Abstract
The identification and cloning of the two major cannabinoid (CB1 and CB2) receptors together with the discovery of their endogenous ligands in the late 80s and early 90s, resulted in a major effort aimed at understanding the mechanisms and physiological roles of the endocannabinoid system (ECS). Due to its expression and localization in the central nervous system (CNS), the CB1 receptor together with its endogenous ligands (endocannabinoids (eCB)) and the enzymes involved in their synthesis and degradation, has been implicated in multiple pathophysiological events ranging from memory deficits to neurodegenerative disorders among others. In this review, we will provide a general overview of the ECS with emphasis on the CB1 receptor in health and disease. We will describe our current understanding of the complex aspects of receptor signaling and trafficking, including the non-canonical signaling pathways such as those mediated by β-arrestins within the context of functional selectivity and ligand bias. Finally, we will highlight some of the disorders in which CB1 receptors have been implicated. Significant knowledge has been achieved over the last 30 years. However, much more research is still needed to fully understand the complex roles of the ECS, particularly in vivo and to unlock its true potential as a source of therapeutic targets.
Introduction
The endocannabinoid system (ECS) plays key modulatory roles during synaptic plasticity and homeostatic processes in the brain. Based on anecdotal evidence obtained from cannabis use, laboratory studies, and from emerging clinical work, modulation of the ECS has been proposed as a promising therapeutic target to treat numerous central nervous system (CNS) disorders including neurodegenerative diseases, epilepsy and cognitive deficits among others (Scotter et al., 2010; Fernández-Ruiz et al., 2011; Bilkei-Gorzo, 2012). However, the widespread expression and complex roles of several components of the ECS in excitatory and inhibitory transmission makes the development of such therapy highly challenging (Di Marzo, 2008). This review will explore some of the relationships between the cannabinoid (CB1 and CB2) receptors and their ligands with the nervous system in health and disease. We will introduce the two major receptors, focusing on the CB1 receptors due to their high expression levels in the CNS; their endogenous ligands or endocannabinoids (eCB) and some synthetic mimetics that activate and modulate their signaling; the signaling pathways that connect this receptor to processes inside the cell; and the role of the CB system in the normally functioning CNS and its alteration or therapeutic modulation in a variety of disease states.
CB1 Receptors
The CB1 receptor is one of the most abundant G protein-coupled receptors (GPCRs) in the CNS and is found in particularly high levels in the neocortex, hippocampus, basal ganglia, cerebellum and brainstem (Herkenham et al., 1991; Marsicano and Kuner, 2008). CB1 receptors are also found on peripheral nerve terminals and some extra-neural sites such as the testis, eye, vascular endothelium and spleen. Interestingly, CB1 receptors are highly enriched at presynaptic and axonal compartments, restricting their function to sites of synaptic activity (Straiker and Mackie, 2005; Wu et al., 2008). In addition to its location on the cell surface, intracellular localization of CB1 receptors has also been reported in heterologous systems and primary cultures (Leterrier et al., 2006; Rozenfeld, 2011). The CB1 receptor binds the main active ingredient of Cannabis sativa (marijuana), Δ9-tetrahydrocannabinol (Δ9-THC) and mediates most of the CNS effects of Δ9-THC (Zimmer et al., 1999). In addition, CB1 receptors bind synthetic cannabimimetic compounds such as CP55940, JWH-015, WIN55212-2 and the endogenous arachidonic acid derivatives arachidonylethanolamine (AEA) and 2-arachidonylglycerol (2-AG; see below; Howlett et al., 2002). Upon ligand binding and receptor activation, CB1 receptors are primarily coupled to pertussis toxin (PTX)-sensitive Gi/o type G proteins which leads to a rapid decrease in levels of cAMP by inhibiting adenylate cyclase activity (Figure 1A; Howlett et al., 2004). Coupling to other G proteins including Gs, albeit with low efficacy, can also stimulate adenylate cyclase (Glass and Felder, 1997; Glass and Northup, 1999; Varga et al., 2008; Bosier et al., 2010) though the extent of accumulation of cAMP is not necessarily a good indicator of G protein coupling (Eldeeb et al., 2016). Evidence of promiscuous coupling to different G proteins, signaling roles mediated by β-arrestins and signaling from intracellular compartments (Figure 1B) adds yet another level of complexity making these receptors, like other GPCRs, pluridimentional (Bosier et al., 2010). For our recent review on the multiple waves of receptor signaling see Nogueras-Ortiz and Yudowski (2016). CB1 receptors exhibit constitutive activity indicative of G protein activation in the absence of agonists and this could mediate their highly polarized localization to axonal and presynaptic compartments (Bouaboula et al., 1997; Nie and Lewis, 2001; Rozenfeld, 2011). The activity associated with this state is reversed by treatment with inverse agonists such as SR141716A (also called rimonabant). The model for GPCR activation has been adapted to include these multiple states (Perez and Karnik, 2005; Park et al., 2008) with distinguishing biochemical characteristics, including extent and selectivity of G protein coupling (Mukhopadhyay and Howlett, 2001; Kenakin, 2004). The recent crystallization of the CB1 receptor bound to the antagonist AM6538, should provide new opportunities for understanding the structure-function relationship of this receptor and help novel drug design (Hua et al., 2016).
Figure 1
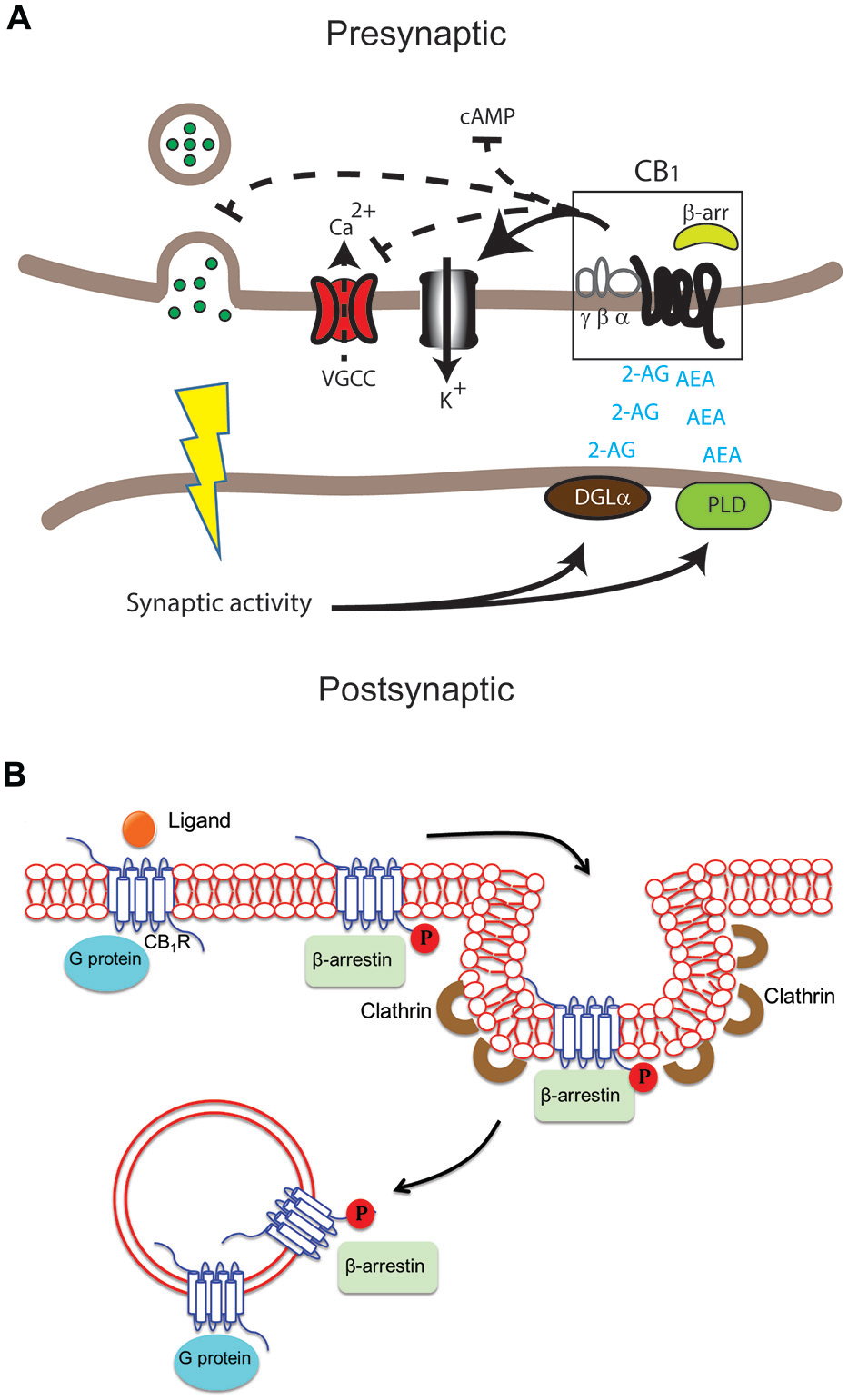
Differential cannabinoid (CB) receptor signaling modalities can impact neuromodulation in health and disease in specific ways. (A) Key enzymes such as diacylglycerol lipase (DGLα) and phospholipase D (PLD) produce the endogenous ligands arachidonylethanolamine (AEA) and 2-arachidonylglycerol (2-AG). These activate the cannabinoid 1 receptor (CB1) receptor in the central nervous system (CNS). The result can include modulation of adenylate cyclase activity to inhibit cAMP accumulation, voltage-gated calcium channels (VGCC), K+ channels and neurotransmitter release in presynaptic excitatory and inhibitory synapses. (B) Following activation of the CB1 receptor by ligand binding, signaling via G protein and/or β-arrestin may occur at the plasma membrane, in endocytic pits or in endosomes after internalization of the receptor. G proteins usually bind the unphosphorylated receptor while β-arrestin binds the receptor phosphorylated by G protein receptor kinases.
CB2 Receptors
The CB2 receptor exhibits a more defined pattern of expression in the brain than CB1 receptors, and is found predominantly in cells and tissues of the immune system (Klein, 2005; Mackie, 2006). In the CNS, CB2 receptor expression is associated with inflammation and it is primarily localized to microglia, resident macrophages of the CNS (Mackie, 2008; Palazuelos et al., 2009). This selective localization together with the modulatory effect of the CB2 receptor on microglia function is particularly relevant since microglial cells have a significant role in Alzheimer’s disease (AD) and other diseases associated with the basal ganglia (Ramírez et al., 2005; Sagredo et al., 2007; Fernández-Ruiz et al., 2011; Yeh et al., 2016). Interestingly, recent work also indicates that CB2 receptors expressed in neurons can control synaptic function and are involved in drug abuse and synaptic plasticity (Xi et al., 2011; Stempel et al., 2016). For example, the selective CB2 receptor agonist JWH133 inhibits dopaminergic firing from the ventral tegmental area and reduced cocaine self-administration (Zhang et al., 2016). Furthermore, neuronal CB2 receptors work independently from CB1 receptors to modulate inhibitory plasticity in the CA2/3 regions of the hippocampus and gamma oscillations in vivo (Stempel et al., 2016). We predict more regulatory roles will be identified for the CB2 receptors expressed in neurons.
Endocannabinoids
eCBs are produced on demand with their synthesis typically triggered via increased intracellular Ca2+ at postsynaptic sites in response to sustained synaptic activity (Figure 1A; Chevaleyre et al., 2006; Mackie, 2006; Heifets and Castillo, 2009). Major eCBs are rapidly deactivated by reuptake mechanisms and degrading enzymes, including fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL; Howlett et al., 2004; Mechoulam and Parker, 2013). Among eCBs, the derivatives of arachidonic acid such as AEA and 2-AG are dominant and orthosteric (Pertwee, 2015). These ligands are agonists for CB1 and CB2 receptors but bind CB1 receptors with higher affinity (AEA Ki = 89 nM and 321 nM for CB1 and CB2 receptors respectively; 2-AG Ki = 472 nM and 1400 nM for CB1 and CB2 receptors respectively; Pertwee et al., 2010). More recently, allosteric eCBs have been identified, including pregnenolone and lipoxin A4 which can modulate CB1 receptor signaling with possible therapeutic value (Pamplona et al., 2012; Vallée et al., 2014; Pertwee, 2015). Further pharmacological characterization is still needed of orthosteric and allosteric modulators to clearly elucidate their physiological roles and modes of action. Nevertheless, the pharmacological manipulation of eCB levels or their actions by allosteric modulators could provide alternative opportunities to regulate the ECS. For a comprehensive review on eCBs see Fonseca et al. (2013).
The Endocannabinoid System in the CNS
The ECS has emerged as one of the key regulatory mechanisms in the brain controlling multiple events such as mood, pain perception, learning and memory among others (Marsicano and Lutz, 2006; Kano et al., 2009). It is also thought to provide a neuroprotective role during traumatic brain injury (TBI) and may be part of the brain’s natural compensatory repair mechanism during neurodegeneration (Pryce et al., 2003; Klein, 2005; Campbell and Gowran, 2007; Bilkei-Gorzo, 2012). New roles for the ECS in drug abuse and dependence are identified almost continuously, further strengthening the relevance of this system not only during cannabis abuse but also other illicit drugs as well (Maldonado et al., 2006; Xi et al., 2011; Parsons and Hurd, 2015). In the CNS, eCBs act as retrograde messengers mediating feedback inhibition modulating synaptic plasticity (Howlett, 2005; Chevaleyre et al., 2006; Katona and Freund, 2012). Specifically, activation of the CB1 receptor leads to activation of inwardly rectifying K+ channel conductance, decreases in N-type and P/Q-type voltage-operated Ca2+ channel conductance and eCB production (Figure 1A; Mackie et al., 1995; Twitchell et al., 1997; Guo and Ikeda, 2004; Demuth and Molleman, 2006). This results in a decrease of neurotransmitter release at excitatory and inhibitory synapses leading to transient effects, as in depolarization-induced suppression of inhibition (DSI) and depolarization-induced suppression of excitation (DSE) or persistent effects as in long-term depression and potentiation (LTP/LTD) during synaptic plasticity (Wilson and Nicoll, 2001; Chevaleyre et al., 2006; Heifets and Castillo, 2009; Kano et al., 2009; Castillo et al., 2012; Soltesz et al., 2015; Maroso et al., 2016). These events make the ECS a key modulator of synaptic plasticity.
Prolonged exposure to CB1 receptor agonists results in rapid attenuation of behavioral responsiveness, termed tolerance, in human and animal models that has been attributed to both a decrease in the ability of the receptor to activate effector pathways (i.e., desensitization) and in the reduction in the number of cell surface-expressed receptors (i.e., internalization; Howlett et al., 2004; Martini et al., 2007). At the molecular level, the agonist-bound GPCR becomes a substrate for G protein coupled receptor kinases (GRKs); these kinases phosphorylate serine and/or threonine residues on GPCR cytoplasmic domains, which then become a high affinity target for β-arrestins (Jin et al., 1999; Delgado-Peraza et al., 2016). Binding of β-arrestins uncouples G-proteins and stimulates receptor internalization and β-arrestin mediated signaling (Jin et al., 1999; Roche et al., 1999).
Ligand induced receptor phosphorylation by GRKs can result in very specific and distinct phosphorylation profiles or “bar-codes” (Butcher et al., 2011; Liggett, 2011; Delgado-Peraza et al., 2016). These bar-codes are finely tuned and define which signaling cascades are activated, thus opening up a spectrum of possibilities frequently defined as functional selectivity or ligand bias (Liggett, 2011; Nobles et al., 2011; Prihandoko et al., 2016). However, careful consideration must be taken when interpreting results obtained from heterologous systems, particularly when signaling can be significantly affected (biased) by the different levels of protein expression across different cell types (Bosier et al., 2010; Atwood et al., 2011; Straiker et al., 2012).
Supporting the bar-code hypothesis and identifying the mechanisms and signaling cascades downstream from the CB1 receptor/β-arrestins, our recent data indicates that receptor and β-arrestin interaction and signaling cascades are dependent on specific phosphorylation sites controlled by unique GRKs (Delgado-Peraza et al., 2016). Mutation of the putative GRK sites from S426/S430 to alanines (rat sequence conserved in human) resulted in reduced β-arrestin 2 recruitment and receptor internalization, but enhanced interaction with β-arrestin 1 and increased β-arrestin 1 mediated signaling (Ahn et al., 2013; Delgado-Peraza et al., 2016). Replacement of series 426/430 to alanines renders the CB1 receptors biased towards β-arrestin 1 signaling and provides an ideal tool to probe the signaling pathways, mechanisms and roles of these cascades. β-arrestin mediated signaling from this biased receptor controls the activation of several cascades including ERK1/2, JNK1/2/3, CREB and P38α. It is important to note that these cascades have been previously linked to the activation of CB1 receptors, but not all to β-arrestins (Rueda et al., 2000; Derkinderen et al., 2001; Hart et al., 2004). Activation of these cascades by CB1 receptors and β-arrestins resulted in the regulation of gene expression and protein synthesis (Delgado-Peraza et al., 2016).
Elucidating the physiological roles of β-arrestins may foster the development of pathway-selective or “biased ligands” with greater therapeutic benefit. Investigating signaling from biased CB1 receptors such as S426A/S430A and the DRY mutant (Asp-Arg-Tyr) together with the identification of biased ligands and the crystal structure of CB1 receptors should provide important tools to elucidate the mechanisms and roles of CB1 receptor signaling (Gyombolai et al., 2015; Delgado-Peraza et al., 2016; Hua et al., 2016).
The subcellular localization and trafficking of CB1 receptors is highly dynamic, with significant effects on receptor signaling (Leterrier et al., 2004; Brailoiu et al., 2011; Rozenfeld, 2011; Dudok et al., 2015). CB1-G protein mediated signaling occurs at the cell surface and at intracellular compartments (Rozenfeld and Devi, 2008; Brailoiu et al., 2014). At the cell surface, CB1 receptor ligands modulate the interaction between receptors and β-arrestin as a mechanism to influence β-arrestin mediated signaling (Flores-Otero et al., 2014). This interaction is initiated at the plasma membrane and can continue into intracellular compartments (Delgado-Peraza et al., 2016). Interestingly, these location-specific signaling events appear to be widespread among several GPCRs. For example, the LH receptor, β2 adrenergic receptor and the CB2 receptor can signal from intracellular compartments either by β-arrestins or G proteins via a “super-complex” ultimately resulting in three different spatio-temporal signaling waves (Brailoiu et al., 2014; Irannejad and von Zastrow, 2014; Lyga et al., 2016; Nogueras-Ortiz and Yudowski, 2016; Thomsen et al., 2016). Constitutive activation also plays a role in their trafficking (Leterrier et al., 2006; McDonald et al., 2007). CB1 receptor location and trafficking are highly dynamic events that are intimately intertwined with their signaling (Dudok et al., 2015). What is the role and relevance of this compartment selective signaling event? Considering the restrictive location of CB1 receptors to presynaptic sites, a possible role could be the local modulation of gene and protein expression after chronic receptor activation. Where do these intracellularly active receptors go and when do they stop signaling are intriguing questions that should provide clues to their physiological roles.
Δ9-THC
The cannabis plant contains more than 60 different active synthetic ligands for CB1/2 (CBs) with Δ9-THC being the major psychoactive molecule among them (Brenneisen, 2007). Exposure to Δ9-THC leads to pleiotropic and sometimes paradoxical effects in humans including analgesic responses, relaxation, dysphoria, tolerance and dependence (Mechoulam and Parker, 2013). Most of these effects are blocked with SR141716, a selective blocker of CB1 receptors (Huestis et al., 2001). In rodents, repetitive administration of Δ9-THC results not only in tolerance but characteristically in a “tetrad” response which includes antinociception, hypothermia, hypoactivity and catalepsy (Little et al., 1988; Fride et al., 2006; Nguyen et al., 2012). However, lack of behavioral sensitization has also been described in mice chronically exposed to Δ9-THC (Varvel et al., 2007). At the molecular level, Δ9-THC acts as a partial agonist of the CB1 receptor, at the G protein level and as a potent activator of β-arrestin 2 recruitment and signaling in heterologous systems (Pertwee et al., 2010; Laprairie et al., 2014, 2016). Perhaps the complex behavioral responses to Δ9-THC could be mediated by the selective activation of these different signaling cascades. Interestingly, β-arrestins mediate some of the behaviors associated with long-term exposure to Δ9-THC (Breivogel et al., 2008; Wu et al., 2008). β-arrestin 2 KO mice display enhanced antinociceptive response to acute Δ9-THC and a decrease in tolerance, indicating the relevance of classical roles of β-arrestin (i.e., receptor desensitization) during G protein signaling (Nguyen et al., 2012). However, recent work on β-arrestin 1 KO mice indicates divergent roles of β-arrestin 1/2 and proposed that β-arrestin 1 regulates receptor sensitivity in an agonist dependent manner, with no significant effects regulating CB tolerance (Breivogel and Vaghela, 2015). Interestingly, our work and others also suggest β-arrestin 1 as the “signaling” arrestin for CB1 receptor. This divergence could be exploited to design compounds that are biased towards G protein signaling with less receptor desensitization and decreased tolerance as recently demonstrated for pain modulation with the mu opioid receptor (Manglik et al., 2016).
CB1 Receptors in Disease
CB1 receptors are indicated in many disorders that impact the CNS including several neurodegenerative disorders such as Huntington’s disease (HD), multiple sclerosis (MS) and AD (Fernández-Ruiz et al., 2011; Di Marzo et al., 2014).
Multiple Sclerosis
MS is a major immune-related neurodegenerative disease characterized by demyelinization with axonal and neuronal loss. Several clinical trials present positive effects of either cannabis, Δ9-THC or other CB agonist on spasticity, spasms and pain among other signs of MS (Croxford, 2003; Pertwee, 2007; Rog, 2010; Notcutt et al., 2012). Use of Sativex® (Nabiximol) an oromucosal spray of cannabis extract containing fixed concentrations of Δ9-THC and cannabidiol (CBD), results in symptomatic improvement in patients with MS. There is a reduction in motor dysfunction and pain, observed in meta-analysis of several clinical studies. However, an increased incidence of non-serious side effects was also reported (Wade et al., 2010; Otero-Romero et al., 2016). Importantly, a review by the National Institute for Health and Care Excellence in the United Kingdom, recommended against the use of Sativex® to treat spasticity in people with MS because it is not a cost effective treatment (Multiple sclerosis in adults: management | 1-recommendations | Guidance and guidelines | NICE, 2014). For a recent and comprehensive analysis of clinical studies see the work of Otero-Romero et al. (2016).
At the molecular level, these improvements are generally linked to the activation of both CB1 receptors and CB2 receptors by agonist, resulting in their dual anti-inflammatory and neuroprotective effects throughout the CNS (Baker et al., 2000; Maresz et al., 2007). These effects include up-regulation of prosurvival molecules such as interleukines in astroglia, and the reduction of cytotoxic factors such as nitric oxide, reactive oxygen species and proinflammatory cytokines in microglia (Fernández-Ruiz et al., 2011). The precise mechanisms by which receptors exert their neuroprotective activity might include activation of phosphatidylinositol 3-kinase/mammalian target of rapamycin complex 1 (mTOR1) pathway and brain-derived neurotrophic factor (BDNF; Ozaita et al., 2007; Blázquez et al., 2015).
Consistent with the clinical data, using synthetic CBs lead to a reduction in inflammation and neuropathic pain in the Experimental Autoimmune Encephalomyelitis (EAE) mouse model (Pryce et al., 2003; Maresz et al., 2007; Fu and Taylor, 2015). Similar results were observed with systemic treatment with the agonists, WIN55212-2, ACEA and JWH-015 of mice with established Theiler’s Murine Encephalomyelitis Virus-induced Demyelinating Disease, a mouse model of chronic progressive MS. Mouse motor function was improved by modulating microglia and lymphocyte infiltration into the spinal cord (Arévalo-Martín et al., 2003). In contrast, when an inverse agonist of the CB1 receptor (SR141716A) was applied, the EAE was worsened likely by releasing pro-inflammatory cytokines in the mouse brain and spinal cord (Saito et al., 2012). Underlying the role of CB1 receptors during neuromodulation and inflammation, work on CB1 receptor−/− mice suggest that these animals are more susceptible to neurotoxicity and damage when compared to wild-type mice (Jackson et al., 2005; Pertwee, 2007). Taken together these results suggest that in MS, the neuroprotective roles of CB1 and CB2 receptors might be impaired and their enhancement could provide new therapeutic approaches. For a comprehensive review of the literature of MS from model systems to clinical studies see Pertwee (2007) and Rog (2010).
Huntington’s Disease
Dysregulation of the ECS is also reported in experimental models and patients with HD. The CB1 receptor expression is reduced, at least somewhat (e.g., 27% decrease in the striatum of the CB1 receptor mRNA), prior to symptoms of neurodegenerative HD in mice (McCaw et al., 2004). Losing the CB1 receptor expression decreases motor performance and increases the amount of aggregates in the striatum of HD mice (Mievis et al., 2011). Major loss of CB1 receptors is also reported in patients with HD (Glass et al., 2000). Interestingly, activation of the CB1 receptor may help reduce the progression of HD. For example, preclinical evidence suggested the use of CBs such as Sativex® for neuroprotection in patients with progressive neurodegenerative conditions like HD (Valdeolivas et al., 2012). Furthermore, selected receptor agonists have neuroprotective potential in a cell culture model of HD (Scotter et al., 2010; Laprairie et al., 2016). Interestingly, ligands biased to β-arrestin mediated signaling such as Δ9-THC, reduced cellular function and viability in these models, suggesting a potential pharmacological profile for therapeutic agonists (Laprairie et al., 2014, 2016). These events are mediated in part by the activation of Gαi/o mediated pathways and might limit glutamate release from cortical neurons and GABA from striatal medium spiny neurons (Dowie et al., 2010; Laprairie et al., 2016). Results obtained investigating the R6/2 mouse model of HD, indicate that CB1 receptor activation parallels BDNF expression leading to neuroprotection (Blázquez et al., 2015). In general, the in vivo and in vitro data suggest that CB agonist with specific pharmacological profiles (biased towards BDNF upregulation and release) could be developed to treat or ameliorate HD.
Alzheimer’s Disease
CB1 receptors have also been the focus of intense research as a potential target in AD. This work has been performed in vitro, animal models and post-mortem samples. Changes in the expression levels of several components of the ECS in post-mortem samples from AD patients have been identified, although their role in the pathophysiology of the disorder is still unknown. For example, CB1 receptors in hippocampus from patients with AD were not different from aged-matched controls. However, the levels of MAGLs, the degradative enzyme of 2-AG, were reduced at their site of action in these patients, suggesting an altered eCB signaling and architecture (Mulder et al., 2011). Limited positive behavioral results have been observed in small clinical trials and pilot studies using analogs of Δ9-THC (Aso and Ferrer, 2014). Analysis of the studies and trials available, suggest significant benefits from synthetic CBs on some of the behavioral and psychological symptoms of dementia (Liu et al., 2015). However, these conclusions were based on short and limited studies; further work will be needed to assess the safety and efficacy of CBs in AD. In experimental models of AD, several findings indicate that the activation of both CB1 receptors and CB2 receptors might have beneficial effects mainly through neuroprotection against Aβ toxicity as previously noted for other neurodegenerative disorders. For example, by crossbreeding the AD mouse model (APP23) with the CB1 receptor-deficient mouse, enhanced cognitive impairment was observed while presenting a reduced amyloid deposition (Stumm et al., 2013). Tau protein phosphorylation is also reduced by CBD in PC12 cells, providing a different neuroprotective mechanism during AD (Esposito et al., 2006). Since CB1 receptors are not likely directly activated by CBD, the impact on Tau phosphorylation may be via the antioxidant effect of CBD or perhaps as a CB receptor independent effect. A reduction in harmful β-amyloid peptide and tau phosphorylation, while promoting intrinsic CNS repair mechanisms may take place consecutively due to activation of the immune and CNS CB system in AD (Aso and Ferrer, 2014). For example, recent work on the TREM2 receptor in microglia, where CB2 receptors are expressed and control cellular responses, also provides an immune related mechanism to control AD (Yeh et al., 2016).
Aging is a major risk factor for neurodegenerative diseases and neuronal progenitor cell proliferation is greatly reduced in the process. Remarkably, CBs can stimulate embryonic and adult neurogenesis (Jiang et al., 2005; Trazzi et al., 2010). Axonal guidance, cell migration, synapse formation and cell survival are also modulated during development. Dysregulation of these processes during development and aging could significantly contribute to multiple disorders of the CNS. For an extensive and thorough review of this topic see the work of Di Marzo et al. (2014) and Maccarrone et al. (2014).
Traumatic Brain Injury
There is good agreement that the CB1/2 receptors are involved in TBI and that 2-AG increases after TBI in animal models (Panikashvili et al., 2001; Mechoulam and Shohami, 2007). There is an “on-demand” signal to generate eCB following TBI that can decrease brain edema and inflammation (Shohami et al., 2011; Gruenbaum et al., 2016). These events may be neuroprotective and prevent excitotoxicity, inhibit inflammatory cytokine production and augment stem cell migration and differentiation. Furthermore, CB1 receptor and CB2 receptor antagonists prevent drug-induced neuroprotection in a mouse mode of TBl (Lopez-Rodriguez et al., 2015). However, as indicated previously for other disorders, limited clinical data is available to support efficacy and safety of CBs during TBI (Gruenbaum et al., 2016).
Future Studies
The modulation of the ECS has great therapeutic potential in many neuropsychiatric and neurodegenerative disorders. Our understanding of the in vivo and in vitro pharmacology of the CB1 receptors and CB2 receptors has significantly increased over the last decades, with new insights into the pathways controlled and the roles of these receptors, enzymes and ligands emerging regularly in the literature. However, this knowledge has not made a complete transition into drug development yet. Complicating this progression, is the mounting anecdotal evidence obtained from cannabis use, which contains over 60 CBs plus other relevant compounds at different concentrations. This variability, together with limited information from clinical trials makes it difficult to scientifically assess the multiple claims associated with cannabis use. Careful investigation of defined molecular entities, in randomized double blind, placebo controlled and multicentric studies should be implemented to clearly move the field forward. At the same time, further work should be performed utilizing cellular and animal models to clearly identify the desired mechanisms and signaling pathways to be therapeutically targeted.
Statements
Author contributions
DAK and GAY wrote and revised this article.
Acknowledgments
We offer our apologies to all authors whose work was not included in this review. The authors thank Yu-Hsien Liao who contributed to Figure 1. This work was supported by a grant from The National Institutes of Health to DAK and GAY (DA040920) and (DA037924) to GAY.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
- AD
Alzheimer’s Disease
- AEA
arachidonylethanolamine
- 2-AG
2-arachidonoylglycerol
- CB1 receptors
cannabinoid 1 receptor
- CB2 receptor
cannabinoid 2 receptor
- CBD
cannabidiol
- CBs
synthetic ligands for CB1/2
- CNS
central nervous system
- eCBs
endocannabinoids
- GPCR
G protein-coupled receptor
- HD
Huntington’s disease
- MS
multiple sclerosis
- TBI
traumatic brain injury.
Abbreviations
References
1
Ahn K. H. Mahmoud M. M. Shim J.-Y. Kendall D. A. (2013). Distinct roles of β-arrestin 1 and β-arrestin 2 in ORG27569-induced biased signaling and internalization of the cannabinoid receptor 1 (CB1). J. Biol. Chem.288, 9790–9800. 10.1074/jbc.M112.438804
2
Arévalo-Martín A. Vela J. M. Molina-Holgado E. Borrell J. Guaza C. (2003). Therapeutic action of cannabinoids in a murine model of multiple sclerosis. J. Neurosci.23, 2511–2516.
3
Aso E. Ferrer I. (2014). Cannabinoids for treatment of Alzheimer’s disease: moving toward the clinic. Front. Pharmacol.5:37. 10.3389/fphar.2014.00037
4
Atwood B. K. Lopez J. Wager-Miller J. Mackie K. Straiker A. (2011). Expression of G protein-coupled receptors and related proteins in HEK293, AtT20, BV2 and N18 cell lines as revealed by microarray analysis. BMC Genomics12:14. 10.1186/1471-2164-12-14
5
Baker D. Pryce G. Croxford J. L. Brown P. Pertwee R. G. Huffman J. W. et al . (2000). Cannabinoids control spasticity and tremor in a multiple sclerosis model. Nature404, 84–87. 10.1038/35003583
6
Bilkei-Gorzo A. (2012). The endocannabinoid system in normal and pathological brain ageing. Philos. Trans. R. Soc. Lond. B Biol. Sci.367, 3326–3341. 10.1098/rstb.2011.0388
7
Blázquez C. Chiarlone A. Bellocchio L. Resel E. Pruunsild P. García-Rincón D. et al . (2015). The CB1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ.22, 1618–1629. 10.1038/cdd.2015.11
8
Bosier B. Muccioli G. G. Hermans E. Lambert D. M. (2010). Functionally selective cannabinoid receptor signalling: therapeutic implications and opportunities. Biochem. Pharmacol.80, 1–12. 10.1016/j.bcp.2010.02.013
9
Bouaboula M. Perrachon S. Milligan L. Canat X. Rinaldi-Carmona M. Portier M. et al . (1997). A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1: evidence for a new model of receptor/ligand interactions. J. Biol. Chem.272, 22330–22339. 10.1074/jbc.272.35.22330
10
Brailoiu G. C. Deliu E. Marcu J. Hoffman N. E. Console-Bram L. Zhao P. et al . (2014). Differential activation of intracellular versus plasmalemmal CB2 cannabinoid receptors. Biochemistry53, 4990–4999. 10.1021/bi500632a
11
Brailoiu G. C. Oprea T. I. Zhao P. Abood M. E. Brailoiu E. (2011). Intracellular cannabinoid type 1 (CB1) receptors are activated by anandamide. J. Biol. Chem.286, 29166–29174. 10.1074/jbc.M110.217463
12
Breivogel C. S. Lambert J. M. Gerfin S. Huffman J. W. Razdan R. K. (2008). Sensitivity to Δ9-tetrahydrocannabinol is selectively enhanced in beta-arrestin2−/– mice. Behav. Pharmacol.19, 298–307. 10.1097/FBP.0b013e328308f1e6
13
Breivogel C. S. Vaghela M. S. (2015). The effects of beta-arrestin1 deletion on acute cannabinoid activity, brain cannabinoid receptors and tolerance to cannabinoids in mice. J. Recept. Signal Transduct. Res.35, 98–106. 10.3109/10799893.2014.1003659
14
Brenneisen R. (2007). “Chemistry and analysis of phytocannabinoids and other cannabis constituents,” in Forensic Science And Medicine, ed. ElSohlyM. A. (Totowa, NJ: Humana Press), 17–49.
15
Butcher A. J. Prihandoko R. Kong K. C. McWilliams P. Edwards J. M. Bottrill A. et al . (2011). Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem.286, 11506–11518. 10.1074/jbc.M110.154526
16
Campbell V. A. Gowran A. (2007). Alzheimer’s disease; taking the edge off with cannabinoids?Br. J. Pharmacol.152, 655–662. 10.1038/sj.bjp.0707446
17
Castillo P. E. Younts T. J. Chávez A. E. Hashimotodani Y. (2012). Endocannabinoid signaling and synaptic function. Neuron76, 70–81. 10.1016/j.neuron.2012.09.020
18
Chevaleyre V. Takahashi K. A. Castillo P. E. (2006). Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci.29, 37–76. 10.1146/annurev.neuro.29.051605.112834
19
Croxford J. L. (2003). Therapeutic potential of cannabinoids in CNS disease. CNS Drugs17, 179–202. 10.2165/00023210-200317030-00004
20
Delgado-Peraza F. Ahn K. H. Nogueras-Ortiz C. Mungrue I. N. Mackie K. Kendall D. A. et al . (2016). Mechanisms of biased β-arrestin-mediated signaling downstream from the cannabinoid 1 receptor. Mol. Pharmacol.89, 618–629. 10.1124/mol.115.103176
21
Demuth D. G. Molleman A. (2006). Cannabinoid signalling. Life Sci.78, 549–563. 10.1016/j.lfs.2005.05.055
22
Derkinderen P. Ledent C. Parmentier M. Girault J. A. (2001). Cannabinoids activate p38 mitogen-activated protein kinases through CB1 receptors in hippocampus. J. Neurochem.77, 957–960. 10.1046/j.1471-4159.2001.00333.x
23
Dowie M. J. Howard M. L. Nicholson L. F. B. Faull R. L. M. Hannan A. J. Glass M. (2010). Behavioural and molecular consequences of chronic cannabinoid treatment in Huntington’s disease transgenic mice. Neuroscience170, 324–336. 10.1016/j.neuroscience.2010.06.056
24
Dudok B. Barna L. Ledri M. Szabó S. I. Szabadits E. Pintér B. et al . (2015). Cell-specific STORM super-resolution imaging reveals nanoscale organization of cannabinoid signaling. Nat. Neurosci.18, 75–86. 10.1038/nn.3892
25
Eldeeb K. Leone-Kabler S. Howlett A. C. (2016). CB1 cannabinoid receptor-mediated increases in cyclic AMP accumulation are correlated with reduced Gi/o function. J. Basic Clin. Physiol. Pharmacol.27, 311–322. 10.1515/jbcpp-2015-0096
26
Esposito G. De Filippis D. Carnuccio R. Izzo A. A. Iuvone T. (2006). The marijuana component cannabidiol inhibits β-amyloid-induced tau protein hyperphosphorylation through Wnt/β-catenin pathway rescue in PC12 cells. J. Mol. Med. (Berl)84, 253–258. 10.1007/s00109-005-0025-1
27
Fernández-Ruiz J. Moreno-Martet M. Rodríguez-Cueto C. Palomo-Garo C. Gómez-Cañas M. Valdeolivas S. et al . (2011). Prospects for cannabinoid therapies in basal ganglia disorders. Br. J. Pharmacol.163, 1365–1378. 10.1111/j.1476-5381.2011.01365.x
28
Flores-Otero J. Ahn K. H. Delgado-Peraza F. Mackie K. Kendall D. A. Yudowski G. A. (2014). Ligand-specific endocytic dwell times control functional selectivity of the cannabinoid receptor 1. Nat. Commun.5:4589. 10.1038/ncomms5589
29
Fonseca B. M. Costa M. A. Almada M. Correia-da-Silva G. Teixeira N. A. (2013). Endogenous cannabinoids revisited: a biochemistry perspective. Prostaglandins Other Lipid Mediat.102-103, 13–30. 10.1016/j.prostaglandins.2013.02.002
30
Fride E. Perchuk A. Hall F. S. Uhl G. R. Onaivi E. S. (2006). Behavioral methods in cannabinoid research. Methods Mol. Med.123, 269–290. 10.1385/1-59259-999-0:269
31
Fu W. Taylor B. K. (2015). Activation of cannabinoid CB2 receptors reduces hyperalgesia in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Neurosci. Lett.595, 1–6. 10.1016/j.neulet.2015.04.002
32
Glass M. Dragunow M. Faull R. L. M. (2000). The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABAA receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience97, 505–519. 10.1016/s0306-4522(00)00008-7
33
Glass M. Felder C. C. (1997). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J. Neurosci.17, 5327–5333.
34
Glass M. Northup J. K. (1999). Agonist selective regulation of G proteins by cannabinoid CB1 and CB2 receptors. Mol. Pharmacol.56, 1362–1369.
35
Gruenbaum S. E. Zlotnik A. Gruenbaum B. F. Hersey D. Bilotta F. (2016). Pharmacologic neuroprotection for functional outcomes after traumatic brain injury: a systematic review of the clinical literature. CNS Drugs30, 791–806. 10.1007/s40263-016-0355-2
36
Guo J. Ikeda S. R. (2004). Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol. Pharmacol.65, 665–674. 10.1124/mol.65.3.665
37
Gyombolai P. Tóth A. D. Timár D. Turu G. Hunyady L. (2015). Mutations in the “DRY” motif of the CB1 cannabinoid receptor result in biased receptor variants. J. Mol. Endocrinol.54, 75–89. 10.1530/JME-14-0219
38
Hart S. Fischer O. M. Ullrich A. (2004). Cannabinoids induce cancer cell proliferation via tumor necrosis factor α-converting enzyme (TACE/ADAM17)-mediated transactivation of the epidermal growth factor receptor. Cancer Res.64, 1943–1950. 10.1158/0008-5472.can-03-3720
39
Heifets B. D. Castillo P. E. (2009). Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol.71, 283–306. 10.1146/annurev.physiol.010908.163149
40
Herkenham M. Lynn A. B. Johnson M. R. Melvin L. S. de Costa B. R. Rice K. C. (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J. Neurosci.11, 563–583.
41
Howlett A. C. (2005). Cannabinoid receptor signaling. Handb. Exp. Pharmacol.168, 53–79. 10.1007/3-540-26573-2_2
42
Howlett A. C. Barth F. Bonner T. I. Cabral G. Casellas P. Devane W. A. et al . (2002). International union of pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev.54, 161–202. 10.1124/pr.54.2.161
43
Howlett A. C. Breivogel C. S. Childers S. R. Deadwyler S. A. Hampson R. E. Porrino L. J. (2004). Cannabinoid physiology and pharmacology: 30 years of progress. Neuropharmacology47, 345–358. 10.1016/j.neuropharm.2004.07.030
44
Hua T. Vemuri K. Pu M. Qu L. Won Han G. Wu Y. et al . (2016). Crystal structure of the human cannabinoid receptor CB1. Cell167, 750.e14–762.e14. 10.1016/j.cell.2016.10.004
45
Huestis M. A. Gorelick D. A. Heishman S. J. Preston K. L. Nelson R. A. Moolchan E. T. et al . (2001). Blockade of effects of smoked marijuana by the CB1-selective cannabinoid receptor antagonist SR141716. Arch. Gen. Psychiatry58, 322–328. 10.1001/archpsyc.58.4.322
46
Irannejad R. von Zastrow M. (2014). GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol.27, 109–116. 10.1016/j.ceb.2013.10.003
47
Jackson S. J. Pryce G. Diemel L. T. Cuzner M. L. Baker D. (2005). Cannabinoid-receptor 1 null mice are susceptible to neurofilament damage and caspase 3 activation. Neuroscience134, 261–268. 10.1016/j.neuroscience.2005.02.045
48
Jiang W. Zhang Y. Xiao L. Van Cleemput J. Ji S. P. Bai G. et al . (2005). Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J. Clin. Invest.115, 3104–3116. 10.1172/jci25509
49
Jin W. Brown S. Roche J. P. Hsieh C. Celver J. P. Kovoor A. et al . (1999). Distinct domains of the CB1 cannabinoid receptor mediate desensitization and internalization. J. Neurosci.19, 3773–3780.
50
Kano M. Ohno-Shosaku T. Hashimotodani Y. Uchigashima M. Watanabe M. (2009). Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev.89, 309–380. 10.1152/physrev.00019.2008
51
Katona I. Freund T. F. (2012). Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci.35, 529–558. 10.1146/annurev-neuro-062111-150420
52
Kenakin T. (2004). Principles: receptor theory in pharmacology. Trends Pharmacol. Sci.25, 186–192. 10.1016/j.tips.2004.02.012
53
Klein T. W. (2005). Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat. Rev. Immunol.5, 400–411. 10.1038/nri1602
54
Laprairie R. B. Bagher A. M. Kelly M. E. M. Denovan-Wright E. M. (2016). Biased type 1 cannabinoid receptor signaling influences neuronal viability in a cell culture model of Huntington disease. Mol. Pharmacol.89, 364–375. 10.1124/mol.115.101980
55
Laprairie R. B. Bagher A. M. Kelly M. E. M. Dupré D. J. Denovan-Wright E. M. (2014). Type 1 cannabinoid receptor ligands display functional selectivity in a cell culture model of striatal medium spiny projection neurons. J. Biol. Chem.289, 24845–24862. 10.1074/jbc.m114.557025
56
Leterrier C. Bonnard D. Carrel D. Rossier J. Lenkei Z. (2004). Constitutive endocytic cycle of the CB1 cannabinoid receptor. J. Biol. Chem.279, 36013–36021. 10.1074/jbc.M403990200
57
Leterrier C. Lainé J. Darmon M. Boudin H. Rossier J. Lenkei Z. (2006). Constitutive activation drives compartment-selective endocytosis and axonal targeting of type 1 cannabinoid receptors. J. Neurosci.26, 3141–3153. 10.1523/JNEUROSCI.5437-05.2006
58
Liggett S. B. (2011). Phosphorylation barcoding as a mechanism of directing GPCR signaling. Sci. Signal.4:pe36. 10.1126/scisignal.2002331
59
Little P. J. Compton D. R. Johnson M. R. Melvin L. S. Martin B. R. (1988). Pharmacology and stereoselectivity of structurally novel cannabinoids in mice. J. Pharmacol. Exp. Ther.247, 1046–1051.
60
Liu C. S. Chau S. A. Ruthirakuhan M. Lanctôt K. L. Herrmann N. (2015). Cannabinoids for the treatment of agitation and aggression in Alzheimer’s disease. CNS Drugs29, 615–623. 10.1007/s40263-015-0270-y
61
Lopez-Rodriguez A. B. Siopi E. Finn D. P. Marchand-Leroux C. Garcia-Segura L. M. Jafarian-Tehrani M. et al . (2015). CB1 and CB2 cannabinoid receptor antagonists prevent minocycline-induced neuroprotection following traumatic brain injury in mice. Cereb. Cortex25, 35–45. 10.1093/cercor/bht202
62
Lyga S. Volpe S. Werthmann R. C. Götz K. Sungkaworn T. Lohse M. J. et al . (2016). Persistent cAMP signaling by internalized LH receptors in ovarian follicles. Endocrinology157, 1613–1621. 10.1210/en.2015-1945
63
Maccarrone M. Guzmán M. Mackie K. Doherty P. Harkany T. (2014). Programming of neural cells by (endo)cannabinoids: from physiological rules to emerging therapies. Nat. Rev. Neurosci.15, 786–801. 10.1038/nrn3846
64
Mackie K. (2006). Mechanisms of CB1 receptor signaling: endocannabinoid modulation of synaptic strength. Int. J. Obes. (Lond).30, S19–S23. 10.1038/sj.ijo.0803273
65
Mackie K. (2008). Cannabinoid receptors: where they are and what they do. J. Neuroendocrinol.20, 10–14. 10.1111/j.1365-2826.2008.01671.x
66
Mackie K. Lai Y. Westenbroek R. Mitchell R. (1995). Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J. Neurosci.15, 6552–6561.
67
Maldonado R. Valverde O. Berrendero F. (2006). Involvement of the endocannabinoid system in drug addiction. Trends Neurosci.29, 225–232. 10.1016/j.tins.2006.01.008
68
Manglik A. Lin H. Aryal D. K. McCorvy J. D. Dengler D. Corder G. et al . (2016). Structure-based discovery of opioid analgesics with reduced side effects. Nature537, 185–190. 10.1038/nature19112
69
Maresz K. Pryce G. Ponomarev E. D. Marsicano G. Croxford J. L. Shriver L. P. et al . (2007). Direct suppression of CNS autoimmune inflammation via the cannabinoid receptor CB1 on neurons and CB2 on autoreactive T cells. Nat. Med.13, 492–497. 10.1038/nm1561
70
Maroso M. Szabo G. G. Kim H. K. Alexander A. Bui A. D. Lee S.-H. et al . (2016). Cannabinoid control of learning and memory through HCN channels. Neuron89, 1059–1073. 10.1016/j.neuron.2016.01.023
71
Marsicano G. Kuner R. (2008). “Anatomical distribution of receptors, ligands and enzymes in the brain and in the spinal cord: circuitries and neurochemistry,” in Cannabinoids and The Brain, ed. KöfalviA. (Boston, MA: Springer US), 161–201.
72
Marsicano G. Lutz B. (2006). Neuromodulatory functions of the endocannabinoid system. J. Endocrinol. Invest.29, 27–46.
73
Martini L. Waldhoer M. Pusch M. Kharazia V. Fong J. Lee J. H. et al . (2007). Ligand-induced down-regulation of the cannabinoid 1 receptor is mediated by the G-protein-coupled receptor-associated sorting protein GASP1. FASEB J.21, 802–811. 10.1096/fj.06-7132com
74
Di Marzo V. (2008). Targeting the endocannabinoid system: to enhance or reduce?Nat. Rev. Drug Discov.7, 438–455. 10.1038/nrd2553
75
Di Marzo V. Stella N. Zimmer A. (2014). Endocannabinoid signalling and the deteriorating brain. Nat. Rev. Neurosci.16, 30–42. 10.1038/nrn3876
76
McCaw E. A. Hu H. Gomez G. T. Hebb A. L. O. Kelly M. E. M. Denovan-Wright E. M. (2004). Structure, expression and regulation of the cannabinoid receptor gene (CB1) in Huntington’s disease transgenic mice. Eur. J. Biochem.271, 4909–4920. 10.1111/j.1432-1033.2004.04460.x
77
McDonald N. A. Henstridge C. M. Connolly C. N. Irving A. J. (2007). An essential role for constitutive endocytosis, but not activity, in the axonal targeting of the CB1 cannabinoid receptor. Mol. Pharmacol.71, 976–984. 10.1124/mol.106.029348
78
Mechoulam R. Parker L. A. (2013). The endocannabinoid system and the brain. Annu. Rev. Psychol.64, 21–47. 10.1146/annurev-psych-113011-143739
79
Mechoulam R. Shohami E. (2007). Endocannabinoids and traumatic brain injury. Mol. Neurobiol.36, 68–74. 10.1007/s12035-007-8008-6
80
Mievis S. Blum D. Ledent C. (2011). Worsening of Huntington disease phenotype in CB1 receptor knockout mice. Neurobiol. Dis.42, 524–529. 10.1016/j.nbd.2011.03.006
81
Mukhopadhyay S. Howlett A. C. (2001). CB1 receptor-G protein association. Subtype selectivity is determined by distinct intracellular domains. Eur. J. Biochem.268, 499–505. 10.1046/j.1432-1327.2001.01810.x
82
Mulder J. Zilberter M. Pasquaré S. J. Alpár A. Schulte G. Ferreira S. G. et al . (2011). Molecular reorganization of endocannabinoid signalling in Alzheimer’s disease. Brain134, 1041–1060. 10.1093/brain/awr046
83
Multiple sclerosis in adults: management | 1-recommendations | Guidance and guidelines | NICE . (2014). NICE. Available online at: https://www.nice.org.uk/guidance/CG186
84
Nguyen P. T. Schmid C. L. Raehal K. M. Selley D. E. Bohn L. M. Sim-Selley L. J. (2012). β-arrestin2 regulates cannabinoid CB1 receptor signaling and adaptation in a central nervous system region-dependent manner. Biol. Psychiatry71, 714–724. 10.1016/j.biopsych.2011.11.027
85
Nie J. Lewis D. L. (2001). Structural domains of the CB1 cannabinoid receptor that contribute to constitutive activity and G-protein sequestration. J. Neurosci.21, 8758–8764.
86
Nobles K. N. Xiao K. Ahn S. Shukla A. K. Lam C. M. Rajagopal S. et al . (2011). Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal.4:ra51. 10.1126/scisignal.2001707
87
Nogueras-Ortiz C. Yudowski G. A. (2016). The multiple waves of cannabinoid 1 receptor signaling. Mol. Pharmacol.90, 620–626. 10.1124/mol.116.104539
88
Notcutt W. Langford R. Davies P. Ratcliffe S. Potts R. (2012). A placebo-controlled, parallel-group, randomized withdrawal study of subjects with symptoms of spasticity due to multiple sclerosis who are receiving long-term Sativex® (nabiximols). Mult. Scler.18, 219–228. 10.1177/1352458511419700
89
Otero-Romero S. Sastre-Garriga J. Comi G. Hartung H.-P. Soelberg Sørensen P. Thompson A. J. et al . (2016). Pharmacological management of spasticity in multiple sclerosis: systematic review and consensus paper. Mult. Scler.22, 1386–1396. 10.1177/1352458516643600
90
Ozaita A. Puighermanal E. Maldonado R. (2007). Regulation of PI3K/Akt/GSK-3 pathway by cannabinoids in the brain. J. Neurochem.102, 1105–1114. 10.1111/j.1471-4159.2007.04642.x
91
Palazuelos J. Aguado T. Pazos M. R. Julien B. Carrasco C. Resel E. et al . (2009). Microglial CB2 cannabinoid receptors are neuroprotective in Huntington’s disease excitotoxicity. Brain132, 3152–3164. 10.1093/brain/awp239
92
Pamplona F. A. Ferreira J. Menezes de Lima O. Jr. Duarte F. S. Bento A. F. Forner S. et al . (2012). Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proc. Natl. Acad. Sci. U S A109, 21134–21139. 10.1073/pnas.1202906109
93
Panikashvili D. Simeonidou C. Ben-Shabat S. Hanuš L. Breuer A. Mechoulam R. et al . (2001). An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature413, 527–531. 10.1038/35097089
94
Park P. S.-H. S.-H. Lodowski D. T. Palczewski K. (2008). Activation of G protein-coupled receptors: beyond two-state models and tertiary conformational changes. Annu. Rev. Pharmacol. Toxicol.48, 107–141. 10.1146/annurev.pharmtox.48.113006.094630
95
Parsons L. H. Hurd Y. L. (2015). Endocannabinoid signalling in reward and addiction. Nat. Rev. Neurosci.16, 579–594. 10.1038/nrn4004
96
Perez D. M. Karnik S. S. (2005). Multiple signaling states of G-protein-coupled receptors. Pharmacol. Rev.57, 147–161. 10.1124/pr.57.2.2
97
Pertwee R. G. (2007). Cannabinoids and multiple sclerosis. Mol. Neurobiol.36, 45–59. 10.1007/s12035-007-0005-2
98
Pertwee R. G. (2015). Endocannabinoids and their pharmacological actions. Handb. Exp. Pharmacol.231, 1–37. 10.1007/978-3-319-20825-1_1
99
Pertwee R. G. Howlett A. C. Abood M. E. Alexander S. P. H. Di Marzo V. Elphick M. R. et al . (2010). International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol. Rev.62, 588–631. 10.1124/pr.110.003004
100
Prihandoko R. Alvarez-Curto E. Hudson B. D. Butcher A. J. Ulven T. Miller A. M. et al . (2016). Distinct phosphorylation clusters determine the signaling outcome of free fatty acid receptor 4/G protein-coupled receptor 120. Mol. Pharmacol.89, 505–520. 10.1124/mol.115.101949
101
Pryce G. Ahmed Z. Hankey D. J. R. Jackson S. J. Croxford J. L. Pocock J. M. et al . (2003). Cannabinoids inhibit neurodegeneration in models of multiple sclerosis. Brain126, 2191–2202. 10.1093/brain/awg224
102
Ramírez B. G. Blázquez C. Gómez del Pulgar T. G. Guzmán M. de Ceballos M. L. (2005). Prevention of Alzheimer’s disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J. Neurosci.25, 1904–1913. 10.1523/JNEUROSCI.4540-04.2005
103
Roche J. P. Bounds S. Brown S. Mackie K. (1999). A mutation in the second transmembrane region of the CB1 receptor selectively disrupts G protein signaling and prevents receptor internalization. Mol. Pharmacol.56, 611–618. 10.1124/mol.56.3.611
104
Rog D. J. (2010). Cannabis-based medicines in multiple sclerosis–a review of clinical studies. Immunobiology215, 658–672. 10.1016/j.imbio.2010.03.009
105
Rozenfeld R. (2011). Type I cannabinoid receptor trafficking: all roads lead to lysosome. Traffic12, 12–18. 10.1111/j.1600-0854.2010.01130.x
106
Rozenfeld R. Devi L. A. (2008). Regulation of CB1 cannabinoid receptor trafficking by the adaptor protein AP-3. FASEB J.22, 2311–2322. 10.1096/fj.07-102731
107
Rueda D. Galve-Roperh I. Haro A. Guzmán M. (2000). The CB1 cannabinoid receptor is coupled to the activation of c-Jun N-terminal kinase. Mol. Pharmacol.58, 814–820. 10.1124/mol.58.4.814
108
Sagredo O. García-Arencibia M. de Lago E. Finetti S. Decio A. Fernández-Ruiz J. (2007). Cannabinoids and neuroprotection in basal ganglia disorders. Mol. Neurobiol.36, 82–91. 10.1007/s12035-007-0004-3
109
Saito V. M. Rezende R. M. Teixeira A. L. (2012). Cannabinoid modulation of neuroinflammatory disorders. Curr. Neuropharmacol.10, 159–166. 10.2174/157015912800604515
110
Scotter E. L. Abood M. E. Glass M. (2010). The endocannabinoid system as a target for the treatment of neurodegenerative disease. Br. J. Pharmacol.160, 480–498. 10.1111/j.1476-5381.2010.00735.x
111
Shohami E. Cohen-Yeshurun A. Magid L. Algali M. Mechoulam R. (2011). Endocannabinoids and traumatic brain injury. Br. J. Pharmacol.163, 1402–1410. 10.1111/j.1476-5381.2011.01343.x
112
Soltesz I. Alger B. E. Kano M. Lee S.-H. Lovinger D. M. Ohno-Shosaku T. et al . (2015). Weeding out bad waves: towards selective cannabinoid circuit control in epilepsy. Nat. Rev. Neurosci.16, 264–277. 10.1038/nrn3937
113
Stempel A. V. Stumpf A. Zhang H. Y. Özdogan T. Pannasch U. Theis A. K. et al . (2016). Cannabinoid type 2 receptors mediate a cell type-specific plasticity in the hippocampus. Neuron90, 795–809. 10.1016/j.neuron.2016.03.034
114
Straiker A. Mackie K. (2005). Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J. Physiol.569, 501–517. 10.1113/jphysiol.2005.091918
115
Straiker A. Wager-Miller J. Hutchens J. Mackie K. (2012). Differential signalling in human cannabinoid CB1 receptors and their splice variants in autaptic hippocampal neurones. Br. J. Pharmacol.165, 2660–2671. 10.1111/j.1476-5381.2011.01744.x
116
Stumm C. Hiebel C. Hanstein R. Purrio M. Nagel H. Conrad A. et al . (2013). Cannabinoid receptor 1 deficiency in a mouse model of Alzheimer’s disease leads to enhanced cognitive impairment despite of a reduction in amyloid deposition. Neurobiol. Aging34, 2574–2584. 10.1016/j.neurobiolaging.2013.05.027
117
Thomsen A. R. B. Plouffe B. Cahill T. J. III. Shukla A. K. Tarrasch J. T. Dosey A. M. et al . (2016). GPCR-G protein-β-arrestin super-complex mediates sustained G protein signaling. Cell166, 907–919. 10.1016/j.cell.2016.07.004
118
Trazzi S. Steger M. Mitrugno V. M. Bartesaghi R. Ciani E. (2010). CB1 cannabinoid receptors increase neuronal precursor proliferation through AKT/glycogen synthase kinase-3β/β-catenin signaling. J. Biol. Chem.285, 10098–10109. 10.1074/jbc.M109.043711
119
Twitchell W. Brown S. Mackie K. (1997). Cannabinoids inhibit N-and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol.78, 43–50.
120
Valdeolivas S. Satta V. Pertwee R. G. Fernández-Ruiz J. Sagredo O. (2012). Sativex-like combination of phytocannabinoids is neuroprotective in malonate-lesioned rats, an inflammatory model of Huntington’s disease: role of CB1 and CB2 receptors. ACS Chem. Neurosci.3, 400–406. 10.1021/cn200114w
121
Vallée M. Vitiello S. Bellocchio L. Hébert-Chatelain E. Monlezun S. Martin-Garcia E. et al . (2014). Pregnenolone can protect the brain from cannabis intoxication. Science343, 94–98. 10.1126/science.1243985
122
Varga E. V. Georgieva T. Tumati S. Alves I. Salamon Z. Tollin G. et al . (2008). Functional selectivity in cannabinoid signaling. Curr. Mol. Pharmacol.1, 273–284. 10.2174/1874-470210801030273
123
Varvel S. A. Martin B. R. Lichtman A. H. (2007). Lack of behavioral sensitization after repeated exposure to THC in mice and comparison to methamphetamine. Psychopharmacology (Berl)193, 511–519. 10.1007/s00213-007-0811-2
124
Wade D. T. Collin C. Stott C. Duncombe P. (2010). Meta-analysis of the efficacy and safety of Sativex (nabiximols), on spasticity in people with multiple sclerosis. Mult. Scler.16, 707–714. 10.1177/1352458510367462
125
Wilson R. I. Nicoll R. A. (2001). Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature410, 588–592. 10.1038/35069076
126
Wu D.-F. Yang L.-Q. Goschke A. Stumm R. Brandenburg L.-O. Liang Y.-J. et al . (2008). Role of receptor internalization in the agonist-induced desensitization of cannabinoid type 1 receptors. J. Neurochem.104, 1132–1143. 10.1111/j.1471-4159.2007.05063.x
127
Xi Z.-X. Peng X.-Q. Li X. Song R. Zhang H.-Y. Liu Q.-R. et al . (2011). Brain cannabinoid CB2 receptors modulate cocaine’s actions in mice. Nat. Neurosci.14, 1160–1166. 10.1038/nm.2483
128
Yeh F. L. Wang Y. Tom I. Gonzalez L. C. Sheng M. (2016). TREM2 binds to apolipoproteins, including APOE and CLU/APOJ and thereby facilitates uptake of amyloid-β by microglia. Neuron91, 328–340. 10.1016/j.neuron.2016.06.015
129
Zhang H.-Y. Gao M. Shen H. Bi G.-H. Yang H.-J. Liu Q.-R. et al . (2016). Expression of functional cannabinoid CB2 receptor in VTA dopamine neurons in rats. Addict. Biol. [Epub ahead of print]. 10.1111/adb.12367
130
Zimmer A. Zimmer A. M. Hohmann A. G. Herkenham M. Bonner T. I. (1999). Increased mortality, hypoactivity and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc. Natl. Acad. Sci. U S A96, 5780–5785. 10.1073/pnas.96.10.5780
Summary
Keywords
CB1 receptors, signaling, endocannabinoid system, neuromodulation, Δ9-THC
Citation
Kendall DA and Yudowski GA (2017) Cannabinoid Receptors in the Central Nervous System: Their Signaling and Roles in Disease. Front. Cell. Neurosci. 10:294. doi: 10.3389/fncel.2016.00294
Received
29 August 2016
Accepted
08 December 2016
Published
04 January 2017
Volume
10 - 2016
Edited by
Hansen Wang, University of Toronto, Canada
Reviewed by
Carla Cannizzaro, University of Palermo, Italy; Eilís Dowd, National University of Ireland, Galway, Ireland
Updates
Copyright
© 2017 Kendall and Yudowski.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution and reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guillermo A. Yudowski guillermo.yudowski@upr.edu
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.