 Kenneth A. Jacobson
Kenneth A. Jacobson Dilip K. Tosh
Dilip K. Tosh Shanu Jain
Shanu Jain- Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD, United States
Adenosine receptors (ARs) function in the body’s response to conditions of pathology and stress associated with a functional imbalance, such as in the supply and demand of energy/oxygen/nutrients. Extracellular adenosine concentrations vary widely to raise or lower the basal activation of four subtypes of ARs. Endogenous adenosine can correct an energy imbalance during hypoxia and other stress, for example, by slowing the heart rate by A1AR activation or increasing the blood supply to heart muscle by the A2AAR. Moreover, exogenous AR agonists, antagonists, or allosteric modulators can be applied for therapeutic benefit, and medicinal chemists working toward that goal have reported thousands of such agents. Thus, numerous clinical trials have ensued, using promising agents to modulate adenosinergic signaling, most of which have not succeeded. Currently, short-acting, parenteral agonists, adenosine and Regadenoson, are the only AR agonists approved for human use. However, new concepts and compounds are currently being developed and applied toward preclinical and clinical evaluation, and initial results are encouraging. This review focuses on key compounds as AR agonists and positive allosteric modulators (PAMs) for disease treatment or diagnosis. AR agonists for treating inflammation, pain, cancer, non-alcoholic steatohepatitis, angina, sickle cell disease, ischemic conditions and diabetes have been under development. Multiple clinical trials with two A3AR agonists are ongoing.
Introduction
Adenosine receptors (ARs) are G protein-coupled receptors (GPCRs) that sense an imbalance of demand and supply of energy/oxygen/nutrients. Extracellular adenosine concentrations rise in response to hypoxia and other stress, to act upon four subtypes of ARs (A1AR, A2AAR, A2BAR, and A3AR) (Fredholm et al., 2001; Chen et al., 2013). As shown with mice lacking all four AR subtypes, extracellular adenosine is mainly a sensor of tissue damage or danger, rather than a homeostatic regulator under baseline conditions (Xiao et al., 2019). Elevated adenosine can correct an energy imbalance during distress of an organ, for example by slowing the heart rate by A1AR activation or increasing the blood supply to heart muscle by the A2AAR (Borea et al., 2016). However, there are conditions in which chronic adenosine overproduction can be harmful, leading to increased inflammation, fibrosis, cytokine release, brain dopamine depletion, and kidney damage (Borea et al., 2017). Moreover, exogenous AR agonists, antagonists, or allosteric modulators can be applied for therapeutic benefit, and thousands of such agents have been reported by medicinal chemists working toward that goal (Jacobson and Gao, 2006; Kiesman et al., 2009). Clinically important effects of adenosine also include suppression of the immune response, glomerular filtration, seizures and pain (Fredholm et al., 2001; Cekic and Linden, 2016; Antonioli et al., 2018). Adenosine 5′-triphosphate (ATP) released from cells under stress conditions or damage is the source of much of the extracellular adenosine. There is typically a basal level of AR stimulation, especially for A1AR, A2AAR and A3AR at low nM concentrations, while A2BAR activation generally occurs at higher (μM) adenosine concentrations (Fredholm et al., 2001). Therefore, AR antagonists have distinct biological effects in vivo. Purinergic signaling is also to be considered in the larger context of ligand (ATP)-gated P2X receptors or G protein-coupled P2Y receptors that respond to extracellular purine and pyrimidine mono- and di-nucleotides (Burnstock and Boeynaems, 2014).
There are many approaches to small molecule therapeutics based on experimental modulators of ARs (Chen et al., 2013; Borea et al., 2018). Numerous subtype-selective agonists and antagonists have been introduced, both as pharmacological probes and as clinical candidate molecules, and representative compounds are described here. AR knockout (KO) mice are increasingly used to validate in vivo results with agonists and antagonists, which can actually have variable selectivity (Carlin et al., 2018). This review covers both directly acting AR modulators, i.e., agonists, and several positive allosteric modulators (PAMs). It describes compounds that have been in human trials, for both therapeutics and diagnostics, and some compounds for which clinical trials have only been contemplated. Other reviews cover the use of enzymatic or transport inhibitors to increase – such as inhibitors of adenosine kinase or adenosine deaminase that deplete adenosine levels, or uptake inhibitors – or to decrease, such as inhibitors of ectonucleotidases, the levels of endogenous nucleosides (Boison, 2013; Allard et al., 2017; Vuerich et al., 2019). These enzymes are often upregulated in inflammatory states (Boison, 2013; Haskó et al., 2018).
AR Agonists for Clinical Development
Agonists of the A1AR, A2AAR and A3AR have been the subject of preclinical and clinical evaluation (Figures 1, 2 and Tables 1, 2). A2BAR agonist development is the most limited among the ARs, and agonists for this AR subtype have not yet entered clinical trials. There is also much controversy about whether A2BAR agonists or antagonists would be more useful clinically (Eisenstein et al., 2015; Sun and Huang, 2016; Gao and Jacobson, 2017).
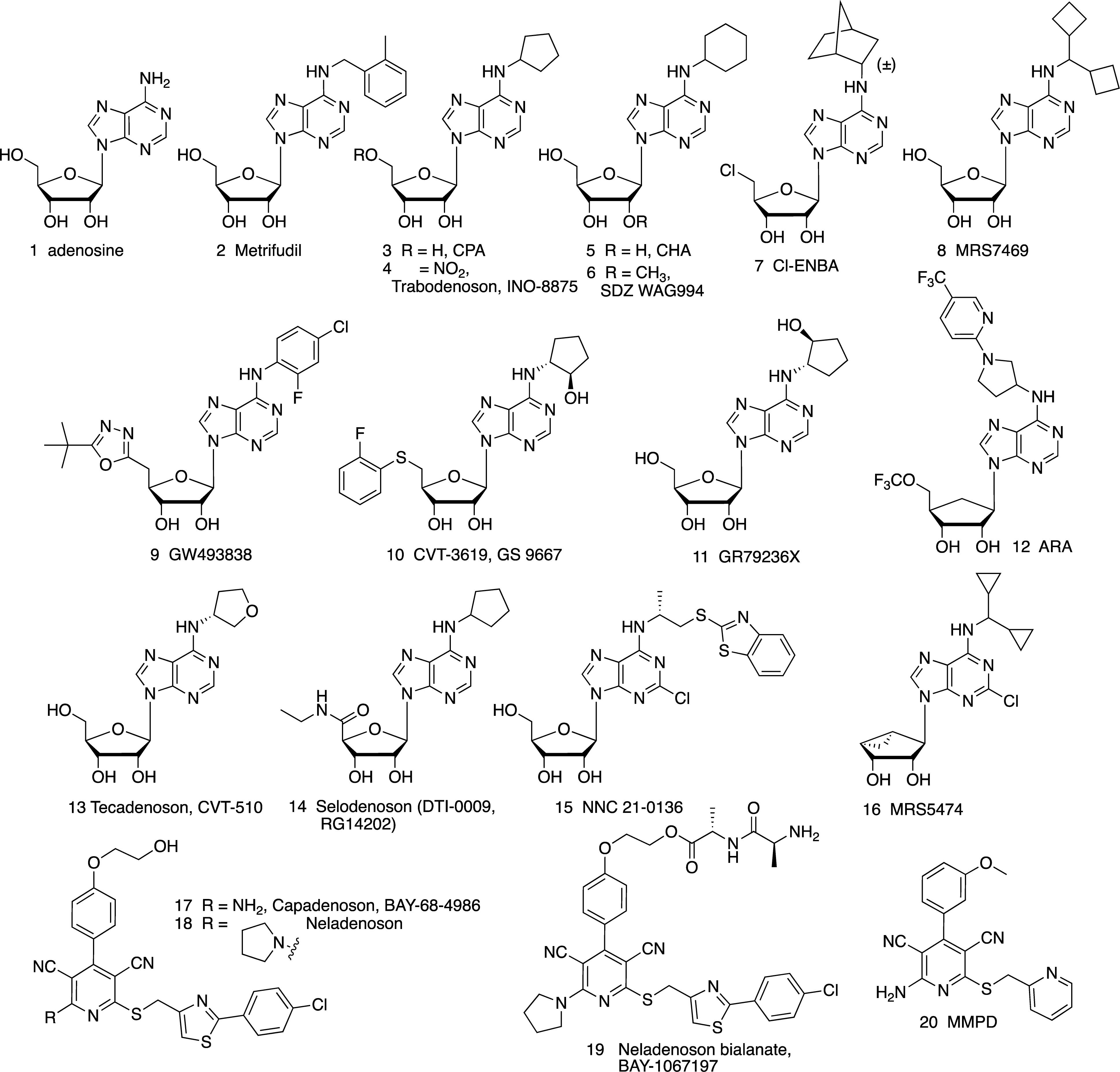
Figure 1. Adenosine (1), a non-selective AR agonist, and its derivatives as A1AR-selective agonists, including nucleosides (2–16) and non-nucleosides (17–20).
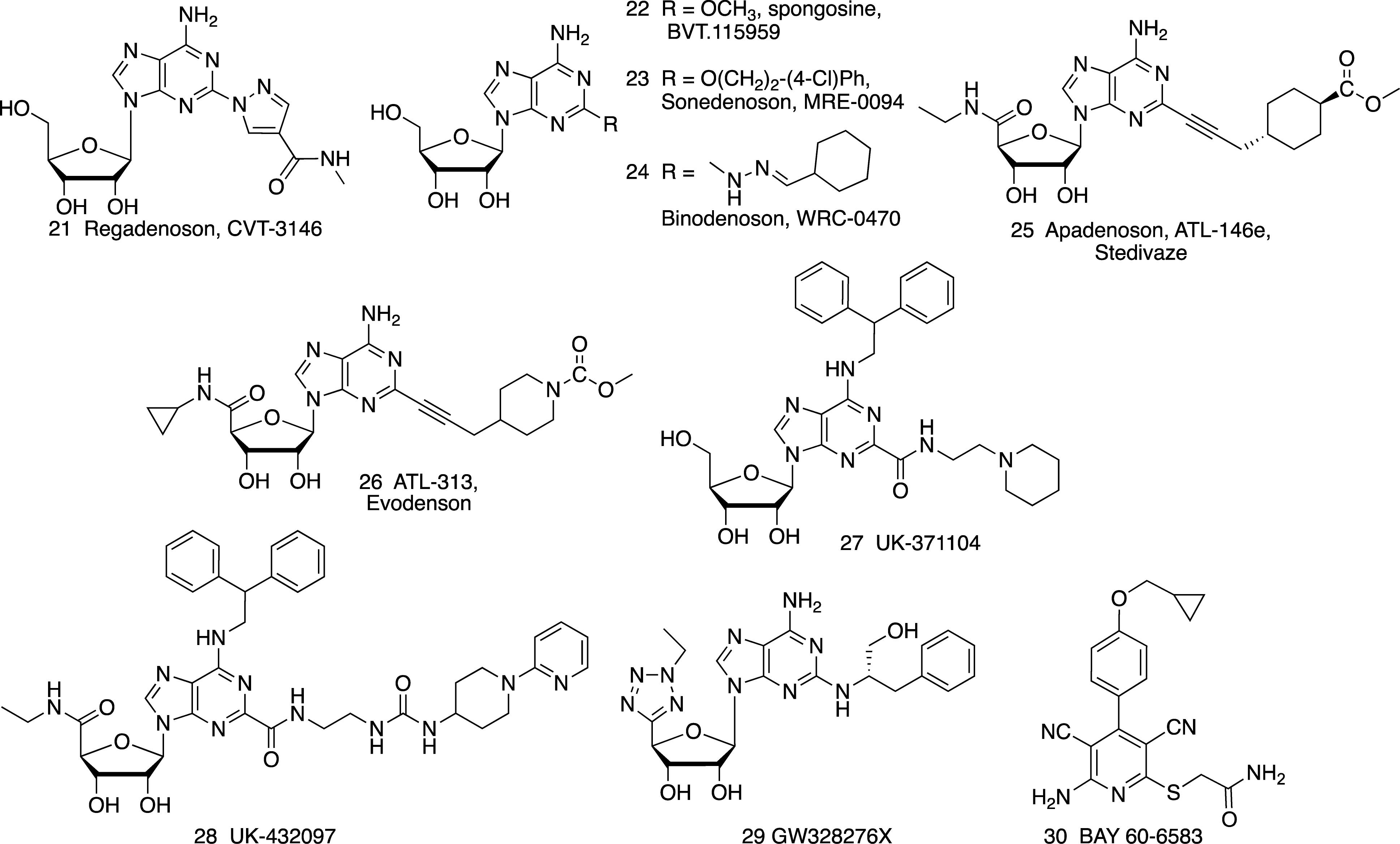
Figure 2. A2AAR- (21–29) and A2BAR- (30) selective agonists.
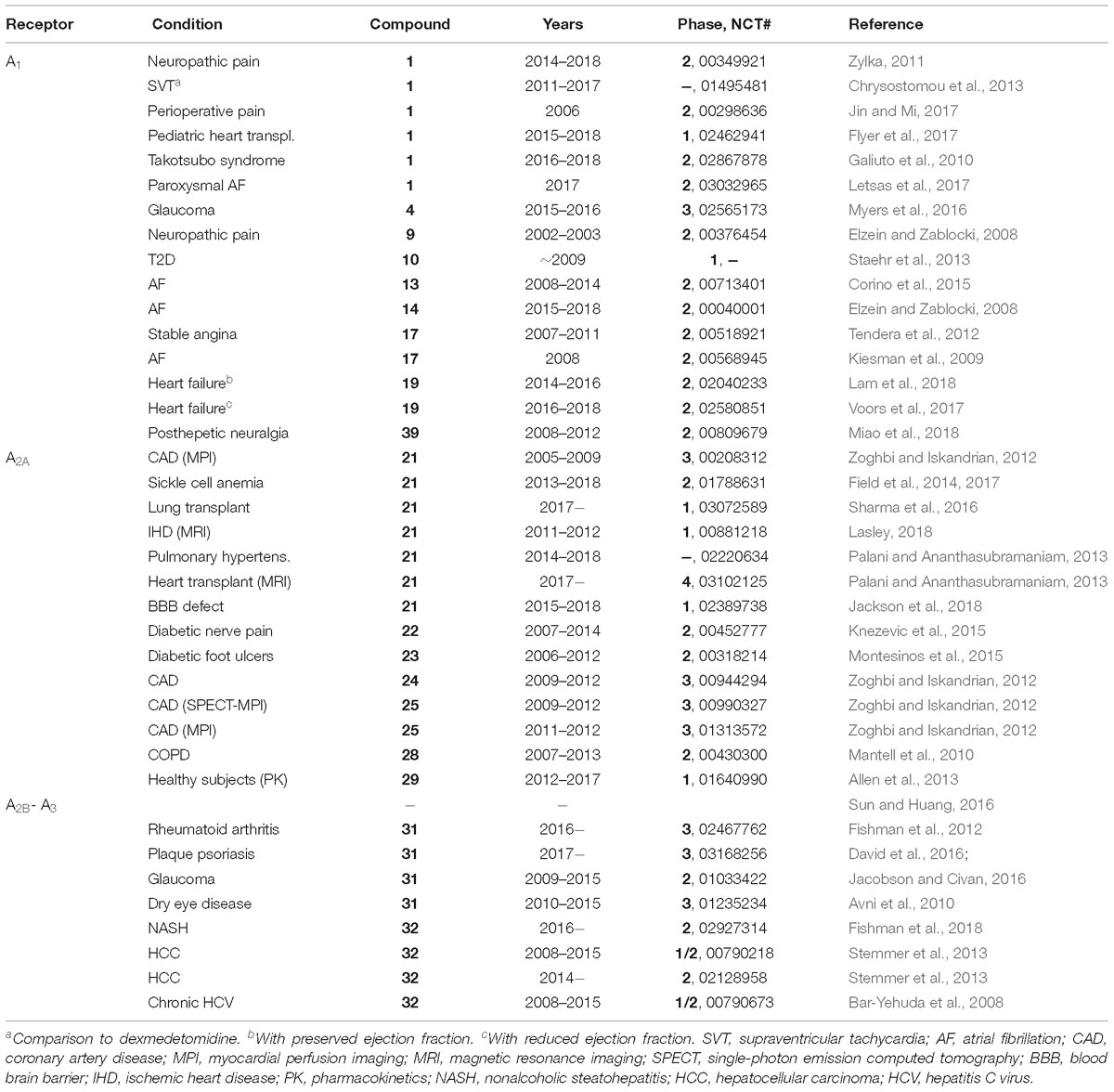
Table 1. Representative recent clinical trials of AR agonists and an A1AR PAM (data from ClinicalTrials.gov, accessed 12-28-2018).
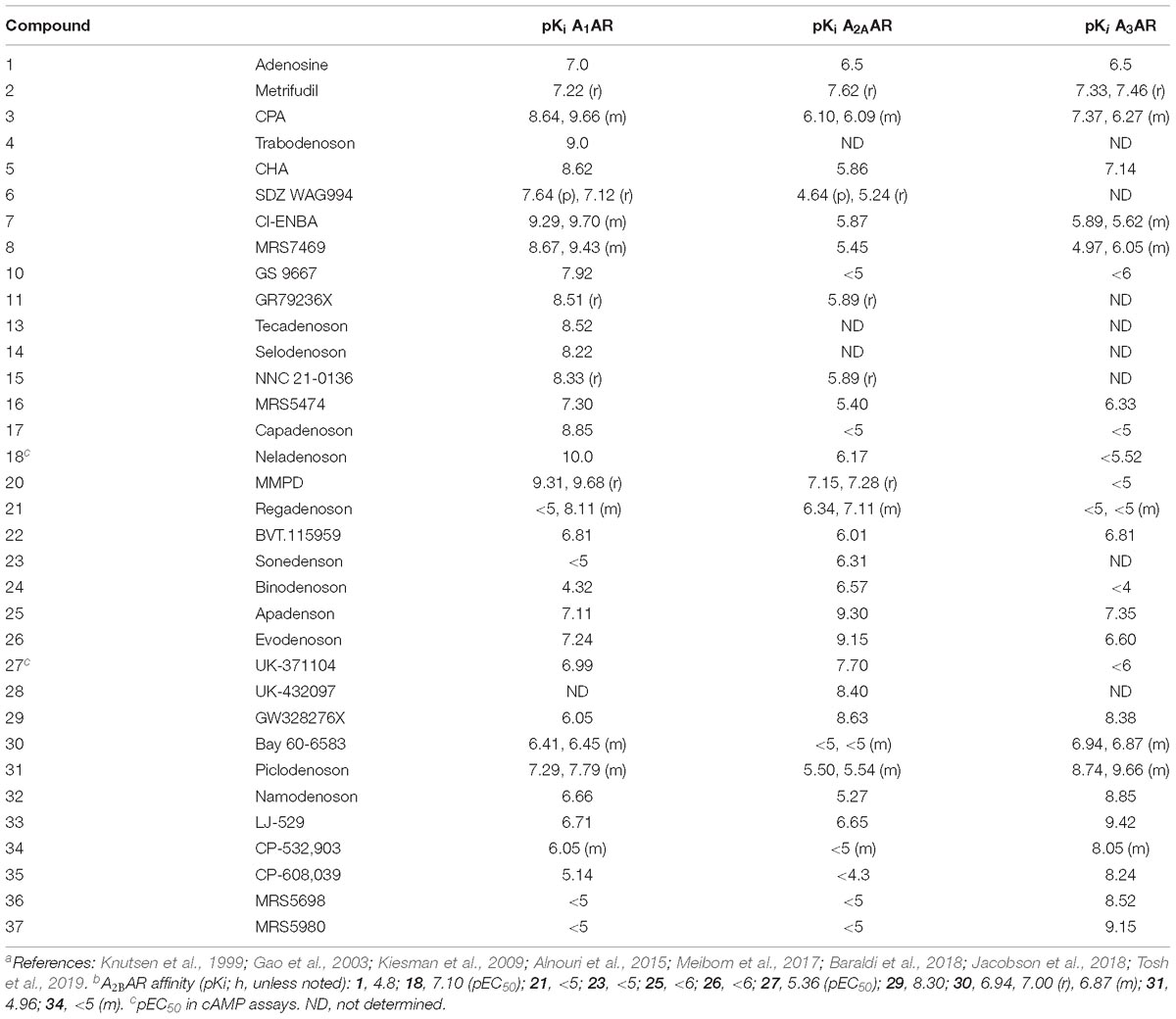
Table 2. AR Binding affinity of selected AR agonists described here (human, if not specified; p, pig; r, rat; m, mouse).a,b
Each AR can signal through multiple pathways, and different agonists may show a signaling preference, i.e., bias. For example, the A2BAR acts through Gs, Gq, or Gi, depending on the tissue, receptor density and final measure of activity (Gao et al., 2018). The possibility of biased signaling of AR agonists or PAMs could provide a means to increase selectivity for a particular tissue or condition (Gao et al., 2011; Baltos et al., 2017; Gao et al., 2018; Vecchio et al., 2018).
One limitation of AR agonists for human therapeutics is that their target GPCRs might be subject to agonist-induced desensitization as shown for all four ARs in cell lines (Mundell and Kelly, 2011). However, in other cases, depending on multiple pharmacological factors, a prolonged agonist exposure did not lead to in vivo desensitization of the desired effect (Little et al., 2015), even with a full A3AR agonist. Two possible approaches to circumvent GPCR desensitization are the use of partial agonists in cases where the desired effect is maintained, such as in A1AR antiarrhythmic activity (Elzein and Zablocki, 2008; Voors et al., 2017), and the use of PAMs (Vincenzi et al., 2014). Partial agonists are also known to display tissue or organ selectivity, because of the differential receptor expression level (Van der Graaf et al., 1999). Thus, when spare receptors are present, i.e., overexpression or high level of endogenous expression, a partial agonist could be fully efficacious in tissues where there is a receptor reserve and activation of only a fraction of the receptors suffices.
Positron emission tomography (PET) for in vivo imaging of ARs has been extensively explored (van Waarde et al., 2018). PET imaging could potentially be essential in drug discovery and clinical development of AR modulators, to determine the target engagement and to assess the role of endogenous adenosine. Although most high affinity ligands designed for in vivo AR imaging by PET have been antagonists, some agonists have also been labeled with positron-emitting 18F, 76Br or 11C isotopes (Kiesewetter et al., 2009; Guo et al., 2018).
A1AR and A2AAR are examples of the growing list of GPCRs for which physically determined three-dimensional structures with a bound agonist are known (Lebon et al., 2011; Xu et al., 2011). This understanding facilitates the structure-based design of novel AR agonists (Tosh et al., 2019). Several structures determined by X-ray crystallography and cryo-electron microscopy contain a G-protein or G-protein fragment, which is more representative of the active state AR structures (Draper-Joyce et al., 2018; García-Nafría et al., 2018). NMR studies of assigned A2AAR Trp and Gly residues are informative about microswitches involved in the signal propagation from bound agonist toward the intracellular region for G protein binding (Eddy et al., 2018). There can be multiple active conformations of a given GPCR, and agonist-induced changes are nuanced with respect to both structure and signaling. Thus, direct biophysical methods for receptor characterization can impact agonist design.
The following examples of AR drugs and molecular probes are arranged according to their target receptor: A1 (1 – 20, 39, 40), A2A (21 – 29), A2B (30), A3 (31 – 38, 41). It has also been suggested that coactivation of two AR subtypes might be beneficial, such as both A1AR and A3AR in cardioprotection (Jacobson et al., 2005).
Early AR Agonists
Adenosine 1
Arrhythmias
Many pathophysiological conditions including hypoxia and ischemia may cause arrhythmias. Adenosine is considered as an endogenous antiarrhythmic agent partly due to its endogenous anti-ischemic property (Kiesman et al., 2009; Szentmiklosi et al., 2015). As described earlier, infused adenosine (under the name of Adenocard, approved in 1989, Olsson, 2003) and its precursor ATP have long been used for the treatment of paroxysmal supraventricular tachycardia (PSVT) (Pelleg et al., 2012; Szentmiklosi et al., 2015). The antiarrhythmic action of adenosine has been suggested to occur via the A1AR in the sinoatrial and atrioventricular nodes, which leads to modification of AV nodal conduction (Kiesman et al., 2009). A1AR activation is known to induce opening of ATP-sensitive potassium channels (Fredholm et al., 2001; Jacobson and Gao, 2006). Adenosine infusion (under the name of Adenoscan, approved in 1995) is used for myocardial perfusion imaging (MPI), through its short-lived A2AAR activation, to dilate the coronary artery (Olsson, 2003). Many recent clinical trials in various cardiovascular and ischemic conditions have utilized adenosine as an approved drug being tested for new uses (Table 1). For example, a clinical trial of adenosine to reverse left ventricular impairment in Takotsubo syndrome (Galiuto et al., 2010) was initiated, but terminated in 2018.
Dermatological conditions
Adenosine applied topically has been used to promote hair growth and skin health (Abella, 2006; Faghihi et al., 2013). For the treatment of androgenetic alopecia, adenosine (0.75% solution) displayed efficacy similar to minoxidil but was preferred by patients because of the response speed and its quality. Applied in cosmetic preparations (0.1% cream or 1% dissolvable film) for 2 months, adenosine significantly improved skin smoothness and reduced facial wrinkles.
Epilepsy
Adenosine is considered an endogenous antiseizure agent and also attenuates epileptogenesis by an epigenetic mechanism, i.e., nuclear adenosine reduces DNA methylation (Boison, 2013). Elevated adenosine indirectly inhibits DNA methyltransferases through two stages of enzymatic product inhibition, beginning with S-adenosylhomocysteine hydrolase (SAH). In the epileptic brain there is an overexpression of adenosine kinase in astrocytes, which depletes adenosine both inside and outside the cell. Because peripheral adenosine itself is rapidly removed from circulation in seconds, drug delivery systems are needed to raise its brain concentration. Experimental use of an adenosine-releasing silk brain implant was found to be an efficacious form of adenosine augmentation therapy (AAT) for treating refractory epilepsy in a rat model of kindling epileptogenesis (Szybala et al., 2009). Adenosine was encapsulated in microspheres, which were embedded into nanofilm-coated silk fibroin scaffolds. The polymer was introduced surgically into the infrahippocampal cleft, and when monitored for 10 days, demonstrated a lack of convulsive seizures. This protective effect was antagonized by an A1AR-selective antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX).
AMP (Adenosine 5′-Monophosphate) and ATP (Adenosine 5′-Triphosphate)
Asthmatic drug testing
Ectonucleotidases readily cleave the naturally occurring 5′-phosphoesters of adenosine 5′-phosphates to produce adenosine in situ. Adenosine 5′-monophosphate (AMP) is used diagnostically in inhalation challenge testing (Basoglu et al., 2005; Isogai et al., 2017). AMP readily forms adenosine in situ and thus most of its actions occur through ARs, but not all of its in vivo effects arise from AR activation (Carlin et al., 2018; Xiao et al., 2019).
Cancer
Adenosine 5′-triphosphate plays a physiological role in obstructive airway disease to increase inflammation and to induce bronchoconstriction and cough (Pelleg et al., 2016). ATP has been administered in humans intravenously in clinical trials for the treatment of both cachexia in cancer and the cancer itself (Rapaport et al., 2015). The working hypothesis was that by increasing the intracellular ATP pools in erythrocytes, the energy balance could be restored. Alternatively, an action on P2 receptors was considered, but the extracellular ATP pools were elevated only briefly in patients with advanced solid tumors. Nevertheless, with the ubiquitous presence of ectonucleotidases, one main action of ATP would be through increased adenosine acting at ARs.
Metrifudil 2
Glomerulonephritis
Many adenosine derivatives found in early studies (1960s and 1970s) to be biologically active at what were later termed the A1AR and A2AAR, contain bulky, hydrophobic substitution at the adenine N6 position (Olsson, 2003; Jacobson and Gao, 2006; Mantell et al., 2010). This describes two potent agonists, Metrifudil 2 and R-PIA (R-N6-phenylisopropyladenosine (structure not shown), which are moderately A1AR-selective. Metrifudil (60 mg oral dose) was one of the earliest synthetic adenosine agonists to be studied in humans (Schaumann and Kutscha, 1972; Wilbrandt et al., 1972), along with R-PIA (Schaumann et al., 1972). Following the discovery that vasodilator dipyridamole (equilibrative adenosine transporter ENT1 inhibitor) was active against glomerulonephritis by an adenosinergic mechanism, Metrifudil was applied to its treatment, which resulted in limited improvement in three patients. However, no subsequent trials of Metrifudil or R-PIA were reported, probably because of their cardiovascular side effects.
A1AR-Selective Agonists
Trabodenoson (INO-8875, PJ-875) 4
Glaucoma
A1AR partial agonist Trabodenoson (INO-8875) 4, which is a 5′-nitrate ester derived from CPA 3, was in advanced clinical trials as an ophthalmic formulation for primary open-angle glaucoma and ocular hypertension. However, the development was terminated in 2017 following a Phase 3 trial due to failure to achieve its primary endpoint (Jacobson and Civan, 2016). Trabodenoson and its congeners were also considered for treatment of arrhythmias (Elzein and Zablocki, 2008; see below). Another analog containing a 5′-nitrate ester, but with a N6-3-aminotetrahydrofuryl group was found to lose its nitro group in vivo to form the parent full agonist, which was associated with side effects.
CHA 5
Hypothermia
The use of A1AR agonist CHA 5 for inducing therapeutic hypothermia has been proposed (Jinka et al., 2015). However, it is likely that the hypothermic effect of CHA in rodents may be through both a peripheral A3AR, and a central A1AR (Carlin et al., 2017).
SDZ WAG994 6
Diabetes and obesity
A1AR is highly expressed in adipose tissue and involved in triglyceride (TG) storage. Breakdown of TG and the subsequent increase in plasma free fatty acids (FFA) results in development of insulin resistance in peripheral organs. Insulin resistance is associated with obesity and development of type 2 diabetes (Antonioli et al., 2015). Adenosine-mediated A1AR activation or A1AR overexpression in adipocytes results in suppression of lipolysis and reduction of plasma FFA. Various adenosine analogs have been developed for their potential intervention in diabetes. N6-Cyclopentyladenosine (CPA, 3) reduced FFA and cholesterol levels and increased glycogen synthesis in skeletal muscle in streptozotocin (STZ) diabetic rats (Cheng et al., 2000). Thus, A1AR agonists may have beneficial effects on glucose utilization by peripheral tissues to lower plasma glucose.
Typically, both 2′- and 3′-hydroxyl groups are important for nucleoside recognition in the AR binding sites. SDZ WAG994 6 is a structurally unusual 2′-O-methyl A1AR agonist that did not display hemodynamic side effects in humans. However, SDZ WAG994 increased PR interval, consistent with A1AR activation in the AV node and its suggested use in tachycardia (Jacobson and Knutsen, 2001).
SDZ WAG994 6 was also under development as an antilipolytic agent for treating diabetes (Ishikawa et al., 1998). Oral administration of SDZ WAG994 and another A1AR agonist Selodenoson (RG14202, 14) in STX-treated rats decreased FFA, TG levels and heart rate in a dose-dependent manner (Cox et al., 1997). A human study revealed that RPR749 (structure not shown), was capable of decreasing circulating FFA levels and thus may be beneficial in treating hyperlipidemia (Shah et al., 2004). These compounds have therapeutic potential for the treatment of cardiovascular and metabolic disorders.
Cl-ENBA 7 and MRS7469 8
Hypothermia and pain
Several A1AR agonists, Cl-ENBA 7 and MRS7469 8, were recently reported to activate in the mouse A1AR in the brain when administered peripherally, without comparable peripheral A3AR activation (Carlin et al., 2017; Tosh et al., 2019). Cl-ENBA 7 is a highly A1AR-selective adenosine agonist that is used as a pharmacological probe (Tosh et al., 2019), and its efficacy in pain models demonstrated (Luongo et al., 2012). It consists of a mixture of two diastereoisomers, and thus its in vivo A1AR target engagement is complicated. However, when administered intraperitoneally in mice, it activated the central A1AR preferentially over the peripheral A3AR in mast cells, due partly its being a full agonist for the A1AR but a partial agonist of low efficacy or an antagonist at the A3AR (Carlin et al., 2017). This is consistent with a report that S-N6-endo-norbornyl-adenosine (S-ENBA) is a full agonist at the A1AR but low-efficacy partial agonist at the A3AR (Gao et al., 2003).
Like Cl-ENBA, MRS7469 8 is a highly selective agonist that activated the central A1AR preferentially when administered peripherally, leading to A1AR-dependent hypothermia and locomotor depression (Tosh et al., 2019). When administered icv. (52 μg/kg) it caused intense hypothermia. Thus, it crosses the blood brain barrier (BBB) sufficiently, given its high affinity (Ki 0.37 nM at mouse A1AR), to activate the A1AR. Moreover, MRS7469 with a non-chiral N6 group is a pure diastereoisomer, which is advantageous for in vivo studies.
GW493838 9
Pain
The A1AR is involved in depressant and protective functions in the brain and spinal cord, including suppressing pain (Sawynok, 1998; Zylka, 2011; Giorgi and Nieri, 2013). Intrathecal opioids induce local adenosine release (Eisenach et al., 2004), and exogenously applied adenosine and other A1AR agonists and PAMs reduce pain (Eisenach et al., 2013; Vincenzi et al., 2014). A1AR agonist GW493838 9, which is an adenosine analog highly modified at N6 and 5′ positions, showed efficacy in animal models of pain (Imlach et al., 2015). However, GW493838 (50 mg oral dose) failed to show significant efficacy in a clinical trial for treatment of chronic pain (diabetic pain). Also, a meta-analysis of patient postoperative pain in clinical data showed no analgesic effect of adenosine (Jin and Mi, 2017).
CVT-3619 (GS-9667) 10, GR79236 11 and ARA 12
Diabetes
Increasing evidence indicates a crucial role of A1AR in the regulation of insulin sensitivity and glucose homeostasis especially in metabolically active organs such as adipose tissue, liver and skeletal muscle, which are related to diabetes mellitus (Peleli and Carlstrom, 2017). It has been convincingly demonstrated that A1AR is critical for regulation of lipid metabolism, and thus A1AR agonists have been proposed for the treatment of type II diabetes (T2D) and obesity (Antonioli et al., 2018). The white adipocyte A1AR inhibits lipolysis. Curiously, a functional A2AAR activates thermogenic brown adipose tissue (BAT) as indicated using human PET imaging (Lahesmaa et al., 2018), and A2AAR agonists might prove beneficial in metabolic conditions (Tozzi and Novak, 2017). Cold exposure in human subjects reduced PET ligand binding in BAT, indicative of elevated local adenosine release.
Several A1AR agonists, GR79236 11, ARA 12, and CVT-3619 10, have been in clinical trials for T2D due to their ability to increase insulin sensitivity (Bigot et al., 2004; Kiesman et al., 2009; Staehr et al., 2013). However, development of full agonists, such as GR79236 and ARA, was not successful due to cardiovascular side effects (Elzein and Zablocki, 2008). Although both full and partial agonists may lower non-esterified fatty acid levels, it is suggested that partial agonists may improve insulin sensitivity without producing severe cardiovascular side effects (Elzein and Zablocki, 2008). A single dose of ARA given to healthy individuals during a phase I clinical trial reduced plasma FFA levels, but individuals developed tolerance to the drug (Zannikos et al., 2001). The A1AR partial agonist CVT-3619 (GS-9667) has been reported to lower FFA in both healthy and obese subjects without showing evidence of A1AR desensitization (Staehr et al., 2013), but oral doses of ≥ 300 mg were required to see the FFA effect. The individual benefits of GS-9667 and sitagliptin [Januvia, an inhibitor of DPP4 (dipeptidyl peptidase 4)] on glucose and lipid homeostasis were enhanced in combination (Ning et al., 2011).
Tecadenoson 13 and Selodenoson 14
Arrhythmias
Despite its demonstrated A1AR-dependent beneficial effect in PSVT, adenosine is known to cause atrial fibrillation (AF) in about 15% of patients by decreasing the refractory period of the atrium and causes other adverse effects related to the activation of other AR subtypes (Glatter et al., 1999). Thus, extensive efforts have been made in developing selective A1AR agonists as anti-arrhythmic agents (Mason and DiMarco, 2009). A1AR full agonists Tecadenoson 13 (Corino et al., 2015), Selodenoson 14 and Trabodenoson 4 (Kiesman et al., 2009; Mason and DiMarco, 2009) have been under development. However, full agonists are known to cause tachyphylaxis, presumably due to A1AR desensitization. Tecadenoson has been in a Phase 3 trial for the termination of supraventricular tachycardia (SVT) (Elzein and Zablocki, 2008; Mason and DiMarco, 2009), but its development was discontinued in 2009. A clinical safety study of Tecadenoson for the treatment of AF was performed, but its clinical development was also curtailed.
NNC-21-0136 15
Stroke
NNC-21-0136 15 is an A1AR selective agonist that was designed for neuroprotection. Its hemodynamic effects were minimal, revealing a brain-protective effect in stroke models (Knutsen et al., 1999; Jacobson and Knutsen, 2001), but it did not enter human testing.
MRS5474 16
Seizures and depression
A1AR agonists are of interest in the CNS for their anxiolytic, antinociceptive, antidepressant and antiseizure properties, and behavioral results with A1AR KO mice support the use of A1AR in this context. Unfortunately, many adenosine derivatives display minimal ability to cross the BBB (Schaddelee et al., 2005; Tosh et al., 2019). 4′-Truncated nucleosides, thionucleosides and methanocarba-nucleosides (containing a [3.1.0]bicyclohexyl ring system) were originally characterized as A3AR low-efficacy, selective partial agonists. However, an N6-dicyclopropylmethyl group present in MRS5474 16 substantially shifts the selectivity toward A1AR, especially in mouse (204-fold compared to mouse A3AR), compared to other a-branched N6 groups (Tosh et al., 2012, 2019; Carlin et al., 2017). The small molecular weight (376), polar surface area (93 Å2) and number of H-bond donor groups (3) positioned MRS5474 for potential brain application. MRS5474 showed antidepressant activity in a mouse model that was mediated by homer1 protein in the medial prefrontal cortex, and upregulation of homer1 by an AR agonist was lost in A1AR KO mice (Serchov et al., 2015). Nevertheless, it also activated a peripheral mA3AR (Carlin et al., 2017).
Capadenoson 17 and Neladenoson 18
Angina
Anti-ischemic effect of A1AR agonists has been demonstrated in animal studies, but clinical successes are lacking, and more relevant clinical models are needed (Borea et al., 2016; Lasley, 2018).
Most known AR agonists are adenosine derivatives, but two classes of pyridine-derived agonists are known (Guo et al., 2018). The non-nucleoside A1AR agonist Capadenoson 17 (BAY68-4986), having an atypical 3,5-dicyanopyridine structure, was evaluated in patients with stable angina using an oral dose of 4 mg, once daily (Kiesman et al., 2009; Tendera et al., 2012). However, Capadenoson was withdrawn from clinical trials for angina and for AF.
Heart failure
Rather than full agonists, an A1AR partial agonist Neladenoson 18 (in the form of a dipeptide ester prodrug 19) is now being tested in patients with chronic heart failure (Greene et al., 2016; Dinh et al., 2017; Meibom et al., 2017; Voors et al., 2017). Compared with Capadenoson (Baltos et al., 2017), Neladenoson is a more selective partial agonist for A1AR. Neladenoson has been shown to improve cardiac function without producing bradycardia, atrioventricular blocks, or undesirable effect on blood pressure (Meibom et al., 2017; Voors et al., 2017). The rationale for using partial A1AR agonists is based on the observation that the activation of myocardial A1ARs by partial agonists protects cardiac function related to ischemia and reperfusion injury without producing severe side effects (Albrecht-Küpper et al., 2012; Voors et al., 2017). A multiple dose study of Neladenoson (BAY 1067197) (ParSiFAL, 5 – 40 mg oral dose, once daily) in heart failure is ongoing.
MMPD 20
Imaging
Numerous A1AR ligands have been in development for potential use in diagnosis of various conditions, such as depression, Parkinson’s disease, Alzheimer’s disease, epilepsy, ischemia, and sleep disorders. The A1AR is highly expressed in many brain regions, such as the hippocampus, neocortex, thalamus and basal ganglia (Fredholm et al., 2001). In vivo imaging of A1AR in the human brain is therefore an attractive approach for diagnosis, and various AR agonists and antagonists have been developed for PET brain imaging (van Waarde et al., 2018). Although varied AR subtype selectivities and agonist efficacies are seen with the class of atypical 3,5-dicyanopyridine ligands, partial agonist MMPD 20 was recently shown to be highly A1AR selective, with 16% maximal human A1AR activation, and suitable for the PET imaging in the rat brain (Guo et al., 2018). It has a relatively low molecular weight (373) and polar surface area (108 Å2), which allows it to cross the BBB. Typical ribose-containing A1AR agonists have limited utility to be administered therapeutically for CNS treatment due to their low degree of brain uptake from the periphery (Schaddelee et al., 2005).
A2AAR-Selective Agonists
Regadenoson 21
Imaging
Regadenoson 21 (CVT-3146, Lexiscan) is a moderately selective, short acting A2AAR agonist that is administered i.v. for MPI (Palani and Ananthasubramaniam, 2013). It was first approved as a pharmacologic stress agent in 2008. At present, it is the only synthetic AR agonist that is approved for human use, although it is not highly potent or selective for the A2AAR. Nevertheless, Regadenoson’s A2AAR selectivity is higher in human than in mouse, in which it is actually 10-fold A1AR selective compared to A2AAR (Carlin et al., 2018). The availability of an FDA-approved new chemical entity (NCE) allows it to be tested in diverse clinical trials for cardiovascular treatment and diagnosis (>60 currently listed in ClinicalTrials.gov, accessed 12-31-2018).
Sickle cell disease
In addition to their diagnostic application in MPI, A2AAR agonists have been considered for treatment of inflammation (Cekic and Linden, 2016) and sickle cell disease (SCD, Field et al., 2014). A2AAR activation in natural killer T (iNKT) cells is anti-inflammatory as demonstrated in a transgenic mouse model of SCD. However, A2BAR activation in erythrocytes is predicted to have a harmful effect in SCD. The effects of Regadenoson 21 as an A2AAR agonist in SCD patients were evaluated in a clinical trial, but there was no statistically significant benefit (Field et al., 2017).
Lung transplantation
Murine lung ischemia reperfusion injury occurring via NADPH oxidase 2 (NOX2) and IL-17 is also attenuated by A2AAR agonist 26 (Sharma et al., 2016), which has led to an ongoing clinical trial of Regadenoson 21 in lung transplantation. The safety of using Regadenoson for MPI in patients with mild to moderate COPD and asthma was established (Golzar and Doukky, 2014).
Glioblastoma
A2AAR agonists transiently increase BBB permeability (Kim and Bynoe, 2016), and this is being evaluated as a novel pharmacological approach to drug delivery to the brain. Regadenoson was tested clinically in an attempt to raise the concentration of the anticancer drug temozolomide in the brain interstitium, determined using microdialysis in glioblastoma patients (Jackson et al., 2018).
Spongosine (BVT.115959, CBT-1008) 22 and Other Naturally Occurring AR Agonists
Pain
Numerous other A2AAR agonists were studied in preclinical testing or clinical trials prior to the approval of Regadenoson. Among the first such agonists was the simple 2-methoxy derivative of adenosine, spongosine (BVT.115959) 22, a marine natural product (García et al., 2018). Actually, spongosine is slightly selective for and equipotent at the human A1AR and A3AR. It was shown to be effective in a clinical trial for diabetic neuropathic pain (7 mg oral dose, 3X daily), which was terminated because the company discontinued small molecule research (Knezevic et al., 2015).
Inflammation
Activation of the A2AAR by endogenous adenosine provides benefit in animal models of inflammation and rheumatic disease, for example in rat models of osteoarthritis (Cronstein and Sitkovsky, 2017; Haskó et al., 2018). Other naturally occurring adenosine or deoxyadenosine derivatives have been applied as AR agonists. Polydeoxyribonucleotide (PDRN, structure not shown), of molecular weight 80–200 KD and extracted from trout or salmon sperm, is degraded by plasma DNA nucleases or cell membrane-bound nucleases giving rise to nucleosides and nucleotides. It is asserted that the degraded products pharmacologically activate the A2AAR, based on antagonism by the relatively weak and non-selective A2AAR antagonist 3,7-dimethyl-1-propargylxanthine (DMPX). PRDN’s therapeutic effects include tissue repairing, anti-ischemic, and anti-inflammatory, making it suitable in regenerative medicine and for treating diabetic foot ulcers. Topically applied PDRN was in a clinical trial for reducing inflammation to promote wound healing in cases of diabetic foot ulcers (Squadrito et al., 2017). Also, topically applied PDRN significantly reduced pain and increased joint function in an animal model of osteoarthritis and increased neurogenesis in a spinal cord injury model (Irrera et al., 2018).
Sonedenoson (MRE-0094) 23 and Binodenoson (WRC-0470, MRE-0470) 24
Imaging
Many adenosine derivatives that proved to be A2AAR-selective agonists have bulky, hydrophobic substitution at the C2 position of adenine (Jacobson and Gao, 2006). A2AAR agonist Binodenoson 24 (≤ 1.5 μg/kg, i.v.) administered for MPI of patients with coronary artery disease did not cause the side effect of bronchoconstriction (Murray et al., 2009).
Wound healing
Sonedenoson (MRE-0094) 23 was effective in the treatment of poorly healing wounds in animal models (Victor-Vega et al., 2002), an A2AAR-agonist effect later found to be dependent on tissue plasminogen activator (Montesinos et al., 2015). A Phase 2 clinical trial of Sonedenoson administered as a topical gel for diabetic foot ulcers had poor enrollment and was terminated in 2008.
Apadenoson (ATL-146e, BMS 068645) 25 and Evodenoson (ATL-313, DE-112) 26
Imaging
Apadenoson 25 (Rieger et al., 2001; Zoghbi and Iskandrian, 2012) was in several clinical trials for MPI and SCD, which has a component of hypoxia. Apadenoson contains a labile ester moiety, which is cleaved in vivo to limit its duration of action. Its more stable, urethane-containing congener Evodenoson (ATL-313) 26, was developed as a candidate drug for treating multiple myeloma (Rickles et al., 2010; van Waarde et al., 2018).
UK-371104 27 and UK-432097 28
Pulmonary inflammation
Intratracheal administration of A2AAR agonist UK-371104 27, with sterically bulky N6 and C2 substituents, in anesthetized guinea pig, inhibited the capsaicin-induced bronchoconstriction without affecting blood pressure (Trevethick et al., 2008). Thus, additional lung-focused A2AAR agonists were explored for treating lung inflammation.
UK-432097 28 is a selective A2AAR agonist that was in a failed clinical trial for COPD (Mantell et al., 2010), although its pharmacology is comparable to its preceding congener, UK-371104 27. UK-432097 as an inhaled dry powder was not efficacious in human trials (discontinued in 2008), possibly due to its agonist activity at the A1 and A3ARs, and/or its high MW (778), and multiple H-bond donor (7) and acceptor (13) groups reduced its bioavailability, even when administered directly in the lungs by inhalation. However, it displays a favorably slow off-rate from the receptor, which has been suggested to contribute to its sustained agonist effects (Hothersall et al., 2017). The extended N6 and C2 substituents of UK-432097 and its congeners interact with A2AAR extracellular regions to impede their dissociation. The bulky N6 and C2 substitutions of UK-432097 enabled the structural determination of its A2AAR complex (Xu et al., 2011).
GW328267X 29
Asthma and allergy
A dual A2AAR agonist and A3AR antagonist GW328267X 29 failed to show efficacy in a clinical trial for asthma (inhaled) and allergic rhinitis (intranasal) (Trevethick et al., 2008), despite its anti-inflammatory efficacy in animal models. Its structure is unusual in that it contains an ethyl-tetrazole group at the ribose 4′ position, thus contributing to its dual action at the two AR subtypes (Trevethick et al., 2008). Its side effects (hypotension, tachycardia) even when administered by inhalation were dose-limiting in the clinical trials. However, intravenous infusion of GW328267X in humans (52 μg/kg, over 5.5 h) resulted in a mechanism-related tachycardia that was ascribed to A2AAR activation in the carotid bodies, which was not alleviated upon prolonged agonist exposure. The lack of tachyphylaxis leading to prolonged tachycardia was not acceptable (Allen et al., 2013), and its clinical testing was discontinued. The translational failure of A2AAR agonists is likely due to their limited selectivity, especially their agonist activity at the A1AR. It has been suggested that A1AR antagonists and A2AAR agonists may have beneficial effects for asthma (Gao and Jacobson, 2017).
A2BAR-Selective Agonist
BAY 60-6583 30
Ischemia, inflammation, diabetes, asthma, and cancer
Although there are no A2BAR agonists currently in clinical evaluation, animal models suggest its activation might result in beneficial effects in acute lung injury, ischemia and vascular leakage (Eltzschig et al., 2003; Eltzschig, 2009). The non-nucleoside (3,5-dicyanopyridine) agonist BAY 60-6583 30 has been used as an in vitro and in vivo pharmacological probe, although its degree of efficacy and its species dependence of affinity/selectivity can vary (Gao et al., 2014). In some models, including insulin release in MIN6 mouse insulinoma cells, the compound was reported to act as an A2BAR antagonist. Therefore, highly selective and reliably efficacious A2BAR agonists are still lacking. Moreover, the signaling pathways activated or inhibited by the nominally Gs-coupled A2BAR are complex and involve multiple G proteins (Gao et al., 2018).
Mast cell A2BAR activation might be useful in the treatment of asthma (Gao and Jacobson, 2017). In the intestines, kidney and other organs, this receptor has an anti-ischemic effect (Grenz et al., 2008; Hart et al., 2011). A2BAR activation is predicted to have beneficial cardiovascular effects and maintain the endothelial cell barrier (Eltzschig et al., 2003). BAY 60-6583 was shown to have protective effects in a model of myocardial reperfusion injury (Tian et al., 2015). A2BAR activation leading to the PI3K/Akt pathway is anti-inflammatory by shifting macrophages to an M2 phenotype. Pre-ischemic administration of BAY 60-6583 stimulated leukocyte PI3K/Akt in the mouse spleen to reduce myocardial reperfusion injury in an IL-10-dependent manner (Ni et al., 2018).
A2BAR activation might also be useful in treating T2D and atherosclerosis, and preventing vascular lesions due to smooth muscle cell proliferation after angioplasty (Koupenova et al., 2012; Sun and Huang, 2016). A2BAR KO mice displayed increased fatty liver pathology, tissue inflammation and insulin resistance due to the lack of this receptor in macrophages (Johnston-Cox et al., 2014). A2BAR activation reduced inflammation and macrophage activation resulting from FFA (Csóka et al., 2014). The receptor was highly upregulated in mice subjected to a high-fat diet (HFD), and A2BAR KO mice on this diet developed obesity and insulin resistance. BAY 60-6583 administered for 4 weeks HFD restored endocrine function and reduced inflammation. However, A2BAR gene expression was found to be elevated in cases of human gestational diabetes, but this observation did not establish whether an A2BAR agonist or antagonist would be more beneficial (Wojcik et al., 2014).
Although blocking the A2BAR is considered a target in conjunction with cancer immunotherapy, its activation also has been reported to reduce proliferation of cancer cells (Koussémou et al., 2018). A2BAR activation led to ERK1/2 dephosphorylation and reduced cell proliferation through inhibition of the MAPK signaling pathway in the MDA-MB-231 breast cancer cell line.
A3AR-Selective Agonists
IB-MECA 31
Autoimmune inflammatory diseases
A3AR agonists display anti-inflammatory and anticancer effects in various in vivo disease models (Cronstein and Sitkovsky, 2017; Jacobson et al., 2018). A3AR agonists stimulate chemotaxis in neutrophils through the leading edge, which could be pro-inflammatory. However, systemic A3AR agonist administration could actually have an anti-inflammatory effect by inhibiting neutrophil chemotaxis because of the non-directional agonist exposure (Chen et al., 2006).
IB-MECA (CF101, Piclodenoson) 31, the first moderately selective A3 agonist (Gallo-Rodriguez et al., 1994), is being developed for the treatment of autoimmune anti-inflammatory diseases, including rheumatoid arthritis (RA) and psoriasis (both in Phase 3) (Fishman et al., 2012). In Phase 2 trials, its action in RA and psoriasis compared favorably to existing treatments for those conditions, but it did not display serious adverse effects, as do the current treatments. In a comparison of 1, 2, and 4 mg oral IB-MECA doses in a 12-week Phase 2 psoriasis trial, the greatest patient improvement was observed with the 2 mg dose (Fishman et al., 2012). Similarly in a Phase 2 RA trial, the middle (1 mg, compared to 0.1 and 4 mg) oral dose achieved the highest responses. Peripheral blood mononuclear cells (PBMCs) from psoriasis patients showed elevated A3AR expression. IB-MECA inhibited proliferation and formation of IL-17 and IL-23 in a human keratinocyte cell line (Cohen et al., 2018). IB-MECA was previously in Phase 2 clinical trials for dry eye disease and glaucoma, 1 mg and 2 mg, respectively (oral, twice daily), which failed to demonstrate efficacy (Avni et al., 2010; Jacobson and Civan, 2016).
Cl-IB-MECA 32
Liver diseases
Cl-IB-MECA 32, was initially shown to display a higher A3AR agonist selectivity than IB-MECA at the rat ARs. However, at the mA3AR, IB-MECA is more potent and selective than Cl-IB-MECA (Carlin et al., 2017). Cl-IB-MECA (CF102, Namodenoson) is being developed for the treatment of liver conditions, including hepatocellular carcinoma (HCC) and non-alcoholic steatohepatitis (NASH) (Fishman et al., 2018). A3AR agonists have apoptotic and anticancer effects in vivo induced by Wnt signaling deregulation (Bar-Yehuda et al., 2008). The US Food and Drug Administration and the European Medicines Agency granted fast track designation to 32 for the treatment of liver cancer. Cl-IB-MECA (up to a 25 mg oral dose) increased the median overall survival in patients with advanced HCC by 7.8 months in patients (Stemmer et al., 2013), which was improved over the current treatment. There were no serious adverse effects or dose-limiting toxicity. Secondarily, the trial examined using the A3AR as a predictive marker of the CF102 clinical response. The use of A3AR to prevent cytokine release syndrome in cancer immunotherapy has been proposed (Cohen and Fishman, 2019).
An anti-steatotic effect of Cl-IB-MECA in an HFD mouse model of NASH, induced by STZ administered 2 days after birth, was mediated via a molecular mechanism leading to decreased α-smooth muscle actin (αSMA, a marker of pathological fibroblasts) and cytokeratin 18 (CK-18, a predictor of NASH severity). A Phase 2 trial of CF-102 (12.5 and 25 mg oral doses, twice daily) for NASH treatment is underway.
Skeletal muscle protection
A3AR agonists, including Cl-IB-MECA, protect skeletal muscle in ischemic models in a phospholipase C-b2/b3-dependent manner (Zheng et al., 2007).
CP-608,039 34 and CP-608,039 35
Cardioprotection
DeNinno et al. (2003) reported that CP-608,039 35 is a highly A3AR selective and water-soluble agonist that was being evaluated for the prevention of perioperative myocardial ischemic injury. This follows numerous other reports showing that A3AR activation protects ischemic cardiomyocytes by preconditioning (Lasley, 2018).
Recently, selective A3AR deletion in mouse cardiomyocytes was used to demonstrate that activation by selective agonist CP-532,903 (34) of a myocardial A3AR provides ischemic tolerance that is dependent on KATP channels (Wan et al., 2019). This study resolves a long-standing controversy by showing that a protective A3AR is present in adult ventricular cardiomyocytes, although expressed at very low levels (copy number of 85 per 100 ng total RNA versus 12,830 for the A1AR).
MRS5698 36
Pain
As noted above, A1AR agonists and PAMs are already under consideration for pain treatment. The efficacy of A3AR agonists for chronic pain was first explored depth in 2011 (Chen et al., 2012). The approach of using A3AR agonists for pain treatment was initially controversial, as A3AR activation had been described in earlier review papers as an uninteresting target or even an anti-target for pain relief (Nascimento et al., 2012; Janes et al., 2016). Reasons for this premature characterization were: truly selective A3AR agonists were not initially available and the A3AR causes release of inflammatory mediators, e.g., histamine and serotonin, from peripheral mast cells in rodents, but not human and other species (Auchampach et al., 1997; Leung et al., 2014; Gao and Jacobson, 2017). These mediators can contribute to inflammation in mouse and rat, and Sawynok (1998) concluded that A3AR activation induces pain and paw oedema. However, consistent with the now well-documented action of A3AR activation in various chronic pain models, A3AR KO mice had a lower pain threshold in the hind-paw hot-plate test (40% greater latency, Fedorova et al., 2003). Curiously, there was no difference between A3AR KO and WT mice in the acute pain response in the tail-flick test.
The two selective A3AR agonists in already clinical trials, IB-MECA and Cl-IB-MECA, at high doses in vivo might interact with other ARs, as has been observed in experimental models (Fozard, 2010). A new series of C2-extended (N)-methanocarba analogs displayed even greater A3AR selectivity, estimated to be in the range of at least 10,000-fold in comparison to other ARs (Jacobson et al., 2018). Among these agonists is MRS5698 33, which has been shown to reduce chronic neuropathic pain, including oxaliplatin-induced neuropathic pain (Little et al., 2015; Wahlman et al., 2018). MRS5698 has many drug-like properties – it is non-toxic and relatively stable in vivo, except that its oral bioavailability in the rat is only 5%F (Tosh et al., 2015). Nevertheless, by various modes of administration it is efficacious in pain models in vivo, including chronic constriction injury-induced, chemotherapy-induced and cancer-induced. Its ability to reduce chronic hyperalgesia is not affected by the A3AR-induced histamine release observed in rodent but not human mast cells (Carlin et al., 2016).
MRS5980 37
Pain
MRS5980 37 is a highly selective C2-arylethynyl (N)-methanocarba A3AR agonist, which has been demonstrated to be highly efficacious in in vivo pain models following administered by oral gavage, with a protective effect lasting up to 3 h (Tosh et al., 2014; Janes et al., 2016), and its metabolomics has been studied (Fang et al., 2015). The 2-chlorothienyl group is stable in vivo, and the aryl alkyne group was shown to be not highly reactive. MRS5980 and other A3AR agonist were shown to indirectly block a pro-nociceptive N-type Ca2+ calcium channels, a proven target in controlling pain, and cell excitability in the spinal cord dorsal horn (Coppi et al., 2019). Thus, therapeutic application of A3AR agonists appears to be a promising approach for treating pain of different etiologies.
LJ-529 33 and MRS4322 38
Stroke
von Lubitz et al. (1999) found that both A1AR and A3AR agonists have cerebroprotective properties in a model of gerbil forebrain ischemia. An (N)-methanocarba nucleoside MRS4322 38 was proposed in a patent application (Korinek et al., 2018) as a treatment for stroke and traumatic brain injury. The nucleoside appears to act through the A3AR, to which it binds in the μM range, because a selective A3AR antagonist propyl 6-ethyl-5-((ethylthio)carbonyl)-2-phenyl-4-propylnicotinate (MRS1523) diminished the benefit of reduced stroke lesions. LJ-529 33 (4′-thio-Cl-IB-MECA) is a selective A3 adenosine agonist that was shown to be protective in a rat stroke model and inhibited brain migration of inflammatory cells (Choi et al., 2011). However, platelet A2AAR activation by LJ-529 increased the bleeding risk.
AR Allosteric Enhancers (PAMs)
PAMs of the A1AR and A3AR have been the subject of preclinical and clinical evaluation (Figure 3 and Table 2). PAMs, in principle, may remain silent until a large rise in the extracellular adenosine occurs, at which time the PAM would amplify adenosine’s action at a particular AR subtype. Thus, PAMs are described as temporally and spatially specific modulators (Gao et al., 2011). Also, PAMs tend to be more subtype selective than orthosteric agonists, because their non-canonical binding regions on the GPCRs are those that have the most diverse sequences within the receptor family (Vecchio et al., 2018).
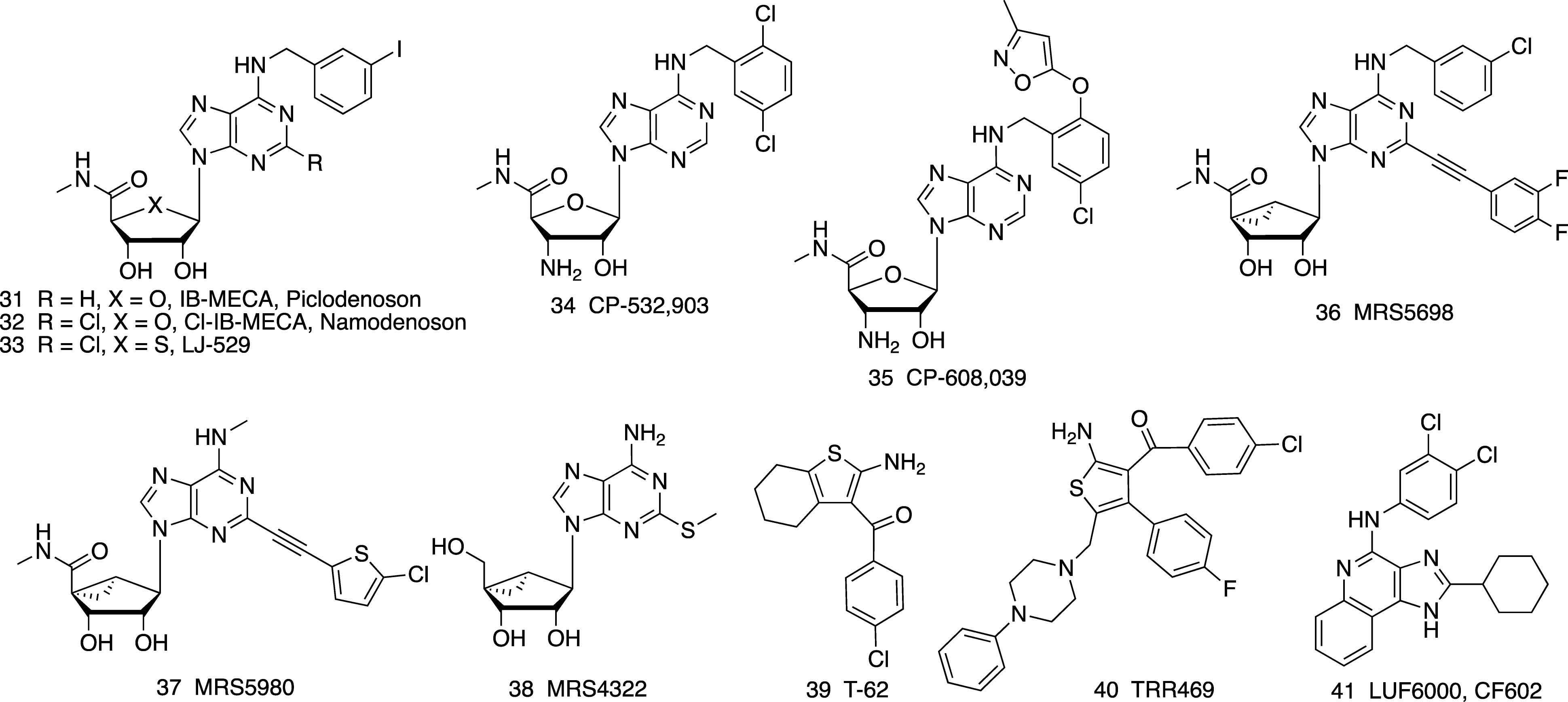
Figure 3. A3AR- (31-38) selective agonists and allosteric enhancers (PAMs) of the A1AR (39, 40) and A3AR (41).
T-62 39
Benzoylthiophenes are the earliest and most extensively studied class of A1AR PAMs, and they have allosteric agonist properties as well as enhancing the effect of other A1AR agonists (Vincenzi et al., 2014; Jacobson and Gao, 2017). The benzoylthiophenes Just as A1AR agonists have been considered for pain treatment, a representative benzoylthiophene T-62 39 entered a clinical trial for postherpetic neuropathic pain in 2008 that was discontinued because of its lack of efficacy (Giorgi and Nieri, 2013). Also, some patients displayed transient, elevated liver transaminases. Nevertheless, T-62 reduced hypersensitivity in animal models of neuropathic pain. The structural basis for recognition of benzoylthiophene PAMs involving A1AR extracellular loops (ELs) has been predicted using Gaussian accelerated molecular dynamics (Miao et al., 2018).
TRR469 40
Pain
A later generation benzoylthiophene TRR469 40 was shown to be an A1AR PAM that increases the affinity of A1AR agonists (Vincenzi et al., 2014, 2016). It reduced pain, comparably to morphine, in writhing and formalin tests and in chronic STZ-induced diabetic neuropathy, and it displayed fewer behavioral side effects than an A1AR orthosteric agonist. Furthermore, the same A1AR PAM was anxiolytic in four behavioral models in the mouse, in a manner comparable to the anxiolytic drug diazepam (Vincenzi et al., 2016). This activity of 40, which was blocked by a selective A1AR antagonist (DPCPX), is consistent with the previously noted anxiolytic activity of A1AR agonists.
LUF6000 41
Inflammation and erectile dysfunction
LUF6000 41 is a imidazoquinolinamine A3AR allosteric enhancer (PAM). It enhanced the maximal efficacy of A3AR agonists but had no agonism on its own (Gao et al., 2011). Its interaction with the A3AR was species-dependent (Du et al., 2018). Although it was more efficacious in human, canine and rabbit than in rodent species, it was shown to produce an anti-inflammatory effect in rat models of adjuvant-induced arthritis and iodoacetate-induced osteoarthritis and in a mouse model of concanavalin A-induced liver inflammation (Cohen et al., 2014). The molecule is termed CF602 and is on a translational path for treatment of erectile dysfunction (Cohen et al., 2016).
Conclusion
The structural and pharmacological features of key AR agonists and positive allosteric modulators (PAMs) have been summarized, with an emphasis on molecules that have been in humans or that were considered for human testing. From the thousands of selective AR agonists or allosteric enhancers reported, there are few translational successes. Many AR agonists have been in clinical trials for disease treatment or diagnosis, but only two are approved for human use, i.e., short-acting agonists adenosine and Regadenoson. However, new concepts and compounds are currently being developed and applied toward preclinical and clinical evaluation, and initial results are encouraging. AR agonists for treating inflammation, pain, cancer, NASH, angina, sickle cell disease, ischemic conditions and diabetes are under development. Multiple clinical trials with two A3AR agonists are ongoing.
Author Contributions
KJ organized the outline and wrote most of the text. DT contributed to the writing and researching the topic. SJ contributed to the writing and researching the topic. Z-GG contributed to the writing and researching the topic.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the NIDDK Intramural Research Program for funding (ZIA DK-31117).
References
Abella, M. L. (2006). Evaluation of anti-wrinkle efficacy of adenosine-containing products using the FOITS technique. Int. J. Cosmetic Sci. 28, 447–451. doi: 10.1111/j.1467-2494.2006.00349.x
Albrecht-Küpper, B. E., Leineweber, K., and Nell, P. G. (2012). Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal. 8(Suppl. 1), 91–99. doi: 10.1007/s11302-011-9274-3
Allard, B., Longhi, M. S., Robson, S. C., and Stagg, J. (2017). The ectonucleotidases CD39 and CD73: novel checkpoint inhibitor targets. Immunol. Rev. 276,121–144. doi: 10.1111/imr.12528
Allen, A., Koch, A., Garman, N., Cahn, A., Dewitt, O. E., and Rambaran, C. N. (2013). A PK/PD study of a selective A2a agonist, (GW328267X) a potential IV therapeutic for acute lung injury: no tachyphylaxis to the heart rate effect. Abstract PC028. Br. Pharmacol. Soc. Available at: https://bps.conference-services.net/resources/344/3654/pdf/PHARM13_0008.pdf
Alnouri, M. W., Jepards, S., Casari, A., Schiedel, A. C., Hinz, S., and Müller, C. E. (2015). Selectivity is species-dependent: characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal. 11, 389–407. doi: 10.1007/s11302-015-9460-9
Antonioli, L., Blandizzi, C., Csóka, B., Pacher, P., and Haskó, G. (2015). Adenosine signalling in diabetes mellitus–pathophysiology and therapeutic considerations. Nat. Rev. Endocrinol. 11, 228–241. doi: 10.1038/nrendo.2015.10
Antonioli, L., Fornai, M., Blandizzi, C., and Haskó, G. (2018). “Adenosine Regulation of the Immune System,” in BT - The Adenosine Receptors, eds P. A. Borea, K. Varani, S. Gessi, S. Merighi, and F. Vincenzi (Cham: Springer International Publishing), 499–514. doi: 10.1007/978-3-319-90808-3_20
Auchampach, J. A., Jin, X., Wan, T. C., Caughey, G. H., and Linden, J. (1997). Canine mast cell adenosine receptors: cloning and expression of the A3 receptor and evidence that degranulation is mediated by the A2B receptor. Mol. Pharmacol. 52, 846–860. doi: 10.1124/mol.52.5.846
Avni, I., Garzozi, H. J., Barequet, I. S., Segev, F., Varssano, D., Sartani, G., et al. (2010). Treatment of dry eye syndrome with orally-administered CF101: data from a Phase 2 clinical trial. Ophthalmology 117, 1287–1293. doi: 10.1016/j.ophtha.2009.11.029
Baltos, J. A., Vecchio, E. A., Harris, M. A., Qin, C. X., Ritchie, R. H., Christopoulos, A., et al. (2017). Capadenoson, a clinically trialed partial adenosine A1 receptor agonist, can stimulate adenosine A2B receptor biased agonism. Biochem. Pharmacol. 135, 79–89. doi: 10.1016/j.bcp.2017.03.014
Baraldi, S., Baraldi, P. G., Oliva, P., Toti, K. S., Ciancetta, A., and Jacobson, K. A. (2018). “Chapter 5. A2A adenosine receptor: structures, modeling and medicinal chemistry,” in The Adenosine Receptors, The Receptors, Vol. 34, ed. K. Varani (Berlin: Springer), 91–136.
Bar-Yehuda, S., Stemmer, S. M., Madi, L., Castel, D., Ochaion, A., Cohen, S., et al. (2008). The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-kappaB signal transduction pathways. Int. J. Oncol. 33, 287–295.
Basoglu, O. K., Pelleg, A., Essilfie-Quaye, S., Brindicci, C., Barnes, P. J., and Kharitonov, S. A. (2005). Effects of aerosolized adenosine 5′-triphosphate vs adenosine 5′-monophosphate on dyspnea and airway caliber in healthy nonsmokers and patients with asthma. Chest 128, 1905–1909. doi: 10.1378/chest.128.4.1905
Bigot, A., Stengelin, S., Jähne, G., Herling, A., Müller, G., Hock, F. J., et al. (2004). Aventis Pharma Deutschland. Novel Adenosine Analoguexs and their use as Pharmaceutical Agents. US Patent WO04003002.
Boison, D. (2013). Adenosine kinase: exploitation for therapeutic gain. Pharmacol. Rev. 65, 906–943. doi: 10.1124/pr.112.006361
Borea, P. A., Gessi, S., Merighi, S., and Varani, K. (2016). Adenosine as a multi-signalling guardian angel in human diseases: When, where and how does it exert its protective effects? Trends Pharmacol. Sci. 37, 419–434. doi: 10.1016/j.tips.2016.02.006
Borea, P. A., Gessi, S., Merighi, S., Vincenzi, F., and Varani, K. (2017). Pathological overproduction: the bad side of adenosine. Br. J. Pharmacol. 174, 1945–1960. doi: 10.1111/bph.13763
Borea, P. A., Gessi, S., Merighi, S., Vincenzi, F., and Varani, K. (2018). Pharmacology of adenosine receptors: the state of the art. Physiol. Rev. 98, 1591–1625. doi: 10.1152/physrev.00049.2017
Burnstock, G., and Boeynaems, J. M. (2014). Purinergic signalling and immune cells. Purinergic Signal. 10, 529–564. doi: 10.1007/s11302-014-9427-2
Carlin, J. L., Jain, S., Duroux, R., Suresh, R. R., Xiao, C., Auchampach, J. A., et al. (2018). Activation of adenosine A2A or A2B receptors causes hypothermia in mice. Neuropharmacology 139, 268–278. doi: 10.1016/j.neuropharm.2018.02.035
Carlin, J. L., Jain, S., Gizewski, E., Wan, T. C., Tosh, D. K., Xiao, C., et al. (2017). Hypothermia in mouse is caused by adenosine A1 and A3 receptor agonists and AMP via three distinct mechanisms. Neuropharmacology 114, 101–113. doi: 10.1016/j.neuropharm.2016.11.026
Carlin, J. L., Tosh, D. K., Xiao, C., Piñol, R. A., Chen, Z., Salvemini, D., et al. (2016). Peripheral adenosine A3 receptor activation causes regulated hypothermia in mice that is dependent on central histamine H1 receptors. J. Pharmacol. Exp. Ther. 356, 474–482. doi: 10.1124/jpet.115.229872
Cekic, C., and Linden, J. (2016). Purinergic regulation of the immune system. Nat. Rev. Immunol. 16, 177–192. doi: 10.1038/nri.2016.4
Chen, J. F., Eltzschig, H. K., and Fredholm, B. B. (2013). Adenosine receptors as drug targets - What are the challenges? Nat. Rev. Drug Discov. 12, 265–286. doi: 10.1038/nrd3955
Chen, Y., Corriden, R., Inoue, Y., Yip, L., Hashiguchi, N., Zinkernagel, A., et al. (2006). ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science 314, 1792–1795. doi: 10.1126/science.1132559
Chen, Z., Janes, K., Chen, C., Doyle, T., Tosh, D. K., Jacobson, K. A., et al. (2012). Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 26, 1855–1865. doi: 10.1096/fj.11-201541
Cheng, J. T., Chi, T. C., and Liu, I. M. (2000). Activation of adenosine A1 receptors by drugs to lower plasma glucose in streptozotocin-induced diabetic rats. Auton. Neurosci. 83, 127–133. doi: 10.1016/S0165-1838(00)00106-5
Choi, I.-Y., Lee, J.-C., Ju, C., Hwang, S., Cho, G.-S., Lee, H. W., et al. (2011). A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. Am. J. Pathol. 179, 2042–2052. doi: 10.1016/j.ajpath.2011.07.006
Chrysostomou, C., Morell, V. O., Wearden, P., Sanchez-de-Toledo, J., Jooste, E. H., and Beerman, L. (2013). Dexmedetomidine: therapeutic use for the termination of reentrant supraventricular tachycardia. Congenit. Heart Dis. 8, 48–56. doi: 10.1111/j.1747-0803.2012.00669.x
Cohen, S., Barer, F., Bar-Yehuda, S., IJzerman, A. P., Jacobson, K. A., and Fishman, P. (2014). A3 adenosine receptor allosteric modulator induces an anti-inflammatory effect: In vivo studies and molecular mechanism of action. Mediators Inflamm. 2014:708746. doi: 10.1155/2014/708746
Cohen, S., Barer, F., Itzhak, I., Silverman, M. H., and Fishman, P. (2018). Inhibition of IL-17 and IL-23 in human keratinocytes by the A3 adenosine receptor agonist piclidenoson. J. Immunol. Res. 2018:2310970. doi: 10.1155/2018/2310970
Cohen, S., and Fishman, P. (2019). Targeting the A3 adenosine receptor to treat cytokine release syndrome in cancer immunotherapy. Drug Des. Dev. Ther. 13, 491–497. doi: 10.2147/DDDT.S195294
Cohen, S., Fishman, P., and Tikva, P. (2016). CF602 improves erectile dysfunction in diabetic rats. J. Urol. 195:e1138. doi: 10.1016/j.juro.2016.02.2465
Coppi, E., Cherchi, F., Fusco, I., Failli, P., Vona, A., Dettori, I., et al. (2019). Adenosine A3 receptor activation inhibits pro-nociceptive N-type Ca2+ currents in dorsal root ganglion neurons. Pain doi: 10.1097/j.pain.0000000000001488
Corino, V. D. A., Sandberg, F., Mainardi, L. T., Platonov, P. G., and Sörnmo, L. (2015). Noninvasive characterization of atrioventricular conduction in patients with atrial fibrillation. J. Electrocardiol. 48, 938–942. doi: 10.1016/j.jelectrocard.2015.08.010
Cox, B. F., Clark, K. L., Perrone, M. H., Welzel, G. E., Greenland, B. D., Colussi, D. J., et al. (1997). Cardiovascular and metabolic effects of adenosine A1-receptor agonists in streptozotocin-treated rats. J. Cardiovasc. Pharmacol. 29, 417–426. doi: 10.1097/00005344-199703000-00017
Cronstein, B. N., and Sitkovsky, M. (2017). Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat. Rev. Rheumatol. 13, 41–51. doi: 10.1038/nrrheum.2016.178
Csóka, B., Koscsó, B., Törö, G., Kókai, E., Virág, L., Németh, Z. H., et al. (2014). A2B adenosine receptors prevent insulin resistance by inhibiting adipose tissue inflammation via maintaining alternative macrophage activation. Diabetes 63, 850–866. doi: 10.2337/db13-0573
David, M., Gospodinov, D. K., Gheorghe, N., Mateev, G. S., Rusinova, M. V., Hristakieva, E., et al. (2016). Treatment of plaque-type psoriasis with oral CF101: data from a phase II/III multicenter, randomized, controlled trial. J. Drugs Dermatol. 15, 931–938.
DeNinno, M. P., Masamune, H., Chenard, L. K., DiRico, K. J., Eller, C., Etienne, J. B., et al. (2003). 3′-Aminoadenosine-5′-uronamides: discovery of the first highly selective agonist at the human adenosine A3 receptor. J. Med. Chem. 46, 353–355. doi: 10.1021/jm0255724
Dinh, W., Albrecht-Küpper, B., Gheorghiade, M., Voors, A. A., van der Laan, M., and Sabbah, H. N. (2017). Partial adenosine A1 agonist in heart failure. Handb. Exp. Pharmacol. 243, 177–203. doi: 10.1007/164_2016_83
Draper-Joyce, C. J., Khoshouei, M., Thal, D. M., Liang, Y.-L., Nguyen, A. T. N., Furness, S. G. B., et al. (2018). Structure of the adenosine-bound human adenosine A1 receptor–Gi complex. Nature 558, 559–563. doi: 10.1038/s41586-018-0236-6
Du, L., Gao, Z. G., Paoletta, S., Wan, T. C., Barbour, S., van Veldhoven, J. P., et al. (2018). Species differences and mechanism of action of A3 adenosine receptor allosteric modulators. Purinergic Signal. 14, 59–71. doi: 10.1007/s11302-017-9592-1
Eddy, M. T., Lee, M. Y., Gao, Z. G., White, K. L., Didenko, T., Horst, R., et al. (2018). Allosteric coupling of drug binding and intracellular signaling in the A2A adenosine receptor. Cell 172, 68–80. doi: 10.1016/j.cell.2017.12.004
Eisenach, J. C., Hood, D. D., Curry, R., Sawynok, J., Yaksh, T. L., and Li, X. (2004). Intrathecal but not intravenous opioids release adenosine from the spinal cord. J. Pain 5, 64–68. doi: 10.1016/j.jpain.2003.10.001
Eisenach, J. C., Rauck, R. L., and Curry, R. (2013). Intrathecal, but not intravenous adenosine reduces allodynia in patients with neuropathic pain. Pain 105, 65–70. doi: 10.1016/S0304-3959(03)00158-1
Eisenstein, A., Patterson, S., and Ravid, K. (2015). The many faces of the A2b adenosine receptor in cardiovascular and metabolic diseases. J. Cell. Physiol. 230, 2891–2897. doi: 10.1002/jcp.25043
Eltzschig, H. K. (2009). Adenosine: an old drug newly discovered. Anesthesiology 111, 904–915. doi: 10.1097/ALN.0b013e3181b060f2
Eltzschig, H. K., Ibla, J. C., Furuta, G. T., Leonard, M. O., Jacobson, K. A., Enjyoji, K., et al. (2003). Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in post-hypoxic endothelium: role of ectonucleotidases and adenosine A2B-receptors. J. Exp. Med. 198, 783–796. doi: 10.1084/jem.20030891
Elzein, E., and Zablocki, J. (2008). A1 adenosine receptor agonists and their potential therapeutic applications. Expert Opin. Invest. Drugs 17, 1901–1910. doi: 10.1517/13543780802497284
Faghihi, G., Iraji, F., Rajaee Harandi, M., Nilforoushzadeh, M. A., and Askari, G. (2013). Comparison of the efficacy of topical minoxidil 5% and adenosine 0.75% solutions on male androgenetic alopecia and measuring patient satisfaction rate. Acta Dermatovenerol. Croat. 21, 155–159.
Fang, Z. Z., Tosh, D. K., Tanaka, N., Wang, H., Krausz, K. W., O’Connor, R., et al. (2015). Metabolic mapping of A3 adenosine receptor agonist MRS5980. Biochem. Pharmacol. 97, 215–223. doi: 10.1016/j.bcp.2015.07.007
Fedorova, I. M., Jacobson, M. A., Basile, A., and Jacobson, K. A. (2003). Behavioral characterization of mice lacking the A3 adenosine receptor: sensitivity to hypoxic neurodegeneration. Cell Mol. Neurobiol. 23, 431–447. doi: 10.1023/A:1023601007518
Field, J. J., Majerus, E., Gordeuk, V. R., Gowhari, M., Hoppe, C., Heeney, M. M., et al. (2017). Randomized phase 2 trial of regadenoson for treatment of acute vaso-occlusive crises in sickle cell disease. Blood Adv. 1, 1645–1649. doi: 10.1182/bloodadvances.2017009613
Field, J. J., Nathan, D. G., and Linden, J. (2014). The role of adenosine signaling in sickle cell therapeutics. Hematol. Oncol. Clin. North Am. 28, 287–299. doi: 10.1016/j.hoc.2013.11.003
Fishman, P., Bar-Yehuda, S., Liang, B. T., and Jacobson, K. A. (2012). Pharmacological and therapeutic effects of A3 adenosine receptor (A3AR) agonists. Drug Discov. Today 17, 359–366. doi: 10.1016/j.drudis.2011.10.007
Fishman, P., Salhab, A., Cohen, S., Amer, J., Itzhak, I., Barer, F., et al. (2018). The anti-inflammatory and anto-fibrogenic effects of namodenoson in NAFLD/NASH animal models. Abstract Thu-487. J. Hepatol. 68:S349. doi: 10.1016/S0168-8278(18)30921-8
Flyer, J. N., Zuckerman, W. A., Richmond, M. E., Anderson, B. R., Mendelsberg, T. G., McAllister, J. M., et al. (2017). Prospective study of adenosine on atrioventricular nodal conduction in pediatric and young adult patients after heart transplantation. Circulation 135, 2485–2493. doi: 10.1161/CIRCULATIONAHA.117.028087
Fozard, J. R. (2010). “From hypertension (+) to asthma: interactions with the adenosine A3 receptor from a personal perspective,” in A3 Adenosine Receptors from Cell Biology to Pharmacology and Therapeutics, ed. P. A. Borea (Dordrecht: Springer Science+Business Media B.V), 3–26. doi: 10.1007/978-90-481-3144-0_1
Fredholm, B. B., IJzerman, A. P., Jacobson, K. A., Klotz, K. N., and Linden, J. (2001). International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552.
Galiuto, L., De Caterina, A. R., Porfidia, A., Paraggio, L., Barchetta, S., Locorotondo, G., et al. (2010). Reversible coronary microvascular dysfunction: a common pathogenetic mechanism in apical ballooning or tako-tsubo syndrome. Eur. Heart J. 31, 1319–1327. doi: 10.1093/eurheartj/ehq039
Gallo-Rodriguez, C., Ji, X.-D., Melman, N., Siegman, B. D., Sanders, L. H., Orlina, J., et al. (1994). Structure-activity relationships of N6-benzyladenosine-5(-uronamides as A3-selective adenosine agonists. J. Med. Chem. 37, 636–646. doi: 10.1021/jm00031a014
Gao, Z. G., Balasubramanian, R., Kiselev, E., Wei, Q., and Jacobson, K. A. (2014). Probing biased/partial agonism at the G protein-coupled A2B adenosine receptor. Biochem. Pharmacol. 90, 297–306. doi: 10.1016/j.bcp.2014.05.008
Gao, Z. G., Blaustein, J., Gross, A. S., Melman, N., and Jacobson, K. A. (2003). N6-Substituted adenosine derivatives: selectivity, efficacy, and species differences at A3 adenosine receptors. Biochem. Pharmacol. 65, 1675–1684. doi: 10.1016/S0006-2952(03)00153-9
Gao, Z. G., Inoue, A., and Jacobson, K. A. (2018). On the G protein-coupling selectivity of the native A2B adenosine receptor. Biochem. Pharmacol. 151, 201–213. doi: 10.1016/j.bcp.2017.12.003
Gao, Z. G., and Jacobson, K. A. (2017). Purinergic signaling in mast cell degranulation and asthma. Front. Pharmacol. 8:947. doi: 10.3389/fphar.2017.00947
Gao, Z. G., Verzijl, D., Zweemer, A., Ye, K., Göblyös, A., IJzerman, A. P., et al. (2011). Functionally biased modulation of A3 adenosine receptor agonist efficacy and potency by imidazoquinolinamine allosteric enhancers. Biochem. Pharmacol. 82, 658–668. doi: 10.1016/j.bcp.2011.06.017
García, P. A., Valles, E., Díez, D., and Castro, M. -Á (2018). Marine alkylpurines: a promising group of bioactive marine natural products. Mar. Drugs 16:E6. doi: 10.3390/md16010006
García-Nafría, J., Lee, Y., Bai, X., Carpenter, B., and Tate, C. G. (2018). Cryo-EM structure of the adenosine A2A receptor coupled to an engineered heterotrimeric G protein. Elife 7:e35946. doi: 10.7554/eLife.35946
Giorgi, I., and Nieri, P. (2013). Adenosine A1 modulators: a patent update (2008 to present). Expert Opin. Ther. Pat. 23, 1109–1121. doi: 10.1517/13543776.2013.799142
Glatter, K. A., Cheng, J., and Dorostkar, P. (1999). Electrophysiologic effects of adenosine in patients with supraventricular tachycardia. Circulation 99, 1034–1040. doi: 10.1161/01.CIR.99.8.1034
Golzar, Y., and Doukky, R. (2014). Regadenoson use in patients with chronic obstructive pulmonary disease: the state of current knowledge. Int. J. Chron. Obstruct. Pulmon. Dis. 9, 129–137. doi: 10.2147/COPD.S56879
Greene, S. J., Sabbah, H. N., Butler, J., Voors, A. A., Albrecht-Küpper, B., Düngen, H. D., et al. (2016). Partial adenosine A1 receptor agonism: a potential new therapeutic strategy for heart failure. Heart Fail. Rev. 21, 95–102. doi: 10.1007/s10741-015-9522-7
Grenz, A., Osswald, H., Eckle, T., Yang, D., Zhang, H., Tran, Z. V., et al. (2008). The reno-vascular A2B adenosine receptor protects the kidney from ischemia. PLoS Med. 5:e137. doi: 10.1371/journal.pmed.0050137
Guo, M., Gao, Z.-G., Tyler, R., Stodden, T., Wang, G.-J., Wiers, C., et al. (2018). Preclinical evaluation of the first adenosine A1 receptor partial agonist radioligand for positron emission tomography (PET) imaging. J. Med. Chem. 61, 9966–9975. doi: 10.1021/acs.jmedchem.8b01009
Hart, M. L., Grenz, A., Gorzolla, I. C., Schittenhelm, J., Dalton, J. H., and Eltzschig, H. K. (2011). Hypoxia-inducible factor-1α-dependent protection from intestinal ischemia/reperfusion injury involves ecto-5′-nucleotidase (CD73) and the A2B adenosine receptor. J. Immunol. 186, 4367–4374. doi: 10.4049/jimmunol.0903617
Haskó, G., Antonioli, L., and Cronstein, B. N. (2018). Adenosine metabolism, immunity and joint health. Biochem. Pharmacol. 151, 307–313. doi: 10.1016/j.bcp.2018.02.002
Hothersall, J. D., Guo, D., Sarda, S., Sheppard, R. J., Chen, H., Keur, W., et al. (2017). Structure-activity relationships of the sustained effects of adenosine A2A receptor agonists driven by slow dissociation kinetics. Mol. Pharmacol. 91, 25–38. doi: 10.1124/mol.116.105551
Imlach, W. L., Bhola, R. F., May, L. T., Christopoulos, A., and Christie, M. J. (2015). A positive allosteric modulator of the adenosine A1 receptor selectively inhibits primary afferent synaptic transmission in a neuropathic pain model. Mol. Pharmacol. 88, 460–468. doi: 10.1124/mol.115.099499
Irrera, N., Arcoraci, V., Mannino, F., Vermiglio, G., Pallio, G., Minutoli, L., et al. (2018). Activation of A2A receptor by PDRN reduces neuronal damage and stimulates WNT/β-catenin driven neurogenesis in spinal cord injury. Front. Pharmacol. 9:506. doi: 10.3389/fphar.2018.00506
Ishikawa, J., Mitani, H., Bandoh, T., Kimura, M., Totsuka, T., and Hayashi, S. (1998). Hypoglycemic and hypotensive effects of 6-cyclohexyl-2′-O-methyl-adenosine, an adenosine A1 receptor agonist, in spontaneous hypertensive rat complicated with hyperglycemia. Diabetes Res. Clin. Pract. 39, 3–9. doi: 10.1016/S0168-8227(97)00116-2
Isogai, S., Niwa, Y., Yatsuya, H., Hayashi, M., Yamamoto, N., Okamura, T., et al. (2017). Increased airway hyperresponsiveness to adenosine in patients with aspirin intolerant asthma. Allergol. Int. 66, 360–362. doi: 10.1016/j.alit.2016.10.001
Jackson, S., Weingart, J., Nduom, E. K., Harfi, T. T., George, R. T., McAreavey, D., et al. (2018). The effect of an adenosine A2A agonist on intra-tumoral concentrations of temozolomide in patients with recurrent glioblastoma. Fluids Barriers CNS 15:2. doi: 10.1186/s12987-017-0088-8
Jacobson, K. A., and Civan, M. M. (2016). Ocular purine receptors as drug targets in the eye. J. Ocular Pharmacol. Ther. 32, 534–547. doi: 10.1089/jop.2016.0090
Jacobson, K. A., and Gao, Z. G. (2006). Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 5, 247–264. doi: 10.1038/nrd1983
Jacobson, K. A., and Gao, Z. G. (2017). “Allosteric modulators of adenosine, P2Y and P2X receptors,” in Chapter 11 in Allosterism in Drug Discovery (RSC Drug Discovery Series No. 56), ed. D. Doller (London: Royal Society of Chemistry), 247–270. doi: 10.1039/9781782629276
Jacobson, K. A., Gao, Z. G., Tchilibon, S., Duong, H. T., Joshi, B. V., Sonin, D., et al. (2005). Semirational design of (N)-methanocarba nucleosides as dual acting A1 and A3 adenosine receptor agonists: novel prototypes for cardioprotection. J. Med. Chem. 48, 8103–8107. doi: 10.1021/jm050726b
Jacobson, K. A., and Knutsen, L. J. S. (2001). P1 and P2 purine and pyrimidine receptors. Handb. Exp. Pharmacol. 151, 129–175.
Jacobson, K. A., Merighi, S., Varani, K., Borea, P. A., Baraldi, S., Tabrizi, M. A., et al. (2018). A3 adenosine receptors as modulators of inflammation: from medicinal chemistry to therapy. Med. Res. Rev. 38, 1031–1072. doi: 10.1002/med.21456
Janes, K., Symons-Liguori, A. M., Jacobson, K. A., and Salvemini, D. (2016). Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br. J. Pharmacol. 173, 1253–1267. doi: 10.1111/bph.13446
Jin, X., and Mi, W. (2017). Adenosine for postoperative analgesia: a systematic review and meta-analysis. PLoS One 12:e0173518. doi: 10.1371/journal.pone.0173518
Jinka, T. R., Combs, V. M., and Drew, K. L. (2015). Translating drug-induced hibernation to therapeutic hypothermia. ACS Chem. Neurosci. 6, 899–904. doi: 10.1021/acschemneuro.5b00056
Johnston-Cox, H., Eisenstein, A. S., Koupenova, M., Carroll, S., and Ravid, K. (2014). The macrophage A2B adenosine receptor regulates tissue insulin sensitivity. PLoS One 9:e98775. doi: 10.1371/journal.pone.0098775
Kiesewetter, D. O., Lang, L., Ma, Y., Bhattacharjee, A. K., Gao, Z. G., Joshi, B. V., et al. (2009). Synthesis and characterization of [76Br]-labeled high affinity A3 adenosine receptor ligands for positron emission tomography. Nucl. Med. Biol. 36, 3–10. doi: 10.1016/j.nucmedbio.2008.10.003
Kiesman, W. F., Elzein, E., and Zablocki, J. (2009). A1 Adenosine receptor antagonists, agonists, and allosteric enhancers. Handb. Exp. Pharmacol. 193, 25–58. doi: 10.1007/978-3-540-89615-9_2
Kim, D.-G., and Bynoe, M. S. (2016). A2A adenosine receptor modulates drug efflux transporter P-glycoprotein at the blood-brain barrier. J. Clin. Invest. 126, 1717–1733. doi: 10.1172/JCI76207
Knezevic, N. N., Cicmil, N., Knezevic, I., and Candido, K. D. (2015). Discontinued neuropathic pain therapy between 2009–2015. Exp. Opin. Invest. Drugs 24, 1631–1646. doi: 10.1517/13543784.2015.1099627
Knutsen, L. J. S., Lau, J., Petersen, H., Thomsen, C., Weis, J. U., Shalmi, M., et al. (1999). N-Substituted adenosines as novel neuroprotective A1 agonists with diminished hypotensive effects. J. Med. Chem. 42, 3463–3477. doi: 10.1021/jm960682u
Korinek, W. S., Lechleiter, J. D., and Liston, T. E. (2018). Compounds and Methods for Treating Neurological and Cardiovascular Conditions. Washington, DC: U.S. Patent and Trademark Office.
Koupenova, M., Johnston-Cox, H., Vezeridis, A., Gavras, H., Yang, D., Zannis, V., et al. (2012). A2b adenosine receptor regulates hyperlipidemia and atherosclerosis. Circulation 125, 354–363. doi: 10.1161/CIRCULATIONAHA.111.057596
Koussémou, M., Lorenz, K., and Klotz, K.-N. (2018). The A2B adenosine receptor in MDA-MB-231 breast cancer cells diminishes ERK1/2 phosphorylation by activation of MAPK-phosphatase-1. PLoS One 13:e0202914. doi: 10.1371/journal.pone.0202914
Lahesmaa, M., Oikonen, V., Helin, S., Luoto, P., U Din, M., Pfeifer, A., et al. (2018). Regulation of human brown adipose tissue by adenosine and A2A receptors – studies with [15O]H2O and [11C]TMSX PET/CT. Eur. J. Nucl. Med. Mol. Imaging 46, 743–750. doi: 10.1007/s00259-018-4120-2
Lam, C. S. P., Voors, A. A., de Boer, R. A., Solomon, S. D., and van Veldhuisen, D. J. (2018). Heart failure with preserved ejection fraction: from mechanisms to therapies. Eur. Heart J. 39, 2780–2792. doi: 10.1093/eurheartj/ehy301
Lasley, R. D. (2018). Adenosine receptor-mediated cardioprotection - current limitations and future directions. Front. Pharmacol. 9:310. doi: 10.3389/fphar.2018.00310
Lebon, G., Warne, T., Edwards, P. C., Bennett, K., Langmead, C. J., Leslie, A. G. W., et al. (2011). Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature 474, 521–525. doi: 10.1038/nature10136
Letsas, K. P., Georgopoulos, S., Efremidis, M., Liu, T., Bazoukis, G., Vlachos, K., et al. (2017). Adenosine-guided radiofrequency catheter ablation of atrial fibrillation: a meta-analysis of randomized control trials. J. Arrhythm. 33, 247–255. doi: 10.1016/j.joa.2017.02.002
Leung, C. T., Li, A., Banerjee, J., Gao, Z. G., Kambayashi, T., Jacobson, K. A., et al. (2014). The role of activated adenosine receptors in degranulation of human LAD2 mast cells. Purinergic Signal. 10, 465–475. doi: 10.1007/s11302-014-9409-4
Little, J. W., Ford, A., Symons-Liguori, A. M., Chen, Z., Janes, K., Doyle, T., et al. (2015). Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain 138, 28–35. doi: 10.1093/brain/awu330
Luongo, L., Petrelli, R., Gatta, L., Giordano, C., Guida, F., Vita, P., et al. (2012). 5′-Chloro-5′-deoxy-( ± )-ENBA, a potent and selective adenosine A1 receptor agonist, alleviates neuropathic pain in mice through functional glial and microglial changes without affecting motor or cardiovascular functions. Molecules 17, 13712–13726. doi: 10.3390/molecules171213712
Mantell, S., Jones, R., and Trevethick, M. (2010). Design and application of locally delivered agonists of the adenosine A2A receptor. Expert Rev. Clin. Pharmacol. 3, 55–72. doi: 10.1586/ecp.09.57
Mason, P. K., and DiMarco, J. P. (2009). New pharmacological agents for arrhythmias. Circ. Arrhythm. Electrophysiol. 2, 588–597. doi: 10.1161/CIRCEP.109.884429
Meibom, D., Albrecht-Küpper, B., Diedrichs, N., Hübsch, W., Kast, R., Krämer, T., et al. (2017). Neladenoson Bialanate hydrochloride: a prodrug of a partial adenosine A1 receptor agonist for the chronic treatment of heart diseases. Chem. Med. Chem. 12, 728–737. doi: 10.1002/cmdc.201700151
Miao, Y., Bhattarai, A., Nguyen, A. T. N., Christopoulos, A., and May, L. T. (2018). Structural basis for binding of allosteric drug leads in the adenosine A1 receptor. Sci. Rep. 8:16836. doi: 10.1038/s41598-018-35266-x
Montesinos, M. C., Desai-Merchant, A., and Cronstein, B. N. (2015). Promotion of wound healing by an agonist of adenosine A2A Receptor is dependent on tissue plasminogen activator. Inflammation 38, 2036–2041. doi: 10.1007/s10753-015-0184-3
Mundell, S., and Kelly, E. (2011). Adenosine receptor desensitization and trafficking. Biochim. Biophys. Acta 1808, 1319–1328. doi: 10.1016/j.bbamem.2010.06.007
Murray, J. J., Weiler, J. M., Schwartz, L. B., Busse, W. W., Katial, R. K., Lockey, R. F., et al. (2009). Safety of binodenoson, a selective adenosine A2A receptor agonist vasodilator pharmacological stress agent, in healthy subjects with mild intermittent asthma. Circ. Cardiovasc. Imaging 2, 492–498. doi: 10.1161/CIRCIMAGING.108.817932
Myers, J. S., Sall, K. N., DuBiner, H., Slomowitz, N., McVicar, W., Rich, C. C., et al. (2016). A dose-escalation study to evaluate the safety, tolerability, pharmacokinetics, and efficacy of 2 and 4 weeks of twice-daily ocular trabodenoson in adults with ocular hypertension or primary open-angle glaucoma. J. Ocul. Pharmacol. Ther. 32, 555–562. doi: 10.1089/jop.2015.0148
Nascimento, F. P., Macedo, S.T., and Santos, A. R. (2012). “The involvement of purinergic system in pain: adenosine receptors and inosine as pharmacological tools in future treatments,” in Chap. 28 in Pharmacology, eds S. J. Macedo Jr. and L. Gallelli (London: InTech).
Ni, Y., Liang, D., Tian, Y., Kron, I. L., French, B. A., and Yang, Z. (2018). Infarct-sparing effect of adenosine A2B receptor agonist is primarily due to its action on splenic leukocytes via a PI3K/Akt/IL-10 pathway. J. Surg. Res. 232, 442–449. doi: 10.1016/j.jss.2018.06.042
Ning, Y., Jiang, J., Belardinelli, L., and Dhalla, A. K. (2011). Short-Term Treatment of GS-9667 in Combination with Sitagliptin Improves Glucose and Lipid Homeostasis in ZDF rats. Arlington, VI: American Diabetes Association.
Olsson, R. A. (2003). Robert Berne: his place in the history of purine research. Drug Dev. Res. 58, 296–301. doi: 10.1002/ddr.10197
Palani, G., and Ananthasubramaniam, K. (2013). Regadenoson: review of its established role in myocardial perfusion imaging and emerging applications. Cardiol. Rev. 21, 42–48. doi: 10.1097/CRD.0b013e3182613db6
Peleli, M., and Carlstrom, M. (2017). Adenosine signaling in diabetes mellitus and associated cardiovascular and renal complications. Mol. Aspects Med. 55, 62–74. doi: 10.1016/j.mam.2016.12.001
Pelleg, A., Kutalek, S. P., Flammang, D., and Benditt, D. (2012). ATPaceTM: injectable adenosine 5′-triphosphate: diagnostic and therapeutic indications. Purinergic Signal. 8(Suppl. 1), 57–60. doi: 10.1007/s11302-011-9268-1
Pelleg, A., Schulman, E. S., and Barnes, P. J. (2016). Extracellular adenosine 5′-triphosphate in obstructive airway diseases. Chest 150, 908–915. doi: 10.1016/j.chest.2016.06.045
Rapaport, E., Salikhova, A., and Abraham, E. H. (2015). Continuous intravenous infusion of ATP in humans yields large expansions of erythrocyte ATP pools but extracellular ATP pools are elevated only at the start followed by rapid declines. Purinergic Signal. 11, 251–262. doi: 10.1007/s11302-015-9450-y
Rickles, R. J., Padval, M., Giordano, T., Rieger, J. M., and Lee, M. S. (2010). ATL313, a potent, and selective A2A agonist as a novel drug candidate for the treatment of multiple myeloma. Blood 116:2990.
Rieger, J. M., Brown, M. L., Sullivan, G. W., Linden, J., and Macdonald, T. L. (2001). Design, synthesis, and evaluation of novel A2A adenosine receptor agonists. J. Med. Chem. 44, 531–539. doi: 10.1021/jm0003642
Sawynok, J. (1998). Adenosine receptor activation and nociception. Eur. J. Pharmacol. 347, 1–11. doi: 10.1016/S0014-2999(97)01605-1
Schaddelee, M. P., Read, K. D., Cleypool, C. G., IJzerman, A. P., Danhof, M., and de Boer, A. G. (2005). Brain penetration of synthetic adenosine A1 receptor agonists in situ: role of the rENT1 nucleoside transporter and binding to blood constituents. Eur. J. Pharm. Sci. 24, 59–66. doi: 10.1016/j.ejps.2004.09.010
Schaumann, E., and Kutscha, W. (1972). Clinical-pharmacological studies with a new orally active adenosine derivative. Drug Res. 22, 783–790.
Schaumann, E., Schlierf, G., Ptleiderer, T., and Weber, E. (1972). Effect of repeated doses of phenylisopropyladenosine on lipid and carbohydrate metabolism in healthy fasting subjects. Arzneim. Forsch. 22, 593–596.
Serchov, T., Clement, H.-W., Schwarz, M. K., Iasevoli, F., Tosh, D. K., Idzko, M., et al. (2015). Increased signaling via adenosine A1 receptors, sleep deprivation, imipramine, and ketamine inhibit depressive-like behavior via induction of homer1a. Neuron 87, 549–562. doi: 10.1016/j.neuron.2015.07.010
Shah, B., Rohatagi, S., Natarajan, C., Kirkesseli, S., Baybutt, R., and Jensen, B. K. (2004). Pharmacokinetics, pharmacodynamics, and safety of a lipid-lowering adenosine A1 agonist, RPR749, in healthy subjects. Am. J. Ther. 11, 175–189. doi: 10.1097/00045391-200405000-00005
Sharma, A. K., LaPar, D. J., Stone, M. L., Zhao, Y., Mehta, C. K., Kron, I. L., et al. (2016). NOX2 activation of natural killer T cells is blocked by the adenosine A2A receptor to inhibit lung ischemia-reperfusion injury. Am. J. Respir. Crit. Care Med. 193, 988–999. doi: 10.1164/rccm.201506-1253OC
Squadrito, F., Bitto, A., Irrera, N., Pizzino, G., Pallio, G., Minutoli, L., et al. (2017). Pharmacological activity and clinical use of PDRN. Front. Pharmacol. 8:224. doi: 10.3389/fphar.2017.00224
Staehr, P. M., Dhalla, A. K., Zack, J., Wang, X., Ho, Y. L., Bingham, J., et al. (2013). Reduction of free fatty acids, safety, and pharmacokinetics of oral GS-9667, an A1 adenosine receptor partial agonist. J. Clin. Pharmacol. 53, 385–392. doi: 10.1002/jcph.9
Stemmer, S. M., Benjaminov, O., Medalia, G., Ciuraru, N. B., Silverman, M. H., Bar-Yehuda, S., et al. (2013). CF102 for the treatment of hepatocellular carcinoma: a phaseI/II, open- label, dose-escalation study. Oncologist 18, 25–26. doi: 10.1634/theoncologist.2012-0211
Sun, Y., and Huang, P. (2016). Adenosine A2B receptor: from cell biology to human diseases. Front. Chem. 4:37. doi: 10.3389/fchem.2016.00037
Szentmiklosi, A. J., Galajda, Z., Cseppento, A., Gesztelyi, R., Susan, Z., Hegyi, B., et al. (2015). The Janus face of adenosine: antiarrhythmic and proarrhythmic actions. Curr. Pharm. Des. 21, 965–976. doi: 10.2174/1381612820666141029100346
Szybala, C., Pritchard, E. M., Lusardi, T. A., Li, T., Wilz, A., Kaplan, D. L., et al. (2009). Antiepileptic effects of silk-polymer based adenosine release in kindled rats. Exp. Neurol. 219, 126–135. doi: 10.1016/j.expneurol.2009.05.018
Tendera, M., Gaszewska-Żurek, E., Parma, Z., Ponikowski, P., Jankowska, E., Kawecka-Jaszcz, K., et al. (2012). The new oral adenosine A1 receptor agonist capadenoson in male patients with stable angina. Clin. Res. Cardiol. 101, 585–591. doi: 10.1007/s00392-012-0430-8
Tian, Y., Piras, B. A., Kron, I. L., French, B. A., and Yang, Z. (2015). Adenosine 2B receptor activation reduces myocardial reperfusion injury by promoting anti-inflammatory macrophages differentiation via PI3K/Akt pathway. Oxid. Med. Cell. Longev. 2015:585297. doi: 10.1155/2015/585297
Tosh, D. K., Finley, A., Paoletta, S., Moss, S. M., Gao, Z. G., Gizewski, E., et al. (2014). In vivo phenotypic screening for treating chronic neuropathic pain: modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J. Med. Chem. 57, 9901–9914. doi: 10.1021/jm501021n
Tosh, D. K., Padia, J., Salvemini, D., and Jacobson, K. A. (2015). Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signal. 11, 371–387. doi: 10.1007/s11302-015-9459-2
Tosh, D. K., Paoletta, S., Deflorian, F., Phan, K., Moss, S. M., Gao, Z. G., et al. (2012). Structural sweet spot for A1 adenosine receptor activation by truncated (N)-methanocarba nucleosides: receptor docking and potent anticonvulsant activity. J. Med. Chem. 55, 8075–8090. doi: 10.1021/jm300965a
Tosh, D. K., Rao, H., Bitant, A., Salmaso, V., Mannes, P., Lieberman, D. I., et al. (2019). Design and in vivo characterization of A1 adenosine receptor agonists in the native ribose and conformationally-constrained (N)-methanocarba series. J. Med. Chem. 62, 1502–1522. doi: 10.1021/acs.jmedchem.8b01662
Tozzi, M., and Novak, I. (2017). Purinergic receptors in adipose tissue as potential targets in metabolic disorders. Front. Pharmacol. 8:878. doi: 10.3389/fphar.2017.00878
Trevethick, M. A., Mantell, S. J., Stuart, E. F., Barnard, A., Wright, K. N., and Yeadon, M. (2008). Treating lung inflammation with agonists of the adenosine A2A receptor: promises, problems and potential solutions. Br. J. Pharmacol. 155, 463–474. doi: 10.1038/bjp.2008.329
Van der Graaf, P. H., Van Schaick, E. A., Visser, S. A. G., De Greef, H. J. M. M., IJzerman, A. P., and Danhof, M. (1999). Mechanism-based pharmacokinetic-pharmacodynamic modeling of antilipolytic effects of adenosine A1 receptor agonists in rats: prediction of tissue-dependent efficacy in vivo. J. Pharmacol. Exp. Ther. 290, 702–709.
van Waarde, A., Dierckx, R. A. J. O., Zhou, X., Khanapur, S., Tsukada, H., Ishiwata, K., et al. (2018). Potential therapeutic applications of adenosine A2A receptor ligands and opportunities for A2A receptor imaging. Med. Res. Rev. 38, 5–56. doi: 10.1002/med.21432
Vecchio, E. A., Baltos, J. A., Nguyen, A. T. N., Christopoulos, A., White, P. J., and May, L. T. (2018). New paradigms in adenosine receptor pharmacology: allostery, oligomerization and biased agonism. Br. J. Pharmacol. 175,4036–4046. doi: 10.1111/bph.14337
Victor-Vega, C., Victor-Vega, C., Desai, A., Montesinos, M. C., and Cronstein, B. N. (2002). Adenosine A2A receptor agonists pro- mote more rapid wound healing than recombinant human platelet- derived growth factor (Becaplermin gel). Inflammation 26, 19–24. doi: 10.1023/A:1014417728325
Vincenzi, F., Ravani, A., Pasquini, S., Merighi, S., Gessi, S., Romagnoli, R., et al. (2016). Positive allosteric modulation of A1 adenosine receptors as a novel and promising therapeutic strategy for anxiety. Neuropharmacology 111, 283–292. doi: 10.1016/j.neuropharm.2016.09.015
Vincenzi, F., Targa, M., Romagnoli, R., Merighi, S., Gessi, S., Baraldi, P. G., et al. (2014). TRR469, a potent A1 adenosine receptor allosteric modulator, exhibits antinociceptive properties in acute and neuropathic pain models in mice. Neuropharmacology 82, 6–14. doi: 10.1016/j.neuropharm.2014.01.028
von Lubitz, D. K. J. E., Lin, R.-C., Boyd, M., Bischofberger, N., and Jacobson, K. A. (1999). Chronic administration of adenosine A3 receptor agonist and cerebral ischemia: neuronal and glial effects. Eur. J. Pharmacol. 367, 157–163. doi: 10.1016/S0014-2999(98)00977-7
Voors, A. A., Düngen, H. D., Senni, M., Nodari, S., Agostoni, P., Ponikowski, P., et al. (2017). Safety and tolerability of Neladenoson Bialanate, a novel oral partial adenosine A1 receptor agonist, in patients with chronic heart failure. J. Clin. Pharmacol. 57, 440–451. doi: 10.1002/jcph.828
Vuerich, M., Harshe, R. P., Robson, S. C., and Longhi, M. S. (2019). Dysregulation of adenosinergic signaling in systemic and organ-specific autoimmunity. Int. J. Mol. Sci. 20:528. doi: 10.3390/ijms20030528
Wahlman, C., Doyle, T., Little, J. W., Luongo, L., Janes, K., Chen, Z., et al. (2018). Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels via astrocyte-dependent mechanisms. Pain 159, 1025–1034. doi: 10.1097/j.pain.0000000000001177
Wan, T. C., Tampob, A., Kwokb, W. M., and Auchampach, J. A. (2019). Ability of CP-532,903 to protect mouse hearts from ischemia/reperfusion injury is dependent on expression of A3 adenosine receptors in cardiomyoyctes. Biochem. Pharmacol. 163, 21–31. doi: 10.1016/j.bcp.2019.01.022
Wilbrandt, R., Frotscher, U., Freyland, M., Messerschmidt, W., Richter, R., Schulte-Lippern, M., et al. (1972). Zur Behandlung der Glomerulonephritis mit Metrifudil. Medizinische Klinik 67, 1138–1140.
Wojcik, M., Zieleniak, A., Mac-Marcjanek, K., Wozniak, L. A., and Cypryk, K. (2014). The elevated gene expression level of the A2B adenosine receptor is associated with hyperglycemia in women with gestational diabetes mellitus. Diabetes Metab. Res. Rev. 30, 42–53. doi: 10.1002/dmrr.2446
Xiao, C., Liu, N., Jacobson, K. A., Gavrilova, O., and Reitman, M. L. (2019). Physiology and effects of nucleosides in mice lacking all four adenosine receptors. PLoS Biol. 17:e3000161. doi: 10.1371/journal.pbio.3000161
Xu, F., Wu, H., Katritch, V., Han, G. W., Jacobson, K. A., Gao, Z. G., et al. (2011). Structure of an agonist-bound human A2A adenosine receptor. Science 332, 322–327. doi: 10.1126/science.1202793
Zannikos, P. N., Rohatagi, S., and Jensen, B. K. (2001). Pharmacokinetic-pharmacodynamic modeling of the antilipolytic effects of an adenosine receptor agonist in healthy volunteers. J. Clin. Pharmacol. 41, 61–69. doi: 10.1177/00912700122009845
Zheng, J., Wang, R., Zambraski, E., Wu, D., Jacobson, K. A., and Liang, B. T. (2007). Protective roles of adenosine A1, A2A, and A3 receptors in skeletal muscle ischemia and reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 293, 3685–3691. doi: 10.1152/ajpheart.00819.2007
Zoghbi, G. J., and Iskandrian, A. E. (2012). Selective adenosine agonists and myocardial perfusion imaging. J. Nucl. Cardiol. 19, 126–141. doi: 10.1007/s12350-011-9474-9
Keywords: purinergic signaling, adenosine receptors, inflammation, pain, CNS
Citation: Jacobson KA, Tosh DK, Jain S and Gao Z-G (2019) Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development. Front. Cell. Neurosci. 13:124. doi: 10.3389/fncel.2019.00124
Received: 08 January 2019; Accepted: 13 March 2019;
Published: 28 March 2019.
Edited by:
David Blum, INSERM U1172 Centre de Recherche Jean-Pierre Aubert, FranceReviewed by:
Sergi Ferre, Intramural Research Program (NIDA), United StatesFrancisco Ciruela, University of Barcelona, Spain
Patrizia Popoli, Istituto Superiore di Sanità (ISS), Italy
Copyright © 2019 Jacobson, Tosh, Jain and Gao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kenneth A. Jacobson, a2VubmV0aEpAbmlkZGsubmloLmdvdg==; a2FqYWNvYnNAaGVsaXgubmloLmdvdg==