 Rodrigo O. Kuljiš1,2,3*
Rodrigo O. Kuljiš1,2,3*
- 1 The Brain-Mind Project, Inc, Encephalogistics, Inc, University of Miami Miami, FL, USA
- 2 Departments of Neurology, Neuroscience and Cell Biology, and Pathology, The University of Texas Medical Branch Galveston, TX, USA
- 3 Mitchell Center for Neurodegenerative Disease, The University of Texas Medical Branch Galveston, TX, USA
The biological substrate for cognition remains a challenge as much as defining this function of living beings. Here, we examine some of the difficulties to understand normal and disordered cognition in humans. We use aspects of Alzheimer’s disease and related disorders to illustrate how the wealth of information at many conceptually separate, even intellectually decoupled, physical scales – in particular at the Molecular Neuroscience versus Systems Neuroscience/Neuropsychology levels – presents a challenge in terms of true interdisciplinary integration towards a coherent understanding. These unresolved dilemmas include critically the as yet untested quantum brain hypothesis, and the embryonic attempts to develop and define the so-called connectome in humans and in non-human models of disease. To mitigate these challenges, we propose a scheme incorporating the vast array of scales of the space and time (space–time) manifold from at least the subatomic through cognitive-behavioral dimensions of inquiry, to achieve a new understanding of both normal and disordered cognition, that is essential for a new era of progress in the Generative Sciences and its application to translational efforts for disease prevention and treatment.
Introduction
The term “cognition” is notorious among many in the Generative Sciences for the elusive and unresolved definitional difficulties. Cognition is used in the scientific context to address “the process of thought,” or “the mental processes involved in gaining knowledge and comprehension” (Anonymous, 2009). It can also refer to different aspects of such processes among different fields of study. For example, in some areas of Psychology and the Cognitive Sciences it most commonly refers to an information-processing view of psychological functions – without necessarily paying attention to the neural mechanisms involved. In Clinical Neurology this term often denotes a patient’s ability to carry out relatively short-term and somewhat basic tasks, such as memorizing simple data sets, and carrying out relatively simple visuospatial tasks and calculations. In this latter context, one of the critical aims is to identify as precisely as possible the regions of the brain that mediate the task in question. The main purpose being to ascertain and treat a variety of causes that can interfere with the proper functioning of those regions, both in isolation as well as together with other regions affected by disease. Other relevant formulations, such as the “Society of Mind,” postulate that non-intelligent or “mindless” agents interact to achieve intelligent (cognitive) outcomes such as forming concepts, which could hypothetically be implemented (artificially) and occurs also without the indispensable intervention of intention-endowed external agents, both in biological and (theoretically) in inert systems (Minsky, 1985). Other formulations much less grounded in “classical” physics or materialistic philosophy allege that components of such systems are “proto conscious” and that somehow their aggregate functioning can lead to more elaborate and complex states of consciousness (Vimal, 2008).
One of the central problems to a true understanding of cognition is: precisely how do assemblies of components such as electronic elements, molecules (e.g., neurotransmitters and neuromodulators), parts of the signaling machinery of neurons (e.g., synapses) and possibly also non-neuronal components of the neural tissue, neuronal assemblies – and even selected parts of the brain – mediate such a complex and mysterious product as cognition? The now “traditional” – and as yet unsurpassed – answer is to adduce the principle of Emergence, which in Philosophy and Systems Theory is the tenet that a multiplicity of relatively simple interactions among components in Complex Systems results in “emergent properties.” This has been suggested to be the case in Nervous Systems, and, hypothetically also, for future artificial information-processing systems (e.g., Artificial Intelligence), in which the interactions of their parts somehow (e.g., “emergence”) results in a variety of products such as cognition. The general idea is not new, since it appears in the works of Aristotle (“…the whole is greater than the sum of the parts…”), Mill (1843) (“…The chemical combination of two substances produces, as is well known, a third substance with properties different from those of either of the two substances separately, or of both of them taken together…” 1843) and Julian Huxley (“…now and again there is a sudden rapid passage to a totally new and more comprehensive type of order or organization, with quite new emergent properties, and involving quite new methods for further evolution…” Huxley and Huxley, 1947). Lewes (1875) is most often credited with the first use of the term “emergent” in the sense most relevant to our discussion:
“…Every resultant is either a sum or a difference of the co-operant forces: their sum, when their directions are the same – their difference, when their directions are contrary. Further, every resultant is clearly traceable it its components, because these are homogeneous and commensurable. It is otherwise with emergents, when, instead of adding measurable motion to measurable motion, or things of one kind to other individuals of their kind, there is a co-operation of things of unlike kinds. The emergent is unlike its components insofar as these are incommensurable, and it cannot be reduced to their sum or their difference.”
The aim of this essay is not to eliminate the challenges of defining the term “cognition” more precisely, but to explore the obstacles to integrate the rapidly increasing amounts of experimental data on the presumed “machinery” for cognition in the normal and diseased human brain, to eventually achieve Purpura’s goal:
“What we require now are approaches that can unite basic neurobiology and behavioral sciences into a single operational network” (Purpura, 1975).
To do so, we use some of the information obtained in the process of attempting to understand Alzheimer’s disease and related neurodegenerative disorders, easily the most common “cause” of disordered cognition world-wide (Kuljiš, 2010) and thus potentially a valuable tool to understand the mechanisms that mediate it (Kuljiš, 2009a,b; Anderson and Kuljiš, 2008). This approach may serve as a model to better understand the challenges to integrate information obtained by practitioners of widely disparate disciplines, that have attempted to integrate the data for several decades with limited success. Important challenges include conceptual limitations that preclude the integration among practitioners of disciplines that focus on different components of the biological (and not merely neural) “machinery” that presumably excels at generating emergent properties, including cognition (Kaneko, 1998). We focus especially on the challenge of developing a harmonious, consistent, and truly useful understanding of the processes that mediate cognition while spanning the continuum of the subatomic (quantum; e.g., quantum brain hypotheses), molecular, cellular, cellular assemblies (e.g., the connectome), cognitive/behavioral and evolutionary levels. This perceived – but as yet unproven – continuum is arguably best highlighted by disorders that result in a disrupted “neural machinery,” but that cannot be explained adequately at present by integrating even large amounts of data from two, and even less so, more than two of these levels. This conundrum clearly points to the necessity for both new tools and, even more importantly, for a new conceptual framework in order to facilitate true advances in the understanding of cognition.
In the present formulation, for simplicity on a topic that is notably bereft of such property, we address mainly cognitive processes that are conceivably mediated by interactions among molecules in the Nervous System and among neural cells (both neuronal and non-neuronal). In that sense, we restrict the potentially considerably broad meaning of the term “cognitive”, with full awareness that this may be an oversimplification when considering Maturana and Varela’s Santiago Theory of Cognition, which holds that “Living systems are cognitive systems, and living as a process is a process of cognition. This statement is valid for all organisms, with or without a nervous system” (Capra, 1997).
A Broad Array of Time Scales Relevant to the Assembly of the Biological “Machinery” for Cognition and Its Disassembly in Disease
Figure 1 depicts only a part of the spatial and temporal scales relevant to the understanding of cognitive phenomena, as well as the approximate portions of those scales that may be approachable by existing empirical methods. Most students of neurodegenerative conditions are familiar with the phenomena of action potential transmission (millisecond scale), embryonic cerebral development (weeks-to-months scale), and clinically evident cerebrocortical degeneration (months-to-years scale, perhaps even decades), but few seem to have an appreciation of the far longer time frame that appears to be necessary for the entire process leading to most neuron-targeting degenerative disorders of the brain (decades to well beyond the life span in the feral condition of primates). To these temporal levels of resolution one would have to add the Biological Evolution time scale (millions of years), which is necessary to account completely for the processes that led to the existence of specific organisms and their specializations for cognition. This is in contrast with an entirely different category of acutely developing conditions that are not neuron-selective, and result in the abrupt cessation of neuronal function (e.g., head injury and cerebrovascular insults). Even conditions that appear to cause dementia invariably if the individual survives long enough (e.g., familial Alzheimer’s disease (AD) and severe cases of Down’s syndrome), the clinically evident manifestations usually appear only after several decades of life, although the “cause” (i.e., DNA mutations) exists since conception, and certainly during the histogenesis of the cerebral cortex. What are the factors responsible for the delay in clinical manifestations although the “cause” of the cognitive disorder is present since conception? Although this delay in the appearance of genetically caused dementia is often attributed to “aging,” this remains profoundly unsatisfactory because of the lack of an understanding of precisely what aging is, and exactly how does this poorly conceptualized phenomenon apply to the brain and to each of its myriad components that appear to be affected differently by the ill defined process of “aging.” Furthermore, the parameters responsible for the delay between the onset of the causative factor(s) and the clinical manifestations are even more elusive in the case of sporadic AD – since the cause(s) remain (or remains) unknown – but many postulate that the scale is in the realm of decades which is in contrast with the time span of the clinical disorder that usually runs its course in a few years or less than two decades (Anderson and Kuljiš, 2008). Thus, although most are accustomed to accept that there is a considerable delay between the onset of the cause’s action and its ultimate clinical expression, few seem to consider precisely what may be the reasons for such a delay in the context of the millisecond-to-minutes scale of energy supply and biochemical/molecular renewal and function essential for the maintenance of the integrity and function of the cerebral cortex – and other considerably shorter-scale phenomena – than the far longer time course of neurodegenerative disorders (Kuljiš, 2009a). The mere mention of the fact that complete anoxia results in irreversible cessation of cortical function in a few minutes (Dugan and Kim-Han, 2006) seems to be in stark contrast with the much longer time scale for the “conventional” (and obviously incomplete) conceptualization for the pathogenesis of neuron-targeting neurodegenerative disorders (at least several decades) in the spectrum of AD-like conditions (Anderson and Kuljiš, 2008).
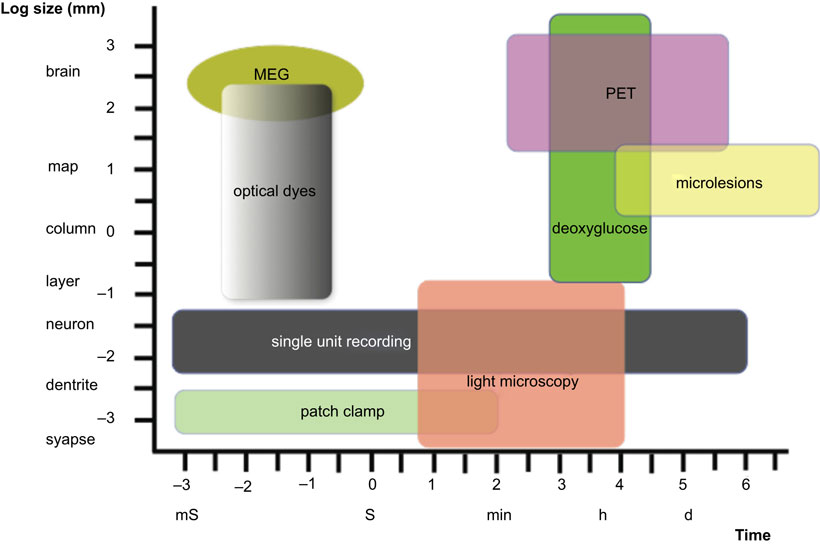
Figure 1. Sketch of some of the spatial (ordinate) and temporal (abscissa) scales demonstrated so far to be relevant to normal and altered cognition, and the role of selected experimental methods to address inevitably limited aspects of these levels of organization and function. Note the considerable extent of the gaps in the manifold that are not covered by the technology represented, which are considerable larger if we take into account the subatomic (no less than 10−10 m and 10−44 s) and the evolutionary (astronomical distances and millions of years) scales. MEG, magnetoencephalography; PET, positron emission tomography. Modified from Churchland and Sejnowski (1988).
This has recently been made even more complicated by the recognition that the cerebral manifestations of sporadic AD – both macroscopic and microscopic, and perhaps also biochemical – are affected to a considerable extent by the age of the individuals manifesting the disorder (Savva et al., 2009). This does support our view that both the “normal” changes in the continuum of cognition both in the course of the normal life span, and, as a result of disease occurring at different stages in life, are best understood with a conceptual framework that recognizes that “causes” may act differently at different stages of the life of the individual. We argue below that a wide array of physical (i.e., time–space) scales must be considered concurrently to understand this type of phenomena.
A Broad Array of Spatial Scales Relevant to the Assembly of the Biological Substrate for Normal and Disordered Cognition
Another rarely recognized conundrum is that, apart from the vast time scale required for the full course of neuron-targeting neurodegenerative disorders, these generally affect neurons in a remarkably selective fashion. This is not unique to AD, since, for example, in amyotrophic lateral sclerosis (which can be associated with dementia), motor neurons are predominantly targeted although very specific local circuit cerebrocortical neurons seem to be affected as well (Kuljiš and Schelper, 1996). Likewise, in the heredodegenerative ataxias, selected cerebellar neurons are the main target (Butterworth, 1992; Schols et al., 2004) and in Parkinson’s disease the dopaminergic nigrostriatal projection neurons are targeted early in the disease process (Riederer and Wuketich, 2005) that very often leads to dementia as well. Perhaps the best example of this remarkable selectivity is AD, in which – despite the seemingly widespread distribution of the lesions – even in the late stages of the disease specific neurons and neuronal assemblies appear to be selectively (and stereotypically) targeted whereas immediately adjacent ones are spared (Kuljiš, 1997, 2009a,b; Kuljiš and Tikoo, 1997).
In fact, in sporadic AD, quick and merely cursory inspection of most regions of the cerebral cortex appears to indicate a seemingly extensive, random involvement. However, a slightly closer examination armed both with a familiarity with the microscopic anatomy of this region, and methodology to reveal aspects of its regional, areal, laminar, cellular, and supra-cellular assembly (a.k.a. modular) organization reveals that the involvement – although spread among many functionally different regions – is remarkably selective both in space and in time throughout the cerebral cortex (Kuljiš, 1994, 1997; Kuljiš and Tikoo, 1997). This region-, area-, layer- cell-, and cortical module component-selective targeting (Figures 2 and 3) defies to this day even the most highly imaginative molecular hypotheses of the etiology and pathogenesis of the condition, since virtually all such approaches are forced to treat the brain as a “black box” (e.g., Figure 4) essentially ignoring even basic elements of its anatomical and functional organization that have been known in some instances since the 19th Century (Kuljiš, 1997, 2009b). Thus, since they ignore key aspects of brain organization and function, strictly molecular explanations of neurodegenerative disorders may be condemned to remain incomplete (e.g., Fernández et al., 2008; Figure 4), but may succeed in a small number of cases – due to fortuitously useful oversimplification – in being applied to effective treatment and prevention (Kuljiš, 2009a, 2010).
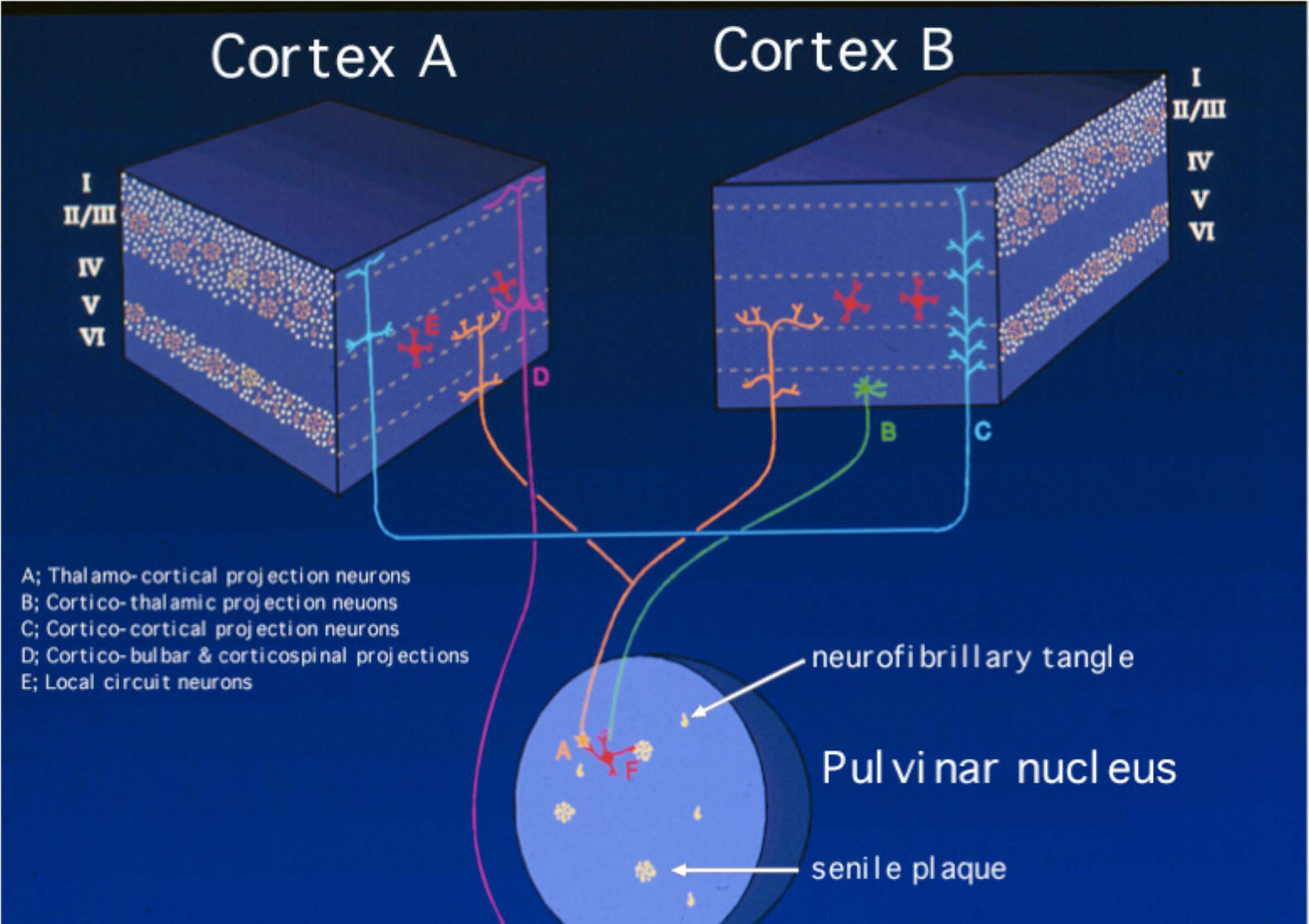
Figure 2. Schematic diagram of the selective laminar distribution of senile plaques and neurofibrillary tangles in various regions of the cortex and the pulvinar nucleus of the thalamus, a structure highly interconnected with a vast array of so-called association cortices targeted heavily and early by sporadic Alzheimer’s disease (sAD). This view of the pathobiology of AD focuses on the circuitry affected and is limited to the resolution of the light microscopy. Thus, while it helps to understand the connections that are affected and to establish their correlation with the clinical manifestations of the disorder, it does not convey information about the ultrastructural, electrophysiological, functional imaging, and molecular domains, among others.
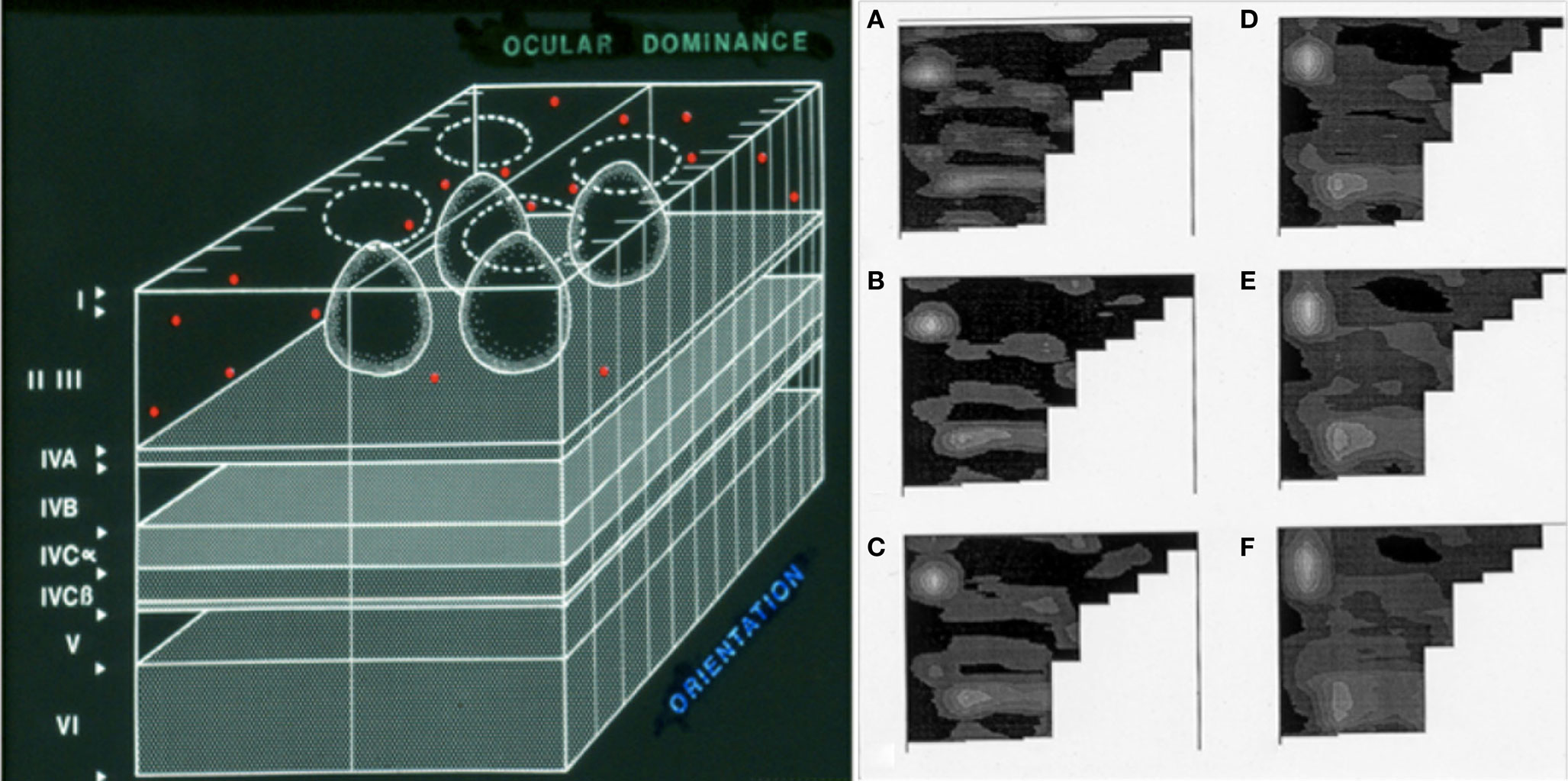
Figure 3. A light microscopy-level representation of selective circuit targeting in sporadic Alzheimer’s disease. Our work in the primary visual cortex is based on the understanding of its modular organization in primates (summarized in the drawing on the left, from Kuljiš and Rakic, 1990). The hypercolumns that make this region contain iterated circuits in layers II/III that are disposed in “rows” along the tangential plane (i.e., parallel to the pial surface, shown in the left panel). Periodograms (right panel, labeled A–F) of the distribution of senile plaques in computer-assisted two-dimensional maps of layers II/III of the primary visual (striate) cortex in a patient with Alzheimer’s disease. The non-uniform distribution of the lesions is apparent with virtually all “windowing” parameters, from the highest (A) to the lowest (B) spatial resolutions. The row-like distribution of the lesions along the “tangential” domain (i.e., parallel to the pial surface and the white-gray matter interface) of the layers affected is strongly reminiscent of the eye dominance columns, and thus suggests that components of the “hypercolumns” that compose this region are affected, while immediately neighboring components remain unaffected even after the eventual demise of the patient years after the clinical onset of the disease. Modified from Kuljiš (1997, 2009b) and Kuljiš and Tikoo (1997). Similar phenomena are known in the entorhinal–perirhinal interface, and may occur in less well understood cortices (Kuljiš, 2009b). The molecular/biochemical basis is unknown, as well as correlates at other levels.
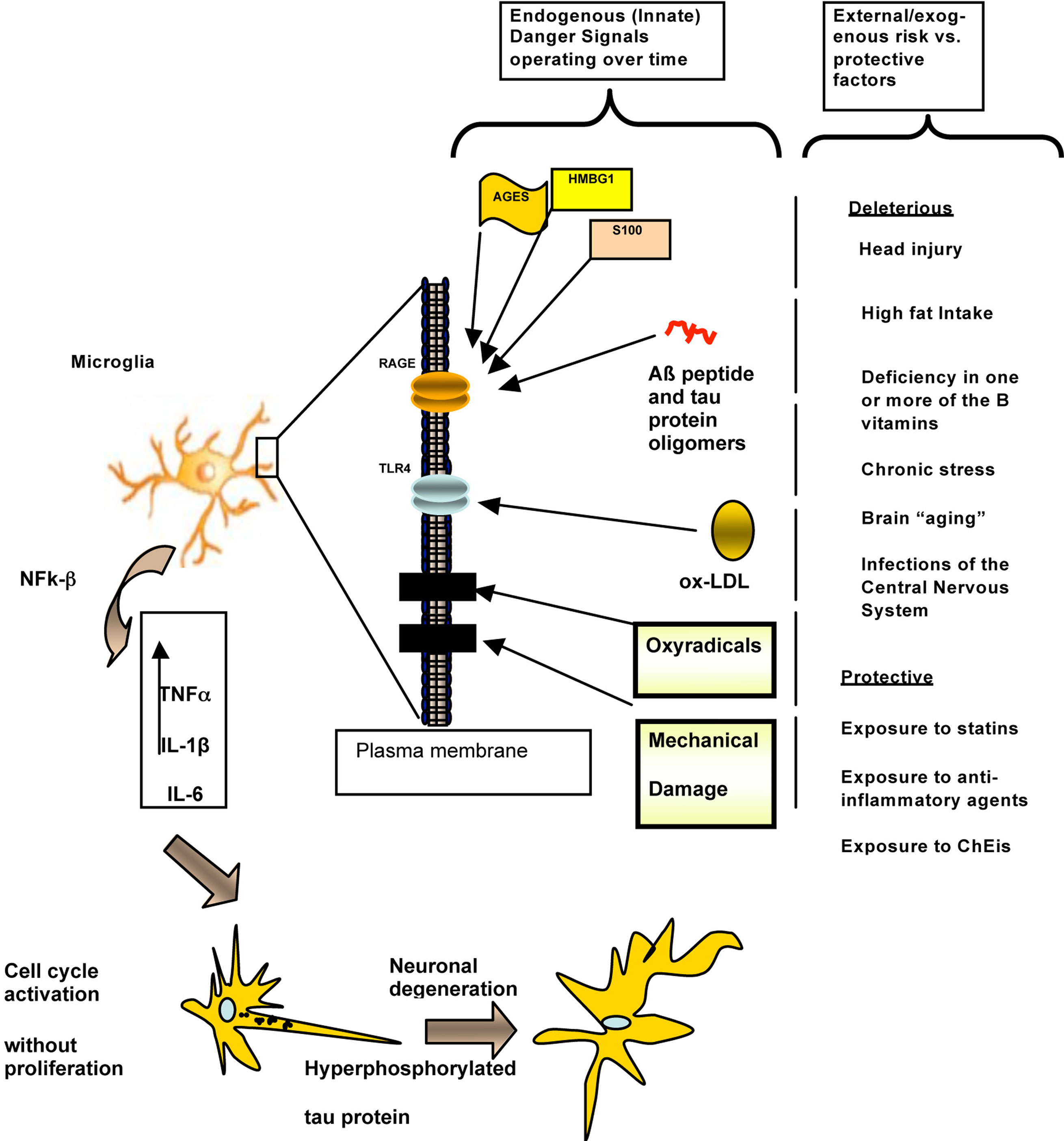
Figure 4. A mainly biochemical/molecular-level schematic representation of the hypothetical roles of endogenous “Danger/alarms Signals” that activate the Innate Immune System in the earliest stage of the pathogenesis of AD. A host of environmental/lifestyle-associated factors both “protective” (such as long-term exposure to cholinesterase inhibitors: ChEIs), as well as deleterious (a.k.a. “risk factors”) such as advancing age, combined with endogenous risk factors, trigger Danger Signals such as advanced glycation end products (AGES) resulting from diabetes mellitus, high mobility box group 1 (HMBG1), S-100 proteins, and Aß peptide oligomers (but not β-pleated fibrillar aggregates) and/or tau protein oligomers, activate microglia through the AGES receptor (RAGE; shown here as a transmembrane protein in a cartoon of a lipid bilayer). Separately, oxidized low-density lipoproteins (oxLDL) activate toll-like receptors (TLRs), and, in particular, TLR4. Additional danger signals, such as trauma and oxyradical damage, possibly acting on separate receptors (black boxes inserted in the membrane) as well as by inducing the production of additional Aß peptide oligomers, AGES and S-100 protein could also contribute to this process. Separately and in various combinations (which would apply differentially to different individuals), these danger signals would trigger the Innate Immune System response to these alarm mechanisms, resulting in the production of tumor necrosis factor alpha (TNFα), interleukin 1-beta (IL1-ß), and interleukin-six (IL-6). These signals would then mediate neuronal damage directly, reflected in alterations such as tau protein hyperphosphorylation and paired helical filaments formation, which eventually result in neuronal degeneration, Aß peptide aggregation in a beta-pleated configuration, and progressively more severe clinical manifestations of cognitive and behavioral decline unrelated to amyloid aggregation. Modified from Fernández et al. (2008). In this view, converging risk factors (countered by “protective” factors) trigger alterations in the Innate Immune System that converge on RAGE, TLR, and possibly other receptors in the microglial membrane, which in turn trigger well-known events that lead to neuronal degeneration and clinically demonstrable manifestations in AD. While this predominantly molecular-level hypothetical explanation of AD pathogenesis is useful as a framework for the eventual development of “Personalized Medicine” approaches to dementia prevention and treatment, it does not account for the region-, area-, layer, circuit-, and neuron ensemble-specific aspects of AD shown by microscopic analysis (Figures 2 and 3), among many other known levels and scales of involvement (Figure 1). This challenge awaits experimental elucidation to begin to approach the multi-dimensional interpretation we advocate here towards an integrative – and presumably more powerful and far-reaching – understanding of dementia.
The Irreducibility of Systems-Level Alterations to Molecular Events (and Vice Versa)
The focus at the molecular level of virtually all contemporary research on AD and related disorders has been very productive over the past three decades, and has led to many influential insights that fuel the search for improved treatments for these formerly untreatable conditions. While it is not the aim of this presentation to review such an extensive field, it is worth mentioning as examples the cholinergic hypothesis of AD (Francis et al., 1999), the amyloid hypothesis of AD (Hardy and Allsop, 1991) and the tau hypothesis of AD (Goedert et al., 1991; Maccioni et al., 2009) to highlight the power of this approach. However, purely molecular/biochemical explanations of dementia cannot possibly provide a complete and satisfactory biological- and cognitive-level account of the conditions of interest, because the anatomical, histopathological and cognitive dimensions (Figure 1) – among others – are irreducible to molecules and biochemistry. In fact, it is quite intriguing that while all of the above hypotheses of AD pathogenesis have their origin in anatomo-pathological observations (e.g., loss of neurons subsequently found to be cholinergic, amyloid-containing senile plaques, and tau-containing neurofibrillary tangles), virtually all contemporary research motivated by such hypotheses is conducted with increasing detachment from its anatomical underpinnings. There are aspects of this detachment that are highly desirable in fulfillment of a strong and rigorous “reductionist” approach, which has proven highly influential in contemporary Biology, including the Neurobiology of Disease. However, one so-far unmitigated casualty of this approach has been an increasing disconnection from the actual human conditions that are modeled by employing animals that only rarely replicate adequately actual human neurodegenerative conditions (Sarasa and Pesini, 2009), or in deliberately (and, in fact, desirably) simplified assays in vitro that bear little resemblance to what may happen in the multi-dimensional manifold of the human brain that includes a much longer time span and spatial frame than those models. Yet the merits of such reductionistic approach will reach its fullest potential only when the insights gained are put back into the context of a manifold, and thus integrated with the anatomical and physiological (e.g., cerebrocortical development and plasticity, specific neurotransmitter system networks, circuitry mediating functions that are selectively targeted versus those essentially spared) as well as the cognitive and behavioral levels (e.g., symptoms attributable to dysfunction at the molecular/biochemical or anatomical or physiological levels, or combinations thereof), among other dimensions. Needless to say, the conceptual framework for such undertaking does not exist, and it is essential to begin formulating it. Thus the need for the space–time manifold approach, which is perhaps most evident when considering AD, given that the relative wealth of information available on this (these) condition(s) today fails to provide an obvious path for its complete understanding, especially when considering any of the levels of inquiry in isolation. This is a challenge not only because most experts in the field have been trained mainly in one of the relevant dimensions/levels of study, but mainly because the conceptual framework to address this challenge is – at best – in its infancy (Kuljiš, 2009a, 2010).
Toward a Multi-Dimensional Formulation of the “Emergent” Properties that Mediate Normal and Disordered Cognition
Albeit vague notions of this integrative approach may be relatively common, yet hardly if ever voiced, an increasingly rigorous formulation of the cerebral space–time manifold is necessary to begin to construct models of the histogenesis and the degeneration of the brain to apprehend and correlate the structural, temporal, biochemical, behavioral and cognitive dimensions that continue to be studied in virtual isolation from one another. To the extent that the manifold remains insufficiently formulated, the vague correlations between “cause” and “effect” will continue to be elusive, especially in conditions such as sporadic AD since this clinico-pathological and biochemical construct appears to result not from a single “cause,” but from inadequately understood individual converging risk factors presumably ameliorated by a host of protective factors (Fernández et al., 2008; Figure 4). This defies conventional notions of causality, for which there is little in the way of an intellectual framework to allow a translation/conversion of concepts from one level of inquiry to another, and much less to integrate these coherently and meaningfully between two domains. The situation is even more precarious for integration/correlation among many of the pertinent domains (Kuljiš, 2009a).
Precisely what is the manifold? The present hypothesis is part of an attempt to take into consideration the temporal and spatial domains (from the microscopic to the macroscopic) together with the molecular/biochemical, cellular, systems, behavioral and cognitive levels at which degenerative phenomena and their clinical manifestations occur, in order to integrate them into an improved explanatory continuum that is better able to account for the highly selective nature of many degenerative conditions in multiple domains. Such hypothetical integrative approach could have powerful implications not only for normal cognition, but also for the understanding of many disorders, including degenerative disorders of the brain, and would likely propel substantial progress in their prevention, diagnosis, and treatment.
The increasingly atomized nature of reductionist research, while necessary and successful in elucidating considerable levels of molecular detail, is ill adapted to put these dramatic achievements into a Systems or Integrative level context. Other experts are relatively powerless to help molecular explorers from treating the brain disorders as a cognitive, behavioral, anatomical, and Systems-level “Black Box”. The manifold approach embraces the presumed interconnected, multi-dimensional nature of the structural, electrophysiological and biochemical/molecular domains, and time–space – among others – and helps to begin integrating these levels with a hopefully realistic expectation to develop a truly useful and innovative understanding of both normal and disordered brain function. This may permit us to eventually understand how the “higher” integrative properties (and their disorders) emerge/result and even possibly interact with the other domains (e.g., molecular). This is essential to approach a truly mechanistic understanding of normal and disordered cognition beyond the simplistic and definitely unsatisfactory notion that they are “emergent properties” of a rather mysterious “Black Box” (at one or another level) susceptible of breaking down, and that this can be completely, or at least sufficiently explained at the molecular/biochemical level, or any other level in essential isolation from the others.
While it would seem that the multi-dimensional aspect of this hypothesis might be agreeable to most readers, the use of the terms “manifold” or “matrix,” among others, could be objectionable and even misleading. This requires important clarifications: (a) these words are not used in their perhaps more common esoteric, supernatural, or literary/fictional sense (e.g., science fiction), (b) the term “matrix” in this presentation does not refer to previous formulations in Logic, Mathematics, Geometry, or Physics, and is unrelated to previous uses of this term and similar terms by Neuroscientists in the past, and (c) the latter class of unrelated terms includes the matrix formulations for language production (Goldrick et al., 2008), the highly influential two-dimensional formulations of Saaty (1977; as opposed to the present and critically multi-dimensional formulation), and the Holonomic brain theory (Pribram, 1987). Thus, the potentially provisional use of the words “matrix” and “manifold” in our formulation should be treated as devoid of prior intellectual or conceptual “baggage,” and, rather, understood as a temporary device to bind together different conceptual and methodological levels of understanding, that would otherwise remain disconnected. Unfortunately, a mathematical formulation is unlikely until the incomplete correlations between the gravitational and quantum mechanical descriptions of the universe are reconciled, including the so-called “decoupling” among length scales in physics. According to the latter principle, “physical phenomena of different length scales cannot affect each other and are said to decouple” (Wikipedia contributors, 2009) which constitutes only one among the many levels of the challenge towards understanding the phenomenon of Emergence.
Implications for Quantum Brain Hypotheses and the Connectome
The challenge of multiple scales/dimensions relevant to cognition – in many cases spanning several orders of magnitude (Figure 1) – their apparent decoupling, and the hypothesized emergent phenomena (despite the contradiction posed by decoupling) applies critically also to two relatively new pursuits pivotal to the development of innovative inquiry in Neuroscience: (a) Quantum Brain (a.k.a. “Quantum Mind” or “QM” hypotheses that postulate that quantum physics-level phenomena are fundamental not only for cognition, but also to ascertain the existence of Free Will (or lack thereof) and to explain it mechanistically (Stapp, 1993; Vitiello, 2009), and (b) the proposal to pursue the Connectome Project – in a way akin to the Human Genome Project – to obtain a complete map of the connections of the human brain (Sporns et al., 2005, 2007; Livet et al., 2007; Lichtman et al., 2008; Bohland et al., 2009; Lu et al., 2009; Sporns, 2009), which is mysteriously claimed to correlate with so-called “hubs” in the brain reportedly visualized by functional brain imaging technology (Izikevich and Edelman, 2008; Hagmann, 2005; Wedeen et al., 2005, 2008; Murayama et al., 2006; He et al., 2007; Hagmann et al., 2007, 2008; Sporns et al., 2007; Iturria-Medina et al., 2008;). The latter represents an unbridled extension beyond the reasonable “proof of principle” obtained some time ago in Caenorhabditis elegans (White et al., 1986), of the claim that the entire synaptic connectivity of a far more complex organism can be mapped in its entirety, and that such information can be interpreted in a meaningful and useful way.
Quantum Brain (a.k.a. QM) hypotheses are relevant to this discussion because – if valid – would represent the smallest/lowest level that needs to be considered among the physical scales necessary for an integrative understanding of brain function. However, QM hypotheses have been challenged both on the grounds of the impossibility of quantum coherence at body temperature (Tegmark, 2000) – since this phenomenon necessitates temperatures very close to absolute zero (i.e., incompatible with life on Earth) – as well as on incisive criticism of its insufficiently articulated formulation (Bourget, 2004). In fact, Bourget has exposed numerous weakness in the hypothesis of Stapp, which render it untestable scientifically as well as implausible due to a lack of a firm correlation with brain mechanisms. Current defenders have been unable to overcome these fundamental objections, and tend to hold on to discredited or outdated concepts about brain oscillations attributed to mass action (Vitiello, 2009) and the thoroughly refuted and thus archaic concept of a lack of cerebral localization of function (Lashley, 1950). As a result, “Quantum theories of mind are routinely derided as having the explanatory power of ‘pixie in the synapses’” (Bourget, 2004, attributed to Rosenblaum and Kuttner, 1999) and are felt to represent the unsubstantiated, untenable, and even implausible opinion of a minority. In addition, QM hypotheses lack even a minimal correlation with the considerably distinct regional architecture and function of the brain, and it is therefore not surprising that, in many cases, it continues to be held on the basis of mass action principles disconnected from the rest of the machinery of the brain (Vitiello, 2009) that many feel – without compelling proof – is clearly not operating by quantum mechanical principles. In particular, the precise mechanisms by which the postulated quantum mechanical phenomena interact with specific brain regions to generate cognition, let alone “Free Will” and consciousness, have not been precisely defined, and, as a consequence, are not amenable yet to experimental testing. This is relevant to our multi-dimensional formulation in that the correlations of quantum and “subquantum” with mesoscale phenomena – all the way from molecules to cognition, behavior and emergent phenomena in communities of living beings, including human civilization, may be mediated by interactions or at least correlations among levels. However, interactions among these phenomena have not been formulated in a way that has ever been scientifically testable. However, although no existing QM hypothesis seems viable, taking Vedral’s (among others’) intriguing proposal that the universe is a giant quantum computer (Vedral, 2010) seriously may eventually necessitate formulating cognitive phenomena in quantum mechanical terms again. This may prove the most powerful argument to redevelop QM hypotheses, and thus necessarily extend the range of scales relevant to cognition to the subquantum level (not represented in Figure 1). Recent empirical observations support this possibility, such as the discovery that quantum effects (e.g., entanglement) can occur at “room temperature” and at even higher temperatures in inert materials (Ghosh et al., 2003; Vedral, 2004), and that quantum effects can be observable macroscopically (Vedral, 2008). These genuine breakthroughs in experimental observation, plus strong indications that key phenomena in living organisms such as photosynthesis can only be explained by invoking (albeit poorly understood) quantum mechanical phenomena (Fleming and Scholes, 2004; Jang et al., 2004) suggest that it may be possible to conceive plausible QM hypotheses in the future. Therefore, this level of analysis cannot be excluded among the range of scales necessary to consider in order to develop an all-encompassing understanding of normal and disordered cognition. It is less than clear, however, how and whether this is a viable or even necessary goal, as the validity of Digital Physics remains in question since the original formulation by Zuse (1969) following up on the influential linking of information theory, statistical thermodynamics and quantum mechanics by Edwin Jaynes (1957). The main tenet of this field is that the universe is describable as information and is therefore computable, and, thus variably held to be essentially information, digital, itself a colossal computer, or the output of a simulated reality exercise. This is a very intriguing and all-encompassing view that has been challenged compellingly (Floridi, 2009) and is therefore far from established, even in terms of plausibility. Therefore, its potentially far-reaching implications for our conceptualization of the physical basis of cognition, consciousness, and emergent phenomena relevant to normal and disordered neural function cannot yet be fully assessed.
The evolving concept of the Connectome is vaguely postulated on multiple scales (Kötter, 2007), that “…corresponding to the levels of spatial resolution…” (Sporns, 2009), and conceived as implemented as a static description of connections despite the well-established fact that two-photon imaging in vivo reveals the rapid appearance and disappearance of key substrates for connectivity such as dendritic spines (Bonhoefer and Yuste, 2002). This difficulty is compounded by the rapidly increasing number of imaging studies in vivo, that purport to reveal functional “networks” and “nodes” at very low resolution (Hagmann et al., 2005, 2007, 2008), i.e., several orders of magnitude lower than indispensable to resolve dendritic spines, or any type of synapse. Unfortunately, the technology to accomplish the latter in vivo, especially in humans, does not exist. Furthermore, the conceptual framework to convert such low-resolution maps to higher-resolution ones capable of translating these vague “nodes” and “networks” into synaptic or at least individual neuronal function does not exist either. There is also no paradigm to interpret the resulting connectivity maps in a way that will help understand the mechanisms of normal and disordered cognition. Therefore, it is unclear precisely what is being accomplished by increasingly lower resolution “mapping” that ignores most of what has been learned about cerebral circuitry employing light- and electron microscopy in the past four decades. This situation obviously calls for a true conceptual framework and matching methodology to ensure that this essential integration and cross-correlation among substantially different levels of magnitude is meaningful. In this regard, the recent proposal to map “…brainwide neuroanatomical connectivity…at the mesoscopic scale…” (Bohland et al., 2009) while laudable in general principle, is probably very far from achieving the goal we propose here, given the low resolution inherent to mesoscopic projects, and, above all, due to the lack of a conceptual framework to move on beyond the mesoscopic level even if current methodology would allow so. This is relevant in terms of elucidating the connection between, at least, the molecular through the cognitive and behavioral domains. Otherwise, there is a considerable risk of creating a New Phrenology – as Noë (2009) among others have already warned us – since it is a well established fact that merely mapping “nodes” in networks does not explain precisely how they work, and, furthermore, that only a few of the “nodes” (which are increasingly multiplying in specialty journals) are likely to convey and/or contain both important and relevant information (Barabasi and Bonabeau, 2003). The latter cannot be overemphasized, since, in virtue of the latter extraordinary principle, increasingly detailed maps may become increasingly less informative and actually distract precious focused attention away from the fundamental goal of elucidating the Connectome in a truly significant and informative way (e.g., with a higher resolution than “mesoscopically,” and guided by an as yet not existing framework for meaningful interpretation). In summary, before embarking in massive low-resolution mapping towards the hoped-for Connectome, both a conceptual scheme to help make the results meaningful, and the deliberate design and selection of the methodology for this purpose is essential for this to become a truly useful enterprise. This challenge is not unique to the Connectome, since proteomic “maps” – among others – are now depicted in a way closely resembling the low-resolution, so-called “Connectome maps” (e.g., Hagmann et al., 2008). As a result, the problem of identifying the truly relevant “nodes” in the “network” and the likelihood of truly correlating the molecular (e.g., proteomic) and connectional (e.g., synaptic networks) domains may become more elusive rather than feasible. This challenge will be compounded further when studies of alterations in the Connectome in conditions such as AD are studied, a task that has not been approached yet at the cellular, subcellular, or any other level of resolution. Another formidable challenge to this task is exemplified by the field of Mesoscopic Physics (not to be confused with mesoscopic brain mapping), which is concerned with key problems such as the fact that the description of objects at the macroscopic level obey the laws of classical mechanics, whereas the mesoscopic descriptions are appear in conflict with the laws of quantum mechanics (Wikipedia contributors, 2010). This illustrates powerfully the potentially greater challenges to address this type of conundrum in Neuroscience towards understanding cognition.
Conclusions
In this embryonic formulation, the proposed multi-dimensional “manifold” or “matrix” approach we posit may allow us to move on beyond little more than paying lip service to multi-disciplinary undertakings in Neuroscience that fail to accomplish most of the purported linking of disparate methodologies, and rarely allow a true conceptual integration in the so far equally hypothetical molecular (and perhaps even “subquantal”; Boyd and Klein, 2007; Vimal, 2008) through cognitive continuum. To move forward in this regard, we are as interested as most authors in the development of a sound foundation in physics for the concept of quantum computing (Vedral, 2010), or an alternative but viable approach, and its application to Biology – and most especially Neuroscience – as well as in the resolution of the relatively recent (if incompletely formulated) challenge to the long-dominant concept that brains are information-processing systems (Noë, 2009).
To tackle the above unresolved conflicts, we propose that a manifold model of dementia (including Alzheimer’s disease) “causation” and pathophysiology theoretically is applicable also to virtually all neurodegenerative disorders, as well as to many disorders of complex non-neural tissue (e.g., the Immune System) and normal cognition. However, it could be argued at this point that in its current formulation the concept is most readily intelligible and can perhaps be postulated most rigorously for disorders that exhibit a predilection for the cerebral cortex and other regions mediating higher intellectual functions. The wealth of information on this region of the brain and its involvement in extensively investigated, commonly occurring conditions such as Alzheimer’s disease, offers a greater conceptual and empirical knowledge platform to develop scientifically testable hypotheses that will help support or refute the competing views, and specific proposals about the mechanisms underlying cognition and dementia summarized above. This could even have a decisive impact on challenging problems in physics such as the impact of observer involvement in strictly material phenomena. This is an example of how the understanding of dementia could lead the way towards the resolution of seemingly incompatible views of brain function and pathophysiology, such as the proposal that brains are not information-processing organs (e.g., Noë, 2009) versus the hypothesis that biological organisms – as well as anything else in the Universe – can be reduced to, or conceptualized as (i.e., be treated as isomorphic to), information (Suze, 1967; Vedral, 2010).
It has not escaped our attention that the multi-dimensional approach to both normal and disordered cognition is a distinctly polytelic task, as it would address a major, long-standing impasse that jeopardizes the solving of closely related problems such as the mechanisms of consciousness, the nature of emotions, and the mechanisms that underlie the development of the notion of the Self (Kuljiš, 2010) and the overarching problem of Emergence in Nature.
Conflicts of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The author is indebted to Drs. Deborah Serda and Marshall Gilula (both members of The Brain-Mind Project), Luis Colom (University of Texas, Brownsville), Larry J. Stensaas (University of Utah), and the Editorial Board of the Frontiers Organization for comments on earlier versions of the manuscript, and to Dr. Harvey J. Karten for stimulating discussions on the mesoscopic approach to brain mapping and the Connectome Project. The experimental work of the author on which part of this formulation is based was supported by The Brain-Mind Project, Inc., the United Through Knowledge Fund’s Research Cooperability Program (Government of Croatia, in collaboration with Professor Melita Šalković-Petrišić from the University of Zagreb School of Medicine), and received prior support from the United States Public Health Service (grant NS 29856) and the United States Department of Veterans Affairs (grant 5065.01 and Special Emphasis Program grant).
References
Anderson, H., and Kuljiš, R. O. (2008). Alzheimer’s disease. eMedicine.com (emedicine.medscape.com/article/11348) accessed 12/28/2008.
Anonymous. (2009). What is cognition?, www.About.com: Psychology, accessed 06/22/2009.
Bohland, J. W., Wu, C., Barbas, H. Bokil, H., Bota, M., Breiter, H. C., Cline, H. T., Doyle, J. C., Freed, P. J., Greenspan, R. J., Haber, S. N., Hawrylycz, M., Herrera, D. G., Hilgetag, C. C., Huang, Z. J., Jones, A., Jones, E. G., Karten, H. J., Kleinfeld, D., Kötter, R., Lester, H. A., Lin, J. M., Mensh, B. D., Mikula, S., Panksepp, J., Price, J. L., Safdieh, J., Saper, C. B., Schiff, N. D., Schmahmann, J. D., Stillman, B. W., Svoboda, K., Swanson, L. W., Toga, A. W., Van Essen, D. C., Watson, J. D. and Mitra, P. P. (2009). A proposal for a coordinated effort for the determination of brainwide neuroanatomical connectivity in model organisms at a mesoscopic scale. PLoS Comput. Biol. 5, e10000334. doi:10.1371/journal.pcbi.1000334.
Bonhoefer, T., and Yuste, R. (2002). Spine motility. Phenomenology, mechanisms, and function. Neuron 35, 1019–1027.
Bourget, D. (2004). Quantum leaps in philosophy of mind: a critique of Stapp’s theory. J. Conscious. Stud. 11, 17–42.
Boyd, R. N., and Klein, A. (2007). Toward a new subquantum integration approach to sentient reality. Paper presented at the Quantum Mind 2007, The University of Salzburg, Salzburg, Austria. Consciousness Research Abstracts: a service from the Journal of consciousness Studies; pp. 33–34 (abstract number 33). Toward a new subquantum integration approach to sentient reality by Klein and Boyd; manuscript in preparation:YWRyaWFuMTBAYmV6ZXFpbnQubmV0. see http://www.rialian.com/rnboyd/what-is-aether.htm for ‘bhutatma’.
Butterworth, R. F. (1992). “Animal models of the cerebellar ataxias,” in Neuromethods, Animal Models of Neurological Disease, Vol. 21, eds A. Boulton, G. Baker, and R. Butterworth (The Humana Press, New York), 275–294.
Capra, F. (1997). The Web of Life: A new Scientific Understanding of Living Systems. New York, NY: Anchor Books.
Churchland, P., and Sejnowski, T. (1988). Perspectives in cognitive neuroscience. Science 242, 741–745.
Dugan, L. L., and Kim-Han, J. S. (2006). “Hypoxic-ischemic brain injury and oxidative stress,” in Basic Neurochemistry, Molecular, Cellular and Medical Aspects, 7th Edn. eds G. Siegel, R. W. Albers, S. Brady, and D. Price D. (Burlington, MA: Elsevier Academic Press), 559–574.
Fernández, J., Rojo, L., Kuljiš, R. O., and Maccioni, R. B. (2008). The damage signals hypothesis of Alzheimer’s disease. J. Alzheimer’s Dis. 14, 329–333.
Francis, P. T., Palmer, A. M., Snape, M., and Wilcock, G. K. (1999). The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol. Neurosurg. Psychiat. 66, 137–147.
Ghosh, S., Rosenbaum, T. F., Aeppli, G., and Coppersmith, S. N. (2003). Entangled quantum state of magnetic dipoles. Nature 425, 48–51.
Goedert, M., Spillantini, M. G., and Crowther, R. A. (1991). Tau proteins and neurofibrillary degeneration. Brain Pathol. 1, 279–286.
Goldrick, M., Costa, A., and Schiller, N. O. (2008). Situating language production within the matrix of human cognition: the state of the art in language production research. J. Lang. Cogn. Proc. 23, 489–494.
Hagmann, P. (2005). From Diffusion MRI to Brain Connectomics. Ph.D. Thesis, Ecole Polytechnique Fédérale de Lausanne (EPFL),. Lausanne, Switzerland, 127 p.
Hagmann, P., Cammoun, L., Gigandet, X., Meuli, R., Honey, C. J., Wedeen, V. J., and Sporns, O. (2008). Mapping the structural core of human cerebral cortex. PLoS Biol. 6, e159 doi:10.1371/journal.pbio.0060159.
Hagmann, P., Kurant, M., Gigandet, X., Thiran, P., Wedeen, V. J., Meuli, R., and Thiran, J. P. (2007). Mapping human whole-brain structural networks with diffusion MRI. PLoS One 2, e597. doi:10.1371/journal.pone.0000597.
Hardy, J., and Allsop, D. (1991). Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 12, 383–388.
He, Y., Chen, Z. J., and Evans, A. C. (2007). Small-world anatomical networks in the human brain revealed by cortical thickness from MRI. Cereb. Cortex 17, 2407–2419.
Huxley, J. S., and Huxley, T. H. (1947). Evolution and Ethics: 1893–1943. London: The Pilot Press, p. 120.
Iturria-Medina, Y., Sótero, R. C., Canales-Rodríguez, E. J., Alemán-Gómez, Y., and Melie-García, L. (2008). Studying the human brain anatomical network via diffusion-weighted MRI and graph theory. Neuroimage 40, 1064–1076.
Izhikevich, E. M., and Edelman, G. M. (2008). Large-scale models of mammalian thalamocortical systems. Proc. Natl. Acad. Sci. USA. 105, 3593–3598.
Jang, S., Newton, M. D., and Silbey, R. J. (2004). Multichromophoric Förster resonance energy transfer. Phys. Rev. Lett. 92, 218301.
Kaneko, K. (1998). Life as complex system: viewpoint from intra-inter dynamics. Complexity, 6, 53–63.
Kötter, R. (2007). “Anatomical concepts of brain connectivity,” in Handbook of Brain Connectivity Series, Understanding Complex Systems, eds V. K. Jirsa and A. R. McIntosh (Springer, Berlin), 149–166.
Kuljiš, R. O. (1994). Lesions in the pulvinar in patients with Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 53, 202–211.
Kuljiš, R. O. (1997). “Modular corticocerebral pathology in Alzheimer’s disease,” in Dementia: A Multidisciplinary Approach, eds C. A. Mangone, R. F. Allegri, R. L. Arizaga, and J. A. Ollari (Buenos Aires, Argentina: Editorial Sagitario), 143–155.
Kuljiš, R. O. (2009a). Toward a multi-dimensional formulation of the pathogenesis and pathophysiology of the Alzheimer dementia-like syndrome applicable to a variety of degenerative disorders and normal cognition. Med. Hypotheses 73, 315–318.
Kuljiš, R. O. (2009b). “Selective cerebrocortical regional, laminar, modular and cellular vulnerability and sparing in Alzheimer’s disease: unexplored clues to pathogenesis, pathophysiology, molecular- and systems-level hypothesis generation and experimental testing,” in Current Hypotheses and Research Milestones in Alzheimer’s Disease, eds R. B. Maccioni and G. Perry (New York: Springer Science and Business Media), 191–204.
Kuljiš, R. O. (2010). Grand challenges in dementia 2010. Front. Dement. 1:4. doi: 103389/fneur.2010.00004
Kuljiš and Rakić, P. (1990). Hypercolumns in primate visual cortex can develop in the absence of cues from photoreceptors. Proc. Natl. Acad. Sci. U.S.A. 87, 5303–5306.
Kuljiš, R. O., and Schelper, R. L. (1996). Alterations in nitrogen monoxide-synthesizing cortical neurons in amyotrophic lateral sclerosis with dementia. J. Neuropathol. Exp. Neurol. 55, 25–35.
Kuljiš, R. O., and Tikoo, R. K. (1997). Discontinuous distribution of lesions in striate cortex hypercolumns in Alzheimer’s disease. Vision Res. 37, 3573–3591.
Lichtman, J. W., Livet, J., and Sanes, J. R. (2008). A technicolour approach to the connectome. Nat. Rev. Neurosci. 9, 417–422.
Livet, J., Weissman, T. A., Kang, H., Draft, R. W., Lu, J., Bennis, R. A., Sanes, J. R., and Lichtman, J. W. (2007). Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 450, 56–62.
Lu, J., Tapia, J. C., White, O. L., and Lichtman, J. W. (2009). The interscutularis muscle connectome. PLoS Biol. 7, e1000032. doi:10.1371/journal.pbio.1000032.
Maccioni, R. B., Farías, G. A., Rojo, L. E., Sekler, M. A., and Kuljiš, R. O. (2009). “What have we learned from the tau hypothesis?,” in Hypotheses and Research Milestones in Alzheimer’s Disease. Current Hypotheses and Research Milestones in Alzheimer’s Disease, eds. R. B. Maccioni and G. Perry (New York: Springer Science and Business Media), 49–62.
Murayama, Y., Weber, B., Saleem, K. S., Augath, M., and Logothetis, N. K. (2006). Tracing neural circuits in vivo with Mn-enhanced MRI. Magn. Reson Imaging 24, 349–358.
Noë, A. (2009). Out of Our Heads. Why You Are not Your Brain, and Other Lessons From the Biology of Consciousness. New York: Hill and Wang.
Pribram, K. H. (1987). “The implicate brain,” in Quantum Implications: Essays in Honour of David Bohm, eds B. J. Hiley and F. D. Peat (London: Routledge and Kegan Paul), 365–371.
Purpura, D. P. (1975). “Introduction and perspectives,” in Golgi Centennial Symposium: Perspectives in Neurobiology, ed. M. Santini (New York: Raven Press), 13–17.
Riederer, P., and Wuketich, St. (2005). Time course of nigrostriatal degeneration in Parkinson’s disease. J. Neural Transm. 38, 277–301.
Rosenblaum, B., and Kuttner, F. (1999). Consciousness and quantum mechanics: the connections and analogies. J. Mind Behav. 21, 229–256.
Saaty, T. L. (1977). A scaling method for priorities in hierarchical structures. J. Math. Psychol. 15, 234–281.
Sarasa, M., and Pesini, P. (2009). Natural non-transgenic animal models for research in Alzheimer’s disease. Curr. Alzheimer Res. 6, 171–178.
Savva, G. M., Wharton, S. B., Ince, P. G., Forster, G., Matthews, F. E., and Brayne, C. (2009). Age, neuropathology, and dementia. New Engl. J. Med. 360, 2303–2309.
Schöls, L., Bauer, P., Schmidt, T., Schulte, T., and Riess, O. (2004). Autosomal dominant cerebellar ataxias: clinical features, genetics, and pathogenesis. Lancet Neurol. 3, 291–304.
Sporns, O., Honey, C. J., and Kötter, R. (2007). Identification and classification of hubs in brain networks. PLoS One 2, e1049. doi:10.1371/journal.pone.0001049.
Sporns, O., Tononi, G., and Kötter, R. (2005). The human connectome: a structural description of the human brain. PLoS Comput. Biol. 1, e42. doi:10.1371/journal.pcbi.0010042.
Tegmark, M. (2000). Importance of quantum decoherence in brain processes. Phys. Rev. E. 61, 4194–4206.
Vedral, V. (2010). Decoding Reality: The Universe as Quantum Information. Oxford: Oxford University Press.
Vimal, R. L. P. (2008). Proto-experiences and subjective experiences: integration of classical quantum, and subquantum concepts. http://www.geocities.com/rlpvimal/PE-SE-Vimal-Short-AR-PE.pdf, accessed 06/29/2009.
Vitiello, G. (2009). Coherent states, fractals and brain waves. arXiv:0906.0564 New Math. Nat. Comput. 5, 245–264.
Wedeen, V. J., Wang, R. P., Schmahmann, J. D., Benner, T., Tseng, W. Y., Dai, G., Pandya, D. N., Hagmann, P., D’Arceuil, H., and de Crespigny, A. J. (2008). Diffusion spectrum magnetic resonance imaging (DSI) tractography of crossing fibers. Neuroimage 41, 1267–1277.
Wikipedia contributors (2009). Length scale. Wikipedia The Free Encyclopedia, 12/17/2009. Accessed 05/12/2010, available at http://en.wikipedia.org/w/index.php?title=Length_scale&oldid=332178064.
Wikipedia contributors (2010). Mesoscopic physics. Wikipedia, The Free Encyclopedia 18 April 2010, accessed 12 May 2010, available at http://en.wikipedia.org/w/index.php?title=Mesoscopic_physics&oldid=356793416
Keywords: Alzheimer’s disease, connectome, quantum brain, mesoscale, cognition
Citation: Kuljiš RO (2010) Integrative understanding of emergent brain properties, quantum brain hypotheses, and connectome alterations in dementia are key challenges to conquer Alzheimer’s disease. Front. Neur. 1:15. doi: 10.3389/fneur.2010.00015
Received: 11 March 2010;
Paper pending published: 19 April 2010;
Accepted: 17 June 2010;
Published online: 26 August 2010.
Edited by:
Walter E. Kaufmann, Johns Hopkins School of Medicine, USAReviewed by:
Dean M. Hartley, Rush Medical Center, USAChuang-Kuo Wu, Texas Tech University Health Sciences Center, USA
Copyright: © 2010 Kuljiš. This is an open-access article subject to an exclusive license agreement between the authors and the Frontiers Research Foundation, which permits unrestricted use, distribution, and reproduction in any medium, provided the original authors and source are credited.
*Correspondence: Rodrigo O. Kuljiš, The Brain-Mind Project, 17 Compass Circle, Galveston, TX 77554-2919, USA. e-mail:cm9kcmlnby5rdWxqaXNAZ21haWwuY29t