 Angélica Vega-García1
Angélica Vega-García1 Iris Feria-Romero2
Iris Feria-Romero2 Anais García-Juárez3Ana Ch. Munguia-Madera2
Anais García-Juárez3Ana Ch. Munguia-Madera2 Alexia V. Montes-Aparicio2Esli Zequeida-Muñoz4Estefany Garcia-Albavera5
Alexia V. Montes-Aparicio2Esli Zequeida-Muñoz4Estefany Garcia-Albavera5 Sandra Orozco-Suárez2*
Sandra Orozco-Suárez2*- 1Departamento de Fisiología, Facultad de Medicina, Universidad Nacional Autónoma de México, Ciudad de México, Mexico
- 2Unidad de Investigación Médica en Enfermedades Neurológicas, Hospital de Especialidades, “Dr. Bernardo Sepúlveda”, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social, IMSS, Ciudad de México, Mexico
- 3División de Ciencias Biológicas y Ambientales, Centro Universitario de Ciencias Biológicas y Agropecuarias, Universidad de Guadalajara, Guadalajara, Mexico
- 4Facultad de Medicina, Universidad Autónoma de Guerrero, Acapulco, Mexico
- 5Facultad de Biología, Universidad Autónoma del Estado de Morelos, Cuernavaca, Mexico
Neural hyperexcitability in the event of damage during early life, such as hyperthermia, hypoxia, traumatic brain injury, status epilepticus, or a pre-existing neuroinflammatory condition, can promote the process of epileptogenesis, which is defined as the sequence of events that converts a normal circuit into a hyperexcitable circuit and represents the time that occurs between the damaging event and the development of spontaneous seizure activity or the establishment of epilepsy. Epilepsy is the most common neurological disease in the world, characterized by the presence of seizures recurring without apparent provocation. Cannabidiol (CBD), a phytocannabinoid derived from the subspecies Cannabis sativa (CS), is the most studied active ingredient and is currently studied as a therapeutic strategy: it is an anticonvulsant mainly used in children with catastrophic epileptic syndromes and has also been reported to have anti-inflammatory and antioxidant effects, supporting it as a therapeutic strategy with neuroprotective potential. However, the mechanisms by which CBD exerts these effects are not entirely known, and the few studies on acute and chronic models in immature animals have provided contradictory results. Thus, it is difficult to evaluate the therapeutic profile of CBD, as well as the involvement of the endocannabinoid system in epileptogenesis in the immature brain. Therefore, this review focuses on the collection of scientific data in animal models, as well as information from clinical studies on the effects of cannabinoids on epileptogenesis and their anticonvulsant and adverse effects in early life.
Introduction
Seizure disorders are common during childhood; they are causes of morbidity (Glass et al., 2018), and a large percentage of them have a poor response to current first-line anticonvulsant drugs (ADs; Glass et al., 2012). In the postnatal neurodevelopmental period, neuronal excitability is predominantly mediated by the glutamatergic and GABAergic activity system, and it promotes the processes of growth, plasticity, synaptogenesis, and organization of neural networks essential for the adult stage (Ben-Ari, 2002; Ben-Ari and Holmes, 2006; Rakhade and Jensen, 2009). Thus, the immature brain is highly susceptible to developing neuronal hyperexcitability under pathological conditions such as hyperthermia, hypoxia-ischemia, traumatic brain injury (TBI), or a pre-existing neuroinflammatory condition that in turn facilitates the development of seizure activity and the establishment of status epilepticus (SE; Pitkänen et al., 2015; Suchomelova et al., 2015).
Additionally, the early exposure to many ADs, is a significant risk for brain development such as PB, DFH, and valproate, and has been related to developmental disorders (Bittigau et al., 2002; Forcelli et al., 2011; Kaushal et al., 2016; Al-Muhtasib et al., 2018). Furthermore, clinical evidence indicates that gestational exposure to ADs can also lead to deficits in cognitive function (Meador et al., 2012). For this reason, the need to identify new drugs, Cannabidiol (CBD), and its propyl cannabidivarin (CBDV) analog, has aroused interest in the treatment of epilepsy in early life. While CBD was recently approved for the treatment of refractory childhood epilepsies (Abu-Sawwa et al., 2020), little is known about the efficacy and safety of compounds derived from Cannabis sativa (CS) in the early stage of development (Rosenberg et al., 2017). Here we address this issue through a systematic evaluation of the cannabinoid literature investigating multiple therapeutic targets, some of which were tested in early developmental seizure models, including data from our laboratory as well as clinical evidence.
Cannabinoids
CS is an herbaceous plant native to central Asia that is widely distributed in a variety of habitats and altitudes and is a unique species of its kind. Some authors recognize C. ruderalis and C. indica as separate species or subspecies of CS; however, a monospecific criterion has been adopted in many of the nontaxonomic publications, since all the groups of plants that have been included within the genus are interfertile and their morphological diversity shows a diffuse and continuous pattern (Etienne, 2014). This plant has been known for its medicinal and textile uses that date back to more than 5,000 years ago. Currently, a total of 545 constituents of cannabis have been identified, of which 104 are phytocannabinoids classified into 11 types: (−)-Δ9-trans-tetrahydrocannabinol (Δ9 THC) type, (−)-Δ8-trans-tetrahydrocannabinol (Δ8-THC) type, cannabigerol (CBG) type, cannabichrome (CBC) type, cannabidiol (CBD) type, cannabidiol (CBND) type, cannabielsoin (CBE) type, cannabicyclol (CBL) type, cannabinol (CBN) type, cannabitriol (CBT) type, and miscellaneous cannabinoid type. Additional compounds include flavonoids, steroids, phenanthrenes, xanthones, and sugars, among others (Pertwee, 2008; El Sohly et al., 2016). Phytocannabinoids are terpenophenolic products that exhibit a 21- to a 22-carbon skeleton, some of which are breakdown products of other cannabinoids. The predominant compounds in the plant are tetrahydrocannabinol acid (THCA), cannabidiolic acid (CBDA), and cannabinolic acid (CBNA), followed by cannabigerolic acid (CBGA), cannabichromenic acid (CBCA), and cannabinodiolic acid (CBNDA). Although THCA is the most important compound in drug-type cannabis and CBDA in fiber-type cannabis, it is also worth noting that CBCA predominates in young plants and decreases with maturation (Andre et al., 2016). Nevertheless, CS is not the only natural source of cannabinoids, as, in the Radula and Helichrysum gender, the presence of cannabinoid-type terpenophenolics has also been reported; however, little is known about these compounds (Mahmoud and Waseem, 2014; Andre et al., 2016). Currently, the pharmacological actions of the psychotropic cannabinoids Δ9-THC, Δ8-THC, CBN, and Δ9-THCV and the non-psychotropic phytocannabinoids CBC, CBD, CBDA, CBG, THCA, THCVA, CBGA, CBDV and CBGV have been well documented. Also, pharmacological actions of the nonphytocannabinoid component (E)-β-caryophyllene, which is a sesquiterpene, have also been described (Mechoulam and Gaoni, 1965; Pertwee, 2008; Mechoulam and Parker, 2013; Pertwee and Cascio, 2014).
Endocannabinoid System
The endocannabinoid system (eCBs) was discovered by Mechoulam and Gaoni (1965), who isolated the active compound (Δ9-THC); in 1990, they discovered the binding site of this cannabinoid, oriented their search for additional receptors, and described the abundant CB1 receptor (CB1R) in the central nervous system (CNS) with a similar density to the GABA and glutamate receptors. Years later, the CB2 receptor (CB2R), abundant in tissues of the immune system, was described, followed by the functioning of its endogenous ligands, the endocannabinoids 2-arachidonoyl glycerol (2-AG) and N-arachidonoyl ethanolamide or anandamide (AEA; Andre et al., 2016). Similarly, how the endocannabinoids 2-AG and anandamide or AEA are synthesized from fatty acids from the remodeling of the cell lipid membrane and are produced according to the individual’s bodily demands was described shortly thereafter (Di Marzo and Piscitelli, 2015). Anandamide is produced by the action of the enzyme N-acyltransferase (NAT), which produces N-arachidonoyl phosphatidylethanolamine (NArPE). Phospholipase D generates a family of compounds, the arachidonoyl glycerol ethanolamine (FAE) family, which includes 2-AG and anandamide. When these compounds are released, their degradation occurs through metabolism by enzymatic hydrolysis, where fatty acid amide hydrolase (FAAH) intervenes in the degradation of anandamide and monoacylglycerol lipase (MAGL). Finally, 2-AG is metabolized into glycerol and arachidonic acid during postsynaptic cell recapture. Anandamide and 2-AG can also bind to plasma-circulating albumin and have distant effects (Patel et al., 2017). NAT is regulated by calcium and cAMP cyclic adenosine monophosphate and is selectively stimulated by cellular depolarization or by the action of the metabotropic receptors for glutamic acid, dopamine, or acetylcholine (Lu and Mackie, 2016; Patel et al., 2017).
The abovementioned endocannabinoids act as agonists of these receptors and are G protein-coupled to the endocannabinoid CB1R and CB2R. G proteins inhibit adenylate cyclase activation by reducing cAMP concentrations and interfering with the activity of cAMP-dependent protein kinase (PKA). The same G protein activates mitogen activation-dependent protein kinase (MAPK); both PKA and MAPK are involved in the selective expression of genes. The activation of the CB1R and CB2R inhibits L-, N-, P-, and Q-type voltage-dependent Ca2+ channels, which reduces the entry of Ca2+ and stimulates the endogenous K+ rectifier channels at the neuronal level by allowing the release of ions, resulting in hyperpolarization. Interactions with other receptors, such as GPR55, GPCR, and TRPV1 (in this case acting as an antagonist), have also been described, in which the endocannabinoids act as neuromodulators (Castillo et al., 2012; Alexandre et al., 2020; Figure 1). The CB1R is expressed throughout the CNS, particularly in the hippocampus on mossy fibers of the granule cells of the dentate gyrus (DG), wherein the mature stage, eCBs regulate the efficiency of inhibitory synapses during periods of sustained depolarization at the postsynaptic level through increased intracellular Ca2+, which activates the synthesis of endocannabinoids, mainly in pyramidal neurons of the hippocampus (Herkenham et al., 1990, 1991; Katona et al., 1999, 2006). These eCB are fat-soluble, which allows their diffusion through the plasma membrane as well as the synaptic cleft in a retrograde manner towards the neighboring presynaptic terminals, favoring the inhibitory activity mediated by the GABAergic system (Katona et al., 1999, 2006; Kawamura et al., 2006; Monory et al., 2006).
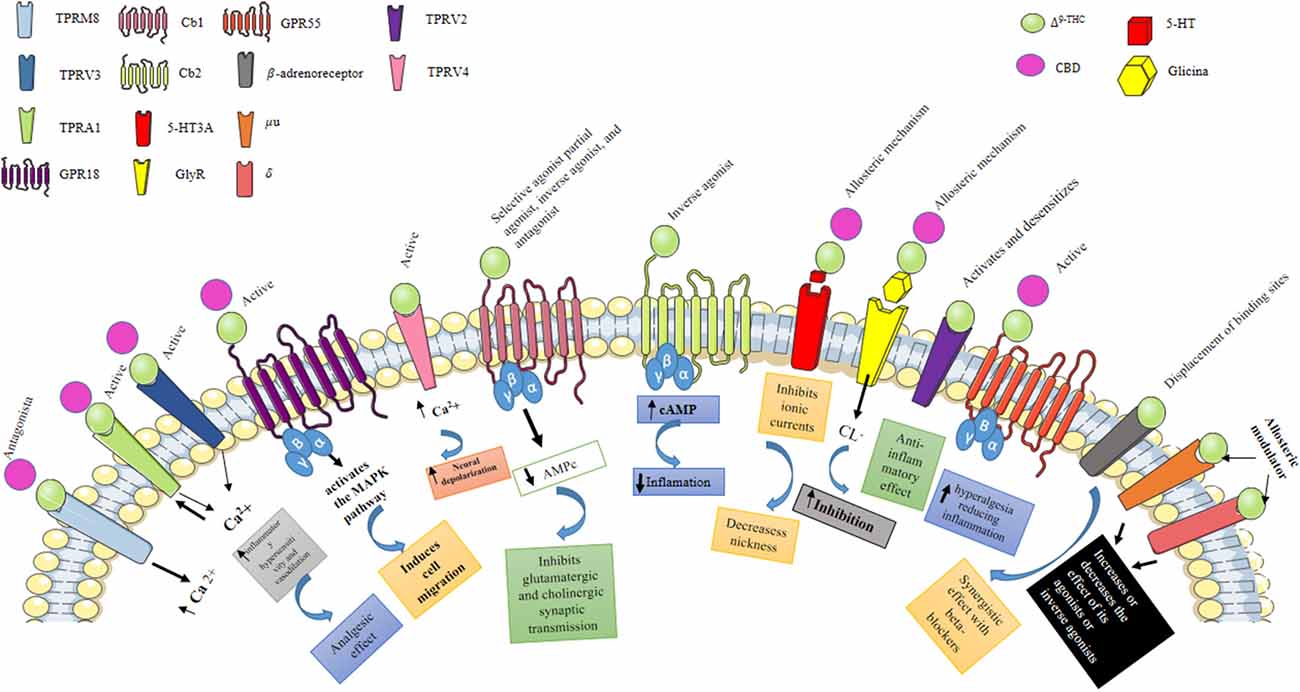
Figure 1. Pharmacological effects of Δ9-THC and CBD. Δ9-THC is the main psychoactive component of C. sativa, which can behave as a selective agonist, partial agonist, inverse agonist, and antagonist of the Cb1 receptor, while when activating the Cb2 receptor it behaves as an inverse agonist. Activation of the Cb1 and Cb2 receptors stimulates GTPγS binding to cell membranes and inhibits cyclic AMP production. Also, Δ9-THC can inhibit 5HT3A receptor-mediated currents induced by 5-hydroxytryptamine (5HT); antagonizing receptor activation, possibly through an allosteric mechanism, by this same mechanism, Δ9-THC and CBD can enhance the activation of GlyR expressed in central tegmental area (ATV) neurons. Additionally, Δ9-THC activates the TRPV3 and TRPV4 receptors, which are nonselective calcium-permeable cation channels that, when activated, raise intracellular Ca2+ and consequently cause neuronal depolarization. Transient receptor potential (TRP) channels are a group of membrane proteins involved in the transduction of a large number of stimuli. Unlike Δ9-THC, CBD does not activate CB1 and CB2 receptors, which likely accounts for its lack of psychotropic activity. However, CBD interacts with many other, nonendocannabinoid signaling systems. It is a “multi-target” drug. At low micromolar to submicromolar concentrations, CBD is a blocker of the equilibrative nucleoside transporter (ENT), the orphan G-protein-coupled receptor GPR55, and the TRP of melastatin type 8 (TRPM8) channel. At higher micromolar concentrations, CBD activates the TRP of vanilloid type 1 (TRPV1) and 2 (TRPV2) channels while also inhibiting cellular uptake and fatty acid amide hydrolase—catalyzed degradation of anandamide.
Endocannabinoid System in Postnatal Neurodevelopment
It has been shown that during critical periods of neurodevelopment, eCBs participate in the maturation of corticolimbic circuits, where excitatory neuronal activity is essential for the processes of synaptogenesis and neuronal plasticity (Harkany et al., 2008), directed by the glutaminergic system, which constitutes most synapses; likewise, ion channels and glutamate transporters and receptors are expressed at levels that promote the activity of excitatory networks (Sanchez et al., 2001; Jensen, 2009). In rodents, during the first week, from 0 to 7 post natal (PN) days, a period analogous to a 36-week, premature human, the GABAergic system participates in excitatory neurotransmission, mediated by an increase in the K+/Cl cotransporter NKCC1 and downregulation of KCC2. The neuronal depolarization is generated by intracellular Cl− increases and activates L-type voltage-sensitive Ca2+ channels (L-VSCC) and NMDARs, which increases the flux of intracellular Ca2+ and activates signaling cascades directed at intracellular trophic activity, neuritic growth, neural network formation, and synaptogenesis (Ben-Ari, 2002, 2014; Galanopoulou, 2008). During this period of neurodevelopment, CB1R is located in the terminals of mossy fibers in the CA3 subfield of the hippocampus, mainly in GABAergic and cholecystokinin (CCK) circuits, reaching a maximum concentration at the day of 4 PN, where CB1Rs are coupled to G proteins. In these terminals, CB1Rs suppresses the release of GABA, reducing the excitatory activity of GABA and exerting a role as a regulator of excitatory neuronal activity important for the development of neuronal circuits (Bernard et al., 2005; Fernández-Ruiz et al., 2005).
The eCBs modulate neuronal activity by regulating retrograde signaling, which participates in short-term synaptic plasticity, also known as suppression of depolarization-induced inhibition (DSI) and excitation (DSE). It is estimated that eCB levels, specifically those of AEA during development, determine the direction of synaptic plasticity (Kreitzer and Regehr, 2001; Castillo et al., 2012). From PN day 8, GABA begins to exert its inhibitory activity due to the increase in expression of the KCC2 cotransporter, which decreases intracellular Cl− levels, favoring neuronal hyperpolarization and the inhibitory activity of eCBs (Ben-Ari, 2002, 2014; Galanopoulou, 2008). Then, from PN days 10–14 in the rat, which corresponds to the first 36 months of human life, GluRs are regulated by glia, and ionotropic glutamate receptors (iGluRs) are linked to ion channels that allow the flow of sodium (Na+), potassium (K+) and Ca2+ ions to different degrees depending on the receptor subunit. The main GluRs are NMDARs, AMPARs, and KARs (Benítez-Diaz et al., 2003; Herlenius and Lagercrantz, 2004; Rakhade and Jensen, 2009). During this period, NMDARs are expressed differently in the brain, with an increase in the expression of NR2B and NR3A subunits, which are not very sensitive to Mg+2 ions, which increases the activation of the receptor and increases intracellular Ca2+ flux, thereby promoting neuronal hyperexcitability and long-term potentiation (LTP; Ben-Ari, 2002, 2014; Qu et al., 2003; Coulter, 2006).
The NMDARs distributed in the immature brain are mainly found in the hippocampus at the postsynaptic level, but some are also present at the presynaptic level and in astrocytes (Lee et al., 2010; Szczurowska and Mareš, 2013; Skowrońska et al., 2019). The glutamate receptors AMPARs and KARs are permeable to Ca2+ ions when GluR2 expression is relatively low or absent (Romjin et al., 1991; Tyzio et al., 1999; Jensen, 2009); KARs are also permeable to Ca2+ when the GluR5 and GluR6 subunits are absent. Additionally, metabotropic glutamate receptors (mGluRs) are coupled to a G protein (GTP) and mediate slow synaptic responses (Jensen, 2009; Ben-Ari, 2014), which favor activation of CB1R expressed in glutamatergic neurons to modulate excitatory and inhibitory neurotransmission. This process is essential in the remodeling of inhibitory circuits during adolescence, a period from PN day 30 in rodents, where the regulation of glutamatergic and GABAergic activity are of vital importance for the formation, maturity, and elimination of synapses, mainly at the level of the prefrontal cortex (PFC), which has greater activity compared with the adult stage, a period from PN day 60 in rodents (Clancy et al., 2001; Auvin and Dupuis, 2014; Dow-Edwards and Silva, 2017). During adolescence, eCBs participate in the formation of neural networks in the prefrontal cortex and maintain the interaction between the amygdala, hippocampus, and hypothalamus, which is responsible for cognitive and emotional development since endocannabinoids are responsible for the normal response to stress and the regulation of neuronal excitation and inhibition (Schonhofen et al., 2018; Cheung et al., 2019).
The expression of CB2R in cells of the immune system at the peripheral level has been reported in CD4+, CD8+ T and B lymphocytes, natural killer cells, monocytes, and polymorphonuclear neutrophils (Atwood and Mackie, 2010). CB2R is also expressed in the microglia, which during postnatal neurodevelopment maintain amoeboid forms that allow them to actively participate in the phagocytosis of apoptotic cell debris, in the induction of apoptosis in other cells during the formation of functional neural circuits, and the removal of residual myelin, as well as in the formation and expansion of neural networks that imply the pruning of synapses at the level of the cerebral cortex, hippocampus, cerebellum, and amygdala (Berdyshev, 2000; Bessis et al., 2007; Atwood and Mackie, 2010; Onaivi et al., 2012). In adult rats, however, the expression of CB2R decreases as the microglia become inactive (Stella, 2004; Fernández-Ruiz et al., 2005; Caiati et al., 2012). Additionally, the expression of CB2R has been reported in glutamatergic and GABAergic neurons in the DG and CA1 subfield of the hippocampus at the level of the pyramidal stratum and stratum radiatum to a lesser degree than CB1R, which is observed mainly in the cerebral cortex, hippocampus, amygdala, and cerebellum (Brusco et al., 2008; Onaivi et al., 2012).
Astrocytes and microglia are responsible for the surveillance and modulation of the immune response and are the main cytokine producers in the CNS (Baud and Saint-Faust, 2019). Here, CB2R has been reported to participate as a neuromodulator of the neuroactive molecules ON, glutamate, PGs, and neurotrophins in the glia and vascular endothelium, which participate in neurodevelopmental processes, homeostatic mechanisms such as sleep, and activation of signaling pathways involved in neuronal plasticity and synaptogenesis; however, the mechanisms responsible for its participation in eCBs during postnatal neurodevelopmental processes are not yet entirely clear (Devinsky et al., 2013; Schonhofen et al., 2018; Cheung et al., 2019).
Endocannabinoid System in Epileptogenesis in the Immature Brain
The term epileptogenesis is defined as a sequence of events that convert a normal circuit into a hyperexcitable circuit (Pitkänen et al., 2015). Epileptogenesis refers to the development of tissue capable of generating spontaneous seizures, which result in an epileptic condition or in the progression of established epilepsy (Pitkänen and Engel, 2014), whereby it is a continuous and multifactorial process in which the eCBs participate in the modulation of neuronal activity, neuronal migration, axonal growth and guidance, synaptic plasticity, and the neuroinflammatory response (Schonhofen et al., 2018; Cheung et al., 2019). It has been established that damage to eCB signaling in the early stages of neurodevelopment can favor the process of epileptogenesis and the establishment of epilepsy in later stages (Figure 2).
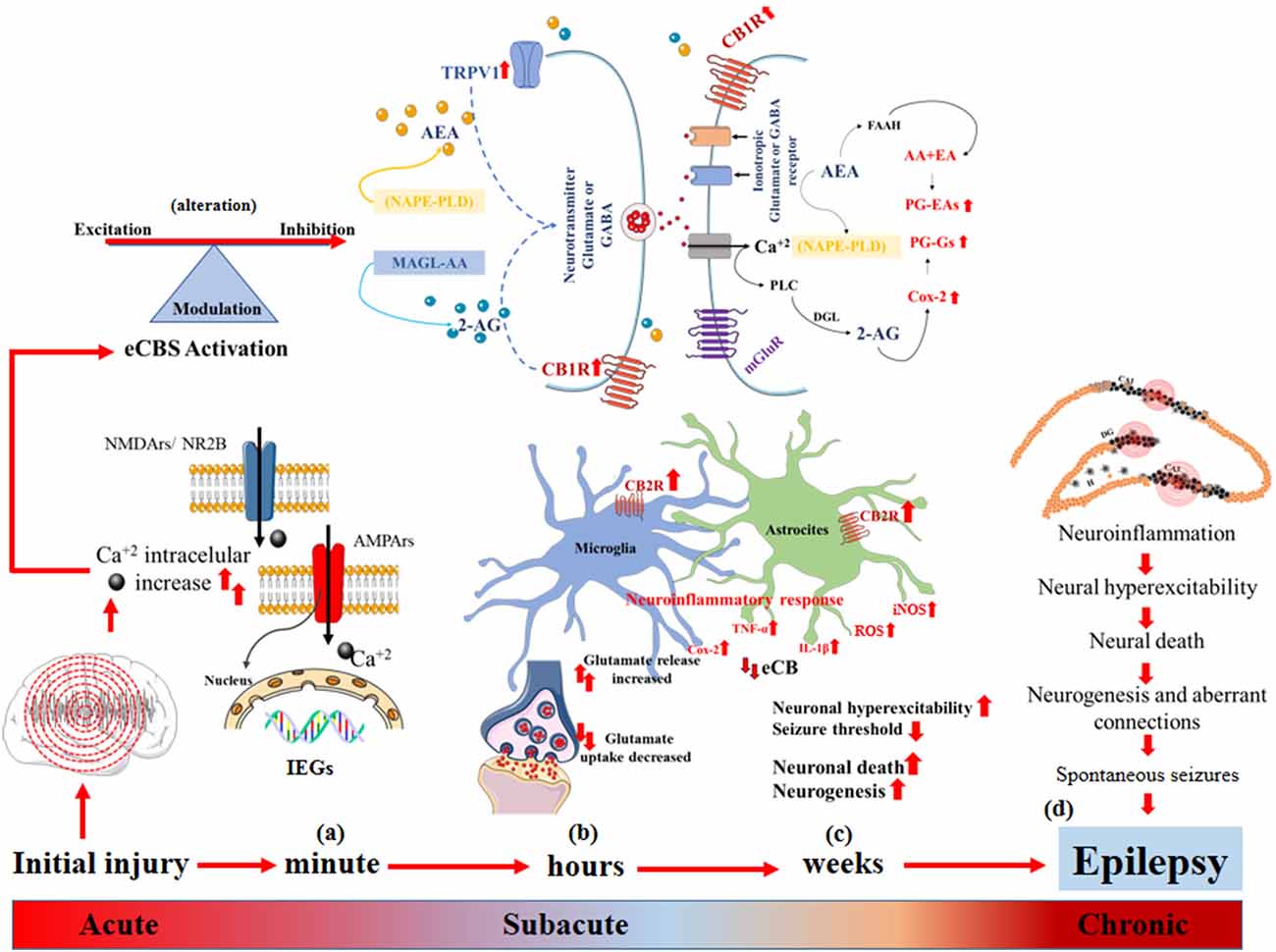
Figure 2. Epileptogenesis and endocannabinoid system (eCBs). (A) Acute changes: increased intracellular Ca2+ flux, induction of early genes (IEGs) that alter synaptic function, decreased threshold to neuronal hyperexcitability, alteration of eCBs that modulates the balance between excitatory and inhibitory neurotransmission. (B) Sub-acute changes: synthesis of endocannabinoids (eCB) mediated by the increase in intracellular Ca2+, neuronal hyperexcitability and the demand for membrane phospholipids diacylglycerol lipase (DAGL) and N-acyl phosphatidyl ethanolamine-hydrolyzing phospholipase D (NAPE-PLD) to form 2-AG and AEA, respectively, and their degradation after the dissociation of CB1R and the activation of TRPTV1 by AEA, which triggers greater glutamate release and increases in intracellular Ca2+. The eCBs degrade rapidly, and 2-AG and AEA are catabolized by the enzymes monoacylglycerol lipase (MAGL) and fatty acid amide hydrolase (FAAH), respectively, which generate AA, PGs-Eas, PG-Gs, and Cox-2 to increase convulsive susceptibility, activation of astrocytes and microglia and increases in CB2R that promote the release of pro-inflammatory proteins IL1-β, Cox-2, TNF-α and iNOS, generating neuroinflammation, neuronal hyperexcitability, and neuronal death. (C,D) Chronic changes: neuronal death, the perpetuation of the neuroinflammatory response and dysregulation of eCBs that promote neurogenesis, formation of aberrant connections, expression of spontaneous seizures, and epilepsy.
During the latent period, the participation of eCBs has been described in different cascades of events defined as acute, subacute, and chronic (Kadam et al., 2010). The induction of early genes (IEGs), including Fos, Jun, Egr4, Egr1, Homer 1, Nurr77, and Arc, occurs during acute changes as a result of intense and repeated synaptic activity during seizure activity, which promotes increased intracellular Ca2+ flux by NMDARs (Herdegen and Leah, 1998). Upregulation of IEGs can pathologically modulate synaptic function, lowering the threshold to neuronal hyperexcitability (Rakhade and Jensen, 2009). The levels of intracellular Ca2+ immediately alters synaptic efficiency mechanisms, such as increasing the postsynaptic density and dendritic spines, grouping NMDARs and AMPARs, and increasing glutamate synthesis (Haglid et al., 1994; Sanchez et al., 2001), where the participation of eCBs plays an important pathophysiological role in modifying excitatory and inhibitory synaptic neurotransmission in the brain (Rosenberg et al., 2017). During postnatal neurodevelopment, CB1R located in presynaptic cells, mainly in the granule cells of the hippocampus, regulates excitatory activity; additionally, endocannabinoids can also act through interactions with other types of receptors, mainly on G protein-coupled receptors, such as GPRSS and the Transient receptor potential (TRP) cation channel subfamily V member 1" or “vanilloid receptor 1” (TRPV1), which participate in neuronal hyperexcitability, favoring intracellular Ca2+ flux (Bhaskaran and Smith, 2010; Schonhofen et al., 2018; Cheung et al., 2019).
As we have indicated, the synthesis of endocannabinoids is mediated by an increase in intracellular Ca2+ generated by neuronal hyperexcitability and the demand for membrane phospholipid diacylglycerol lipase (DAGL) and for glutamate and acetylcholine (Wallace et al., 2003; Rosenberg et al., 2017). The eCBs passively diffuse through presynaptic cells in a retrograde manner to bind orthosterically to and activate CBR1 acylphosphatidyl ethanolamine-hydrolyzing phospholipase D (NAPE-PLD) to form 2-AG and AEA, in the same way, endocannabinoids are also synthesized from the activation of metabotropic receptors and thus inhibit the release of glutamate or GABA, a process defined as depolarization-induced suppression of inhibition (DSI) or excitation (DSE), respectively (Armstrong et al., 2009). In models of febrile seizures at PN day 10, alterations to these DSI and DSE mechanisms have been reported, which are estimated to interfere with the maturation of the GABAergic system and persist in later stages through the generation of hyperexcitable circuits (Bernard et al., 2005).
Similarly, it has been reported that eCB signaling has an important role in epileptogenesis, because, after the dissociation from CB1R, eCB 2-AG and AEA are catabolized by the enzymes monoacylglycerol lipase (MAGL) and alpha-beta hydrolase domain containing 6 (CABHD6) or through fatty acid amide hydrolase (FAAH), respectively, and activation of TRPV1 by AEA can trigger increased glutamate release after intracellular Ca2+ concentrations are increased (Wallace et al., 2003; Rosenberg et al., 2017). This process activates calcineurin (PPP3C or serine-threonine protein phosphatase 2B), which, during intense seizure activity, activates calcium-calmodulin-phosphatase to promote the dephosphorylation and subsequent endocytosis of GABAA (Blair et al., 2004). These phenomena reduce the inhibitory potential and lead to the gradual loss of GABAergic inhibitory networks (Kurz et al., 2001; Blair et al., 2004; Rakhade and Jensen, 2009; Semple et al., 2020). In the immature brain, calcineurin activation increases the phosphorylation of Kv2.1 ion channels (also known as KCNB1) and promotes their expression in the postsynaptic membrane, allowing prolonged neuronal depolarization (Kurz et al., 2001; Rakhade and Jensen, 2009).
Similarly, protein kinase C, which is dependent on protein kinase type II and calcium-calmodulin-phosphatase, increases within minutes after the induction of SE in immature rats, which allows an increase in the phosphorylation of serine 831 of GluR1 and serine 880 of GluR2. This process promotes the endocytosis of GluR2 and increases the permeability to Ca2+ in AMPARs, which generates an increase in seizure susceptibility in later or adult stages (Rice and De Lorenzo, 1998; Sanchez et al., 2001; Rakhade and Jensen, 2009). Additionally, it has been reported that astrocyte activation, immediate to neuronal damage, increases extracellular K+ concentrations, which in turn facilitates continuous neuronal hyperexcitability, a triggering factor for epileptogenesis (Jabs et al., 1997; Jensen, 2009).
Subacute changes in the latent period are established from hours to days after neuronal damage (Figure 2), in which a rapid increase occurs in NGF and BDNF, which together with Trk alter the modulation of the maturation of the KCC2 cotransporter, promoting GABA-mediated inhibitory activity mainly in DG granular cells, which in turn alters the modulating function of eCBs on the GABAergic system to favor long-term neuronal hyperexcitability (Chen et al., 2003, 2007). However, the cellular mechanisms by which CB1R and CB2R act on short- and long-term changes in the seizure threshold are still unknown, particularly in mesiotemporal structures consisting of mainly the hippocampus and the cerebral cortex in the immature brain (Chen et al., 2007).
The neuroinflammatory response activated by microglia and astrocytes, the main response responsible for the synthesis and release of pro-inflammatory cytokines such as IL1-β, activates the IL-1/TLR signaling pathway, where an increase in pro-inflammatory proteins occur. These proteins include Cox-2, an enzyme that catalyzes the production of PGs from AA and increases seizure susceptibility (Chen et al., 2001, 2007; Jiang et al., 2004; Feng et al., 2016), and TNF-α, which generates an increase in the expression of AMPAR and a decrease in GluR2 through the expression of TNF-R1 on the neuronal membrane surface, which in turn causes increased permeability of immature neurons to Ca2+ (Beattie et al., 2002; Stellwagen et al., 2005). The activity of TNF-α has been shown to result in the endocytosis of GABAA receptors, mainly their β 2/3 subunits, in the pyramidal neurons of CA1 and interneurons of the DG, promoting neuronal excitation (Wang et al., 2000; Balosso et al., 2005; Stellwagen et al., 2005; Shimada et al., 2014) and leading to neurodegeneration and neuronal death in the CA1 and CA3 regions of the immature hippocampus (Rizzi et al., 2003; Kawaguchi et al., 2005).
In the same way, the rapid increase in Cox-2 mRNA expression after SE induced by KA itself induces neurodegeneration and neuronal death in the immature hippocampus, mainly the CA1 and CA3 subfields, at 12 and 24 h post SE (De Simoni et al., 2000; Rizzi et al., 2003; Kawaguchi et al., 2005). This finding supports that the neuroinflammatory response precedes neuronal damage (Ravizza et al., 2005; Joseph and Levine, 2006), and this neuronal damage is associated with an increase in Cox-2 and the subsequent production of PGs and ROS, activation of the oxidative stress mechanism, and mitochondrial dysfunction (Gobbo and O’Mara, 2004); thus, it has been reported that the eCBs participate as a substrate of the Cox-2 enzyme for the synthesis of PGs (Nomura et al., 2011; Ruhaak et al., 2011). Similarly, experimental models of SE induced by KA and Li-Pilo in PN 12 day rats have reported a rapid increase in ROS and mitochondrial markers of oxidative damage, including NT-3, 4-HNE and carbonylated proteins, as well as the formation of free radicals 24 h post-SE, which are responsible for neurodegeneration in the subfields of CA1, CA3 and DG of the hippocampus, cerebral cortex and thalamus (Patel and Li, 2003; Folbergrová et al., 2010).
The contribution of mitochondrial dysfunction on complex I of the respiratory chain NADH (Cock, 2002; Kudin et al., 2002) favor the development of neurodegenerative disorders, such as epilepsy, due to the excessive release of glutamate, which causes an alteration in the reuptake mechanisms of this neurotransmitter and an increase in intracellular Ca2+ due to the saturation of the regulatory mechanisms mediated by the Na+/Ca2+ exchanger and the Ca2+ buffer proteins (Tretter et al., 2004).This causes the mitochondria to capture the excess Ca2+ in the mitochondrial matrix, which induces the depolarization of the membrane by the partial inhibition of the chemiosmotic potential and by the accumulation of positive charges in the mitochondrial matrix (Tretter et al., 2004). This sustained overload produces an irreversible depolarization through the activation of the mitochondrial transition pore, a pathway through which Ca2+ returns to the cytosol, causing the collapse of the mitochondrial chemiosmotic potential and a reduction in the synthesis of ATP (Murchison and Griffith, 2000). The decrease in ATP generates metabolic dysfunction, ROS production, activation of proteases, phospholipases, iNOS, and endonucleases, and inhibition of protein synthesis. Furthermore, NO can act as a retrograde messenger, enhancing the excitotoxic effect of glutamate and increasing its release from presynaptic terminals (Lorigados et al., 2013). Likewise, it has been reported that complex I dysfunction may also be the result of an increase in carbonylated proteins during the subacute phase post-SE induced by Li-Pilo and that this alteration persists up to 5 weeks, a period in which spontaneous convulsive activity is observed (Folbergrová et al., 2010).
Likewise, Cox-2 and TNF-α act directly on receptors and ion channels and indirectly by modulating extracellular glutamate reuptake systems via the reduction of selective transporters to glutamate GLT-1, mediated by astrocytes, mainly in CA1 and DG of the hippocampus. Additionally, the activation of iNOS, which promotes the synthesis of NO, increases the release of glutamate and substance P, which preserves the activation of cytokines together with the increase in the affinity of NMDARs and AMPARs, which supports the finding that the activation of the neuroinflammatory response by microglia and astrocytes increases the vulnerability of the immature hippocampus to neuronal hyperexcitability and that these changes depend on the age of development (De Simoni et al., 2000; Rizzi et al., 2003; Viviani et al., 2003; Ravizza et al., 2005; Stellwagen et al., 2005; Bessis et al., 2007; Chen et al., 2013). The participation of CB2R, which is overexpressed by microglia in the neuroinflammatory response in response to neuronal damage and the infiltration of cells of the immune system into the brain parenchyma (Sagredo et al., 2009; Bouchard et al., 2012), induces chronic CB2R activation, which increases excitatory neurotransmission (Li and Kim, 2015) and decreases inhibitory neurotransmission (Morgan et al., 2009). The CB2R, which are mostly expressed in microglia, have been reported to participate in the neuroinflammatory response in experimental models in rats, where they increase 2-AG and CB1R in the hippocampus after SE (Wallace et al., 2003); however, the increase in 2-AG in the seizure model in mice shows a decrease in seizure activity (Sugaya et al., 2016).
Chronic changes (Figure 2) can be observed over 2–12 weeks in rodents and from months to years in humans. It has been reported that during the first 2 weeks of life, the resistance to excitotoxic damage in the immature brain is relative since the amount of Ca2+ that enters a pyramidal neuron is directly related to the age of PN development, where in the first 3 days of life, glutamate minimally increases intracellular Ca2+. Conversely, between PN days 10 and 25, intracellular Ca2+ increases markedly due to recurrent seizure activity, leading to neuronal death in the CA1 subfield of the hippocampus, the area most vulnerable to excitotoxic damage, due to an increase in NMDARs and AMPARs (Kubová et al., 2001, 2012; Kubová and Mareš, 2013). Additionally, an alteration in the immature glutamatergic system that results in the overexpression of GluR2 and its activity on the intracellular Ca2+ flux through AMPARs and KARs, a condition that pathologically persists during the adolescent stage mainly in the prefrontal cortex, favors the formation of hyperexcitable circuits. Similarly, prolonged positive regulation has been reported in the expression of the CB1R and in the suppression of the inhibition induced by depolarization, which can cause alterations in neuronal hyperexcitability throughout life (Chen et al., 2003; Bernard et al., 2005).
Anticonvulsant and Neuroprotective Effect of CBD on Experimental Models of Status Epilepticus and Epilepsy
It has been reported that CBD has a low affinity for CB1R and CB2, and at high levels, it can act as an indirect CB1R antagonist as evaluated by a wide range of experimental models of seizures and epilepsy in adult rats. The evaluation of high doses of CBD, up to 300 mg/kg i.p., in the hippocampal kindling model reveals a reduction in amplitude of the discharge and an increase in the after-discharge threshold (ADT; Turkanis et al., 1979; Ghovanloo et al., 2018). However, in the lamotrigine drug-resistant tonsillar kindling model, CBD does not show anticonvulsant or neuroprotective effects (Klein et al., 2017). Though, in both models, potential neuroprotective effects were identified at low vs. high doses of CBD, which increased the neurotoxic effects of this active ingredient (Patra et al., 2019). Conversely, no anticonvulsant effects of repeated CBD administration have been reported (Rosenberg et al., 2017). The neuroprotective effects of CBD have been identified when administered after SE induced by intrahippocampal pilocarpine microinjection and in in vitro models in rat hippocampal slices, in which CBD decreases the amplitude and duration of the epileptiform activity induced by 4-aminopyridine (Jones et al., 2010; Franco and Perucca, 2019). However, despite the experimental existence of the anticonvulsant effects of CBD, the responsible mechanisms remain unclear, and few studies have examined its anticonvulsant effect in the immature stage (Table 1).

Table 1. Effect of cannabinoids in experimental models of epileptic seizures.
Therefore, we decided to carry out a pilot study to evaluate the anticonvulsant effect of CBD for 12 days PN on the model of SE induced by KA 3 mg/kg delivered via an intraperitoneal route, which showed that the oral route of low doses (20 and 25 mg/kg) of CBD 30 min before KA caused behavioral arrest; however, administration of higher doses (30 and 35 mg/kg) of CBD resulted in changes in motor behavior such as startles, wet dog shakes and lower limb myoclonus. During the evaluation of the anticonvulsant effect, compared with the KA group, the 20 and 25 mg/kg CBD groups showed a significant increase in SE initiation latency (p < 0.0001), while the 30 and 35 mg/kg CBD groups showed significant reductions in SE initiation latency (p < 0.01 and p < 0.001, respectively). Conversely, in the high-dose groups of 40, 60, 80, and 90 mg/kg CBD administered 30 min before KA, chewing, wet dog shaking, and unilateral myoclonus as well as scratching movements with the lower extremities were observed. Although CBD induced these behaviors, the anticonvulsant effect in the 40 and 60 mg/kg groups was efficient in significantly reducing the SE initiation latency (p < 0.01) relative to the KA group; however, in the 80 and 100 mg/kg of CBD groups, an increase in the severity of SE was observed (Figure 3). Motor behavior has been previously reported in experimental models of seizures and KA-induced SE in 10–14-day-old immature PN rats (Vega-García et al., 2020), which indicates that in SE models, an increase in the dose has opposite effects to those in adults (Jones et al., 2010; Hill et al., 2014; Klein et al., 2017). Agreeing with this, Anderson et al. (2020) in the Scn1a+/– mouse model of Dravet syndrome, detected that subchronic oral administration of Δ9-THC or CBD alone did not affect spontaneous seizure frequency or mortality while, surprisingly, their co-administration (70 mg Δ9-THC + 3,500 mg CBD/kg chow) increased the severity of spontaneous seizures and overall mortality. The authors point out the detrimental outcome might simply be explained by a significant pharmacokinetic interaction between CBD and Δ9-THC, as plasma and brain concentrations of both Δ9-THC and CBD were dramatically increased, which has been demonstrated in mice and rats in other epilepsy models (Sofia et al., 1976; Chan et al., 1996).
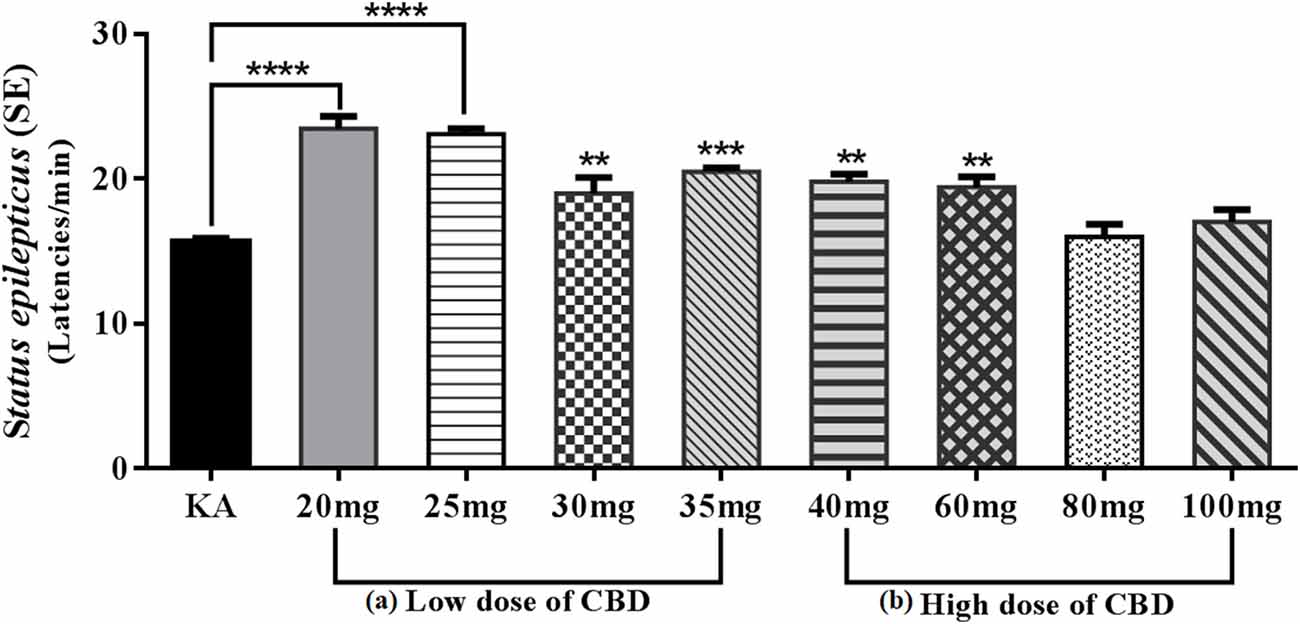
Figure 3. Graph showing the mean ± SE of the latencies of Status epilepticus (SE) induced by KA. (A) Low doses: the 20 and 25 mg/kg CBD groups showed an increase of SE latencies with a significant difference ****p < 0.0001 compared with the KA group. However, the 30 and 35 mg/kg groups of CBD showed a reduction in SE latencies though with a significant difference, **p < 0.01 and ***p < 0.001, respectively, compared with the KA group. (B) The high-dose 40 and 60 mg/kg groups of CBD showed an increase in the latencies of SE with a significant difference, **p < 0.01 compared with the KA group. The 80 and 100 mg/kg CBD groups did not show significant differences compared with the KA group. One-way ANOVA followed by Bonferroni’s post hoc test, p < 0.05 (unpublished data).
Additionally, the anticonvulsant effect was examined in a Dravet syndrome model in voltage-gated sodium channel Nav1-knockout mice. Treatment with CBD from PN days 21–27 decreased the severity and number of seizures induced by hyperthermia as well as the number of spontaneous seizures (Kaplan et al., 2017). However, the mechanisms responsible for the anticonvulsant effect of CBD remain unclear. Previous experimental studies have reported that the anticonvulsant effect of CBD is linked to the modulating effects on neuronal hyperexcitability through the antagonism of G protein-coupled receptor 55 (GPR55), of which CBD is a negative allosteric modulator (Laprairie et al., 2015), desensitization of TRPV1 (Chen and Hackos, 2015) and inhibition of adenosine reuptake by blocking equilibrative nucleotide transporter 1 (ENT1), which increases the concentration of extracellular adenosine (Pandolfo et al., 2011). These mechanisms decrease intracellular Ca2+ flux and regulate neuronal hyperexcitability. These effects have also been tested in GPR55- and TRPV1-knockout mice (Klein et al., 2017; Stott et al., 2018; Franco and Perucca, 2019).
Moreover, the activity of CBD has been identified in blocking voltage-gated sodium channels and T-type Ca2+ channels and in the modulation of VDAC1 (Ghovanloo et al., 2018). Similarly, the interaction of CBD with voltage-gated potassium channels has been shown to have an effect on glycine α1 and α3 receptors and on the modulation of TNF-α (Gaston and Friedman, 2017; Gaston and Szafarski, 2018). It has been reported that CBD modulates the activity of the immune system in a concentration-dependent manner (Pertwee, 2008), showing that the anti-inflammatory, antioxidant and neuroprotective effects of CBD are attributed to its agonist activity on CB1R and CB2R (Ibeas et al., 2015). Also, CBD has been shown to act as an inhibitor of fatty acid amide hydrolase (FAAH), an enzyme involved in the degradation of eCB (Capasso et al., 2008). Likewise, CBD acts as a competitive inhibitor of adenosine uptake via ENT1 in microglia and increases exogenous adenosine, which activates the A2A receptor (Liou et al., 2008; Haskó et al., 2009); nevertheless, the mechanism of action of the A2A receptor on the modulation of the inflammatory response activated by microglia is unclear (Haskó et al., 2009; Rosenberg et al., 2017).
Pretreatment with CBD attenuates the formation of ROS and the mitochondrial damage generated by the increase in glutamate. Similarly, in models of artery sclerosis, treatment with CBD inhibits the pro-inflammatory pathway of NF-kB and decreases nitrotyrosine and iNOS, while in the endothelial cells of human coronary arteries, it decreases adhesion molecules ICAM-1 and VECAM-1. In the same way, CBD attenuates the transendothelial migration of monocytes (Rajesh et al., 2007). The antioxidant effect of CBD has been reported in a model of nephropathy induced by cisplatin, reducing ROS and generating NAPDH oxidases, iNOS and NT-3, which reduce cell death and improve kidney function (Pan et al., 2009). Therefore, it is suggested that the antioxidant effect of CBD acts at the mitochondrial level (Booz, 2011). Additionally, CBD has been reported to act as an antagonist of the GPR55 receptor, which decreases intracellular Ca2+ flux and modulates neuronal hyperexcitability (Rosenberg et al., 2017).
There are few reports on the neuroprotective effects of CBD in developing epilepsy models. In newborn Wistar rats that underwent IH injury (10% oxygen for 120 min after carotid artery electrocoagulation), CBD (1 mg/kg single dose, 1 mg/kg every 24 h for 72 h, or 1 mg/kg every 8 h for 72 h) modulated the expression of oxidative stress and inflammation, which reduced neuronal death and produced greater functional recovery (Pazos et al., 2012). Similarly, in a sciatic nerve section model of 2 PN day rats that received treatment with a single dose of 15 or 30 mg/kg CBD, 30% synaptic preservation was observed by immunohistochemical analysis (obtained by synaptophysin staining). The administration of CBD decreased astroglial and microglial reactions by 30 and 27%, respectively, and reduced the number of apoptotic cells mainly in the intermediate zone of the spinal cord (Pérez et al., 2013).
Clinical Studies
Cannabis-Derived Approved Drugs
While there are many descriptive and anecdotal reports on the benefits of cannabinoids, few cannabinoid drugs are regulated for clinical use. The most important licensed cannabis-based drugs are described herein: (a) dronabinol (Marinol®) was approved by the FDA on May 31, 1985, for nausea and vomiting control, associated with cancer chemotherapy that had not responded adequately to conventional antiemetic treatments. First marketed in 1986, on December 22, 1992, Marinol® was approved for the relief of anorexia associated with weight loss in patients with HIV-AIDS (human immunodeficiency virus/acquired immunodeficiency syndrome). At the time of its approval, the FDA recognized that dronabinol Δ9-THC was considered to be the psychoactive component of marijuana. Marinol® is also licensed for use in other countries, such as Canada and Germany. Generic forms of dronabinol (first approved in 2011) have now been approved in the US (Wright and Guy, 2014); (b) nabilone (Cesamet® dl-3-(1,1-dimethyl heptyl)-6,6ab 7, 8, 10, 10a alpha-hexahydro-1-hydroxy-6,6-dimethyl-9H-dibenzo pyran-9-(one) is a synthetic analog of Δ9 THC that was developed in the 1970s long before the target receptor was identified. It has a low nanomolar affinity for the CB1 receptor and a somewhat reduced affinity for the CB2 receptor. It was initially approved by the FDA in 1985 for chemotherapy-induced nausea and vomiting that do not respond to conventional antiemetics. The drug was marketed in the United States until 2006 (Wright and Guy, 2014); (c) nabiximols (Sativex®) differs from the other authorized cannabinoids insofar as it comprises an extract of the cannabis plant. It is formulated as a sublingual/oromucosal spray, every 100 μl of which provides 2.7 mg of Δ9 THC and 2.5 mg of CBD. It was approved for the first time in Canada in 2005 under legislation that allows the conditional approval of a new drug in areas of high unmet medical need and in the presence of very promising clinical data. Its approval in the USA was delayed until the end of 2010 and early 2011 when Savitex was approved by the UK and Spain following a decentralized procedure. It is licensed as second-line therapy for the relief of spasticity in multiple sclerosis (MS); in some other countries, it may also be prescribed for the treatment of neuropathic pain in people with MS and for the treatment of cancer-related pain, where it is used as an opioid supplement (Wright and Guy, 2014); and (d) epidiolex, is the first plant-derived, purified pharmaceutical-grade CBD medication. It was approved in the USA by the FDA on June 25, 2018 (Sekar and Pack, 2019). Its approval for patients ≥2 years of age with DS or LGS markedly altered the treatment of medically refractory seizures in these disorders. Epidiolex is a CBD only component with no Δ9THC, and the psychoactive component of cannabis responsible for appetite stimulation and euphoria sensation (Abu-Sawwa et al., 2020; Specchio et al., 2020).
Pharmacokinetics of Cannabinoids
To date, few clinical studies have been conducted in pediatric patients with difficult-to-control epilepsy and oral administration with cannabidiol solutions (CBD) to determine the pharmacokinetic parameters. In this review, data were obtained from a population with an age range between 0–20 years (Devinsky et al., 2018a; Wheless et al., 2019). The pharmacokinetics of cannabinoids is divided into four phases: absorption, distribution, metabolism, and excretion, but they are modified by interactions with other drugs. The pharmacokinetics of these compounds are divided into four phases: absorption, distribution, metabolism, and excretion, but they are modified by interactions with other drugs.
Absorption
The absorption rate of cannabinoids is determined by the route of administration of the drug and its formulation. Administration through oral formulations of any cannabinoid or food presents variable absorption and extensive first-pass hepatic metabolism. The administration of oral formulations presents a variable absorption depending on whether it is a single dose or repeated doses, as well as an extensive first-pass hepatic metabolism. The geometric mean of the time to maximum plasma concentration (Tmax) of cannabidiol ranged from 2 to 4 h after the administration of a single dose in a concentration range from 5–20 mg/kg without previous consumption of food. Thus, the route of administration of this pharmaceutical form has a slow absorption with a wide absorption phase. Concerning the administration of repeated doses (7 days), the Tmax of cannabidiol was detected between 2–3 h in a concentration range between 10–40 mg/kg/day (Wheless et al., 2019). Sublingual administration is very similar to oral administration. Oro-mucosal or spray preparations of Δ9THC—CBD (1:1) allow rapid absorption, whereas higher plasma concentrations are reached compared with oral administrations, but inhaled Δ9 THC leads to lower concentrations in the plasma (Karschner et al., 2011; Pertwee and Cascio, 2014).
Distribution
Once absorbed, CBD can be detected in plasma. The geometric means of the maximum plasma concentration (Cmax) were 29.12, 47.19, and 103.7 ng/ml with a single dose administration of 5, 10, and 20 mg/kg, respectively. Also, an increase in the geometric means of Cmax of 3.12, 2.67, and 3.03 times was observed in repeated doses of 10, 20, and 40 mg/kg for 7 days compared with the single dose. The geometric averages of the area under the plasma concentration curve at time 0–12 h [AUC (0–12 h)] were 122, 243.6, and 473.5 ng·h/ml with the administration of a single dose of 5, 10, and 20 mg/kg, respectively. Additionally, an increase of 4.15, 3.43, and 4.45 times in AUC(0-τ) was observed with the administration of repeated doses of 10, 20, and 40 mg/kg for 7 days compared with the single-dose (Wheless et al., 2019).
Metabolism
Both CBD and its metabolites 7-OH CBD and 7-COOH-CBD are metabolized in the liver and can induce the expression of CYP1A1, CYP1A2, CYP2C9, and CYP2D6 during prolonged periods of administration (Greene and Saunders, 1974; Bornheim et al., 1992; Watanabe et al., 2007). Pharmacokinetic parameters of 7-OH CBD and the geometric averages of Tmax 7-OH CBD were similar to the Tmax of CBD after the administration of a single dose of CBD in a concentration range from 5–20 mg/kg, with repeated dosing for 7 days. Nevertheless, the Tmax of 7-OH CBD was detected at 2 h. The geometric means of Cmax of 7-OH CBD were 22.03, 34.56, and 71.7 ng/ml with the administration of a single dose of 5, 10, and 20 mg/kg of CBD, respectively. However, the Cmax of 7-OH CBD increased to 2.97, 2.81, and 3.04 with repeated doses of 10, 20, and 40 mg/kg for 7 days compared with the single dose. Additionally, a decrease in the percentage of the coefficient of variation (CV%) was observed compared with a single dose. The geometric averages of AUC (0–12 h) of 7-OH CBD were 104, 202.4, and 381.9 ng h/ml with the administration of a single dose of 5, 10, and 20 mg/kg of CBD, respectively. The AUC(0-τ) of 7-OH CBD increased 4.12, 3.25, and 4.42 times with the administration of repeated doses of 10, 20, and 40 mg/kg CDB for 7 days compared with the single dose. Finally, the geometric averages of the metabolite to parent (7-OH cannabidiol/cannabidiol) ratio of AUC(0-τ) was 0.8, 0.75, and 0.76 with the administration of repeated doses of 10, 20, and 40 mg/kg for 7 days, respectively (Wheless et al., 2019).
In another study, the mean plasma concentrations of CBD and its metabolites 6-OH-CBD and 7-COOH-CBD were detected after the administration of a single dose (1.25 mg/kg) or multiple doses (5, 10, or 20 mg/kg/day) of CBD. The most abundant circulating metabolite was 7-COOH-CBD. At the end of the treatment, the AUC(0-τ) of 7-COOH-CBD was 13–17 times higher than the AUC(0-τ) of CBD. The CV% was considered to be high to moderate (20–121%; Devinsky et al., 2018b).
Elimination
The geometric averages of the apparent terminal half-life (t1/2) of CBD were 26.4, 29.6, and 19.5 h with the administration of a single dose of 5, 10, and 20 mg/kg, respectively. The geometric averages of the apparent total body clearance after oral administration at steady state (CLss/F) were 9.9, 12.3, and 9.5 L/h/kg, with the administration of repeated doses of 10, 20, and 40 mg/kg for 7 days, respectively (Wheless et al., 2019). Regarding the pharmacokinetic parameters of its metabolite 7-OH CBD, the geometric means of t1/2 were 18.4, 25.6, and 14.2 h with the administration of a single dose of 5, 10, and 20 mg/kg, respectively (Wheless et al., 2019). The metabolites of 7-OH-CBD are excreted in the feces and, to a lesser extent, the urine (Ohlsson et al., 1986).
During interactions with other drugs, the pharmacokinetic parameters of CBD change significantly with the administration of clobazam (CBZ). Therefore, it is often necessary to reduce clobazam due to excessive sedation. Pediatric patients who received repeated doses of 40 mg/kg for 7 days of CBD and CBZ had a 2.36-fold increase in CBD AUC(0-τ) and a 2.5-fold increase in CL/F compared with those who did not receive CBZ (Wheless et al., 2019). Conversely, CBD inhibits CYP2C19, and CYP3A4, which catalyze the metabolism of norclobazam (nCBZ). nCBZ is detected at high plasma concentrations (500% 300% mean increase) compared with the increase detected in CBZ (60% 80%) in a concentration range from 20–25 mg/kg/day of CBD and 0.18–2.24 mg/kg/day of CBZ (Geffrey et al., 2015).
Clinical Evidence of Cannabinoids in the Treatment of Pediatric Epilepsy
Clinical studies reported below assess the safety and/or efficacy of CBD in addition to common AEDs. Most of these studies enrolled pediatric patients (0.5–17 years) with diagnoses of genetically based epilepsy, Dravet syndrome, Lennox Gastaut, febrile seizures, and seizures associated with tuberous sclerosis are the most difficult epilepsies to treat, and the latest generation of antiepileptic drugs offers limited control for these diseases (Devinsky et al., 2017; Lattanzi et al., 2018, 2019). The use of CS in an empirical way has been documented for a long time for the treatment of epilepsy, and in most cases, reports provide a subjective perception of benefit. Despite the positive effects of the use of medicinal cannabis CBD for the control of epileptic seizures, there is controversy in the use of CBD at the pediatric level. The first trials examining purified CBD (Epidiolex) were launched as an expanded access program (EAP) in 2014 for patients with refractory epilepsy. Szaflarski et al. (2018) published provisional data for 600 patients who used CBD during 96 weeks, revealing a reduction of seizure events by 51% mainly in cases related to Dravet syndrome, Lennox Gastaut, and tuberous sclerosis complex (reviewed by Silvestro et al., 2019).
Given the increase in the use of handcrafted cannabinoids in pediatric epilepsy and the lack of studies providing data on their safety and usefulness, a prospective investigation was carried out in 32 children with refractory epilepsy. CBD was administered at a dose of 10 mg/kg/day for 12 weeks, and the frequency of seizures and serum CBD levels were evaluated. There was a 50% reduction in the frequency of seizures during the final weeks compared with the initial ones (Knupp et al., 2019). In another open comparative study on the use of CBD to treat epilepsy, 55 patients aged 1 and 30 years with CDKL5 deficiency disorder and Aicardi, Doose, and Dup15q syndromes were included. The patients were treated in 11 institutions from January 2014 to December 2016, and a decrease in the frequency of seizures was reported from the start of CBD treatment. The dose used ranged between 10 and 20 mg/kg/day orally, and this open-label trial provided class III evidence for the long-term safety and efficacy of CBD administration in patients with refractory epilepsy (Devinsky et al., 2014, 2016, 2018a,b; Elliott et al., 2018).
In a clinical trial of children aged 1–17 years with refractory epilepsy (60), an oral solution of CBD at doses between 5, 10, and 20 mg/kg/day was administered for 12 weeks as a complement to their regimen of antiepileptic drugs. A 50% decrease in seizure frequency was observed, no serious adverse effects were found, and CBD was well tolerated (Wheless et al., 2019). In most trials, CBD is administered orally as an oil solution. In open trials, maximum doses of 25 mg/kg/day have been used in controlled studies, and higher doses of up to 50 mg/kg/day were also used; however, studies on Lennox Gastaut syndrome have shown that a significant proportion of children respond to doses of 10 mg/kg/day. Therefore, a “slow start” and “scale as appropriate” strategy is recommended, beginning with 5 mg/kg/day, increasing to 10 mg/kg/day after 2 weeks, reviewing the clinical response and adverse effects, remaining with the dose if it is effective, and otherwise increasing the dose of 5 mg/kg/day according to tolerance up to a maximum of 20–25 mg/kg/day (Treat et al., 2017; Devinsky et al., 2018b; Arzimanoglou et al., 2020).
In the previous systematic review, published in August 2017, examined 22 clinical trials, only five of which were controlled clinical trials that included a total of 795 children. The greatest evidence was for its use as an antiemetic, analgesic, and antiepileptic agent (Wong and Wilens, 2017). Currently, only two synthetic products derived from cannabis are approved by the FDA of the United States: dronabinol and nabilone. Both contain Δ9–THC as the main cannabinoid and are specified for the treatment of cancer comorbidities and anorexia associated with patients with HIV/AIDS (Pertwee and Cascio, 2014; Wong and Wilens, 2017).
To date, in Uruguay, the only pharmaceutical specialty registered by the MS is epifatan as 2% or 5% oral solution (drops), which contains 2 g or 5 g of CBD, respectively, and less than 0.1% THC and tetrahydrocannabinolic acid (THCA) every 100 ml. Most reports concerning the therapeutic effect of CBD in drug-resistant epilepsy have been conducted in children and adolescents (Wong and Wilens, 2017), thus providing the greatest amount of available scientific evidence. In recent decades, various investigations have been carried out on the use of cannabis in the treatment of refractory epilepsy in children, especially in epileptic syndromes such as DS, Doose, and LGS (Wong and Wilens, 2017).
In the same way, Tzadok et al. (2016) in a retrospective review of a cohort of 74 children and adolescents with drug-resistant epilepsy reported that CBD reduced the frequency of seizures in 89% of patients. In a survey of 19 parents of children with treatment-resistant epilepsy, Porter and Jacobson (2013) found that CBD treatment of 117 children with drug-resistant epilepsy reduced the seizure frequency in 84% of the patients. Conversely, in a case series of six children with drug-resistant epilepsy, dronabinol has also been reported to reduce seizures in two patients. However, most of the studies have shown inconsistency in terms of the control of the variables and absence of a placebo group, making it difficult to generate an accurate conclusion, in addition to secondary effects such as drowsiness, diarrhea, and decreased appetite (Tzadok et al., 2016). Additionally, in controlled trials, Δ9THC most commonly led to side effects of drowsiness and dizziness, with greater severity associated with higher doses (Wang et al., 2008). Likewise, cannabis overdose has been reported with multiple adverse effects, among which are reports of seizures among young children, which may be due to the toxicity of high-dose Δ9 THC (Wang et al., 2008; Mechoulam and Parker, 2013).
However, clinical investigations have methodological limitations, such as the small population of children included, the short follow-up period, and the methodological design. Nevertheless, they have reported a reduction between 57% and 84% in the frequency and intensity of epileptic seizures, with a greater impact on DS and LGS (Wong and Wilens, 2017). Few studies have examined medical cannabinoids for the treatment of seizures in children and adolescents, and they have reported that CBD reduces the frequency of seizures in the pediatric population with resistance to initial treatment in epilepsy of different etiologies (Devinsky et al., 2017; Huntsman et al., 2020). Additionally, since 2014, CBD has been administered in a continuously EAP. In a review of 119 pediatric epilepsy patients, Treat et al. (2017) reported that oral CS extracts improved seizures in 49% of the cohort. In a second study, the oral CS extracts reduced the frequency and intensity of seizures in 57% of 75 patients with drug-resistant epilepsy (Treat et al., 2017).
In addition to the lack of studies on CS, the long-term risks associated with medical CBD in pediatric patients limits our understanding of the mechanism of action and its secondary effects on neurodevelopment and epileptogenesis (Mechoulam and Parker, 2013). However, evidence has shown that onset of CS use before the age of 16 years has a relationship with cognitive and verbal learning deficits and poor psychomotor performance and attention, similar to later onset of CS, which have been associated with poorer attention, executive functioning, memory and verbal performance (Solowij et al., 2011). Similarly, frequent and recreational CS use before the age of 15 years is associated with an increased risk of depression, confusion, and subsequent suicidal tendencies. In contrast, cannabis use in early adolescence shows a greater relationship with the early onset of psychotic disorders (Hayatbakhsh et al., 2007; Claudet et al., 2017). When using CBD as an adjuvant with other antiepileptic drugs, there was an increase in liver enzymes, jaundice, and thrombocytopenia, but these alterations were resolved when the antiepileptic was discontinued. An interaction is observed between the use of CBD and clobazam, and the main adverse reactions to the use of CBD are fatigue and diarrhea; however, an increase in appetite has been documented in some patients (Geffrey et al., 2015). There is evidence of the efficacy of a large percentage of CBD in epilepsy of different etiologies despite the few studies conducted to date. However, some of them have a risk of bias or poor follow-up by caregivers of the patients. Therefore, more protocols or experimental studies are suggested with internationally approved administration doses according to age, weight, and specific indication. Such studies will provide a broader picture to not only test the efficacy of drugs in epilepsy but also study the adverse effects of drugs derived from cannabis.
Conclusions
Evidence describing the effects of the major cannabinoids that do not act as CBR ligands, in particular cannabidiol and cannabidivarin, reveals consistently beneficial therapeutic effects in preclinical models of seizures, epilepsy, epileptogenesis, and neuroprotection. The emerging results of clinical trials in humans have methodological limitations, such as the small population of children included, the short follow-up period, and the methodological design, although, in most of them, they have reported a reduction in the frequency and intensity of seizures, in the drug-resistant pediatric population of different etiologies. What is clear is that the beneficial effects in epilepsy for the immature brain vary compared to the adult brain, due to neurodevelopmental processes. Therefore, more protocols or experimental studies should be performed with a stricter, broader, internationally approved administration dose according to age, weight, and specific indication.
Ethics Statement
All procedures, including the use, management and care of animals, adhered to the guidelines of and were approved by the Ethics and Research Committee of the Hospital of Specialties in the National Medical Center Century XXI, Mexican Social Security Institute (IMSS) and Mexican Official Standard [1999]; NOM-062-ZOO, 1999.
Author Contributions
AV-G developed the topic of epileptogenesis, performed the experiments with kainic acid and evaluated the latencies with the support of AM-M. IF-R, AM-A, and EZ-M developed the clinical studies. AG-J reviewed the derivatives of Cannabis and participated in the images. EG-A developed with AV-G the effects of CD on neurodevelopment. SO-S developed the idea, the topics to be considered, organized and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the IMSS health research fund, FIS/IMSS/PROT/PRIO/19/107 number grant for the included experimental part and the correction of the English style.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CNS, central nervous system; ABHD6, α-β-hydrolase domain 6; ACEA, arachidonyl-2′-chloroethylamide; AEA, anandamide; eCBS, endocannabinoid system; eCB-endocannabinoide CBD, cannabidiol; CBDV, cannabidivarin; CB1R, cannabinoid type 1 receptor; CB2R, cannabinoid type 2; GluR, glutamate receptor; DAGL, diacylglycerol lipase; Δ9-THC, Δ9-tetrahydrocannabinol; DSE, depolarization-induced suppression of excitation; DSI, depolarization-induced suppression of inhibition; FAAH, fatty acid amide hydrolase; GABA γ, aminobutyric acid; GAD, glutamate decarboxylase; GPR, G- protein-coupled receptor; KA, kainic acid; MAGL, monoacylglycerol lipase; MDA, maximal dentate activation; MES, maximal electroshock; NAPE-PLD, N-acylphosphatidylethanolamine-hydrolyzing phospholipase D; PMSF, phenylmethane sulfonyl fluoride; PTZ, pentylenetetrazole; TRPV1, transient receptor potential vanilloid receptor (type 1); NMDArs, N-methyl-d-aspartate receptor; AMPArs, AMPA-type glutamate receptors; FS, febrile seizures; SE, Status epilepticus; TBI, traumatic brain injury; PN, post natal; eCB, endocannabinoid system; CCK, colecistoquina; Ca2+, calcium; K+, potassium; Cl−, chlorine; Cox-2, cyclooxigenase 2; 2-AG, 2-arachidonoylglycerol; AA, arachidonic acid; AEA, N-arachidonoyl-ethanolamine or anandamide; CNS, central nervous system; Cox, cyclooxygenase; IL-1β, interleukin-1β; LOX, lipoxygenase.
References
Abu-Sawwa, R., Scutt, B., and Park, Y. (2020). Emerging use of epidiolex (Cannabidiol) in epilepsy. J. Pediatr. Pharmacol. Ther. 25, 485–499. doi: 10.5863/1551-6776-25.6.485
Alexandre, J., Carmo, H., Carvalho, F., and Pedro Silva, J. (2020). Synthetic cannabinoids and their impact on neurodevelopmental processes. Addict. Biol. 25:e12824. doi: 10.1111/adb.12824
Al-Muhtasib, N., Sepulveda-Rodriguez, A., Vicini, S., and Forcelli, P. A. (2018). Neonatal phenobarbital exposure disrupts GABAergic synaptic maturation in rat CA1 neurons. Epilepsia 59, 333–344. doi: 10.1111/epi.13990
Anderson, L. L., Low, I. K., McGregor, I. S., and Arnold, J. C. (2020). Interactions between cannabidiol and Δ9-tetrahydrocannabinol in modulating seizure susceptibility and survival in a mousae model of Dravet syndrome. Br. J. Pharmacol. 177, 4261–4274. doi: 10.1111/bph.15181
Andre, C. M., Hausman, J. F., and Guerriero, G. (2016). Cannabis sativa: the plant of the thousand one molecules. Front. Plant Sci. 7:19. doi: 10.3389/fpls.2016.00019
Armstrong, C., Morgan, R. J., and Soltesz, I. (2009). Pursuing paradoxical proconvulsant prophylaxis for epileptogenesis. Epilepsia 50, 1657–1669. doi: 10.1111/j.1528-1167.2009.02173.x
Arzimanoglou, A., Bland, U. B., Cross, J. H., Gil-Nagel, A., Lagae, L., Landmark, C. J., et al. (2020). Epilepsy and cannabidiol: a guide to treatment. Epileptic Disord. 22, 1–14. doi: 10.1684/epd.2020.1141
Atwood, B. K., and Mackie, K. (2010). CB2: a cannabinoid receptor with an identity crisis. Br. J. Pharmacol. 160, 467–479. doi: 10.1111/j.1476-5381.2010.00729.x
Auvin, S., and Dupuis, N. (2014). Outcome of status epilepticus. What do we learn from animal data? Prolonged epileptic seizures: identification and treatment. Epileptic Disord. 16, S37–S43. doi: 10.1684/epd.2014.0670
Balosso, S., Ravizza, T., Perego, C., Peschon, J., Campbell, I. L., De Simoni, M. G., et al. (2005). Tumor necrosis factor-alpha inhibits seizures in mice via p75 receptors. Ann. Neurol. 57, 804–812. doi: 10.1002/ana.20480
Baud, O., and Saint-Faust, M. (2019). Neuroinflammation in the developing brain: risk factors, involvement of microglial cells, and implication for early anesthesia. Anesth. Analg. 128, 718–725. doi: 10.1213/ANE.0000000000004032
Beattie, E. C., Stellwagen, D., Morishita, W., Bresnahan, J. C., Ha, B. K., Von Zastrow, M., et al. (2002). Control of synaptic strength by glial TNF α. Science 295, 2282–2285. doi: 10.1126/science.1067859
Ben-Ari, Y. (2002). Excitatory actions of GABA during development: the nature of the nurture. Nat. Rev. Neurosci. 3, 728–739. doi: 10.1038/nrn920
Ben-Ari, Y. (2014). The GABA excitatory/inhibitory developmental sequence: a personal journey. Neuroscience 279, 187–219. doi: 10.1016/j.neuroscience.2014.08.001
Ben-Ari, Y., and Holmes, G. L. (2006). Effects of seizures on developmental processes in the immature brain. Lancet Neurol. 5, 1055–1063. doi: 10.1016/S1474-4422(06)70626-3
Benítez-Diaz, P., Miranda-Contreras, L., Mendoza-Briceño, R. V., Peña-Contreras, Z., and Palacios-Prü, E. (2003). Prenatal and postnatal contents of amino acid neurotransmitters in mouse parietal cortex. Dev. Neurosci. 25, 366–374. doi: 10.1016/j.ridd.2020.103793
Berdyshev, E. V. (2000). Cannabinoid receptors and the regulation of immune response. Chem. Phys. Lipids. 108, 169–190. doi: 10.1016/s0009-3084(00)00195-x
Bernard, C., Milh, M., Morozov, Y. M., Ben-Ari, Y., Freund, T. F., and Gozlan, H. (2005). Altering cannabinoid signaling during development disrupts neuronal activity. Proc. Natl. Acad. Sci. U S A 102, 9388–9393. doi: 10.1073/pnas.0409641102
Bessis, A., Béchade, C., Bérnard, D., and Roumier, A. (2007). Microglial control of neuronal death and synaptic properties. Glia 55, 233–238. doi: 10.1002/glia.20459
Bhaskaran, M. D., and Smith, B. N. (2010). Effects of TRPV1 activation on synaptic excitation in the dentate gyrus of a mouse model of temporal lobe epilepsy. Exp. Neurol. 223, 529–536. doi: 10.1016/j.expneurol.2010.01.021
Bittigau, P., Sifringer, M., Genz, K., Reith, E., Pospischil, D., Govindarajalu, S., et al. (2002). Antiepileptic drugs and apoptotic neurodegeneration in the developing brain. Proc. Natl. Acad. Sci. U S A 99, 15089–15094. doi: 10.1073/pnas.222550499
Blair, R. E., Sombati, S., Lawrence, D. C., McCay, B. D., and De Lorenzo, R. J. (2004). Epileptogenesis causes acute and chronic increases in GABAA receptor endocytosis that contributes to the induction and maintenance of seizures in the hippocampal culture model of acquired epilepsy. J. Pharmacol. Exp. Ther. 310, 871–880. doi: 10.1124/jpet.104.068478
Booz, G. W. (2011). Cannabidiol as an emergent therapeutic strategy for lessening the impact of inflammation on oxidative stress. Free Radic. Biol. Med. 51, 1054–1061. doi: 10.1016/j.freeradbiomed.2011.01.007
Bornheim, L. M., Lasker, J. M., and Raucy, J. L. (1992). Human hepatic microsomal metabolism of δ1-tetrahydrocannabinol. Drug Metab. Dispos. 20, 241–246.
Bouchard, J., Truong, J., Bouchard, K., Dunkelberger, D., Desrayaud, S., Moussaoui, S., et al. (2012). Cannabinoid receptor 2 signaling in peripheral immune cells modulates disease onset and severity in mouse models of Huntington’s disease. J. Neurosci. 32, 18259–18268. doi: 10.1523/JNEUROSCI.4008-12.2012
Bouchet, C. A., and Ingram, S. L. (2020). Cannabinoids in the descending pain modulatory circuit: role in inflammation. Pharmacol. Ther. 209:107495. doi: 10.1016/j.pharmthera.2020.107495
Brusco, A., Tagliaferro, P., Saez, T., and Onaivi, E. S. (2008). Postsynaptic localization of CB2 cannabinoid receptors in the rat hippocampus. Synapse 62, 944–949. doi: 10.1002/syn.20569
Burstein, S. (2015). Cannabidiol (CBD) and its analogs: a review of their effects on inflammation. Bioorg. Med. Chem. 23, 1377–1385. doi: 10.1016/j.bmc.2015.01.059
Caiati, M. D., Sivakumaran, S., Lanore, F., Mulle, C., Richard, E., Verrier, D., et al. (2012). Developmental regulation of CB1-mediated spike-time dependent depression at immature mossy fiber-CA3 synapses. Sci. Rep. 2:285. doi: 10.1038/srep00285
Capasso, R., Borrelli, F., Aviello, G., Romano, B., Scalisi, C., Capasso, F., et al. (2008). Cannabidiol, extracted from Cannabis sativa, selectively inhibits inflammatory hypermotility in mice. Br. J. Pharmacol. 154, 1001–1008. doi: 10.1038/bjp.2008.177
Castillo, P. E., Younts, T. J., Chávez, A. E., and Hashimotodani, Y. (2012). Endocannabinoid signaling and synaptic function. Neuron 76, 70–81. doi: 10.1016/j.neuron.2012.09.020
Chan, P. C., Sills, R. C., Braun, A. G., Haseman, J. K., and Bucher, J. R. (1996). Toxicity and carcinogenicity of Δ9-tetrahydrocannabinol in Fischer rats and B6C3F1 mice. Fundam. Appl. Toxicol. 30, 109–117. doi: 10.1006/faat.1996.0048
Chen, J., and Hackos, D. H. (2015). TRPA1 as a drug target—promise and challenges. Naunyn. Schmiedebergs. Arch. Pharmacol. 388, 451–463. doi: 10.1007/s00210-015-1088-3
Chen, K., Aradi, I., Thon, N., Eghbal-Ahmadi, M., Baram, T. Z., and Soltesz, I. (2001). Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat. Med. 7, 331–337. doi: 10.1038/85480
Chen, K., Neu, A., Howard, A. L., Foldy, C., Echegoyen, J., Hilgenberg, L., et al. (2007). Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J. Neurosci. 27, 46–58. doi: 10.1523/JNEUROSCI.3966-06.2007
Chen, K., Ratzliff, A., Hilgenberg, L., Gulyas, A., Freund, T. F., Smith, M., et al. (2003). Long-term plasticity of endocannabinoid signaling induced by developmental febrile seizures. Neuron 39, 599–611. doi: 10.1016/s0896-6273(03)00499-9
Chen, Y.-H., Kuo, T.-T., Chu, M.-T., Ma, H.-I., Chiang, Y.-H., and Huang, E. Y.-K. (2013). Postnatal systemic inflammation exacerbates impairment of hippocampal synaptic plasticity in an animal seizure model. Neuroimmunomodulation 20, 223–232. doi: 10.1159/000348440
Cheung, K. A., Peiris, H., Wallace, G., Holland, O. J., and Mitchell, M. D. (2019). The interplay between the endocannabinoid system, epilepsy and cannabinoids. Int. J. Mol. Sci. 20:6079. doi: 10.3390/ijms20236079
Clancy, B., Darlington, R. B., and Finlay, B. L. (2001). Translating developmental time across mammalian species. Neuroscience 105, 7–17. doi: 10.1016/s0306-4522(01)00171-3
Claudet, I., Le Breton, M., Bréhin, C., and Franchitto, N. (2017). A 10-year review of cannabis exposure in children under 3-years of age: do we need a more global approach? Eur. J. Pediatr. 176, 553–556. doi: 10.1007/s00431-017-2872-5
Cock, H. R. (2002). The role of mitochondria and oxidative stress in neuronal damage after brief and prolonged seizures. Prog. Brain Res. 135, 187–196. doi: 10.1016/S0079-6123(02)35018-0
Correa, F., Docagne, F., Mestre, L., Loría, F., Hernangómez, M., Borrell, J., et al. (2007). Cannabinoid system and neuroinflammation: implications for multiple sclerosis. Neuroimmunomodulation 14, 182–187. doi: 10.1159/000110644
Coulter, D. A. (2006). Endocannabinoids: a critical regulator of activity in the developing brain. Epilepsy Curr. 6, 18–19. doi: 10.1111/j.1535-7511.2005.00082.x
De Simoni, M. G., Perego, C., Ravizza, T., Moneta, D., Conti, M., Marchesi, F., et al. (2000). Inflammatory cytokines and related genes are induced in the rat hippocampus by limbic status epilepticus. Eur. J. Neurosci. 12, 2623–2633. doi: 10.1046/j.1460-9568.2000.00140.x
Devinsky, O., Cilio, M. R., Cross, H., Fernandez-Ruiz, J., French, J., Hill, C., et al. (2014). Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia 55, 791–802. doi: 10.1111/epi.12631
Devinsky, O., Cross, J. H., Laux, L., Marsh, E., Miller, I., Nabbout, R., et al. (2017). Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N. Engl. J. Med. 376, 2011–2020. doi: 10.1056/NEJMoa1611618
Devinsky, O., Marsh, E., Friedman, D., Thiele, E., Laux, L., Sullivan, J., et al. (2016). Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol. 15, 270–278. doi: 10.1016/S1474-4422(15)00379-8
Devinsky, O., Patel, A. D., Thiele, E. A., Wong, M. H., Appleton, R., Harden, C. L., et al. (2018a). Randomized, dose-ranging safety trial of cannabidiol in Dravet syndrome. Neurology 90, e1204–e1211. doi: 10.1212/WNL.0000000000005254
Devinsky, O., Verducci, C., Tiele, E., Laux, L., Patel, A. D., Filloux, F., et al. (2018b). Open-label use of highly purified CBD (Epidiolex®) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav. 86, 131–137. doi: 10.1016/j.yebeh.2018.05.013
Devinsky, O., Vezzani, A., Najjar, S., De Lanerolle, N. C., and Rogawski, M. A. (2013). Glia and epilepsy: excitability and inflammation. Trends Neurosci. 36, 174–184. doi: 10.1016/j.tins.2012.11.008
Di Marzo, V., and Piscitelli, F. (2015). The endocannabinoid system and its modulation by phytocannabinoids. Neurotherapeutics 12, 692–698. doi: 10.1007/s13311-015-0374-6
Dow-Edwards, D., and Silva, L. (2017). Endocannabinoids in brain plasticity: cortical maturation, HPA axis function and behavior. Brain Res. 1654, 157–164. doi: 10.1016/j.brainres.2016.08.037
Elliott, J., DeJean, D., Clifford, T., Coyle, D., Potter, B., Skidmore, B., et al. (2018). Cannabis for pediatric epilepsy: protocol for a living systematic review. Syst. Rev. 7:95. doi: 10.1186/s13643-018-0761-2
El-Remessy, A. B., Tang, Y., Zhu, G., Matragoon, S., Khalifa, Y., Liu, E. K., et al. (2008). Neuroprotective effects of cannabidiol in endotoxin-induced uveitis: critical role of p38 MAPK activation. Mol. Vis. 14, 2190–2203.
El Sohly, E. A., Mehmedic, Z., Foster, S., Gon, C., Chandra, S., and Church, J. C. (2016). Changes in cannabis potency over the last 2 decades (1995–2014): analysis of current data in the united states. Biol. Psychiatry 79, 613–619. doi: 10.1016/j.biopsych.2016.01.004
Etienne, M. (2014). “The chemical phenotypes (chemotypes) of cannabis,” in Hand Book of Cannabis, ed R. Pertwee (University Press), 144–165.
Feng, B., Tang, Y., Chen, B., Xu, C., Wang, Y., Dai, Y., et al. (2016). Transient increase of interleukin-1β after prolonged febrile seizures promotes adult epileptogenesis through long-lasting upregulating endocannabinoid signaling. Sci. Rep. 6:21931. doi: 10.1038/srep21931
Fernández-Ruiz, J., Berrendero, F., Hernández, M. L., and Ramos, J. A. (2005). The endogenous cannabinoid system and brain development. Trends Neurosci. 23, 14–20. doi: 10.1016/s0166-2236(99)01491-5
Folbergrová, J., Jesina, P., Haugvicová, R., Lisý, V., and Houstek, J. (2010). Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem. Int. 56, 394–403. doi: 10.1016/j.neuint.2009.11.011
Forcelli, P. A., Kim, J., Kondratyev, A., and Gale, K. (2011). Pattern of antiepileptic drug-induced cell death in limbic regions of the neonatal rat brain. Epilepsia 52, e207–e211. doi: 10.1111/j.1528-1167.2011.03297.x
Franco, V., and Perucca, E. (2019). Pharmacological and therapeutic properties of cannabidiol for epilepsy. Drugs 79, 1435–1454. doi: 10.1007/s40265-019-01171-4
Galanopoulou, A. S. (2008). GABAA receptors in normal development and seizures: friends or foes? Curr. Neuropharmacol. 6, 1–20. doi: 10.2174/157015908783769653
Gaston, T. E., and Friedman, D. (2017). Pharmacology of cannabinoids in the treatment of epilepsy. Epilepsy Behav. 70, 313–318. doi: 10.1016/j.yebeh.2016.11.016
Gaston, T. E., and Szafarski, J. P. (2018). Cannabis for the treatment of epilepsy: an update. Curr. Neurol. Neurosci. Rep. 8:73. doi: 10.1007/s11910-018-0882-y
Geffrey, A. L., Pollack, S. F., Bruno, P. L., and Thiele, E. A. (2015). Drug-drug interaction between clobazam and cannabidiol in children with refractory epilepsy. Epilepsia 56, 1246–1251. doi: 10.1111/epi.13060
Ghovanloo, M. R., Shuart, N. G., Mezeyova, J., Dean, R. D., Ruben, P. C., and Goodchild, S. J. (2018). Inhibitory effects of cannabidiol on voltage-dependent sodium currents. J. Biol. Chem. 293, 16546–16558. doi: 10.1074/jbc.RA118.004929
Glass, H. C., Grinspan, Z. M., and Shellhaas, R. A. (2018). Outcomes after acute symptomatic seizures in neonates. Semin. Fetal Neonatal Med. 23, 218–222. doi: 10.1016/j.siny.2018.02.001
Glass, H. C., Kan, J., Bonifacio, S. L., and Ferriero, D. (2012). Neonatal seizures: treatment practices among term and preterm infants. Pediatr. Neurol. 46, 111–115. doi: 10.1016/j.pediatrneurol.2011.11.006
Gobbo, O. L., and O’Mara, S. M. (2004). Post-treatment, but not pre-treatment, with the selective cyclooxygenase-2 inhibitor celecoxib markedly enhances functional recovery from kainic acid-induced neurodegeneration. Neuroscience 125, 317–327. doi: 10.1016/j.neuroscience.2004.01.045
Greene, M. L., and Saunders, D. R. (1974). Metabolism of tetrahydrocannabinol by the small intestine. Gastroenterology 66, 365–372. doi: 10.1016/S0016-5085(74)80135-6
Haglid, K. G., Wang, S., Qiner, Y., and Hamberger, A. (1994). Excitotoxicity. Experimental correlates to human epilepsy. Mol Neurobiol. 9, 259–263. doi: 10.1007/BF02816125
Harkany, T., Keimpema, E., Barabás, K., and Mulder, J. (2008). Endocannabinoid functions controlling neuronal specification during brain development. Mol. Cell. Endocrinol. 286, S84–S90. doi: 10.1016/j.mce.2008.02.011
Haskó, G., Csóka, B., Németh, Z. H., Vizi, E. S., and Pacher, P. (2009). A2B adenosine receptors in immunity and inflammation. Trends Immunol. 30, 263–270. doi: 10.1016/j.it.2009.04.001
Hayakawa, K., Irie, K., Sano, K., Watanabe, T., Higuchi, S., Enoki, M., et al. (2009). Therapeutic time window of cannabidiol treatment on delayed ischemic damage via high-mobility group box1-inhibiting mechanism. Biol. Pharm. Bull. 32, 1538–1544. doi: 10.1248/bpb.32.1538
Hayatbakhsh, M. R., Najman, J. M., Jamrozik, K., Mamun, A. A., Alati, R., and Bor, W. (2007). Cannabis and anxiety and depression in young adults: a large prospective study. J. Am. Acad. Child Adolesc. Psychiatry 46, 408–417. doi: 10.1097/chi.0b013e31802dc54d
Herdegen, T., and Leah, J. D. (1998). Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox and CreB/ATF proteins. Brain Res. Rev. 28, 370–490. doi: 10.1016/s0165-0173(98)00018-6
Herkenham, M., Lynn, A. B., Johnson, M. R., Melvin, L. S., de Costa, B. R., and Rice, K. C. (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 11, 563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991
Herkenham, M., Lynn, A. B., Little, M. D., Johnson, M. R., Melvin, L. S., de Costa, B. R., et al. (1990). Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. U S A 87, 1932–1936. doi: 10.1073/pnas.87.5.1932
Herlenius, E., and Lagercrantz, H. (2004). Development of neurotransmitter systems during critical periods. Exp. Neurol. 190, S8–S21. doi: 10.1016/j.expneurol.2004.03.027
Hill, A. J., Jones, N. A., Smith, I., Hill, C. L., Williams, C. M., Stephens, G. J., et al. (2014). Voltage-gated sodium (NaV) channel blockade by plant cannabinoids does not confer anticonvulsant effects per se. Neurosci. Lett. 566, 269–274. doi: 10.1016/j.neulet.2014.03.013
Huntsman, R. J., Wai, R., and Shackelford, A. E. (2020). Cannabis for Pediatric Epilepsy. J. Clin. Neurophysiol. 37, 2–8. doi: 10.1097/WNP.0000000000000641
Ibeas, B. C., Chen, T., Nunn, A. V., Bazelot, M., Dallas, M., and Whalley, B. J. (2015). Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics 12, 699–730. doi: 10.1007/s13311-015-0377-3
Jabs, R., Paterson, I. A., and Walz, W. (1997). Qualitative analysis of membrane currents in glial cells from normal and gliotic tissue in situ: down-regulation of Na+ current and lack of P2 purinergic responses. Neuroscience 81, 847–860. doi: 10.1016/s0306-4522(97)00207-8
Jensen, E. F. (2009). Neonatal seizures: an update on mechanisms and management. Clin. Perinatol. 36:881. doi: 10.1016/j.clp.2009.08.001
Jiang, J., Borisenko, G. G., Osipov, A., Martin, I., Chen, R., Shvedova, A. A., et al. (2004). Arachidonic acid-induced carbon-centered radicals and phospholipid peroxidation in cyclo-oxygenase-2-transfected PC12 cells. J. Neurochem. 90, 1036–1049. doi: 10.1111/j.1471-4159.2004.02577.x
Jones, N. A., Hill, A. J., Smith, I., Bevan, S. A., Williams, C. M., Whalley, B. J., et al. (2010). Cannabidiol displays antiepileptiform and antiseizure properties in vitro and in vivo. J. Pharmacol. Exp. Ther. 332, 569–577. doi: 10.1124/jpet.109.159145
Joseph, E. K., and Levine, J. D. (2006). Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain 121, 105–114. doi: 10.1016/j.pain.2005.12.010
Kadam, S. D., White, A. M., Staley, K. J., and Dudek, F. E. (2010). Continuous electro encephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. J. Neurosci. 30, 404–415. doi: 10.1523/JNEUROSCI.4093-09.2010
Kaplan, J. S., Stella, N., Catterall, W. A., and Westenbroek, R. E. (2017). Cannabidiol attenuates seizures and social defcits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U S A 114, 11229–11234. doi: 10.1073/pnas.1711351114
Karschner, E. L., Darwin, W. D., Goodwin, R. S., Wright, S., and Huestis, M. A. (2011). Plasma cannabinoid pharmacokinetics following controlled oral Δ9-tetrahydrocannabinol and oromucosal cannabis extract administration. Clin. Chem. 57, 66–75. doi: 10.1373/clinchem.2010.152439
Katona, I., Sperlagh, B., Sik, A., Kafalvi, A., Vizi, E. S., Mackie, K., et al. (1999). Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J. Neurosci. 19, 4544–4558. doi: 10.1523/JNEUROSCI.19-11-04544.1999
Katona, I., Urban, G. M., Wallace, M., Ledent, C., Jung, K. M., Piomelli, D., et al. (2006). Molecular composition of the endocannabinoid system at glutamatergic synapses. J. Neurosci. 26, 5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006
Kaushal, S., Tamer, Z., Opoku, F., and Forcelli, P. A. (2016). Anticonvulsant drug-induced cell death in the developing white matter of the rodent brain. Epilepsia 57, 727–734. doi: 10.1111/epi.13365
Kawaguchi, K., Hickey, R. W., Rose, M. E., Zhu, L., Chen, J., and Graham, S. H. (2005). Cyclooxygenase-2 expression is induced in rat brain after kainate-induced seizures and promotes neuronal death in CA3 hippocampus. Brain Res. 1050, 130–137. doi: 10.1016/j.brainres.2005.05.038
Kawamura, Y., Fukaya, M., Maejima, T., Yoshida, T., Miura, E., Watanabe, M., et al. (2006). The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J. Neurosci. 26, 2991–3001. doi: 10.1523/JNEUROSCI.4872-05.2006
Klein, B. D., Jacobson, C. A., Metcalf, C. S., Smith, M. D., Wilcox, K. S., Hampson, A. J., et al. (2017). Evaluation of cannabidiol in animal seizure models by the Epilepsy Therapy Screening Program (ETSP). Neurochem. Res. 42, 1939–1948. doi: 10.1007/s11064-017-2287-8
Knupp, K. G., Rice, J. D., Helmkamp, L. J., Galinkin, J., Sempio, C., Jost, K., et al. (2019). Prospective evaluation of oral cannabis extracts in children with epilepsy. Seizure 72, 23–27. doi: 10.1016/j.seizure.2019.09.007
Kreitzer, A. C., and Regehr, W. G. (2001). Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 29, 717–727. doi: 10.1016/s0896-6273(01)00246-x
Kubová, H., Druga, R., Lukasiuk, K., Suchomelová, L., Haugvicová, R., Jirmanová, I., et al. (2001). Status epilepticus causes necrotic damage in the mediodorsal nucleus of the thalamus in immature rats. J. Neurosci. 21, 3593–3599. doi: 10.1523/JNEUROSCI.21-10-03593.2001
Kubová, H., Lukasiuk, K., and Pitkänen, A. (2012). New insight on the mechanisms of epileptogenesis in the developing brain. Adv. Tech. Stand. Neurosurg. 39, 3–44. doi: 10.1007/978-3-7091-1360-8_1
Kubová, H., and Mareš, P. (2013). Are morphologic and functional consequences of status epilepticus in infant rats progressive? Neuroscience 235, 232–249. doi: 10.1016/j.neuroscience.2012.12.055
Kudin, A., Tatiana, A., Jan, K., Seyfried, S. V., Beck, H., Elger, C. E., et al. (2002). Seizure-dependent modulation of mitochondrial oxidative phosphorylation in rat hippocampus. Eur. J. Neurosci. 15, 1105–1114. doi: 10.1046/j.1460-9568.2002.01947.x
Kurz, J. E., Sheets, D., Parsons, J. T., Rana, A., Delorenzo, R. J., and Churn, S. B. (2001). A significant increase in both basal and maximal calcineurin activity in the rat pilocarpine model of status epilepticus. J. Neurochem. 78, 304–315. doi: 10.1046/j.1471-4159.2001.00426.x
Laprairie, R. B., Bagher, A. M., Kelly, M. E. M., and Denovan-Wright, E. M. (2015). Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 172, 4790–4805. doi: 10.1111/bph.13250
Lattanzi, S., Brigo, F., Trinka, E., Zaccara, G., Cagnetti, C., Del Giovane, C., et al. (2018). Efficacy and safety of cannabidiol in epilepsy: a systematic review and meta-analysis. Drugs 78, 1791–1804. doi: 10.1007/s40265-018-0992-5
Lattanzi, S., Trinka, E., Russo, E., Striano, P., Citraro, R., Silvestrini, M., et al. (2019). Cannabidiol as adjunctive treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome. Drugs Today 55, 177–196. doi: 10.1358/dot.2019.55.3.2909248
Lee, M. C., Ting, K. K., Adams, S., Brew, B. J., Chung, R., and Guillemin, G. J. (2010). Characterization of the expression of NMDA receptors in human astrocytes. PLoS One 5:e14123. doi: 10.1371/journal.pone.0014123
Leo, A., Russo, E., and Elia, M. (2016). Cannabidiol and epilepsy: rationale and therapeutic potential. Pharmacol. Res. 107, 85–92. doi: 10.1016/j.phrs.2016.03.005
Li, Y., and Kim, J. (2015). Neuronal expression of CB2 cannabinoid receptor mRNAs in the mouse hippocampus. Neuroscience 311, 253–267. doi: 10.1016/j.neuroscience.2015.10.041
Liou, G. I., Auchampach, J. A., Hillard, C. J., Zhu, G., Yousufzai, B., Mian, S., et al. (2008). Mediation of cannabidiol anti-inflammation in the retina by equilibrative nucleoside transporter and A2A adenosine receptor. Invest. Ophthalmol. Vis. Sci. 49, 5526–5531. doi: 10.1167/iovs.08-2196
Lorigados, L., Orozco, S., Morales, L., Estupiñán, B., García, I., and Rocha, L. (2013). Excitotoxicidad y muerte neuronal en la epilepsia. Biotecnología Aplicad. 30, 1–8. doi: 10.2174/1381612811319380009
Lu, H. C., and Mackie, K. (2016). An introduction to the endogenous cannabinoid system. Biol. Psychiatry. 79, 516–525. doi: 10.1016/j.biopsych.2015.07.028
Mahmoud, E., and Waseem, G. (2014). “Constituents of cannabis sativa,” in Hand Book of Cannabis, ed R. Pertwee (University Press Oxford Scholarship), 46–66.
Malfait, A. M., Gallily, R., Sumariwalla, P. F., Malik, A. S., Andreakos, E., Mechoulam, R., et al. (2000). The nonpsychoactive cannabis constituent cannabidiol is an oral anti-arthritic therapeutic in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. U S A. 97, 9561–9566. doi: 10.1073/pnas.160105897
Meador, K. J., Baker, G. A., Browning, N., Cohen, M. J., Bromley, R. L., Clayton-Smith, J., et al. (2012). Effects of fetal antiepileptic drug exposure: outcomes at age 4.5 years. Neurology 78, 1207–1214. doi: 10.1212/WNL.0b013e318250d824
Mecha, M., Carrillo-Salinas, F. J., Feliú, A., Mestre, L., and Guaza, C. (2016). Microglia activation states and cannabinoid system: therapeutic implications. Pharmacol. Ther. 166, 40–55. doi: 10.1016/j.pharmthera.2016.06.011
Mechoulam, R., and Gaoni, Y. (1965). The isolation and structure of cannabinolic cannabidiolic and cannabigerolic acids. Tetrahedron 21, 1223–1229. doi: 10.1016/0040-4020(65)80064-3
Mechoulam, R., and Parker, L. A. (2013). The endocannabinoid system and the brain. Annu. Rev. Psychol. 64, 21–47. doi: 10.1146/annurev-psych-113011-143739
Monory, K., Massa, F., Egertová, M., Eder, M., Blaudzun, H., Westenbroek, R., et al. (2006). The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron 51, 455–466. doi: 10.1016/j.neuron.2006.07.006
Morgan, N. H., Stanford, I. M., and Woodhall, G. L. (2009). Functional CB2 type cannabinoid receptors at CNS synapses. Neuropharmacology 57, 356–368. doi: 10.1016/j.neuropharm.2009.07.017
Mori, M. A., Meyer, E., Soares, L. M., Milani, H., Guimarães, F. S., and de Oliveira, R. M. W. (2017). Cannabidiol reduces neuroinflammation and promotes neuroplasticity and functional recovery after brain ischemia. Prog. Neuropsychopharmacol. Biol. Psychiatry 75, 94–105. doi: 10.1016/j.pnpbp.2016.11.005
Murchison, D., and Griffith, W. H. (2000). Mitochondria buffer non-toxic calcium loads and release calcium through the mitochondrial permeability transition pore and sodium/calcium exchanger in rat basal forebrain neurons. Brain Res. 854, 139–151. doi: 10.1016/s0006-8993(99)02297-0
Nomura, D. K., Morrison, B. E., Blankman, J. L., Long, J. Z., Kinsey, S. G., Marcondes, M. C. G., et al. (2011). Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334, 809–813. doi: 10.1126/science.1209200
Ohlsson, A., Lindgren, J. E., Andersson, S., Agurell, S., Gillespie, H., Hollister, L. E., et al. (1986). Single-dose kinetics of deuterium-labelled cannabidiol in man after smoking and intravenous administration. Biomed. Environ. Mass Spectrom. 13, 77–83. doi: 10.1002/bms.1200130206
Onaivi, E. S., Ishiguro, H., Gu, S., and Qing-Rong, L. (2012). CNS effects of CB2 cannabinoid receptors: beyond neuro-immuno-cannabinoid activity. J. Psychopharmacol. 26, 92–103. doi: 10.1177/0269881111400652
Pan, H., Mukhopadhyay, P., Rajesh, M., Patel, V., Mukhopadhyay, B., Gao, B., et al. (2009). Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation and cell death. J. Pharmacol. Exp. Ther. 328, 708–714. doi: 10.1124/jpet.108.147181
Pandolfo, P., Silveirinha, V., dos Santos-Rodrigues, A., Venance, L., Ledent, C., Takahashi, R. N., et al. (2011). Cannabinoids inhibit the synaptic uptake of adenosine and dopamine in the rat and mouse striatum. Eur. J. Pharmacol. 655, 38–45. doi: 10.1016/j.ejphar.2011.01.013
Patel, S., Hill, M. N., Cheer, J. F., Wotjak, C. T., and Holmes, A. (2017). The endocannabinoid system as a target for novel anxiolytic drugs. Neurosci. Biobehav. Rev. 76, 56–66. doi: 10.1016/j.neubiorev.2016.12.033
Patel, M., and Li, Q. Y. (2003). Age dependence of seizure-induced oxidative stress. Neuroscience 118, 431–437. doi: 10.1016/s0306-4522(02)00979-x
Patra, P. H., Barker-Haliski, M., White, H. S., Whalley, B. J., Glyn, S., Sandhu, H., et al. (2019). Cannabidiol reduces seizures and associated behavioral comorbidities in a range of animal seizure and epilepsy models. Epilepsia 60, 303–314. doi: 10.1111/epi.14629
Pazos, M. R., Cinquina, V., Gómez, A., Layunta, R., Santos, M., Fernández-Ruiz, J., et al. (2012). Cannabidiol administration after hypoxia-ischemia to newborn rats reduces long-term brain injury and restores neurobehavioral function. Neuropharmacology 63, 776–783. doi: 10.1016/j.neuropharm.2012.05.034
Pérez, M., Benitez, S. U., Cartarozzi, L. P., Del Bel, E., Guimarães, F. S., and Oliveira, A. L. R. (2013). Neuroprotection and reduction of glial reaction by cannabidiol treatment after sciatic nerve transection in neonatal rats. Eur. J. Neurosci. 38, 3424–3434. doi: 10.1111/ejn.12341
Pertwee, R. G. (2008). The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 153, 199–215. doi: 10.1038/sj.bjp.0707442
Pertwee, R. G., and Cascio, M. G. (2014). “Known pharmacological actions of delta-9-tetrahydrocannabinol and of four other chemical constituents of cannabis that activate cannabinoid receptors,” in Hand Book of Cannabis, ed R. Pertwee (UK: University of Aberdeen) 167–185.
Pitkänen, A., and Engel, J., Jr. (2014). Review Past and present definitions of epileptogenesis and its biomarkers. Neurotherapeutics 11, 231–241. doi: 10.1007/s13311-014-0257-2
Pitkänen, A., Lukasiuk, K., Dudek, F. E., and Staley, K. J. (2015). Epileptogenesis. Cold Spring Harb. Perspect. Med. 5:a022822. doi: 10.1101/cshperspect.a022822
Porter, B. E., and Jacobson, C. (2013). Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav. 29, 574–577. doi: 10.1016/j.yebeh.2013.08.037
Qu, Y., Vadivelu, S., Choi, L., Liu, S., Lu, A., Lewis, B., et al. (2003). Neurons derived from embryonic stem (ES) cells resemble normal neurons in their vulnerability to excitotoxic death. Exp. Neurol. 184, 326–336. doi: 10.1016/j.expneurol.2003.07.001
Rajesh, M., Mukhopadhyay, P., Bátkai, S., Haskó, G., Liaudet, L., Drel, V. R., et al. (2007). Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am. J. Physiol. Heart Circ. Physiol. 293, H610–H619. doi: 10.1152/ajpheart.00236.2007
Rakhade, S. N., and Jensen, F. E. (2009). Epileptogenesis in the immature brain: emerging mechanisms. Nat. Rev. Neurol. 5:380. doi: 10.1038/nrneurol.2009.80
Ravizza, T., Rizzi, M., Perego, C., Richichi, C., Veliskova, J., Moshe, S. L., et al. (2005). Inflammatory response and glia activation in developing rat hippocampus after status epilepticus. Epilepsia 5, 113–117. doi: 10.1111/j.1528-1167.2005.01006.x
Rice, A. C., and De Lorenzo, R. J. (1998). NMDA receptor activation during status epilepticus is required for the development of epilepsy. Brain Res. 782, 240–247. doi: 10.1016/s0006-8993(97)01285-7
Rizzi, M., Perego, C., Aliprandi, M., Richichi, C., Ravizza, T., Colella, D., et al. (2003). Glia activation and cytokine increase in rat hippocampus by kainic acidinduced status epilepticus during postnatal development. Neurobiol. Dis. 14, 494–503. doi: 10.1016/j.nbd.2003.08.001
Romjin, H., Hofman, M., and Gramsberg, A. (1991). At what age is the developing cerebral cortex of the rat comparable to that the full-term newborn baby? Early Human dev. 26, 61–67. doi: 10.1016/0378-3782(91)90044-4
Rosenberg, E., Patra, P. H., and Whalley, B. J. (2017). Therapeutic effects of cannabinoids in animal models of seizures, epilepsy, epileptogenesis and epilepsy-related neuroprotection. Epilepsy Behav. 70, 319–327. doi: 10.1016/j.yebeh.2016.11.006
Ruhaak, L. R., Felth, J., Karlsson, P. C., Rafter, J. J., Verpoorte, R., and Bohlin, L. (2011). Evaluation of the cyclooxygenase inhibiting effects of six major cannabinoids isolated from Cannabis sativa. Biol. Pharm. Bull. 34, 774–778. doi: 10.1248/bpb.34.774
Sagredo, O. S., González, I., Aroyo, M. R., Pazos, C., Benito, I., Lastres-Becker, J. P., et al. (2009). Cannabinoid CB2 receptor agonists protect the striatum against malonate toxicity: relevance for Huntington’s disease. Glia 57, 1154–1167. doi: 10.1002/glia.20838
Sanchez, R. M., Koh, S., Rio, C., Wang, C., Lamperti, E. D., Sharma, D., et al. (2001). Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J. Neurosci. 21, 8154–8163. doi: 10.1523/JNEUROSCI.21-20-08154.2001
Schonhofen, P., Bristot, I. J., Crippa, J. A., Hallak, J. E. C., Zuardi, W. Z., Parsons, R. B., et al. (2018). Cannabinoid–based therapies and brain development: potential harmful efect of early modulation of the endocannabinoid system. CNS Drugs 32, 697–712. doi: 10.1007/s40263-018-0550-4
Sekar, K., and Pack, A. (2019). Epidiolex as adjunct therapy for treatment of refractory epilepsy: a comprehensive review with a focus on adverse effects. F1000Res. 8:234. doi: 10.12688/f1000research.16515.1
Semple, B. D., Dill, L. K., and O’Brien, T. J. (2020). Immune challenges and seizures: how do early life insults influence epileptogenesis? Front. Pharmacol. 11:2. doi: 10.3389/fphar.2020.00002
Shimada, T., Takemiya, T., Sugiura, H., and Yamagata, K. (2014). Role of inflammatory mediators in the pathogenesis of epilepsy. Mediators Inflamm. 2014:901902. doi: 10.1155/2014/901902
Silvestro, S., Mammana, S., Cavalli, E., Bramanti, P., and Mazzon, E. (2019). Use of cannabidiol in the treatment of epilepsy: efficacy and security in clinical trials. Molecules 24:1459. doi: 10.3390/molecules24081459
Skowrońska, K., Obara-Michlewska, M., Zielińska, M., and Albrecht, J. (2019). NMDA receptors in astrocytes: in search for roles in neurotransmission and astrocytic homeostasis. Int. J. Mol. Sci. 20:309. doi: 10.3390/ijms20020309
Sofia, R. D., Solomon, T. A., and Barry, H., 3rd (1976). Anticonvulsant activity of Δ9-tetrahydrocannabinol compared with three other drugs. Eur. J. Pharmacol. 35, 7–16. doi: 10.1016/0014-2999(76)90295-8
Solowij, N., Jones, K. A., Rozman, M. E., Davis, S. M., Ciarrochi, J., Heaven, C. L., et al. (2011). Verbal learning and memory in adolescent cannabis users, alcohol users and non-users. Psychopharmacology 216, 131–144. doi: 10.1007/s00213-011-2203-x
Specchio, N., Pietrafusa, N., and Cross, H. (2020). Source of cannabinoids: what is available, what is used, and where does it come from? Epileptic Disord. 22, 1–9. doi: 10.1684/epd.2019.1121
Stellwagen, D., Beattie, E. C., Seo, J. Y., and Malenka, R. C. (2005). Differential regulation of AMPA receptor and GABA receptor trafficking by tumor necrosis factor-alpha. J. Neurosci. 25, 3219–3228. doi: 10.1523/JNEUROSCI.4486-04.2005
Stott, N. K., Jones, C., Bazelot, N., and Whalley, B. J. (2018). “The proposed multimodal mechanism of action of cannabidiol in epilepsy: modulation of intracellular calcium and adenosine-mediated signaling”, American Epilepsy Society Annual Meeting, New Orleans, LA. Available online at: https://www.aesnet.org/meetings_events/annual_meeting_abstracts/view/554059.
Suchomelova, L., Lopez-Meraz, M. L., Niquet, J., Kubova, H., and Wasterlain, C. G. (2015). Hyperthermia aggravates status epilepticus-induced epileptogenesis and neuronal loss in immature rats. Neuroscience 305, 209–224. doi: 10.1016/j.neuroscience.2015.08.006
Sugaya, Y., Yamazaki, M., Uchigashima, M., Kobayashi, K., Watanabe, M., Sakimura, K., et al. (2016). Crucial roles of the endocannabinoid 2-arachidonoylglycerol in the suppression of epileptic seizures. Cell Rep. 16, 1405–1415. doi: 10.1016/j.celrep.2016.06.083
Szaflarski, J. P., Bebin, E. M., Comi, A. M., Patel, A. D., Joshi, C., Checketts, D., et al. (2018). Long-term safety and treatment effects of cannabidiol in children and adults with treatment-resistant epilepsies: expanded access program results. Epilepsia 59, 1540–1548. doi: 10.1111/epi.14477
Szczurowska, E., and Mareš, P. (2013). NMDA and AMPA receptors: development and status epilepticus. Physiol. Res. 1, 21–38. doi: 10.33549/physiolres.932662
Treat, L., Chapman, K. E., Colborn, K. L., and Knupp, K. G. (2017). Duration of use of oral cannabis extract in a cohort of pediatric epilepsy patients. Epilepsia 58, 123–127. doi: 10.1111/epi.13617
Tretter, L., Sipos, I., and Vizi, A. V. (2004). Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson’s disease. Neurochem. Res. 29, 569–577. doi: 10.1023/b:nere.0000014827.94562.4b
Turkanis, S. A., Smiley, K. A., Borys, H. K., Olsen, D. M., and Karler, R. (1979). An electrophysiological analysis of the anticonvulsant action of cannabidiol on limbic seizures in conscious rats. Epilepsia 20, 351–363. doi: 10.1111/j.1528-1157.1979.tb04815.x
Tyzio, R., Represa, A., Jorquera, I., Ben-Ari, Y., Gozlan, H., and Anikszejn, L. (1999). The establishment of GABAergic and Glutamatergic synapses on CA1 pyramidal neurons is sequential and correlates whit the development of the apical dendrite. J. Neurosci. 19, 10372–10382. doi: 10.1523/JNEUROSCI.19-23-10372.1999
Tzadok, M., Uliel-Siboni, S., Linder, I., Kramer, U., Epstein, O., Menascu, S., et al. (2016). CBD-enriched medical cannabis for intractable pediatric epilepsy: the current Israeli experience. Seizure 35, 41–44. doi: 10.1016/j.seizure.2016.01.004
Vega-García, A., Rocha, L., Guevara-Guzmán, R., Guerra-Araiza, C., Feria-Romero, I., Gallardo, J. M., et al. (2020). Magnolia officinalis reduces inflammation and damage induced by recurrent status epilepticus in immature rats. Curr. Pharm. Des. 26, 1388–1401. doi: 10.2174/1381612826666200320121813
Viviani, B., Bartesaghi, S., Gardoni, F., Vezzani, A., Behrens, M. M., Bartfai, T., et al. (2003). Interleukin-1β enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci. 23, 8692–8700. doi: 10.1523/JNEUROSCI.23-25-08692.2003
Wallace, M. J., Blair, R. E., Falenski, K. W., Martin, B. R., and DeLorenzo, R. J. (2003). The endogenous cannabinoid system regulates seizures frequency and duration in a model of temporal lobe epilepsy. J. Pharmacol. Exp. Ther. 307, 129–137. doi: 10.1124/jpet.103.051920
Wang, S., Cheng, Q., Malik, S., and Yang, J. (2000). Interleukin-1beta inhibits gamma-aminobutyric acid type A (GABAA) receptor current in cultured hippocampal neurons. J. Pharmacol. Exp Ther. 292, 497–504.
Wang, T., Collet, J. P., Shapiro, S., and Ware, M. A. (2008). Adverse effects of medical cannabinoids: a systematic review. CMAJ 178, 1669–1678. doi: 10.1503/cmaj.071178
Watanabe, K., Yamaori, S., Funahashi, T., Kimura, T., and Yamamoto, I. (2007). Cytochrome P450 enzymes involved in the metabolism of tetrahydrocannabinols and cannabinol by human hepatic microsomes. Life Sci. 80, 1415–1419. doi: 10.1016/j.lfs.2006.12.032
Wheless, J. W., Dlugos, D., Miller, I., Oh, D. A., Parikh, N., Phillips, S., et al. (2019). Pharmacokinetics and tolerability of multiple doses of pharmaceutical-grade synthetic cannabidiol in pediatric patients with treatment-resistant epilepsy. CNS Drugs 33, 593–604. doi: 10.1007/s40263-019-00624-4
Keywords: cannabinoids, epileptogenesis, neurodevelopment, neuroprotection, anti-inflammatory, pharmacokinetics
Citation: Vega-García A, Feria-Romero I, García-Juárez A, Munguia-Madera ACh, Montes-Aparicio AV, Zequeida-Muñoz E, Garcia-Albavera E and Orozco-Suárez S (2021) Cannabinoids: A New Perspective on Epileptogenesis and Seizure Treatment in Early Life in Basic and Clinical Studies. Front. Behav. Neurosci. 14:610484. doi: 10.3389/fnbeh.2020.610484
Received: 26 September 2020; Accepted: 26 November 2020;
Published: 12 January 2021.
Edited by:
Dolores E. López García, University of Salamanca, SpainReviewed by:
Orlando Jorge Castellano, University of Salamanca, SpainAlan Talevi, National University of La Plata, Argentina
Copyright © 2021 Vega-García, Feria-Romero, García-Juárez, Munguia-Madera, Montes-Aparicio, Zequeida-Muñoz, Garcia-Albavera and Orozco-Suárez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Orozco-Suárez, c29yb3pjbzVAaG90bWFpbC5jb20=