrenia patients vs. controls (Chuang et al., 2016). These findings indicate that altered genetic and epigenetic regulation of ARC expression, specifically that reduces ARC function, may increase risk for schizophrenia.
Role in LTD
Like many of the upstream proteins in the proposed pathway, ARC is also required for hippocampal LTD. NMDA-mediated LTD deficits in Schaffer collateral-CA1 synapses were found in slices from Arc−/− mice compared to wildtype mice (Plath et al., 2006). In addition, mGLuR-mediated LTD was shown to depend on rapid translation of ARC, and ARC-mediated AMPAR endocytosis, in hippocampal neurons (Waung et al., 2008).
Neuronal activity stimulates expression of ARC, which then accumulates at the spines of inactive synapses, effectively “tagging” these synapses for remodeling. This process is dependent on Ca2+/calmodulin-dependent protein kinase II (CamKIIβ), and results in ARC-mediated AMPA receptor endocytosis (Okuno et al., 2012). Minatohara et al. (2016) suggest that ARC expression and localization during LTD may increase the strength of the active synapses by causing endocytosis of AMPARs at the inactive synapses. Arc−/− mice also show decreased spine density and increased spine width in CA1 pyramidal neurons and DG cells (Peebles et al., 2010), suggesting that ARC plays a critical role in maintenance of spine morphology that may impact learning and behavior. These data suggest that decreased ARC expression results in deficits in LTD.
Relationship to EGR3
Previous research has shown that Arc is a transcriptional target of EGR3 (Li et al., 2005). Our proposed biological pathway (Gallitano-Mendel et al., 2007, 2008) combines environment stress-responsive proteins EGR3, CN, and NMDARs, each of which are associated with schizophrenia susceptibility. Since mice deficient for these proteins share phenotypes of LTD and memory deficits, similar to mice lacking Arc, we hypothesized that the ARC gene should likewise be a schizophrenia candidate gene.
To test this hypothesis, we used next generation sequencing to resequence the ARC gene, and flanking regions, from schizophrenia patient and control subjects from two separate ethnic groups. These studies revealed association between the ARC SNP and schizophrenia in both ethnic groups (Huentelman et al., 2015). In a separate study, we found that the ARC SNP was associated with response to cognitive remediation therapy in a cohort of new onset psychosis patients (Breitborde et al., 2017). Recently, a heritable chromosomal microdeletion that encompasses several genes, including ARC, was also shown to be associated with several neurodevelopmental psychiatric disorders such as attention deficit hyperactivity disorder and autism spectrum disorder (Hu et al., 2015) in addition to schizophrenia. Overall, substantial evidence suggests that ARC, an important regulator of synaptic function, memory, and LTD, influences risk for schizophrenia, and possibly other neuropsychiatric illnesses. As a direct target of EGR3, ARC represents an important output element of our proposed biological pathway.
Additional Genes That Interact with the Pathway
Neuregulin-1
NRG1 was one of the first schizophrenia candidate genes to be identified using molecular genetic methods at a genomic locus defined by linkage analysis studies in family pedigrees (Stefansson et al., 2002). It has subsequently been validated in numerous populations, and supported by preclinical studies (recently reviewed in, Mostaid et al., 2016). NRG1 regulates EGR3 in human muscle and breast cancer cell lines (Sweeney et al., 2001; Jacobson et al., 2004). In mice NRG1 regulates Egr3 expression in developing muscle cells (Jacobson et al., 2004), and this regulation is required to maintain development of muscle spindles. These studies demonstrate a functional link between these two schizophrenia-associated genes in two types of human cell lines, and in vivo in mice (Hippenmeyer et al., 2002; Jacobson et al., 2004; Herndon et al., 2013). Although this regulatory relationship has not yet been validated in the brain, we hypothesize that a similar regulatory relationship may functionally link these two schizophrenia-associated genes in the central nervous system as well.
No studies investigating LTD in NRG1 deficient mice have been reported. However, the fact that NRG1 regulation of Egr3 in the muscle spindle is mediated by SRF (Jacobson et al., 2004; Herndon et al., 2013), and loss of either Srf or Egr3 results in memory and LTD deficits (Etkin et al., 2006; Gallitano-Mendel et al., 2007), leads us to predict that NRG1 should also play a critical role in this form of hippocampal synaptic plasticity. Indeed, NRG1 impairs endocannabinoid 2-arachidonoylglycerol (2-AG)-mediated LTD in rat hippocampal slices (Du et al., 2013).
Serum Response Factor (SRF)
We have previously highlighted SRF as a protein in the proposed biological pathway for schizophrenia susceptibility based on its requirement for both novelty memory and LTD in mice (Etkin et al., 2006), as well its regulatory interactions with other proteins in the pathway (Etkin et al., 2006; Gallitano-Mendel et al., 2007). SRF is activated downstream of NRG1 in mouse muscle cells (Herndon et al., 2013, 2014). In vitro studies demonstrate that NRG1 is capable of stimulating SRF expression in human HeLa cells expressing the NRG1 receptor ErbB4, and that this is mediated by mitogen-activated protein kinase (MAPK; Eto et al., 2010). SRF is also activated by CN/NFAT, in combination with other factors, in lymphocytes (Lin et al., 2002; Hao et al., 2003). In turn, SRF regulates expression of Egr1 and Egr2 in the brain in response to novelty, and mediates the regulation of Egr3 by NRG1 in muscle spindle development (Hao et al., 2003; Ramanan et al., 2005; Etkin et al., 2006; Herndon et al., 2013). SRF has also been shown to regulate Egr3 expression in the hippocampus (personal communication from Naren Ramanan).
Together with another transcription factor Elk-1, SRF regulates expression of Egr1 in response to chemically-induced LTD (by 3,5-dihydroxyphenylglycine, DHPG; Lindecke et al., 2006). And Srf-deficient mice have deficits in LTD (Etkin et al., 2006). Interestingly two NRG1 schizophrenia-associated SNPs from the original Icelandic haplotype occur in regions that show predicted binding sites for SRF that are abolished by presence of the SNPs (Law et al., 2006). Although we are unaware of studies demonstrating that genetic polymorphisms in SRF are directly associated with risk for schizophrenia at this time, we hypothesize that the positioning of SRF in this pathway, and its requirement for hippocampal LTD, will lead to identification of such roles in the future.
EGR1
Egr1 is the founding member of the Egr family of immediate early gene transcription factors. It was identified as a gene activated in response to application of NGF to PC-12 cells in culture, and thus named “NGF inducible A” NGFI-A (O’Donovan et al., 1999). Egr1 was independently identified by other laboratories, which accounts for its numerous aliases (zif-268, Krox-24, NGFI-A, TIS8, ZIF-268, ZNF225). Egr1 is required for long-term memory formation and late-phase LTP (Cole et al., 1989; Worley et al., 1993).
While a number of studies have identified associations between EGR1 and schizophrenia, the most significant is that of the 2014 Schizophrenia Working Group of the Psychiatric Genomics Consortium GWAS, in which EGR1 resides in region 69 of the 108 loci associated with the illness, and contains the Index SNP for that region (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). In a follow-up study examining the consortium dataset, Pouget et al. (2016) identified six independent regions from the 108 loci where the SNP with the lowest association p-value (the Index SNP) was in an immune gene. EGR1 was one of the six.
Postmortem brain tissue studies have also identified EGR1 as one of a small number of genes differentially expressed in schizophrenia patients compared with controls (Pérez-Santiago et al., 2012). Studies have also identified differences in mRNA levels of EGR1 in peripheral blood cells (Cattane et al., 2015; Xu et al., 2016; Liu et al., 2017), and one study in fibroblasts (Cattane et al., 2015), of schizophrenia patients compared with controls Interestingly, the differences in EGR1 mRNA levels identified by the two research groups were in opposite directions, which may be due to methodological differences. In particular, the two studies reporting reduced peripheral blood cell levels of EGR1 were examining schizophrenia patients during an acute psychotic episode, while the other study did not specify the patients’ current symptom status (Cattane et al., 2015; Xu et al., 2016; Liu et al., 2017).
Studies in mice deficient for Egr1 demonstrate that both long-term memory and LTP require function of this immediate early gene (Jones et al., 2001; Bozon et al., 2003). Furthermore, over-expression of Egr1 facilitates hippocampal LTP and enhances long-term memory of spatial location (Penke et al., 2013). Although Egr1 is upregulated in response to LTD induction by DHPG (Lindecke et al., 2006), loss of Egr1 has not been reported to result in LTD deficits.
As the founding member of the EGR family, EGR1 shares gene sequence homology, and DNA binding element recognition, with EGR3. EGR1 and EGR3 are activated by many of the same stimuli and are expressed in the same cells in the brain (Yamagata et al., 1994), though their proteins follow different temporal patterns of expression and perdurance (O’Donovan et al., 1998).
Together EGR1 and EGR3 bind to the promoter, and induce expression of, their coregulatory factor NAB2 in cells of neuroectodermal origin (Kumbrink et al., 2010). NAB2 protein, via interaction with the EGRs, subsequently feedback inhibits its own expression (Kumbrink et al., 2010). These regulatory relationships that EGR1 shares with EGR3 and other proteins in the proposed pathway, as well as with NAB2, another GWAS-implicated gene, strengthen the likelihood that the position of EGR1 as an Index SNP in the 108 loci indicates an actual role for EGR1 in schizophrenia susceptibility.
NGFI-A Binding Protein 2 (NAB2)
NAB2 is an activity-dependent immediate early gene that functions as a transcriptional coregulatory protein by binding to a specific recognition domain on EGR family transcription factors EGR1, EGR2 and EGR3. In cells of neuroectodermal origin EGR1 and EGR3 bind in concert to the promoter of the NAB2 gene to induce its expression (Kumbrink et al., 2010). NAB2 protein has both co-activation and co-repression actions. Once bound to the EGRs, NAB2 feedback inhibits its own expression, as well as that of EGRs (Svaren et al., 1996; Mechta-Grigoriou et al., 2000; Kumbrink et al., 2010).
The regulatory relationships with schizophrenia-associated EGRs (EGR1 and EGR3) suggest a role for NAB2 in neuropsychiatric illness. Direct support for possible association between NAB2 and schizophrenia comes from the 2014 Schizophrenia Working Group of the Psychiatric Genomics Consortium GWAS. NAB2 is located at region 20 of the 108 loci, and is one of the genes at the site of the locus’ Index SNP (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014).
The majority of studies involving NAB2 have investigated its role in the immune system and cancer biology (Collins et al., 2006, 2008; Hastings et al., 2017). The role of NAB2 in the nervous system is much less well investigated, and the functions of NAB2 in the brain are still largely unknown. As an immediate early gene, Nab2 expression is induced in the brain in response to stimuli (Jouvert et al., 2002). Among the stimuli that activate Nab2 expression in neurons is BDNF (Chandwani et al., 2013). In the peripheral nervous system loss of both Nab2, and its family member Nab1, results in severe myelination defects (Le et al., 2005).
No studies have yet been published examining the role of NAB2 in memory or hippocampal synaptic plasticity, or in behavior. However, unpublished data from our laboratory indicate significant behavioral abnormalities in mice lacking only Nab2 (Gallitano, unpublished observation). Despite the paucity of published work on the role of NAB2 in the brain, the regulatory relationships between NAB2, EGR1 and EGR3 link this gene not only to other specific schizophrenia-associated genes, but also to the proposed pathway for illness association.
Brain-Derived Neurotrophic Factor (BDNF)
Numerous studies have associated BDNF with mental illnesses, including schizophrenia (Islam et al., 2017). The requirement for BDNF, particularly its precursor (pro) form, for synaptic plasticity and memory, suggests a mechanism linking BDNF to neuropsychiatric disorders (reviewed in, Carlino et al., 2013). Specifically, the human BDNF polymorphism that converts the 66 position amino acid valine to a methionine (Val66Met) has been associated not only with mental illness risk, but also with memory (Egan et al., 2003; Hariri et al., 2003; Hashimoto et al., 2008; van Wingen et al., 2010).
BDNF is included in the proposed pathway as it has been shown to induce expression of Egr3 in the mouse brain as an essential step in regulating expression of GABAA receptor alpha 4 subunit (Roberts et al., 2006; Kim et al., 2012). In a separate article submitted to this Research Topic issue (Meyers et al., 2017a), we demonstrate that Egr3 is required for expression of Bdnf in the mouse dentate gyrus 1 h following seizure, a stimulus that induces high level Bdnf expression in this hippocampal region. Thus, BDNF may function both upstream, as well as downstream, of Egr3.
It was recently reported that BDNF-deficient mice display defects in hippocampal LTD (Novkovic et al., 2015; Bukalo et al., 2016). In addition, exogenous application of the precursor (pro) peptide of BDNF facilitates development of LTD, a process that requires function of GLUN2B containing NMDARs (Mizui et al., 2015; Kojima and Mizui, 2017). Notably, the hippocampal Schaffer collateral neurons in Egr3−/− mice, that fail to develop LTD, are unresponsive to the GLUN2B selective antagonist ifenprodil (Gallitano-Mendel et al., 2007), suggesting that GLUN2B-containing NMDARs are critical for both BDNF- and Egr3-, mediated LTD. These findings support a position for BDNF in our proposed pathway for neuropsychiatric illness risk.
Discussion
In this article, we review evidence supporting an activity-dependent biological pathway that incorporates numerous schizophrenia candidate genes with critical roles in the regulation of memory and LTD, and that culminates in activation of the immediate early gene transcription factor EGR3. The unique position of immediate early gene transcription factors, at the nexus between environmental events and regulation of the neuronal response to activity, makes them ideally suited to account for both the genetic and environmental contributions to schizophrenia.
Additional proteins implicated in risk for schizophrenia, but that have not yet been reported to affect LTD, interact with this pathway either as upstream activators (e.g., NRG1) or as transcriptional regulators (e.g., EGR1 and NAB2). Individual genes in this pathway mediate neuroprotective functions including myelination, vascularization, growth factor regulation, and synapse formation, abnormalities in which are found in schizophrenia (Milbrandt, 1987; Mechtcheriakova et al., 1999; Jones et al., 2001; Jessen and Mirsky, 2002; Fahmy et al., 2003; Fahmy and Khachigian, 2007; Gallitano-Mendel et al., 2007). Dysfunction in any of the genes of this pathway would result in disruption of these processes, and may thereby account for the neuropathologic and clinical features of schizophrenia, including deficiencies white matter, brain volumes, and cerebral blood flow, reduced synaptic spine density, and deficits in memory and cognitive processing (Ingvar and Franzen, 1974; Saykin et al., 1991; Glantz and Lewis, 2000; Moises et al., 2002; Davis et al., 2003; Nabavi et al., 2014).
Moreover, numerous genes in the pathway have critical functions in the immune system, particularly in T-cell activation, underscoring a longstanding recognition of a relationship between immune system and risk for schizophrenia. Finally, numerous genes that are part of the originally-defined pathway, as well as others that interact with proteins in the pathway, have subsequently been identified in the 108 loci GWAS (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). Notably, each of the pathway genes that is at one of the 108 schizophrenia loci is, in fact, the Index SNP at that locus. Together these findings strongly support the biological relevance of our proposed pathway in schizophrenia.
In the “Introduction” section, we highlighted that there are two unanswered questions at the forefront of psychiatric genetics: (1) how can so many genes contribute susceptibility to schizophrenia? and (2) how do genes implicated in risk for schizophrenia interact with environmental factors to give rise to the disorder? The concept of a biological pathway addresses the first unanswered question by hypothesizing that numerous genes share roles in a much smaller number of critical functional processes. Disruption in these key biological processes create risk for schizophrenia.
To account for the effect of environment, we propose a model whereby genetic variations that decreased function of any protein in the pathway will result in increased risk for schizophrenia in a manner that is dependent upon the stress history of an individual. Stress is commonly thought of as detrimental. Specific negative consequences of stress demonstrated in animal and human studies include stress-hormone mediated decreases in dendritic arborization and spine density and reduced regional brain volumes (reviewed in Lupien et al., 2009). However, despite the pervasive experience of stress throughout life, most people do not develop severe mental illness. This is presumably because stress also activates molecular and cellular processes that are protective. The balance of damaging and protective responses to stress creates resilience and maintain homeostasis. This is shown in Figure 2A.
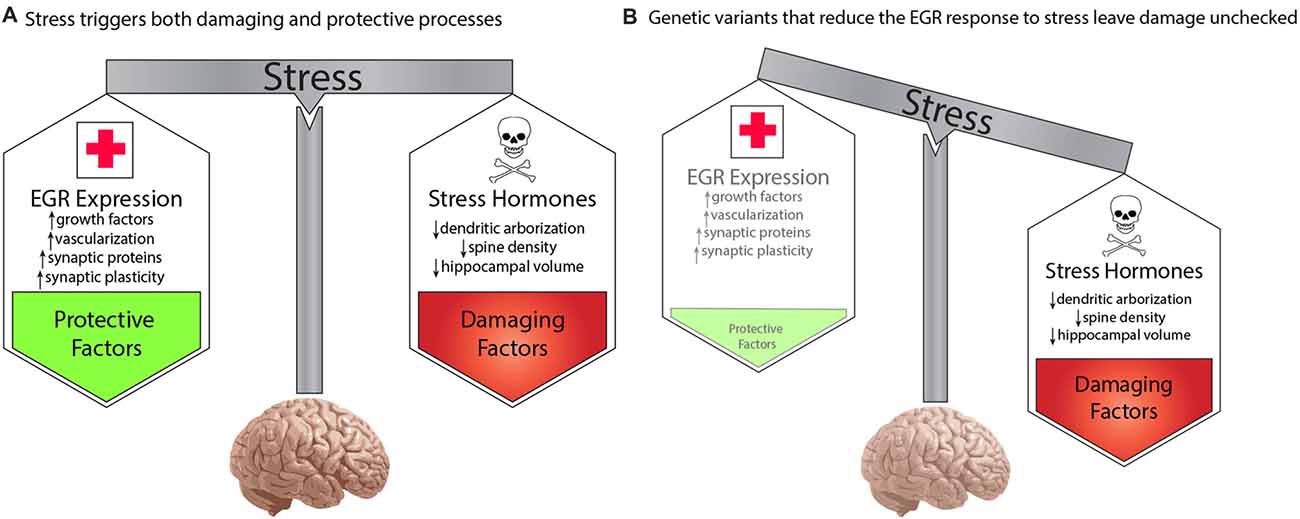
Figure 2. A developmental model for genetic predisposition to stress susceptibility. (A) Stress hormones cause detrimental effects on the brain, such as decreased dendritic arborization, reduced density of synaptic spines, and decreased regional brain volumes, which are seen in schizophrenia. Stress also activates EGR family immediate early genes, which mediate numerous beneficial processes, such as growth factor response, vascularization, synaptic protein expression, and synaptic plasticity. The balance between these processes protects the brain from damage under stressful conditions. (B) Individuals carrying genetic variants that impair the activation of pathway proteins may be fine under normal conditions. However, in the context of stress, the damaging effects override the insufficient protective response, resulting in neurotoxic insults which, if sustained or repeated, may result in the neuropathology that gives rise to symptoms of schizophrenia. In this manner, genetic variations in genes of the proposed pathway create a predisposition to develop schizophrenia in a manner dependent upon the stress history of an individual.
We propose that the activity-dependent biological pathway we have described here represents a component of the healthy biological response to stress. The normal activation of the pathway in response to stress triggers molecular and cellular processes that protects the brain from the harmful effects of stress hormones, and other detrimental elements of the stress response. While extreme, or unremitting, stress conditions may over-ride the buffering abilities of this protective arm of the stress response, it is sufficient to withstand typical episodic stress that occurs in daily life.
However, if an individual carries a genetic variation that decreases the responsiveness of a critical protein in our proposed pathway (Figure 2B), then exposure to stress will result in the detrimental effects of stress that are not sufficiently balanced by the protective elements. This preponderance of damaging effects over time is hypothesized to cause the neuropathologic features of schizophrenia that presumably give rise to symptoms of the illness. In the absence of sufficient stress, however, the lower level of function of the EGR pathway may be sufficient to prevent neuropathological consequences. This can explain how two individuals may carry the same genetic variation, but be discordant for schizophrenia.
In this manner, the proposed model would account for the dual genetic and environmental contributions to schizophrenia susceptibility, a crucial feature that has not been accounted for by previous gene or pathway models. We hope that this pathway and model provide the structure for investigation of additional components that may contribute to the neuropathology underlying schizophrenia and other neuropsychiatric disorders, advancing the field toward identification of more effective therapies, and perhaps one day a cure, for these illnesses.
We have focused on the role of our proposed pathway in schizophrenia due to the large body of supporting studies from human genetic, post-mortem, and basic science animal studies. However, we hypothesize that this pathway is relevant for other neuropsychiatric and neurodegenerative disorders in which stress plays a role and memory is affected. Indeed, EGR genes, and EGR3 in particular, has been implicated in bipolar disorder, depression and Alzheimer’s disease (Hokama et al., 2014; Pfaffenseller et al., 2016; Francis et al., 2017). Although in-depth discussion of these disorders is beyond the scope of this review, we look forward to application of our proposed pathway to these illnesses by others in the future.
Author Contributions
ALG conceived the hypothesis that is the focus of the manuscript. KKM and ALG were both responsible for collecting literature, review and writing of the manuscript.
Funding
This work was supported by a National Alliance for Research on Schizophrenia and Depression (NARSAD)/Sidney R. Baer Jr. Foundation Young Investigator Award, and National Institutes of Health (NIH) grants R01 MH097803 and R21MH113154 to ALG.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Shenfeng Qiu for providing generous and expert assistance with design and execution of the figures.
Footnotes
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fnbeh.2018.00023/full#supplementary-material
TABLE S1 | Supplemental table contains details regarding human variant studies discussed in the review. For each study discussed the associated gene, protein, variant, p value, effect size (if reported), and major findings have been summarized.
References
Abdul, H. M., Furman, J. L., Sama, M. A., Mathis, D. M., and Norris, C. M. (2010). NFATs and Alzheimer’s disease. Mol. Cell. Pharmacol. 2, 7–14.
Abraham, W. C., Christie, B. R., Logan, B., Lawlor, P., and Dragunow, M. (1994). Immediate early gene expression associated with the persistence of heterosynaptic long-term depression in the hippocampus. Proc. Natl. Acad. Sci. U S A 91, 10049–10053. doi: 10.1073/pnas.91.21.10049
American Psychiatric Association. (2013). Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: American Psychiatric Association.
Amitai, N., and Markou, A. (2010). Disruption of performance in the five-choice serial reaction time task induced by administration of N-methyl-D-aspartate receptor antagonists: relevance to cognitive dysfunction in schizophrenia. Biol. Psychiatry 68, 5–16. doi: 10.1016/j.biopsych.2010.03.004
Aramburu, J., Garcia-Cózar, F., Raghavan, A., Okamura, H., Rao, A., and Hogan, P. G. (1998). Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol. Cell 1, 627–637. doi: 10.1016/s1097-2765(00)80063-5
Artegiani, B., de Jesus Domingues, A. M., Bragado Alonso, S., Brandl, E., Massalini, S., Dahl, A., et al. (2015). Tox: a multifunctional transcription factor and novel regulator of mammalian corticogenesis. EMBO J. 34, 896–910. doi: 10.15252/embj.201490061
Artola, A., von Frijtag, J. C., Fermont, P. C., Gispen, W. H., Schrama, L. H., Kamal, A., et al. (2006). Long-lasting modulation of the induction of LTD and LTP in rat hippocampal CA1 by behavioural stress and environmental enrichment. Eur. J. Neurosci. 23, 261–272. doi: 10.1111/j.1460-9568.2005.04552.x
Awadalla, P., Gauthier, J., Myers, R. A., Casals, F., Hamdan, F. F., Griffing, A. R., et al. (2010). Direct measure of the de novo mutation rate in autism and schizophrenia cohorts. Am. J. Hum. Genet. 87, 316–324. doi: 10.1016/j.ajhg.2010.07.019
Balu, D. T. (2016). The NMDA receptor and schizophrenia: from pathophysiology to treatment. Adv. Pharmacol. 76, 351–382. doi: 10.1016/bs.apha.2016.01.006
Bartlett, T. E., Lu, J., and Wang, Y. T. (2011). Slice orientation and muscarinic acetylcholine receptor activation determine the involvement of N-methyl D-aspartate receptor subunit GluN2B in hippocampal area CA1 long-term depression. Mol. Brain 4:41. doi: 10.1186/1756-6606-4-41
Bear, M. F., and Malenka, R. C. (1994). Synaptic plasticity: LTP and LTD. Curr. Opin. Neurobiol. 4, 389–399. doi: 10.1016/0959-4388(94)90101-5
Beazely, M. A., Lim, A., Li, H., Trepanier, C., Chen, X., Sidhu, B., et al. (2009). Platelet-derived growth factor selectively inhibits NR2B-containing N-methyl-D-aspartate receptors in CA1 hippocampal neurons. J. Biol. Chem. 284, 8054–8063. doi: 10.1074/Jbc.M805384200
Bechstein, W. O. (2000). Neurotoxicity of calcineurin inhibitors: impact and clinical management. Transpl. Int. 13, 313–326. doi: 10.1007/s001470050708
Bjarnadottir, M., Misner, D. L., Haverfield-Gross, S., Bruun, S., Helgason, V. G., Stefansson, H., et al. (2007). Neuregulin1 (NRG1) signaling through Fyn modulates NMDA receptor phosphorylation: differential synaptic function in NRG1+/– knock-outs compared with wild-type mice. J. Neurosci. 27, 4519–4529. doi: 10.1523/JNEUROSCI.4314-06.2007
Bozon, B., Davis, S., and Laroche, S. (2002). Regulated transcription of the immediate-early gene Zif268: mechanisms and gene dosage-dependent function in synaptic plasticity and memory formation. Hippocampus 12, 570–577. doi: 10.1002/hipo.10100
Bozon, B., Davis, S., and Laroche, S. (2003). A requirement for the immediate early gene zif268 in reconsolidation of recognition memory after retrieval. Neuron 40, 695–701. doi: 10.1016/s0896-6273(03)00674-3
Braff, D. L., Grillon, C., and Geyer, M. A. (1992). Gating and habituation of the startle reflex in schizophrenic patients. Arch. Gen. Psychiatry 49, 206–215. doi: 10.1001/archpsyc.1992.01820030038005
Bramham, C. R., Alme, M. N., Bittins, M., Kuipers, S. D., Nair, R. R., Pai, B., et al. (2010). The Arc of synaptic memory. Exp. Brain Res. 200, 125–140. doi: 10.1007/s00221-009-1959-2
Breitborde, N. J. K., Maple, A. M., Bell, E. K., Dawson, S. C., Woolverton, C., Harrison-Monroe, P., et al. (2017). Activity-regulated cytoskeleton-associated protein predicts response to cognitive remediation among individuals with first-episode psychosis. Schizophr. Res. 184, 147–149. doi: 10.1016/j.schres.2016.12.005
Brigman, J. L., Wright, T., Talani, G., Prasad-Mulcare, S., Jinde, S., Seabold, G. K., et al. (2010). Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J. Neurosci. 30, 4590–4600. doi: 10.1523/JNEUROSCI.0640-10.2010
Brown, A. S. (2011). The environment and susceptibility to schizophrenia. Prog. Neurobiol. 93, 23–58. doi: 10.1016/j.pneurobio.2010.09.003
Brown, A. S., and Derkits, E. J. (2010). Prenatal infection and schizophrenia: a review of epidemiologic and translational studies. Am. J. Psychiatry 167, 261–280. doi: 10.1176/appi.ajp.2009.09030361
Bukalo, O., Lee, P. R., and Fields, R. D. (2016). BDNF mRNA abundance regulated by antidromic action potentials and AP-LTD in hippocampus. Neurosci. Lett. 635, 97–102. doi: 10.1016/j.neulet.2016.10.023
Butterick, T. A., Igbavboa, U., Eckert, G. P., Sun, G. Y., Weisman, G. A., Müller, W. E., et al. (2010). Simvastatin stimulates production of the antiapoptotic protein Bcl-2 via endothelin-1 and NFATc3 in SH-SY5Y cells. Mol. Neurobiol. 41, 384–391. doi: 10.1007/s12035-010-8122-8
Caetano, M. S., Vieira-de-Abreu, A., Teixeira, L. K., Werneck, M. B., Barcinski, M. A., and Viola, J. P. (2002). NFATC2 transcription factor regulates cell cycle progression during lymphocyte activation: evidence of its involvement in the control of cyclin gene expression. FASEB J. 16, 1940–1942. doi: 10.1096/fj.02-0282fje
Calabrese, F., Riva, M. A., and Molteni, R. (2016). Synaptic alterations associated with depression and schizophrenia: potential as a therapeutic target. Expert. Opin. Ther. Targets 20, 1195–1207. doi: 10.1080/14728222.2016.1188080
Carlén, M., Meletis, K., Siegle, J. H., Cardin, J. A., Futai, K., Vierling-Claassen, D., et al. (2012). A critical role for NMDA receptors in parvalbumin interneurons for γ rhythm induction and behavior. Mol. Psychiatry 17, 537–548. doi: 10.1038/mp.2011.31
Carlino, D., De Vanna, M., and Tongiorgi, E. (2013). Is altered BDNF biosynthesis a general feature in patients with cognitive dysfunctions? Neuroscientist 19, 345–353. doi: 10.1177/1073858412469444
Carvajal, F. J., Mattison, H. A., and Cerpa, W. (2016). Role of NMDA receptor-mediated glutamatergic signaling in chronic and acute neuropathologies. Neural Plast. 2016:2701526. doi: 10.1155/2016/2701526
Cattane, N., Minelli, A., Milanesi, E., Maj, C., Bignotti, S., Bortolomasi, M., et al. (2015). Altered gene expression in schizophrenia: findings from transcriptional signatures in fibroblasts and blood. PLoS One 10:e0116686. doi: 10.1371/journal.pone.0116686
Chalasani, L., Kant, R., and Chengappa, K. N. (2001). Clozapine impact on clinical outcomes and aggression in severely ill adolescents with childhood-onset schizophrenia. Can. J. Psychiatry 46, 965–968. doi: 10.1177/070674370104601010
Chandwani, S., Keilani, S., Ortiz-Virumbrales, M., Morant, A., Bezdecny, S., and Ehrlich, M. E. (2013). Induction of DARPP-32 by brain-derived neurotrophic factor in striatal neurons in vitro is modified by histone deacetylase inhibitors and Nab2. PLoS One 8:e76842. doi: 10.1371/journal.pone.0076842
Chávez, A. E., Chiu, C. Q., and Castillo, P. E. (2010). TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat. Neurosci. 13, 1511–1518. doi: 10.1038/nn.2684
Chen, W. S., and Bear, M. F. (2007). Activity-dependent regulation of NR2B translation contributes to metaplasticity in mouse visual cortex. Neuropharmacology 52, 200–214. doi: 10.1016/j.neuropharm.2006.07.003
Chengappa, K. N., Vasile, J., Levine, J., Ulrich, R., Baker, R., Gopalani, A., et al. (2002). Clozapine: its impact on aggressive behavior among patients in a state psychiatric hospital. Schizophr. Res. 53, 1–6. doi: 10.1016/s0920-9964(00)00175-4
Chowdhury, S., Shepherd, J. D., Okuno, H., Lyford, G., Petralia, R. S., Plath, N., et al. (2006). Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459. doi: 10.1016/j.neuron.2006.08.033
Chuang, Y. A., Hu, T. M., Chen, C. H., Hsu, S. H., Tsai, H. Y., and Cheng, M. C. (2016). Rare mutations and hypermethylation of the ARC gene associated with schizophrenia. Schizophr. Res. 176, 106–113. doi: 10.1016/j.schres.2016.07.019
Cole, A. J., Saffen, D. W., Baraban, J. M., and Worley, P. F. (1989). Rapid increase of an immediate early gene messenger RNA in hippocampal neurons by synaptic NMDA receptor activation. Nature 340, 474–476. doi: 10.1038/340474a0
Collingridge, G. L., Peineau, S., Howland, J. G., and Wang, Y. T. (2010). Long-term depression in the CNS. Nat. Rev. Neurosci. 11, 459–473. doi: 10.1038/nrn2867
Collins, S., Lutz, M. A., Zarek, P. E., Anders, R. A., Kersh, G. J., and Powell, J. D. (2008). Opposing regulation of T cell function by Egr-1/NAB2 and Egr-2/Egr-3. Eur. J. Immunol. 38, 528–536. doi: 10.1002/eji.200737157
Collins, S., Wolfraim, L. A., Drake, C. G., Horton, M. R., and Powell, J. D. (2006). Cutting Edge: TCR-induced NAB2 enhances T cell function by coactivating IL-2 transcription. J. Immunol. 177, 8301–8305. doi: 10.4049/jimmunol.177.12.8301
Corcoran, C., Gallitano, A., Leitman, D., and Malaspina, D. (2001). The neurobiology of the stress cascade and its potential relevance for schizophrenia. J. Psychiatr. Pract. 7, 3–14. doi: 10.1097/00131746-200101000-00002
Corcoran, C., Walker, E., Huot, R., Mittal, V., Tessner, K., Kestler, L., et al. (2003). The stress cascade and schizophrenia: etiology and onset. Schizophr. Bull. 29, 671–692. doi: 10.1093/oxfordjournals.schbul.a007038
Courtwright, A., Siamakpour-Reihani, S., Arbiser, J. L., Banet, N., Hilliard, E., Fried, L., et al. (2009). Secreted frizzle-related protein 2 stimulates angiogenesis via a calcineurin/NFAT signaling pathway. Cancer Res. 69, 4621–4628. doi: 10.1158/0008-5472.CAN-08-3402
Crabtree, G. W., and Gogos, J. A. (2014). Synaptic plasticity, neural circuits, and the emerging role of altered short-term information processing in schizophrenia. Front. Synaptic. Neurosci. 6:28. doi: 10.3389/fnsyn.2014.00028
Cui, Z., Feng, R., Jacobs, S., Duan, Y., Wang, H., Cao, X., et al. (2013). Increased NR2A:NR2B ratio compresses long-term depression range and constrains long-term memory. Sci. Rep. 3:1036. doi: 10.1038/srep01036
Curran, T., and Morgan, J. I. (1995). Fos: an immediate-early transcription factor in neurons. J. Neurobiol. 26, 403–412. doi: 10.1002/neu.480260312
Cutler, N. R. (2001). Pharmacokinetic studies of antipsychotics in healthy volunteers versus patients. J. Clin. Psychiatry 62, 10–13; discussion 23–14.
Davis, K. L., Stewart, D. G., Friedman, J. I., Buchsbaum, M., Harvey, P. D., Hof, P. R., et al. (2003). White matter changes in schizophrenia: evidence for myelin-related dysfunction. Arch. Gen. Psychiatry 60, 443–456. doi: 10.1001/archpsyc.60.5.443
Demontis, D., Nyegaard, M., Buttenschøn, H. N., Hedemand, A., Pedersen, C. B., Grove, J., et al. (2011). Association of GRIN1 and GRIN2A-D with schizophrenia and genetic interaction with maternal herpes simplex virus-2 infection affecting disease risk. Am. J. Med. Genet. B Neuropsychiatr. Genet. 156B, 913–922. doi: 10.1002/ajmg.b.31234
Donai, H., Sugiura, H., Ara, D., Yoshimura, Y., Yamagata, K., and Yamauchi, T. (2003). Interaction of Arc with CaM kinase II and stimulation of neurite extension by Arc in neuroblastoma cells expressing CaM kinase II. Neurosci. Res. 47, 399–408. doi: 10.1016/j.neures.2003.08.004
Drexhage, R. C., van der Heul-Nieuwenhuijsen, L., Padmos, R. C., van Beveren, N., Cohen, D., Versnel, M. A., et al. (2010). Inflammatory gene expression in monocytes of patients with schizophrenia: overlap and difference with bipolar disorder. A study in naturalistically treated patients. Int. J. Neuropsychopharmacol. 13, 1369–1381. doi: 10.1017/s1461145710000799
Du, H., Kwon, I. K., and Kim, J. (2013). Neuregulin-1 impairs the long-term depression of hippocampal inhibitory synapses by facilitating the degradation of endocannabinoid 2-AG. J. Neurosci. 33, 15022–15031. doi: 10.1523/JNEUROSCI.5833-12.2013
Dudek, S. M., and Bear, M. F. (1992). Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-D-aspartate receptor blockade. Proc. Natl. Acad. Sci. U S A 89, 4363–4367. doi: 10.1073/pnas.89.10.4363
Egan, M. F., Kojima, M., Callicott, J. H., Goldberg, T. E., Kolachana, B. S., Bertolino, A., et al. (2003). The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 112, 257–269. doi: 10.1016/s0092-8674(03)00035-7
Etkin, A., Alarcón, J. M., Weisberg, S. P., Touzani, K., Huang, Y. Y., Nordheim, A., et al. (2006). A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron 50, 127–143. doi: 10.1016/j.neuron.2006.03.013
Eto, K., Hommyo, A., Yonemitsu, R., and Abe, S. (2010). ErbB4 signals Neuregulin1-stimulated cell proliferation and c-fos gene expression through phosphorylation of serum response factor by mitogen-activated protein kinase cascade. Mol. Cell. Biochem. 339, 119–125. doi: 10.1007/s11010-009-0375-z
Fahmy, R. G., Dass, C. R., Sun, L. Q., Chesterman, C. N., and Khachigian, L. M. (2003). Transcription factor Egr-1 supports FGF-dependent angiogenesis during neovascularization and tumor growth. Nat. Med. 9, 1026–1032. doi: 10.1038/nm905
Fahmy, R. G., and Khachigian, L. M. (2007). Suppression of growth factor expression and human vascular smooth muscle cell growth by small interfering RNA targeting EGR-1. J. Cell. Biochem. 100, 1526–1535. doi: 10.1002/jcb.21145
Flynn, S. W., Lang, D. J., Mackay, A. L., Goghari, V., Vavasour, I. M., Whittall, K. P., et al. (2003). Abnormalities of myelination in schizophrenia detected in vivo with MRI and post-mortem with analysis of oligodendrocyte proteins. Mol. Psychiatry 8, 811–820. doi: 10.1038/sj.mp.4001337
Francis, T. C., Chandra, R., Gaynor, A., Konkalmatt, P., Metzbower, S. R., Evans, B., et al. (2017). Molecular basis of dendritic atrophy and activity in stress susceptibility. Mol. Psychiatry 22, 1512–1519. doi: 10.1038/mp.2017.178
Freed, W. J., Bing, L. A., and Wyatt, R. J. (1984). Effects of neuroleptics on phencyclidine (PCP)-induced locomotor stimulation in mice. Neuropharmacology 23, 175–181. doi: 10.1016/s0028-3908(84)80011-8
Fromer, M., Pocklington, A. J., Kavanagh, D. H., Williams, H. J., Dwyer, S., Gormley, P., et al. (2014). De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184. doi: 10.1038/nature12929
Fujimoto, T., Tanaka, H., Kumamaru, E., Okamura, K., and Miki, N. (2004). Arc interacts with microtubules/microtubule-associated protein 2 and attenuates microtubule-associated protein 2 immunoreactivity in the dendrites. J. Neurosci. Res. 76, 51–63. doi: 10.1002/jnr.20056
Gallitano, A. L. (2008). “Dysfunction of immediate early gene transcription factor Egr3 is implicated in schizophrenia abstract number,” 303.5 in Society for Neuroscience (Washington, DC).
Gallitano-Mendel, A., Izumi, Y., Tokuda, K., Zorumski, C. F., Howell, M. P., Muglia, L. J., et al. (2007). The immediate early gene early growth response gene 3 mediates adaptation to stress and novelty. Neuroscience 148, 633–643. doi: 10.1016/j.neuroscience.2007.05.050
Gallitano-Mendel, A., Wozniak, D. F., Pehek, E. A., and Milbrandt, J. (2008). Mice lacking the immediate early gene Egr3 respond to the anti-aggressive effects of clozapine yet are relatively resistant to its sedating effects. Neuropsychopharmacology 33, 1266–1275. doi: 10.1038/sj.npp.1301505
Gao, X. M., Sakai, K., Roberts, R. C., Conley, R. R., Dean, B., and Tamminga, C. A. (2000). Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am. J. Psychiatry 157, 1141–1149. doi: 10.1176/appi.ajp.157.7.1141
Ge, Y., Dong, Z., Bagot, R. C., Howland, J. G., Phillips, A. G., Wong, T. P., et al. (2010). Hippocampal long-term depression is required for the consolidation of spatial memory. Proc. Natl. Acad. Sci. U S A 107, 16697–16702. doi: 10.1073/pnas.1008200107
Gerber, D. J., Hall, D., Miyakawa, T., Demars, S., Gogos, J. A., Karayiorgou, M., et al. (2003). Evidence for association of schizophrenia with genetic variation in the 8p21.3 gene, PPP3CC, encoding the calcineurin γ subunit. Proc. Natl. Acad. Sci. U S A 100, 8993–8998. doi: 10.1073/pnas.1432927100
Glantz, L. A., and Lewis, D. A. (2000). Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73. doi: 10.1001/archpsyc.57.1.65
Gómez-Sintes, R., and Lucas, J. J. (2010). NFAT/Fas signaling mediates the neuronal apoptosis and motor side effects of GSK-3 inhibition in a mouse model of lithium therapy. J. Clin. Invest. 120, 2432–2445. doi: 10.1172/JCI37873
Groth, R. D., Coicou, L. G., Mermelstein, P. G., and Seybold, V. S. (2007). Neurotrophin activation of NFAT-dependent transcription contributes to the regulation of pro-nociceptive genes. J. Neurochem. 102, 1162–1174. doi: 10.1111/j.1471-4159.2007.04632.x
Guillozet-Bongaarts, A. L., Hyde, T. M., Dalley, R. A., Hawrylycz, M. J., Henry, A., Hof, P. R., et al. (2014). Altered gene expression in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 19, 478–485. doi: 10.1038/mp.2013.30
Guo, A. Y., Sun, J., Jia, P., and Zhao, Z. (2010). A novel microRNA and transcription factor mediated regulatory network in schizophrenia. BMC Syst. Biol. 4:10. doi: 10.1186/1752-0509-4-10
Guzowski, J. F., Setlow, B., Wagner, E. K., and McGaugh, J. L. (2001). Experience-dependent gene expression in the rat hippocampus after spatial learning: a comparison of the immediate-early genes Arc, c-fos, and zif268. J. Neurosci. 21, 5089–5098.
Hall, J., Trent, S., Thomas, K. L., O’Donovan, M. C., and Owen, M. J. (2015). Genetic risk for schizophrenia: convergence on synaptic pathways involved in plasticity. Biol. Psychiatry 77, 52–58. doi: 10.1016/j.biopsych.2014.07.011
Hao, S., Kurosaki, T., and August, A. (2003). Differential regulation of NFAT and SRF by the B cell receptor via a PLCγ-Ca2+-dependent pathway. EMBO J. 22, 4166–4177. doi: 10.1093/emboj/cdg401
Hariri, A. R., Goldberg, T. E., Mattay, V. S., Kolachana, B. S., Callicott, J. H., Egan, M. F., et al. (2003). Brain-derived neurotrophic factor val66met polymorphism affects human memory-related hippocampal activity and predicts memory performance. J. Neurosci. 23, 6690–6694.
Hashimoto, R., Moriguchi, Y., Yamashita, F., Mori, T., Nemoto, K., Okada, T., et al. (2008). Dose-dependent effect of the Val66Met polymorphism of the brain-derived neurotrophic factor gene on memory-related hippocampal activity. Neurosci. Res. 61, 360–367. doi: 10.1016/j.neures.2008.04.003
Hastings, K. T., Elizalde, D., Muppana, L., Levine, S., Kamel, C. M., Ingram, W. M., et al. (2017). Nab2 maintains thymus cellularity with aging and stress. Mol. Immunol. 85, 185–195. doi: 10.1016/j.molimm.2017.02.019
Hector, R. I. (1998). The use of clozapine in the treatment of aggressive schizophrenia. Can. J. Psychiatry 43, 466–472. doi: 10.1177/070674379804300503
Hernández, G. L., Volpert, O. V., Iñiguez, M. A., Lorenzo, E., Martínez-Martínez, S., Grau, R., et al. (2001). Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J. Exp. Med. 193, 607–620. doi: 10.1084/jem.193.5.607
Herndon, C. A., Ankenbruck, N., and Fromm, L. (2014). The Erk MAP kinase pathway is activated at muscle spindles and is required for induction of the muscle spindle-specific gene Egr3 by neuregulin1. J. Neurosci. Res. 92, 174–184. doi: 10.1002/jnr.23293
Herndon, C. A., Ankenbruck, N., Lester, B., Bailey, J., and Fromm, L. (2013). Neuregulin1 signaling targets SRF and CREB and activates the muscle spindle-specific gene Egr3 through a composite SRF-CREB-binding site. Exp. Cell Res. 319, 718–730. doi: 10.1016/j.yexcr.2013.01.001
Hippenmeyer, S., Shneider, N. A., Birchmeier, C., Burden, S. J., Jessell, T. M., and Arber, S. (2002). A role for neuregulin1 signaling in muscle spindle differentiation. Neuron 36, 1035–1049. doi: 10.1016/s0896-6273(02)01101-7
Hogan, P. G., Chen, L., Nardone, J., and Rao, A. (2003). Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232. doi: 10.1101/gad.1102703
Hokama, M., Oka, S., Leon, J., Ninomiya, T., Honda, H., Sasaki, K., et al. (2014). Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb. Cortex 24, 2476–2488. doi: 10.1093/cercor/bht101
Holtzman, C. W., Trotman, H. D., Goulding, S. M., Ryan, A. T., Macdonald, A. N., Shapiro, D. I., et al. (2013). Stress and neurodevelopmental processes in the emergence of psychosis. Neuroscience 249, 172–191. doi: 10.1016/j.neuroscience.2012.12.017
Horiuchi, Y., Ishiguro, H., Koga, M., Inada, T., Iwata, N., Ozaki, N., et al. (2007). Support for association of the PPP3CC gene with schizophrenia. Mol. Psychiatry 12, 891–893. doi: 10.1038/sj.mp.4002019
Horne, E. A., and Dell’Acqua, M. L. (2007). Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J. Neurosci. 27, 3523–3534. doi: 10.1523/JNEUROSCI.4340-06.2007
Hu, J., Sathanoori, M., Kochmar, S., Azage, M., Mann, S., Madan-Khetarpal, S., et al. (2015). A novel maternally inherited 8q24.3 and a rare paternally inherited 14q23.3 CNVs in a family with neurodevelopmental disorders. Am. J. Med. Genet. A 167A, 1921–1926. doi: 10.1002/ajmg.a.37110
Huentelman, M. J., Muppana, L., Corneveaux, J. J., Dinu, V., Pruzin, J. J., Reiman, R., et al. (2015). Association of SNPs in EGR3 and ARC with schizophrenia supports a biological pathway for schizophrenia risk. PLoS One 10:e0135076. doi: 10.1371/journal.pone.0135076
Husi, H., Ward, M. A., Choudhary, J. S., Blackstock, W. P., and Grant, S. G. (2000). Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 3, 661–669. doi: 10.1038/76615
Ingvar, D. H., and Franzen, G. (1974). Abnormalities of cerebral blood flow distribution in patients with chronic schizophrenia. Acta Psychiatr. Scand. 50, 425–462. doi: 10.1111/j.1600-0447.1974.tb09707.x
Islam, F., Mulsant, B. H., Voineskos, A. N., and Rajji, T. K. (2017). Brain-derived neurotrophic factor expression in individuals with schizophrenia and healthy aging: testing the accelerated aging hypothesis of schizophrenia. Curr. Psychiatry Rep. 19:36. doi: 10.1007/s11920-017-0794-6
Jacobson, C., Duggan, D., and Fischbach, G. (2004). Neuregulin induces the expression of transcription factors and myosin heavy chains typical of muscle spindles in cultured human muscle. Proc. Natl. Acad. Sci. U S A 101, 12218–12223. doi: 10.1073/pnas.0404240101
Javitt, D. C., and Zukin, S. R. (1991). Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 148, 1301–1308. doi: 10.1176/ajp.148.10.1301
Jayanthi, S., Deng, X., Ladenheim, B., McCoy, M. T., Cluster, A., Cai, N. S., et al. (2005). Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc. Natl. Acad. Sci. U S A 102, 868–873. doi: 10.1073/pnas.0404990102
Jessen, K. R., and Mirsky, R. (2002). Signals that determine Schwann cell identity. J. Anat. 200, 367–376. doi: 10.1046/j.1469-7580.2002.00046.x
Jones, M. W., Errington, M. L., French, P. J., Fine, A., Bliss, T. V., Garel, S., et al. (2001). A requirement for the immediate early gene Zif268 in the expression of late LTP and long-term memories. Nat. Neurosci. 4, 289–296. doi: 10.1038/85138
Jones, E. A., Jang, S. W., Mager, G. M., Chang, L. W., Srinivasan, R., Gokey, N. G., et al. (2007). Interactions of Sox10 and Egr2 in myelin gene regulation. Neuron Glia Biol. 3, 377–387. doi: 10.1017/S1740925X08000173
Jouvert, P., Dietrich, J. B., Aunis, D., and Zwiller, J. (2002). Differential rat brain expression of EGR proteins and of the transcriptional corepressor NAB in response to acute or chronic cocaine administration. Neuromolecular. Med. 1, 137–151. doi: 10.1385/nmm:1:2:137
Kahn, R. S., Sommer, I. E., Murray, R. M., Meyer-Lindenberg, A., Weinberger, D. R., Cannon, T. D., et al. (2015). Schizophrenia. Nat. Rev. Dis. Primers 1:15067. doi: 10.1038/nrdp.2015.67
Kao, S. C., Wu, H., Xie, J., Chang, C. P., Ranish, J. A., Graef, I. A., et al. (2009). Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science 323, 651–654. doi: 10.1126/science.1166562
Kemp, A., and Manahan-Vaughan, D. (2007). Hippocampal long-term depression: master or minion in declarative memory processes? Trends Neurosci. 30, 111–118. doi: 10.1016/j.tins.2007.01.002
Kenny, E. M., Cormican, P., Furlong, S., Heron, E., Kenny, G., Fahey, C., et al. (2014). Excess of rare novel loss-of-function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol. Psychiatry 19, 872–879. doi: 10.1038/mp.2013.127
Kiani, A., Rao, A., and Aramburu, J. (2000). Manipulating immune responses with immunosuppressive agents that target NFAT. Immunity 12, 359–372. doi: 10.1016/s1074-7613(00)80188-0
Kim, J. J., Foy, M. R., and Thompson, R. F. (1996). Behavioral stress modifies hippocampal plasticity through N-methyl-D-aspartate receptor activation. Proc. Natl. Acad. Sci. U S A 93, 4750–4753. doi: 10.1073/pnas.93.10.4750
Kim, J. H., Roberts, D. S., Hu, Y., Lau, G. C., Brooks-Kayal, A. R., Farb, D. H., et al. (2012). Brain-derived neurotrophic factor uses CREB and Egr3 to regulate NMDA receptor levels in cortical neurons. J. Neurochem. 120, 210–219. doi: 10.1111/j.1471-4159.2011.07555.x
Kim, S. H., Song, J. Y., Joo, E. J., Lee, K. Y., Ahn, Y. M., and Kim, Y. S. (2010). EGR3 as a potential susceptibility gene for schizophrenia in Korea. Am. J. Med. Genet. B Neuropsychiatr. Genet. 153B, 1355–1360. doi: 10.1002/ajmg.b.31115
Kipanyula, M. J., Kimaro, W. H., and Seke Etet, P. F. (2016). The emerging roles of the calcineurin-nuclear factor of activated T-lymphocytes pathway in nervous system functions and diseases. J. Aging Res. 2016:5081021. doi: 10.1155/2016/5081021
Kirov, G., Pocklington, A. J., Holmans, P., Ivanov, D., Ikeda, M., Ruderfer, D., et al. (2012). De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153. doi: 10.1038/mp.2011.154
Klein, M., Klein-Hessling, S., Palmetshofer, A., Serfling, E., Tertilt, C., Bopp, T., et al. (2006). Specific and redundant roles for NFAT transcription factors in the expression of mast cell-derived cytokines. J. Immunol. 177, 6667–6674. doi: 10.4049/jimmunol.177.10.6667
Kojima, M., and Mizui, T. (2017). BDNF propeptide: a novel modulator of synaptic plasticity. Vitam. Horm. 104, 19–28. doi: 10.1016/bs.vh.2016.11.006
Kozlovsky, N., Matar, M. A., Kaplan, Z., Kotler, M., Zohar, J., and Cohen, H. (2008). The immediate early gene Arc is associated with behavioral resilience to stress exposure in an animal model of posttraumatic stress disorder. Eur. Neuropsychopharmacol. 18, 107–116. doi: 10.1016/j.euroneuro.2007.04.009
Kraepelin, E., and Robertson, G. M. (1919). Dementia Praecox and Paraphrenia. Edinburgh: Livingstone.
Krakowski, M. I., Czobor, P., Citrome, L., Bark, N., and Cooper, T. B. (2006). Atypical antipsychotic agents in the treatment of violent patients with schizophrenia and schizoaffective disorder. Arch. Gen. Psychiatry 63, 622–629. doi: 10.1001/archpsyc.63.6.622
Kumbrink, J., Gerlinger, M., and Johnson, J. P. (2005). Egr-1 induces the expression of its corepressor nab2 by activation of the nab2 promoter thereby establishing a negative feedback loop. J. Biol. Chem. 280, 42785–42793. doi: 10.1074/jbc.M511079200
Kumbrink, J., Kirsch, K. H., and Johnson, J. P. (2010). EGR1, EGR2, and EGR3 activate the expression of their coregulator NAB2 establishing a negative feedback loop in cells of neuroectodermal and epithelial origin. J. Cell. Biochem. 111, 207–217. doi: 10.1002/jcb.22690
Kurosaka, M., Ogura, Y., Funabashi, T., and Akema, T. (2017). Early growth response 3 (Egr3) contributes a maintenance of C2C12 myoblast proliferation. J. Cell. Physiol. 232, 1114–1122. doi: 10.1002/jcp.25574
Kutsuwada, T., Sakimura, K., Manabe, T., Takayama, C., Katakura, N., Kushiya, E., et al. (1996). Impairment of suckling response, trigeminal neuronal pattern formation, and hippocampal LTD in NMDA receptor ε2 subunit mutant mice. Neuron 16, 333–344. doi: 10.1016/s0896-6273(00)80051-3
Lahti, A. C., Weiler, M. A., Tamara Michaelidis, B. A., Parwani, A., and Tamminga, C. A. (2001). Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology 25, 455–467. doi: 10.1016/s0893-133x(01)00243-3
Law, A. J., Lipska, B. K., Weickert, C. S., Hyde, T. M., Straub, R. E., Hashimoto, R., et al. (2006). Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc. Natl. Acad. Sci. U S A 103, 6747–6752. doi: 10.1176/foc.4.3.350
Le, N., Nagarajan, R., Wang, J. Y., Svaren, J., LaPash, C., Araki, T., et al. (2005). Nab proteins are essential for peripheral nervous system myelination. Nat. Neurosci. 8, 932–940. doi: 10.1038/nn1490
Li, L., Carter, J., Gao, X., Whitehead, J., and Tourtellotte, W. G. (2005). The neuroplasticity-associated arc gene is a direct transcriptional target of early growth response (Egr) transcription factors. Mol. Cell. Biol. 25, 10286–10300. doi: 10.1128/mcb.25.23.10286-10300.2005
Li, S., Miao, T., Sebastian, M., Bhullar, P., Ghaffari, E., Liu, M., et al. (2012). The transcription factors Egr2 and Egr3 are essential for the control of inflammation and antigen-induced proliferation of B and T cells. Immunity 37, 685–696. doi: 10.1016/j.immuni.2012.08.001
Lin, K., Wang, D., and Sadee, W. (2002). Serum response factor activation by muscarinic receptors via RhoA. Novel pathway specific to M1 subtype involving calmodulin, calcineurin and, Pyk2. J. Biol. Chem. 277, 40789–40798. doi: 10.1074/jbc.M202745200
Lindecke, A., Korte, M., Zagrebelsky, M., Horejschi, V., Elvers, M., Widera, D., et al. (2006). Long-term depression activates transcription of immediate early transcription factor genes: involvement of serum response factor/Elk-1. Eur. J. Neurosci. 24, 555–563. doi: 10.1111/j.1460-9568.2006.04909.x
Link, W., Konietzko, U., Kauselmann, G., Krug, M., Schwanke, B., Frey, U., et al. (1995). Somatodendritic expression of an immediate early gene is regulated by synaptic activity. Proc. Natl. Acad. Sci. U S A 92, 5734–5738. doi: 10.1073/pnas.92.12.5734
Lipska, B. K., and Weinberger, D. R. (2000). To model a psychiatric disorder in animals: schizophrenia as a reality test. Neuropsychopharmacology 23, 223–239. doi: 10.1016/s0893-133x(00)00137-8
Liu, Y. L., Fann, C. S., Liu, C. M., Chang, C. C., Yang, W. C., Hung, S. I., et al. (2007). More evidence supports the association of PPP3CC with schizophrenia. Mol. Psychiatry 12, 966–974. doi: 10.1038/sj.mp.4001977
Liu, L., Wong, T. P., Pozza, M. F., Lingenhoehl, K., Wang, Y., Sheng, M., et al. (2004). Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science 304, 1021–1024. doi: 10.1126/science.1096615
Liu, S., Zhang, F., Shugart, Y. Y., Yang, L., Li, X., Liu, Z., et al. (2017). The early growth response protein 1-miR-30a-5p-neurogenic differentiation factor 1 axis as a novel biomarker for schizophrenia diagnosis and treatment monitoring. Transl. Psychiatry 7:e998. doi: 10.1038/tp.2016.268
Lohoff, F. W., Weller, A. E., Bloch, P. J., Buono, R. J., Doyle, G. A., Ferraro, T. N., et al. (2008). Association between polymorphisms in the vesicular monoamine transporter 1 gene (VMAT1/SLC18A1) on chromosome 8p and schizophrenia. Neuropsychobiology 57, 55–60. doi: 10.1159/000129668
Lupien, S. J., McEwen, B. S., Gunnar, M. R., and Heim, C. (2009). Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat. Rev. Neurosci. 10, 434–445. doi: 10.1038/nrn2639
Lyford, G. L., Yamagata, K., Kaufmann, W. E., Barnes, C. A., Sanders, L. K., Copeland, N. G., et al. (1995). Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron 14, 433–445. doi: 10.1016/0896-6273(95)90299-6
Macian, F. (2005). NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 5, 472–484. doi: 10.1038/nri1632
Malenka, R. C., and Bear, M. F. (2004). LTP and LTD: an embarrassment of riches. Neuron 44, 5–21. doi: 10.1016/j.neuron.2004.09.012
Malhotra, A. K., Pinals, D. A., Adler, C. M., Elman, I., Clifton, A., Pickar, D., et al. (1997). Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 17, 141–150. doi: 10.1016/s0893-133x(97)00036-5
Managò, F., Mereu, M., Mastwal, S., Mastrogiacomo, R., Scheggia, D., Emanuele, M., et al. (2016). Genetic disruption of Arc/Arg3.1 in mice causes alterations in dopamine and neurobehavioral phenotypes related to schizophrenia. Cell Rep. 16, 2116–2128. doi: 10.1016/j.celrep.2016.07.044
Manahan-Vaughan, D., and Braunewell, K. H. (1999). Novelty acquisition is associated with induction of hippocampal long-term depression. Proc. Natl. Acad. Sci. U S A 96, 8739–8744. doi: 10.1073/pnas.96.15.8739
Maple, A., Lackie, R. E., Elizalde, D. I., Grella, S. L., Damphousse, C. C., Xa, C., et al. (2017). Attenuated late-phase arc transcription in the dentate gyrus of mice lacking Egr3. Neural Plast. 2017:6063048. doi: 10.1155/2017/6063048
Maple, A. M., Zhao, X., Elizalde, D. I., McBride, A. K., and Gallitano, A. L. (2015). Htr2a expression responds rapidly to environmental stimuli in an Egr3-dependent manner. ACS Chem. Neurosci. 6, 1137–1142. doi: 10.1021/acschemneuro.5b00031
Marrone, D. F., Satvat, E., Shaner, M. J., Worley, P. F., and Barnes, C. A. (2012). Attenuated long-term Arc expression in the aged fascia dentata. Neurobiol. Aging 33, 979–990. doi: 10.1016/j.neurobiolaging.2010.07.022
Martinez, M., Calvo-Torrent, A., and Herbert, J. (2002). Mapping brain response to social stress in rodents with c-fos expression: a review. Stress 5, 3–13. doi: 10.1080/102538902900012369
McGue, M., and Gottesman, I. I. (1991). The genetic epidemiology of schizophrenia and the design of linkage studies. Eur. Arch. Psychiatry Clin. Neurosci. 240, 174–181. doi: 10.1007/bf02190760
Mechta-Grigoriou, F., Garel, S., and Charnay, P. (2000). Nab proteins mediate a negative feedback loop controlling Krox-20 activity in the developing hindbrain. Development 127, 119–128.
Mechtcheriakova, D., Wlachos, A., Holzmüller, H., Binder, B. R., and Hofer, E. (1999). Vascular endothelial cell growth factor-induced tissue factor expression in endothelial cells is mediated by EGR-1. Blood 93, 3811–3823.
Messaoudi, E., Kanhema, T., Soule, J., Tiron, A., Dagyte, G., da Silva, B., et al. (2007). Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J. Neurosci. 27, 10445–10455. doi: 10.1523/JNEUROSCI.2883-07.2007
Mexal, S., Frank, M., Berger, R., Adams, C. E., Ross, R. G., Freedman, R., et al. (2005). Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. Mol. Brain Res. 139, 317–332. doi: 10.1016/j.molbrainres.2005.06.006
Meyers, K., Marballi, K. K., Brunwasser, S., Renda, B., Charbel, M., Marrone, D. F., et al. (2017a). The immediate early gene Egr3 is required for activity-dependent induction of hippocampal Bdnf. Front. Behav. Neurosci.
Meyers, K., Marballi, K. K., Campbell, J. M., and Gallitano, A. L. (2017b). “Induction of BDNF by Electroconvulsive stimulation requires the immediate early gene early growth response 3 (Egr3),”472.06 in Society for Neuroscience (Washington, DC).
Milbrandt, J. (1987). A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science 238, 797–799. doi: 10.1126/science.3672127
Minatohara, K., Akiyoshi, M., and Okuno, H. (2016). Role of immediate-early genes in synaptic plasticity and neuronal ensembles underlying the memory trace. Front. Mol. Neurosci. 8:78. doi: 10.3389/fnmol.2015.00078
Mittal, V. A., Ellman, L. M., and Cannon, T. D. (2008). Gene-environment interaction and covariation in schizophrenia: the role of obstetric complications. Schizophr. Bull. 34, 1083–1094. doi: 10.1093/schbul/sbn080
Mittelstadt, P. R., and Ashwell, J. D. (1998). Cyclosporin A-sensitive transcription factor Egr-3 regulates Fas ligand expression. Mol. Cell. Biol. 18, 3744–3751. doi: 10.1128/mcb.18.7.3744
Mittelstadt, P. R., and Ashwell, J. D. (1999). Role of Egr-2 in up-regulation of Fas ligand in normal T cells and aberrant double-negative lpr and gld T cells. J. Biol. Chem. 274, 3222–3227. doi: 10.1074/jbc.274.5.3222
Miyakawa, T., Leiter, L. M., Gerber, D. J., Gainetdinov, R. R., Sotnikova, T. D., Zeng, H., et al. (2003). Conditional calcineurin knockout mice exhibit multiple abnormal behaviors related to schizophrenia. Proc. Natl. Acad. Sci. U S A 100, 8987–8992. doi: 10.1073/pnas.1432926100
Mizui, T., Ishikawa, Y., Kumanogoh, H., Lume, M., Matsumoto, T., Hara, T., et al. (2015). BDNF pro-peptide actions facilitate hippocampal LTD and are altered by the common BDNF polymorphism Val66Met. Proc. Natl. Acad. Sci. U S A 112, E3067–E3074. doi: 10.1073/pnas.1422336112
Mohn, A. R., Gainetdinov, R. R., Caron, M. G., and Koller, B. H. (1999). Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98, 427–436. doi: 10.1016/s0092-8674(00)81972-8
Moises, H. W., Zoega, T., and Gottesman, I. I. (2002). The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. BMC Psychiatry 2:8. doi: 10.1186/1471-244x-2-8
Morris, M. E., Viswanathan, N., Kuhlman, S., Davis, F. C., and Weitz, C. J. (1998). A screen for genes induced in the suprachiasmatic nucleus by light. Science 279, 1544–1547. doi: 10.1126/science.279.5356.1544
Mostaid, M. S., Lloyd, D., Liberg, B., Sundram, S., Pereira, A., Pantelis, C., et al. (2016). Neuregulin-1 and schizophrenia in the genome-wide association study era. Neurosci. Biobehav. Rev. 68, 387–409. doi: 10.1016/j.neubiorev.2016.06.001
Mulkey, R. M., Endo, S., Shenolikar, S., and Malenka, R. C. (1994). Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature 369, 486–488. doi: 10.1038/369486a0
Mulkey, R. M., and Malenka, R. C. (1992). Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9, 967–975. doi: 10.1016/0896-6273(92)90248-c
Nabavi, S., Fox, R., Proulx, C. D., Lin, J. Y., Tsien, R. Y., and Malinow, R. (2014). Engineering a memory with LTD and LTP. Nature 511, 348–352. doi: 10.1038/nature13294
Nagarajan, R., Svaren, J., Le, N., Araki, T., Watson, M., and Milbrandt, J. (2001). EGR2 mutations in inherited neuropathies dominant-negatively inhibit myelin gene expression. Neuron 30, 355–368. doi: 10.1016/s0896-6273(01)00282-3
Neria, F., del Carmen Serrano-Perez, M., Velasco, P., Urso, K., Tranque, P., and Cano, E. (2013). NFATc3 promotes Ca2+ -dependent MMP3 expression in astroglial cells. Glia 61, 1052–1066. doi: 10.1002/glia.22494
Ning, Q. L., Ma, X. D., Jiao, L. Z., Niu, X. R., Li, J. P., Wang, B., et al. (2012). [A family-based association study of the EGR3 gene polymorphisms and schizophrenia]. Yi. Chuan 34, 307–314. doi: 10.3724/sp.j.1005.2012.00307
Nishimura, Y., Takizawa, R., Koike, S., Kinoshita, A., Satomura, Y., Kawasaki, S., et al. (2014). Association of decreased prefrontal hemodynamic response during a verbal fluency task with EGR3 gene polymorphism in patients with schizophrenia and in healthy individuals. Neuroimage 85, 527–534. doi: 10.1016/j.neuroimage.2013.08.021
Novkovic, T., Mittmann, T., and Manahan-Vaughan, D. (2015). BDNF contributes to the facilitation of hippocampal synaptic plasticity and learning enabled by environmental enrichment. Hippocampus 25, 1–15. doi: 10.1002/hipo.22342
O’Donovan, K. J., Tourtellotte, W. G., Millbrandt, J., and Baraban, J. M. (1999). The EGR family of transcription-regulatory factors: progress at the interface of molecular and systems neuroscience. Trends Neurosci. 22, 167–173. doi: 10.1016/s0166-2236(98)01343-5
O’Donovan, K. J., Wilkens, E. P., and Baraban, J. M. (1998). Sequential expression of Egr-1 and Egr-3 in hippocampal granule cells following electroconvulsive stimulation. J. Neurochem. 70, 1241–1248. doi: 10.1046/j.1471-4159.1998.70031241.x
Okamura, H., Aramburu, J., Garcia-Rodríguez, C., Viola, J. P., Raghavan, A., Tahiliani, M., et al. (2000). Concerted dephosphorylation of the transcription factor NFAT1 induces a conformational switch that regulates transcriptional activity. Mol. Cell 6, 539–550. doi: 10.1016/s1097-2765(00)00053-8
Okuno, H., Akashi, K., Ishii, Y., Yagishita-Kyo, N., Suzuki, K., Nonaka, M., et al. (2012). Inverse synaptic tagging of inactive synapses via dynamic interaction of Arc/Arg3.1 with CaMKIIβ. Cell 149, 886–898. doi: 10.1016/j.cell.2012.02.062
Olney, J. W., Newcomer, J. W., and Farber, N. B. (1999). NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 33, 523–533. doi: 10.1016/S0022-3956(99)00029-1
O’Neill, M. F., and Shaw, G. (1999). Comparison of dopamine receptor antagonists on hyperlocomotion induced by cocaine, amphetamine, MK-801 and the dopamine D1 agonist C-APB in mice. Psychopharmacology 145, 237–250. doi: 10.1007/s002130051055
Oukka, M., Ho, I. C., de la Brousse, F. C., Hoey, T., Grusby, M. J., and Glimcher, L. H. (1998). The transcription factor NFAT4 is involved in the generation and survival of T cells. Immunity 9, 295–304. doi: 10.1016/s1074-7613(00)80612-3
Papadia, S., and Hardingham, G. E. (2007). The dichotomy of NMDA receptor signaling. Neuroscientist 13, 572–579. doi: 10.1177/1073858407305833
Park, S., Uesugi, M., and Verdine, G. L. (2000). A second calcineurin binding site on the NFAT regulatory domain. Proc. Natl. Acad. Sci. U S A 97, 7130–7135. doi: 10.1073/pnas.97.13.7130
Paulsen, J. S., Heaton, R. K., Sadek, J. R., Perry, W., Delis, D. C., Braff, D., et al. (1995). The nature of learning and memory impairments in schizophrenia. J. Int. Neuropsychol. Soc. 1, 88–99. doi: 10.1017/S135561770000014X
Peebles, C. L., Yoo, J., Thwin, M. T., Palop, J. J., Noebels, J. L., and Finkbeiner, S. (2010). Arc regulates spine morphology and maintains network stability in vivo. Proc. Natl. Acad. Sci. U S A 107, 18173–18178. doi: 10.1073/pnas.1006546107
Penke, Z., Morice, E., Veyrac, A., Gros, A., Chagneau, C., LeBlanc, P., et al. (2013). Zif268/Egr1 gain of function facilitates hippocampal synaptic plasticity and long-term spatial recognition memory. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130159. doi: 10.1098/rstb.2013.0159
Pérez-Santiago, J., Diez-Alarcia, R., Callado, L. F., Zhang, J. X., Chana, G., White, C. H., et al. (2012). A combined analysis of microarray gene expression studies of the human prefrontal cortex identifies genes implicated in schizophrenia. J. Psychiatr. Res. 46, 1464–1474. doi: 10.1016/j.jpsychires.2012.08.005
Pfaffenseller, B., da Silva Magalhães, P. V., De Bastiani, M. A., Castro, M. A., Gallitano, A. L., Kapczinski, F., et al. (2016). Differential expression of transcriptional regulatory units in the prefrontal cortex of patients with bipolar disorder: potential role of early growth response gene 3. Transl. Psychiatry 6:e805. doi: 10.1038/tp.2016.78
Plath, N., Ohana, O., Dammermann, B., Errington, M. L., Schmitz, D., Gross, C., et al. (2006). Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444. doi: 10.1016/j.neuron.2006.08.024
Porteous, D. (2008). Genetic causality in schizophrenia and bipolar disorder: out with the old and in with the new. Curr. Opin. Genet. Dev. 18, 229–234. doi: 10.1016/j.gde.2008.07.005
Pouget, J. G., Gonçalves, V. F., Schizophrenia Working Group of the Psychiatric Genomics Consortium, Spain, S. L., Finucane, H. K., Raychaudhuri, S., et al. (2016). Genome-wide association studies suggest limited immune gene enrichment in schizophrenia compared to 5 autoimmune diseases. Schizophr. Bull. 42, 1176–1184. doi: 10.1093/schbul/sbw059
Purcell, S. M., Moran, J. L., Fromer, M., Ruderfer, D., Solovieff, N., Roussos, P., et al. (2014). A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190. doi: 10.1038/nature12975
Ramanan, N., Shen, Y., Sarsfield, S., Lemberger, T., Schütz, G., Linden, D. J., et al. (2005). SRF mediates activity-induced gene expression and synaptic plasticity but not neuronal viability. Nat. Neurosci. 8, 759–767. doi: 10.1038/nn1462
Ramirez-Amaya, V., Angulo-Perkins, A., Chawla, M. K., Barnes, C. A., and Rosi, S. (2013). Sustained transcription of the immediate early gene Arc in the dentate gyrus after spatial exploration. J. Neurosci. 33, 1631–1639. doi: 10.1523/jneurosci.2916-12.2013
Rao, A., Luo, C., and Hogan, P. G. (1997). Transcription factors of the NFAT family: regulation and function. Annu. Rev. Immunol. 15, 707–747. doi: 10.1146/annurev.immunol.15.1.707
Rengarajan, J., Mittelstadt, P. R., Mages, H. W., Gerth, A. J., Kroczek, R. A., Ashwell, J. D., et al. (2000). Sequential involvement of NFAT and Egr transcription factors in FasL regulation. Immunity 12, 293–300. doi: 10.1016/s1074-7613(00)80182-x
Rison, R. A., and Stanton, P. K. (1995). Long-term potentiation and N-methyl-D-aspartate receptors: foundations of memory and neurologic disease? Neurosci. Biobehav. Rev. 19, 533–552. doi: 10.1016/0149-7634(95)00017-8
Roberts, D. S., Hu, Y., Lund, I. V., Brooks-Kayal, A. R., and Russek, S. J. (2006). Brain-derived neurotrophic factor (BDNF)-induced synthesis of early growth response factor 3 (Egr3) controls the levels of type A GABA receptor α 4 subunits in hippocampal neurons. J. Biol. Chem. 281, 29431–29435. doi: 10.1074/jbc.c600167200
Rompala, G. R., Zsiros, V., Zhang, S., Kolata, S. M., and Nakazawa, K. (2013). Contribution of NMDA receptor hypofunction in prefrontal and cortical excitatory neurons to schizophrenia-like phenotypes. PLoS One 8:e61278. doi: 10.1371/journal.pone.0061278
Rowan, M. J., Anwyl, R., and Xu, L. (1998). Stress and long-term synaptic depression. Mol. Psychiatry 3, 472–474. doi: 10.1038/sj.mp.4000429
Rusnak, F., and Mertz, P. (2000). Calcineurin: form and function. Physiol. Rev. 80, 1483–1521. doi: 10.1152/physrev.2000.80.4.1483
Sanderson, J. L., Gorski, J. A., and Dell’Acqua, M. L. (2016). NMDA receptor-dependent LTD requires transient synaptic incorporation of Ca2+-permeable AMPARs mediated by AKAP150-anchored PKA and calcineurin. Neuron 89, 1000–1015. doi: 10.1016/j.neuron.2016.01.043
Saykin, A. J., Gur, R. C., Gur, R. E., Mozley, P. D., Mozley, L. H., Resnick, S. M., et al. (1991). Neuropsychological function in schizophrenia. Selective impairment in memory and learning. Arch. Gen. Psychiatry 48, 618–624. doi: 10.1001/archpsyc.1991.01810310036007
Scharf, M. T., Woo, N. H., Lattal, K. M., Young, J. Z., Nguyen, P. V., and Abel, T. (2002). Protein synthesis is required for the enhancement of long-term potentiation and long-term memory by spaced training. J. Neurophysiol. 87, 2770–2777. doi: 10.1152/jn.2002.87.6.2770
Schizophrenia Working Group of the Psychiatric Genomics Consortium. (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427. doi: 10.1038/nature13595
Schulze, C., Büchse, T., Mikkat, S., and Bittorf, T. (2008). Erythropoietin receptor-mediated Egr-1 activation: structural requirements and functional implications. Cell. Signal 20, 1848–1854. doi: 10.1016/j.cellsig.2008.06.013
Schulz, R. A., and Yutzey, K. E. (2004). Calcineurin signaling and NFAT activation in cardiovascular and skeletal muscle development. Dev. Biol. 266, 1–16. doi: 10.1016/j.ydbio.2003.10.008
Schwartz, N., Schohl, A., and Ruthazer, E. S. (2009). Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron 62, 655–669. doi: 10.1016/j.neuron.2009.05.007
Selvaraj, S., Arnone, D., Cappai, A., and Howes, O. (2014). Alterations in the serotonin system in schizophrenia: a systematic review and meta-analysis of postmortem and molecular imaging studies. Neurosci. Biobehav. Rev. 45, 233–245. doi: 10.1016/j.neubiorev.2014.06.005
Senba, E., and Ueyama, T. (1997). Stress-induced expression of immediate early genes in the brain and peripheral organs of the rat. Neurosci. Res. 29, 183–207. doi: 10.1016/s0168-0102(97)00095-3
Serrano-Pérez, M. C., Fernández, M., Neria, F., Berjón-Otero, M., Doncel-Pérez, E., Cano, E., et al. (2015). NFAT transcription factors regulate survival, proliferation, migration and differentiation of neural precursor cells. Glia 63, 987–1004. doi: 10.1002/glia.22797
Serrano-Pérez, M. C., Martín, E. D., Vaquero, C. F., Azcoitia, I., Calvo, S., Cano, E., et al. (2011). Response of transcription factor NFATc3 to excitotoxic and traumatic brain insults: identification of a subpopulation of reactive astrocytes. Glia 59, 94–107. doi: 10.1002/glia.21079
Shin, S. Y., Kim, J. H., Baker, A., Lim, Y., and Lee, Y. H. (2010). Transcription factor Egr-1 is essential for maximal matrix metalloproteinase-9 transcription by tumor necrosis factor α. Mol. Cancer Res. 8, 507–519. doi: 10.1158/1541-7786.mcr-09-0454
Shipton, O. A., and Paulsen, O. (2014). GluN2A and GluN2B subunit-containing NMDA receptors in hippocampal plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130163. doi: 10.1098/rstb.2013.0163
Srinivasan, R., Jang, S. W., Ward, R. M., Sachdev, S., Ezashi, T., and Svaren, J. (2007). Differential regulation of NAB corepressor genes in Schwann cells. BMC Mol. Biol. 8:117. doi: 10.1186/1471-2199-8-117
Stefansson, H., Sigurdsson, E., Steinthorsdottir, V., Bjornsdottir, S., Sigmundsson, T., Ghosh, S., et al. (2002). Neuregulin 1 and susceptibility to schizophrenia. Am. J. Hum. Genet. 71, 877–892. doi: 10.1086/342734
Suarez, B. K., Duan, J., Sanders, A. R., Hinrichs, A. L., Jin, C. H., Hou, C., et al. (2006). Genomewide linkage scan of 409 European-ancestry and African American families with schizophrenia: suggestive evidence of linkage at 8p23.3–p21.2 and 11p13.1–q14.1 in the combined sample. Am. J. Hum. Genet. 78, 315–333. doi: 10.1086/500272
Suehiro, J., Hamakubo, T., Kodama, T., Aird, W. C., and Minami, T. (2010). Vascular endothelial growth factor activation of endothelial cells is mediated by early growth response-3. Blood 115, 2520–2532. doi: 10.1182/blood-2009-07-233478
Svaren, J., Sevetson, B. R., Apel, E. D., Zimonjic, D. B., Popescu, N. C., and Milbrandt, J. (1996). NAB2, a corepressor of NGFI-A (Egr-1) and Krox20, is induced by proliferative and differentiative stimuli. Mol. Cell. Biol. 16, 3545–3553. doi: 10.1128/mcb.16.7.3545
Sweeney, C., Fambrough, D., Huard, C., Diamonti, A. J., Lander, E. S., Cantley, L. C., et al. (2001). Growth factor-specific signaling pathway stimulation and gene expression mediated by ErbB receptors. J. Biol. Chem. 276, 22685–22698. doi: 10.1074/jbc.m100602200
Tabarés-Seisdedos, R., and Rubenstein, J. L. (2009). Chromosome 8p as a potential hub for developmental neuropsychiatric disorders: implications for schizophrenia, autism and cancer. Mol. Psychiatry 14, 563–589. doi: 10.1038/mp.2009.2
Tamminga, C. A., Lahti, A. C., Medoff, D. R., Gao, X. M., and Holcomb, H. H. (2003). Evaluating glutamatergic transmission in schizophrenia. Ann. N Y Acad. Sci. 1003, 113–118. doi: 10.1196/annals.1300.062
Taylor, D. L., Tiwari, A. K., Lieberman, J. A., Potkin, S. G., Meltzer, H. Y., Knight, J., et al. (2016). Genetic association analysis of N-methyl-D-aspartate receptor subunit gene GRIN2B and clinical response to clozapine. Hum. Psychopharmacol. 31, 121–134. doi: 10.1002/hup.2519
Tourtellotte, W. G., Keller-Peck, C., Milbrandt, J., and Kucera, J. (2001). The transcription factor Egr3 modulates sensory axon-myotube interactions during muscle spindle morphogenesis. Dev. Biol. 232, 388–399. doi: 10.1006/dbio.2001.0202
Tourtellotte, W. G., and Milbrandt, J. (1998). Sensory ataxia and muscle spindle agenesis in mice lacking the transcription factor Egr3. Nat. Genet. 20, 87–91. doi: 10.1038/1757
Ulrich, J. D., Kim, M. S., Houlihan, P. R., Shutov, L. P., Mohapatra, D. P., Strack, S., et al. (2012). Distinct activation properties of the nuclear factor of activated T-cells (NFAT) isoforms NFATc3 and NFATc4 in neurons. J. Biol. Chem. 287, 37594–37609. doi: 10.1074/jbc.p112.365197
van den Buuse, M. (2010). Modeling the positive symptoms of schizophrenia in genetically modified mice: pharmacology and methodology aspects. Schizophr. Bull. 36, 246–270. doi: 10.1093/schbul/sbp132
van Os, J., Kenis, G., and Rutten, B. P. (2010). The environment and schizophrenia. Nature 468, 203–212. doi: 10.1038/nature09563
van Wingen, G., Rijpkema, M., Franke, B., van Eijndhoven, P., Tendolkar, I., Verkes, R. J., et al. (2010). The brain-derived neurotrophic factor Val66Met polymorphism affects memory formation and retrieval of biologically salient stimuli. Neuroimage 50, 1212–1218. doi: 10.1016/j.neuroimage.2010.01.058
van Winkel, R., Stefanis, N. C., and Myin-Germeys, I. (2008). Psychosocial stress and psychosis. A review of the neurobiological mechanisms and the evidence for gene-stress interaction. Schizophr. Bull. 34, 1095–1105. doi: 10.1093/schbul/sbn101
Vihma, H., Luhakooder, M., Pruunsild, P., and Timmusk, T. (2016). Regulation of different human NFAT isoforms by neuronal activity. J. Neurochem. 137, 394–408. doi: 10.1111/jnc.13568
Vihma, H., Pruunsild, P., and Timmusk, T. (2008). Alternative splicing and expression of human and mouse NFAT genes. Genomics 92, 279–291. doi: 10.1016/j.ygeno.2008.06.011
Waung, M. W., Pfeiffer, B. E., Nosyreva, E. D., Ronesi, J. A., and Huber, K. M. (2008). Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97. doi: 10.1016/j.neuron.2008.05.014
Weickert, C. S., Fung, S. J., Catts, V. S., Schofield, P. R., Allen, K. M., Moore, L. T., et al. (2013). Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry 18, 1185–1192. doi: 10.1038/mp.2012.137
Weigelt, K., Carvalho, L. A., Drexhage, R. C., Wijkhuijs, A., de Wit, H., van Beveren, N. J., et al. (2011). TREM-1 and DAP12 expression in monocytes of patients with severe psychiatric disorders. EGR3, ATF3 and PU.1 as important transcription factors. Brain Behav. Immun. 25, 1162–1169. doi: 10.1016/j.bbi.2011.03.006
Weinstock, M. (2017). Prenatal stressors in rodents: effects on behavior. Neurobiol. Stress 6, 3–13. doi: 10.1016/j.ynstr.2016.08.004
Williams, A. A., Ingram, W. M., Levine, S., Resnik, J., Kamel, C. M., Lish, J. R., et al. (2012). Reduced levels of serotonin 2A receptors underlie resistance of Egr3-deficient mice to locomotor suppression by clozapine. Neuropsychopharmacology 37, 2285–2298. doi: 10.1038/npp.2012.81
Winslow, M. M., Pan, M., Starbuck, M., Gallo, E. M., Deng, L., Karsenty, G., et al. (2006). Calcineurin/NFAT signaling in osteoblasts regulates bone mass. Dev. Cell 10, 771–782. doi: 10.1016/j.devcel.2006.04.006
Wong, T. P., Howland, J. G., Robillard, J. M., Ge, Y., Yu, W., Titterness, A. K., et al. (2007). Hippocampal long-term depression mediates acute stress-induced spatial memory retrieval impairment. Proc. Natl. Acad. Sci. U S A 104, 11471–11476. doi: 10.1073/pnas.0702308104
Worley, P. F., Bhat, R. V., Baraban, J. M., Erickson, C. A., McNaughton, B. L., and Barnes, C. A. (1993). Thresholds for synaptic activation of transcription factors in hippocampus: correlation with long-term enhancement. J. Neurosci. 13, 4776–4786.
Xiong, W., Yang, Y., Cao, J., Wei, H., Liang, C., Yang, S., et al. (2003). The stress experience dependent long-term depression disassociated with stress effect on spatial memory task. Neurosci. Res. 46, 415–421. doi: 10.1016/s0168-0102(03)00120-2
Xu, L., Anwyl, R., and Rowan, M. J. (1997). Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature 387, 497–500. doi: 10.1038/387497a0
Xu, Y., Yue, W., Yao Shugart, Y., Li, S., Cai, L., Li, Q., et al. (2016). Exploring transcription factors-micrornas co-regulation networks in schizophrenia. Schizophr. Bull. 42, 1037–1045. doi: 10.1093/schbul/sbv170
Yamada, K., Gerber, D. J., Iwayama, Y., Ohnishi, T., Ohba, H., Toyota, T., et al. (2007). Genetic analysis of the calcineurin pathway identifies members of the EGR gene family, specifically EGR3, as potential susceptibility candidates in schizophrenia. Proc. Natl. Acad. Sci. U S A 104, 2815–2820. doi: 10.1073/pnas.0610765104
Yamagata, K., Kaufmann, W. E., Lanahan, A., Papapavlou, M., Barnes, C. A., Andreasson, K. I., et al. (1994). Egr3/Pilot, a zinc finger transcription factor, is rapidly regulated by activity in brain neurons and colocalizes with Egr1/zif268. Learn. Mem. 1, 140–152.
Yan, H. Q., Shin, S. S., Ma, X., Li, Y., and Dixon, C. E. (2014). Differential effect of traumatic brain injury on the nuclear factor of activated T Cells C3 and C4 isoforms in the rat hippocampus. Brain Res. 1548, 63–72. doi: 10.1016/j.brainres.2013.12.028
Zeng, H., Chattarji, S., Barbarosie, M., Rondi-Reig, L., Philpot, B. D., Miyakawa, T., et al. (2001). Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell 107, 617–629. doi: 10.1016/s0092-8674(01)00585-2
Keywords: schizophrenia, immediate early gene, long-term depression, EGR3, ARC, NFATC3, EGR1, Nab2
Citation: Marballi KK and Gallitano AL (2018) Immediate Early Genes Anchor a Biological Pathway of Proteins Required for Memory Formation, Long-Term Depression and Risk for Schizophrenia. Front. Behav. Neurosci. 12:23. doi: 10.3389/fnbeh.2018.00023
Received: 30 September 2017; Accepted: 29 January 2018;
Published: 19 February 2018.
Edited by:
Johannes Gräff, École Polytechnique Fédérale de Lausanne, SwitzerlandReviewed by:
Esther Walton, University of Bristol, United KingdomWen-Jun Gao, College of Medicine, Drexel University, United States
Copyright © 2018 Marballi and Gallitano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amelia L. Gallitano, YW1lbGlhQGVtYWlsLmFyaXpvbmEuZWR1