 Lian-Shun Zheng1,2
Lian-Shun Zheng1,2 Naoko Kaneko
Naoko Kaneko Kazunobu Sawamoto
Kazunobu Sawamoto- 1Department of Developmental and Regenerative Biology, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan
- 2Institute of Anatomy and Cell Biology, School of Medicine, Zhejiang University, Hangzhou, China
Interferon-alpha (IFN-α) is a proinflammatory cytokine that is widely used for the treatment of chronic viral hepatitis and malignancy, because of its immune-activating, antiviral, and antiproliferative properties. However, long-term IFN-α treatment frequently causes depression, which limits its clinical utility. The precise molecular and cellular mechanisms of IFN-α-induced depression are not currently understood. Neural stem cells (NSCs) in the hippocampus continuously generate new neurons, and some evidence suggests that decreased neurogenesis plays a role in the neuropathology of depression. We previously reported that IFN-α treatment suppressed hippocampal neurogenesis and induced depression-like behaviors via its receptors in the brain in adult mice. However, it is unclear how systemic IFN-α administration induces IFN-α signaling in the hippocampus. In this study, we analyzed the role of microglia, immune cells in the brain, in mediating the IFN-α-induced neurogenic defects and depressive behaviors. In vitro studies demonstrated that IFN-α treatment induced the secretion of endogenous IFN-α from microglia, which suppressed NSC proliferation. In vivo treatment of adult mice with IFN-α for 5 weeks increased the production of proinflammatory cytokines, including IFN-α, and reduced neurogenesis in the hippocampus. Both effects were prevented by simultaneous treatment with minocycline, an inhibitor of microglial activation. Furthermore, minocycline treatment significantly suppressed IFN-α-induced depressive behaviors in mice. These results suggest that microglial activation plays a critical role in the development of IFN-α-induced depression, and that minocycline is a promising drug for the treatment of IFN-α-induced depression in patients, especially those who are low responders to conventional antidepressant treatments.
Introduction
Interferon-alpha is a proinflammatory cytokine that is widely used for the treatment of chronic viral hepatitis and malignancy, because of its immune-activating, antiviral, and antiproliferative properties (Tagliaferri et al., 2005; Deutsch and Hadziyannis, 2008; Papatheodoridis et al., 2008). However, the use of IFN-α can result in the development of various side effects including digestive, skin, and neuropsychiatric symptoms (Dieperink et al., 2000; Malek-Ahmadi, 2001). Depression is the most prevalent and severe side effect, affecting approximately 30–45% of patients receiving IFN-α treatment, thus limiting its clinical utility (Bonaccorso et al., 2001; Lieb et al., 2006). While numerous studies have suggested the involvement of neurochemical and/or neuroendocrinological pathways in the development of IFN-α-induced depression (Schiepers et al., 2005; Reyes-Vazquez et al., 2012; Hoyo-Becerra et al., 2014), the underlying molecular and cellular mechanisms remained to be clarified.
In the adult mammalian brain, NSCs, which are located in the SGZ of the DG and in the ventricular–subventricular zone (V-SVZ) located at the lateral wall of lateral ventricles, generate new neurons daily throughout life (Altman and Das, 1965; Doetsch and Alvarez-Buylla, 1996; Jankovski and Sotelo, 1996; Eriksson et al., 1998; Gould et al., 1999; Petreanu and Alvarez-Buylla, 2002). Although the functional significance of adult-born neurons is not completely clear, there is growing evidence for their involvement in mood regulation (Samuels and Hen, 2011; Eisch and Petrik, 2012). We previously reported, using brain-specific IFN receptor knockout mice and hippocampal NSC cultures, that IFN-α directly suppresses proliferation of the hippocampal and V-SVZ NSCs and the generation of new neurons, and that it induces depressive behavioral phenotypes via the type 1 IFN receptor (IFNAR), expressed in the central nervous system (Kaneko et al., 2006; Zheng et al., 2014). These findings suggest that IFN-α may induce depression by directly affecting neural cells in the brain. However, in addition to the small fraction of circulating IFN-α passing through the blood–brain barrier, IFN-α is also produced locally in the brain (Dafny and Yang, 2005). Therefore, it is still unclear how systemic IFN-α administration induces IFNAR signaling in NSCs in the brain.
Inflammation has been implicated in the pathophysiology of depression (Dantzer et al., 2008; Miller et al., 2009). Consistent with this notion, higher levels of proinflammatory cytokines and inflammatory markers are found in the peripheral blood of depressed individuals, compared to controls (Maes, 1999; Schiepers et al., 2005; Dowlati et al., 2010). In the brain, the activation of microglia by various inflammatory stimuli (Garden and Moller, 2006; Dheen et al., 2007) leads to the release of excess proinflammatory cytokines, which contribute to the pathophysiology of neurological and psychiatric disorders, including depression (Hanisch and Kettenmann, 2007; Maes, 2011). Notably, several proinflammatory cytokines directly affect NSC proliferation and neurogenic functions (Das and Basu, 2008; Gonzalez-Perez et al., 2012), and microglial activation inhibits hippocampal neurogenesis (Ekdahl et al., 2003; Monje et al., 2003). In addition, IFN-α stimulates the production of a series of proinflammatory cytokines not only in the periphery, but also within the central nervous system (Raison et al., 2009; Felger et al., 2013). On the basis of these studies, we hypothesized that proinflammatory cytokines secreted by activated microglia are involved in IFN-α-induced depression.
Here, we found that chronic IFN-α treatment increased the production of proinflammatory cytokines, including endogenous IFN-α, and reduced neurogenesis in the adult mouse hippocampus, and that simultaneous treatment with minocycline, an inhibitor of microglial activation, prevented these effects. Furthermore, minocycline treatment significantly suppressed IFN-α-induced depression-like behaviors in mice. These results suggest that microglial activation mediates the development of IFN-α-induced depression.
Materials and Methods
Animals
Male 8-week-old C57BL/6J mice were purchased from SLC (Shizuoka, Japan). All experiments using live animals were performed in accordance with the guidelines and regulations of Nagoya City University.
Administration of IFN-α, Minocycline, and Bromodeoxyuridine
Mice were given daily intraperitoneal (i.p.) injections of either phosphate-buffered saline (PBS) or mouse IFN-α (Miltenyi Biotec, Auburn, CA, USA) diluted with PBS at a dose of 4 × 105 IU/kg, for 4 or 5 weeks; this dosage is reported to induce depression-like behavioral changes (Zheng et al., 2014). Minocycline (Sigma, Saint Louis, MO, USA) diluted with PBS was injected i.p. at a dose of 50 mg/kg (Li et al., 2013) for 2 d prior to and throughout the IFN-α-treatment period.
To label newly generated cells, at the beginning of the 5th week of IFN-α treatment, mice were injected with bromodeoxyuridine (BrdU; Sigma, 50 mg/kg, i.p., in PBS) once every 8 h for a total of six injections.
Cell Culture and Treatments
The murine microglia cell line BV-2 (Blasi et al., 1990) was cultured at 37°C in 5% CO2 in the following maintenance medium: Dulbecco’s modified Eagle’s medium/Nutrient mixture F12 (DMEM/F12, Gibco, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS, Gibco) and 1% penicillin–streptomycin. The cells were plated into 24-well culture plates (1 × 105 cells/well) for ELISA or 100-mm culture dishes (1.2 × 106 cells/dish) for collecting conditioned media (CM). For preparation of the CM the BV-2 microglia were treated with IFN-α (1 × 103 IU/ml) or PBS (unstimulated controls) for 6 h, and then incubated in fresh medium without FBS. CM samples were collected after 6, 12, 18, 24, and 48 h of incubation.
Adult hippocampal NSC cultures were derived from adult female Fischer 344 rats (Gage et al., 1995) and maintained in DMEM/F12 medium (Invitrogen, Carlsbad, CA, USA) containing 1% N2 supplement (Invitrogen) and 20 ng/ml fibroblast growth factor-2 (FGF-2, PeproTech, Rocky Hill, NJ, USA). The NSCs were plated into 24-well plates at a density of 3 × 104 cells/well. After 24 h, the microglial CM (collected after 24 h of incubation), with or without 1 μg/ml anti-IFN-α neutralizing antibody, (R&D Systems, Piscataway, NJ, USA), was added to the NSC culture. To label proliferating cells, the NSCs were treated with 10 μM BrdU (dissolved in cell culture medium) for the last 4 h of the 48 h incubation period. The results reported here are based on data obtained from three independent experiments.
Real-Time PCR
To examine the mRNA levels of proinflammatory cytokines, mice were deeply anesthetized, sacrificed, and their hippocampal tissues collected at 2, 6, and 24 h after the final injection of IFN-α and/or minocycline. Total RNA was extracted from the hippocampal tissues with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and cDNA synthesis was performed using the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen). Quantitative SYBR Green real time PCR was carried out as follows. Briefly, each 25 μl SYBR Green reaction consisted of 5 μl of cDNA (50 ng/μl), 12.5 μl of 2× Universal SYBR Green PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA), and 3.75 μl each of 50 nM forward and reverse primers. Optimization was performed for each gene-specific primer prior to the experiment, to confirm that the 50 nM primer concentrations did not produce non-specific primer-dimer amplification signals in the no-template control tubes. Primer sequences were designed using Primer Express Software (Applied Biosystems). Quantitative RT-PCR was performed on an ABI 7500 Fast Real-Time PCR instrument (Applied Biosystems) by using the following 3-stage program parameters provided by the manufacturer: 2 min at 50°C, 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. Each sample was tested in duplicate, and the results obtained from three independent experiments were used to calculate the means and SD. The data are expressed as the fold change in gene expression relative to the PBS control group. The following primers were used: 5′-AAGGACAGGAAGGATTTTGGATT-3′ and 5′-GAGCCTTCTGGATCTGTTGGTT-3′, which amplify a 64-bp IFN-α product; 5′-TTGACGGACCCCAAAAGAT-3′ and 5′-GAAGCTGGATGCTCTCATCAG-3′, which amplify a 75-bp IL-1β product; 5′-TGATGGATGCTACCAAACTGGA-3′ and 5′-TGGTACTCCAGAAGACCAGAGG-3′, which amplify a 75-bp IL-6 product; 5′-CACAAGATGCTGGGACAGTGA-3′, and 5′-TCCTTGATGGTGGTGCATGA-3′, which amplify a 58-bp TNF-α product; and 5′-CATGGCCTTCCGTGTTCCTA-3′ and 5′-CACGTCAGATCCA-3′, which amplify a 55-bp GAPDH product.
Elisa Analysis
BV-2 microglia were incubated with IFN-α (103 IU/ml) for 6 h, washed carefully with PBS to remove IFN-α from the cultures, and then incubated in the maintenance medium for 6, 12, 18, 24, or 48 h. The levels of IFN-α, IL-1β, IL-6, and TNF-α released into the media were measured using ELISA kits (Boster Biological Technology, Wuhan, China), according to the manufacturer’s instructions. The data are presented as fold changes, relative to the control sample levels.
Immunohistochemistry
Immediately following the treatment period, the mice were deeply anesthetized and fixed by transcardiac perfusion with 4% paraformaldehyde in 0.1 M phosphate buffer. Brain sections were prepared and stained as previously described (Kaneko et al., 2010). Briefly, following post-fixation with 4% paraformaldehyde in 0.1 M phosphate buffer overnight, the brains were cut into 50-μm-thick coronal sections on a vibratome (VT1200S, Leica, Wetzlar, Germany). The sections were incubated for 40 min in 1% H2O2 in PBS, 1 h in blocking solution (10% donkey serum and 0.2% Triton X-100 in PBS), overnight at 4°C with the primary antibodies, and 2 h at room temperature with Alexa Fluor-conjugated secondary antibodies (1:500, Invitrogen). Signal amplification was performed with biotinylated secondary antibodies (1:500, Jackson Laboratory, West Grove, PA, USA) and the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA), and the signals were visualized using the TSA Fluorescence System (PerkinElmer, Waltham, MA, USA). For BrdU staining, the sections were pretreated with 2 N HCl for 40 min at 60°C before H2O2 incubation. The following primary antibodies were used: rat anti-BrdU (1:200, Abcam, Cambridge, MA, USA); rabbit anti-Iba1 (1:2000, Wako, Osaka, Japan); rabbit anti-Ki67 (1:500, Leica); rabbit anti-TBR2 (1:200, Abcam); goat anti-doublecortin (DCX; 1:100, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Nuclei were stained with Hoechst 33342 (1 μg/ml, Sigma).
Immunocytochemistry
As described previously (Palmer et al., 1999), cells on coverslips were rinsed in PBS and fixed with 4% PFA in 0.1 M phosphate buffer at room temperature for 15 min. After a 1-h pre-incubation in blocking solution (10% donkey serum and 0.2% Triton X-100 in PBS), the cells were incubated with primary antibodies at 4°C overnight, and at room temperature for 2 h with Alexa Fluor-conjugated secondary antibodies (1:500, Invitrogen). For BrdU staining, the cells were pretreated with 2 N HCl for 30 min at 37°C before blocking. The following primary antibodies were used: rat anti-BrdU (1:1000) and chicken anti-Nestin (1:2000, Aves Labs, Tigard, OR, USA).
Imaging Quantification
The hippocampal sections were processed and stained as described above. To visualize and quantify the Ki67- and TBR2-immunoreactive cells, images of the labeled samples were captured using a fluorescence microscope (BX-51, Olympus, Tokyo, Japan) with a 20× objective. To visualize and quantify BrdU+DCX+ cells, confocal z-stack images with a step size of 1 μm were captured using a confocal laser microscope LSM700 (Carl Zeiss, Jena, TH, Germany) with a 40× objective, and only the cells with a BrdU+ nucleus completely surrounded by DCX+ cytoplasm were counted. To visualize and quantify Iba1+ cells, confocal z-stack images with a step of 4 μm were captured using a confocal laser microscope LSM700 (Carl Zeiss) with a 20× objective. In all of the histological analyses, the actual number of cells in every sixth 50-μm-thick coronal section was counted bilaterally, and the obtained cell number was multiplied by six to obtain the total number of cells per DG.
For analysis of the in vitro stimulated cells, the double-labeled cells were examined with a confocal laser microscope (LSM700, Carl Zeiss). Three optical fields were randomly chosen under a 20× objective from each well for quantification. The percentage of BrdU+ NSCs was calculated by dividing the number of BrdU+Nestin+ cells by the total number of Nestin+ cells in the same field.
Tail Suspension Test
Immediately after the 5-week IFN-α/minocycline treatment, the mice were subjected to the tail suspension test as described previously (Steru et al., 1985). Briefly, the mice were suspended 35 cm above the floor in a visually isolated area by adhesive tape placed 1–1.5 cm from the tip of the tail. The escape movement and duration of immobility were recorded over a 6-min test period. Immobility lasting for less than 2 s was not included in the analysis. The time spent in an immobile posture was measured as an index of depression-like behavior.
Porsolt Forced Swim Test
The Porsolt forced swim test was performed 24 h after the tail suspension test, as described previously (Porsolt et al., 1977). Briefly, the mice were placed in a vertical glass cylinder (35 cm height × 15 cm diameter) filled with 25 cm water at 23 ± 1°C. The escape movement and duration of immobility were recorded over a 6-min test period. Immobility was defined as only those movements required to keep the mouse afloat, and immobility lasting for less than 2 s was not included in the analysis. The time spent in an immobile posture was measured as an index of depression-like behavior.
Statistical Analysis
All data were expressed as the mean ± SEM. Differences between the means were determined by the two-tailed Student’s t-test, one-way ANOVA, or two-way repeated measures ANOVA, followed by a Tukey–Kramer multiple comparison test, unless specified otherwise. A P-value of <0.05 was considered significant.
Results
Minocycline Attenuates IFN-α-Induced Production of Proinflammatory Cytokines in the Hippocampus
Previously, we found that mice subjected to peripheral IFN-α treatment (4 × 105 IU/kg, i.p., once daily for 5 weeks) exhibited suppressed neurogenesis (Zheng et al., 2014). To determine if peripheral IFN-α treatment leads to hippocampal inflammation, mice were subjected to the same IFN-α treatment regimen, the hippocampal tissues were collected at 2, 6, and 24 h after the final injection of IFN-α, and the expression of proinflammatory cytokines was quantified by real-time PCR (Figure 1A). IFN-α treatment was previously shown to increase the IL-1β, IL-6, and TNF-α levels in the brain (Kaneko et al., 2006; Raison et al., 2009). Consistent with these reports, the mRNA levels of these cytokines in the hippocampus were significantly upregulated by IFN-α administration, exhibiting ∼2-, 3-, and 8-fold increases compared with the control PBS-treated group at 6 h after treatment (Figure 1A), suggesting that our treatment protocol efficiently stimulated hippocampal inflammation in the mouse. We also found that our IFN-α treatment protocol increased the IFN-α mRNA level in the hippocampus, resulting in a greater than fourfold increase over background levels after 6 h of treatment, suggesting that peripherally administered IFN-α can enhance IFN-α signaling in the hippocampus.
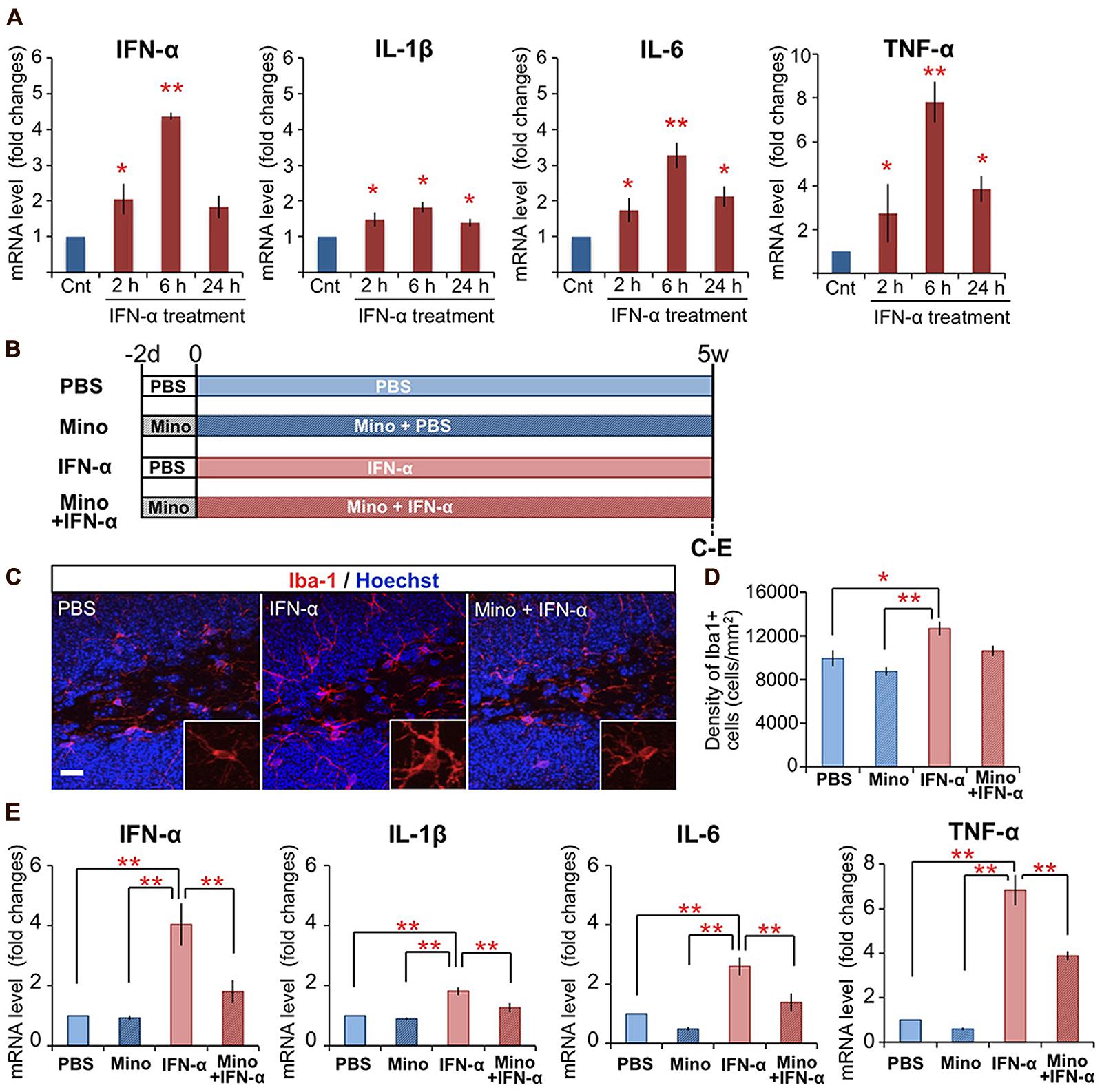
FIGURE 1. Chronic treatment with minocycline inhibits IFN-α-induced proinflammatory cytokine expression in the hippocampus. (A) Time-dependent alteration of IFN-α, IL-1β, IL-6, and TNF-α mRNA levels in the hippocampus at 2, 6, and 24 h following the final IFN-α injection of the 5-week treatment. The mRNAs were quantified by real-time PCR, and the results are expressed as relative values, compared to the PBS-treated control group. n = 3 mice per group. *P < 0.05, **P < 0.01 versus the PBS-treated group. (B) Experimental design of the minocycline study. (C–E) Effect of minocycline on IFN-α-induced microglial activation in the hippocampus. Coronal brain sections prepared after the 5-week treatment with IFN-α in the presence or absence of minocycline, immunostained for Iba1, a microglial marker (red) with Hoechst nuclear staining (blue) (C). Density of Iba1+ microglia in the DG and hilus (D). Relative IFN-α, IL-1β, IL-6, and TNF-α mRNA levels in the hippocampus quantified by real-time PCR (E). n = 5 mice per group. *P < 0.05, **P < 0.01. Error bars: means ± SEM; Scale bar, 25 μm.
As microglia are the primary immune cells that produce proinflammatory cytokines in response to inflammatory stimuli in the brain, we examined the activation of microglia in IFN-α-treated mice by immunostaining for Iba1, a microglial marker (Figures 1B–D). In the DG and hilus of the hippocampus, in contrast to the ramified microglia in the control PBS-treated group, which had elongated thin processes extending from a small soma (Figure 1C, left), microglia in the IFN-α treatment group exhibited a hypertrophic morphology with a larger soma and thicker processes (Figure 1C, middle), typical of activated microglia (Schiepers et al., 2005; Giunti et al., 2014). IFN-α treatment also significantly increased the density of Iba1+ microglia in the hippocampus, including the DG and hilus (Figure 1D) and CA3 (Supplementary Figure 1A), but not in other brain regions implicated in the pathology of depression (Belzung et al., 2014; Figure 1D; Supplementary Figure 1A). These findings suggested that the 4-week IFN-α treatment resulted in microglial activation in the hippocampus.
Minocycline, a tetracycline-type antibiotic with anti-inflammatory properties, readily crosses the blood–brain barrier and suppresses microglial activation (Garrido-Mesa et al., 2013). To investigate its ability to suppress the IFN-α-induced microglial activation, minocycline treatment was begun 2 days before the 5-week IFN-α treatment, and continued for the duration of the experiment (Figure 1B). The minocycline treatment suppressed both the IFN-α-induced hypertrophic change in microglial morphology (Figure 1C, right) and the increase in Iba1+ cell density in the DG and hilus (Figure 1D). Moreover, minocycline treatment significantly suppressed the IFN-α-induced up-regulation of proinflammatory cytokine mRNAs in the hippocampus (Figure 1E). Taken together, these results suggest that IFN-α increases the expression of endogenous proinflammatory cytokines, including IFN-α, via microglial activation in the hippocampus.
IFN-α-Treated Microglia Secrete Proinflammatory Cytokines and Suppress Proliferation of Adult Hippocampal NSCs
To investigate the direct effects of IFN-α on microglia in vitro, microglia-derived BV-2 cells were treated with IFN-α (1 × 103 IU/ml) for 6 h. After being carefully washed to remove IFN-α, the cells were cultured in fresh medium without FBS (Figure 2A). To determine the levels of IFN-α, IL-1β, IL-6, and TNF-α released from the microglia, culture supernatants were collected at 6, 12, 18, 24, and 48 h after IFN-α removal, and analyzed by ELISA (Figure 2B). The concentrations of these cytokines were significantly increased at all time points in the IFN-α-treated group, compared with those in the PBS-treated control group, indicating that IFN-α stimulates the microglial production of proinflammatory cytokines.
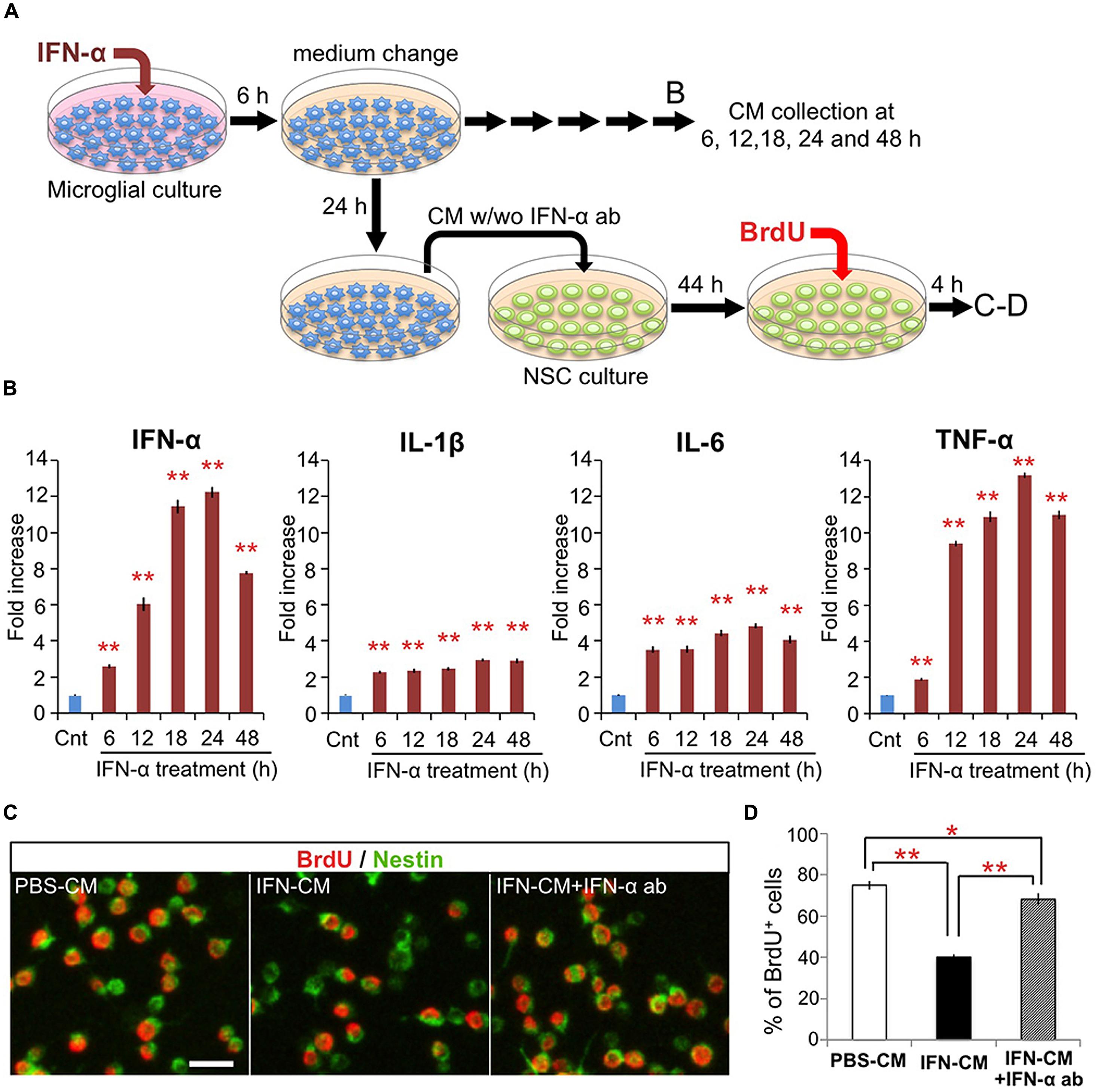
FIGURE 2. IFN-α-stimulated microglia suppress proliferation of hippocampal NSCs by production and release of IFN-α. (A) Experimental design. (B) Proinflammatory cytokine levels in the conditioned media (CM) of microglia following incubation with IFN-α. The CM samples were collected before (control) or 6, 12, 18, 24, and 48 h after the 6 h incubation with IFN-α. The concentrations of IFN-α, IL-1β, IL-6, and TNF-α were quantified by ELISA and are expressed as relative values compared to the control. n = 6 cultures per group; Cnt, control. (C,D) Proliferation of the cultured hippocampal NSCs incubated with the CM from PBS or IFN-α-stimulated microglia (PBS-CM or IFN-CM, respectively), or with IFN-CM supplemented with an IFN-α neutralizing antibody (IFN-CM + IFN-α ab). NSCs were immunostained for Nestin and BrdU (C), then the percentage of BrdU+ cells in the Nestin+ population was quantified (D). n = 6 cultures per group. *P < 0.05, **P < 0.01. Error bars: means ± SEM, Scale bar, 20 μm.
We next examined the effects of the microglial conditioned medium (CM) on cultured adult hippocampal NSCs. The microglial CM, collected 24 h after the 6 h treatment with IFN-α (IFN-CM) or PBS (PBS-CM; Figure 2A), was added to the NSC cultures, and the cells were incubated for 48 h. During the last 4 h of the incubation, BrdU was added to label the proliferating NSCs. The percentage of BrdU+ cells in the Nestin+ population of the IFN-CM-treated culture was significantly reduced compared with that in the PBS-CM-treated culture (Figures 2C,D). These data suggest that secreted factors from the IFN-α-stimulated microglia suppressed the proliferation of the hippocampal NSCs.
We recently reported that IFN-α decreases the proliferation of cultured hippocampal NSCs without affecting their differentiation (Zheng et al., 2014). Therefore, we examined whether the IFN-α produced by the IFN-α-stimulated microglia was responsible for suppressing NSC proliferation, by adding a neutralizing anti-IFN-α antibody (1 μg/ml) to the IFN-CM just before its incubation with NSCs. The impaired proliferation of NSCs in response to IFN-CM was significantly, although not completely, restored by the anti-IFN-α antibody (Figure 2D), suggesting that IFN-α present in the microglial CM played an important role in suppressing NSC proliferation. Taken together, these results suggest that the in vivo IFN-α treatment promotes the microglial production and secretion of IFN-α, which suppresses NSC proliferation.
Minocycline Ameliorates IFN-α-Induced Neurogenic Defects and Depression-Like Behaviors
We finally tested whether the IFN-α-induced defects in hippocampal neurogenesis and induction of depressive behaviors (Zheng et al., 2014) could be ameliorated by an anti-microglial agent. Mice were treated with minocycline (50 mg/kg) for 2 d prior to and during the IFN-α (4 × 105 IU/kg) treatment (Figure 3A). After the 4-week treatment, brain sections were immunostained for the proliferation marker Ki67 (Figure 3B) and the neuronal progenitor marker TBR2 (Figure 3C). To quantify newly generated neurons, the mice were injected with BrdU once every 8 h for a total of six injections, at the beginning of the 5th week of the IFN-α/minocycline treatment, and the brain sections were immunostained for BrdU and DCX, a marker of immature neurons. The decreased numbers of Ki67+cells (Figure 3D), TBR2+ cells (Figure 3E), and BrdU+DCX+ cells in the IFN-α-treated group (Figures 3F,G) were significantly restored by the simultaneous treatment with minocycline.
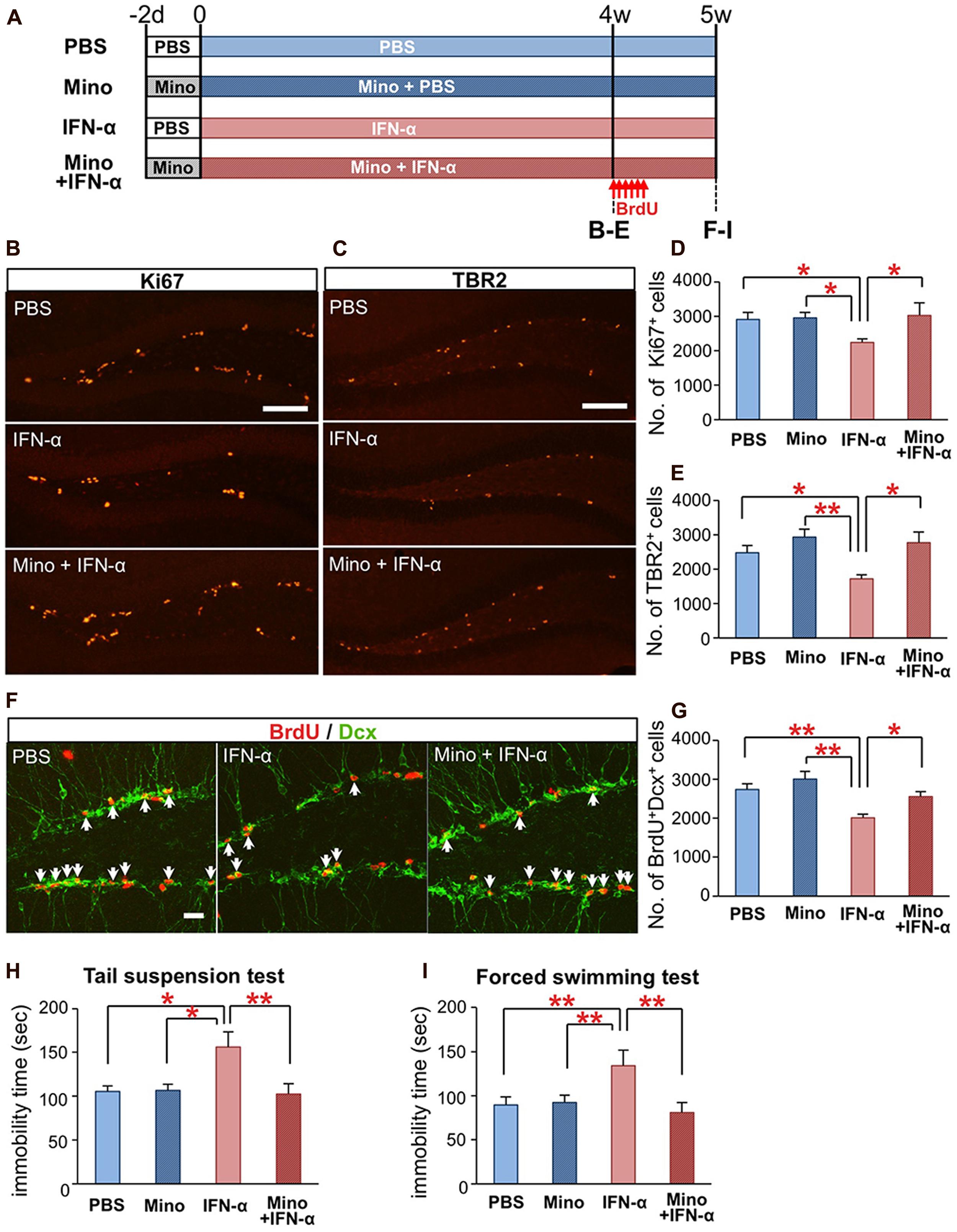
FIGURE 3. Minocycline treatment ameliorates IFN-α-induced neurogenic defects and depressive behaviors. (A) Experimental design. (B–E) Quantification of proliferating cells in the SGZ following the 4-week treatment with PBS or IFN-α, in the absence or presence of minocycline (Mino). The brain sections were immunostained for the proliferation marker, Ki67 (B) and neuronal progenitor marker, TBR2 (C), and then the Ki67+ and TBR2+ cells in the SGZ were counted and compared among the treatment groups (Ki67, D; TBR2, E). n = 7 mice per group. (F,G) Quantification of newly generated neurons in the DG after the 5-week IFN-α treatment. The new neurons were labeled with BrdU administered at the beginning at the 5th week of the IFN-α treatment and visualized by immunostaining for BrdU and DCX, a marker of immature neurons (F) The number of BrdU+DCX+ cells in the DG was counted and compared among the groups. n = 5 mice per group. (H,I) The effects of IFN-α and minocycline on depression-like behaviors in mice. After the final injection of IFN-α and/or minocycline, the mice were subjected to the tail suspension (H) and forced swimming test (I). The immobility times observed in these tests were quantified and compared among the groups. n = 10 mice per group. *P < 0.05; **P < 0.01; Error bars: means ± SEM; Scale bars: (B,C) = 100 μm; (F) = 25 μm.
Interferon-alpha-treated mice exhibit increased immobility in the tail-suspension (Steru et al., 1985) and forced swimming tests (Porsolt et al., 1977), typical depression-like behavioral phenotypes in rodents (Zheng et al., 2014). Simultaneous minocycline administration significantly reduced the observed immobility times in these tests (Figures 3H,I). Minocycline did not alter neurogenesis or behavior in mice that did not receive the IFN-α treatment (Figures 3D,E,G–I). Taken together, these results suggest that minocycline treatment ameliorated IFN-α-induced defects in hippocampal neurogenesis and depression-like behaviors via the suppression of microglial activation.
Discussion
Depression is a major and serious side effect of IFN-α that limits its use as an antiviral and antitumor drug; however, the underlying mechanism of IFN-α-induced depression remains unclear. In this study, we demonstrated that systemic IFN-α treatment caused microglial activation and cytokine production in the hippocampus. The IFN-α-induced decrease in hippocampal neurogenesis and increase in depression-like behaviors were significantly improved by co-administration of the anti-microglial agent minocycline. These results suggest that microglia play an important role in the development of depression during IFN-α treatment.
Although new neuron production was responsive to IFN-α and minocycline, the density of NeuN+ mature neurons in the granule cell layer in the DG was not affected by these treatments, similar to those in the CA3, amygdala, prefrontal cortex, and cingulate cortex, non-neurogenic areas implicated in the pathology of depression (Hulvershorn et al., 2011; Belzung et al., 2014; Supplementary Figure 1B). This lack of effect might be due to the small contribution of new neurons to the neuronal network in the DG (Ninkovic et al., 2007; Imayoshi et al., 2008). Consistent with this possibility, we did not find any differences in the expression of MAP2, PSD95, or Synaptophysin1, markers of synaptic plasticity (VanGuilder et al., 2010), in the projection area of the new neurons (hilus and CA3) among the treatment groups (Supplementary Figure 1C). Together with our previous findings that IFN-α suppresses the proliferation and survival of NSCs, but not those of differentiated cells (Zheng et al., 2014), these data support our hypothesis that NSCs are susceptible to IFN-α, an effect that may play a role in the pathology of IFN-induced depression.
Microglia are activated by various inflammatory stimuli; however, the effect of IFN-α treatment on these cells has been unclear. Our in vitro experiments revealed that microglia exposed to IFN-α exhibited increased proinflammatory cytokine release (Figure 2B), suggesting that IFN-α can directly activate microglia. Interestingly, the density of microglia in the hippocampus, but not in other regions implicated in the pathology of depression (Hulvershorn et al., 2011; Belzung et al., 2014), was significantly increased (Figure 1D; Supplementary Figure 1A). These data suggest that microglia in the hippocampus are more sensitive to IFN-α than the microglia in other brain regions. It is also possible that the hippocampal microglia were exposed to a higher concentration of IFN-α due to their location close to the ventricle, although the precise pharmacological kinetics of IFN-α in the brain is unknown. Previous studies indicate that microglia have diverse context-dependent effects on NSCs (Garden and Moller, 2006; Gonzalez-Perez et al., 2012; Gemma and Bachstetter, 2013). They modulate the proliferation, survival, and neuronal differentiation of NSCs and their progeny by releasing cytokines (Aarum et al., 2003; Monje et al., 2003; Butovsky et al., 2006; Iosif et al., 2006; Choi et al., 2008; Vukovic et al., 2012; Shigemoto-Mogami et al., 2014), and remove apoptotic cells by phagocytosis (Sierra et al., 2010). Here we found that the CM of IFN-α-stimulated microglia reduced the proliferation of cultured hippocampal NSCs. Notably, this effect was significantly suppressed in the presence of an IFN-α neutralizing antibody (Figure 2D). Considering that IFN-α inhibits NSC proliferation (Zheng et al., 2014), it is likely that the in vivo administration of IFN-α induces microglial IFN-α expression, which suppresses NSC proliferation.
We recently reported that the IFN-α-induced decrease in hippocampal neurogenesis and depressive behavior was mediated by IFNAR expressed in Nestin+ NSCs and their progeny (Zheng et al., 2014). In addition to a small fraction of peripheral IFN-α that enters the brain (Vitkovic et al., 2000), it is possible that the activation of IFNAR signaling in NSCs involves endogenous IFN-α expressed in the brain. In IFN-α treated patients, elevated plasma concentrations of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α were correlated with the induction of depressive symptoms (Bonaccorso et al., 2001; Raison et al., 2009, 2010). If these cytokines or other proinflammatory factors/cells reach the hippocampus, they may also cause microglial activation. Thus, hippocampal inflammation may be involved in mediating microglial activation induced by peripherally administered IFN-α, resulting in the induction of depression.
Minocycline is a tetracycline derivative that has powerful anti-inflammatory, anti-apoptotic, and antioxidant properties that are independent of its antibacterial activity. Because of its high lipid solubility, minocycline easily crosses the blood–brain barrier (Brogden et al., 1975), where it efficiently suppresses microglial activation and protects neurons and glial cells in various brain disease models in which the inflammatory process has been implicated, including ischemic stroke, traumatic injury, and neurodegenerative diseases (Plane et al., 2010; Garrido-Mesa et al., 2013). Minocycline treatment also improves depression-like behaviors in animals and depressive symptoms in patients (Kim and Suh, 2009; Soczynska et al., 2012), although the underlying molecular and cellular mechanisms by which minocycline functions remain largely unknown. Here, we demonstrated that minocycline significantly improved IFN-α-induced depression-like behaviors while concomitantly suppressing microglial activation and restoring hippocampal neurogenesis (Figure 3). Minocycline treatment has been reported to protect NSCs, neuronal progenitors, and new neurons, resulting in increased neurogenesis in the hippocampus (Ekdahl et al., 2003; Liu et al., 2007). Therefore, minocycline treatment can promote neurogenesis both directly by impacting NSCs and their progeny, and indirectly via microglial suppression. As reduced hippocampal neurogenesis is thought to be involved in the neuropathology of depression, minocycline’s antidepressant-like action in the IFN-α-treated mice may be due to its ability to promote neurogenesis.
In conclusion, the results of our study implicate a microglia-mediated mechanism in the development of IFN-induced depression. Further studies are needed to determine the precise mechanism by which minocycline improves depressive behavior. Although treatment with antidepressants, including the selective serotonin reuptake inhibitors (SSRIs) can reduce the development and symptoms of IFN-induced depression (Musselman et al., 2001; Capuron et al., 2002; Hauser et al., 2002; Raison et al., 2007; McNutt et al., 2012), some patients are refractory to the conventional antidepressant treatment. Our data suggest that minocycline may be a promising drug for the treatment of these patients.
Author Contributions
Lian-Shun Zheng and Naoko Kaneko performed experiments and analyzed data. Naoko Kaneko and Kazunobu Sawamoto designed the study. Lian-Shun Zheng, Naoko Kaneko, and Kazunobu Sawamoto wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Seiji Hitoshi (Shiga University of Medical Science) and Hiroaki Wake (National Institute for Physiological Sciences) for their valuable comments on this manuscript, Dr. Ichiro Miyoshi (Nagoya City University) for technical support, and members of the Sawamoto Laboratory for valuable discussions. This work was supported by a Research grant from the Japanese Ministry of Health, Labour and Welfare (to Naoko Kaneko, and Kazunobu Sawamoto), the Funding Program for the Next Generation World-Leading Researchers Grant Number LS104 (to Kazunobu Sawamoto), and JSPS KAKENHI Grant Numbers 23680041 and 25111727 (to Naoko Kaneko), and 26250019 (to Kazunobu Sawamoto), grants from Takeda Science Foundation (to Naoko Kaneko and Kazunobu Sawamoto), Terumo Life Science Foundation (to Kazunobu Sawamoto), Brain Science Foundation (to Naoko Kaneko), and Zhejiang Provincial Natural Science Foundation of China (No. LQ12H09004 to Lian-Shun Zheng).
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fncel.2015.00005/abstract
Abbreviations
IFN-α, interferon-alpha; NSC, neural stem cell; SGZ, subgranular zone; DG, dentate gyrus; IFNAR, type 1 interferon receptor; IL, interleukin; TNF, tumor necrosis factor.
References
Aarum, J., Sandberg, K., Haeberlein, S. L., and Persson, M. A. (2003). Migration and differentiation of neural precursor cells can be directed by microglia. Proc. Natl. Acad. Sci. U.S.A. 100, 15983–15988. doi: 10.1073/pnas.2237050100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Altman, J., and Das, G. D. (1965). Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J. Comp. Neurol. 124, 319–335. doi: 10.1002/cne.901240303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belzung, C., Willner, P., and Philippot, P. (2014). Depression: from psychopathology to pathophysiology. Curr. Opin. Neurobiol. 30C, 24–30. doi: 10.1016/j.conb.2014.08.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blasi, E., Barluzzi, R., Bocchini, V., Mazzolla, R., and Bistoni, F. (1990). Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J. Neuroimmunol. 27, 229–237. doi: 10.1016/0165-5728(90)90073-V
Bonaccorso, S., Puzella, A., Marino, V., Pasquini, M., Biondi, M., Artini, M.,et al. (2001). Immunotherapy with interferon-alpha in patients affected by chronic hepatitis C induces an intercorrelated stimulation of the cytokine network and an increase in depressive and anxiety symptoms. Psychiatry Res. 105, 45–55. doi: 10.1016/S0165-1781(01)00315-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brogden, R. N., Speight, T. M., and Avery, G. S. (1975). Minocycline: a review of its antibacterial and pharmacokinetic properties and therapeutic use. Drugs 9, 251–291. doi: 10.2165/00003495-197509040-00005
Butovsky, O., Ziv, Y., Schwartz, A., Landa, G., Talpalar, A. E., Pluchino, S.,et al. (2006). Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci. 31, 149–160. doi: 10.1016/j.mcn.2005.10.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Capuron, L., Gumnick, J. F., Musselman, D. L., Lawson, D. H., Reemsnyder, A., Nemeroff, C. B.,et al. (2002). Neurobehavioral effects of interferon-alpha in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 26, 643–652. doi: 10.1016/S0893-133X(01)00407-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Choi, S. H., Veeraraghavalu, K., Lazarov, O., Marler, S., Ransohoff, R. M., Ramirez, J. M.,et al. (2008). Non-cell-autonomous effects of presenilin 1 variants on enrichment-mediated hippocampal progenitor cell proliferation and differentiation. Neuron 59, 568–580. doi: 10.1016/j.neuron.2008.07.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dafny, N., and Yang, P. B. (2005). Interferon and the central nervous system. Eur. J. Pharmacol. 523, 1–15. doi: 10.1016/j.ejphar.2005.08.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dantzer, R., O’Connor, J. C., Freund, G. G., Johnson, R. W., and Kelley, K. W. (2008). From inflammation to sickness and depression: when the immune system subjugates the brain. Nat. Rev. Neurosci. 9, 46–56. doi: 10.1038/nrn2297
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Das, S., and Basu, A. (2008). Inflammation: a new candidate in modulating adult neurogenesis. J. Neurosci. Res. 86, 1199–1208. doi: 10.1002/jnr.21585
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deutsch, M., and Hadziyannis, S. J. (2008). Old and emerging therapies in chronic hepatitis C: an update. J. Viral. Hepat. 15, 2–11.
Dheen, S. T., Kaur, C., and Ling, E. A. (2007). Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 14, 1189–1197. doi: 10.2174/092986707780597961
Dieperink, E., Willenbring, M., and Ho, S. B. (2000). Neuropsychiatric symptoms associated with hepatitis C and interferon alpha: a review. Am. J. Psychiatry 157, 867–876. doi: 10.1176/appi.ajp.157.6.867
Doetsch, F., and Alvarez-Buylla, A. (1996). Network of tangential pathways for neuronal migration in adult mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 93, 14895–14900. doi: 10.1073/pnas.93.25.14895
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dowlati, Y., Herrmann, N., Swardfager, W., Liu, H., Sham, L., Reim, E. K.,et al. (2010). A meta-analysis of cytokines in major depression. Biol. Psychiatry 67, 446–457. doi: 10.1016/j.biopsych.2009.09.033
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eisch, A. J., and Petrik, D. (2012). Depression and hippocampal neurogenesis: a road to remission? Science 338, 72–75. doi: 10.1126/science.1222941
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ekdahl, C. T., Claasen, J. H., Bonde, S., Kokaia, Z., and Lindvall, O. (2003). Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. U.S.A. 100, 13632–13637. doi: 10.1073/pnas.2234031100
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Eriksson, P. S., Perfilieva, E., Bjork-Eriksson, T., Alborn, A. M., Nordborg, C., Peterson, D. A.,et al. (1998). Neurogenesis in the adult human hippocampus. Nat. Med. 4, 1313–1317. doi: 10.1038/3305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Felger, J. C., Li, L., Marvar, P. J., Woolwine, B. J., Harrison, D. G., Raison, C. L.,et al. (2013). Tyrosine metabolism during interferon-alpha administration: association with fatigue and CSF dopamine concentrations. Brain Behav. Immun. 31, 153–160. doi: 10.1016/j.bbi.2012.10.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gage, F. H., Coates, P. W., Palmer, T. D., Kuhn, H. G., Fisher, L. J., Suhonen, J. O.,et al. (1995). Survival and differentiation of adult neuronal progenitor cells transplanted to the adult brain. Proc. Natl. Acad. Sci. U.S.A. 92, 11879–11883. doi: 10.1073/pnas.92.25.11879
Garden, G. A., and Moller, T. (2006). Microglia biology in health and disease. J. Neuroimmune Pharmacol. 1, 127–137. doi: 10.1007/s11481-006-9015-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Garrido-Mesa, N., Zarzuelo, A., and Galvez, J. (2013). Minocycline: far beyond an antibiotic. Br. J. Pharmacol. 169, 337–352. doi: 10.1111/bph.12139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gemma, C., and Bachstetter, A. D. (2013). The role of microglia in adult hippocampal neurogenesis. Front. Cell Neurosci. 7:229. doi: 10.3389/fncel.2013.00229
Giunti, D., Parodi, B., Cordano, C., Uccelli, A., and Kerlero De Rosbo, N. (2014). Can we switch microglia’s phenotype to foster neuroprotection? Focus on multiple sclerosis. Immunology 141, 328–339. doi: 10.1111/imm.12177
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gonzalez-Perez, O., Gutierrez-Fernandez, F., Lopez-Virgen, V., Collas-Aguilar, J., Quinones-Hinojosa, A., and Garcia-Verdugo, J. M. (2012). Immunological regulation of neurogenic niches in the adult brain. Neuroscience 226, 270–281. doi: 10.1016/j.neuroscience.2012.08.053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gould, E., Reeves, A. J., Fallah, M., Tanapat, P., Gross, C. G., and Fuchs, E. (1999). Hippocampal neurogenesis in adult old world primates. Proc. Natl. Acad. Sci. U.S.A. 96, 5263–5267. doi: 10.1073/pnas.96.9.5263
Hanisch, U. K., and Kettenmann, H. (2007). Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 10, 1387–1394. doi: 10.1038/nn1997
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hauser, P., Khosla, J., Aurora, H., Laurin, J., Kling, M. A., Hill, J.,et al. (2002). A prospective study of the incidence and open-label treatment of interferon-induced major depressive disorder in patients with hepatitis C. Mol. Psychiatry 7, 942–947. doi: 10.1038/sj.mp.4001119
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoyo-Becerra, C., Schlaak, J. F., and Hermann, D. M. (2014). Insights from interferon-alpha-related depression for the pathogenesis of depression associated with inflammation. Brain Behav. Immun. 42C, 222–231. doi: 10.1016/j.bbi.2014.06.200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hulvershorn, L. A., Cullen, K., and Anand, A. (2011). Toward dysfunctional connectivity: a review of neuroimaging findings in pediatric major depressive disorder. Brain Imaging Behav. 5, 307–328. doi: 10.1007/s11682-011-9134-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Imayoshi, I., Sakamoto, M., Ohtsuka, T., Takao, K., Miyakawa, T., Yamaguchi, M.,et al. (2008). Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 11, 1153–1161. doi: 10.1038/nn.2185
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Iosif, R. E., Ekdahl, C. T., Ahlenius, H., Pronk, C. J., Bonde, S., Kokaia, Z.,et al. (2006). Tumor necrosis factor receptor 1 is a negative regulator of progenitor proliferation in adult hippocampal neurogenesis. J. Neurosci. 26, 9703–9712. doi: 10.1523/JNEUROSCI.2723-06.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jankovski, A., and Sotelo, C. (1996). Subventricular zone-olfactory bulb migratory pathway in the adult mouse: cellular composition and specificity as determined by heterochronic and heterotopic transplantation. J. Comp. Neurol. 371, 376–396. doi: 10.1002/(SICI)1096-9861(19960729)371:3<376::AID-CNE3>3.0.CO;2-#
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaneko, N., Kudo, K., Mabuchi, T., Takemoto, K., Fujimaki, K., Wati, H.,et al. (2006). Suppression of cell proliferation by interferon-alpha through interleukin-1 production in adult rat dentate gyrus. Neuropsychopharmacology 31, 2619–2626. doi: 10.1038/sj.npp.1301137
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kaneko, N., Marin, O., Koike, M., Hirota, Y., Uchiyama, Y., Wu, J. Y.,et al. (2010). New neurons clear the path of astrocytic processes for their rapid migration in the adult brain. Neuron 67, 213–223. doi: 10.1016/j.neuron.2010.06.018
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, H. S., and Suh, Y. H. (2009). Minocycline and neurodegenerative diseases. Behav. Brain Res. 196, 168–179. doi: 10.1016/j.bbr.2008.09.040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, C., Yuan, K., and Schluesener, H. (2013). Impact of minocycline on neurodegenerative diseases in rodents: a meta-analysis. Rev. Neurosci. 24, 553–562. doi: 10.1515/revneuro-2013-0040
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lieb, K., Engelbrecht, M. A., Gut, O., Fiebich, B. L., Bauer, J., Janssen, G.,et al. (2006). Cognitive impairment in patients with chronic hepatitis treated with interferon alpha (IFNalpha): results from a prospective study. Eur. Psychiatry 21, 204–210. doi: 10.1016/j.eurpsy.2004.09.030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Liu, Z., Fan, Y., Won, S. J., Neumann, M., Hu, D., Zhou, L.,et al. (2007). Chronic treatment with minocycline preserves adult new neurons and reduces functional impairment after focal cerebral ischemia. Stroke 38, 146–152. doi: 10.1161/01.STR.0000251791.64910.cd
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maes, M. (1999). Major depression and activation of the inflammatory response system. Adv. Exp. Med. Biol. 461, 25–46. doi: 10.1007/978-0-585-37970-8_2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maes, M. (2011). Depression is an inflammatory disease, but cell-mediated immune activation is the key component of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 35, 664–675. doi: 10.1016/j.pnpbp.2010.06.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Malek-Ahmadi, P. (2001). Mood disorders associated with interferon treatment: theoretical and practical considerations. Ann. Pharmacother. 35, 489–495. doi: 10.1345/aph.10172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
McNutt, M. D., Liu, S., Manatunga, A., Royster, E. B., Raison, C. L., Woolwine, B. J.,et al. (2012). Neurobehavioral effects of interferon-alpha in patients with hepatitis-C: symptom dimensions and responsiveness to paroxetine. Neuropsychopharmacology 37, 1444–1454. doi: 10.1038/npp.2011.330
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miller, A. H., Maletic, V., and Raison, C. L. (2009). Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65, 732–741. doi: 10.1016/j.biopsych.2008.11.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Monje, M. L., Toda, H., and Palmer, T. D. (2003). Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765. doi: 10.1126/science.1088417
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Musselman, D. L., Lawson, D. H., Gumnick, J. F., Manatunga, A. K., Penna, S., Goodkin, R. S.,et al. (2001). Paroxetine for the prevention of depression induced by high-dose interferon alfa. N. Engl. J. Med. 344, 961–966. doi: 10.1056/NEJM200103293441303
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ninkovic, J., Mori, T., and Gotz, M. (2007). Distinct modes of neuron addition in adult mouse neurogenesis. J. Neurosci. 27, 10906–10911. doi: 10.1523/JNEUROSCI.2572-07.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Palmer, T. D., Markakis, E. A., Willhoite, A. R., Safar, F., and Gage, F. H. (1999). Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J. Neurosci. 19, 8487–8497.
Papatheodoridis, G. V., Manolakopoulos, S., Dusheiko, G., and Archimandritis, A. J. (2008). Therapeutic strategies in the management of patients with chronic hepatitis B virus infection. Lancet Infect. Dis. 8, 167–178. doi: 10.1016/S1473-3099(07)70264-5
Petreanu, L., and Alvarez-Buylla, A. (2002). Maturation and death of adult-born olfactory bulb granule neurons: role of olfaction. J. Neurosci. 22, 6106–6113.
Plane, J. M., Shen, Y., Pleasure, D. E., and Deng, W. (2010). Prospects for minocycline neuroprotection. Arch. Neurol. 67, 1442–1448. doi: 10.1001/archneurol.2010.191
Porsolt, R. D., Bertin, A., and Jalfre, M. (1977). Behavioral despair in mice: a primary screening test for antidepressants. Arch. Int. Pharmacodyn. Ther. 229, 327–336.
Raison, C. L., Borisov, A. S., Majer, M., Drake, D. F., Pagnoni, G., Woolwine, B. J.,et al. (2009). Activation of central nervous system inflammatory pathways by interferon-alpha: relationship to monoamines and depression. Biol. Psychiatry 65, 296–303. doi: 10.1016/j.biopsych.2008.08.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raison, C. L., Borisov, A. S., Woolwine, B. J., Massung, B., Vogt, G., and Miller, A. H. (2010). Interferon-alpha effects on diurnal hypothalamic-pituitary-adrenal axis activity: relationship with proinflammatory cytokines and behavior. Mol. Psychiatry 15, 535–547. doi: 10.1038/mp.2008.58
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Raison, C. L., Woolwine, B. J., Demetrashvili, M. F., Borisov, A. S., Weinreib, R., Staab, J. P.,et al. (2007). Paroxetine for prevention of depressive symptoms induced by interferon-alpha and ribavirin for hepatitis C. Aliment. Pharmacol. Ther. 25, 1163–1174. doi: 10.1111/j.1365-2036.2007.03316.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Reyes-Vazquez, C., Prieto-Gomez, B., and Dafny, N. (2012). Interferon modulates central nervous system function. Brain Res. 1442, 76–89. doi: 10.1016/j.brainres.2011.09.061
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Samuels, B. A., and Hen, R. (2011). Neurogenesis and affective disorders. Eur. J. Neurosci. 33, 1152–1159. doi: 10.1111/j.1460-9568.2011.07614.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schiepers, O. J., Wichers, M. C., and Maes, M. (2005). Cytokines and major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 201–217. doi: 10.1016/j.pnpbp.2004.11.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shigemoto-Mogami, Y., Hoshikawa, K., Goldman, J. E., Sekino, Y., and Sato, K. (2014). Microglia enhance neurogenesis and oligodendrogenesis in the early postnatal subventricular zone. J. Neurosci. 34, 2231–2243. doi: 10.1523/JNEUROSCI.1619-13.2014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sierra, A., Encinas, J. M., Deudero, J. J., Chancey, J. H., Enikolopov, G., Overstreet-Wadiche, L. S.,et al. (2010). Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7, 483–495. doi: 10.1016/j.stem.2010.08.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Soczynska, J. K., Mansur, R. B., Brietzke, E., Swardfager, W., Kennedy, S. H., Woldeyohannes, H. O.,et al. (2012). Novel therapeutic targets in depression: minocycline as a candidate treatment. Behav. Brain Res. 235, 302–317. doi: 10.1016/j.bbr.2012.07.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steru, L., Chermat, R., Thierry, B., and Simon, P. (1985). The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85, 367–370. doi: 10.1007/BF00428203
Tagliaferri, P., Caraglia, M., Budillon, A., Marra, M., Vitale, G., Viscomi, C.,et al. (2005). New pharmacokinetic and pharmacodynamic tools for interferon-alpha (IFN-alpha) treatment of human cancer. Cancer Immunol. Immunother. 54, 1–10. doi: 10.1007/s00262-004-0549-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
VanGuilder, H. D., Yan, H., Farley, J. A., Sonntag, W. E., and Freeman, W. M. (2010). Aging alters the expression of neurotransmission-regulating proteins in the hippocampal synaptoproteome. J. Neurochem. 113, 1577–1588. doi: 10.1111/j.1471-4159.2010.06719.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Vitkovic, L., Konsman, J. P., Bockaert, J., Dantzer, R., Homburger, V., and Jacque, C. (2000). Cytokine signals propagate through the brain. Mol. Psychiatry 5, 604–615. doi: 10.1038/sj.mp.4000813
Vukovic, J., Colditz, M. J., Blackmore, D. G., Ruitenberg, M. J., and Bartlett, P. F. (2012). Microglia modulate hippocampal neural precursor activity in response to exercise and aging. J. Neurosci. 32, 6435–6443. doi: 10.1523/JNEUROSCI.5925-11.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zheng, L. S., Hitoshi, S., Kaneko, N., Takao, K., Miyakawa, T., Tanaka, Y.,et al. (2014). Mechanisms for interferon-alpha-induced depression and neural stem cell dysfunction. Stem Cell Rep. 3, 73–84. doi: 10.1016/j.stemcr.2014.05.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: interferon, microglia, depression, neurogenesis, hippocampus
Citation: Zheng L-S, Kaneko N and Sawamoto K (2015) Minocycline treatment ameliorates interferon-alpha-induced neurogenic defects and depression-like behaviors in mice. Front. Cell. Neurosci. 9:5. doi: 10.3389/fncel.2015.00005
Received: 20 September 2014; Accepted: 06 January 2015;
Published online: 28 January 2015.
Edited by:
Jens Christian Schwamborn, University of Luxembourg, LuxembourgReviewed by:
Kinichi Nakashima, Kyushu University, JapanSasidhar Reddy Murikinati, Yale University School of Medicine, USA
Copyright © 2015 Zheng, Kaneko and Sawamoto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Naoko Kaneko and Kazunobu Sawamoto, Department of Developmental and Regenerative Biology, Nagoya City University Graduate School of Medical Sciences, 1-Kawasumi, Mizuho-cho, Mizuho-ku, Nagoya, Aichi 467-8601, Japan e-mail:bmFva29rYUBtZWQubmFnb3lhLWN1LmFjLmpw;c2F3YW1vdG9AbWVkLm5hZ295YS1jdS5hYy5qcA==